

Abstract
Four genes that encode the homologues of plant geranylgeranyl reductase were isolated from a hyperthermophilic archaeon Archaeoglobus fulgidus, which produces menaquinone with a fully saturated heptaprenyl side chain, menaquinone-7(14H). The recombinant expression of one of the homologues in Escherichia coli led to a distinct change in the quinone profile of the host cells, although the homologue is the most distantly related to the geranylgeranyl reductase. The new compounds found in the profile had successively longer elution times than those of ordinary quinones from E. coli, i.e., menaquinone-8 and ubiquinone-8, in high-performance liquid chromatography on a reversed-phase column. Structural analyses of the new compounds by electron impact-mass spectrometry indicated that their molecular masses progressively increase relative to the ordinary quinones at a rate of 2 U but that they still contain quinone head structures, strongly suggesting that the compounds are quinones with partially saturated prenyl side chains. In vitro assays with dithionite as the reducing agent showed that the prenyl reductase is highly specific for menaquinone-7, rather than ubiquinone-8 and prenyl diphosphates. This novel enzyme noncovalently binds flavin adenine dinucleotide, similar to geranylgeranyl reductase, but was not able to utilize NAD(P)H as the electron donor, unlike the plant homologue.
Saturated prenyl groups are widely found in archaea (6, 10). The most characteristic example is the membrane lipids of all archaea. The archaeal lipids contain phytanyl groups (or dimerized phytanyl chains in case of tetraether lipids) as their hydrocarbon moieties, whereas those from other organisms possess acyl groups, and their characteristic structures could serve as an aid in distinguishing these organisms from others (Fig. 1A). Diphytanyl glycerol phosphate, a typical type of the membrane lipid, has also been reported to be utilized in the modification of the cell surface glycoprotein of Halobacterium halobium (13). In addition, many respiratory quinones in archaea, i.e., menaquinone (MK), caldariellaquinone, sulfolobusquinone, thermoplasmaquinone, etc., also contain fully or partially saturated polyprenyl chains (Fig. 1B). Such features, i.e., saturated prenyl moieties, found in the archaeal isoprenoid compounds are considered to be very important for their biological functions because the saturation of double bonds in the hydrocarbon chains would confer chemical and biological stability on the lipoid compounds and would have a significant effect on lipid-lipid interactions in the membrane. The former effect appears to be important especially for archaea, which generally inhabit extreme environments.
FIG. 1.

Archaeal isoprenoid compounds with saturated prenyl chains. (A) Typical structure of a “diether” archaeal membrane lipid. X represents a polar head group. (B) Structure of respiratory quinone from A. fulgidus, MK-7(14H).
The saturation of prenyl moieties is not process specific for archaea. The process also occurs in other organisms, although it takes place on a relatively limited biomolecules, compared to the archaea. For example, phylloquinones (vitamin K1) in plants have a phytyl group as its side chain. MKs from a small number of bacterial species contain partially saturated prenyl side chains, although those from other bacteria usually contain nonsaturated ones (5). Chlorophylls and bacteriochlorophylls, which are produced in plants and photosynthetic bacteria, respectively, generally possess a phytyl group as well. Tocopherols, which are also produced by plants, possess a prenyl moiety that are saturated and partially cyclized. Saturated prenyl moieties are also found in dolichol, cholesterol, vitamin D3, etc. During the past several years, the genes responsible for the reductive conversion of a geranylgeranyl group into a phytyl group have been identified in some higher plants and bacteria. The chlP genes from plants (12, 17) and cyanobacteria (1) encode multifunctional geranylgeranyl reductase (GGR), which is involved in the biosyntheses of chlorophyll, tocopherol, and probably phyloquinone, whereas the bacterial homologue gene, bchP, encodes an enzyme that catalyzes the saturation of the geranylgeranyl moiety in bacteriochlorophyll and/or bacteriopheophytin (2, 3). The amino acid sequences of these reductases have a high similarity with those of FAD/NAD(P)H-dependent oxidoreductases, and these coenzymes have been shown to be sufficient for the activity of GGR from Arabidopsis thaliana (12).
In the present study, we report the isolation of four genes encoding homologues of GGR from an anaerobic, hyperthermophilic archaeon, Archaeoglobus fulgidus, which is known to produce MK-7 with a fully saturated heptaprenyl side chain, i.e., MK-7(14H) (18) (Fig. 1B), and the expression of each of them in Escherichia coli. As a result, E. coli expressing one of the GGR homologous, although it is the most distantly related to GGR among the four homologues, produced respiratory quinones with partially saturated prenyl side chains. In vitro assays with an affinity-purified recombinant enzyme showed that the enzyme specifically reduced the prenyl side chain of MK, whereas the prenyl moieties of ubiquinone (UQ) and prenyl diphosphates were relatively resistant to reduction. The prenyl reductase (PR) appeared to contain noncovalently bonded flavin adenine dinucleotide (FAD), similar to A. thaliana GGR. However, in contrast to the plant homologue, the enzyme requires sodium dithionite for activity and might be highly oxygen sensitive.
MATERIALS AND METHODS
Materials.
MK-7 and (All-E) geranylgeranyl diphosphate (GGPP) were donated by K. Ogura and T. Koyama, Tohoku University. UQ-8 was donated by M. Tsujii, Eisai Chemical Co., Ltd. Isopentenyl diphosphate (IPP) and dimethylallyl diphosphate were donated by C. Ohto, Toyota Motor Co. [1-14C]IPP was purchased from Amersham Biosciences. Precoated reversed-phase thin-layer chromatography (TLC) plates (LKC-18F) were purchased from Whatman Chemical Separation, Inc. Cellulose TLC plates (aluminum sheets) were purchased from Merck. All other chemicals were of analytical grade.
General procedures.
