

Abstract
Parathyroid hormone (PTH) binds to a family B G protein coupled receptor, parathyroid hormone 1 receptor (PTH1R). One of its functions is to regulate Ca2+ homeostasis in bone remodeling, during which Ca2+ can reach up to 40 mM. A truncated version of PTH, PTH(1–34), can fully activate PTH1R and has been used for osteoporosis treatments. Here, we used fluorescence anisotropy to examine the binding of PTH(1–34) to PTH1R purified in nanodiscs (PTH1R-ND) and found that the affinity increases 5-fold in the presence of 15 mM Ca2+. However, PTHrP(1–36), another truncated endogenous agonist for PTH1R, does not show this Ca2+ effect. Mutations of Glu19 and Glu22 in PTH(1–34) that are not conserved in PTHrP(1–36) largely abolished the Ca2+ effect. The results support that PTH(1–34) not only activates PTH1R but can also uniquely sense Ca2+. This dual function of a peptide hormone is a novel observation that couples changes in extracellular environment with endocrine signaling. Understanding this can potentially reveal the complex role of PTH signaling in bone remodeling and improve the PTH(1–34) treatment for osteoporosis.
Family B G protein coupled receptors (GPCRs) are members of the very large GPCR gene family of over 800 different receptors.1 There are 15 family B GPCRs with characteristically large extracellular domains that bind peptide hormone ligands (Scheme 1), implicating these receptors in metabolic diseases, such as diabetes, obesity and osteoporosis.2–5 Among them, parathyroid hormone 1 receptor (PTH1R) is primarily expressed on bone and kidney cells and mediates cell signaling in response to extracellular stimuli, including two endogenous peptide hormones, parathyroid hormone (PTH) and parathyroid hormone-related peptide (PTHrP).6 PTH1R regulates serum calcium and phosphate homeostasis via PTH, and tissue growth during embryonic development via PTHrP.7 Truncated versions of these two hormones, PTH(1–34) and PTHrP(1–36), are full agonists of PTH1R.8 Both truncated ligands bind PTH1R following the family B GPCR two domain binding model (Scheme 1), where the C-terminus of the peptide binds with high affinity to the receptor’s extracellular domain and the N-terminus of the peptide interacts with the receptor’s juxtamembrane domain to activate receptor signaling (Scheme 1).4, 9–11 Indeed, crystal structures show high affinity binding of the two ligands’ C-terminal domain to the receptor’s N-terminal extracellular domain and fluorescence studies show receptor activation, following the two domain-binding model.12, 13 However, this model is not sufficient to explain differences in how PTH(1–34) and PTHrP(1–36) interact with and signal through PTH1R.14, 15
Scheme 1: PTH(1–34) bound to PTH1R shown in a nanodisc.
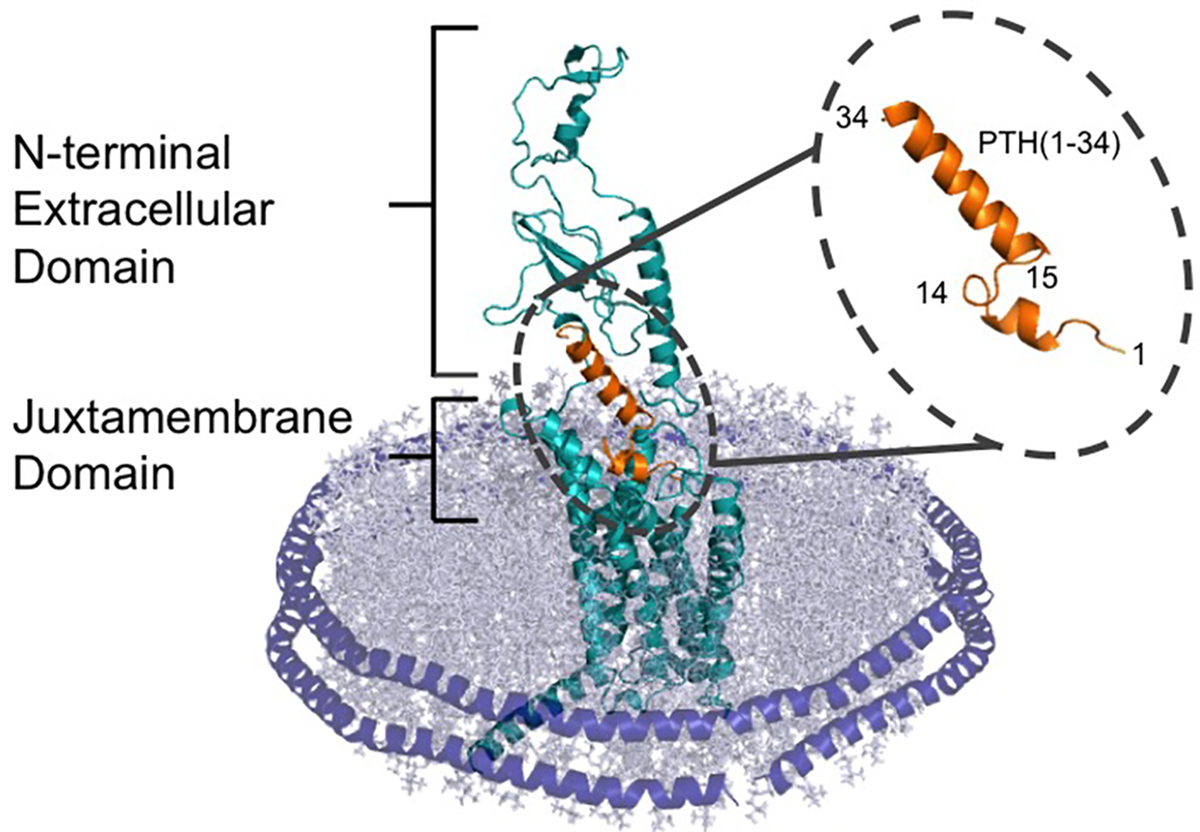
The C terminus of PTH(1–34) (orange) interacts with the extracellular domain of the receptor, while the N terminus of the peptide hormone interacts with the juxtamembrane domain. The receptor is surround by a lipid bilayer (grey), which is held together with the alpha helical membrane scaffold protein MSP1E3D1 (purple).
Sequence alignment shows that PTH(1–34) and PTHrP(1–36) share 70% similarity and 30% identity (Scheme 2). Crystal structures show that the C-terminus of both peptides, PTH(15–34) and PTHrP(15–36) that are bound to the extracellular domain structurally overlap with backbone root-mean-square deviation (RMSD) of 0.357 Å.12, 13 Many studies have mutated residues of PTH(1–34) and PTHrP(1–36) in order to develop antagonists, agonists, or biased agonists for specific signaling pathways.15–19 For instance, the N-terminally truncated peptide PTH(7–34) has been developed as a potent antagonist of PTH1R.20 In addition, although PTH(1–14) and PTHrP(1–14) show very little binding affinity and efficacy, chemical modifications, such as incorporation of unnatural amino acids or covalent linkage to a lipid molecule, were shown to significantly enhance the efficacy of PTH(1–14) or improve the binding affinity to PTH1R.18, 19, 21–24
Scheme 2: Sequence alignment of the PTH and PTHrP ligands under study.
