

Abstract
Introduction:
Recent genome-wide association studies identified new dementia-associated variants. We assessed the performance of updated polygenic risk scores (PRSs) using these variants in an independent cohort.
Methods:
We used Cox models and area under the curve (AUC) to validate new PRSs (PRS-83SNP, PRS-SBayesR, and PRS-CS) compared with an older PRS-23SNP in 12,031 initially-healthy participants ≥70 years of age. Dementia was rigorously adjudicated according to Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) criteria.
Results:
PRS-83SNP, PRS-SBayesR, and PRS-CS were associated with incident dementia, with fully adjusted (including apolipoprotein E [APOE] ε4) hazard ratios per standard deviation (SD) of 1.35 (1.23–1.47), 1.37 (1.25–1.50), and 1.42 (1.30–1.56), respectively. The AUC of a model containing conventional/non-genetic factors and APOE was 74.7%. This was improved to 75.7% (p = 0.007), 76% (p = 0.004), and 76.1% (p = 0.003) with addition of PRS-83SNP, PRS-SBayesR, and PRS-CS, respectively. The PRS-23SNP did not improve AUC (74.7%, p = 0.95).
Conclusion:
New PRSs for dementia significantly improve risk-prediction performance, but still account for less risk than APOE genotype overall.
Keywords: APOE gene, incident dementia, longitudinal study, polygenic risk score
1 |. BACKGROUND
Dementia results in tremendous medical, economic, and social impact with an increasing global burden.1,2 Although lifestyle factors form a large part of all-cause dementia risk, the genetic heritability of Alzheimer’s disease (AD), the most common form of dementia, is estimated at 58%–79% from twin studies.3,4 This opens a potential avenue for preventing and treating dementia by targeting high-risk individuals based on genetic predisposition. The apolipoprotein E (APOE) gene is the strongest known genetic determinant of common dementia.5–7 Recent genome-wide association studies (GWASs) have identified many additional dementia-associated risk variants.8–14 A recent GWAS of AD and related dementias by Bellengeuz et al.14 involving 39,106 clinically diagnosed cases, 46,828 proxy cases, and 401,577 controls of European ancestry, identified 83 independent dementia-associated variants (excluding the APOE region), more than doubling the number of previously identified variants.15 Updated polygenic risk scores (PRSs) incorporating these variants can now be calculated, which may improve dementia risk prediction.14 Performance of dementia risk-prediction models, including using these newly associated variants from outside the APOE region, may help identify high-risk individuals with more accuracy.16–18 This, in turn, may facilitate more targeted prevention and early intervention strategies for individuals or families, and facilitate recruitment into disease-modifying or prevention intervention trials.
There is a requirement to independently assess and validate the performance of newly derived genomic risk scores in external cohorts. Here, we independently assess the performance of three newly derived PRSs19–21 based on Bellengeuz et al.14 in the ASPirin in Reducing Events in the Elderly (ASPREE) clinical trial, with extended observational follow-up through the eXTension (ASPREE-XT) study.22–25 Together, these studies comprise a population of older individuals ≥70 years of age with no personal history of diagnosed cardiovascular disease events, dementia, or severe physical disability at enrollment, who were followed prospectively with all-cause dementia adjudicated as a primary endpoint. All incident dementia cases in ASPREE were adjudicated by expert panels and used the same standardized protocols. Our study helps assess the future potential utility of genomic risk prediction for all-cause dementia.
2 |. METHODS
2.1 |. Study population
ASPREE was a randomized double-blind, placebo-controlled clinical trial (2010–2017) to determine whether daily 100-mg aspirin extended disability-free survival in 19,114 healthy adults aged ≥70 years (≥65 years for US minorities) with no prior diagnosed cardiovascular diseases, dementia, physical disability, or other life-threatening illness (based on their medical records at the clinics and confirmed with general practitioners) at enrolment. Afterward, an ongoing observational follow-up study (ASPREE-XT), which was established in 2018, continues to collect data from ASPREE participants and investigates long-lasting effects of low-dose aspirin on diseases, such as cancer. In addition, it investigates a broad range of factors that contribute to the maintenance of physical and cognitive health in older adults. The design and protocol of the ASPREE trial and ASPREE-XT study have been reported previously.22–26 All participants provided written informed consent. The study was approved by the Human Research Ethics Committees at Monash University and Alfred Hospital in Australia and site-specific institutional review boards in the United States.
In this genetic study we included 12,031 genotyped participants from the ASPREE trial and excluded participants with non-European ancestry, who were younger than 70 years at enrollment, and close relatives based on a coefficient of relatedness >0.05 (Figure 1). We excluded ASPREE participants with non-European ancestry from our study because (1) the GWAS performed by Bellengeuz et al.14 used to derive the PRSs was based predominately on individuals with European ancestry,27 and (2) there were relatively few participants with non-European ancestry enrolled in the ASPREE trial. The sample size of genotyped non-European participants in the ASPREE trial was n < 535 (across multiple ancestries, see Figure 1). This provided insufficient power to test the performance of PRSs in an ancestry-specific manner. All phenotypic data used in this study were collected in person from participants at the time of enrollment.22
FIGURE 1.