Restriction enzyme digestions, transformations, and other standard molecular biology techniques were carried out as described by Sambrook et al. (16).
Cultivation of archaea.
A. fulgidus Stetter DSM 4304 was anaerobically cultured in ATCC 1775 medium at 80°C for 120 h and harvested to prepare genomic DNA.
Isolation of the genes of GGR homologues from A. fulgidus.
A homologous search of the entire genome sequence of A. fulgidus was conducted with the webserver of the Microbial Genome Database (MBGD; http://mbgd.genome.ad.jp/) with the amino acid sequence of A. thaliana GGR as a probe. Using the GENETYX-MAC software (GENETYX Co.), the similarities and identities between the amino acid sequences of the proteins were obtained, and hydrophobicity analyses of the proteins were conducted. Multiple alignment of the amino acid sequences was conducted by using the CLUSTAL W program on the EBI website (http://www.ebi.ac.uk/clustalw/). All parameters used in the program were at the default settings. The open reading frames (ORFs) encoding the four GGR homologues searched were amplified by PCR with the following primers: AF0464, 5′-GATGACATATGCACCGGCTGCGGAATC-3′ and 5′-ATTAAGGATCCTTAGAAAAGGTCCTTGAGGTC-3′; AF1637, 5′-TTCCACATATGAAGTTTGGTGAAAACTTTAAAA-3′ and 5′-TAAATGGATCCTCAGATTAGCTTCTGGACTATTG-3′; AF1023, 5′-GGCAGCATATGGATGCTGCAGTAGTCGG-3′ and 5′-TTGATGGATCCTTAGAGAAACAGGCTCTTCAC-3′; and AF0648, 5′-CAGTCCATATGATTCAGATTTACGGCGC-3′ and 5′-CTTGAGGATCCTCATAGCCCTATGAGCTTC-3′. The genome of A. fulgidus, as the template, and EX Taq DNA polymerase (TaKaRa) were used for the reaction. The restriction sites that were introduced in the forward and reverse primers, the NdeI and BamHI sites, respectively, are indicated by underlining. The amplified fragments, extracted from a 0.8% agarose gel after electrophoresis, were digested with NdeI and BamHI and then ligated into the NdeI-BamHI sites of the pET-3a vector (Novagen).
Expression and purification of the recombinant enzyme.
E. coli BL21(DE3) transformed with each of the resultant plasmids was aerobically cultivated in Luria-Bertani broth supplemented with 50 mg of ampicillin/liter. When required, the media were not shaken during the cultivation to create semianaerobic conditions. When the optical density at 600 nm of the culture reached 0.6, the transformed bacteria were induced by treatment with 1.0 mM IPTG (isopropyl-β-d-thiogalactopyranoside). After 4 h of additional cultivation, the cells were harvested and disrupted by sonication in 50 mM morpholinepropanesulfonic acid-KOH buffer (pH 7.5). The homogenate was centrifuged at 15,000 × g for 15 min, and the supernatant was recovered as a crude extract. The crude extract was heated at 55°C for 1 h, and the denatured proteins were removed by centrifugation at 15,000 × g for 15 min. The supernatant fraction was recovered as a heat-treated enzyme. The level of protein expression was determined by 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
Quinone profile analysis.
Using the method of Bligh and Dyer (4), total lipids were extracted from the transformed E. coli cultured, as described above. A total of 29 volumes of acetone were added to the lipid extract, and the mixture was allowed to stand at 4°C for 3 h. After centrifugation, the supernatant was recovered and then evaporated to dryness. The residual lipid was dissolved in n-pentane, and partitioned against the same volume of water. The upper n-pentane layer was recovered and evaporated to dryness, and the resulting residual lipid was dissolved with 2-propanol for use in high-performance liquid chromatography (HPLC) analyses. The sample was loaded on a YMC Pack ODS-A column (4.6 by 250 mm, 5 μm) and isocratically eluted with 0.8 ml of 2-propanol-methanol (3:2)/min. The elution of respiratory quinones was detected at 243 nm, the absorption maximum of MKs, and all peaks were fractionated for analysis by electron impact-mass spectrometry (EI-MS).
Cloning of a gene encoding prenyl diphosphate synthase from A. fulgidus.
The ORF AF1551, encoding a protein annotated as (all-E) octaprenyl diphosphate synthase, was amplified by PCR with the primers 5′-GAACCCATATGATTGATACCTGGG-3′ and 5′-GTTCAGGATCCTCAGAAAATAGCC-3′, and the genome of A. fulgidus as the template. The newly introduced recognition sites for NdeI and BamHI, respectively, are underlined. The amplified DNA fragment was digested at the sites and then inserted into the pET-3a vector. The resulting plasmid was introduced in E. coli BL21(DE3), and the transformed E. coli was cultivated in Luria-Bertani broth containing 50 mg of ampicillin/liter and then induced by treatment with IPTG. The cells were harvested and disrupted, and the enzyme solution was heat treated according to the same procedures used for the GGR homologues.
Preparation of hypothetical substrates for A. fulgidus PR.
The reaction mixture for the production of 14C-labeled (all-E) polyprenyl diphosphate contained, in a final volume of 1 ml, 25 nmol of [1-14C]IPP, 10 nmol of GGPP, 50 μmol of MOPS-KOH buffer (pH 7.5), 5 μmol of MgCl2, and 50 μl of the heat-treated enzyme solution containing the hypothetical prenyltransferase from A. fulgidus. The mixture was incubated at 55°C for 30 min, and the reaction was stopped by adding 200 μl of a cold, saturated solution of NaCl. The products extracted with 600 μl of 1-butanol saturated with H2O were concentrated and used as the substrates for PR. The products were analyzed by reversed-phase TLC, with LKC-18F (Whatman) developed with acetone-H2O (19:1), after a phosphatase treatment as described by Fujii et al. (9). To synthesize [14C]GGPP, we replaced the enzyme with purified Sulfolobus acidocaldarius GGPP synthase prepared as described in our earlier study (15). At the same time, the buffer and allylic substrate were replaced with potassium phosphate (pH 5.8) and dimethylally diphosphate, respectively.
