


Abstract
Small exons are pervasive in transcriptomes across organisms, and their quantification in RNA isoforms is crucial for understanding gene functions. Although long-read RNA-seq based on Oxford Nanopore Technologies (ONT) offers the advantage of covering transcripts in full length, its lower base accuracy poses challenges for identifying individual exons, particularly microexons (≤ 30 nucleotides). Here, we systematically assess small exons quantification in synthetic and human ONT RNA-seq datasets. We demonstrate that reads containing small exons are often not properly aligned, affecting the quantification of relevant transcripts. Thus, we develop a local-realignment method for misaligned exons (MisER), which remaps reads with misaligned exons to the transcript references. Using synthetic and simulated datasets, we demonstrate the high sensitivity and specificity of MisER for the quantification of transcripts containing small exons. Moreover, MisER enabled us to identify small exons with a higher percent spliced-in index (PSI) in neural, particularly neural-regulated microexons, when comparing 14 neural to 16 non-neural tissues in humans. Our work introduces an improved quantification method for long-read RNA-seq and especially facilitates studies using ONT long-reads to elucidate the regulation of genes involving small exons.
Graphical Abstract
Graphical Abstract.

Introduction
Small exons refer to short exons present in the RNA transcripts of eukaryotic organisms, with microexons being a subset often characterized by their size not exceeding 30 nucleotides (nt) (1). Alternative splicing of these small exons often causes regulatory or structural function changes in genes involved in various cellular functions and neurological diseases. Currently, short-reads RNA-seq has been applied to examine the expression of small exons in multiple species, including humans, mice, flies, nematodes, and plants (2–5). A major bottleneck in studying small exons is the complexity of analysis. Current approaches to map small exons in short-reads include Microexonator, GMAP, VAST-TOOLS and ATMap (2,6–8), but methods for identifying small exons in long-reads has still been lacking.
Long-read sequencing technology has greatly improved the analysis of RNA transcripts as it produces reads over 10kb which usually covers the whole RNA molecules (9). This advantage in read length enables more precise identifications of splicing events. Numerous novel transcripts have been detected using cDNA and direct RNA sequencing with Oxford Nanopore technology (ONT) (10). However, due to fluctuations in the speed of target molecules passing through the nanopore, the raw reads have an error rate of around 1–5% (11–14). The current analysis workflow in long-read RNA-seq typically requires mapping reads to the reference genome. Several alignment tools were specifically developed for long-reads, with optimisation for low-quality reads. Among them, minimap2 is the most adopted method in the ONT community because of its high performance for spliced reads alignment (15,16). However, artefacts for exons mapping have been observed in alignments generated by minimap2 (17,18), resulting in misquantification of transcript isoforms. Researchers have started addressing this problem using long-read sequencing platforms with higher accuracy, such as the PacBio platform. For instance, Ray et al. comprehensively profiled microexon-containing transcript isoforms during retinal development using lrCaptureSeq, a method that enriches target sequences using long-read capture sequencing (19). Nevertheless, it is still not clear which exons would be misaligned and how this misalignment impact isoform quantification, especially in the ONT sequencing data. Therefore, there is a clear need for a systematic assessment of how small exons are mapped in ONT datasets.
To this end, we explored the properties of exons that are misaligned in ONT reads and developed a method to address the alignment issue. Our method MisER (Misaligned Exon Realignment), an annotation-based approach to identify misaligned reads and then locally correct the alignments in the regions containing misaligned exons. At first, MisER identifies all intron regions with potential alignment issues that overlap annotated exons. These regions will be marked as potential misaligned regions (PMRs). Then, MisER attempts to realign the read sequences within these PMRs to the reference sequences of annotated exons, calculates a new alignment score. The new alignment will replace the existing alignment if the alignment score improves after realignment. We estimated the accuracy and sensitivity of MisER using long-read sequencing data for sequencing spike-ins (sequins), a set of synthetic RNA standards, that represents full-length spliced mRNA isoforms (20). Additionally, we applied MisER to estimate the misalignment ratio of small exons in ONT cDNA and direct RNA-seq, using a dataset for a human B lymphocyte cell line (21). Our findings revealed that exons shorter than 50 nt faced the risk of misalignment while exons less than 20 nt were almost entirely lost during the annotation-free alignment. To demonstrate real-wold utility, we further applied MisER to a comprehensive ONT cDNA dataset consisting of 84 sequencing libraries for 14 neural and 16 non-neural tissues (22). MisER significantly improved the read alignments of small exons and facilitated the detection of more alternative spliced-in exons, particularly neural-regulated microexons. These results for the first time systematically characterized the features of misdetection of small exons and provide a robust realign approach to identify and quantify the RNA isoforms for genes with small exons in long-read RNA-seq datasets.
Materials and methods
Analysis of constitutive exon missing in the ONT cDNA sequencing data of sequins
Sequins ONT cDNA sequencing data was downloaded from GEO under accession number GSE151984 (23). Briefly, it contains four sequins cDNA libraries (mix A and B with two replicates of each), which were constructed with SQK-PCS109 cDNA and SQK-PBK004 barcoding kit. The libraries were sequenced by R9.4.1 MinION flow cell and base-called by Guppy (version 4.0.11). Reads were mapped to sequins chromosome (version 2.4) by minimap2 (version 2.17, with parameters: -ax splice -k10 - -secondary=no - -splice-flank=yes) and processed by MisER (default parameters). We also used -k15 and -k20 to examine the influence of kmer size on misaligned exons. We only used the spliced reads in our analysis.
Constitutive exons were defined based on sequins official gene annotations (version 2.4). A constitutive exon is an exon that presents in all transcript isoforms of its belonging gene. In total, 163 constitutive exons were determined in 43 genes (Genes overlapping with others and constitutive exons with read count less than 10 were removed). We defined the constitutive exon and its upstream and downstream exon as a triplet. Reads aligned to all three exons in the triplet were assigned as correct-mapped reads, while reads aligned to upstream and downstream exons but not the constitutive exons were assigned as missed reads. The sum of correct-mapped reads and missed reads were defined as total reads. Exons with total read count of < 10 were removed. The miss ratio of the constitutive exon was defined as the percentage of missed reads.
Analysis of constitutive exon missing in human ONT cDNA and direct RNA sequencing data
ONT cDNA and direct RNA sequencing data of the human B lymphocyte cell line (GM12878) was downloaded from its published data resource (21). There are 30 runs of direct RNA sequencing data constructed with SQK-RNA001 kit and 12 runs of cDNA sequencing data constructed with SQK-PCS108 kit, which were base-called by Guppy (version 4.2.2). Reads are mapped to the human GRCh38 genome by minimap2 (version 2.17) with parameters: -ax splice -ub -k15 - -secondary=no (for cDNA) and -ax splice -uf -k15 - -secondary=no (for direct RNA). Alignment results were further processed by MisER (default parameters). Only the spliced reads are used in further analysis. Constitutive exons were defined based on Ensembl human gene annotations (GRCh38 version 97) and 11 025 constitutive exons are detected in total. The miss ratio of constitutive exons is calculated as we previously described in sequins analysis.
Misaligned exons realignment (MisER)
MisER first scanned and identified potential misaligned regions (PMRs) on all reads which were overlapped but not aligned to annotated exons, and then realigned the potential misaligned regions to the exons and judged whether the exons were missed or not.