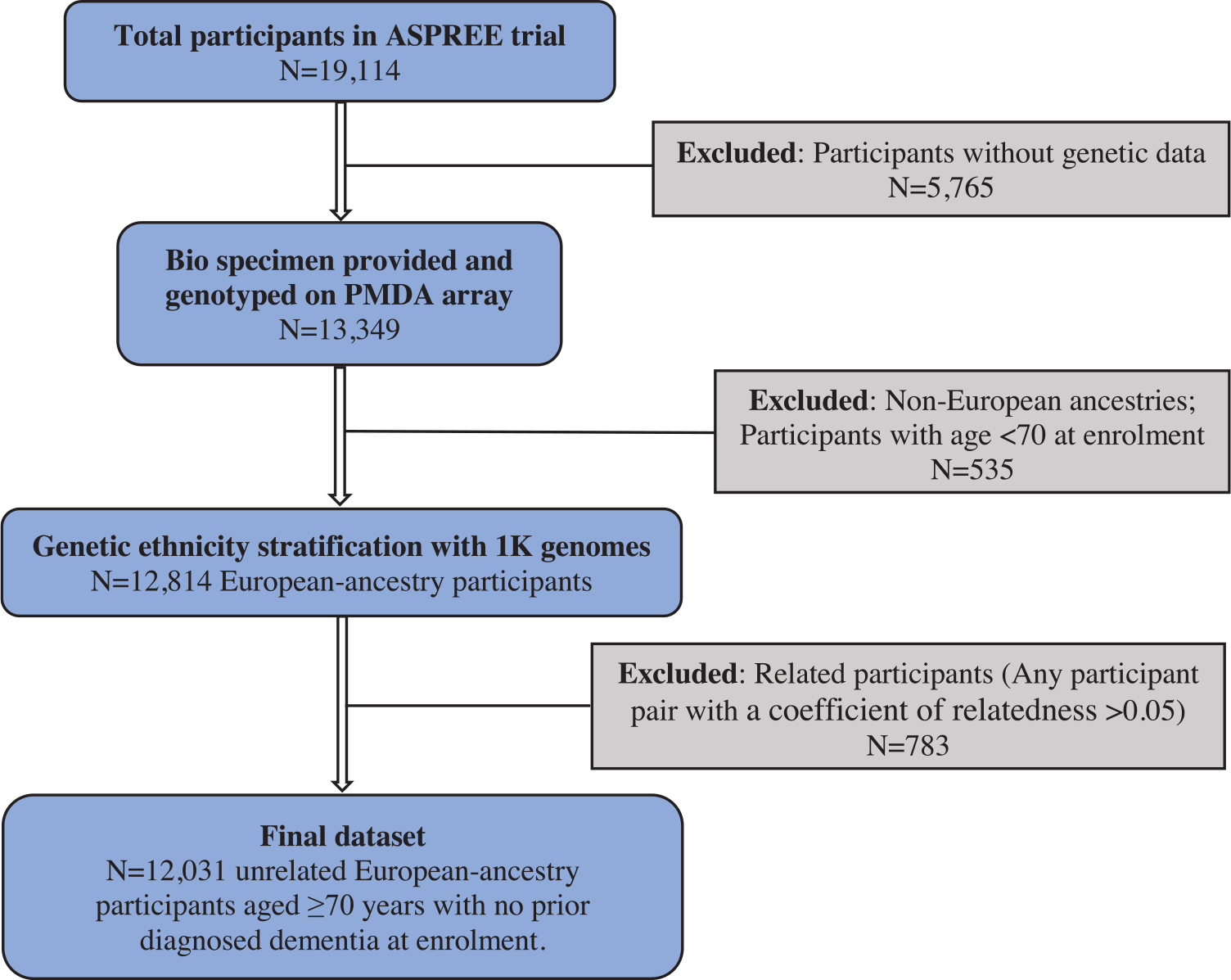
Flow chart of selection of ASPREE participants included in this genetic study.
2.2 |. Incident dementia diagnosis
Cognitive assessments were administered by trained and accredited staff at baseline and year 1, and then biennially over the follow-up period. The cognitive battery included the Modified Mini-Mental State (3MS)28 test to measure global cognition, the Hopkins Verbal Learning Test-Revised (HVLT-R)29 for episodic memory, the single letter (F) Controlled Oral Word Association Test (COWAT)30 for language and executive function, and the Symbol Digit Modalities Test (SDMT)31 to measure psychomotor speed. Participants who had a suspected dementia diagnosis (“trigger”) were referred for further standardized cognitive and functional evaluations. Triggers for dementia were defined as a 3MS score <78,32 which explained ≈50% of the triggers; a drop of >10.15 points from the predicted score based on their own baseline 3MS adjusted for age and education33; a report of memory concerns or other cognitive problems to a specialist; clinician diagnosis of dementia as indicated in the participant’s medical records; and prescription of a cholinesterase inhibitor.34–36
To reduce the likelihood of delirium contributing to the cognitive profile, participants were administered a battery of cognitive tests at least 6 weeks following the initial dementia trigger. These tests included the Alzheimer’s Disease Assessment Scale-Cognitive (ADAS-Cog) subscale,37 Color Trails,38 Lurian overlapping figures,39 and the Alzheimer’s Disease Cooperative Study–Activities of Daily Living (ADCS-ACL) scale,40 which was completed by the participant and study partner. Other information relevant to the dementia assessment included laboratory tests, brain imaging (computed tomography or magnetic resonance imaging [CT or MRI]), and clinical case notes. A committee of geriatricians and neurologists assessed each dementia trigger case following the ASPREE methodology for clinical adjudication. The Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) criteria were used for dementia diagnosis. The dementia diagnosis date was taken as the date of the trigger that resulted in a confirmed dementia diagnosis by the adjudication committee.34
2.3 |. Genotyping
The peripheral blood samples provided by ASPREE participants at the time of study enrollment were processed to buffy coat within 4 hours of collection and then stored at −80°C. DNA was later purified from the buffy coat via magnetic bead extraction41 and genotyped using the Axiom 2.0 Precision Medicine Diversity Research Array (Thermo Fisher Scientific, CA). Variant calling used a custom pipeline aligned to the human reference genome GRCh38. The imputation was performed based on the Haplotype Reference Consortium panel for European samples using the Michigan imputation server.42 Post-imputation quality control removed variants with r2 < 0.3. APOE genotype was measured directly at two single-nucleotide polymorphisms (SNPs), rs7412 and rs429358, which were included in the Axiom array.
2.4 |. Polygenic risk scores
We calculated four different PRSs. Our goal was to investigate the contribution of polygenic risk based on variants outside the APOE locus. Therefore, all four PRSs excluded variants in the APOE region (44,000,000–46,000,000 bp on chromosome 19 in GRCh38).14
PRS-83SNP is a new PRS comprising 83 independent genome-wide significant variants (p < 5 × 10−8) at 75 loci from a GWAS of AD and related dementias performed by Bellengeuz et al.14 The PRS was calculated as the sum of an individual’s risk alleles weighted by effect sizes taken from the GWAS summary statistics (Table S1).
PRS-SBayesR is a new PRS derived from the SBayesR method that models the genetic architecture with linkage disequilibrium (LD) in a Bayesian framework.19 The LD matrices provided by SBayesR developers were calculated based on ≈1.1 million HapMap3 common SNPs in a random sample of 50,000 unrelated individuals of European ancestry in the UK Biobank data set.19 We performed SBayesR (with default parameters that the software recommended19,21) using these LD matrices on summary statistics from the aforementioned GWAS14 and obtained improved effect sizes for 1,081,756 HapMap3 SNPs. The PRS was then calculated using the newly derived effect sizes of the 1,081,756 SNPs.
PRS-CS is a new PRS derived from the PRS-CS (continuous shrinkage) method,20 which infers posterior effect sizes of SNPs using GWAS summary statistics and an external LD reference panel. PRS-CS developers provided the LD reference panel, which was calculated based on ≈1.1 million HapMap3 SNPs in 375,120 unrelated individuals of European ancestry in the UK Biobank data set.20 We performed PRS-CS (with default parameters that the software recommended20,21) using the LD reference panel on summary statistics from the aforementioned GWAS14 and obtained posterior (improved) effect sizes for 1,103,008 HapMap3 SNPs. The PRS was then calculated using new effect sizes of the 1,103,008 SNPs.
