

Abstract
PyrR is a protein that regulates the expression of genes and operons of pyrimidine nucleotide biosynthesis (pyr genes) in many bacteria. PyrR acts by binding to specific sequences on pyr mRNA and causing transcriptional attenuation when intracellular levels of uridine nucleotides are elevated. PyrR from Bacillus subtilis has been purified and extensively studied. In this work, we describe the purification to homogeneity and characterization of recombinant PyrR from the thermophile Bacillus caldolyticus and the crystal structures of unliganded PyrR and a PyrR-nucleotide complex. The B. caldolyticus pyrR gene was previously shown to restore normal regulation of the B. subtilis pyr operon in a pyrR deletion mutant. Like B. subtilis PyrR, B. caldolyticus PyrR catalyzes the uracil phosphoribosyltransferase reaction but with maximal activity at 60°C. Crystal structures of B. caldolyticus PyrR reveal a dimer similar to the B. subtilis PyrR dimer and, for the first time, binding sites for nucleotides. UMP and GMP, accompanied by Mg2+, bind specifically to PyrR active sites. Nucleotide binding to PyrR is similar to other phosphoribosyltransferases, but Mg2+ binding differs. GMP binding was unexpected. The protein bound specific sequences of pyr RNA 100 to 1,000 times more tightly than B. subtilis PyrR, depending on the RNA tested and the assay method; uridine nucleotides enhanced RNA binding, but guanosine nucleotides antagonized it. The new findings of specific GMP binding and its antagonism of RNA binding suggest cross-regulation of the pyr operon by purines.
Expression of genes encoding the enzymes of pyrimidine nucleotide biosynthesis (pyr genes) in many diverse bacteria is regulated by transcriptional attenuation, a mechanism in which the termination of transcription, rather than its initiation, is regulated (25). In the case of pyr genes, one or more transcription terminators, which are formed by stem-loops, followed by a poly(U) tract in the mRNA, lie upstream of the structural genes being regulated. Another upstream stem-loop, called the antiterminator, prevents formation of the terminator stem-loop. The regulatory protein PyrR binds specifically to pyr mRNA when intracellular levels of uridine nucleotides are high and interferes with formation of the antiterminator; this permits the terminator to form. Experimental evidence supporting this description has been summarized previously (25).
Regulation of pyr transcription by PyrR has been studied most extensively with Bacillus subtilis, in which methods for molecular genetic analysis are well developed. PyrR from B. subtilis has been purified to homogeneity and extensively characterized (26, 27). PyrR regulates attenuation of B. subtilis pyr genes in response to uridine nucleotides during transcription in vitro (13), and it binds to pyr mRNA at the predicted sites with high specificity (3, 27).
The 1.6-Å crystal structure of unliganded B. subtilis PyrR (26) showed that PyrR is a member of the homologous PRT family, most of whose members are phosphoribosyltransferase (PRTase) enzymes of nucleotide synthesis and salvage (22). PyrR is easily recognized as a PRT protein at the sequence level by virtue of a 10-residue motif for phosphoribosylpyrophosphate (PRPP) binding. B. subtilis PyrR also has uracil PRTase catalytic activity (uracil + PRPP → UMP + PPi), although its primary biological function is regulation of transcriptional attenuation (27). Another Bacillus uracil PRTase is encoded by the upp gene (14). By both sequence and structural similarity, PyrR appears to have evolved from a hypoxanthine guanine PRTase and not from a upp-encoded uracil PRTase (26). Although the details of molecular recognition of RNA by PyrR have been studied by examining the binding of numerous RNA sequence variants (3) and by site-directed mutagenesis of the putative RNA-binding site of PyrR (21), a good understanding of the elements involved in recognition demands the determination of the structure of a protein-RNA complex at high resolution. However, B. subtilis PyrR has poor solubility at the high concentrations required for crystallization, and repeated attempts to crystallize the protein with a bound uridine nucleotide or in a complex with pyr RNA have not been successful. Structures of PyrR homologs from Thermus thermophilus (Protein Data Bank [PDB] code 1UFR) and from Mycobacterium tuberculosis (PDB code 1W30) have also been reported, but none of the reported structures is of a nucleotide or RNA complex.
The studies of Ghim and Neuhard (7) of the thermophilic bacterium Bacillus caldolyticus have identified an excellent candidate for structural studies of PyrR. B. caldolyticus grows optimally at 72°C (10), and its pyr operon is organized and regulated in the same fashion as the B. subtilis pyr operon (7, 8). The amino acid sequences of PyrR from B. subtilis and B. caldolyticus are 73% identical and 85% similar. The B. caldolyticus pyrR gene has been shown to complement an Escherichia coli upp mutant, to encode thermostable uracil PRTase activity, and to restore normal regulation of the pyr operon to a B. subtilis mutant strain from which the pyrR gene had been deleted (7).
In this communication, we describe the facile purification, characterization, and crystal structure of the recombinant B. caldolyticus PyrR protein. The properties of the isolated protein demonstrate that it is indeed an excellent functional homologue of B. subtilis PyrR, is quite thermostable, and binds B. subtilis pyr RNA tightly. In its unliganded state and the nucleotide-bound form, B. caldolyticus PyrR is a tetramer. Nucleotides and divalent cations bind synergistically, and both influence RNA binding.
MATERIALS AND METHODS
Construction of plasmid pSHCO2.
The B. caldolyticus pyrR gene was amplified by PCR with DNA from the plasmid pHPSY2 (7) as the template and the deoxyoligonucleotide primers 5′-CGGGATCCCATATGCAAAAGGCGGTCGTGATGGACG-3′ (in which the N-terminal coding region is underlined and tandem BamH1 and NdeI restriction sites are italicized) and 5′-CGGAATTCTACTATTTTTCATGAATGGAGACTTGATCGATGCCG-3′ (in which the complement to the C-terminal coding region is underlined and two tandem stop codons and a noncomplementary EcoRI site are italicized). The PCR product (550 bp) was inserted between EcoRI and NdeI sites in plasmid pET20b (Novagen) to create the expression plasmid pSHCO2. The structure of pSHCO2 was confirmed by restriction enzyme analysis and by dideoxy sequencing of both strands of the insert.
Overexpression of pyrR and purification of recombinant B. caldolyticus PyrR.