We scanned potentially missed exons when their upstream and downstream exons are mapped, but the missed ones are not. Thus, we used part of the mapped upstream and downstream exons as anchors to define the potential misaligned regions (PMR). We defined the splice sites of the upstream and downstream exons on the reference as 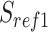 ,
, 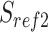 , and on the read as
, and on the read as 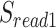 ,
, 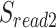 . The boundaries of splice sites were extended
. The boundaries of splice sites were extended 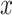 (20 nt as default) to determine the PMR. Specifically, the start (
(20 nt as default) to determine the PMR. Specifically, the start (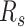 ) and end (
) and end (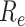 ) of PMR are defined as follows.
) of PMR are defined as follows.
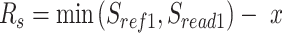 |
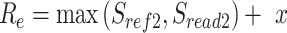 |
Then we calculated the length of reads sequences aligned to PMR (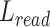 ) and the length of reference upstream and downstream exons in PMR (
) and the length of reference upstream and downstream exons in PMR (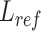 ). The difference between (
). The difference between (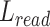 ) and reference (
) and reference (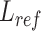 ), and defined as delta length (
), and defined as delta length (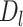 ):
):
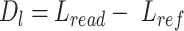 |
Intuitively, 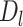 is the length of the sequences in the extra protruding region on misaligned reads.
is the length of the sequences in the extra protruding region on misaligned reads. 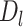 is compared to the length of the misaligned exon (
is compared to the length of the misaligned exon (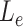 ). General idea is that
). General idea is that 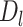 should be close to
should be close to 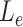 if it comes from the misaligned exon. The delta ratio (
if it comes from the misaligned exon. The delta ratio (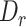 ) between
) between 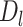 and
and 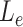 is calculated as follows:
is calculated as follows:
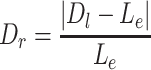 |
In the default setup, MisER selected the potential misaligned regions with 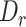 no more than 0.5 and
no more than 0.5 and 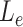 no more than 80 nt.
no more than 80 nt. 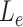 in the above equation would be replaced by the sum of misaligned exons sizes if there were multiple misaligned exons, and each misaligned exon should be less or equal to 80 nt in length. Users can tune these two parameters to adjust the potential misaligned regions MisER detected.
in the above equation would be replaced by the sum of misaligned exons sizes if there were multiple misaligned exons, and each misaligned exon should be less or equal to 80 nt in length. Users can tune these two parameters to adjust the potential misaligned regions MisER detected.
Next, MisER realigned the sequences in potential misaligned regions of reads to the reference sequences of annotated exons and used the alignment result to identify the real misaligned exons. To accelerate the realignment, MisER used SIMD C library Parasail (24) to align the partial sequences in the region of reads to the joint sequences of annotated exons. We proposed the alignment score 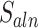 to evaluate the matching status of the region.
to evaluate the matching status of the region. 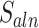 is calculated as follows, where
is calculated as follows, where 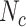 ,
, 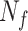 ,
, 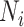 and
and 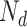 represent the number of correct matches, mismatches, insertions, and deletions in the region.
represent the number of correct matches, mismatches, insertions, and deletions in the region.
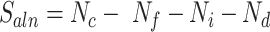 |
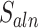 were calculated before and after realignment. If the alignment score improved, MisER assigned the regions as real misaligned regions, and exons inside as misaligned exons. The improved alignments were used and written into a new BAM file for further analysis.
were calculated before and after realignment. If the alignment score improved, MisER assigned the regions as real misaligned regions, and exons inside as misaligned exons. The improved alignments were used and written into a new BAM file for further analysis.
Estimation of the sensitivity and specificity of MisER
We used misaligned constitutive exons in sequins cDNA sequencing data to estimate the sensitivity of MisER. We tested the sensitivity of MisER using different 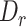 thresholds. We only used the region misaligned single exon in this analysis. There were 161 misaligned constitutive exons ranging from 25 nt to 532 nt, and 17 989 reads regions misaligned these constitutive exons in all sequins cDNA data. We used these misaligned regions to estimate the sensitivity of MisER.
thresholds. We only used the region misaligned single exon in this analysis. There were 161 misaligned constitutive exons ranging from 25 nt to 532 nt, and 17 989 reads regions misaligned these constitutive exons in all sequins cDNA data. We used these misaligned regions to estimate the sensitivity of MisER.
For specificity, we constructed a set of simulated triplet-exons annotations, for which we created a simulated middle exon between any two continuous constitutive exons in sequins. To do so, we annotated the intron sequences in the middle position of the two constitutive exons as a simulated exon. Simultaneously, the two constitutive exons were also truncated in the total length equal to the size of the middle exon, to make 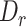 close to 0. We truncated the constitutive exons because the most value of
close to 0. We truncated the constitutive exons because the most value of 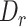 of misaligned microexons is 0. We required the size of the constitutive exon larger than 70 nt, while the intron between these two exons larger than 200 nt. In total, we detected 123 continuous constitutive exon pairs in sequins and added simulated middle exons with a length range from 5 to 50 nt. There were 387 335 reads regions were identified when aligning to the simulated exons triplets in sequins mixA replicate 1 cDNA data. We used these regions to estimate the specificity of MisER.
of misaligned microexons is 0. We required the size of the constitutive exon larger than 70 nt, while the intron between these two exons larger than 200 nt. In total, we detected 123 continuous constitutive exon pairs in sequins and added simulated middle exons with a length range from 5 to 50 nt. There were 387 335 reads regions were identified when aligning to the simulated exons triplets in sequins mixA replicate 1 cDNA data. We used these regions to estimate the specificity of MisER.
Analysis of simulated reads with various sequencing accuracies
We constructed a simulated dataset based on exon triplets (consecutively occurred three exons) derived from human transcript annotations. We required the length of the middle exons range from 1 nt to 80 nt and the length of the exons on both ends should larger than 50 nt. For each length of the middle exon, we selected up to 50 triplets to ensure the uniformity of exon length distribution in our simulated dataset. The middle exon of each triplet was designated as the alternative exon, while the exon triplet was divided into two isoforms: iso1 containing the middle exons and iso2 lacking them. This simulated dataset contained a total of 4622 alternative exons, among which 1222 were microexons with a length of ≤ 30 nt. Notably, 240 microexons had a length no longer than 10 nt. Each isoform was simulated to generate 500 reads at sequencing accuracies ranging from 80% to 100%. Sequencing errors were simulated as random distributed substitutions, deletions, and insertions with equal probabilities (33.3% for each type). Following this, we aligned the simulated reads to the human genome using minimap2 (parameters: -ax splice -k15 - -secondary=no - -splice-flank=yes -G 1000k) and applied MisER (default parameters) to detect and realign any misaligned small exons.
Furthermore, to assess MisER’s performance on more complex splicing isoforms that include tandem alternative exons, we created another simulated dataset based on exon quadruplets from human transcript annotations. For each permutation of the length of the two middle exons, we selected up to 50 quadruplets to ensure a uniform distribution of exon lengths in our simulated dataset. This dataset comprised 12 669 quadruplets, including 870 quadruplets with alternative microexons (≤ 30 nt), with 70 quadruplets containing microexons no longer than 10 nt. In this dataset, the middle two exons of each quadruplet were designated as alternative exons, and the exon quadruplet was divided into four isoforms: iso1 containing both middle exons, iso2 and iso3 containing one middle exon each, and iso4 lacking any middle exons. Each isoform generated 500 simulated reads at sequencing accuracies ranging from 80% to 100%. We aligned the simulated reads to the human genome using minimap2 (parameters: -ax splice -k15 - -secondary=no - -splice-flank=yes -G 1000k) and applied MisER (default parameters) to detect and realign any misaligned small exons.
