

Abstract
Xenorhabdus spp. and Photorhabdus spp., entomopathogenic bacteria symbiotically associated with nematodes of the families Steinernematidae and Heterorhabditidae, respectively, were shown to produce different lipases when they were grown on suitable nutrient agar. Substrate specificity studies showed that Photorhabdus spp. exhibited a broad lipase activity, while most of the Xenorhabdus spp. secreted a specific lecithinase. Xenorhabdus spp. occur spontaneously in two variants, phase I and phase II. Only the phase I variants of Xenorhabdus nematophilus and Xenorhabdus bovienii strains produced lecithinase activity when the bacteria were grown on a solid lecithin medium (0.01% lecithin nutrient agar; 24 h of growth). Five enzymatic isomers responsible for this activity were separated from the supernatant of a X. nematophilus F1 culture in two chromatographic steps, cation-exchange chromatography and C18 reverse-phase chromatography. The substrate specificity of the X. nematophilus F1 lecithinase suggested that a phospholipase C preferentially active on phosphatidylcholine could be isolated. The entomotoxic properties of each isomer were tested by injection into the hemocoels of insect larvae. None of the isomers exhibited toxicity with the insects tested, Locusta migratoria, Galleria mellonella, Spodoptera littoralis, and Manduca sexta. The possible role of lecithinase as either a virulence factor or a symbiotic factor is discussed.
Bacterial symbionts of entomopathogenic nematodes in the families Steinernematidae and Heterorhabditidae are members of the family Enterobacteriaceae and belong to the genera Xenorhabdus and Photorhabdus, respectively (7, 29). These bacteria are carried in an intestinal vesicle of the nonfeeding infective stage of members of the Steinernematidae (5) and throughout the whole intestine of infective juveniles of members of the Heterorhabditidae (14). The nematodes release their bacterial symbionts into the hemocoels of the insects, where growth induces a lethal septicemia and contributes to the symbiotic relationship by providing nutrients required by nematode partners during reproduction in insect cadavers (23).
All Xenorhabdus strains spontaneously produce two distinct physiological states in vitro (2), phase I and II variants (6). Phase I variants absorb dyes on agar plates, produce several antibiotics, secrete a variety of proteins (e.g., lipases and proteases), and produce fimbriae and flagella, while these properties are either apparently absent or greatly reduced in phase II variants (6, 17, 21). Both types are pathogens of insect larvae, but phase I variants are associated only with infective nematodes that naturally parasitize insects (2). The entomotoxicity mechanisms of these bacteria and the benefits provided by the bacteria to their host nematodes are not well-documented, but it has been suggested that extracellular molecules produced by Xenorhabdus spp. may participate in both virulence and symbiosis with nematodes (3, 15).
Production of phosphatidylcholine-hydrolyzing phospholipases (or lecithinase) is detected on solid media as opalescent zones surrounding colonies grown on agar supplemented with egg yolk (the egg yolk test) (30). A wide variety of gram-positive and gram-negative bacteria have been found to produce lecithinases when this reaction is used as an assay (30). Many of these lecithinases have been purified and characterized as single secreted-polypeptide proteins. Lecithinases are toxic determinants, as well as a means of securing bacterial supplies of phosphates (9, 30). They may also have an important role in the induction of pathogenicity in host organisms (27). For example, the ability of Bacillus thuringiensis to develop is thought to be due in part to its high-level production of phospholipases (11). Because phase I variants of Photorhabdus and Xenorhabdus species have been reported to give positive results in the egg yolk test (6), it was apposite to define any toxic property of this enzymatic activity. More recently, it was found that some Tn5-induced Xenorhabdus bovienii lecithinase mutants exhibited reduced virulence for Galleria mellonella (22).
Phospholipases which hydrolyze the glycerophospholipids belong to the lipase subgroup (glycerol ester hydrolases; EC 3.1.1.3) of esterases (carboxyl esterases; EC 3.1.1.1). Phospholipases A1, A2, B, C, and D are characterized by the sites of cleavage of the phospholipids (18, 19). Phospholipases A1, A2, and B are carboxyl esterases, while phospholipases C and D are phosphoryl esterases. It was shown previously that bacterial symbionts of entomopathogenic nematodes produced positive reactions on both Tween agar and egg yolk agar, suggesting that these bacteria exhibit lipase activities (6). The variability of the lipase activities noticed during phase variation suggested that at least two different lipases were produced by the bacterial symbionts.
To elucidate the nature of lipase production by symbiotic bacteria of entomopathogenic nematodes, we compared the enzymatic activities of several symbionts on Tween and lecithin (phosphatidylcholine) agar plates. The enzymatic activities were determined for a range of Xenorhabdus sp. and Photorhabdus sp. strains from our worldwide laboratory collection. After comparing the lipase activities of members of both genera, we concentrated our efforts on Xenorhabdus nematophilus in order to characterize the lipases of this species biochemically and pathologically. The X. nematophilus F1 lecithinase was purified and partially characterized in a first step, and then the entomotoxic and cytotoxic activities of the purified molecules were studied.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All of the bacterial strains used in this study are listed in Table 1. For each subculture, the phase status was determined by culturing on NBTA and measuring antibacterial activity against Micrococcus luteus (8). Phase I colonies are blue on NBTA and produce agar-diffusible antibiotics, while phase II colonies are red and produce reduced or no antibacterial activity. Phases I and II of strains are indicated as suffixes (/1 and /2, respectively) attached to strain designations. Bacterial cells were grown in Luria-Bertani broth for liquid cultures and on nutrient agar (NA) manufactured by Pasteur Institute or on Luria-Bertani agar for solid cultures.