PRS-23SNP is an older PRS comprising 23 genome-wide significant (p < 5 × 10−8) AD-associated SNPs, which have recently been used by van der Lee et al.16 and Riaz et al.17 These SNPs were identified in previous studies.8,43,44 The PRS was calculated as the sum of an individual’s risk alleles weighted by effect sizes derived by a metaanalysis.16
2.5 |. Statistical analysis
We used Cox proportional hazard (PH) regression models to estimate the association (hazard ratio [HR] and 95% confidence interval [CI]) between the PRS as a continuous variable (per standard deviation, SD) and the risk of progression to all-cause dementia. In Model 1, we adjusted for age at recruitment, sex, and the first 20 principal components (PCs) of genetic ancestry to account for population stratification. Model 2 was additionally adjusted for the number of APOE ε4 and APOE ε2 alleles as done previously.14 We evaluated a third model (Model 3) with adjustment for baseline smoking status, alcohol drinking status, body mass index, living status (alone vs with others), years of education, depressive symptoms (Center for Epidemiological Studies-Depression-10 [CES-D]scale ), diabetes status, systolic blood pressure, diastolic blood pressure, dyslipidemia, and dementia family history (father, mother, or siblings) (Table 1). We did not include aspirin treatment as a covariate as we found no evidence that aspirin was associated with risk of dementia in the ASPREE cohort.34 The PRS included in the models was also investigated by categorical groups (deciles). As a sensitivity analysis, we also used the Fine-Gray sub-distribution hazard model45 accounting for a competing risk of mortality, to estimate the association between the PRS as a continuous variable and the risk of progression to all-cause dementia, with the same covariates used in Models 1–3. The results were considered to be significant after Bonferroni correction for multiple comparisons.
TABLE 1.
Baseline characteristics of ASPREE participants included in this genetic study.
| Baseline categories | Overall (n = 12,031) |
|---|---|
| Age, mean (SD), years | 75.1 (4.2) |
| Sex | |
| Male, n (%) | 5425 (45.1%) |
| Female, n (%) | 6606 (54.9%) |
| Smoking status | |
| Current, n (%) | 367 (3.1%) |
| Former, n (%) | 4990 (41.5%) |
| Never, n (%) | 6674 (55.5%) |
| Alcohol drinking status | |
| Current, n (%) | 9608 (79.9%) |
| Former, n (%) | 584 (4.9%) |
| Never, n (%) | 1839 (15.3%) |
| Years of education | |
| <9 years, n (%) | 1840 (15.3%) |
| 9–11 years, n (%) | 3781 (31.4%) |
| 12 years, n (%) | 1343 (11.2%) |
| 13–15 years, n (%) | 1898 (15.8%) |
| 16 years, n (%) | 1105 (9.2%) |
| 17–21 years, n (%) | 2064 (17.2%) |
| Living status | |
| At home alone, n (%) | 3813 (31.7%) |
| At home with family, friends, or others, n (%) | 8218 (68.3%) |
| CES-D-10,a mean (SD) | 3.1 (3.2) |
| BMIb, mean (SD), kg/m2 | 28.0 (4.5) |
| Systolic blood pressure, mean (SD), mm Hg | 139.5 (16.3) |
| Diastolic blood pressure, mean (SD), mm Hg | 77.2 (9.9) |
| Dyslipidemia | |
| Yes, n (%) | 7987 (66.4%) |
| No, n (%) | 4044 (33.6%) |
| Diabetes status | |
| Yes, n (%) | 1108 (9.2%) |
| No, n (%) | 10,923 (90.8%) |
| Dementia family historyc, n (%) | 3064 (25.5%) |
| APOE genotyped | |
| ε1/ε3: ε2/ε4, n (%) | 440 (3.7%) |
| ε 1/ε4, n (%) | 2 (<0.1%) |
| ε2/ε2, n (%) | 64 (<0.1%) |
| ε2/ε3, n (%) | 1642 (13.6%) |
| ε3/ε3, n (%) | 7214 (60.0%) |
| ε3/ε4, n (%) | 2459 (20.4%) |
| ε4/ε4, n (%) | 185 (1.5%) |
CES-D-10, Center for Epidemiologic Studies Depression Scale, 10-item version.
BMI, body mass index, calculated as weight in kilograms divided by height in meters squared.
Family history of dementia in father, mother, or sibling.
Twenty-five participants had missing APOE genotypes due to quality control of genotyping at rs7412 or rs429358.
To evaluate whether the relevant covariates mentioned above may modify the effect of PRSs on dementia progression, we also used Cox PH models to investigate interactions between PRSs (as continuous variables) and sex, APOE genotypes, and other covariates used in Model 3. We then performed subgroup analyses for covariates with significant interactions.
To assess the effects of PRSs on dementia risk prediction, we estimated the area under the receiver-operating characteristic (ROC) curve (AUC) after a mean of 6.5 years follow-up. The baseline model (logistic regression) contained the conventional/non-genetic variables mentioned above (Model 3). Improvement in AUC was assessed after addition of APOE genotypes to the baseline model, and then again after addition of the PRSs to the baseline + APOE model, using the R package “pROC.”46 The DeLong test was used for comparing AUCs of nested models.47 Statistical analyses were performed using R version 4.0.3.
3 |. RESULTS
3.1 |. Baseline characteristics and polygenic score calculation
The baseline characteristics of the study population are shown in Table 1. The mean age of the participants was 75.1 years, and 54.9% were female. Only a small proportion were current smokers (3.1%) or had diabetes (9.2%) at baseline.
The distributions of four PRSs (standardized to a mean of 0 and variance of 1) of the 12,031 participants are shown in Figure S1. These distributions are all approximately Gaussian (Kolmogorov–Smirnov test p-values for four PRS distributions are all >0.05) but moderately correlated (Table S2).
3.2 |. PRS and incident dementia risk
During a mean follow-up of 6.5 years, 505 incident all-cause dementia events were observed. The newly derived PRS-83SNP, PRS-SBayesR, and PRS-CS were significantly associated with incident dementia risk in all models (fully adjusted model, Model 3: HR = 1.35 per SD [95% CI: 1.23–1.47] for PRS-83SNP, HR = 1.37 per SD [1.25–1.50] for PRS-SBayesR, and HR = 1.42 per SD [1.30–1.56] for PRS-CS) (Table 2). The older PRS-23SNP was associated with incident dementia risk, but with a lower effect size (Model 3: HR = 1.12 per SD [1.02–1.22], p = 0.02). This association did not remain significant after Bonferroni correction (p < 0.05/12 = 0.004). After adjusting for the competing risk of death using the Fine-Gray model, these results remained unchanged (Table S3).
TABLE 2.