Due to the relative scarcity of microexons with lengths <10 nt in human transcript annotations, we additionally built simulated exon triplets and quadruplets, setting the middle exons to specific lengths. For exon triplets, the length of the middle alternative exon was set to range from 1 to 80 nt, with 50 triplets designed for each length. For exon quadruplets, the middle alternative exon pairs were given equal lengths, ranging from 1 to 80 nt, and each length pair generated 50 quadruplets. Overall, we generated 4000 simulated exon triplets and quadruplets individualy, each containing middle exons with lengths ranging from 1 to 80 nt. The combinations of the middle alternative exons resulted in two isoforms for each exon triplet and four isoforms for each exon quadruplet. Each isoform generated 500 simulated reads at sequencing accuracies ranging from 80% to 100%. Subsequently, we aligned the simulated reads to decoy chromosomes (random ATCG sequences, containing canonical splice site GT-AG patterns) using minimap2 (parameters: -ax splice -k15 - -secondary=no - -splice-flank=yes) and applied MisER (parameters: - -allTranscripts) to detect and realign any misaligned small exons.
After reads alignment, we quantified the read counts for each isoform (only the reads mapped to all exons of their belonging annotated isoform were counted) in all simulated datasets, and calculated recall and precision rates before and after MisER realignment.
Quantification of sequins exons and transcript isoforms
We quantified the read count of exons with lengths no more than 80 nt in sequins gene annotations (version 2.4). To do so, we only considered the reads mapping to the annotated exons, and where the difference on splice sites in the alignments was no longer than 10 nt on either side. We quantified each annotated sequins transcript isoform based on the reads mapping on the genome. Only the reads mapped to all exons of the annotated transcript were counted. The quantifications of exons and transcript isoforms were calculated before and after MisER realignment. The transcript ratio was calculated as the ratio of each transcript's quantification to the sum of all transcripts within a gene.
Calculation of exon misalignment ratio in ONT cDNA and direct RNA sequencing data
We quantified the read count of exons with length no more than 80 nt in human Ensembl gene annotation (GRCh38 version 97). To do so, we only considered the reads mapping to the annotated exons, and where the difference on splice sites in the alignments was no longer than 10 nt on either side. We compared the read counts of exons before and after MisER processing. The increased reads were defined as misaligned reads. The misalignment ratio is defined as the number of misaligned reads divided by the total read counts after MisER processing. Exons with a read count of < 10 or a few exon cases whose read counts decreased after MisER realignment were excluded from the calculation.
Analysis of alternative spliced-in exons in ONT cDNA sequencing data of human tissues
ONT cDNA sequencing datasets of 30 human tissues, including 84 ONT cDNA sequencing libraries (using R9.4.1 and R10.3 flow cells, SQK-LSK109 sequencing kit), were downloaded from GEO (accession number: GSE192955) (22). Raw data (fast5 files) were base-called by Guppy (version 6.4.6, with hac model). The Reads were further mapped to the human GRCh38 genome by minimap2 (version 2.17) with parameters: -ax splice -ub -k15 - -secondary=no, and then processed with MisER. We used Bambu (25) (version 3.0.8) to quantify the expression of transcripts based on the human Ensembl gene annotation (GRCh38 version 97). Only the full-length reads of annotated transcript isoforms were counted and used for further analysis. The expression level of transcript isoforms was normalized as transcripts per million (TPM). We used SUPPA (26) (version 2.3) to calculate the percent spliced-in indexs (PSIs) of cassette exon (exon skipping) events. The alternative spliced-in exons in cassette exons events were defined as ASI exons. The difference in percent spliced-in index (PSI) between neural and non-neural samples was defined as delta PSI. ASI exons with adjusted P-value < 0.05 (Wilcoxon rank sum test, Bonferroni correction) and delta PSI > 25% were considered as neural up-regulated exons, while ASI exons with adjusted P-value < 0.05 and delta PSI < −25% were considered as neural down-regulated exons.
To estimate the conservation of sequences around splice sites of exons, we used the phastCons scores generated from multiple alignments of 100 vertebrate species (UCSC, https://hgdownload.soe.ucsc.edu/goldenPath/hg38/phastCons100way/hg38.phastCons100way.bw). Average phastCons scores were calculated for bases located in exonic and flanking intronic regions of constitutive exons or alternative spliced-in exons.
Analysis of alternative spliced-in exons in GTEx datasets of human tissues
We obtained quantifications of transcript isoforms generated by short-read sequencing data across 54 human tissue regions from the GTEx datasets (dbGaP Accession phs000424.v8.p2, https://storage.googleapis.com/gtex_analysis_v8/rna_seq_data/GTEx_Analysis_2017-06-05_v8_RSEMv1.3.0_transcript_expected_count.gct.gz). PSIs of cassette exon (exon skipping) events were calculated utilizing SUPPA. The difference in percent spliced-in index (PSI) between neural and non-neural samples was defined as delta PSI.
Analysis of alternative spliced-in exons in ONT cDNA sequencing data of neuroblastoma differentiation
ONT cDNA sequencing data of samples representing the differentiation of neuroblastoma (human SH-SY5Y cell line, 5 samples) into neuron-like cells (5 samples) were download from ENA database (PRJEB44502) (27). The libraries were constructed with sequencing kit SQK-PCS109 and base-called by Guppy (version 3.2.2). The reads were mapped to the human GRCh38 genome by minimap2 (version 2.17, parameters: -ax splice -ub -k15 - -secondary=no), and then processed with MisER. We utilized Bambu (version 3.0.8) to quantify the expression of transcripts based on the human Ensembl gene annotation (GRCh38 version 97). Only the full-length reads of annotated transcript isoforms were counted and used for further analysis. The expression levels of transcript isoforms were normalized as transcripts per million (TPM).
Results
Microexons are often mis-mapped in long-read sequencing data
To examine if small exons can be properly mapped in long-read data, we explored the ONT sequencing datasets for synthetic spike-in (sequins), a set of RNA molecules representing spliced gene isoforms (20). There are 163 constitutive exons in sequins. Sequencing reads mapping to a gene are expected to cover all constitutive exons of that gene. We first calculated the proportion of reads missing any constitutive exons in sequins and found small exons are often not aligned (Figure 1A). 9.1% of small exons (≤ 50 nt, 25 exons in sequins) were not correctly aligned, affecting the quantification of 10% (16/160) of sequins transcripts. The percentage of unmapped constitutive exons in reads increased when exon sizes reduced. In fact, 28.6% reads covering the smallest constitutive exon in sequin, a 25 nt exon in R1_83 gene, omitted the exon in their alignments.
Figure 1.