TABLE 1.
Activities of phase I variants of Xenorhabdus and Photorhabdus species grown on NA plates containing 1% (vol/vol) Tween and NA plates containing 0.01% (wt/vol) lecithin after 2 days of incubation at 28°C
| Nematode host | Bacterium
|
Activity witha:
|
||||
|---|---|---|---|---|---|---|
| Taxon | Strain | Lecithin | Tweens 20, 40, and 60 | Tween 80 | Tween 85 | |
| Steinernema sp. | X. beddingii | Q58 | + | + | +w | + |
| S. feltiae | X. bovienii | F5 | + | − | − | + |
| S. affine | X. bovienii | F3 | + | − | − | − |
| S. feltiae | X. bovienii | T228 | + | − | − | − |
| S. intermedium | X. bovienii | Si | + | − | − | − |
| S. kraussei | X. bovienii | SK2 | + | − | − | − |
| S. carpocapsae | X. nematophilus | F1 | + | + | + | − |
| S. carpocapsae | X. nematophilus | A24 | + | + | − | + |
| S. carpocapsae | X. nematophilus | AN6 | +w | + | − | − |
| Steinernema sp. | Xenorhabdus sp. | M16 | + | +w | + | +w |
| S. anomalae | Xenorhabdus sp. | Sav | + | + | − | + |
| S. puertoricense | Xenorhabdus sp. | PR6a | − | + | − | +w |
| S. rarum | Xenorhabdus sp. | K77 | − | + | + | + |
| S. cubanum | Xenorhabdus sp. | Cub | − | + | +w | − |
| S. glaseri | X. poinarii | NC33 | − | + | +w | − |
| S. glaseri | X. poinarii | G6 | − | + | − | − |
| Heterorhabditis bacteriophora | P. luminescens | K80 | − | + | + | + |
| Heterorhabditis bacteriophora | P. luminescens | Hb | − | + | + | + |
| Heterorhabditis bacteriophora | P. luminescens | C1 | − | + | + | + |
| Heterorhabditis bacteriophora | P. luminescens | HP88 | − | + | + | + |
| H. indica | P. luminescens | E20 | − | + | + | + |
| H. indica | P. luminescens | HG21 | − | + | + | + |
| H. indica | P. luminescens | IS5 | + | + | + | + |
| H. indica | P. luminescens | Meg | +w | + | + | + |
| H. megidis | P. luminescens | HW79 | − | + | + | + |
| H. megidis | P. luminescens | XLNach | − | + | +w | + |
| H. zealandica | P. luminescens | NZH3 | − | + | + | + |
| Heterorhabditis sp. | P. luminescens | C8406 | − | + | + | + |
| Heterorhabditis sp. | P. luminescens | K122 | − | + | + | + |
| Heterorhabditis sp. | P. luminescens | ItH211 | − | + | + | + |
| Heterorhabditis sp. | P. luminescens | Hm | − | + | + | + |
| Clinical strain | P. luminescens | 43950 | − | − | − | − |
Activity as measured by the production of a white halo of precipitation surrounding each colony. +, strong production; −, no production; +w, weak production.
Measurements of lipase activity.
Qualitative assays of lipase activity were performed by plating solid bacterial cultures on Tween agar plates as described previously (8). Lipase-releasing colonies were identified by the formation of precipitates of water-insoluble fatty acids from hydrolyzed Tween surrounding them (26).
Glycerol ester hydrolase activity was qualitatively assayed by plating solid bacterial cultures or liquid culture supernatants on triolein solid medium plates; pure triolein (Sigma) was added to NA at 45°C to a final concentration of 1% (vol/vol) and was emulsified by sonication at 125 W for 5 min with a Branson 2210 Sonifier. Lipase-releasing colonies or supernatants were recognized by the formation of precipitates of water-insoluble fatty acids from the hydrolyzed triglyceride.
A photometric assay of lipase activity was performed with p-nitrophenylpalmitate as the substrate, as previously described (28, 32).
Measurements of lecithinase activity.
Lecithinase activity was assayed by spotting on NA containing 0.01% (wt/vol) lecithin prepared as described previously (8). After 48 h of incubation, lecithinase-releasing colonies were recognized by the formation of precipitation zones of water-insoluble fatty acids from hydrolyzed lecitin surrounding them (30). The substrate specificity of the purified X. nematophilus F1 lecithinase was determined by replacing the crude egg yolk lecithin with different highly purified phospholipids from egg yolk (see Table 4) (99% pure; Sigma). Agarose (1%, wt/vol) was dissolved in 0.1 M NaCl–0.02 M Tris buffer (pH 9, 45°C), and each phospholipid was emulsified as described above at a final concentration of 0.01% (wt/vol) before it was spread over colonies grown on NA.