Affinity purification of PR.
The NdeI-BamHI fragment containing the ORF AF0648 was inserted into the pET-15b vector (Novagen). E. coli BL21(DE3) transformed with the resultant plasmid was cultivated in 50 ml of Luria-Bertani broth supplemented with ampicillin. Induction with 1 mM IPTG was conducted when the optical density at 600 nm of the culture reached 0.5. After an additional 6-h cultivation, the cells were harvested and disrupted by sonication in HisTrap (Amersham Biosciences) binding buffer containing 20 mM potassium phosphate (pH 7.6), 0.5 M NaCl, and 10 mM imidazole and prepared according to the manufacturer's instructions. The homogenate was centrifuged at 4,000 × g for 15 min, and the supernatant was recovered as a crude extract. The crude extract, after filtration through a 0.45-μm membrane, was loaded on a HisTrap column, which had been equilibrated with HisTrap binding buffer. The column was washed with the binding buffer, and specifically bound proteins were then eluted with HisTrap elution buffer containing 20 mM potassium phosphate buffer (pH 7.6), 0.5 M NaCl, and 350 mM imidazole and used for characterization as purified PR. The level of purification was determined by SDS-14% PAGE. UV-visible analysis of the purified PR solution (containing 0.59 μg of the enzyme/μl) was conducted with a U-2000 spectrophotometer (Hitachi).
Flavin analysis.
We concentrated 250 μl of the purified PR solution into ca. 100 μl with a Microcon YM-10 spin filter (Millipore), replacing the buffer with water. To the concentrated enzyme solution, 1 ml of methanol was added. The mixture was heated at 100°C for 15 min and then centrifuged at 20,000 × g for 10 min. The recovered supernatant was evaporated to ca. 50 μl, and a 2-μl aliquot was analyzed by TLC with a cellulose plate (an aluminum sheet [Merck]) developed with n-butanol-methanol-5% Na2HPO4 (60:15:30). Authentic flavin mononucleotide (FMN) and FAD were chromatographed on the same plate. Spots corresponding to flavins on the plate were detected by UV illumination.
In vitro assay of PR.
All manipulations for the in vitro assay were carried out in an anaerobic chamber until the reaction ended. The standard reaction mixture contained, in a volume of 150 μl, 10 nmol of MK-7, 0.13% Triton X-100, 50 μmol of morpholinepropanesulfonic acid-KOH buffer (pH 7.5), and 30 μg of purified PR. The stock solutions of the contents, except for the enzyme and detergent, were bubbled with N2 gas to remove oxygen. To the mixture, 50 μmol of Na2S2O4 (sodium dithionite) dissolved in 50 μl of N2-bubbled water was added. The mixture was then incubated at 55°C for 1 h, and the reaction was stopped by adding 200 μl of cold, saturated solution of NaCl. The mixture was extracted with 600 μl of 1-butanol saturated with H2O. The butanol layer was evaporated and partitioned between 4 ml each of n-pentane and H2O. The n-pentane layer was recovered and evaporated to dryness. The residual lipid was dissolved in 100 μl of 2-propanol and used for HPLC analyses with the apparatus used for the quinone profile analysis. To prepare the products for analysis by MS, a reaction mixture with five times the volume of the assay mixture and a YMC Pack ODS-A column (10 by 250 mm, 5 μm), isocratically eluted with 0.5 ml of 2-propanol-methanol (3:2)/min, were used. To determine the product specificity of PR, the substrate was replaced with 10 nmol of UQ-8, 10 nmol of GGPP, 0.1 nmol of [14C]GGPP, or 0.1 mol of [14C]polyprenyl diphosphate (a 7:3 mixture of heptaprenyl diphosphate and octaprenyl diphosphate).
MS.
EI-MS analyses of the concentrated lipid fractions were performed with an MStation JMS-700 MS system (JEOL) in the positive ion mode.
RESULTS
Isolation of archaeal genes encoding homologues of GGR.
As the result of a search for the homologues of A. thaliana GGR using the web server of the MBGD (http://mbgd.genome.ad.jp/), we found that three homologues of GGR (included in homologous ORF cluster O1668) and one hypothetical protein related to them to a certain extent (cluster O6510) were encoded in the genome of A. fulgidus. The homologues encoded in the ORFs, AF0464 (named chlP-1), AF1023 (chlP-2), and AF1637 (chlP-3), have sequence similarities of 62, 62, and 41% (23, 19, and 7% identities), respectively, with A. thaliana GGR. On the other hand, the protein encoded in AF0648 has no detectable whole-length similarity with GGR and the former two GGR homologues, as well as other enzymes whose functions have been confirmed, but has a 48% sequence similarity (8% identity) with the protein encoded in AF1637. Multiple alignment of the amino acid sequences of the four archaeal proteins and GGRs from A. thaliana and Synechocystis sp. strain PCC 6803 shows that some highly conserved amino acids among the GGRs and their homologues are also shared by the AF0648 protein, indicating AF0648 encodes a distantly related homologue of GGR (Fig. 2). The four ORFs from A. fulgidus were amplified by PCR and ligated with the pET-3a vector. E. coli cells were transformed with the resultant plasmids, and the expression levels of the recombinant proteins were verified by SDS-PAGE. As shown in Fig. 3, a strong protein band was observed in the crude extract, even after the heat treatment, from E. coli that had been transformed with the vector for the expression of AF0464. On the other hand, overexpressed protein bands were found in the precipitate fractions from E. coli harboring the vector for AF1637 or AF1023, suggesting that proteins encoded in the ORFs were expressed as inclusion bodies. In the samples from E. coli expressing AF0648, weak but distinct protein bands were found both in the precipitate fraction and in the heated enzyme solution. The molecular masses of the expressed proteins, deduced from the SDS-PAGE analysis, were consistent with those estimated from their DNA sequences, whereas an additional smaller band, which might have arisen from the degradation of the expressed protein, was found in the precipitate fraction from E. coli expressing AF1637.