Missed small exons in the alignment of nanopore sequencing data. (A) Miss ratio of constitutive exons in sequins cDNA sequencing data. Each point represents one constitutive exon in sequins and the point size reflects the sum of correctly mapped reads and missed reads. The points are fitted by a generalized linear model (red line) with a confidence interval colored in grey. Exons with read count of less than 10 are discarded. The points are colored by samples. (B) One example of missed microexons (chrIS:3011619–3011645, gene R2_47 in sequins) in read alignments. Exons in the transcript annotation reference are colored in blue. The misaligned parts on upstream exon are circled and zoomed in. (C) Correlation between delta length and the length of misaligned constitutive exon. Delta length is defined as panel B. Intuitively, it is the sequence length in the extra protruding parts of upstream or downstream exons. (D) Miss ratio of constitutive exons in human cDNA sequencing data. Each box represents the distribution of misalignment ratio of exons on a certain length.
To further investigate whether reads contain missed sequences of small exons, we visually inspected mapping results and found most of the missed exons were caused by alignment errors as illustrated in Figure 1B. The mapped regions beyond annotated splice sites have high indel rates, indicative of alignment issues. We further defined the length of misaligned portion as delta length and compared it with the length of the missed exon. The delta length showed a strong correlation with the length of missed constitutive exons (Figure 1C), suggesting that the misaligned sequences come from missed exons in most cases.
As sequins do not have constitutive microexons smaller than 20 nt, we further calculated the miss ratio of constitutive exons in long-read RNA-seq data for human lymphoblastoid cell line GM12878 (21). We defined 11 025 constitutive exons based on Ensembl gene annotations (version GRCh38.97). We assessed the mapping issues in cDNA and direct RNA reads. Our analysis of constitutive exons revealed that there are numerous small exons not mapped during the alignment process (Figure 1D, Supplementary Figure S1). Exons no longer than 50 nt were likely to be missed, with the cumulative average miss ratio of 25.5% in cDNA and 15.3% in direct RNA (Supplementary Tables S1, S2). Although there are only 6.3% exons less than or equal to 50 nt in the human genome, 27.0% transcripts containing these small exons might be affected in quantification (Supplementary Figure S2). Among them, two smallest constitutive exons (18 nt) showed a high miss ratio of 91.5% and 78.7% for cDNA, 95.9% and 85.5% for direct RNA, respectively. These results demonstrate that small exons are often mis-aligned in ONT-based long-read RNA-seq, both for cDNA and direct RNA reads.
Development and benchmark of MisER
To systematically detect all misalignments of microexons and adjust them, we developed a new method for misaligned exons realignment (MisER). Based on our observation of high correlation of delta length and exon size (Figure 1C), we defined delta ratio (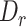 ), which represents the ratio between delta length and the length of the misaligned exon (see Materials and Methods). This delta ratio was utilized to identify possible misaligned read alignments. In brief, MisER first finds all regions in read alignments which overlap but not mapping to annotated exons. Then, based on delta ratio, MisER identifies potential misaligned regions (PMRs) and extracts the corresponding sequences, including partial sequences of upstream and downstream mapped exons (Figure 2A). These read sequences are realigned to the reference sequences of annotated exons and MisER compares the alignment scores before and after realignment. If alignment score improves, the region will be labelled as misaligned region and the alignment will be adjusted using the realignment result. Additionally, to enhance efficiency, MisER is designed as a multi-threaded tool, allowing independent processing of multi alignments in parallel (Supplementary Figure S3).
), which represents the ratio between delta length and the length of the misaligned exon (see Materials and Methods). This delta ratio was utilized to identify possible misaligned read alignments. In brief, MisER first finds all regions in read alignments which overlap but not mapping to annotated exons. Then, based on delta ratio, MisER identifies potential misaligned regions (PMRs) and extracts the corresponding sequences, including partial sequences of upstream and downstream mapped exons (Figure 2A). These read sequences are realigned to the reference sequences of annotated exons and MisER compares the alignment scores before and after realignment. If alignment score improves, the region will be labelled as misaligned region and the alignment will be adjusted using the realignment result. Additionally, to enhance efficiency, MisER is designed as a multi-threaded tool, allowing independent processing of multi alignments in parallel (Supplementary Figure S3).
Figure 2.

The workflow of MisER and sensitivity estimation. (A) MisER identifies potential misaligned regions (PMRs) of reads for realignment. Transcript annotation with small exons as reference are colored in blue and reads are colored in yellow. Misaligned sequences on read are colored in red. 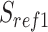 ,
, 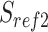 ,
, 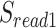 ,
, 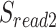 are splice sites on reference and reads.
are splice sites on reference and reads. 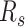 ,
, 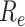 are start and end of the potential misaligned region (PMR). (B) The sensitivity plot of MisER using different delta ratio cut-offs, estimated by constitutive exons of different lengths in sequins data. The red dashed line represents the average sensitivity using different delta ratio thresholds of all exons.
are start and end of the potential misaligned region (PMR). (B) The sensitivity plot of MisER using different delta ratio cut-offs, estimated by constitutive exons of different lengths in sequins data. The red dashed line represents the average sensitivity using different delta ratio thresholds of all exons.
We evaluated MisER’s sensitivity based on alignments for sequins constitutive exons as they should be present in all reads of the corresponding genes. In fact, we found 17 989 read alignments that missed single constitutive exons in sequins ONT cDNA sequencing data. Among them, 14 851 (82.6%) cases could be corrected, while 3138 (17.4%) cases were not. MisER showed high sensitivity (average sensitivity: 96.0% at 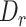 threshold 0.5) for exons shorter than 50 nt, and relative low sensitivity (average sensitivity: 62.4% at
threshold 0.5) for exons shorter than 50 nt, and relative low sensitivity (average sensitivity: 62.4% at 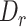 threshold 0.5) for exons longer than 50 nt (Figure 2B). The larger constitutive exons missed were likely caused by bona fide sequencing errors rather than alignment issues. We also tested MisER specificity on simulated triplet-exon annotations (Supplementary Figure S4) and MisER showed very high specificity (average specificity: 99.7% at
threshold 0.5) for exons longer than 50 nt (Figure 2B). The larger constitutive exons missed were likely caused by bona fide sequencing errors rather than alignment issues. We also tested MisER specificity on simulated triplet-exon annotations (Supplementary Figure S4) and MisER showed very high specificity (average specificity: 99.7% at 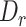 threshold 0.5). These results demonstrate MisER is a robust approach with high sensitivity and specificity to identify and adjust the misalignment of microexons.
threshold 0.5). These results demonstrate MisER is a robust approach with high sensitivity and specificity to identify and adjust the misalignment of microexons.
To further evaluate MisER’s performance on reads with different sequencing accuracies, we simulated reads with sequencing accuracies varying from 80% to 100%, based on exon triplets (4622 in total) or quadruplets (12 669 in total) derived from human transcript annotations (Supplementary Figure S5A, D, see Materials and Methods). The middle exons were set as alternative exons, resulting in two isoforms for each exon triplet and four isoforms for each exon quadruplet. We performed MisER realignment on simulated reads and found that as the accuracy of the reads decreased, the recall of the reads to the corresponding isoforms also decreased, with approximately 15–28% of the reads unable to be accurately recalled to the exons with 90% sequencing accuracy (Supplementary Figure S5B, E). Notably, most of microexons with lengths of ≤ 10 nt were misaligned even at 100% read accuracy before MisER realignment. Moreover, the reads including these alternative exons were often misassigned to isoforms lacking the middle exons, leading to over-estimation of these isoforms, ending up with low precision rates (Supplementary Figure S5C, F). More importantly, after applying MisER realignment, both the recall and the precision rates improved significantly, particularly for isoforms containing microexons no longer than 10 nt. Nevertheless, we additionally built artificial exon triplets and quadruplets using random sequences, performed the same sequencing accuracy simulation analysis, and obtained similar performance metrics of MisER (Supplementary Figure S6A–F, see Materials and Methods).