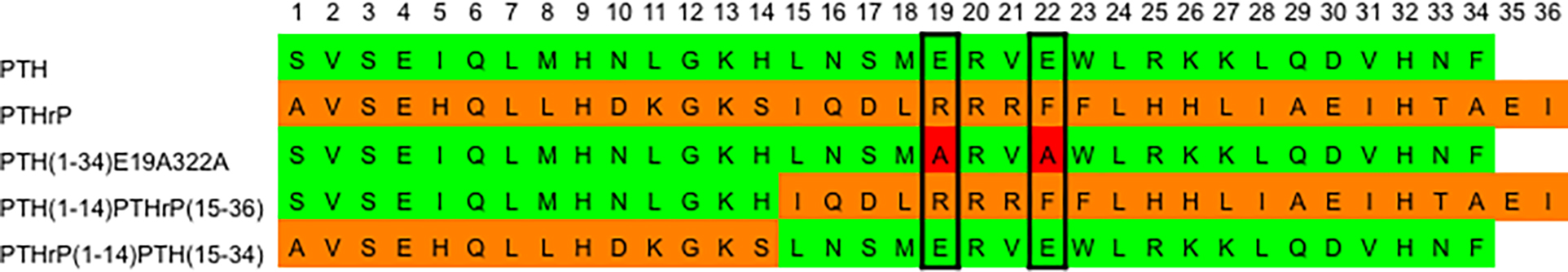
PTH residues (green), PTHrP residues (orange), and E19A and E22A mutations (red).
One specific function of PTH is to regulate bone remodeling, a process that replaces between 5–10% of total bone mass a year and repairs bone fracture and micro-damages to maintain bone health.25, 26 Bone remodeling contains two processes, bone resorption and bone formation, during which the calcium concentration surrounding the bone tissue fluctuates between 1.5 and 40 mM.27 An imbalance in regulation of bone remodeling can result in metabolic bone diseases, such as osteoporosis. Paradoxically, intermittent PTH exposure leads to bone formation while prolonged PTH exposure causes bone resorption.28 Although the exact mechanism of PTH in bone remodeling is not fully understood, intermittent injections of PTH(1–34) have been developed as the only FDA-approved hormone treatment for augmenting bone formation in severe cases of osteoporosis.29–32 It remains largely unexplored how intermittent injection couples with fluctuation of calcium concentrations to achieve the therapeutic effect of PTH(1–34) in modulating bone remodeling for the treatment of osteoporosis.
To understand the therapeutic mechanism of PTH(1–34) and ligand-receptor interactions, PTH1R must be studied at the molecular level. Until recently, studies of PTH1R were primarily carried out in cells or membrane preparations due to the technical difficulty of purifying functional receptors.33–39 We recently introduced a new method for GPCR purification using nanodiscs, also known as nanoscale apoliprotein bound bilayers (NABBs) or high density lipoproteins (HDLs), which are nanometer-sized lipid bilayers surrounded by two α-helical membrane scaffold proteins (MSP) (Scheme 1).40–42 Using this method to purify PTH1R, we previously studied the ligand binding activity using fluorescence anisotropy and observed that the presence of 15 mM Ca2+ increases the binding affinity of PTH(1–34) by one order of magnitude.36
In this study, we explore this Ca2+ dependence by comparing the binding of PTH(1–34) and PTHrP(1–36) to PTH1R purified in nanodiscs and observed Ca2+-enhanced binding for PTH(1–34) but not PTHrP(1–36). We did not observe this enhancement effect by using Mg2+ instead of Ca2+. We also measured the binding affinity of their variants, including a mutant and two chimeras (Scheme 2), to full-length purified PTH1R in nanodiscs, and, further, we examined PTH(1–34) binding to the N-terminal extracellular domain in the absence of the transmembrane region of the receptor. The results provide insight into the mechanism behind the differences in the Ca2+ effect of PTH(1–34) and PTHrP(1–36). The distinct Ca2+ effect on PTH(1–34) binding reveals two functions of PTH(1–34): (1) carrying endocrine signals to activate PTH1R and (2) sensing Ca2+ in the millimolar range in bone tissue during the bone remodeling process. The result therefore introduces a novel concept of dual functionality of PTH, establishing a new paradigm in which PTH has a built-in sensing ability to detect changes in extracellular environments for modulation of endocrine signals. This dual functionality can potentially guide new hypotheses to elucidate the complex signaling processes of bone remodeling and therapeutic mechanism of PTH(1–34) for osteoporosis treatment.
Results and Discussion
Fluorescence Anisotropy.
Figure 1 and Table 1 summarize the results of fluorescence anisotropy assays in studying the effect of 15 mM Ca2+ on binding of various peptides to PTH1R-ND, including wild-type PTH(1–34) and PTHrP(1–36), as well as the E19AE22A mutant of PTH(1–34), and two chimeras, PTH(1–14)PTHrP (15–36) and PTHrP(1–14)PTH(15–34) (Scheme 2). The E19AE22A mutant removes two non-conserved glutamic acids while the two chimeras contains N-terminal (1–14) and C-terminal (15–34 or 15–16) fragments of the two native peptides that bind to the extracellular and transmembrane domains of receptor, respectively, (Scheme 1) according to the two domain binding model. Each peptide was labeled with 5/6-fluorescein (FAM) at Lys13. Crystal structures show that Lys13 is solvent exposed and does not perturb receptor binding12, 13 as supported by previous studies.36 Control experiments show peptides maintain defined secondary structures in circular dichroism spectroscopy43 (SI Figure S1) and are able to activate PTH1R in cell-based cAMP accumulation assays (SI Figure S2). Since free peptides in solution have low anisotropy due to fast rotation and those bound to PTH1R-ND have high anisotropy due to slow rotation, fluorescence anisotropy can be used to study binding. In the experiments presented below, the concentration of peptides was kept constant at 50 nM and titrated with purified PTH1R-ND. The titration curve was fitted into a simple two-state equilibrium model, where the unbound state of the receptor interacts with a free peptide to form the bound state (see the Methods Section). The measured anisotropy values are determined by the size of molecular entities and photochemical properties of fluorescence probe in its local environment.44 Thus, the anisotropy of free peptides were measured independently and the anisotropy of peptides bound to PTH1R-ND were determined using multiple preparations of purified PTH1R-ND for each peptide. The titration curves can therefore be fitted into Eq (1), as described in the Method section, to determine the dissociation constant . Figure 1 shows titration curve of the average anisotropy at each tested [PTH1R-ND] the fitted are summarized in Table 1.
Figure 1: Binding of peptide ligands to PTH1R incorporated in nanodiscs (PTH1R-ND).