FIG. 2.

Alignment of GGRs and their homologues from A. fulgidus. SynChlP, GGR from Synechocystis sp. strain PCC 6803; AtGGR, GGR from A. thaliana. Af1023 (chlP-2), Af0464 (chlP-1), Af1637 (chlP-3), and Af0648 are the GGR homologues from A. fulgidus.
FIG. 3.

Recombinant expression of GGR homologues. Lanes 1, 7, 13, and 19, molecular standard marker; lanes 2 to 6, precipitated fractions after the cell disruption of E. coli BL21(DE3)/pET3a-AF0464, BL21(DE3)/pET3a-AF1637, BL21(DE3)/pET3a-AF1023, BL21(DE3)/pET3a-AF0648, and BL21(DE3)/pET3a, respectively; lanes 8 to 12, crude extracts from E. coli strains are shown above, in the same order; lanes 14 to 18, heat-treated enzyme from each strain, in the same order. Asterisks indicate the bands of highly expressed recombinant enzymes, whose molecular masses, as estimated from their mobility, correspond well to those calculated from their amino acid sequences. Double asterisks indicate a band that would arise from the partial degradation of the recombinant protein.
Quinone profiles of E. coli expressing GGR homologues.
We next analyzed the quinone profiles of E. coli cells expressing the archaeal GGR homologues to verify the in vivo activities of the recombinant proteins. If the proteins catalyze the saturation of double bonds at the prenyl chain of respiratory quinones, they would be expected to affect the quinone profiles of the host cells. Respiratory quinones of E. coli were extracted from the cells and analyzed by HPLC with a C18 reversed-phase column. As reported previously, E. coli cells harboring the pET-3a plasmid were shown to produce UQ-8 and MK-8 and, probably, demethyl MK-8 (Fig. 4A). In contrast, the quinone profile of cells harboring the ORF AF0648, shown in Fig. 4B, was totally different from those of the other transformants, whereas those of E. coli expressing the other homologues of GGR encoded in A. fulgidus chlP genes were almost identical to that in Fig. 4A. Several peaks that eluted sequentially and more slowly than that of MK-8 were observed in the profile. A slower elution indicates that the new compounds are more hydrophobic than MK-8, suggesting that the double bonds in the prenyl side chain of MK-8 have been saturated.
FIG. 4.
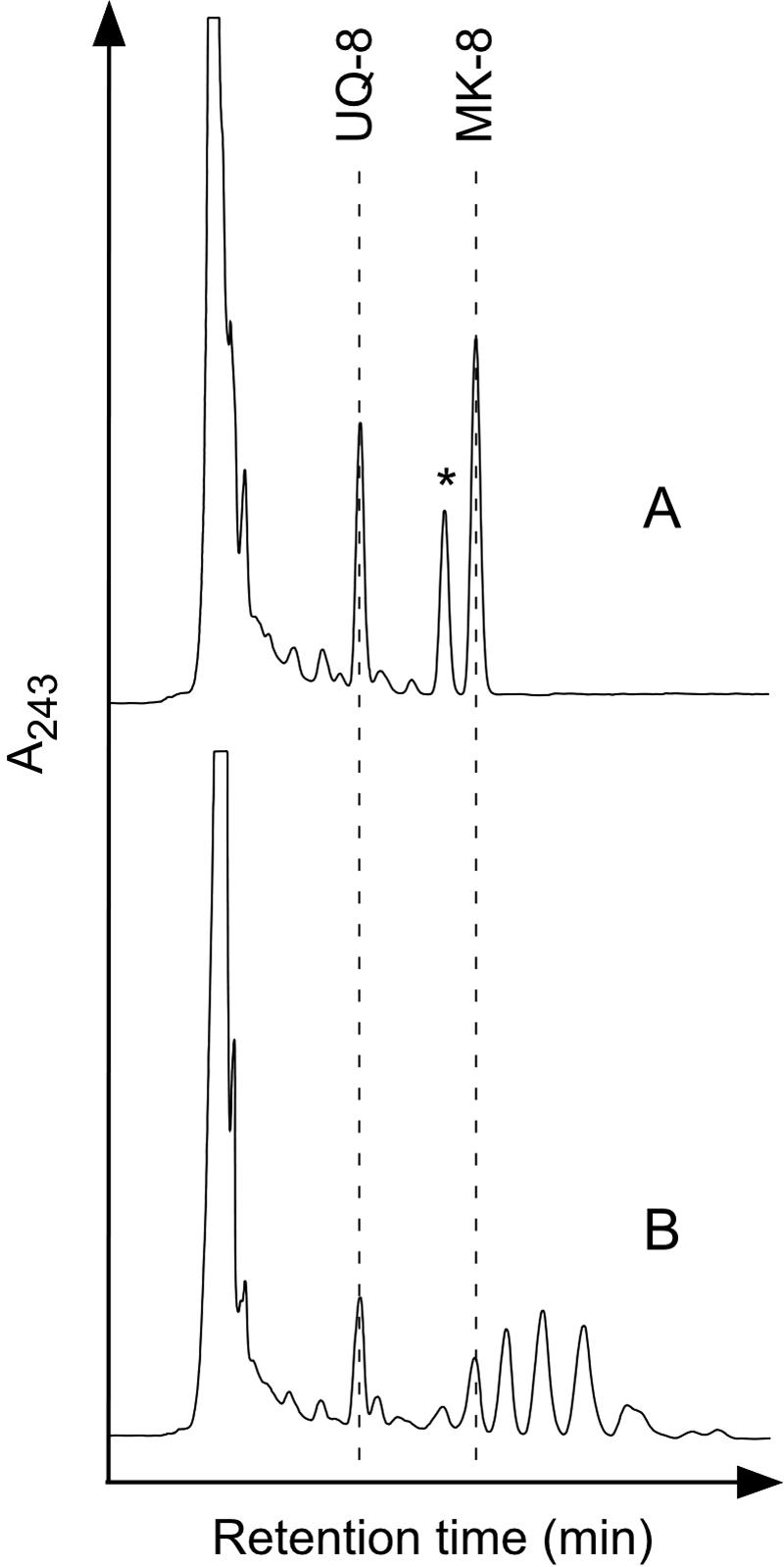
Quinone profiles of E. coli expressing GGR homologues. Respiratory quinones extracted from BL21(DE3)/pET3a (A) and BL21(DE3)/pET3a-AF0648 (B) were analyzed by HPLC. An asterisk indicates the probable peak of demethyl MK-8.