TABLE 4.
Substrate specificity of the purified lecithinase on agarose-lipid layersa
| Substrate of lipase | Results |
|---|---|
| Egg yolk lecithin (60% PC)b | +c |
| Phosphatidylcholine (99%)d | + |
| Lyso-phosphatidylcholine (99%)d | + |
| Phosphatidylinositol (99%)d | + |
| Lyso-phosphatidylinositol (99%)d | + |
| Phosphatidylethanolamine (99%)d | − |
| Sphingomyelin (99%)d | + |
| Diacylglycerol | − |
| Phosphatidic acid (99%)d | + |
| Triolein | − |
| p-Nitrophenylphosphorylcholine | − |
| p-Nitrophenylpalmitate | − |
As measured by the formation of a white halo of precipitation surrounding a hole in an agarose-lipid layer filled with 3 μl of the purified lecithinase.
Containing 60% phosphatidylcholine.
+, strong precipitation; −, no precipitation.
Percent purity.
A semiquantitative lecithinase assay, adapted from the assay described by Giskow et al. (16), was used to monitor lecithinase production. It was performed as a diffusion speed assay with thin 1% agarose gels containing 0.01% lecithin, 0.1 M NaCl, and 0.02 M Tris (pH 9). Three-microliter aliquots were added to wells (diameter, 1 mm) made in the gels with a Pasteur pipette. The presence of lecithinase induced a precipitation-diffusing zone around each hole. We found that there was a linear relationship between the logarithm of a serial dilution of a concentrated sample and the radius of the precipitating zone in the gel (data not shown). After incubation at 28°C, enzymatic activity was determined by recording the radius (in millimeters) of the precipitation zone in each gel at given times. Lecithinase activity was calculated by determining the change in the precipitation radius (in millimeters) per unit of time (24 h), and specific activity was calculated by determining the change in the precipitation radius (in millimeters) per unit of time (24 h) and unit of material (microgram of protein or milligram of dried bacterial material). The protein concentration was measured by the bicinchoninic acid (Mallet SA; Pierce) method with serum albumin as the standard (34).
IEF and blotting of lecithinase activity.
Isoelectric focusing (IEF) was performed by using an LKB apparatus (Pharmacia, Uppsala, Sweden) and broad isoelectric point (pHi) precast gels (pH 3 to 10) according to the manufacturer’s instructions. A broad-pHi calibration kit (pH 3 to 10; Pharmacia) was used to determine the lecithinase pHi. After electrophoresis, the position of lecithinase in the IEF gels was determined by washing the gels five times in 100 ml of Tris-NaCl buffer (0.1 M NaCl, 0.02 M Tris [pH 9]) (30 min each) and overlapping them with a 1% agarose layer containing 0.01% lecithin in Tris-NaCl buffer. After 2 h of incubation at 28°C, the position of lecithinase activity was observed as a white precipitation zone in the agarose-lecithin gel.
Purification of lecithinase.
X. nematophilus F1/1 was cultivated at 28°C in 100 ml of Luria-Bertani broth for 3 days. Cells were removed by low-speed centrifugation (6,000 × g, 10 min, 4°C), and a filter-sterilized (pore size, 0.22 μm) supernatant was dialyzed overnight at 4°C against 10 liters of Tris-NaCl buffer. The dialyzed supernatant (100 ml) was subjected to cation-exchange chromatography on a SP MemSep cartridge column (void volume, 1.4 ml; Millipore) at a flow rate of 1.4 ml · min−1 and was washed with 14 ml of Tris-NaCl buffer. The lecithinase activity was eluted once with 0.5 M NaCl–0.02 M Tris buffer (pH 9). Active fractions were pooled and diluted with 1 volume (1.5 ml) of Tris-NaCl buffer.
This crude lecithinase preparation was acidified with 0.1% (vol/vol) trifluoroacetic acid (TFA) and then subjected to reverse-phase high-performance liquid chromatography (HPLC) on a C18 column (Supelcosil LC318; 25 cm by 4.6 mm; Supelco). Solvent A contained 0.1% TFA in MilliQ-treated water (Millipore), and solvent B contained 90% acetonitrile in 0.1% (vol/vol) TFA–water. Unbound material was removed by washing at a flow rate of 0.5 ml · min−1 with solvent A for 20 min. Proteins were eluted with a 0 to 80% solvent B gradient generated with a Waters delivery system over a period of 30 min. Peptides were detected with a photodiode array detector (model 990; Waters), and the optical density at 220 nm was recorded. Eluted fractions (500 μl) were evaporated in a speed vacuum apparatus, and the dried material was resuspended in 50 μl of Tris-NaCl buffer.
During the purification procedure, lecithinase activity was routinely monitored by spotting on NA plates containing 0.01% lecithin and was quantitatively determined by the radial diffusion assay.
Toxicity assays.