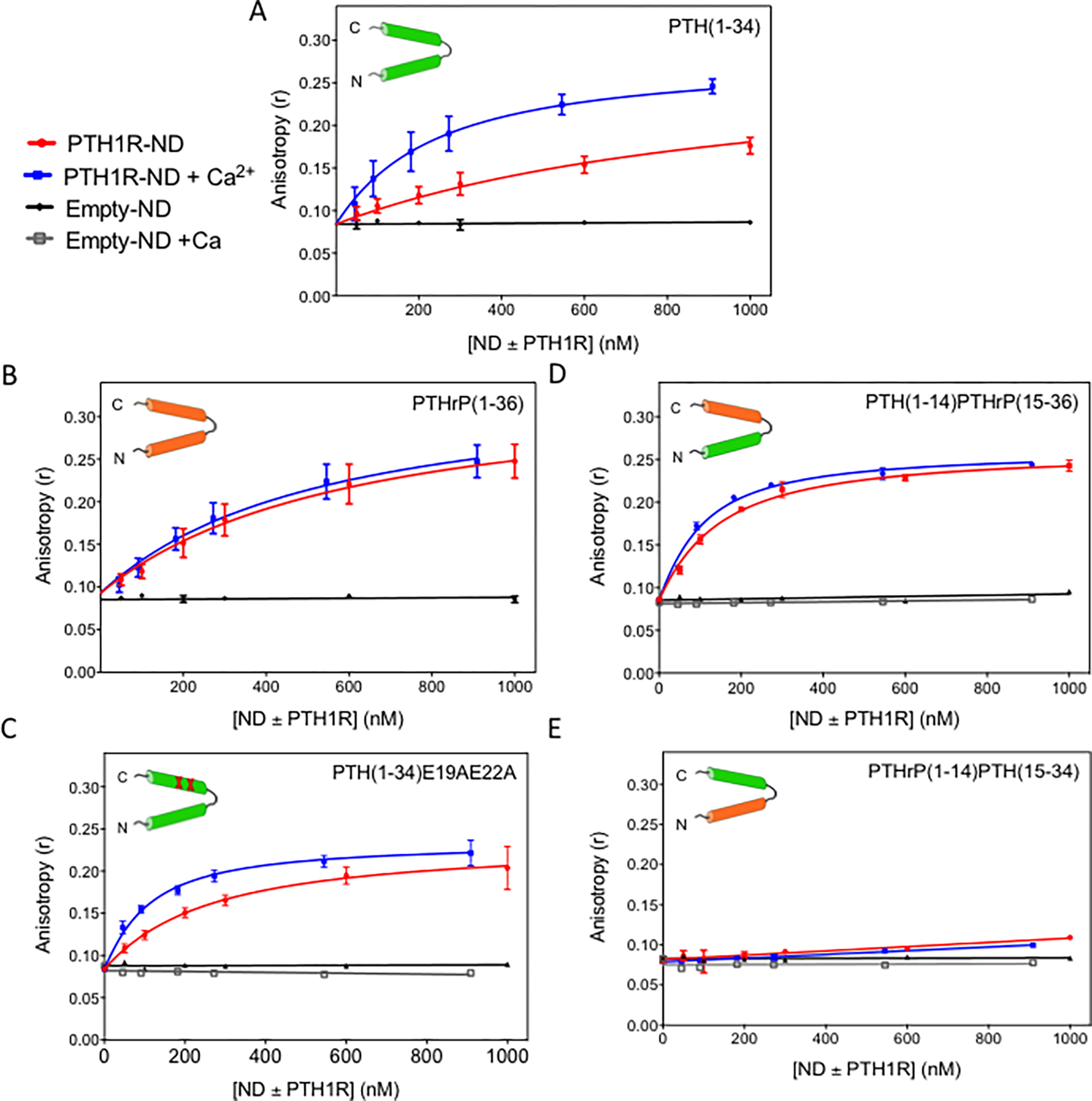
Titrations of (A) PTH(1–34); (B) PTHrP(1–36); (C) PTH(1–34)E19AE22A; (D) PTH(1–14)PTHrP(15–36) and (E) PTHrP(1–14)PTH(15–34) with PTH1R in nanodiscs (PTH1R-ND) with (blue) and without (red) addition of 15 mM Ca2+ and nanodiscs containing no PTH1R (empty-ND) with (grey) and without (black) addition of 15 mM Ca2+. The peptide concentration was kept constant at 50 and 55 nM with and without addition of 15 mM Ca2+, respectively. (Buffer: 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 3 mM MgCl2, and 100 μM EDTA). Each data point in the titration curves is an average of more than three experiments and each experiment was performed using independent preparations of purified PTH1R-ND.
Table 1:
Binding affinity of the tested PTH1R ligands
| Total Charges | KD (nM) | KD (0 mM) / KD (15 mM) | ||
|---|---|---|---|---|
|
| ||||
| Peptide | + / − (net charge) | 0 mM Ca2+ | 15 mM Ca2+ | |
|
| ||||
| PTH(1–34) | 7 / 4 (+3) | 1001 ± 41 | 204 ± 17** | 4.9 ± 0.5 |
| PTHrP(1–36) | 10 / 5 (+5) | 568 ± 37 | 495 ± 31 * | 1.1 ± 0.1 |
| PTH(1–34)E19AE22A | 7 / 2 (+5) | 224 ± 18 | 82 ± 6 ** | 2.7 ± 0.3 |
| PTH(1–14)PTHrP(15–36) | 9/4(+4) | 114 ± 5 | 76 ± 4 | 1.5 ± 0.1 |
| PTHrP(1–14)PTH(15–34) | 9 / 5(+4) | No binding | No binding | NA |
p<0.05 and
p<0.001 indicate significant difference in the fitted KD values for the tested ligand in the presence and absence of 15 mM Ca2+. The ratio KD(0 mM)/KD(15 mM) for PTH(1–34) is significantly different from the same ratio for all of the other tested ligands p < 0.0001.
Wild-type PTH(1–34).
Figure 1A shows the titration of PTH(1–34) with PTH1R-ND. At constant concentration of PTH(1–34) (50 nM), the anisotropy increases with PTH1R-ND concentration. With addition of 15 mM Ca2+, the anisotropy increases more rapidly, and plateaus at a higher maximum anisotropy value (~ 0.25). Fitting the titration curves yields the values of 1001 ± 41 nM with no additional Ca2+ (red), and 204 ± 17 nM with 15 mM Ca2+ (blue), shown also in Table 1. As a control, we titrated PTH(1–34) with nanodiscs containing no receptors (empty-ND) with and without addition of 15 mM Ca2+ and the titrations show no change in anisotropy (black and gray, respectively), suggesting the observed binding is specific to the receptor and the Ca2+ effect is not due to nanodiscs. Hence, 15 mM Ca2+ increases the binding affinity of PTH(1–34) to PTH1R-ND by a factor of 4.9 ± 0.5.
Wild-type PTHrP(1–36).
Figure 1B shows the titration of PTHrP(1–36) at 50 nM with PTH1R-ND. In both the presence and absence of 15 mM Ca2+, addition of PTH1R-ND increases the anisotropy value; however, the enhancement effect by Ca2+ is no longer observed. The control titrations (black and grey) using empty-ND in both the presence and absence of 15 mM Ca2+ do not show an increase in anisotropy, suggesting that the binding of PTHrP(1–36) is receptor specific. Fitting the titration curves yields values of 568 ± 37 nM without additional Ca2+, and 495 ± 31 nM with addition of 15 mM Ca2+ (Table 1). Hence, 15 mM Ca2+ does not show significant effect on the binding of PTHrP(1–36) to PTH1R-ND with a ratio of 1.1 ± 0.1. The combined results in Figures 1A and B suggest that ligand binding to PTH1R-ND can be enhanced by 15 mM Ca2+ for PTH(1–34), but not PTHrP(1–36).
E19AE22A mutant of PTH(1–34).
Figure 1C shows the titrations of the E19AE22A mutant of PTH(1–34). Adding PTH1R-ND without additional Ca2+ (red) or with additional 15 mM Ca2+ (blue) steadily increased anisotropy. Addition of 15 mM Ca2+ leads to a slightly more rapid increase in anisotropy. The fitted values are 224 ± 18 nM with no additional Ca2+, and 82 ± 6 nM with 15 mM Ca2+. As controls, titrating the mutant ligand with empty-ND with (grey) and without (black) additional 15 mM Ca2+ shows no binding. Addition of 15 mM Ca2+ increases the binding affinity of the mutant to PTH1R-ND by 2.7 ± 0.3 fold (Figure 1C, Table 1). The results show that the double mutation partially abolishes the Ca2+ enhancement.
PTH(1–14)PTHrP(15–36) chimera.
Figure 1D shows the titrations of PTH(1–14)PTHrP(15–36), which does not contain the E19 and E22 residues in the C-terminal domain of PTH(1–34) (Scheme 2) that contribute to the Ca2+effect shown in Figure 1C. Both in the presence and absence of 15 mM Ca2+, the anisotropy values increase with the concentration of PTH1R-ND. Fitting the curves yields the values of 114 ± 5 nM without additional Ca2+ and 76 ± 4 nM with the addition of 15 mM Ca2+ (Figure 1D, Table 1). The peptide shows no binding to empty-ND regardless of addition of 15 mM Ca2+ (black and grey, Figure 1D). The binding affinity only slightly increases (1.5 ± 0.1 fold) with 15 mM Ca2+. The results show that the chimera largely eliminates the Ca2+ effect of PTH(1–34), implying that the segment PTH(15–34) likely contains residues responsible for binding to Ca2+, in agreement with the results of the E19AE22A mutant of PTH(1–34) (Figure 1C).