Association of four dementia-related polygenic risk scores (as a continuous variable, per SD) with risk of incident dementia.
| Dementia-related polygenic risk score | Model 1 |
Model 2 |
Model 3 |
|||
|---|---|---|---|---|---|---|
| HR (95% CI) | p-value | HR (95% CI) | p-value | HR (95% CI) | p-value | |
| PRS-83SNP | 1.35 (1.23–1.47) | 2.85E-11 | 1.37 (1.26–1.50) | 3.12E-12 | 1.35 (1.23–1.47) | 5.33E-11 |
| PRS-SBayesR | 1.39 (1.27–1.51) | 6.48E-13 | 1.37 (1.26–1.50) | 3.07E-12 | 1.37 (1.25–1.50) | 9.51E-12 |
| PRS-CS | 1.43 (1.31–1.57) | 3.56E-15 | 1.42 (1.30–1.56) | 1.43E-14 | 1.42 (1.30–1.56) | 1.67E-14 |
| PRS-23SNP | 1.11 (1.02–1.21) | 0.02 | 1.12 (1.03–1.23) | 0.01 | 1.12 (1.02–1.22) | 0.02 |
Note: Cox PH regression models were used to estimate the hazard ratio (HR) of the PRS per standard deviation, with 95% confidence intervals (CIs). Model 1 was adjusted for age at recruitment, sex, and first 20 genetic PCs. Model 2 was additionally adjusted for the number of APOE ε4 and APOE ε2 alleles. Model 3 was further adjusted for baseline smoking status, alcohol drinking status, BMI, living status, years of education, CES-D-10, diabetes status, systolic blood pressure, diastolic blood pressure, dyslipidemia, and dementia family history
Figure 2 presents associations between different categorical groups of the PRSs (deciles of the PRS distributions) with incident dementia risk during follow-up. Compared with the lowest/1st decile group as a reference (0%–10%), the highest/10th decile group (90%–100%) had an ≈3-fold higher risk of incident dementia for PRS-83SNP (HR = 3.14 [95% CI: 1.99–4.96]), PRS-SBayesR (HR = 2.44 [1.65–3.62]), and PRS-CS (HR = 3.90 [2.51–6.07]) in the fully adjusted model (Model 3). However, the comparison between the lowest and highest deciles for the older PRS-23SNP resulted in a far lower effect size that was not statistically significant (HR = 1.32 [0.89–1.96] in Model 3).
FIGURE 2.
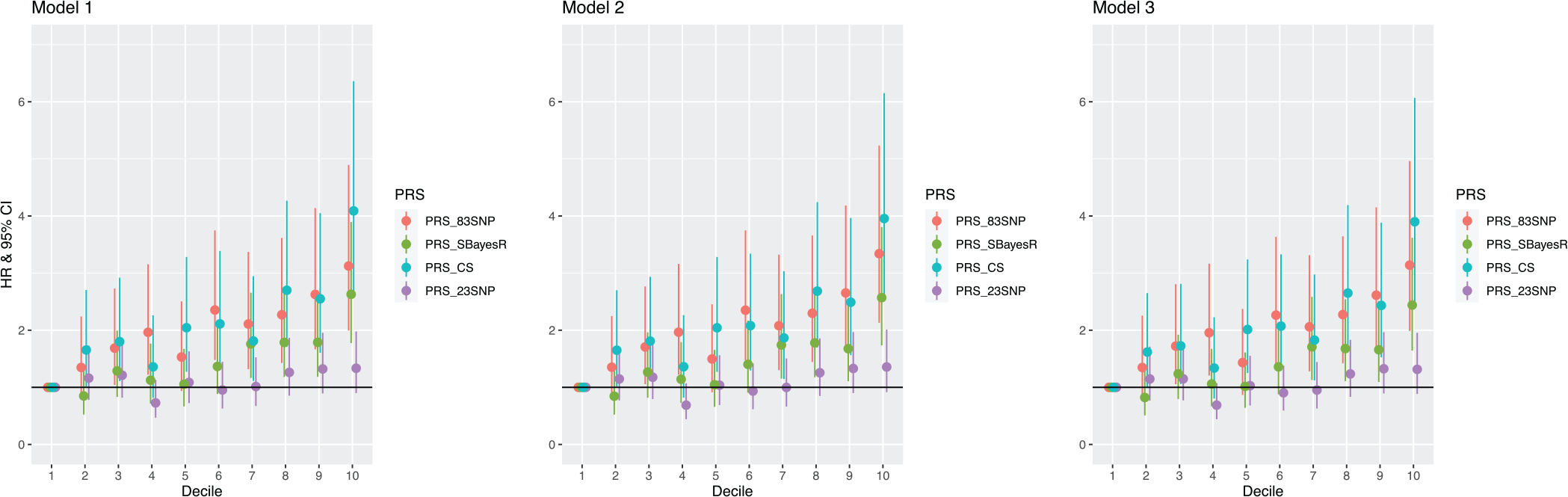
Association of PRS decile groups with incident dementia. Cox proportional hazards (PH) models were used to estimate the hazard ratio (HR) with 95% confidence intervals (CIs). Model 1 was adjusted for age at recruitment, sex, and first 20 genetic PCs. Model 2 was additionally adjusted for the number of APOE ε4 and APOE ε2 alleles. Model 3 was further adjusted for baseline smoking status, alcohol drinking status, BMI, living status, years of education, CES-D-10, diabetes status, systolic blood pressure, diastolic blood pressure, dyslipidemia, and dementia family history.
3.3 |. Subgroup analyses
Testing for PRS-by-sex interactions revealed no statistically significant interactions (p > 0.05, Table S4), suggesting that PRS effects on dementia are not sex specific. In PRS-by-covariate interaction analysis (Table S4), we observed evidence of interaction between the PRSs and alcohol drinking status (p = 0.02 for PRS-83SNP; p = 0.001 for PRS-SBayesR; p = 0.01 for PRS-CS; and p = 0.02 for PRS-23SNP), PRS-SBayesR and the number of APOE ε4 (p = 0.004), and PRS-CS and the number of APOE ε4 (p = 0.01), but no other interaction signals were detected (all p > 0.05). Thus we examined the effects of PRSs on incident dementia for subgroups of participants according to their alcohol drinking status and carrying numbers of APOE ε4 (Table 3). The PRS effects on incident dementia were stronger in individuals who never drank alcohol (HR = 1.74 [1.40–2.16] for PRS-83SNP, HR = 1.95 [1.56–2.43] for PRS-SBayesR, and HR = 1.84 [1.49–2.28] for PRS-CS) relative to current or former alcohol drinkers. For APOE ε4 non-carriers (a large population subgroup), PRS effects on prediction of dementia development were stronger (HR = 1.40 [1.25–1.58] for PRS-83SNP, HR = 1.52 [1.34–1.71] for PRS-SBayesR, and HR = 1.59 [1.40–1.79] for PRS-CS) compared to the general population. However, for participants who carry two copies of APOE ε4, PRSs had no effect on dementia risk, as no association was significant after Bonferroni correction (p < 0.05/12 = 0.004).
TABLE 3.