E. coli strain BL21(DE3) (24) was transformed with pSHCO2 and grown at 37°C on Luria-Bertani medium (16) containing ampicillin (100 μg/ml). Three 1-liter cultures were inoculated with an exponentially growing culture of strain BL21/pSHCO2 (10 ml of inoculum per liter), grown for 5 h at 37°C, and then induced for 3 h with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). Cells were harvested by centrifugation (15,000 × g at 4°C for 15 min), washed once with cold 0.9% NaCl, resuspended in 100 mM Tris-acetate (pH 7.0; 6 ml per gram of cell paste), and ruptured by sonication. After centrifugation (15,000 × g for 15 min) of the lysate, the supernatant was heated at 65°C for 15 min and centrifuged (10,000 × g for 15 min). The supernatant from the heat treatment was dialyzed overnight against two changes of 100 mM Tris-acetate (pH 7.0)-100 mM NaCl.
The dialyzed preparation was loaded onto an 80-ml ion-exchange column (Q-Sepharose FF; Pharmacia, Piscataway, N.J.) preequilibrated with 100 mM Tris-acetate-10 mM sodium phosphate (pH 7.0) containing 100 mM NaCl, and the column was washed with 400 ml of the same buffer. PyrR was eluted with 800 ml of a 100 to 200 mM linear gradient of NaCl in 100 mM Tris-acetate-10 mM sodium phosphate (pH 7.0). PyrR eluted from the column at about 150 mM NaCl. Pooled fractions containing PyrR were concentrated about 15 fold by pressure dialysis, dialyzed against 10 mM Tris-acetate (pH 7.0) for crystallization trials and against 100 mM Tris-acetate (pH 7.0)-10 mM potassium acetate containing 20% glycerol for all other studies, and stored at −80°C. The yield of >98% pure PyrR was about 75 mg/liter of E. coli culture.
Physical characterization.
The native molecular weight of PyrR was determined by molecular sieving (“Gold” Beckman Systems chromatograph with a 125 solvent module and a 168 detector). Diluted PyrR was loaded onto a 300- by 7.8-mm column (Bio-Sil SEC 250; Bio-Rad, Hercules, Calif.) and eluted at room temperature with 0.1 M Na phosphate-0.15 M NaCl-0.01 M Na azide (pH 6.8) at a speed of 1 ml/min. The PyrR protein was analyzed at initial concentrations of 1 and 4 mg/ml. A standard curve for estimating molecular weights from the elution times from the column was determined with gel filtration standards from Bio-Rad.
Thermal denaturation of PyrR was analyzed by differential scanning calorimetry with a Micro Calorimetry Systems unit and Origin software (Microcal, North Hampton, Mass.). Protein samples (1 mg/ml of 100 mM Tris-acetate [pH 7.5]-10 mM potassium acetate-20% glycerol) were scanned from 25 to 80°C at 80°C per h. Transition points (Tm values) for thermal denaturation were reproducible within 2°C. Some protein precipitation occurred during the analyses, and the thermal denaturation observed was not reversible.
Matrix-assisted laser desorption ionization-time of flight mass spectrometry of PyrR was performed with an Applied Biosystems Voyager DE STR instrument in the Protein Science Facility of the University of Illinois Biotechnology Center. The sample was dialyzed against water, redissolved in aqueous 8 M guanidine-HCl in 2.5% trifluoroacetic acid, concentrated by filtration-centrifugation (C4 Ziptip; Millipore), and crystallized with sinapinic acid.
Uracil PRTase assays.
Assays of the uracil PRTase activity of B. caldolyticus PyrR were carried out at 37°C to follow purification and at various other temperatures with the purified protein. The final reaction mixture contained 90 mM serine (pH 8.9), 5 mM MgCl2, 0.4 mM PRPP, and 0.2 mM uracil in a final volume of 50 μl and had a pH of 8.6 at 37°C. [14C]UMP was determined in each reaction mixture at various times by spotting 5-μl samples onto DEAE-cellulose paper, followed by washing and scintillation counting as previously described (27).
Filter-binding assay of RNA binding.
PCR templates for in vitro transcription of B. subtilis BL2 RNA (3) were generated with Pfx polymerase (Invitrogen) and the following primers (the incorporated T7 promoter is indicated by underlining): BSBL2_FOR (5′-GGGGTACCTAATACGACTCACTATAGGGAAAACGAATAAT AGATCACCTTTTTAAGGGC) and BSBL2_REV (5′-CTTTTTGGGCCTTTGTTGTG). 32P-labeled BL2 RNA (79 nucleotides) was generated by in vitro transcription (MaxiScript kit; Ambion) according to the manufacturer's instructions; reaction mixtures included 500 mM (each) unlabeled ATP, UTP, and CTP and 3.1 mM [α-32P]CTP (800 Ci/mmol; MP Biochemical, Inc.). The 702-741CL and 707-736CL RNAs (3) were chemically synthesized by Dharmacon (Lafayette, Colo.) and end labeled with [γ-32P]ATP (800 Ci/mmol; MP Biochemical, Inc.) and T4 polynucleotide kinase (Promega). Labeled RNA was purified with denaturing polyacrylamide gel electrophoresis (15% acrylamide, 8 M urea) as described previously (1), with 1 μg of yeast tRNA (Ambion) as carrier RNA before precipitation. Purified RNA was resuspended in TYKE buffer (25 mM Tris-acetate, 50 mM K acetate, 1 mM EDTA [pH 7.5]). Prior to use in filter binding, the purified RNA was diluted to the proper concentration with TYKE buffer and additional yeast tRNA (at a concentration of 10 μg/ml in the binding reaction mixture). To allow for uniform folding of the PyrR-binding loop, BL2 RNA was denatured at 80°C for 15 min and then allowed to refold for 15 min at 37°C. Mg acetate was added to final concentrations ranging from 0 to 20 mM, and the RNA was placed on ice until use.
Binding assays were performed with a double-filter-binding method (29) with a Minifold II slot blot apparatus (Schleicher and Schuell). Nitrocellulose membrane (Protran BA85; Schleicher and Schuell) and positively charged nylon membrane (Hybond-N+; Amersham Biosciences) were both equilibrated for at least 20 min in binding buffer (25 mM Tris-acetate, 50 mM K acetate, and the appropriate concentration of Mg acetate) prior to use. Final binding reaction mixture conditions per 50 μl of reaction volume were as follows: 25 nM 32P-labeled BL2 RNA, 25 mM Tris-acetate, 50 mM potassium acetate, 10 μg of yeast tRNA/ml, 0.08 U of SUPERase-In (Ambion)/μl, 25 μg of acetylated bovine serum albumin/ml, and various concentrations of PyrR, Mg acetate, and nucleotide effectors. Reaction mixtures were allowed to equilibrate for at least 40 min on ice before 45 μl of each reaction mixture was filtered through both membranes. Wells were washed once with an additional 50 μl of the appropriate binding buffer within 10 s of the application of samples to the slot blot apparatus. When experimental conditions included varying the Mg2+ concentrations in one set of reaction mixtures, filters were equilibrated in binding buffer without Mg acetate, and no wash was performed, to prevent the potential dissociation of bound RNA-protein complexes.