Detect and realign misaligned small exons in sequins long-read sequencing data
We applied MisER to ONT cDNA long-read sequencing data from sequins to investigate the impact of microexon misalignment on reads and isoforms. We observed significant improvements of read alignments for small exons. Of note, not only single alternatively spliced exons (R2_24, 15 nt) but also alternatively spliced exons in tandem (R2_57, 39 and 24 nt) (Figure 3A-B) were rescued. We quantified changes in read counts for exons (≤ 80 nt) within sequins transcripts before and after applying MisER realignment (Figure 3C. Supplementary Figure S7A, see Materials and Methods). MisER improved the correlations between the read counts and the annotated concentrations of these small exons in all samples of sequins. For sequins mixA replicate 2, Pearson correlation between the read counts of MisER resecured small exons and the annotated concentration improved from 0.91 to 0.95 (Figure 3C). The top four exons with the largest changes in relative read counts after realignment were from genes R2_24 (15 nt), R2_57 (39 and 24 nt), and R2_72 (18 nt). Moreover, the number of misaligned reads increased when the kmer size increased in the mapping process (Supplementary Figure S8). We found that 2.4–8.5% spliced reads contained misaligned exons (with MisER 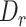 threshold 0.5 and
threshold 0.5 and 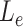 threshold 80) across three different kmer sizes (10, 15 and 20 nt) in minimap2. The misalignment problem has been slightly alleviated by using small kmer, but it may cause unwanted splits in exons and increase the burden of calculation. Specifically, for the mapping using 15 nt kmer, we found the mapping of 3.6% reads on 33 small exons are adjusted after MisER realignment in four sequin samples, involving 41 isoforms.
threshold 80) across three different kmer sizes (10, 15 and 20 nt) in minimap2. The misalignment problem has been slightly alleviated by using small kmer, but it may cause unwanted splits in exons and increase the burden of calculation. Specifically, for the mapping using 15 nt kmer, we found the mapping of 3.6% reads on 33 small exons are adjusted after MisER realignment in four sequin samples, involving 41 isoforms.
Figure 3.

MisER detects and realigns the misaligned exons in sequins ONT cDNA sequencing data. (A) Illustration of a misaligned microexon (chrIS:1144725–1144739) within the R2_24 gene. The misaligned exons (15 nt) are indicated by red arrows. (B) Illustration of two misaligned small exons (chrIS:9964822–9964860 and chrIS:9966507–9966530) within the R2_57 gene. The misaligned exons (39 and 24 nt) are indicated by red arrows. (C) Scatter plot depicting the read counts (x-axis) and annotated concentrations (y-axis) of exons (≤ 80 nt) in sequins mixA replicate 2 (GEO: GSM4594549). Exons that underwent changes in read counts after MisER realignment are highlighted in cyan (before realignment) and red (after realignment) colors, connected by black lines. The cyan and red data points are independently fitted by linear models (cyan and red dash lines). Pearson correlations for the altered exons are computed and denoted in cyan (pre-realignment) and red (post-realignment) text. (D, E) Transcript isoform quantification improvement after MisER realignment comparing with the annotated concentration of sequins RNA isoforms in sequins mixA replicate 2. Transcript isoforms that underwent changes in read counts after MisER realignment are highlighted in cyan (before realignment) and red (after realignment) colors, connected by black lines. The transcript ratio is calculated using the quantification (read count or concentration) of each transcript isoform divided by the sum of all transcripts in one gene. (F) Bar chart representing the percentages of transcript isoforms in genes before (original) and after MisER realignment (realigned), compared to the percentages calculated based on the annotated concentrations of sequins mixA.
Furthermore, we quantified transcript isoforms of sequins before and after MisER adjustment (Supplementary Figure S9), and calculated their quantification changes. Transcript quantifications are compared with annotated concentrations (Figure 3D, Supplementary Figure S7B, Supplementary Table S3). Among the 103 transcript isoforms with read counts of more than 10 in sequins mixA replicate 2, 12 isoforms (11.7%) showed an improvement of > 10% for quantification. The Pearson correlation between the changed isoforms and annotated concentrations increased from 0.85 to 0.90 (Figure 3D). In addition to transcript quantification using read counts, isoform usage (ratio of transcript count over gene count) is often calculated to assess regulation of alternative splicing. Hence, we also calculated the changes in isoform usage as an additional benchmark. Improvements in transcript read counts also enhanced the quantification of isoform usages in genes (Figure 3E, Supplementary Figure S7C). Notably, we focused on three transcript isoforms (R2_24_1, R2_57_2, R2_72_4) that contained the most prominently changed four exons. MisER increased the read counts of these isoforms (R2_24_1: from 0 to 28; R2_57_2: from 40 to 150; R2_72_4: from 2677 to 15 514) and their isoform usages are more concordant with the percentages calculated based on the annotated concentrations after correction (Figure 3F). Taken together, these results demonstrate that MisER improves the quantification of transcript isoforms, especially those containing microexons.
Detect and adjust misaligned small exons in human lymphoblastoid long-read sequencing data
We applied MisER to cDNA and direct RNA ONT sequencing data for the human lymphoblastoid GM12878 cell line (21). We observed significant improvements in read alignments with small exons, for example, the microexons in VPS29 and UCHL1 genes (Figure 4A, B). In fact, 3.1% spliced reads in cDNA and 2.1% in direct RNA were misaligned (with MisER 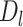 threshold 0.5 and
threshold 0.5 and 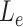 threshold 80). The misalignment ratio was calculated for all exons no longer than 80 nt in gene annotations (Figure 4C, D). Notably, for exons no longer than 50 nt, the cumulative average misalignment ratio reached 50.1% in cDNA and 44.5% in direct RNA, while this value increased to 73.5% (cDNA) and 71.0% (direct RNA) when exons no longer than 30 nt (Supplementary Tables S4, S5). The misalignment ratio further rose to 89.8% in cDNA and 91.3% in direct RNA for exons shorter than 20 nt. These results demonstrated that microexons are pervasive in human transcriptomes. Nevertheless, ONT direct RNA sequencing data displayed relatively lower misalignment ratios compared to ONT cDNA sequencing data (Supplementary Figure S10), which may be explained by the sequence accuracy difference between them. MisER effectively detected and adjusted these misalignments through local realignment, underscoring its importance for accurate quantification and further alternative splicing analyses involving human microexons.
threshold 80). The misalignment ratio was calculated for all exons no longer than 80 nt in gene annotations (Figure 4C, D). Notably, for exons no longer than 50 nt, the cumulative average misalignment ratio reached 50.1% in cDNA and 44.5% in direct RNA, while this value increased to 73.5% (cDNA) and 71.0% (direct RNA) when exons no longer than 30 nt (Supplementary Tables S4, S5). The misalignment ratio further rose to 89.8% in cDNA and 91.3% in direct RNA for exons shorter than 20 nt. These results demonstrated that microexons are pervasive in human transcriptomes. Nevertheless, ONT direct RNA sequencing data displayed relatively lower misalignment ratios compared to ONT cDNA sequencing data (Supplementary Figure S10), which may be explained by the sequence accuracy difference between them. MisER effectively detected and adjusted these misalignments through local realignment, underscoring its importance for accurate quantification and further alternative splicing analyses involving human microexons.