Association of four dementia-related polygenic risk scores (as a continuous variable, per SD) with risk of incident dementia for subgroups according to alcohol drinking status and the number of APOE-ε4.
| Subgroup analysis | PRS-83SNP |
PRS-SBayesR |
PRS-CS |
PRS-23SNP |
|||||
|---|---|---|---|---|---|---|---|---|---|
| HR (95% CI) | p-value | HR (95% CI) | p-value | HR (95% CI) | p-value | HR (95% CI) | p-value | ||
| Alcohol drinking | Current | 1.28 (1.16–1.42) | 2.21E-06 | 1.29 (1.16–1.43) | 2.09E-06 | 1.35 (1.22–1.50) | 2.27E-08 | 1.06 (0.96–1.18) | 0.25 |
| Former | 1.26 (0.85–1.88) | 0.25 | 1.08 (0.73–1.62) | 0.69 | 1.24 (0.81–1.89) | 0.33 | 1.07 (0.72–1.58) | 0.74 | |
| Never | 1.74 (1.40–2.16) | 6.58E-07 | 1.95 (1.56–2.43) | 2.84E-09 | 1.84 (1.49–2.28) | 1.43E-08 | 1.41 (1.14–1.74) | 1.75E-03 | |
| Number of APOE-ε4 | 0 | 1.40 (1.25–1.58) | 1.53E-08 | 1.52 (1.34–1.71) | 1.69E-11 | 1.59 (1.40–1.79) | 1.12E-13 | 1.08 (0.96–1.21) | 0.22 |
| 1 | 1.37 (1.18–1.59) | 3.13E-05 | 1.27 (1.10–1.48) | 1.44E-03 | 1.33 (1.15–1.54) | 1.64E-04 | 1.25 (1.07–1.45) | 4.31E-03 | |
| 2 | 0.59 (0.35–1.02) | 0.06 | 0.67 (0.41–1.09) | 0.11 | 0.53 (0.30–0.92) | 0.02 | 0.82 (0.50–1.34) | 0.43 | |
Note: Cox PH regression models were used to estimate the hazard ratio (HR) of the PRS per standard deviation with 95% confidence intervals (CI), adjusted for age at recruitment, sex, first 20 genetic PCs, the number of APOE-ε4 and APOE-ε2 alleles, baseline smoking status, alcohol drinking status, BMI, living status, years of education, CES-D-10, diabetes status, systolic blood pressure, diastolic blood pressure, dyslipidemia, and dementia family history (Model 3).
3.4 |. AUC estimates and comparisons
Full covariate-adjusted AUC estimates after a mean of 6.5 years of follow-up are shown in Table 4, based on different models examined. This includes changes in the AUC from the baseline model (containing only conventional/non-genetic risk factors), after addition of APOE genotypes and the four PRSs. The AUC of the baseline model containing no genetic information was 71.0% (68.7%–73.3%), which upon addition of APOE genotypes, improved to 74.7% (72.6%–76.8%) (p = 3.27 × 10−6 by DeLong test). When PRS-83SNP was subsequently added to the model containing APOE genotype, the AUC further improved to 75.7% (73.7%–77.8%), a statistically significant improvement (p = 0.007). The addition of PRS-SBayesR to the baseline + APOE model showed a similar AUC improvement to 76.0% (74.0%–78.0%) (p = 0.004). The addition of PRS-CS to the baseline + APOE model also showed a similar AUC improvement to 76.1% (74.1%–78.2%) (p = 0.003). However, the addition of the older PRS-23SNP to the baseline + APOE model showed no evidence of improvement to the AUC, which remained at 74.7% (72.6%–76.8%) (p = 0.95). We also examined larger models by adding any two of PRS-83SNP, PRS-SBayesR, and PRS-CS to the baseline + APOE model (Table 4); however, no significant improvement was found compared to the addition of a single PRS. For example, although the model of baseline + APOE + PRS-83SNP + PRS-CS reached the best estimate of AUC with 76.5% (74.5%–78.5%) and performed a little better than the addition of PRS-83SNP (p = 0.03), it was not significantly better than the baseline + APOE + PRS-CS model (p = 0.13).
TABLE 4.
AUC estimates at a mean of 6.5 years of follow-up and comparisons for predictions of dementia risk.
| Model | AUC (95% CI) | p-value |
| Baseline | 71.0% (68.7%–73.3%) | |
| Baseline + APOE | 74.7% (72.6%–76.8%) | 3.27E-06 (vs Baseline) |
| Baseline + APOE + PRS-83SNP | 75.7% (73.7%–77.8%) | 0.007 (vs Baseline + APOE) |
| Baseline + APOE + PRS-SBayesR | 76.0% (74.0%–78.0%) | 0.004 (vs Baseline + APOE) |
| Baseline + APOE + PRS-CS | 76.1% (74.1%–78.2%) | 0.003 (vs Baseline + APOE) |
| Baseline + APOE + PRS-23SNP | 74.7% (72.6%–76.8%) | 0.95 (vs Baseline + APOE) |
| Baseline + APOE + PRS-83SNP + PRS-SBayesR | 76.2% (74.2%–78.2%) | 0.06 (vs Baseline + APOE + PRS-83SNP) |
| 0.22 (vs Baseline + APOE + PRS-SBayesR) | ||
| Baseline + APOE + PRS-83SNP + PRS-CS | 76.5% (74.5%–78.5%) | 0.03 (vs Baseline + APOE + PRS-83SNP) |
| 0.13 (vs Baseline + APOE + PRS-CS) | ||
| Baseline + APOE + PRS-SBayesR + PRS-CS | 76.2% (74.2%–78.2%) | 0.40 (vs Baseline + APOE + PRS-SBayesR) 0.61 (vs Baseline + APOE + PRS-CS) |
Note: The baseline model includes the predictors in Model 3, excluding APOE genotype variables (the number of ε4 and ε2 alleles) and PRS, that is, age at recruitment, sex, first 20 genetic PCs, smoking status, alcohol drinking status, BMI, living status, years of education, CES-D-10, diabetes status, systolic blood pressure, diastolic blood pressure, dyslipidemia, and dementia family history. p-value was obtained through the DeLong test by comparing AUCs from two nested models.
4 |. DISCUSSION
In this study, we evaluated the performance of three newly derived PRSs, PRS-83SNP, PRS-SBayesR, and PRS-CS, for all-cause dementia in a well-characterized population of older individuals followed prospectively. We found improved performance for the new PRSs, compared to the older PRS-23SNP score, and strong associations between the new PRSs and incident dementia risk during follow-up. The addition of the new PRSs to risk-prediction models containing conventional/non-genetic risk factors and APOE genotype improved the AUC. However, AUC improvements after adding the PRSs were modest (≈1% for PRS-83SNP, ≈1.3% for PRS-SBayesR, and ≈1.4% for PRS-CS). Our study provides evidence that if the sample sizes and power of future GWASs for dementia continue to increase, the performance of resulting PRSs based on greater numbers of dementia-associated variants will continue to improve.
Previous studies have assessed effects of the older PRS-23SNP for risk of progression to dementia,16,17 AD age at onset,16,48 and other cognitive impairments,17,49,50 but found limited performance or practical utility for individual patients. For example, van der Lee et al.16 evaluated the PRS-23SNP on incident dementia in cognitively healthy participants (age >45 years) from the community-based Rotterdam Study. Their results showed that the PRS could significantly modify the risk and age at onset of AD and all-cause dementia beyond the APOE genotype.16 However, this study did not examine the prediction performance of the PRS using AUC or the C-statistic.