Thus, we conducted EI-MS analyses of the compounds that coeluted with the HPLC peaks to confirm whether they are, in fact, derivatives of MK-8, which would contain saturated prenyl side chains. To obtain large amounts of the MK-8 derivatives, host cells were cultivated under semianaerobic conditions, which are known to cause E. coli to increase the production of MK-8 and reduce UQ production, without shaking the flask. The HPLC elution profile of the extracted quinones is shown in Fig. 5. In addition to the appearance of MK-8 derivatives that were more hydrophobic than those observed in the quinone profile of aerobically cultivated E. coli, several peaks successively eluted immediately after the UQ-8 peak, which were expected to arise from UQ-8 derivatives as well, were observed. The compounds corresponding to the peaks numbered from 1 to 10 in Fig. 5 were analyzed by EI-MS (data not shown). The lower intensity of ions and contamination by other unidentified compounds did not allow us to assign the other peaks. In the analysis of peak 1, whose elution time was the same as that of authentic UQ-8, the m/z value of the largest ion, 726, also corresponds to that of UQ-8. In the spectra of peaks 2 and 3, m/z values of 728 and 730 ions were observed, respectively. In all of these three spectra, an ion with an m/z of 235 was present, which corresponds to the fragment ion (Fig. 6A), which has been reported to be typical in the EI-MS analysis of UQ (13). These facts strongly suggest that peak 1 arose from UQ-8 and that peaks 2 and 3 represented derivatives thereof with molecular weights that were increased by 2 and 4 U, respectively. The increase in the molecular weight should arise from the octaprenyl side chain because UQ-8 and derivatives thereof must share the same quinone head structure. Thus, we conclude that the derivatives are UQ-8(2H) and UQ-8(4H), which are produced by the saturation of one and two double bonds in the side chains, respectively. The generation of the commonly observed fragment ion of m/z 235 suggests that the double bond at the α-end of the octaprenyl group remains unsaturated and that the other double bonds might be saturated. However, the possibility that a peak in the HPLC analysis includes multiple derivatives, in which double bonds of the same number, but at different positions (including the α-end) have been saturated, could not be excluded. Thus, the positions of the double bonds that are saturated are unclear. In the EI-MS analyses of peaks 4 to 10, with ions of m/z 720, 722, 724, 724, 726, 726, and 728, respectively, were present, whereas an ion with an m/z of 716 was observed in the analysis of MK-8 extracted from wild-type E. coli. Moreover, an ion of m/z 225 was also commonly observed in these spectra. This ion, which is typical in the EI-MS analysis of MK (8), can be derived from the fragmentation indicated in Fig. 6B. These data strongly suggest that the peaks arise from the MK derivatives with partially saturated side chains, i.e., MK-8(4H), MK-8(6H), MK-8(6H), MK-8(8H), MK-8(8H), and MK-8(10H), respectively. Thus, we conclude that the archaeal GGR homologue encoded in AF0648 has activity for reducing longer-chain prenyl groups in respiratory quinones, and we designated it PR. It is surprising that PR can act on such hydrophobic compounds, which is considered to be incorporated into the lipid bilayer of the membrane, because the enzyme is predicted to have no transmembrane segments, as evidenced by a hydrophobicity analysis (data not shown).
FIG. 5.

HPLC profile of respiratory quinones newly synthesized in vivo. Respiratory quinones were extracted from BL21(DE3)/pET15b-AF0648 cultivated semianaerobically. Numbered peaks were recovered for analysis by EI-MS.
FIG. 6.

Supposed fragmentation of the derivatives of UQ-8 (A) and MK-8 (B) in EI-MS analyses.
Affinity purification and characterization of A. fulgidus PR.
To elucidate the properties of A. fulgidus PR, we reconstructed an expression vector to confer a polyhistidine tag on the enzyme. Recombinant PR was purified from a crude extract from the transformed E. coli by affinity column chromatography (Fig. 7). The UV-visible spectrum of the concentrated, purified enzyme solution is shown in Fig. 8. In this spectrum, a peak at ca. 440 nm is present, which is specific for flavin coenzymes. GGRs from A. thaliana and Rhodobacter sphaeroides would be expected to bind FAD because they have highly conserved motifs for FAD-binding (2, 12), but their specific requirements have not been confirmed yet. The UV-visible spectrum of PR and the fact that the enzyme is related, although distantly, to GGR suggest that A. fulgidus PR is also a flavoprotein. Thus, we attempted to extract the flavin coenzyme from PR by heating a methanol solution of the enzyme. The extracted coenzyme, colored slightly yellow, comigrated with FAD, not with FMN, on a cellulose TLC plate (data not shown). This fact proved that A. fulgidus PR, at least that which is recombinantly expressed in E. coli, contains noncovalently bound FAD.