PTHrP(1–14)PTH(15–34) chimera.
Figure 1E shows the titrations of the second chimera, PTHrP(1–14)PTH(15–34). Surprisingly, adding PTH1R-ND to PTHrP(1–14)PTH(15–34) in both the presence and absence of 15 mM Ca2+ leads to only a slight increase in anisotropy, to ~ 0.10 (red) at the highest concentration PTH1R-ND (1000 nM) used in the titration, which is only slightly higher than (0.084 ± 0.002), indicating insignificant binding. In the context of the two domain ligand binding model (Scheme 1), it is widely accepted that the ligand binding affinity mostly comes from the interaction between the C-terminal domain of the peptide and the extracellular domain of the receptor ( in the 1 μM regime)12, 13 while the binding of the N-terminal domain of the peptide to the juxtamembrane domain of the receptor (Scheme 1) is relatively weak ( in the 102 μM region).22, 45 Hence, it is surprising that the presence of N-terminal domain PTHrP(1–14) almost abolishes the binding of the C-terminal domain PTH(15–34), although this observation is in agreement with observations reported by Gardella and coworkers.46 The results suggest more complicated ligand binding interactions than the widely accepted two domains binding model, implying possible allosteric modulations between receptor’s extracellular and transmembrane domains via interactions with the peptide ligands.
Flow cytometry assays.
To confirm PTH(1–34) binding to PTH1R is calcium dependent in a cell-based environment, we implemented a flow cytometry assay, as reported by Bajaj et al.47–49 We used HEK293S cells overexpressing PTH1R and the PTH(1–34) ligand fluorescently labeled with FAM. In the presence and absence of 15 mM Ca2+, we measured the mean fluorescence intensity of PTH(1–34) bound to individual HEK293S cells at various concentrations of PTH(1–34). The results show that the mean fluorescence intensity increases as the concentration of PTH(1–34) increases for both 0 mM Ca2+ and 15 mM Ca2+ (Figure 2) while control experiments using uninduced HEK293 cells without PTH1R overexpression show a relatively small linear increase in fluorescence intensity. These results suggest binding of PTH(1–34) to PTH1R expressed on the cell surface. The experiments were repeated five times using five separate batches of HEK293S cells grown on different days. The five experiments consistently show that the increase in mean fluorescence intensity as a function of PTH(1–34) concentration occurs with a steeper slope and to a higher maximum intensity for 15 mM Ca2+ than for 0 mM Ca2+. Figure 2 shows a representative set of titration curves while the additional titration curves can be found in the supplementary information (SI Figure S3). The titration curves were fitted with a saturation binding model as described in the Methods section to obtain the average values of 122 ± 50 nM with 0 mM Ca2+ and 34 ± 17 nM with 15 mM Ca2+, suggesting tighter binding in the presence of 15 mM Ca2+ . The results support that PTH(1–34) binding to PTH1R in a cell based environment can be enhanced by 15 mM Ca2+, corroborating the results of anisotropy measurements shown in Figure 1.
Figure 2: Flow cytometry analysis of PTH(1–34)-FAM binding to HEK293S cells expressing PTH1R.
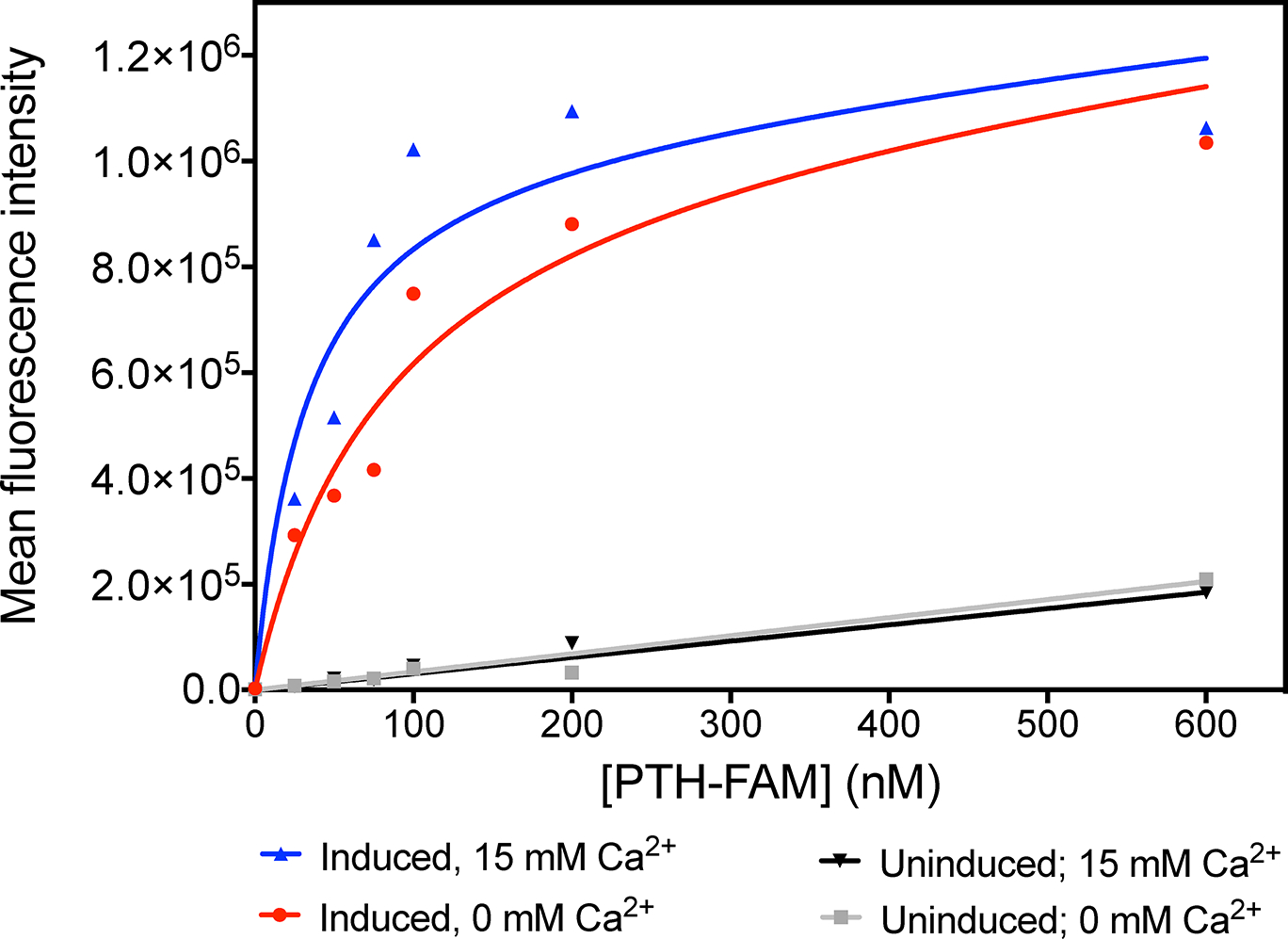
Titrations of HEK293S cells expressing PTH1R with PTH(1–34)-FAM in the presence of 0 mM Ca2+ (red) and 15 mM Ca2+ (blue) and HEK293S cells with no induced PTH1R expression in the presence of 0 mM Ca2+ (grey) and 15 mM Ca2+ (black). Data shown are representative curves from of one of five replicates (SI Figure S3).
Binding of PTH(1–34) to the extracellular domain of PTH1R.