A previous study from our group17 examining the older PRS-23SNP in the ASPREE cohort (using only a mean of 4.5 years of follow-up) found the effect of the PRS on incident dementia risk to be modest, with only a 1.4-fold increase in incident dementia risk between the lowest and the highest PRS-23SNP tertiles. By contrast, in the present study (using the newly derived PRSs and ≈2 additional years of follow-up), the effect sizes per SD increased to HR = 1.41 per SD (PRS-CS), and the increase in risk between the lowest and highest deciles increased to 3.9-fold (PRS-CS). However, the older PRS-23SNP still did not significantly improve dementia risk prediction above the base model (AUC = 74.7%, p = 0.95).
Bellengeuz et al. validated the performance of the new PRS-83SNP using a fixed-effect meta-analysis, presenting the results in the same publication as the GWAS.14 However, the effect size was quite low (HR = 1.08, 95% CI: 1.06–1.09), and the findings required independent assessment in an external cohort. Separately, Stocker et al.51 investigated the performance of a more recent PRS derived using 72 independent SNPs9 and APOE status to predict clinically diagnosed all-cause dementia in a community-based cohort followed over 17 years. The AUC of predicting all-cause dementia using this PRS, with APOE status, age, sex, and education was estimated as 79.2%, which is higher than our results, possibly due to more dementia cases included in a longer follow-up. However, this particular study did not examine the risk of incident dementia using longitudinal analysis. The above studies, taken together with our study, provide evidence that PRS performance for dementia may continue to improve as a function of larger GWAS. This is consistent with our main results indicating a significant improvement to AUC for a model containing conventional/non-genetic factors and APOE (AUC = 74.7%), with the addition of the newer PRS-83SNP (AUC = 75.7%, p = 0.007), PRS-SBayesR (AUC = 76%, p = 0.004), or PRS-CS (AUC = 76.1%, p = 0.003).
However, the concept of PRS performance improving in a linear fashion with more disease-associated variants identified (e.g., by larger GWASs) has been brought into question. A recent study15 compared SNP-based heritability estimates from different AD GWASs and found a downward trend in heritability estimates with larger GWASs. For example, the most recent GWAS by Bellengeuz et. al.14 explained only ≈3% heritability, whereas the previous smaller GWAS by Lambert et. al.8 in 2013 explained ≈9%. Several possible explanations have been provided for this counter-intuitive trend,15 including the lack of clinical screening and younger average age of controls used in the recent GWAS, and the growing use of “proxy dementia cases” rather than clinically diagnosed AD patients. Nonetheless, despite these discrepancies in heritability estimates, we observed clearly improved PRS performance from newly derived PRSs (from large recent GWAS), compared with the older PRS. Despite the improvement we observed in PRS performance, the newly derived PRSs still confer only modest risk when compared with the APOE genotype, and improve risk-prediction models only incrementally (beyond APOE genotype). This raises the question of whether a PRS (containing non-APOE variants) will ever confer sufficient variation and effect to be considered of clinical or epidemiological utility for dementia risk prediction. Given that the newly derived PRSs can be calculated for the same cost and using similar methods as previous PRSs, it is a logical progression to use them. We argue that continued development of improved PRSs (based on more dementia-associated variants other than APOE) is worthwhile, particularly for improved risk prediction and stratification of APOE ε4 non-carriers (e.g., ε3/ε3 homozygotes), who comprise a large population sub-group where many individuals will still go on to develop dementia. Furthermore, there is hope that improved early identification of higher-risk individuals for dementia (based on genetic risk scores and also other factors) will promote prevention and early intervention by individuals or their families to minimize the disease progression, such as modifying lifestyles to reduce risk,52,53 and facilitate more efficient recruitment of clinical trials, especially for new preventive therapies or interventions as they emerge.
In addition, incremental improvements to risk-prediction models (e.g., single-digit AUC) are a common property of PRSs and seen across many disease areas.54–56 In some cases, this small benefit is translating into clinical utility (e.g., coronary heart disease and breast cancer risk prediction and risk stratification). For dementia, a relatively common disease with increasing prevalence due to aging populations, if risk-prediction models are extrapolated to an entire nation’s population, for example, a modest improvement in AUC could result in re-classification of thousands of people into lower- or higher-risk categories. If re-classification determines whether certain interventions will be recommended or not, this may become important, especially if future interventions are particularly expensive or invasive.
Our study found that an updated PRS containing a higher number of dementia-associated common variants identified from GWAS14 has led to the improved prediction of dementia risk. However, beyond the identification of more dementia-associated variants from GWASs (most of them are common variants), more studies are required to better understand the role of rare and structural variants for dementia,57–60 which also need to be incorporated in risk-prediction models. Moreover, the interplay between genes and the environment requires improved understanding with regard to dementia risk.61
Strengths of our study include independent validation of PRS performance in a large sample size of well-characterized, initially-healthy older individuals, followed prospectively over a mean of 6.5 years. All incident dementia cases in ASPREE were adjudicated as primary clinical trial endpoints by expert panels, using standardized protocols for data collection and DSM-IV criteria.
Limitations of our study include analyzing participants of European ancestry only to prevent population stratification biases, meaning our conclusions cannot necessarily be generalized to other ancestries. The ASPREE cohort also has an acknowledged healthy survivorship bias, with reduced prevalence of comorbidities at enrollment, and participants were without diagnosed cardiovascular disease events, major physical disability, or dementia. The ASPREE cohort, overall, has a lower incidence of dementia than the general population would at similar ages. This may result in an underestimation of PRS effects. The molecular processes underlying most of the common variants used in the PRSs are still unknown or uncharacterized. Further functional studies are required to understand the underlying mechanisms between these dementia-associated variants, risk, and clinical phenotypes. Another limitation is that the dementia adjudication did not include clinical phenotyping. Thus, although the majority of dementia endpoints were likely due to AD, the endpoint itself captured all-cause dementia.
5 |. CONCLUSION
We present, to our knowledge, the first external validation of three newly derived PRSs for dementia, in an independent cohort of older individuals followed prospectively. Our study demonstrates that the newly derived PRSs for dementia provide significantly improved performance and improve risk prediction beyond the use of conventional/non-genetic risk factors and APOE genotypes. Our findings have potential implications for the improved risk stratification and prediction of dementia. Further studies will be required to more rigorously assess the clinical utility of genetic risk scores for dementia as new therapeutic and preventive agents become available.
Supplementary Material
RESEARCH IN CONTEXT.
1. Systematic review:
The authors reviewed the literature using PubMed on genome-wide association studies (GWASs) and polygenic risk scores (PRSs) for dementia. Recent GWASs have identified an increased number of dementia-associated variants, and PRSs were updated using these variants. PubMed search did not identify studies on validating these new PRSs in an independent cohort.
2. Interpretation:
Our study showed that three new PRSs for dementia significantly improved risk prediction performance, which suggested a potential clinical use to facilitate more targeted prevention and early intervention strategies for individuals or families and facilitate recruitment into disease-modifying or preventive intervention trials.