Figure 4.

Misalignment ratio of exons in human ONT cDNA and direct RNA sequencing data. (A) A misaligned microexon (chr12:110499535–110499546) within VPS29 gene. The misaligned exon (12 nt) is labeled by a red arrow. (B) A misaligned microexon (chr4:41257115–41257126) within UCHL1 gene. The misaligned exon (12 nt) is labeled by a red arrow. (C) Misalignment ratio in cDNA and (D) direct RNA. Each point represents an annotated exon and its size reflects the total number of reads. Exons with a total read count of < 10 are discarded.
MisER improves the identification and quantification of neural-regulated microexons in human long-read sequencing data
To study microexons that are potentially related to the neural function, we performed MisER approach to a comprehensive human long-read dataset comprising 84 ONT cDNA sequencing libraries from 30 human tissues, encompassing 14 neural and 16 non-neural tissues (22). Our analysis revealed a widespread misalignment of microexons (≤ 30 nt) in all samples (Supplementary Figure S11A). The realignment substantially increased the number of detected alternative spliced-in (ASI) microexons in the cassette exon events, with exons shorter than 10 nt being identified only after realignment (Figure 5A). Notably, these microexons (3∼10 nt) significantly overlapped with the neural-regulated microexons previously identified with short-read sequencing datasets (2) (Figure 5B). Furthermore, the sequences around these microexons, including both the upstream and downstream flanking intronic regions, displayed higher conservation compared to sequences around longer alternative exons (Figure 5C), which is consistent with the previous studies (2,5). In addition, we observed significantly more ASI microexons (≤ 30 nt) in the neural samples, particularly for microexons shorter than 10 nt (Figure 5D, Supplementary Figure S11B). Moreover, we define the difference in percent spliced-in index (PSI) between neural and non-neural samples as delta PSI. We found the values of delta PSI in microexons (≤ 30 nt) were significantly higher than large exons (≥ 51 nt) (Figure 5E), as well as testing across all brain regions individually (Supplementary Figure S11C), which is consistent with the previous report (2). Furthermore, these findings were corroborated by analyzing short-read sequencing data from the GTEx datasets (dbGaP Accession phs000424.v8.p2) across 54 human tissue regions (Supplementary Figure S12).
Figure 5.

Quantifications of microexons in ONT cDNA sequencing data of human tissues. (A) The number of alternative spliced-in exons (y axis) before (original) and after realignment (realigned) for different length of exons. Exons were split into four groups (3–10 nt, 11–30 nt, 31–50 nt, ≥ 51 nt). (B) Overlap between alternative spliced-in microexons (3–10 nt) detected in ONT data and previously reported neural-regulated microexons (Fisher's exact test). The P-value is shown. (C) Average phastCons score (y axis) of sequences (x axis) around alternative exons with different length or constitutive exons (UCSC hg38.phastCons100way). (D) Number of alternative spliced-in exons (y axis) between neural and non-neural samples (x axis), grouped by different length of exons. Neural samples are represented by red boxes and points. The difference in the number of alternative spliced-in exons between all neural and non-neural tissues is assessed using a one-sided Wilcoxon rank sum test (greater). Significant levels are labeled (***P-value ≤ 0.001, **P-value ≤ 0.01, *P-value ≤ 0.05, ns for P-value > 0.05). (E) Difference of the percent spliced in (delta PSI) between neural samples and non-neural samples (y axis) for different length of exons. The delta PSI values of small exons (3–10 nt, 11–30 nt, 31–50 nt) were compared to the delta PSI of large exons (≥ 51 nt) using a one-sided Wilcoxon rank sum test (greater). (F) The significantly up-regulated alternative spliced-in microexons (3–10 nt) in neural samples (red). Gene names and exon lengths are labeled. Two-sided Wilcoxon rank sum test, Bonferroni correction. (G–J) Changes in the expression levels of transcript isoforms of two neural-related genes before (original) and after (realigned) MisER realignment. (G) The splicing junctions of two transcript isoforms of AP1S2 gene (blue tracks, the zoomed-in view is shown below). The microexon (chrX:15828192–15828200) is labeled by a red arrow. Sashimi plots of neural samples (red) and non-neural samples (black) demonstrate the splicing junction changes of read alignments before and after MisER realignment. (H) The expression of two transcript isoforms of AP1S2 gene between neural samples (red) and non-neural samples (black), in units of transcripts per million (TPM), before (original) and after (realigned) MisER realignment. Two-sided Wilcoxon rank sum test. (I) The splicing junctions of three transcript isoforms of APBB1 gene. The microexon (chr11:6401977–6401982) is label by a red arrow. (J) The expression of three transcript isoforms of APBB1 gene.
Through the comparisons of the ASI exons between neural and non-neural samples from ONT cDNA sequencing data, we classified 174 exons as neural up-regulated exons (adjusted P-value < 0.05 and delta PSI > 25%) and 133 exons as neural down-regulated exons (adjusted P-value < 0.05 and delta PSI < –25%) (Supplementary Figure S13). Among them, eight microexons (≤ 10 nt) were neural up-regulated (Figure 5F). Notably, two micoexons in AP1S2 and APBB1 genes were previously reported to be misregulated in autism spectrum disorder (ASD), which might impact neuronal development and synaptic functions (2). The microexon spliced-in event in AP1S2 was further identified in a neuroblastoma differentiation dataset (human SH-SY5Y cell line) (27). The transcript isoform (ENST00000545766), which contains a 9 nt microexon, significantly increased during the transition of neuroblastoma from a neuroblast-like state to a neuronal-like state (Supplementary Figure S14). Read alignments demonstrated that these two microexons were only detected after MisER realignment (Figure 5G, I). Realignment changed the mapping result of these microexons and further influenced the quantifications of the transcript isoforms (Figure 5H, J). In fact, the significant differences of the microexon involving isoforms between neural and non-neural samples were only identified after MisER realignment. More importantly, the two isoforms (ENST00000609360 and ENST00000608704), which included the 6 nt microexon in APBB1 gene, exhibited a distinct level of changes between neural and non-neural samples. These subtle variations in isoform quantification are challenging to detect using short-read sequencing due to the longer length of full transcript isoforms and relatively small differences in splicing patterns. This example shows the advantage of long-read sequencing and the necessity of MisER approach. Therefore, these results underscored the impact of misaligned small exons in ONT sequencing data and highlighted the relevance of our tool in rectifying misalignments specifically associated with microexons. As a result, our tool significantly improved the analysis of microexon alternative usage, as well as the detection and quantification of transcript isoforms containing small exons.