Dissociation constants were determined by curve fitting with SigmaPlot 2001 (Systat Software, Inc.). Apparent dissociation constants were determined by fitting data to a standard binding isotherm equation, Fb = (C*P)/(Kd + P), in which C is the maximum amount of RNA bound, P is the conentration of protein, and Kd is the dissociation constant. Data were also fit to the Hill equation to determine if RNA binding was cooperative. In no case was the value of the Hill coefficient statistically >1. Binding of RNA to PyrR in the presence of GMP gave a Hill coefficient of significantly <1, suggesting negative cooperativity, but additional experiments are required to test that conclusion.
Characterization of UMP binding to PyrR by equilibrium dialysis.
Preparation of [32P]UMP and characterization of its binding to PyrR followed the procedure described previously (9). Dialysis was in 100 mM Tris-acetate, 10 mM K acetate, and 20% glycerol (pH 7.5) at 0°C for 40 to 48 h. Equimolar MgCl2 and UMP were used. The concentration of PyrR was 100 μM. The data were fit to a hyperbolic binding curve with Kaleidagraph 3.0 for Windows.
Crystallization and X-ray diffraction analysis.
Crystals of unliganded PyrR from B. caldolyticus were grown at 20°C by vapor diffusion from a 1:1 mixture of protein solution (9 mg of PyrR/ml, 1 mM UMP, 10 mM Tris [pH 7.0]) and well solution (0.1 M sodium cacodylate [pH 7.4 to 7.6], 200 mM NH4Cl, 2 mM MgCl2, 10 to 12% polyethylene glycol 4000). Diffraction quality crystals grew to a size of 0.2 by 0.1 by 0.1 mm in 2 to 3 weeks. The Mg-sulfate complex was crystallized under identical conditions, except 100 mM (NH4)2SO4 and 2 mM MgSO4 were substituted in the well solution for the chloride salts. Crystals of the nucleotide complex of PyrR were obtained fortuitiously in attempts to cocrystallize PyrR with a 28-nucleotide RNA oligomer with a sequence from BL2 RNA (Dharmacon). The drops contained a 1:1 mixture of PyrR-RNA solution (9 mg of PyrR/ml in 10 mM Tris [pH 7.0], 1 mM UMP, 13 mg of RNA/ml in 10 mM sodium cacodylate [pH 6.5]) and well solution (50 mM sodium cacodylate [pH 7.4 to 7.6], 40 mM MgCl2, 5 to 6% 2-methyl-2,4-pentanediol). The PyrR-RNA solution contained a 1:1 molar ratio of RNA:PyrR monomer. Crystals grew in 7 to 8 weeks to a size of 0.1 by 0.1 by 0.1 mm. Crystals were cryoprotected by being washed in well solution augmented with 1 mM UMP and 35% glycerol and frozen in a gaseous N2 stream at 120 K. Diffraction data for the unliganded structure were collected on an RAXIS-IV imaging plate system with a RU-H2R rotating anode generator. Diffraction data for both the liganded structures were collected at BioCARS beamline ID-14 at the Advanced Photon Source and processed with HKL2000 (Table 1) (19). The structure of unliganded PyrR was solved by molecular replacement from the monomer of B. subtilis PyrR (PDB code 1A3C) with the program AMoRe (17). Crystallographic refinement against all data to 2.4 Å was done with the CNS program (4). The unliganded form of PyrR was used as model for solving structures of the nucleotide and sulfate complexes by molecular replacement. In all structures, residues 73 to 91 of the PRT flexible loop lacked electron density and are missing from the models. Lys39 in all subunits lies in a disallowed region of the Ramachandran plot, but its conformation is well supported by electron density. Model quality is summarized in Table 1.
TABLE 1.
Crystallographic data
| Parameter | Ligand complex
|
||
|---|---|---|---|
| None | Mg2+ nucleotide | Mg2+ sulfate | |
| Crystal component | |||
| Protein solution | Protein + 1 mM UMP | Protein:RNA::1:1 + 1 mM UMP | Protein + 1 mM UMP |
| Mg | 2 mM MgCl2 | 40 mM MgCl2 | 2 mM MgSO4 |
| Diffraction data | |||
| X-ray source | CuKα | APS ID-14 | APS ID-14 |
| Wavelength | 1.54 Å | 1.14 Å | 1.13 Å |
| Space group | C2 | R32 | P41212 |
| Unit cell constants (Å) | 193.6, 60.7, 59.2; β = 97.81° | 128.5, 128.5, 128.8 (hexagonal setting) | 106.5, 106.5, 62.2 |
| Diffraction limit | 2.4 (2.5-2.4)a | 2.8 (2.9-2.8) | 2.27 (2.35-2.27) |
| Average I/σ1 | 19.1 | 8.3 | 20.6 |
| Completeness | 98.2 (92.0) | 99.7 (97.9) | 94.4 (75.2) |
| Average redundancy | 4 | 6 | 10 |
| Rsymb | 4.3 (21.1) | 9.8 (55.7) | 9.4 (31.2) |
| Refinement | |||
| Data range | 50-2.4 | 50-2.8 | 50-2.27 |
| Rwork/Rfreec | 19.2/25.0 | 22.7/30.6 | 22.6/27.3 |
| No. of protein atoms | 5,040 | 2,528 | 2,505 |
| No. of ligand atoms | 0 | 71 | 17 |
| No. of water molecules | 471 | 60 | 95 |
| Average B (Å2) | 40.1 | 40.6 | 49.3 |
| RMSD from ideality | |||
| Covalent bonds (Å) | 0.005 | 0.007 | 0.006 |
| Bond angles (°) | 1.2 | 1.4 | 1.2 |
| Missing residues | 73-90 | 74-90 | 72-90 |
Values in parentheses correspond to the outermost shell of data.