The common cutworm, Spodoptera littoralis, was reared on an artificial diet (24) at 24°C, and the wax moth, G. mellonella, was reared on pollen and wax at 28°C. A locust, Locusta migratoria, was reared on grass at 28°C. Eggs of the tobacco hornworm, Manduca sexta, were obtained from Monika Stengl (University of Regensburg, Regensburg, Germany). M. sexta larvae were reared on an artificial diet (1) at 27°C with light-dark cycles consisting of 16 h of light and 8 h of darkness. Fifth-instar larvae of each insect species were selected and surface sterilized with 70% (vol/vol) ethanol prior to intrahemocoelic injection. The larvae were divided into groups of 12, and each larva was injected with 10 μl of one of the purified HPLC fractions, corresponding to a dose of 0.1 μg per insect, or with phosphate-buffered saline (PBS). The treated larvae were incubated individually for up to 96 h, and then the number of dead insects was recorded.
A liquid hemolysis assay with sheep erythrocytes (25) was used to determine hemolytic activity in purified HPLC fractions. Cytolytic assays were performed with insect hemocytes by collecting hemolymph samples from S. littoralis larvae in an anticoagulant buffer (4). Hemocytes were centrifuged, rinsed in PBS to remove plasmatic factors, and resuspended in the same buffer (2 × 104 hemocytes · ml−1). The suspensions (10 μl) were each mixed with 10 μl of a purified HPLC fraction, corresponding to a 0.1-μg dose, deposited on a slide, and incubated for 20 min at 28°C. Hemocytes with PBS were used as a control. Cell lysis was observed with a light microscope and was recorded.
RESULTS
Lipase- and lecithinase-producing Xenorhabdus and Photorhabdus strains.
No precipitation zones were observed surrounding the Photorhabdus luminescens K122 colonies when they were grown on NA containing 0.01% lecithin even when incubation was prolonged by 1 week. Since this strain is known to secrete a Tween 80 lipase in solid cultures (31), we deduced that the lecithin and Tween assays may be used to discriminate between the two different enzymes (a lecithinase and a lipase, respectively). Screening the lecithinase and lipase activities in the wide range of symbiotic bacteria from entomopathogenic nematodes allowed us to distinguish the following three groups of symbionts: (i) lipase-producing strains which were strongly positive on NA plates containing 1% Tween but failed to generate any precipitates on NA containing 0.01% lecithin; (ii) lecithinase-producing strains which were strongly positive on NA plates containing 0.01% lecithin but failed to generate any precipitates on NA containing 1% Tween; and (iii) strains which produced both lipase and lecithinase (Table 1). Most of the Photorhabdus strains belonged to the first group, while several X. bovienii strains belonged to the second group. X. nematophilus and Xenorhabdus beddingii strains were able to produce both lecithinase and lipase activities.
When both phase I and phase II variants of the lecithinase-producing strains were grown on NA plates containing 0.01% lecithin for 2 days, the phase I variants had large halos around the colonies, while the phase II variants did not. Nevertheless, when incubation was extended by 7 days, the X. nematophilus phase II variants also produced a weak zone of precipitation (Fig. 1). The X. bovienii phase II variants were still negative even after an additional 1 week of incubation. On the other hand, the Tween activities of all of the Tween-positive Xenorhabdus spp. were always found to be greater for the phase II variants than for the phase I variants (Table 2).
FIG. 1.
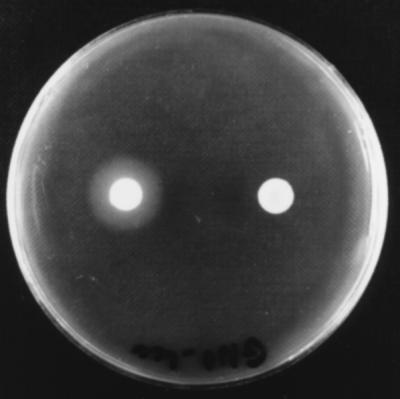
X. nematophilus F1 phase I variant lecithinase-producing colony (left) and phase II variant nonproducing colony (right) grown on an NA plate containing 0.01% lecithin after 2 days of incubation at 28°C.
TABLE 2.
Comparison of the activities of phase I and phase II variants of Xenorhabdus species grown on NA plates containing 1% (vol/vol) Tween and NA plates containing 0.01% (wt/vol) lecithin after 7 days of incubation at 28°C
| Nematode host | Bacterium
|
Activity witha:
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Lecithin
|
Tweens 20, 40, and 60
|
Tween 80
|
Tween 85
|
|||||||
| Taxon | Strain | Phase I variant | Phase II variant | Phase I variant | Phase II variant | Phase I variant | Phase II variant | Phase I variant | Phase II variant | |
| Steinernema sp. | X. beddingii | Q58 | + | − | + | +w | +w | − | + | +w |
| S. feltiae | X. bovienii | F5 | + | − | − | +w | − | − | + | − |
| S. affine | X. bovienii | F3 | + | − | − | +w | − | − | − | − |
| S. feltiae | X. bovienii | T228 | + | − | − | − | − | − | − | − |
| S. intermedium | X. bovienii | Si | + | − | − | +w | − | +w | − | +w |
| S. kraussei | X. bovienii | SK2 | + | − | − | + | − | − | − | − |
| S. carpocapsae | X. nematophilus | F1 | + | +w | + | + | + | + | − | +w |
| S. carpocapsae | X. nematophilus | A24 | + | +w | + | + | − | − | + | + |
| S. carpocapsae | X. nematophilus | AN6 | +w | − | + | + | − | − | − | − |
| Steinernema sp. | Xenorhabdus sp. | M16 | + | − | +w | + | + | + | +w | +w |
| S. anomalae | Xenorhabdus sp. | Sav | + | − | + | + | − | − | + | + |
Activity as measured by the production of a white halo of precipitation surrounding each colony. +, strong production; −, no production; +w, weak production.