Discussion
We systematically evaluated the issues of small exon mis-mapping and found the length of misaligned sequence correlated well with exon size. Thus, we defined the delta ratio to help find regions containing misaligned small exons and further developed a post-processing approach to adjust the alignments around them. Results show that small exons with a size no longer than 50 nt may be lost in mapping procedure and microexons less than 20 nt are almost entirely lost. To our knowledge, this is the first systematic benchmark on how small exons misalign and how transcript isoforms are affected by the misalignment. In fact, these misalignments influence the transcript isoform detection, as one small exon missing, the read would be wrongly assigned to another transcript isoform without the small exon. This raises concerns about the novel transcript isoforms identified in long-read data with small exon skipping, which may be caused by misalignment (28–30). Hence, MisER would be needed to identify bona fide new transcripts with exon skipping events.
For transcripts containing small exons, a transcriptome annotation with splice sites information is typically required in the alignment process to improve read mapping. MisER also needs a transcriptome annotation to perform local realignment in order to adjust the mapping of small exons. Some alignment tools like minimap2 offers option to perform annotation-dependent alignment. Such annotation-dependent alignment may reduce the chance of small exon misalignments. However, it's not suited for novel isoform discovery, especially important for improving transcriptome annotation. In addition, annotation-dependent mapping requires an accurate and complete transcript annotation, which is not always available for organisms under investigation. MisER performs local realignment and compares the alignment scores to determine whether the exon is misaligned or not, which can help to avoid problems caused by incorrect annotations of splice sites on small exons and others. Therefore, MisER is not depending on the quality of small exon annotations, which is very valuable for transcriptome analysis of species in which genome annotation is not as accurate and complete as the human genome. In fact, one could use short-read data to improve the annotation of small exons through assembling or microexon detection specific tools for non-model organisms, then apply MisER to evaluate and improve the alignments around the small exons in long-read data. In addition, MisER is a universal complementary approach after annotation-free mapping approach, as MisER carries out an automatic scan to fix and update the alignment in all reads in BAM format. Therefore, MisER should be a general post-alignment approach that can be run on BAM alignment file to accurately evaluate and revise the alignment of ONT RNA reads. In fact, MisER is suitable for most approaches of long reads isoforms identification and quantifications, such as FLAIR, FulQuant, TALON and Bambu (17,18,25,31), as one step of BAM file refinement for small exons.
MisER shows high sensitivity for alignment issues with microexons, but we also notice that the MisER realignment process does not always fix the missed larger exons. As MisER performed local realignment, the larger constitutive exon missing was likely caused by sequencing errors rather than alignment issues (14). The sequencing errors could generate low quality or big deletion regions in nanopore reads. In fact, direct RNA sequencing data indicates a relatively lower misalignment ratio than cDNA, which may be due to the difference in reads quality (direct RNA median quality 10.6, cDNA median quality 9.1) (21) and the difference in mapping strategy (direct RNA uses -uf parameter and cDNA uses -ub parameter). Misalignment errors are alleviated in read sequences with high accuracy for exons ranging from 20 to 50 nt. However, our analysis of simulated data indicates that microexons with lengths less than 20 nt still experience severe misalignment problems, even with 100% sequence accuracy. This represents a systematic alignment issue, as minimap2 is unable to set anchors on microexons whose lengths are less than or close to the kmer size. High-accuracy sequencing methods, such as Pacbio HiFi sequencing, should also be cautious of the risk of microexons misalignments when using minimap2 without transcript annotations.
In addition to the exon missing problem, there are other kinds of alignment errors around splice sites. Future methods that provide base-level local realignment near the splice sites would further improve the alignment and transcript isoform identification of long-read RNA sequencing data. In fact, there are inherent features of ONT reads around the misaligned exons, such as more mapping errors near the splice sites compared to correct mapped exons. More sophisticated algorithms could be developed to correct the misalignment problems in an annotation-free style in the future, which would be very helpful for non-model organisms with less accurate transcript annotation. Moreover, template switch during the reverse transcription and PCR amplification for cDNA sequencing, may cause the loss of partial sequences, which may aggravate the problem (32,33). Taken together, more comprehensive studies are needed to explore the misalignment issue of larger exons than small exons.
Supplementary Material
Acknowledgements
We thank Xueyi Dong and Michael B Clark for providing sequins sequencing data.
Author contributions: W.W., L.M.S and C.Z. conceived the project. Z. L. and C. Z. developed the MisER approach. Z.L. performed the computational analysis. Z.L., C.Z., L.M.S. and W.W. wrote the manuscript.
Contributor Information
Zhen Liu, Lingang Laboratory, Shanghai, Shanghai 200031, China; CAS Key Laboratory of Computational Biology, Shanghai Institute of Nutrition and Health, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai, Shanghai 200031, China.
Chenchen Zhu, Department of Genetics, School of Medicine, Stanford University, Stanford, CA 94305, USA.
Lars M Steinmetz, Department of Genetics, School of Medicine, Stanford University, Stanford, CA 94305, USA; Stanford Genome Technology Center, Stanford University, Palo Alto, CA 94304, USA.
Wu Wei, Lingang Laboratory, Shanghai, Shanghai 200031, China; CAS Key Laboratory of Computational Biology, Shanghai Institute of Nutrition and Health, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai, Shanghai 200031, China; Center for Biomedical Informatics, Shanghai Children's Hospital, Shanghai Jiao Tong University, Shanghai, Shanghai 200040, China.
Data availability
MisER is an open source python3 software, which is available in the Zenodo depository, at: https://doi.org/10.5281/zenodo.8345036. Our analyses were based on published datasets. Sequins mixes cDNA data is available in the GEO database under accession number GSE151984. The cDNA and direct RNA sequencing data for human lymphoblastoid GM12878 cell line are available from https://github.com/nanopore-wgs-consortium/NA12878. ONT cDNA sequencing datasets of 30 human tissues, including 84 ONT cDNAsequencing libraries (using R9.4.1 and R10.3 flow cells, SQK-LSK109 sequencing kit), are available in the GEO database under accession number GSE192955. ONT cDNA sequencing data of samples representing the differentiation of neuroblastoma (human SH-SY5Y cell line, 5 samples) into neuronlike cells (5 samples) are available in the ENA database under accession number PRJEB44502.
Supplementary data
Supplementary Data are available at NAR Online.
Funding
National Key R&D Program of China [2021YFF0703802]; National Natural Science Foundation of China [81870187, 81911530167]; Major Science and Technology R&D Project of the Science and Technology Department of Jiangxi Province [20213AAG01013] to W.W.
Conflict of interest statement. None declared.