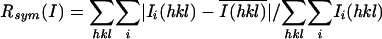
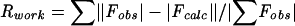 ; Rfree for a 10% subset of reflections.
; Rfree for a 10% subset of reflections.
Protein structure accession numbers.
The final coordinates and structure factors are deposited in the Protein Data Bank with codes 1NON for unliganded PyrR, 1XZN for the Mg-SO4 complex, and 1XZ8 for the nucleotide complex.
RESULTS
Purification and physical properties of recombinant B. caldolyticus PyrR.
The high expression of the B. caldolyticus pyrR gene in E. coli and the stability of the protein when heated to 65°C permitted simple and efficient purification of the native recombinant protein, based on the published procedure for purifying B. subtilis PyrR (27). As expected for a protein from a thermophile, B. caldolyticus PyrR could be heated at 65°C for up to 30 min without loss of uracil PRTase activity, although considerable activity was lost after 60 min at 65°C. Values of 76.3 and 78.7°C were obtained in replicate determinations of the thermal denaturation transition temperature (Tm) for two different preparations of the protein by differential scanning calorimetry. The corresponding Tm value for B. subtilis PyrR ranged between 48 and 50°C (21).
The subunit molecular weight of B. caldolyticus PyrR was determined by mass spectrometry to be 19,952, which agrees well with the value calculated (19,938) from the deduced amino acid sequence (7) and indicates no removal of the initiator methionine residue from the recombinant protein. PyrR eluted from high-performance liquid chromatography (HPLC) molecular sieving chromatography as a single symmetrical peak with an elution time corresponding to a native molecular weight of 77,000. This indicates that the native protein is a tetramer. In contrast, B. subtilis PyrR crystallized in hexameric and dimeric states, corresponding to molecular weights of 120,000 and 40,000, respectively (26), but migrated on gel filtration chromatography as though it had an intermediate molecular weight (27). This has been interpreted as resulting from rapid equilibration between the dimeric and hexameric states (27). Analysis of B. subtilis PyrR by HPLC under the same conditions used for B. caldolyticus PyrR yielded a molecular weight of 110,000 (21). The dimeric state of B. subtilis PyrR crystallized only in the presence of Sm2+, which fortuitously eliminated the hexamer by coordinating amino acid side chains at hexamer subunit contacts. It will be shown below that crystalline B. caldolyticus PyrR is a tetramer.
Functional properties of B. caldolyticus PyrR.
Although the primary physiological function of PyrR is thought to be its role in the regulation of transcriptional attenuation of the pyr operon, we used the protein's uracil PRTase activity to follow the purification and to demonstrate that the purified material is in the native state. Figure 1 shows the uracil PRTase specific activity of purified B. caldolyticus PyrR as a function of the assay temperature. As expected, the maximal catalytic activity (8.8 μmol per min per mg) was observed at 60°C, near the optimal growth temperature for B. caldolyticus. The specific activity at 37°C was about 10-fold lower, 0.9 μmol per min per mg. In contrast, the specific activity of PyrR from the mesophile B. subtilis at 37°C was 15 μmol per min per mg (27).
FIG. 1.

Temperature dependence of the uracil PRTase activity of B. caldolyticus PyrR.
Binding of [32P]UMP to purified B. caldolyticus PyrR at 2 mg per ml was studied by equilibrium dialysis at 0°C. UMP binding obeyed a simple hyperbolic curve, which was described by an apparent dissociation constant of 30 ± 4 μM, and reached saturation at 0.66 ± 0.02 mol of UMP bound per mol of PyrR subunit (Fig. 2). These values are quite similar to those observed with B. subtilis PyrR, which were 27 μM and 0.56 mol per mol of PyrR, respectively (9).
FIG. 2.

UMP binding to PyrR, as evaluated by equilibrium dialysis.
PyrR structure.
Three unrelated crystal forms of B. caldolyticus PyrR were produced under somewhat different conditions in an attempt to crystallize an RNA or nucleotide complex of the protein. We report three crystal structures of PyrR in different ligand states: unliganded, a Mg2+-sulfate complex, and a Mg2+-nucleotide complex (Table 1). The B. caldolyticus PyrR subunit possesses the characteristic PRT fold (22), with the core and hood domains oriented identically in the three crystal structures (Fig. 3). As expected from the size exclusion chromatography experiments described above, B. caldolyticus PyrR is a tetramer, also identical in the three structures. The tetramer is a dimer of dimers and has D2 molecular symmetry.
FIG. 3.

B. caldolyticus PyrR crystal structure. (A) Structure of the unliganded B. caldolyticus PyrR tetramer. The tetramer also occurs in the structures of the Mg2+-nucleotide and Mg2+-sulfate complexes. Subunits are depicted in different colors. (B) Structure of the B. caldolyticus nucleotide-bound PyrR dimer. The violet and the green subunits represent a view perpendicular to that shown in panel A. This dimer is virtually identical to that formed by PyrR from B. subtilis, T. thermophilus, and M. tuberculosis. Bound nucleotides are shown by stick structures; bound Mg2+ is shown by the small blue spheres. The figure was prepared with the programs MolScript (12) and Raster3D (15).
A common dimer occurs in all PyrR homologues of known structure. The B. subtilis PyrR hexamer is a trimer of dimers (26). The PyrR homologue from T. thermophilus (PDB code 1UFR) is a dimer of dimers, as is the homologue from M. tuberculosis (PDB code 1W30). Identical tetramers are formed by PyrR homologues from B. caldolyticus and M. tuberculosis, whereas the tetramer of T. thermophilus PyrR is quite different. We compared the structures by selecting 89 Cα atoms from the subunit core that are common to all structures and superimposing these on the nucleotide complex of B. caldolyticus PyrR. Among the three B. caldolyticus PyrR structures reported here, the 89 Cα atoms superimposed with a root mean square deviation (RMSD) of 0.3 Å, within experimental error. The dimers (178 Cα atoms) superimposed with an RMSD of 0.4 Å. The two Bacillus PyrR proteins superimposed with RMSDs of 0.5 Å for the monomer and 0.6 Å for the dimer. PyrR from T. thermophilus and M. tuberculosis superimposed on B. caldolyticus PyrR with RMSDs of 0.6 Å for the monomers and 0.8 Å for the dimers.
Nucleotide binding to PyrR.