3. Future directions:
Beyond the identification of more dementia-associated variants from GWAS, more studies are required to better understand the role of rare and structural variants for dementia, which also need to be incorporated in risk-prediction models. Future studies will more rigorously assess the clinical utility of these genetic risk scores.
ACKNOWLEDGMENTS
We thank the ASPREE trial staff in Australia and the United States, the participants who volunteered for this trial, and the general practitioners and staff of the medical clinics who cared for the participants. The ASPREE Biobank is supported by a Flagship cluster grant (including the Commonwealth Scientific and Industrial Research Organisation, Monash University, Menzies Research Institute, Australian National University, and University of Melbourne); and grants (U01AG029824 and U19AG062682) from the National Institute on Aging and the National Cancer Institute at the National Institutes of Health, and by grants (334047 and 1127060) from the National Health and Medical Research Council of Australia, and by Monash University and the Victorian Cancer Agency. Dr. Paul Lacaze is supported by a National Heart Foundation Future Leader Fellowship (102604). Dr. John J. McNeil is supported by an National Health and Medical Research Council (NHMRC) Leadership Fellowship (IG1173690).
Open access publishing facilitated by Monash University, as part of the Wiley - Monash University agreement via the Council of Australian University Librarians.
Footnotes
CONFLICT OF INTEREST STATEMENT
Dr. Raj C. Shah reports being the site principal investigator or sub-investigator for Alzheimer’s disease clinical trials for which his institution (Rush University Medical Center) is compensated (Amylyx Pharmaceuticals, Inc., Athira Pharma, Inc., Edgewater NEXT, Eli Lilly & Co., Inc., and Genentech, Inc.). Dr. Trevor Chong has received honoraria for lectures from Roche. All other authors report no conflicts of interest. Author disclosures are available in the supporting information.
CONSENT STATEMENT
All participants in this study provided written informed consent.
SUPPORTING INFORMATION
Additional supporting information can be found online in the Supporting Information section at the end of this article.
REFERENCES
- 1.Sloane PD, Zimmerman S, Suchindran C, et al. The public health impact of Alzheimer’s disease, 2000–2050: potential implication of treatment advances. Annu Rev Public Health. 2002;23:213–231. [DOI] [PubMed] [Google Scholar]
- 2.Takizawa C, Thompson PL, van Walsem A, Faure C, Maier WC. Epidemiological and economic burden of Alzheimer’s disease: a systematic literature review of data across Europe and the United States of America. J Alzheimers Dis. 2015;43(4):1271–1284. [DOI] [PubMed] [Google Scholar]
- 3.Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63(2):168–174. [DOI] [PubMed] [Google Scholar]
- 4.Karlsson IK, Escott-Price V, Gatz M, et al. Measuring heritable contributions to Alzheimer’s disease: polygenic risk score analysis with twins. Brain Commun. 2022;4(1):fcab308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tudorache IF, Trusca VG, Gafencu AV. Apolipoprotein E-a multifunctional protein with implications in various pathologies as a result of its structural features. Comput Struct Biotechnol J. 2017;15:359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qian J, Wolters FJ, Beiser A, et al. APOE-related risk of mild cognitive impairment and dementia for prevention trials: an analysis of four cohorts. PLoS Med. 2017;14(3):e1002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rasmussen KL, Tybjærg-Hansen A, Nordestgaard BG, Frikke-Schmidt R. Absolute 10-year risk of dementia by age, sex and APOE genotype: a population-based cohort study. CMAJ. 2018;190(35):E1033–E1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45(12):1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kunkle BW, Grenier-Boley B, Sims R, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51(3):414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moreno-Grau S, de Rojas I, Hernández I, et al. Genome-wide association analysis of dementia and its clinical endophenotypes reveal novel loci associated with Alzheimer’s disease and three causality networks: the GR@ ACE project. Alzheimer’s Dement. 2019;15(10):1333–1347. [DOI] [PubMed] [Google Scholar]
- 11.Wightman DP, Jansen IE, Savage JE, et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat Genet. 2021;53(9):1276–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kunkle BW, Schmidt M, Klein HU, et al. Novel Alzheimer disease risk loci and pathways in African American individuals using the African genome resources panel: a meta-analysis. JAMA Neurol. 2021;78(1):102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Rojas I, Moreno-Grau S, Tesi N, et al. Common variants in Alzheimer’s disease and risk stratification by polygenic risk scores. Nat Commun. 2021;12(1):3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bellenguez C, Küçükali F, Jansen IE, et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet. 2022;54(4):412–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Escott-Price V, Hardy J. Genome-wide association studies for Alzheimer’s disease: bigger is not always better. Brain Commun. 2022;4(3):fcac125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van der Lee SJ, Wolters FJ, Ikram MK, et al. The effect of APOE and other common genetic variants on the onset of Alzheimer’s disease and dementia: a community-based cohort study. Lancet Neurol. 2018;17(5):434–444. [DOI] [PubMed] [Google Scholar]
- 17.Riaz M, Huq A, Ryan J, et al. Effect of APOE and a polygenic risk score on incident dementia and cognitive decline in a healthy older population. Aging Cell. 2021;20(6):e13384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leonenko G, Baker E, Stevenson-Hoare J, et al. Identifying individuals with high risk of Alzheimer’s disease using polygenic risk scores. Nat Commun. 2021;12(1):4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lloyd-Jones LR, Zeng J, Sidorenko J, et al. Improved polygenic prediction by Bayesian multiple regression on summary statistics. Nat Commun. 2019;10(1):5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ge T, Chen CY, Ni Y, Feng YC, Smoller JW. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat Commun. 2019;10(1):1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ni G, Zeng J, Revez JA, et al. A comparison of ten polygenic score methods for psychiatric disorders applied across multiple cohorts. Biol Psychiatry. 2021;90(9):611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McNeil JJ, Woods RL, Nelson MR, et al. Baseline characteristics of participants in the ASPREE (ASPirin in Reducing Events in the Elderly) study. J Gerontol A Biol Sci Med Sci. 2017;72(11):1586–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McNeil JJ, Woods RL, Nelson MR, et al. Effect of aspirin on disability-free survival in the healthy elderly. N Engl J Med. 2018;379(16):1499–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McNeil JJ, Wolfe R, Woods RL, et al. Effect of aspirin on cardiovascular events and bleeding in the healthy elderly. N Engl J Med. 2018;379(16):1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McNeil JJ, Nelson MR, Woods RL, et al. Effect of aspirin on all-cause mortality in the healthy elderly. N Engl J Med. 2018;379(16):1519–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nunn JS, Sulovski M, Tiller J, Holloway B, Ayton D, Lacaze P. Involving elderly research participants in the co-design of a future multi-generational cohort study. Res Involv Engagem. 2021;7(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin AR, Gignoux CR, Walters RK, et al. Human demographic history impacts genetic risk prediction across diverse populations. Am J Hum Genet. 2017;100(4):635–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ryan J, Woods RL, Britt C, et al. Normative performance of healthy older individuals on the Modified Mini-Mental State (3MS) examination according to ethno-racial group, gender, age, and education level. Clin Neuropsychol. 2021;35(6):1174–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benedict RH, Schretlen D, Groninger L, Brandt J. Hopkins verbal learning test–revised: normative data and analysis of inter-form and test-retest reliability. Clin Neuropsychol. 1998;12(1):43–55. [Google Scholar]