FIG. 7.
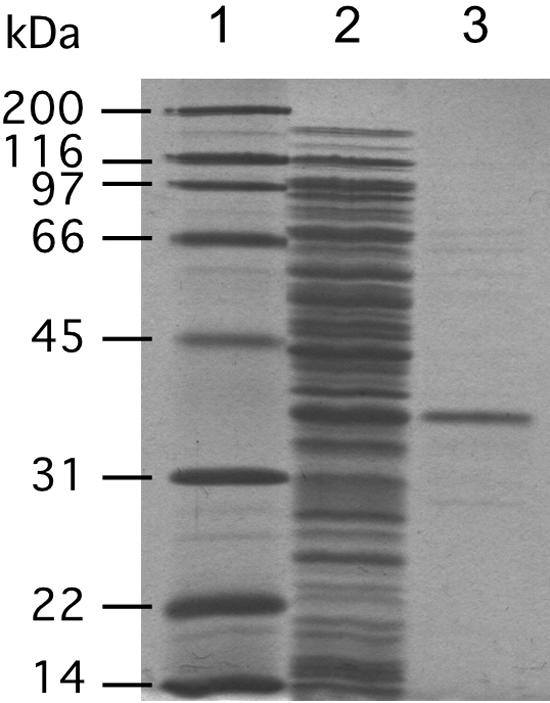
SDS-PAGE of purified A. fulgidus PR. Lane 1, molecular standard marker; lane 2, crude extract from BL21(DE3)/pET15b-AF0648; lane 3, PR purified by Ni-chelating affinity column chromatography.
FIG. 8.

UV-visible spectrum of A. fulgidus PR.
In vitro assay for PR.
We also attempted to assay PR activity in vitro by using MK-7 as the substrate, since A. fulgidus normally produces MK-7(14H). However, the attempt failed, although we added the same cofactors that were used in the previous study of A. thaliana GGR (12), i.e., FAD, NADPH, and Mg2+, in the reaction mixtures. Thus, we hypothesized that the enzyme might be sensitive to oxygen because A. fulgidus is an obligate anaerobe and conducted a second assay under anaerobic conditions. The resulting anaerobic enzyme reaction changed the HPLC elution pattern of MK-7, producing more hydrophobic products, which are likely derivatives of MK-7, such as those observed in the quinone analysis of E. coli expressing PR (Fig. 9). However, interestingly, more detailed studies revealed that the addition of the cofactors mentioned above had no effect on the enzyme activity. FAD could be excluded from the mixture without any problem, because it is still bound to PR after purification. On the other hand, the lack of the requirement of NADPH indicated that dithionite, which was added to the mixture to maintain anaerobic conditions, acted as the actual reducing agent. The replacement of dithionite with high concentrations of NADPH or NADH did not allow the enzyme reaction to proceed, even under anaerobic conditions. As a result, we subsequently used an excess amount of dithionite as the only reducing agent for the in vitro assay. That Mg2+ was not needed was confirmed by the addition of 5 mM EDTA to the solution, which did not affect the enzyme activity as well. We conducted EI-MS analyses of the reaction products corresponding to the numbered peaks in Fig. 9. The analysis of the peak 1, which had been assigned as unreacted MK-7 because of their identical elution times, gave an ion with an m/z of 648, which is in good agreement with that of authentic MK-7. Similarly, the compounds from peaks 2, 3, and 4 gave ions of m/z values of 650, 652, and 654, respectively. In addition to these data, the fact that all of these spectra share a common ion of m/z 225 suggests that the compounds from peaks 2, 3, and 4 are the derivatives of MK-7, i.e., MK-7(2H), MK-7(4H), and MK-7(6H), respectively. On the other hand, in vitro assays with other prenyl substrates—UQ-8, GGPP, and polyprenyl diphosphates—did not yield any detectable saturated products (data not shown). These results clearly indicate that the enzyme is highly specific for MK and strongly suggest that it is involved in the biosynthesis of MK-7(14H) in A. fulgidus, whereas it could also reduce UQ in vivo but to a lower extent than MK.
FIG. 9.