A PyrR complex with UMP and GMP resulted from crystallization of PyrR in the presence of UMP and RNA. Subsequently, we demonstrated that the PyrR preparation had an RNase contaminant. Although the crystallization did not yield the desired RNA complex, it provided the first view of nucleotide binding to any PyrR protein. Crystals of the nucleotide complex had two monomers in the asymmetric unit, one with bound 5′-UMP and the other with bound 5′-GMP. A third nucleotide, 3′-GMP, was bound in a lattice contact between tetramers. The only source of purine 3′ and 5′ nucleotides in the crystallization solution was degraded RNA, as no purine nucleotides were added during purification or crystallization.
UMP binds to PyrR like nucleotides bind to other PRT family members, as expected for the product of the uracil PRTase reaction (22). The 5′ phosphate occupied a positively charged pocket formed by backbone amides and polar side chains at the N terminus of an α-helix (Thr109, Gly110, Arg111, and Thr112 NH), the ribose occupied a loop formed by the PRT sequence motif (residues 100 to 112), and the uracil base was hydrogen bonded with the hood domain (Fig. 4A). We presume this is also the site for UMP enhancement of PyrR-RNA binding.
FIG. 4.

Nucleotide binding to PyrR. (A) Binding in the PRTase active site. The uracil (left) and guanine (center) bases have identical hydrogen bonding partners (I161 backbone amide and R137 NH1 and I161 backbone carbonyl) in B. caldolyticus PyrR. Analogous hydrogen bonds are formed by GMP bound to HGPRTase (T. foetus HGPRTase-PDB 1HGX) (right), illustrating the ancestry of PyrR among the HGPRTases. (B) Mg2+-nucleotide binding. Mg2+ is coordinated by the Asp104-Asp105 dual carboxyl chains. In the subunit with bound UMP (left), one ribose hydroxyl also coordinates Mg2+. The phosphate of 3′-GMP, which is bound in a crystal lattice contact, coordinates Mg2+ in the subunit with bound 5′-GMP (right). Nucleotides and aspartates are represented by atomic colors, C is in grey/black, O is in red, N is in blue, P is in green, and Mg2+ is in magenta; waters are shown as red spheres.
Purine nucleotide binding to the same site in the second subunit was unexpected. This nucleotide was modeled as 5′-GMP based on hydrogen bonding to the protein (Fig. 4A). The 5′ phosphate groups and bases of GMP and UMP bind PyrR identically. The N1 and O6 atoms of guanine were hydrogen bonded to the same protein groups as the N3 and O4 atoms of uracil (Ile161 C=O and NH; Arg137 NH1). The site accommodates both purine and pyrimidine nucleotides by rather loose binding of the UMP and GMP ribose groups, which are not well ordered and are differently positioned within the binding sites. The hydrogen bonds to guanine are analogous to those found in hypoxanthine/guanine PRTases (Ile157 NH, K134 NZ, and Ile157 C=O) (Fig. 4A), and illustrate the probable ancestry of PyrR as an H/G PRTase. Despite the similarities in binding of GMP and UMP, B. caldolyticus PyrR did not exhibit guanine PRTase activity under conditions where it exhibited uracil PRTase activity (data not shown).
A third nucleotide, modeled as 3′-GMP, was bound between PyrR tetramers in a crystal lattice contact such that the 3′-phosphate and guanine base interacted with different tetramers. Guanine N1 contacted the C=O of Phe93, and guanine O6 contacts backbone NHs of Thr96 and Val95. The 3′-phosphate interacted with bound Mg2+ in the subunit that contained 5′-GMP (Fig. 4B).
Divalent cation binding to PyrR.
We found bound Mg2+ in two of the three B. caldolyticus PyrR structures. In the nucleotide and Mg2+-sulfate complexes, Mg2+ was coordinated by the dual carboxyl side chains (Asp 104 and Asp 105) that are a hallmark of the PRT sequence motif (Fig. 4). This binding mode exhibits two novel features compared to Mg2+ binding to other PRT proteins. First, divalent cation binding has been observed previously only in those PRT complexes with phosphate groups proximal to the cation, either the 1′-pyrophosphate of PRPP or the 5′-phosphate of nucleotide in an adjacent site. However, in the PyrR subunit with bound UMP and in the Mg2+-sulfate complex, no phosphate (or sulfate) is proximal to bound Mg2+. In the PyrR subunit with bound 5′-GMP, the 3′-phosphate of 3′-GMP in a crystal lattice contact was proximal to Mg2+ (Fig. 4B). Second, divalent cation coordination by both ribose hydroxyls has been observed previously in all nucleotide complexes of PRT proteins. For example, in PRPP complexes, the cation is coordinated by the 1′-pyrophosphate and ribose hydroxyls of PRPP, and the dual carboxylate side chains form hydrogen bonds with the ribose hydroxyls. However, in the PyrR-nucleotide complex, bound Mg2+ was not coordinated by either ribose hydroxyl group of GMP but rather the 3′-hydroxyl of UMP alone (Fig. 4B). Poorly ordered binding of the ribose groups, as described above, is one consequence of this. Precedent for direct Mg2+ coordination by the dual carboxylates, as seen in PyrR, is found in complexes of glutamine PRPP amidotransferase with the feedback inhibitors AMP or GMP, but in these complexes the ribose hydroxyls also are Mg2+ ligands and the 5′-phosphate of a second nucleotide is proximal to the cation site (5, 11, 23). B. subtilis PurR, another regulatory PRT protein, also has unusual cation-binding properties. PurR binds PRPP, but the PRPP complex does not bind Mg2+ (2).
Effects of nucleotides and Mg2+ on RNA binding to PyrR.
Because UMP, GMP, and Mg2+ bound to PyrR in the crystal structures, we examined further the effects of these ligands on RNA binding by PyrR. The RNA used for most studies was a 90-nucleotide segment of pyr mRNA derived from the pyrR-pyrP intercistronic region (second attenuation region) of the B. subtilis pyr operon, called BL2 RNA, which was previously shown to bind tightly to purified B. subtilis PyrR (3). This RNA bound to B. subtilis PyrR with an apparent dissociation constant of 3 nM at 4°C. Binding was tighter in the presence of 0.5 mM UMP or UTP, with apparent Kd values of 0.7 and 0.02 nM, respectively (3). In preliminary studies, the same electrophoretic gel mobility shift analysis (3) was used to examine the binding of pyr mRNA to purified B. caldolyticus PyrR. BL2 RNA also bound very tightly to B. caldolyticus PyrR, and its apparent affinity was greater in the presence of UMP and UTP (E. Bonner, unpublished data). Determination of the apparent Kd values, which were estimated to be in the picomolar range, required very dilute concentrations of RNA and PyrR.