To explore the role of PTH1R in the Ca2+-dependent binding of PTH(1–34), we used a fluorescence polarization assay to measure the of PTH(1–34) binding to the extracellular domain (ECD) of PTH1R (a.a. 29–187), as used in previous studies.44, 50–52 The peptide was labeled with fluorescein (Flu) at the N-terminus while the ECD of PTH1R was expressed as a soluble maltose-binding protein (MBP) fusion, MBP-PTH1R-ECD (Figure 3). A titration of Flu-PTH(1–34) at 25 nM with MBP-PTH1R-ECD increases the fluorescence polarization as a function of PTH1R-ECD concentration with and without 15 mM Ca2+, yielding a of 977 ± 66 nM, and 958 ± 45 nM, respectively. The results suggest that the ECD in isolation is not sufficient to observe Ca2+-dependent binding of PTH(1–34), suggesting that the transmembrane region of the receptor and/or the lipid membrane is necessary for the observed Ca2+-enhancement effect.
Figure 3: Binding of PTH(1–34) to the extracellular domain (ECD) of PTH1R.
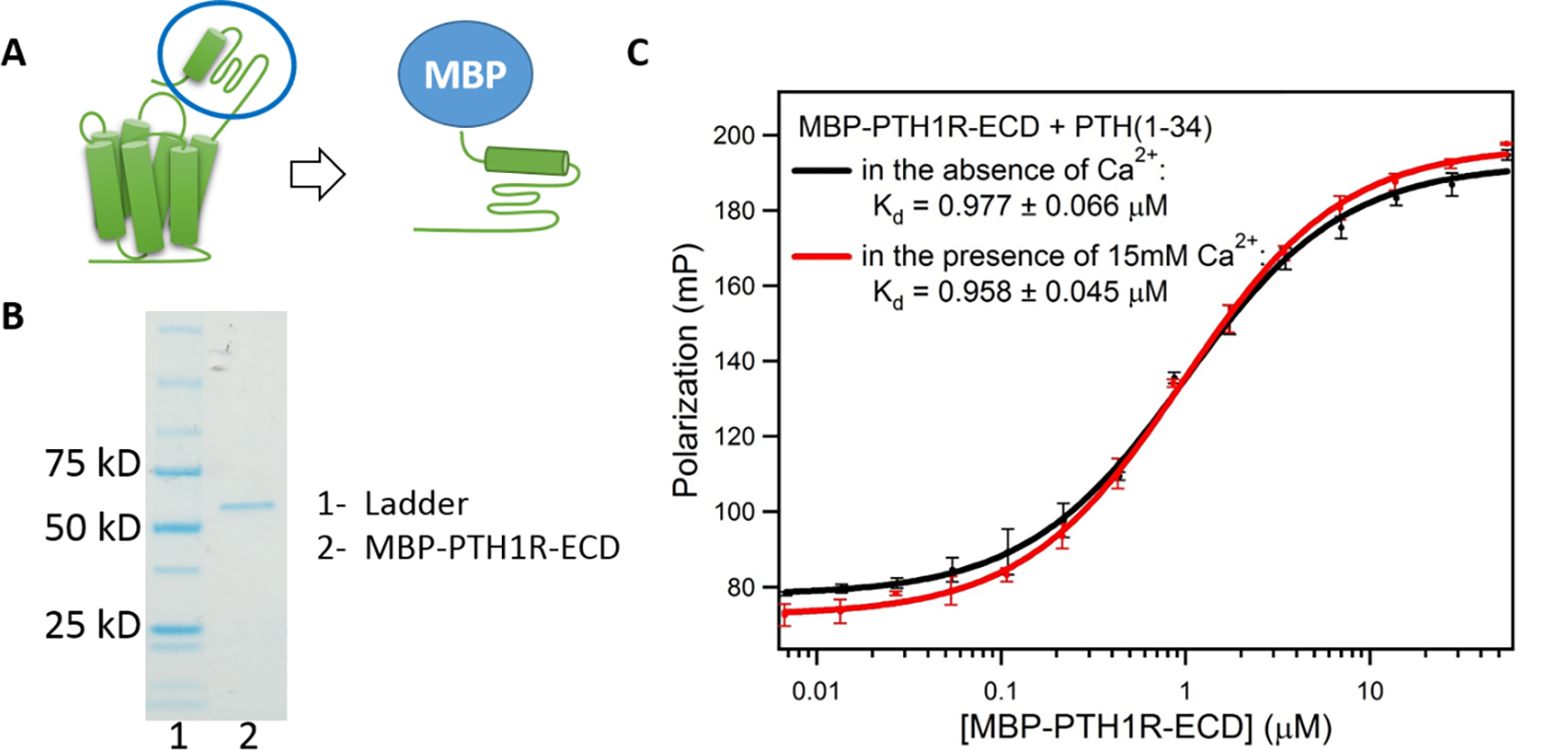
(A) Construction of MBP-PTH1R-ECD. (B) SDS-PAGE gel of MBP-PTH1R-ECD shows Lane 1, markers; lane 2, purified MBP-PTH1R-ECD (62 kD) (C) Binding of PTH(1–34) to MBP-PTH1R-ECD measured by fluorescence polarization. (Buffer: 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA). Each curve shows the average of three fluorescence polarization experiments.
Effect of Mg2+ versus Ca2+ on PTH(1–34) Binding to PTH1R-ND.
In order to directly compare the effect of divalent ions on PTH(1–34) binding to PTH1R-ND, we altered the reaction buffer conditions to remove existing Mg2+ ions (3 mM MgCl2) and titrated 50 nM PTH(1–34) with PTH1R-ND in the new buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, and 100 μM EDTA). We then repeated the titrations to test concentrations of both Ca2+ and Mg2+ from 0 to 100 mM. Figure 4 shows a stronger enhancement of ligand affinity by Ca2+ than Mg2+. Fitting yields the values for each concentration of Ca2+ and Mg2+ (Table 2). In the range of concentrations being tested (up to 100 mM), the decreases less than two fold by addition of Mg2+ while about 10 fold by addition of Ca2+, supporting selectivity of Ca2+ in enhancing binding of PTH(1–34) to PTH1R-ND.
Figure 4: Specific Ca2+ effect on PTH(1–34) binding to PTH1R-ND.
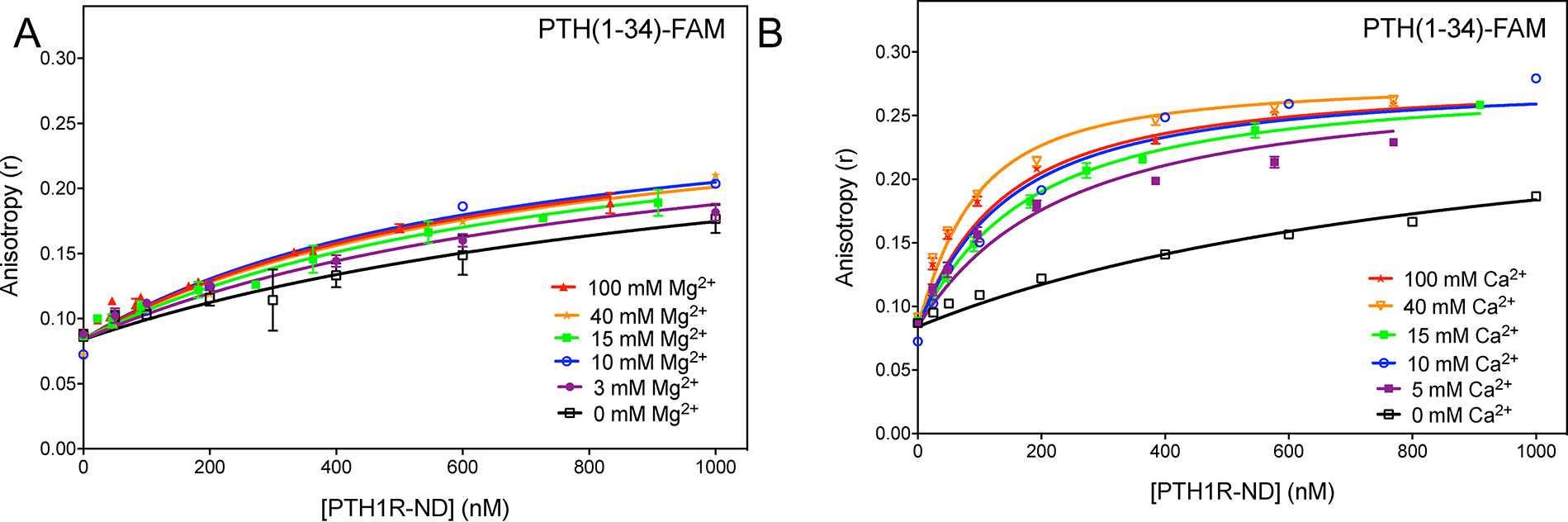
Titration of PTH(1–34)-FAM at 50 nM with PTH1R-ND in Mg2+-depleted buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, and 100 μM EDTA) at various concentration of (A) Mg2+ and (B) Ca2+. Unless specified, each curve shows the average of at least three replicates. The 10 mM Mg2+, 40 mM Mg2+ and 10 mM Ca2+ titrations show a single experiment.