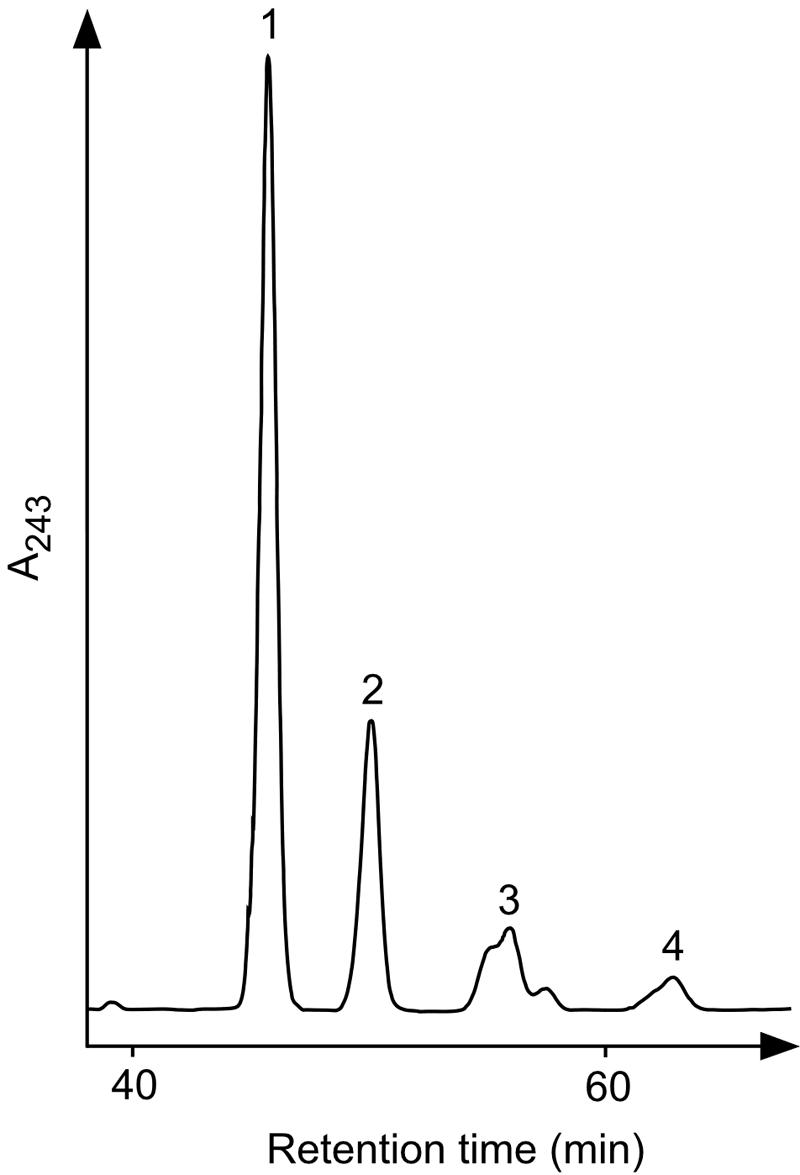
HPLC profile of respiratory quinones reduced in vitro with PR. MK-7 was used as the substrate for the in vitro assay of PR. Numbered peaks were recovered for use in EI-MS analyses.
DISCUSSION
We characterized here the function of an archaeal homologue of GGR for the first time. Unexpectedly, the novel enzyme, MK-specific PR, is more distantly related to known GGRs than the three other GGR homologues from A. fulgidus, whose functions remain unknown. Almost all archaea whose entire genome sequences have been elucidated possess multiple (two to six) genes that are highly homologous to the A. fulgidus chlP genes, and some encode one or two closely related homologues of A. fulgidus PR. Only the symbiotic archaeon Nanoarchaeum equitans, which lacks lipogenic genes in its genome (19), seems to have no homologue of these reductases. The PR homologues would be expected to be involved in the biosynthesis of respiratory quinones. However, in some archaeal species that possess such homologues, such as Sulfolobus sp., one of them might play a different role. As described above, saturated prenyl groups were found not only in respiratory quinones but also in membrane lipids of archaea. All archaea, except for N. equitans, should have biosynthetic pathways for the production of membrane lipids, and some of the GGR homologues are thought to be involved in these pathways. The multiplicity of GGR homologues can be explained by the hypothesis that they might be used to catalyze the saturation of membrane lipids in different states, i.e., diether and tetraether structures, variations in the polar head groups, and so on. However, the possibility that some of the reductases catalyze the saturation of other isoprenoid compounds cannot be excluded. For example, Halobacterium cutirubrum and two Methanobacterium species were reported to produce α-tocopherolquinone and α-tocopherolquinol, both of which also contain a saturated prenyl moiety (11). Although there seem to be no additional reports on these compounds in archaea, some of the archaeal GGR homologues might be involved in the biosynthesis of such compounds. In addition, several crenarchaeota whose entire genomes have been sequenced and Methanosarcina acetivorans C2A additionally possess hypothetical proteins (included in homologous clusters O1395 and O9882, respectively) that are homologous with GGRs but have not been classified into the clusters that include PR or the other GGR homologues from A. fulgidus. These proteins might also catalyze the saturation of prenyl groups in some unknown compounds produced in archaea.
The production of MKs with fully reduced prenyl chains was not observed, either in vivo or in vitro, although such compounds are synthesized by A. fulgidus. The most deeply reduced product observed was MK-8(10H) and was detected in the in vivo assay. We hypothesized in the beginning, from the data of the in vivo assay, that PR might reduce octaprenyl diphosphate, which is an intermediate in the biosynthesis of both MK-8 and UQ-8, and that at least the double bond at the α-end remains to be unsaturated because its presence is necessary for the transfer of prenyl groups to quinone rings. This proposal is consistent with the fact that m/z 235 and 225 ions, which would be derived from the decomposition at the side of the double bond, were detected in all of the EI-MS spectra of the UQs and MKs, respectively. However, the substrate specificity of PR elucidated by means of in vitro assays was in complete contrast to this hypothesis. The saturation of the prenyl chain of MK seems to have occurred after it was transferred to the quinone ring. The insufficient level of saturation observed might simply be due to the difference in the reaction conditions between A. fulgidus and E. coli or the use of an artificial reaction mixture. However, the possibility that the other reductases, which might be a part of GGR homologues mentioned above, are also involved in the biosynthesis of MK-7(14H) to saturate the remaining double bonds cannot be excluded.
Although we used Na2S2O4 as the reducing agent in the in vitro assay for PR, some other biological compounds must be utilized in the cells of A. fulgidus. The true reducing agent does not appear to be NAD(P)H, however. Redox proteins such as ferredoxin, which is conserved both in A. fulgidus and E. coli, represent promising candidates. However, the in vitro PR assay using ferredoxin and ferredoxin:NADP+ oxidoreductase from spinach and an excess amount of NADPH failed to give saturated MK-7 (data not shown). This failure might arise from the low reaction temperature, 30°C, which is appropriate for spinach proteins but might be too low for PR to show any detectable activity. Because the role of ferredoxin as an electron donor was reported in the study on AfpA, an FMN-binding putative reductase from A. fulgidus (7), it is conceivable that PR could also accept electrons from A. fulgidus ferredoxin, although other redox proteins, e.g., rubredoxin and flavodoxin, could serve as a source of electron donors for PR.