Unlike our experience with B. subtilis PyrR, which does not bind quantitatively to hydrophobic filters, we found that RNA binding to B. caldolyticus PyrR could be studied by a very rapid and convenient filter-binding method (29). We used this method for subsequent studies of binding of BL2 RNA to B. caldolyticus PyrR. The apparent affinity of PyrR for BL2 RNA was strongly dependent on the concentrations of Mg2+ and UMP. In the absence of Mg2+, binding of RNA was barely detectable, but Mg2+ and UMP acted synergistically to increase the affinity of PyrR for RNA (Fig. 5A). At 20 mM Mg2+, the apparent Kd for BL2 RNA was 0.1 nM; 0.5 mM UMP decreased the apparent Kd to 0.04 nM. UDP and UTP also stimulated RNA binding (data not shown).
FIG. 5.

Effects of nucleotides and Mg2+ concentration on RNA binding in a filter-binding assay. (A) Stimulation of RNA binding by Mg2+ and UMP. Solid symbols, no UMP; open symbols, 0.5 mM UMP was included; circles, no Mg2+; inverted triangles, 1 mM Mg2+; squares, 20 mM Mg2+. (B) Effects of varying the ratio of UMP to GMP. The total concentration of UMP plus GMP was kept constant at 1 mM; the concentration of Mg2+ was 20 mM. Concentrations of nucleotides were as follows: •, 1 mM UMP only; ○, 0.95 mM UMP and 0.05 mM GMP; ▿, 0.9 mM UMP and 0.1 mM GMP; ▾, 0.75 mM UMP and 0.25 mM GMP; ▪, 0.5 mM UMP and 0.5 mM GMP; □, 0.25 mM UMP and 0.75 mM GMP; ⧫, 0.1 mM UMP and 0.9 mM GMP; ◊, 0.05 mM UMP and 0.95 mM GMP; and ▴, 1.0 mM GMP only.
Comparison of these results with the previously obtained gel shift analyses suggests that B. caldolyticus PyrR binds pyr RNA roughly 100-fold more tightly than PyrR from B. subtilis. However, the magnitude of this difference may depend on the RNA tested; binding of two pyr RNAs that are shorter than BL2 RNA, 701-741CL and 707-736CL (3), to B. caldolyticus PyrR was 1,000-fold tighter than reported for binding of the same RNA species to B. subtilis PyrR, as determined with a previously published gel mobility shift assay (3; C. J. Fields, unpublished data). The effects of nucleotides on the binding of these smaller RNAs were similar to that shown for BL2 RNA (Fig. 5). While a portion of these differences in Kd values probably resulted from the use of different binding assays, we have consistently observed tighter binding of the same RNA to B. caldolyticus PyrR than to B. subtilis PyrR. This may reflect an adaptation to growth at elevated temperatures, which would tend to dissociate protein-RNA complexes.
The unexpected observation of GMP bound to PyrR in the uracil PRTase active site raised the possibility that GMP may play a physiological role by influencing the affinity of PyrR for RNA. We found that 0.5 mM GMP strongly reduced the affinity of BL2 RNA for PyrR (apparent Kd of 1 nM), even at 20 mM Mg2+, a condition in which uridine nucleotides stimulated RNA binding only modestly. GDP and GTP at 0.5 mM had similar effects, but adenosine and cytidine nucleotides did not significantly affect RNA binding at any Mg2+ concentration from 2.5 to 20 mM (data not shown). Uridine nucleotides and guanosine nucleotides had opposing effects on the affinity of PyrR for RNA; various UMP-to-GMP ratios (with the total nucleotide concentration held constant) altered the apparent affinity of PyrR for RNA by several hundred fold (Fig. 5B).
DISCUSSION
Our studies with B. caldolyticus PyrR were undertaken in the expectation that the PyrR homologue from a thermophile would provide a better object for crystallization and structure determination. We have developed a simple procedure for isolating highly purified B. caldolyticus PyrR, which is produced in excellent yield and is stable in solution. B. caldolyticus PyrR was shown in this work to bind tightly to a pyr RNA oligonucleotide to which the B. subtilis PyrR bound well; with both proteins this binding was enhanced by UMP and UTP. The thermostability of the purified PyrR and the avidity with which it bound pyr RNA make it a good candidate for crystallization of the protein and of its RNA complex. The crystal structures of the Mg2+-nucleotide and Mg2+-sulfate complexes pointed out unusual features of divalent cation binding and an unexpected GMP-binding site. We have shown that both of these structural features are relevant to RNA binding by PyrR.
All crystallization experiments with B. caldolyticus PyrR were attempts to form a UMP or UMP/RNA complex of the protein. Yet, nucleotides were bound in only one of the three crystal forms we examined. The three different crystallization conditions were similar in important variables such as buffer (cacodylate), pH (7.4 to 7.6) and ionic strength (100 to 300 mM). However, one crystal contained unliganded PyrR, one was a Mg2+-sulfate complex, and the third was a Mg2+-nucleotide complex. Our working hypothesis, developed from the structural results, is that Mg2+ binds synergistically with an anion in the 5′-phosphate site and does not require a cation-proximal phosphate (or sulfate). In crystallizations leading to the unliganded structure, concentrations of UMP (1 mM) and Mg2+ (2 mM) may have been too low for detectable binding. In crystallizations leading to the Mg2+-sulfate complex, 100 mM sulfate may have competed with 1 mM UMP and also induced Mg2+ (1 mM) binding. We attribute successful crystallization of the nucleotide complex to the significantly higher concentrations of Mg2+ (40 mM) in these experiments. The synergistic binding hypothesis is consistent with our observation that the ribose groups contribute little to nucleotide binding. This is evident in the poor ordering of the ribose groups, the lack of Mg2+ coordination by both the 2′- and 3′-hydroxyl groups, and the lack of hydrogen bonds from ribose hydroxyls to the dual carboxylate side chains.