Lecithinase activity during broth growth of X. nematophilus F1.
Lecithinase production by phase I variants of X. nematophilus occurred after 16 to 24 h of incubation at 28°C in a shaking bath incubator, whereas a phase II variant culture failed to produce any lecithinase activity even after several days. Figure 2 shows that production of lecithinase by X. nematophilus F1/1 was growth phase dependent. No lecithinase activity was observed during the exponential growth phase, but when growth began to slow down and entered the stationary phase, there was a sudden burst of expression, which rapidly increased to the maximum level. Further incubation revealed that the maximum level of lecithinase activity remained constant for several weeks.
FIG. 2.

Relationship between growth in a Luria-Bertani broth culture of X. nematophilus F1/1 and release of lecithinase. Symbols: •, growth, as determined by absorbance at 600 nm; ○, lecithinase specific activity (per milligram of dried material), as determined by measuring the rate of enzyme diffusion (in millimeters per 24 h) on an NA plate containing 0.01% lecithin. Values are the means from three experiments. Error bars indicate standard deviations.
Purification of the X. nematophilus F1 lecithinase.
IEF of a 10-fold-concentrated supernatant sample from a 3-day-old X. nematophilus F1/1 culture was performed on a 5% acrylamide gel containing ampholines ranging from pH 3 to 9.3. After the preparation was blotted with a 0.01% lecithin–1% agarose layer, the lecithinase activity was observed at the cathode, indicating that the pHi was greater than pH 9.3 (data not shown). This basic feature of the lecithinase was used to perform a first chromatographic procedure with an SP-MemSep cation-exchange cartridge at pH 9. Under these basic conditions most proteins from the supernatant (theoretically all proteins with a pHi less than pH 9) were excluded, while the lecithinase was kept on the column. After a single elution with 0.5 M NaCl–0.02 M Tris (pH 9) buffer, the lecithinase was eluted, and the positive fractions were subjected to reverse-phase HPLC on a C18 column. This second chromatographic procedure allowed us to separate five peaks (peaks 5 through 9) containing lecithinase activity (Fig. 3), which eluted at about 50% acetonitrile. Each of these peaks was passed separately a second time through the same C18 column to check its purity and activity (data not shown). Quantification by the diffusion speed assay on the 0.01% lecithin–1% agarose gel showed that peaks 6 and 8 possessed most of the total activity. Nevertheless, the specific activities (in millimeters per 24 h per milligram) were found to be similar in all peaks and increased by a factor of about 350 during the purification procedure (Table 3).
FIG. 3.

C18 chromatographic elution pattern, showing five lecithinase active peaks. Peaks 5 through 9 showed lecithinase activity, as determined by spotting onto an NA plate containing 0.01% lecithin. Solid line, absorbance at 220 nm; dashed line, 0 to 80% acetonitrile gradient in 0.1% (vol/vol) TFA–water.
TABLE 3.
Purification of the extracellular lecithinase from a phase I variant culture of X. nematophilus F1
| Prepn | Vol (ml) | Amt of total protein (mg) | Total activitya | Sp actb | Yield (%) | Purification (fold) |
|---|---|---|---|---|---|---|
| Culture supernatant | 100 | 145 | 580,000 | 4 | 100 | 1 |
| SP chromatography | 3 | 0.36 | 324,000 | 900 | 56 | 225 |
| C18 chromatography | 0.2 | 0.008 | 11,200 | 1,400 | 2 | 350 |
Expressed as speed of diffusion (in millimeters per 24 h).
Expressed as speed of diffusion (in millimeters per 24 h) per microgram of protein.
Biochemical properties and substrate specificity of the HPLC isolates.
Each isolate was very stable, as shown by full recovery of lecithinase activity after long-term storage at room temperature or after 30 min of exposure to 100°C. Moreover, the activity was not affected by preincubation of the purified lecithinase with a commercial lipase or with several proteases. However, treatment with pronase E from Streptomyces griseus (Sigma) completely destroyed the lecithinase. Spectral analysis at wavelengths between 200 and 600 nm showed that the five C18 HPLC peaks exhibited the same narrow absorption spectrum at a λmax of ca. 207 nm (data not shown). The substrate specificity of each of the isolates was assayed separately by spotting aliquots of a purified sample onto 1% agarose gels containing different pure phospholipids. Phosphatidylcholine, lyso-phosphatidylcholine, phosphatidylinositol, lyso-phosphatidylinositol, sphingomyelin, and phosphatidic acid were found to produce an opaque zone with all five lecithinase isolates, whereas phosphatidylethanolamine, diacylglycerol, and triolein did not (Table 4). These data indicate that there was a preferential affinity for polar lipids. Enzymatic assays in liquid failed to reveal any enzymatic reaction with p-nitrophenylphosphorylcholine or p-nitrophenylpalmitate substrates despite the use of several detergents and buffers.