Table 2:
Binding affinity of PTH(1–34) with divalent ion concentrations
| Mg2+ |
Ca2+ |
||
|---|---|---|---|
| Concentration (mM) | KD (nM) | Concentration (mM) | KD (nM) |
|
|
|
||
| 0 | 1132 ± 59 | 0 | 1132 ± 59 |
| 3 | 862 ± 36 | 5 | 200 ± 18 |
| 10 | 600 ± 52 | 10 | 114 ±19 |
| 15 | 732 ± 23 | 15 | 146 ± 6 |
| 40 | 650 ± 65 | 40 | 61 ± 4 |
| 100 | 628 ± 24 | 100 | 105 ± 6 |
Cell based cAMP assays. Ligand binding to PTH1R activates G protein signaling cascades, leading to the production of cAMP. To determine if the calcium dependent binding of PTH(1–34) affects the activation of PTH1R, we performed cAMP assays in the presence and absence of 15 mM Ca2+ (Figure 5). The addition of 15 mM Ca2+ did not affect the potency of PTH(1–34) in activating PTH1R. However, 15 mM Ca2+ significantly decreased the amount of cAMP produced by PTH(1–34) activation of PTH1R. Control experiments to test the effect of 15 mM Ca2+ on cAMP production in the absence of PTH(1–34) show that the cAMP produced decreases from 6.8 ± 2.6 to 0.8 ± 0.2 pmol/mg total protein upon addition of 15 mM Ca2+. Upon PTH(1–34) stimulation of PTH1R, the maximum cAMP produced was 276 ± 67 and 154 ± 42 pmol/mg total protein in the absence and presence of 15 mM Ca2+, respectively. These results suggest 15 mM Ca2+ decreases PTH(1–34) activation of PTH1R; however, it is still important to consider a portion of this decrease could be due to the effect of 15 mM Ca2+ alone on cAMP production of the cells.
Cell based cAMP assays.
Ligand binding to PTH1R activates G protein signaling cascades, leading to the production of cAMP. To determine if the calcium dependent binding of PTH(1–34) affects the activation of PTH1R, we performed cAMP assays in the presence and absence of 15 mM Ca2+ (Figure 5). The addition of 15 mM Ca2+ did not affect the potency of PTH(1–34) in activating PTH1R. However, 15 mM Ca2+ significantly decreased the amount of cAMP produced by PTH(1–34) activation of PTH1R. Control experiments to test the effect of 15 mM Ca2+ on cAMP production in the absence of PTH(1–34) show that the cAMP produced decreases from 6.8 ± 2.6 to 0.8 ± 0.2 pmol/mg total protein upon addition of 15 mM Ca2+. Upon PTH(1–34) stimulation of PTH1R, the maximum cAMP produced was 276 ± 67 and 154 ± 42 pmol/mg total protein in the absence and presence of 15 mM Ca2+, respectively. These results suggest 15 mM Ca2+ decreases PTH(1–34) activation of PTH1R; however, it is still important to consider a portion of this decrease could be due to the effect of 15 mM Ca2+ alone on cAMP production of the cells.
Figure 5: PTH(1–34) activates PTH1R to produce cAMP in the presence and absence of 15 mM Ca2+.
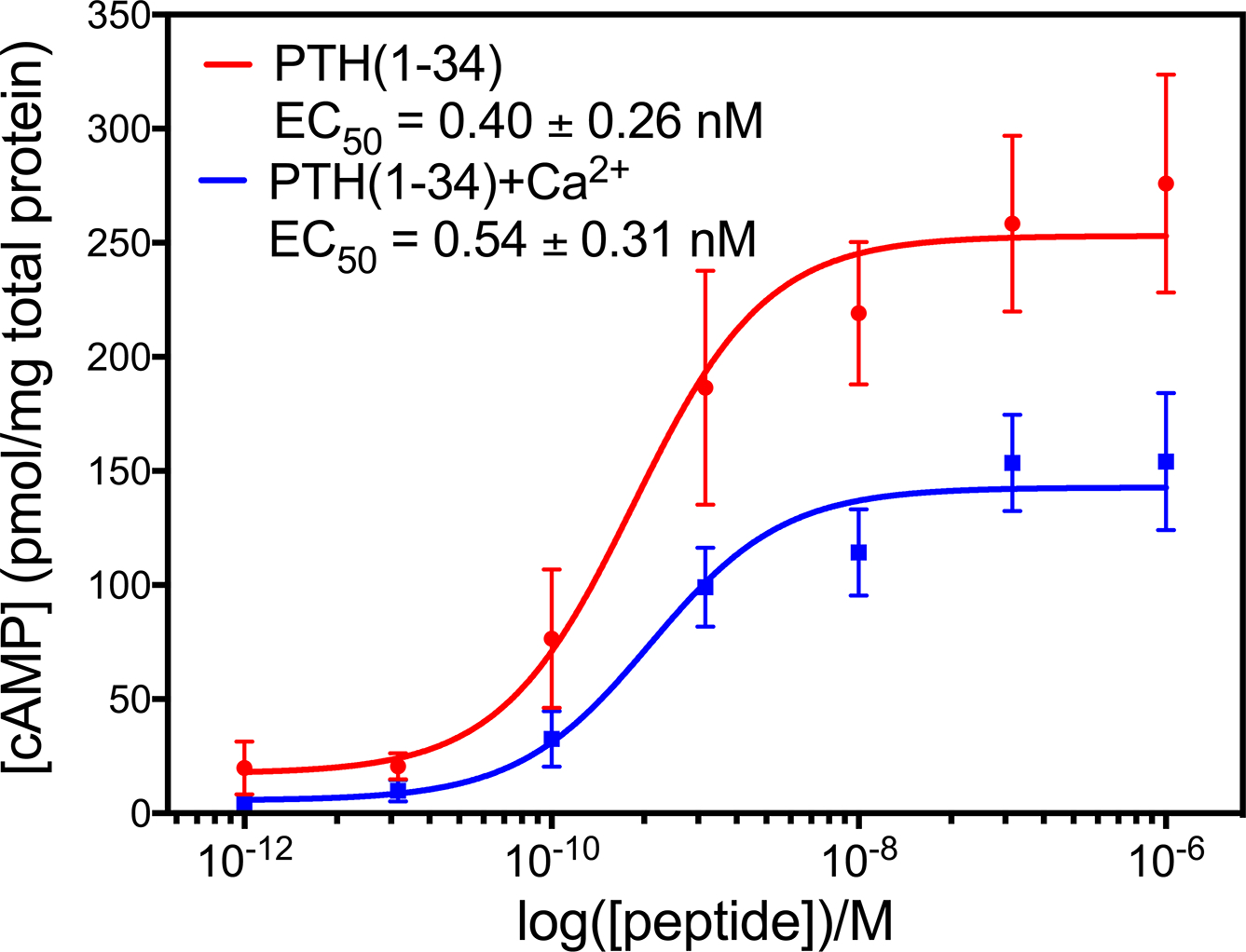
HEK293S cells expressing PTH1R were stimulated with indicated concentrations of PTH(1–34) with 0 mM Ca2+ (red) and 15 mM Ca2+ (blue) for 30 minutes before harvesting to measure the concentration of cAMP. The addition of 15 mM Ca2+ does not change the potency of PTH(1–34) but does decrease the efficacy of PTH(1–34). Titration curves show the average of 3 experiments.