The conundrum of the structures is the lack of observable conformational changes in the protein upon binding of nucleotides that are known to affect affinity for RNA. We assume this is a consequence of the extremely high protein concentration in crystals (∼750 mg/ml), which drives the protein to its most aggregated quaternary structure and may mask nucleotide-induced conformational changes. Nevertheless, important new information comes from the structures. The unusual binding of divalent cation and the unexpected binding of GMP raise the possibility that these ligands may have a physiological role and may influence RNA binding. Binding of GMP to one subunit in a mode nearly identical to UMP binding in the other subunit is consistent with the equilibrium binding results in which saturation was reached with UMP bound to only about half of the subunits. The experiments presented here confirm our previous conclusion that uridine nucleotides greatly stimulate PyrR binding to pyr RNA (3), but the antagonistic effect of uridine nucleotides and guanosine nucleotides on RNA binding was not previously observed. As shown in Fig. 5B, the RNA binding curve could be shifted over a very broad range by varying the UMP/GMP ratio. This echoes observations on the allosteric regulation of B. subtilis carbamyl phosphate synthetase, the pyrimidine-specific isozyme encoded by pyrAA and pyrAB, which is inhibited by UMP and UTP and activated by GMP, GTP, and PRPP (20). The activation by guanine nucleotides provides a mechanism for maintaining the proper balance of purine and pyrimidine nucleotides. GMP inhibition of RNA binding by PyrR suggests that the purine-pyrimidine balance in Bacillus may also be regulated at the level of transcription.
We cannot determine from the data in hand whether UMP-GMP antagonism results from competition for binding at the same sites, or if the UMP-GMP ternary complex is important. Although we do not observe differences in the conformation of nucleotide-bound and -free PyrR and the UMP- and GMP-binding mode appears nearly identical, the conformational consequences of UMP and GMP binding must be different.
There are some discrepancies between the observations of nucleotide effects on RNA binding obtained by the electrophoretic mobility shift method previously used to study binding to B. subtilis PyrR and initial studies with B. caldolyticus PyrR and the filter-binding method (Fig. 5). Using B. caldolyticus PyrR, the apparent dissociation constants obtained with the gel shift method were 50- to 250-fold lower than those observed in filter-binding experiments. Both methods revealed the ability of uridine nucleotides to increase the affinity of PyrR for RNA, but the antagonistic effects of guanosine nucleotides on RNA binding to B. subtilis PyrR were not observed by the gel shift method (3). The reasons for these differences are not understood. However, we believe that the new finding of the effect of guanosine nucleotides is reliable and likely to be physiologically significant. The effects are specific, are highly reproducible with each RNA tested, and occur at low, physiologically relevant concentrations (10 to 500 μM) of the nucleotides. They mirror the specific binding of UMP and GMP to PyrR observed in the crystal structure. The filter-binding method uses conditions closer to true equilibrium binding than those occurring during electrophoretic mobility shift analyses. A more detailed study of the binding of RNA to B. caldolyticus PyrR and the effects of metabolites and divalent cations on RNA binding by different methods is under way in our laboratory. (The antagonistic effect of GMP on PyrR-RNA binding has been confirmed by the electrophoretic mobility shift method.)
PyrR from B. caldolyticus, B. subtilis, T. thermophilus, and M. tuberculosis form identical dimers with identically concave, basic surfaces, presumably the RNA-binding sites (Fig. 6). The PyrR proteins from B. caldolyticus and B. subtilis have a common biological activity, a similar RNA-binding specificity, and 73% identical sequences. Thus, they are expected to have a common molecular mechanism and a common positively charged surface to bind RNA. Genetic experiments with Thermus ZO5, a close relative of T. thermophilus, suggest that this homologue also binds to an RNA similar to that recognized by the Bacillus PyrR proteins (28). The surface charge distributions are similar for the common PyrR dimer from B. caldolyticus (Fig. 6A), B. subtilis (Fig. 6B), T. thermophilus (Fig. 6C), and M. tuberculosis (Fig. 6D). One surface is concave and basic (Fig. 6, right), while the opposite surface is convex and acidic or neutral (Fig. 6, left). Nucleotides bind to the PRTase active sites on the convex face of PyrR (Fig. 6A, left). The common basic surface is the presumed RNA-binding surface, consistent with site-directed mutagenesis results with B. subtilis PyrR (21). However, this surface faces an internal cavity in the B. caldolyticus and M. tuberculosis PyrR tetramers and in the B. subtilis PyrR hexamer, whereas the external surfaces of tetramers and hexamer are negatively charged. Furthermore, none of the internal cavities is large enough to accommodate RNA. The PyrR homologue from T. thermophilus (PDB code 1UFR) is a tetramer, but its basic surface is external. Taken together, the existence of a common dimer structure, the conserved electropositive surface of the dimer, and mutagenesis results with B. subtilis PyrR (21) indicate that PyrR binds pyr RNA as a dimer, not as a tetramer or hexamer. Clearly, a high-resolution structure of the PyrR-RNA complex would greatly advance understanding of the nature of RNA binding and possibly also of the role of nucleotides in modulating the affinity of PyrR for RNA.
FIG. 6.

Electrostatic surface potential of PyrR dimers from B. caldolyticus (PDB 1NON) (A), B. subtilis (1A3C) (B), T. thermophilus (1UFR) (C), and M. tuberculosis (1W30) (D). The images on the left are oriented identically and show the convex, negatively charged surfaces. Nucleotides (green) are shown in the B. caldolyticus PyrR structure. Images on the right show the views from the concave, positively charged side, which is the presumed RNA-binding surface. The figure was made using GRASP (18) and PyMOL (6). Color coding is according to the electrostatic potential of the molecular surface, ranging from approximately −9 kT (red) to +9 kT (blue).
Acknowledgments
J.K.M. was a Howard Hughes Undergraduate Research Fellow at the University of Illinois during the summer of 2001.
National Institutes of Health grants GM47112 to R.L.S. and DK42303 to J.L.S. supported this work.
We thank Eric Bonner for assistance with the preparation of radiolabeled pyr RNA and with gel mobility shift analysis of RNA binding to PyrR, Mark Lies for assistance with HPLC analysis, and Kara Weiss for assistance with differential calorimetry.