Toxic properties.
None of the five isolates exhibited any cytolytic activities against either sheep erythrocytes or insect hemocytes. Although preliminary experiments showed that G. mellonella, S. littoralis, M. sexta, and L. migratoria were very susceptible to intrahemocoelic injection of X. nematophilus F1/1 (16a), injection into the hemocoel of 0.1-μg portions of the purified molecule had no effect on the mortality of fifth-instar larvae of these insects.
DISCUSSION
The previously described lipase-producing organism P. luminescens K122 (31) gave a positive reaction on Tween 80 agar plates but did not produce any precipitate on phospholipid-containing medium, such as the lecithin-NA plates used in our study. The data described here demonstrate that the Tween and lecithin agar assays are enzyme specific and suitable for discriminating lipase-producing strains from lecithinase-producing strains when the bacteria symbiotically associated with the entomopathogenic nematodes are examined. Generally, we found that X. bovienii had lecithinase properties and Photorhabdus spp. had lipase properties. We report here that 13 of 15 Photorhabdus strains were lecithinase negative on lecithin agar, in contrast to previously published results (obtained by using the egg yolk agar test) reported for three Photorhabdus strains formerly considered Xenorhabdus luminescens strains (6). X. nematophilus and X. beddingii formed an intermediate group having both activities. When the bacteria produced only one of the two enzymes, this biochemical characteristic was specific to the phase I variant. The strains that released both enzymes exhibited differential production; the phase I variants were the lecithinase producers, while the phase II variants were the major lipase producers. This last observation provided another demonstration that lecithinase and lipase activities were due to two different enzymes which were conversely regulated during phase variation. P. luminescens K122 lipase was found to possess a triacyl glycerol esterase activity, as indicated by the production of an opaque zone when the organism was grown on a triolein-NA plate. However, this enzyme appeared to be specific to nonpolar lipids because K122 failed to exhibit any reactions with the phospholipids tested. Strains which produced precipitates only on lecithin-NA plates were found to be unable to hydrolyze p-nitrophenylpalmitate, suggesting that a particular lipase activity (lecithinase) was present.
X. nematophilus F1 lecithin precipitate-associated molecules were purified by cation-exchange chromatography and hydrophobic C18 HPLC. Surprisingly, HPLC generated five peaks representing compounds which were capable of inducing precipitates on lecithin-NA plates. These five purified compounds showed the same substrate specificity, forming some precipitates with the polar phospholipids, such as phosphatidylcholine. Moreover, the substrates bearing only carboxyl ester bonds, such as Tweens, diacylglycerol, and triolein, were not precipitated by the five compounds. This substrate specificity indicated that the X. nematophilus F1 lecithin precipitate-associated molecules could correspond to a lecithinase that is able to cleave only phosphoryl ester bonds. Such enzymatic activity could theoretically correspond to phospholipase C or D. However, the latter possibility is unlikely since the purified lecithinase reacted with phosphatidic acid, which cannot be hydrolyzed by phospholipase D. In order to probe phospholipase C activity, we used p-nitrophenylphosphorylcholine, which is a specific substrate of phospholipase C (20). The five purified samples were not able to hydrolyze p-nitrophenylphosphorylcholine. This could reflect a default of the substrate, which is quite different from a true phospholipid (30). The five HPLC-purified molecules might represent five isomers, as expected based on their identical adsorption spectra, their common basic properties, and their identical substrate specificities. Although the protease treatments (pronase E treatments) revealed their peptide character, these proteins share some unusual characteristics, such as the lack of absorption at 280 nm and the inability to be stained by Coomassie blue in sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels (data not shown), indicating that there may be a lack of aromatic amino acids (10, 12). Because the biochemical properties of the five purified molecules remain unclear, we decided to use the generic name lecithinase to describe a molecule which induces a zone of precipitation when it is applied to phospholipid agarose layers.
Several well-described phospholipases C have been found to be involved in bacterial virulence (27). The consequences may be immediate and direct, as is the action of Clostridium perfringens alpha toxin against erythrocytes, or subtle, as is the reaction of Listeria monocytogenes phosphatidylinositol-specific phospholipase C, which allows bacteria to grow in infected cells (27).
Injections of the five purified lecithinases into the hemocoels of different species of insects did not result in increased mortality. Moreover, these molecules did not exhibit cytolytic activities against sheep erythrocytes or insect hemocytes. These results demonstrate that the X. nematophilus lecithinase had no entomotoxic effects. However, although Tn5-induced X. bovienii lecithinase-deficient mutants still killed the insect larvae, they showed reduced virulence for G. mellonella. This was seen clearly in the results of the 50% lethal dose analysis, which showed that the G. mellonella mortality rate was significantly lower with the Tn5-induced lecithinase mutant than with wild-type parent strain T228/1 (22). The Xenorhabdus lecithinase may participate in the virulence of the nematobacterial complex by allowing intracellular bacterial growth in insects in the same way that phospholipase C acts in B. thuringiensis virulence (11).