Discussion.
Using our nanodisc purification method, we observed and investigated the effect of Ca2+ on peptide binding to PTH1R-ND. We showed the binding affinity of PTH(1–34), but not PTHrP(1–36), is enhanced 5-fold by 15 mM Ca2+, indicating a built-in mechanism of PTH(1–34) for sensing Ca2+ in millimolar concentrations. Flow cytometry titrations of HEK293S cells expressing PTH1R show PTH(1–34) binding increases with extracellular Ca2+ concentrations in a cellular context, confirming the results from our nanodisc titrations. Such high Ca2+ concentrations are relevant in the physiological context of bone remodeling, during which extracellular Ca2+ concentrations reach as high as 40 mM.27 We determined that the Ca2+-dependent binding requires the C-terminal residues of PTH(15–34), specifically the negatively charged residues Glu19 and Glu22. In addition, PTH(1–34) shows no Ca2+-dependent binding to the extracellular domain (ECD) of PTH1R in isolation, suggesting that the presence of the full receptor is necessary for PTH(1–34) to sense Ca2+ in millimolar concentrations. Cell-based cAMP assays show that 15 mM Ca2+ decreases the amount of cAMP produced by PTH(1–34) activation of PTH1R. This decrease is greater than that observed when 15 mM Ca2+ is added to the cells in the absence of PTH(1–34), suggesting high calcium concentrations likely attenuate PTH(1–34) activation of PTH1R. Finally, adjusting the reaction buffer to remove divalent ions shows PTH(1–34) binding affinity is affected by Ca2+ concentrations from 5 to 100 mM while the same Mg2+ concentrations do not greatly affect PTH(1–34) binding affinities, demonstrating the effect is specific to Ca2+. Our results show that PTH(1–34) has unique ability to sense Ca2+ in millimolar concentrations. The results highlight the remarkable implication that PTH(1–34) has two signaling functions: one in endocrine signaling for PTH1R activation and one in sensing extracellular Ca2+ concentrations.
We previously reported a 20-fold enhancement in PTH(1–34) binding to PTH1R-ND with the addition of 15 mM Ca2+ based on analysis of titrations under the same conditions but using concentrations of PTH1R-ND only up to 300 nM due to a limited yield of purification.36 In the current study, we optimized the expression level of the inducible cell lines that stably express PTH1R by additional rounds of antibiotic selection.53, 54 The optimized cell lines increased the yield 10-fold, reaching 0.75 μg per 10-cm plate of cell culture. The increased yield allowed us to use higher concentration of PTH1R-ND (~1000 nM) in each titration for more reliable data analysis in determining .
From the results of the double mutant titration and a comparison of the Ca2+ and Mg2+ effect, we propose that ligand binding to PTH1R is modulated by a combination of electrostatic interactions and specific Ca2+ binding. The E19AE22A mutant removed two non-conserved, negatively charged residues of PTH(1–34) (Scheme 2), which decreased the Ca2+-dependent peptide binding to PTH1R-ND from 4.9 ± 0.5 fold to 2.7 ± 0.3 fold. In addition, the binding of PTH(1–34)E19AE22A ( without 15 mM Ca2+ and with 15 mM Ca2+) is tighter than that of wild type PTH(1–34) ( without 15 mM Ca2+ and with 15 mM Ca2+). The enhanced binding of the mutant is attributed to electrostatic interactions with PTH1R, which has a total of 30 negatively charged residues (Glu or Asp) on the extracellular side (red in Scheme 3). The WT PTH(1–34) has a net charge of +3 while the PTH(1–34)E19AE22A mutant has a net charge of +5. Hence, the increase in net positive charges in the mutant can enhance electrostatic interactions with the negatively charged ECD of receptor for a higher binding affinity, which aligns with the Shimizu et al. (2002) report of enhanced binding of PTH(1–34)E19R to PTH1R in competitive, radiolabeled ligand binding assays.55, 56 More importantly, the addition of 15 mM Mg2+ has a much lower impact on the binding affinity of PTH(1–34) than the addition of 15 mM Ca2+ has. Therefore, our report of enhancement effect of PTH(1–34) binding to PTH1R is not merely electrostatic but specific to Ca2+.
Scheme 3: Extracellular domain of PTH1R has a high density of negatively charged residues.
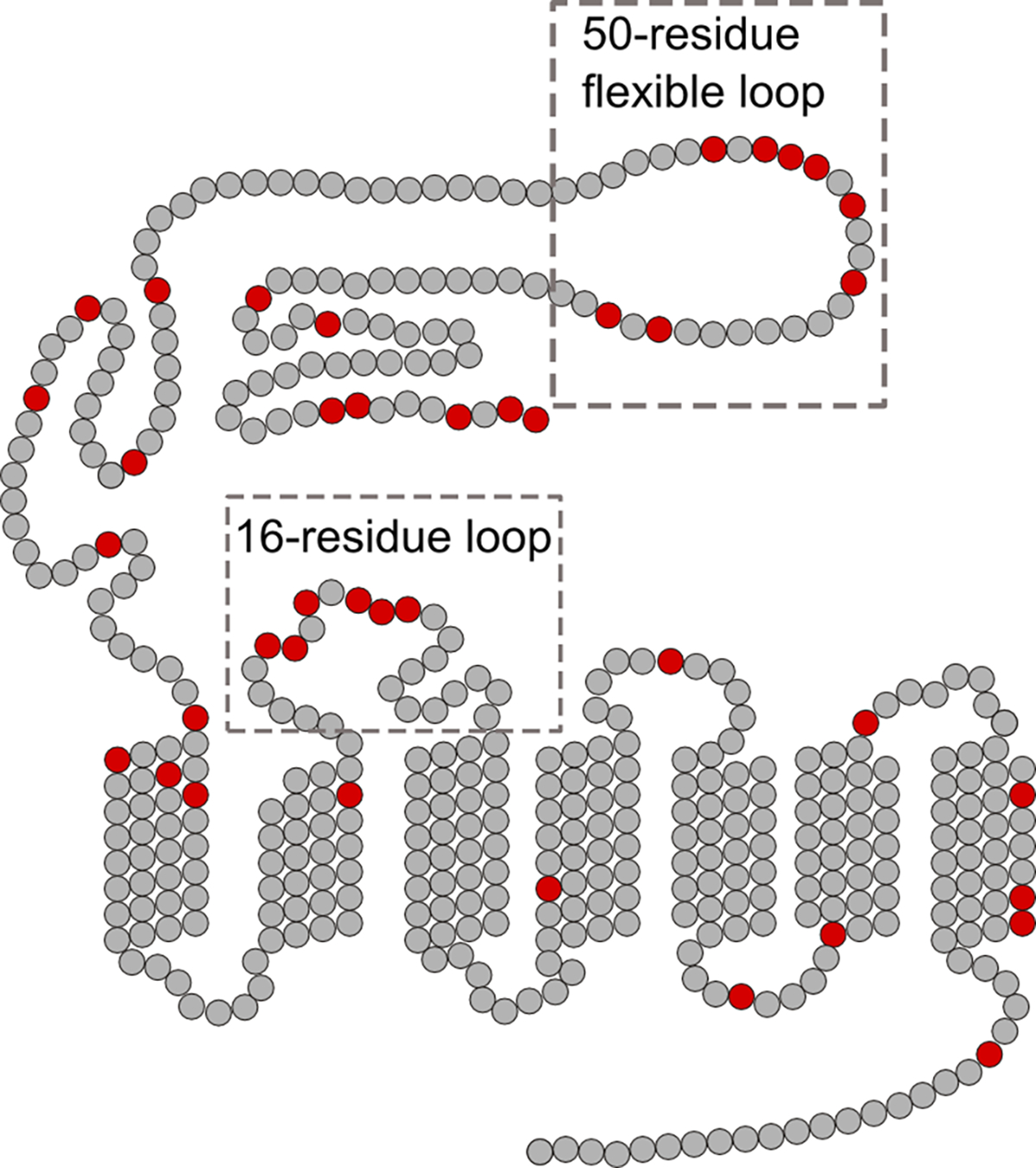
Snake plot of PTH1R shows negatively charged residues in red, which are clustered towards the extracellular side of the receptor. Two regions of PTH1R not conserved in family B GPCRs are boxed: the 50-residue flexible loop contains 8 negatively charged residues and the 16-residue extracellular loop 1 contains 6 negatively charged residues.