REFERENCES
- 1.Babitzke, P., J. Schaak, A. V. Yakhnin, and P. C. Bevilacqua. 2003. Role of RNA structure in transcription attenuation in Bacillus subtilis: the trpEDCFBA operon as a model system. Methods Enzymol. 371:392-404. [DOI] [PubMed] [Google Scholar]
- 2.Bera, A. K., J. Zhu, H. Zalkin, and J. L. Smith. 2003. Functional dissection of the Bacillus subtilis pur operator site. J. Bacteriol. 185:4099-4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonner, E. R., J. N. D'Elia, B. K. Billips, and R. L. Switzer. 2001. Molecular recognition of pyr mRNA by the Bacillus subtilis attenuation regulatory protein PyrR. Nucleic Acids Res. 29:4851-4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brunger, A. T., P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J. S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson, and G. L. Warren. 1998. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D 54:905-921. [DOI] [PubMed] [Google Scholar]
- 5.Chen, S., D. R. Tomchick, D. Wolle, P. Hu, J. L. Smith, R. L. Switzer, and H. Zalkin. 1997. Mechanism of the synergistic end-product regulation of Bacillus subtilis glutamine phosphoribosylpyrophosphate amidotransferase by nucleotides. Biochemistry 36:10718-10726. [DOI] [PubMed] [Google Scholar]
- 6.DeLano, W. L. The PyMOL Molecular Graphics system. DeLano Scientific LLC, San Carlos, Calif. [Online.] http://www.pymol.org.
- 7.Ghim, S.-Y., and J. Neuhard. 1994. The pyrimidine biosynthesis operon of the thermophile Bacillus caldolyticus includes genes for uracil phosphoribosyltransferase and uracil permease. J. Bacteriol. 176:3698-3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghim, S.-Y., P. Nielsen, and J. Neuhard. 1994. Molecular characterization of pyrimidine biosynthesis genes from the thermophile Bacillus caldolyticus. J. Gen. Microbiol. 140:479-491. [DOI] [PubMed] [Google Scholar]
- 9.Grabner, G. K., and R. L. Switzer. 2003. Kinetic studies of the uracil phosphoribosyltransferase reaction catalyzed by the Bacillus subtilis pyrimidine attenuation regulatory protein PyrR. J. Biol. Chem. 278:6921-6927. [DOI] [PubMed] [Google Scholar]
- 10.Jensen, H. K., N. Mikkelsen, and J. Neuhard. 1997. Recombinant uracil phosphoribosyltransferase from the thermophile Bacillus caldolyticus: expression, purification, and partial characterization. Protein Expr. Purif. 10:356-364. [DOI] [PubMed] [Google Scholar]
- 11.Krahn, J. M., J. H. Kim, M. R. Burns, R. R. Parry, H. Zalkin, and J. L. Smith. 1997. Coupled formation of an amidotransferase interdomain ammonia channel and a phosphoribosyltransferase active site. Biochemistry 36:11061-11068. [DOI] [PubMed] [Google Scholar]
- 12.Kraulis, P. 1991. MolScript. A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24:946-950. [Google Scholar]
- 13.Lu, Y., and R. L. Switzer. 1996. Transcriptional attenuation of the Bacillus subtilis pyr operon by the PyrR regulatory protein and uridine nucleotides in vitro. J. Bacteriol. 178:7206-7211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martinussen, J., P. Glaser, P. S. Andersen, and H. H. Saxild. 1995. Two genes encoding uracil phosphoribosyltransferase are present in Bacillus subtilis. J. Bacteriol. 177:271-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Merritt, E. A., and D. J. Bacon. 1997. Raster3D: photorealistic molecular graphics. Methods Enzymol. 277:505-524. [DOI] [PubMed] [Google Scholar]
- 16.Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 17.Navaza, J. 1994. AMoRe—an automated package for molecular replacement. Acta Crystallogr. A 50:157-163. [Google Scholar]
- 18.Nicholls, A., K. A. Sharp, and B. Honig. 1991. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11:281-296. [DOI] [PubMed] [Google Scholar]
- 19.Otwinowski, Z., and W. Minor. 1997. Processing of X-ray diffraction data collected in oscillation mode. Macromol. Crystallogr. A 276:307-326. [DOI] [PubMed] [Google Scholar]
- 20.Paulus, T. J., and R. L. Switzer. 1979. Characterization of pyrimidine-repressible and arginine-repressible carbamyl phosphate synthetases from Bacillus subtilis. J. Bacteriol 137:82-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Savacool, H. K., and R. L. Switzer. 2002. Characterization of the interaction of Bacillus subtilis PyrR with pyr mRNA by site-directed mutagenesis of the protein. J. Bacteriol. 184:2521-2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sinha, S. C., and J. L. Smith. 2001. The PRT protein family. Curr. Opin. Struct. Biol. 11:733-739. [DOI] [PubMed] [Google Scholar]
- 23.Smith, J. L., E. J. Zaluzec, J. P. Wary, L. Niu, R. L. Switzer, H. Zalkin, and Y. Satow. 1994. Structure of the allosteric regulatory enzyme of purine biosynthesis. Science 264:1427-1433. [DOI] [PubMed] [Google Scholar]
- 24.Studier, F. W., and B. A. Moffatt. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189:113-130. [DOI] [PubMed] [Google Scholar]
- 25.Switzer, R. L., R. J. Turner, and Y. Lu. 1999. Regulation of the Bacillus subtilis pyrimidine biosynthetic operon by transcriptional attenuation: control of gene expression by an mRNA-binding protein. Prog. Nucleic Acids Res. Mol. Biol. 62:329-367. [DOI] [PubMed] [Google Scholar]
- 26.Tomchick, D. R., R. J. Turner, R. L. Switzer, and J. L. Smith. 1998. Adaptation of an enzyme to regulatory function: structure of Bacillus subtilis PyrR, a bifunctional pyr RNA-binding attenuation protein and uracil phosphoribosyltransferase. Structure 6:337-350. [DOI] [PubMed] [Google Scholar]
- 27.Turner, R. J., E. R. Bonner, G. K. Grabner, and R. L. Switzer. 1998. Purification and characterization of Bacillus subtilis PyrR, a bifunctional pyr mRNA-binding attenuation protein/uracil phosphoribosyltransferase. J. Biol. Chem. 273:5932-5938. [DOI] [PubMed] [Google Scholar]
- 28.Van de Casteele, M., P. Chen, M. Roovers, C. Legrain, and N. Glansdorff. 1997. Structure and expression of a pyrimidine gene cluster from the extreme thermophile Thermus strain ZO5. J. Bacteriol. 179:3470-3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong, I., and T. M. Lohman. 1993. A double-filter method for nitrocellulose-filter binding: application to protein-nucleic acid interactions. Proc. Natl. Acad. Sci. USA 90:5428-5432. [DOI] [PMC free article] [PubMed] [Google Scholar]