It is commonly accepted that the symbiotic bacteria Xenorhabdus spp. provide some nutrients to their nematode hosts by releasing digestive exoenzymes into insect cadavers (15). Lipidic nutrition of the nematodes is considered a key factor for the reproduction and development of the nematode larvae (33). Most Xenorhabdus spp. possess both a lipase specific for the nonpolar lipids and a lecithinase which hydrolyzes the polar phospholipids. The bacterial activity of the latter may reflect a high level of adaptation of the Xenorhabdus spp. to their nematode hosts. Dunphy and Webster (13) showed that in vitro production of Steinernema carpocapsae on lipid-containing media involves the consumption of choline, a nutrient which is liberated after hydrolysis of phosphatidylcholine by phospholipase C. This fact strongly suggests that the lecithinase of X. nematophilus may play a role in the lipidic metabolism of its nematode host, S. carpocapsae, by providing the choline nutrient to it.
Future work on lecithinases produced by X. nematophilus will focus on enzymatic characterization of the purified molecule in which radiolabeled substrates and protein sequencing are used. Gnotobiological experiments on lipid-rich media involving both purified enzyme and lecithinase mutants should greatly help workers to evaluate the role of this bacterial enzyme in the lipidic metabolism of S. carpocapsae.
ACKNOWLEDGMENTS
We thank Jean Luciani for assistance with the insect pathogenesis assays and Michel Brehélin for assistance with the insect hemocyte experiments. We also thank Trevor Jackson (Agresearch, Lincoln, New Zealand) and Carey Smith (CSIRO-CILBA, Montpellier, France) for revising the manuscript.
REFERENCES
- 1.Ahmad I M, Waldbauer G P, Friedman S. A defined artificial diet for the larvae of Manduca sexta. Entomol Exp Appl. 1989;53:189–191. [Google Scholar]
- 2.Akhurst R J. Morphological and functional dimorphism in Xenorhabdus spp., bacteria symbiotically associated with the insect pathogenic nematodes Neoplectana and Heterorhabditis. J Gen Microbiol. 1980;121:303–309. [Google Scholar]
- 3.Akhurst R J, Boemare N E. Biology and taxonomy of Xenorhabdus spp. In: Gaugler R, Kaya H, editors. Entomopathogenic nematodes in biological control. Boca Raton, Fla: CRC Press; 1990. pp. 75–90. [Google Scholar]
- 4.Anggraeni T, Ratcliffe N A. Studies on cell-cell cooperation during phagocytosis by purified haemocyte populations of the wax moth, Galleria mellonella. J Insect Physiol. 1991;37:453–460. [Google Scholar]
- 5.Bird A F, Akhurst R J. The nature of the intestinal vesicle in nematodes of the family Steinernematidae. Int J Parasitol. 1983;13:599–606. [Google Scholar]
- 6.Boemare N E, Akhurst R J. Biochemical and physiological characterization of colony form variants in Xenorhabdus spp. (Enterobacteriaceae) J Gen Microbiol. 1988;134:1835–1845. doi: 10.1099/00221287-134-7-1835. [DOI] [PubMed] [Google Scholar]
- 7.Boemare N E, Akhurst R J, Mourant R G. DNA relatedness between Xenorhabdus spp. (Enterobacteriaceae), symbiotic bacteria of entomopathogenic nematodes, and a proposal to transfer Xenorhabdus luminescens to a new genus, Photorhabdus gen. nov. Int J Syst Bacteriol. 1993;43:249–255. [Google Scholar]
- 8.Boemare N E, Thaler J-O, Lanois A. Simple bacteriological tests for phenotypic characterization of Xenorhabdus and Photorhabdus phase variants. Symbiosis. 1997;22:167–175. [Google Scholar]
- 9.Borgström B, Brockman H L. Lipases. Amsterdam, The Netherlands: Elsevier Science; 1984. [Google Scholar]
- 10.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 11.Bucher G E. Potential bacterial pathogens of insects and their characteristics. J Insect Pathol. 1960;2:172–195. [Google Scholar]
- 12.Compton S J, Jones C G. Mechanism of dye response and interference in the Bradford protein assay. Anal Biochem. 1985;151:369–374. doi: 10.1016/0003-2697(85)90190-3. [DOI] [PubMed] [Google Scholar]
- 13.Dunphy G B, Webster J M. The monoxenic culture of Neoaplectana carpocapsae DD-136 and Heterorhabditis heliothidis. Rev Nematol. 1988;12:113–123. [Google Scholar]
- 14.Endo B Y, Nickle W R. Ultrastructure of the intestinal epithelium, lumen and associated bacteria in Heterorhabditis bacteriophora. J Helminthol Soc Wash. 1991;58:202–212. [Google Scholar]
- 15.Forst S A, Dowds B, Boemare N E, Stackebrandt E. Xenorhabdus spp. and Photorhabdus spp.: bugs that kill bugs. Annu Rev Microbiol. 1997;51:47–72. doi: 10.1146/annurev.micro.51.1.47. [DOI] [PubMed] [Google Scholar]
- 16.Giskow M, Olsen L, Molin S. Cloning and expression in Escherichia coli of the gene for extracellular phospholipase A1 from Serratia liquefaciens. J Bacteriol. 1988;170:5855–5862. doi: 10.1128/jb.170.12.5855-5862.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16a.Givaudan, A. Unpublished data.