References
- 1. Ustianenko D., Weyn-Vanhentenryck S.M., Zhang C.. Microexons: discovery, regulation, and function. Wiley Interdiscip. Rev. RNA. 2017; 8:e1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Irimia M., Weatheritt R.J., Ellis J.D., Parikshak N.N., Gonatopoulos-Pournatzis T., Babor M., Quesnel-Vallieres M., Tapial J., Raj B., O’Hanlon D.et al.. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell. 2014; 159:1511–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Torres-Mendez A., Pop S., Bonnal S., Almudi I., Avola A., Roberts R.J.V., Paolantoni C., Alcaina-Caro A., Martin-Anduaga A., Haussmann I.U.et al.. Parallel evolution of a splicing program controlling neuronal excitability in flies and mammals. Sci. Adv. 2022; 8:eabk0445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Choudhary B., Marx O., Norris A.D.. Spliceosomal component PRP-40 is a central regulator of microexon splicing. Cell Rep. 2021; 36:109464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yu H., Li M., Sandhu J., Sun G., Schnable J.C., Walia H., Xie W., Yu B., Mower J.P., Zhang C.. Pervasive misannotation of microexons that are evolutionarily conserved and crucial for gene function in plants. Nat. Commun. 2022; 13:820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu T.D., Watanabe C.K.. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005; 21:1859–1875. [DOI] [PubMed] [Google Scholar]
- 7. Li Y.I., Sanchez-Pulido L., Haerty W., Ponting C.P.. RBFOX and PTBP1 proteins regulate the alternative splicing of micro-exons in human brain transcripts. Genome Res. 2015; 25:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Parada G.E., Munita R., Georgakopoulos-Soares I., Fernandes H.J., Kedlian V.R., Metzakopian E., Andres M.E., Miska E.A., Hemberg M.. MicroExonator enables systematic discovery and quantification of microexons across mouse embryonic development. Genome Biol. 2021; 22:1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kono N., Arakawa K.. Nanopore sequencing: review of potential applications in functional genomics. Dev. Growth Differ. 2019; 61:316–326. [DOI] [PubMed] [Google Scholar]
- 10. Oikonomopoulos S., Bayega A., Fahiminiya S., Djambazian H., Berube P., Ragoussis J.. Methodologies for Transcript Profiling Using Long-Read Technologies. 2020; 11: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dohm J.C., Peters P., Stralis-Pavese N., Himmelbauer H.. Benchmarking of long-read correction methods. NAR Genom. Bioinform. 2020; 2:lqaa037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deamer D., Akeson M., Branton D. Three decades of nanopore sequencing. Nat. Biotechnol. 2016; 34:518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jain M., Fiddes I.T., Miga K.H., Olsen H.E., Paten B., Akeson M.. Improved data analysis for the MinION nanopore sequencer. Nat. Methods. 2015; 12:351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Delahaye C., Nicolas J.. Sequencing DNA with nanopores: troubles and biases. PLoS One. 2021; 16:e0257521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018; 34:3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Makalowski W., Shabardina V.. Bioinformatics of nanopore sequencing. J. Hum. Genet. 2020; 65:61–67. [DOI] [PubMed] [Google Scholar]
- 17. Zhu C., Wu J., Sun H., Briganti F., Meder B., Wei W., Steinmetz L.M.. Single-molecule, full-length transcript isoform sequencing reveals disease-associated RNA isoforms in cardiomyocytes. Nat. Commun. 2021; 12:4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tang A.D., Soulette C.M., van Baren M.J., Hart K., Hrabeta-Robinson E., Wu C.J., Brooks A.N.. Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat. Commun. 2020; 11:1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ray T.A., Cochran K., Kozlowski C., Wang J., Alexander G., Cady M.A., Spencer W.J., Ruzycki P.A., Clark B.S., Laeremans A.et al.. Comprehensive identification of mRNA isoforms reveals the diversity of neural cell-surface molecules with roles in retinal development and disease. Nat. Commun. 2020; 11:3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hardwick S.A., Chen W.Y., Wong T., Deveson I.W., Blackburn J., Andersen S.B., Nielsen L.K., Mattick J.S., Mercer T.R.. Spliced synthetic genes as internal controls in RNA sequencing experiments. Nat. Methods. 2016; 13:792–798. [DOI] [PubMed] [Google Scholar]
- 21. Workman R.E., Tang A.D., Tang P.S., Jain M., Tyson J.R., Razaghi R., Zuzarte P.C., Gilpatrick T., Payne A., Quick J.et al.. Nanopore native RNA sequencing of a human poly(A) transcriptome. Nat. Methods. 2019; 16:1297–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gao Y., Wang F., Wang R., Kutschera E., Xu Y., Xie S., Wang Y., Kadash-Edmondson K.E., Lin L., Xing Y.. ESPRESSO: robust discovery and quantification of transcript isoforms from error-prone long-read RNA-seq data. Sci. Adv. 2023; 9:eabq5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dong X., Tian L., Gouil Q., Kariyawasam H., Su S., De Paoli-Iseppi R., Prawer Y.D.J., Clark M.B., Breslin K., Iminitoff M.et al.. The long and the short of it: unlocking nanopore long-read RNA sequencing data with short-read differential expression analysis tools. NAR Genom. Bioinform. 2021; 3:lqab028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Daily J. Parasail: SIMD C library for global, semi-global, and local pairwise sequence alignments. BMC Bioinf. 2016; 17:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen Y., Sim A., Wan Y.K., Yeo K., Lee J.J.X., Ling M.H., Love M.I., Goke J.. Context-aware transcript quantification from long-read RNA-seq data with Bambu. Nat. Methods. 2023; 20:1187–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Trincado J.L., Entizne J.C., Hysenaj G., Singh B., Skalic M., Elliott D.J., Eyras E.. SUPPA2: fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 2018; 19:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wright D.J., Hall N.A., Irish N., Man A.L., Glynn W., Mould A., Angeles A.D.L., Angiolini E., Swarbreck D., Gharbi K.. Long read sequencing reveals novel isoforms and insights into splicing regulation during cell state changes. Bmc Genomics [Electronic Resource]. 2022; 23:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sahlin K., Medvedev P.. Error correction enables use of Oxford Nanopore technology for reference-free transcriptome analysis. Nat. Commun. 2021; 12:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Amarasinghe S.L., Su S., Dong X., Zappia L., Ritchie M.E., Gouil Q.. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020; 21:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hu Y., Fang L., Chen X., Zhong J.F., Li M., Wang K.. LIQA: long-read isoform quantification and analysis. Genome Biol. 2021; 22:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wyman D., Balderrama-Gutierrez G., Reese F., Jiang S., Rahmanian S., Forner S., Matheos D., Zeng W., Williams B., Trout D.et al.. A technology-agnostic long-read analysis pipeline for transcriptome discovery and quantification. 2019; bioRxiv doi:18 June 2019, preprint: not peer reviewed 10.1101/672931. [DOI]
- 32. Cocquet J., Chong A., Zhang G., Veitia R.A.. Reverse transcriptase template switching and false alternative transcripts. Genomics. 2006; 88:127–131. [DOI] [PubMed] [Google Scholar]
- 33. Schulz L., Torres-Diz M., Cortes-Lopez M., Hayer K.E., Asnani M., Tasian S.K., Barash Y., Sotillo E., Zarnack K., Konig J.et al.. Direct long-read RNA sequencing identifies a subset of questionable exitrons likely arising from reverse transcription artifacts. Genome Biol. 2021; 22:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
MisER is an open source python3 software, which is available in the Zenodo depository, at: https://doi.org/10.5281/zenodo.8345036. Our analyses were based on published datasets. Sequins mixes cDNA data is available in the GEO database under accession number GSE151984. The cDNA and direct RNA sequencing data for human lymphoblastoid GM12878 cell line are available from https://github.com/nanopore-wgs-consortium/NA12878. ONT cDNA sequencing datasets of 30 human tissues, including 84 ONT cDNAsequencing libraries (using R9.4.1 and R10.3 flow cells, SQK-LSK109 sequencing kit), are available in the GEO database under accession number GSE192955. ONT cDNA sequencing data of samples representing the differentiation of neuroblastoma (human SH-SY5Y cell line, 5 samples) into neuronlike cells (5 samples) are available in the ENA database under accession number PRJEB44502.