Acknowledgments
This study was supported by grants-in-aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
We are grateful to K. Ogura and T. Koyama, Tohoku University, for providing MK-7 and GGPP. We thank M. Tsujii, Eisai Chemical Co., Ltd., for providing UQ-8. We thank C. Ohto, Toyota Motor Co., for donating IPP and DMAPP. We are grateful to M. Watanabe, Tohoku University, for technical assistance with the EI-MS analyses.
REFERENCES
- 1.Addlesee, H. A., L. C. Gibson, P. E. Jensen, and C. N. Hunter. 1996. Cloning, sequencing and functional assignment of the chlorophyll biosynthesis gene, chlP, of Synechocystis sp. PCC 6803. FEBS Lett. 389:126-130. [DOI] [PubMed] [Google Scholar]
- 2.Addlesee, H. A., and C. N. Hunter. 1999. Physical mapping and functional assignment of the geranylgeranyl-bacteriochlorophyll reductase gene, bchP, of Rhodobacter sphaeroides. J. Bacteriol. 181:7248-7255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Addlesee, H. A., and C. N. Hunter. 2002. Rhodospirillum rubrum possesses a variant of the bchP gene, encoding geranylgeranyl-bacteriopheophytin reductase. J. Bacteriol. 184:1578-1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bligh, E. G., and W. J. Dyer. 1959. A rapid method of total lipid extraction and purification. Can. J. Med. Sci. 37:911-917. [DOI] [PubMed] [Google Scholar]
- 5.Collins, M. D., and D. Jones. 1981. Distribution of isoprenoid quinone structural types in bacteria and their taxonomic implication. Microbiol. Rev. 45:316-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Rosa, M., and A. Gambacorta. 1988. The lipids of archaebacteria. Prog. Lipid Res. 27:153-175. [DOI] [PubMed] [Google Scholar]
- 7.Ding, Y. H., and J. G. Ferry. 2004. Flavin mononucleotide-binding flavoprotein family in the domain Archaea. J. Bacteriol. 186:90-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dunphy, P. J., D. L. Gutnick, P. G. Phillips, and A. F. Brodie. 1968. A new natural naphthoquinone in Mycobacterium phlei: cis-dihydromenaquinone-9 structure and function. J. Biol. Chem. 243:398-407. [PubMed] [Google Scholar]
- 9.Fujii, H., T. Koyama, and K. Ogura. 1982. Efficient enzymatic hydrolysis of polyprenyl pyrophosphates. Biochim. Biophys. Acta 712:716-718. [PubMed] [Google Scholar]
- 10.Gambacorta, A., A. Trincone, B. Nicolaus, L. Lama, and M. De Rosa. 1994. Unique features of lipids of Archaea. Syst. Appl. Microbiol. 16:518-527. [Google Scholar]
- 11.Hughes, P. E., and S. B. Tove. 1982. Occurrence of α-tocopherolquinone and α-tocopherolquinol in microorganisms. J. Bacteriol. 151:1397-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keller, Y., F. Bouvier, A. d'Harlingue, and B. Camara. 1998. Metabolic compartmentation of plastid prenyllipid biosynthesis: evidence for the involvement of a multifunctional geranylgeranyl reductase. Eur. J. Biochem. 251:413-417. [DOI] [PubMed] [Google Scholar]
- 13.Kikuchi, A., H. Sagami, and K. Ogura. 1999. Evidence for covalent attachment of diphytanylglyceryl phosphate to the cell-surface glycoprotein of Halobacterium halobium. J. Biol. Chem. 274:18011-18016. [DOI] [PubMed] [Google Scholar]
- 14.Muraca, R. F., J. S. Whittick, G. D. Daves, Jr., P. Friis, and K. Folkers. 1967. Mass spectra of ubiquinones and ubiquinols. J. Am. Chem. Soc. 89:1505-1508. [DOI] [PubMed] [Google Scholar]
- 15.Ohnuma, S.-i., K. Hirooka, H. Hemmi, C. Ishida, C. Ohto, and T. Nishino. 1996. Conversion of product specificity of archaebacterial geranylgeranyl-diphosphate synthase. Identification of essential amino acid residues for chain length determination of prenyltransferase reaction. J. Biol. Chem. 271:18831-18837. [DOI] [PubMed] [Google Scholar]
- 16.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 17.Tanaka, R., U. Oster, E. Kruse, W. Rudiger, and B. Grimm. 1999. Reduced activity of geranylgeranyl reductase leads to loss of chlorophyll and tocopherol and to partially geranylgeranylated chlorophyll in transgenic tobacco plants expressing antisense RNA for geranylgeranyl reductase. Plant Physiol. 120:695-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tindall, B. J., K. O. Stetter, and M. D. Collins. 1989. A novel, fully saturated menaquinone from the thermophilic, sulphate-reducing archaebacterium Archaeoglobus fulgidus. J. Gen. Microbiol. 135:693-696. [Google Scholar]
- 19.Waters, E., M. J. Hohn, I. Ahel, D. E. Graham, M. D. Adams, M. Barnstead, K. Y. Beeson, L. Bibbs, R. Bolanos, M. Keller, K. Kretz, X. Lin, E. Mathur, J. Ni, M. Podar, T. Richardson, G. G. Sutton, M. Simon, D. Soll, K. O. Stetter, J. M. Short, and M. Noordewier. 2003. The genome of Nanoarchaeum equitans: insights into early archaeal evolution and derived parasitism. Proc. Natl. Acad. Sci. USA 100:12984-12988. [DOI] [PMC free article] [PubMed] [Google Scholar]