- 30.Ross TP. The reliability of cluster and switch scores for the Controlled Oral Word Association Test. Arch Clin Neuropsychol. 2003;18(2):153–164. [PubMed] [Google Scholar]
- 31.Smith A Symbol Digit Modalities Test (SDMT) Manual (revised). Los Angeles, California: Western psychological services; 1982. [Google Scholar]
- 32.Bland RC, Newman SC. Mild dementia or cognitive impairment: the Modified Mini-Mental State examination (3MS) as a screen for dementia. Can J Psychiatry. 2001;46(6):506–510. [DOI] [PubMed] [Google Scholar]
- 33.Tombaugh TN. Test-retest reliable coefficients and 5-year change scores for the MMSE and 3MS. Arch Clin Neuropsychol. 2005;20(4):485–503. [DOI] [PubMed] [Google Scholar]
- 34.Ryan J, Storey E, Murray AM, et al. Randomized placebo-controlled trial of the effects of aspirin on dementia and cognitive decline. Neurology. 2020;95(3):e320–e331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Orchard SG, Polekhina G, Ryan J, et al. Combination of gait speed and grip strength to predict cognitive decline and dementia. Alzheimers Dement (Amst). 2022;14(1):e12353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collyer TA, Murray AM, Woods RL, et al. Association of dual decline in cognition and gait speed with risk of dementia in older adults. JAMA Network Open. 2022;5(5):e2214647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Graham DP, Cully JA, Snow AL, Massman P, Doody R. The Alzheimer’s Disease Assessment Scale-Cognitive subscale: normative data for older adult controls. Alzheimer Dis Assoc Disord. 2004;18(4):236–240. [PubMed] [Google Scholar]
- 38.D’Elia LF, Satz P, Uchiyama CL, White T. Color Trails Test: Professional Manual. Odessa, Florida: Psychological Assessment Resources; 1996. [Google Scholar]
- 39.Reid W, Broe G, Creasey H, et al. Age at onset and pattern of neuropsychological impairment in mild early-stage Alzheimer disease: a study of a community-based population. Arch Neurol. 1996;53(10):1056–1061. [DOI] [PubMed] [Google Scholar]
- 40.Fish J Alzheimer’s Disease Cooperative Study ADL Scale. Encyclopedia of Clinical Neuropsychology. Springer; 2011. [Google Scholar]
- 41.Pinese M, Lacaze P, Rath EM, et al. The Medical Genome Reference Bank contains whole genome and phenotype data of 2570 healthy elderly. Nat Commun. 2020;11(1):435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Das S, Forer L, Schönherr S, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48(10):1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruiz A, Dols-Icardo O, Bullido MJ, et al. Assessing the role of the TREM2 p. R47H variant as a risk factor for Alzheimer’s disease and frontotemporal dementia. Neurobiol Aging. 2014;35(2):444.e1–4. [DOI] [PubMed] [Google Scholar]
- 44.Desikan RS, Fan CC, Wang Y, et al. Genetic assessment of age-associated Alzheimer disease risk: development and validation of a polygenic hazard score. PLoS Med. 2017;14(3):e1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999;94(446):496–509. [Google Scholar]
- 46.Robin X, Turck N, Hainard A, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinf. 2011;12:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeLong ER, DeLong DM, Clarke-Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988:837–45. [PubMed] [Google Scholar]
- 48.Leonenko G, Sims R, Shoai M, et al. Polygenic risk and hazard scores for Alzheimer’s disease prediction. Ann Clin Transl Neurol. 2019;6(3):456–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marden JR, Mayeda ER, Walter S, et al. Using an Alzheimer’s Disease polygenic risk score to predict memory decline in black and white Americans over 14 years of follow-up Running head: AD polygenic risk score predicting memory decline. Alzheimer Dis Assoc Disord. 2016;30(3):195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chaudhury S, Brookes KJ, Patel T, et al. Alzheimer’s disease polygenic risk score as a predictor of conversion from mild-cognitive impairment. Transl Psychiatry. 2019;9(1):154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stocker H, Perna L, Weigl K, et al. Prediction of clinical diagnosis of Alzheimer’s disease, vascular, mixed, and all-cause dementia by a polygenic risk score and APOE status in a community-based cohort prospectively followed over 17 years. Mol Psychiatry. 2021;26(10):5812–5822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kivipelto M, Mangialasche F, Ngandu T. Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat Rev Neurol. 2018;14(11):653–666. [DOI] [PubMed] [Google Scholar]
- 53.McMaster M, Kim S, Clare L, et al. Lifestyle risk factors and cognitive outcomes from the multidomain dementia risk reduction randomized controlled trial, body brain life for cognitive decline (BBL-CD). J Am Geriatr Soc. 2020;68(11):2629–2637. [DOI] [PubMed] [Google Scholar]
- 54.Torkamani A, Wineinger NE, Topol EJ. The personal and clinical utility of polygenic risk scores. Nat Rev Genet. 2018;19(9):581–590. [DOI] [PubMed] [Google Scholar]
- 55.Lambert SA, Abraham G, Inouye M. Towards clinical utility of polygenic risk scores. Hum Mol Genet. 2019;28(R2):R133–R142. [DOI] [PubMed] [Google Scholar]
- 56.O’Sullivan JW, Raghavan S, Marquez-Luna C, et al. Polygenic risk scores for cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2022;146(8):e93–e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Geier EG, Bourdenx M, Storm NJ, et al. Rare variants in the neuronal ceroid lipofuscinosis gene MFSD8 are candidate risk factors for frontotemporal dementia. Acta Neuropathol. 2019;137(1):71–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cuyvers E, Bettens K, Philtjens S, et al. Investigating the role of rare heterozygous TREM2 variants in Alzheimer’s disease and frontotemporal dementia. Neurobiol Aging. 2014;35(3):726.e11–19. [DOI] [PubMed] [Google Scholar]
- 59.Beck J, Pittman A, Adamson G, et al. Validation of next-generation sequencing technologies in genetic diagnosis of dementia. Neurobiol Aging. 2014;35(1):261–265. [DOI] [PubMed] [Google Scholar]
- 60.Qiang W, Yau WM, Lu JX, Collinge J, Tycko R. Structural variation in amyloid-β fibrils from Alzheimer’s disease clinical subtypes. Nature. 2017;541(7636):217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dunn AR, O’Connell KM, Kaczorowski CC. Gene-by-environment interactions in Alzheimer’s disease and Parkinson’s disease. Neurosci Biobehav Rev. 2019;103:73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.