- 17.Givaudan A, Baghdiguian S, Lanois A, Boemare N E. Swarming and swimming changes concomitant with phase variation in Xenorhabdus nematophilus. Appl Environ Microbiol. 1995;61:1408–1413. doi: 10.1128/aem.61.4.1408-1413.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaeger K-E, Ransac S, Dijkstra B W, Colson C, van Heuveul M, Misset O. Bacterial lipases. FEMS Microbiol Rev. 1994;15:29–63. doi: 10.1111/j.1574-6976.1994.tb00121.x. [DOI] [PubMed] [Google Scholar]
- 19.Kok R G, van Thor J J, Nugteren-Roodzant I M, Brouwer M B, Egmond M R, Nudel C B, Vosman B, Hellingwerf K J. Characterization of the extracellular lipase, LipA, of Acinetobacter calcoaceticus BD413 and sequence analysis of the cloned structural gene. Mol Microbiol. 1995;15:803–818. doi: 10.1111/j.1365-2958.1995.tb02351.x. [DOI] [PubMed] [Google Scholar]
- 20.Kurioka S, Matsuda M. Phospholipase C assay using p-nitrophenylphosphorylcholine together with sorbitol and its application to studying the metal and detergent requirement of the enzyme. Anal Biochem. 1976;75:281–289. doi: 10.1016/0003-2697(76)90078-6. [DOI] [PubMed] [Google Scholar]
- 21.Moureaux N, Karjalainen T, Givaudan A, Bourlioux P, Boemare N E. Biochemical characterization and agglutinating properties of Xenorhabdus nematophilus F1 fimbriae. Appl Environ Microbiol. 1995;61:2707–2712. doi: 10.1128/aem.61.7.2707-2712.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pinyon R A, Linedale E C, Webster M A, Thomas C J. Tn5-induced Xenorhabdus bovienii lecithinase mutants demonstrate reduced virulence for Galleria mellonella larvae. J Appl Bacteriol. 1996;80:411–417. [Google Scholar]
- 23.Poinar G O, Thomas G M. Significance of Achromobacter nematophilus Poinar & Thomas (Achromobacteriaceae:Eubacteriales) in the development of the nematode, DD136 (Neoplectana sp. Steinernematidae) Parasitology. 1966;56:385–390. doi: 10.1017/s0031182000070980. [DOI] [PubMed] [Google Scholar]
- 24.Poitout S, Bues R. Elevage de plusieurs espèces de Lépidoptères Noctuidae sur milieu artificiel riche et sur milieu artificiel simplifié. Ann Zool Ecol Anim. 1970;2:79–91. [Google Scholar]
- 25.Rowe G E, Welch R A. Assays of hemolytic toxins. Methods Enzymol. 1994;235:657–667. doi: 10.1016/0076-6879(94)35179-1. [DOI] [PubMed] [Google Scholar]
- 26.Sierra G. A simple method for the detection of lipolytic activity of micro-organisms and some observations on the influence of the contact between cells and fatty substrates. J Microbiol Serol. 1957;23:15–22. doi: 10.1007/BF02545855. [DOI] [PubMed] [Google Scholar]
- 27.Songer J G. Bacterial phospholipases and their role in virulence. Trends Microbiol. 1997;5:156–161. doi: 10.1016/S0966-842X(97)01005-6. [DOI] [PubMed] [Google Scholar]
- 28.Stuer W, Jaeger K E, Winkler U K. Purification of extracellular lipase from Pseudomonas aeruginosa. J Bacteriol. 1986;168:1070–1074. doi: 10.1128/jb.168.3.1070-1074.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas G M, Poinar G O., Jr Xenorhabdus gen. nov., a genus of entomopathogenic and nematophilic bacteria of the family Enterobacteriaceae. Int J Syst Bacteriol. 1979;29:352–360. [Google Scholar]
- 30.Titball R W. Bacterial phospholipases C. Microbiol Rev. 1993;57:347–366. doi: 10.1128/mr.57.2.347-366.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang H, Dowds B C. Phase variation in Xenorhabdus luminescens: cloning and sequencing of the lipase gene and analysis of its expression in primary and secondary phases of the bacterium. J Bacteriol. 1993;175:1665–1673. doi: 10.1128/jb.175.6.1665-1673.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Winkler U K, Stuckman M. Glycogen, hyaluronate, and some other polysaccharides greatly enhance the formation of exolipase by Serratia marcescens. J Bacteriol. 1979;138:663–670. doi: 10.1128/jb.138.3.663-670.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wouts W M. Mass production of the entomogenous nematode Heterorhabditis heliothidis (Nematoda: Heterorhabditidae) on artificial media. J Nematol. 1981;13:467–469. [PMC free article] [PubMed] [Google Scholar]
- 34.Zaweija D. Analysis of picogram quantities of protein in subnanoliter-size samples. Anal Biochem. 1984;142:182–188. doi: 10.1016/0003-2697(84)90535-9. [DOI] [PubMed] [Google Scholar]