In probing the molecular mechanism of Ca2+ binding, we analyzed the sequence of both the ligand and receptor. We found that PTH(1–34) and PTH1R do not contain a canonical Ca2+ binding EF-hand motif, which is typically a sequence of 9 residues providing 7 ligands to coordinate a Ca2+ ion with affinity from nM to μM.57, 58 The weak, non-canonical Ca2+ binding we observe with PTH(1–34) in the mM range involves not only E19 and E22 but also likely other negatively charged or polar residues of PTH1R. This agrees with the observation that PTH(1–34) binding to the PTH1R extracellular domain (ECD) is independent of millimolar Ca2+ (Figure 3C). Moreover, sequence analysis shows that PTH1R contains a high density of glutamic or aspartic acid residues on the extracellular side of the receptor (Scheme 3). In particular, PTH1R contains a 50-residue flexible loop on the ECD, which cannot be resolved in the crystal structures. This loop is not present in other family B GPCRs, but contains 8 negatively charged residues (Scheme 3). In addition, the 16 residues of extracellular loop 1 also include 6 negatively charged residues (Scheme 3). We propose that these negatively charged residues on the extracellular side of PTH1R can potentially coordinate with E19 and E22 of PTH(1–34) in sensing Ca2+ in the millimolar range, a concentration relevant in bone remodeling.27
The titration curves of the two chimeric peptides show that residues 15–34 are needed for the Ca2+-dependent binding of PTH(1–34). The results of the PTH(1–14)PTHrP(15–36) titrations show a 1.5 ± 0.1 -fold change in binding with the addition of 15 mM Ca2+, corroborating the double mutant results by implicating residues 15–34 of PTH for Ca2+ sensing (Figure 1D). Thus, we expected that the chimera PTHrP(1–14)PTH(15–34) containing the PTH(15–34) would retain the Ca2+ sensing ability of PTH(1–34). However, we observed no binding to PTH1R-ND in both the presence and absence of 15 mM Ca2+ (Figure 1E). We performed control experiments using circular dichroism spectroscopy and determined this lack of binding is not due to loss of secondary structure of PTHrP(1–14)PTH(15–34) (SI Figure S1). Gardella et al. (1995) also observed that PTHrP(1–14) is incompatible with the fragment PTH(15–34), likely because the combination of the two fragments disrupts important allosteric interactions between the N- and C-terminus of PTH(1–34) through PTH1R.46 In our studies, we further showed that even the addition of 15 mM Ca2+ cannot rescue the binding of PTHrP(1–14)PTH(15–34). This loss of binding affinity (Figure 1E) highlights the complex nature of the interactions of PTH(1–34) and PTHrP(1–36) with PTH1R. Understanding these interactions may provide a new understanding of the Ca2+ sensing ability of the PTH(1–34) and allostery in family B GPCRs.
After studying the ligand residues important for Ca2+-dependent binding, we investigated components of PTH1R that might contribute to Ca2+-dependent binding. Because the mutant and chimeric ligands show PTH(15–34) is important for the Ca2+ sensing ability, and the two domain binding model suggests that C-terminal fragment interacts with the extracellular domain (ECD) of the receptor, we decided to test the binding of PTH(1–34) to the ECD (Figure 3). Our results show PTH(1–34) binding to the ECD in isolation shows no Ca2+ dependence, which suggests the presence of the transmembrane domain is crucial for the Ca2+ sensing of PTH(1–34). We also obtained experimental data to show that the Ca2+-dependent PTH(1–34) binding to PTH1R does not depend on the lipid bilayer. Using detergent solubilized PTH1R, fluorescence anisotropy assays still show that 15 mM Ca2+ can enhance binding of PTH(1–34) (SI Figure S4). The lack of Ca2+-dependent binding using the ECD of PTH1R adds to the growing evidence of allosteric interactions between the two domains of binding for PTH(1–34), thus expanding our understanding of ligand binding in family B GPCRs.
Prior to our studies, the Ca2+-sensing ability of PTH(1–34) was unobservable likely due to the limitations of cell based assays and other purification methods. To determine if PTH1R activation is affected by 15 mM Ca2+, we performed cell-based cAMP accumulation assays to measure PTH(1–34) activation of PTH1R in the presence and absence of 15 mM Ca2+. We observed that 15 mM Ca2+ does not significantly affect the potency of PTH(1–34); however, it decreases the amount of cAMP produced from 276 ± 67 to 154 ± 42 pmol/mg total protein. The observed decrease in cAMP production in the presence of 15 mM Ca2+ potentially provides an a novel understanding about the physiological relevance of the PTH(1–34)’s Ca2+ sensing ability. Physiologically, PTH(1–34) is released into circulation when Ca2+ levels are low, leading to bone resorption and Ca2+ release. However, previous studies show PTH(1–34) has opposing effects depending on the duration of exposure. Prolonged exposure to PTH(1–34) breaks down bones to release Ca2+ while intermittent exposure leads to increased bone denisty.26, 29, 30 Despite the importance of PTH(1–34) used as a drug for osteoporosis, the molecular mechanisms defining this paradoxical signaling of PTH are unknown. While our results do not shed light on the underlying paradox of PTH(1–34)’s role in bone remodeling, they do provide interesting insights. For instance, our ligand binding results show tighter binding of PTH(1–34) in the presence of very high Ca2+ and our cAMP results show decreased cAMP production of PTH(1–34) activation of PTH1R with 15 mM Ca2+. Thus, it is possible that the observed Ca2+ sensing ability of PTH(1–34) increases its binding affinity to PTH1R while negatively regulating PTH1R activation to decrease cAMP production. This initial hypothesis requires future experiments to determine if 15 mM Ca2+ affects other aspects of PTH1R signaling in more physiologically relevant experimental systems.
While the signaling process of bone remodeling is complicated, it is well established that the duration of PTH exposure determines if the hormone has an anabolic or catabolic effect on bone density.26, 32 Intermittent exposure increases bone density through increased osteoblast numbers while prolonged exposure activates osteoclasts, which break down bones, releasing extracellular Ca2+.28 In addition, PTH(1–34) is an FDA-approved peptide hormone drug for severe osteoporosis, with daily injections leading to increased bone density.28–30 Thus, the increased binding affinity of PTH(1–34) with high extracellular Ca2+ could modulate PTH(1–34) signaling to affect osteoblast or osteoclast activity. Furthermore, interactions with accessory proteins and downstream partners such as NHERFs and β-arrestin may also be affected by the Ca2+-sensing ability of PTH(1–34).59, 60 Understanding the intricacies of the dual functions of PTH(1–34) signaling will also help us to better target the PTH1R pathway in osteoporosis treatment, leading to more efficacious drugs and treatment paradigms.
The proposed Ca2+-sensing ability of PTH(1–34) is unprecedented for a peptide hormone. Our results show PTH(1–34) is able to sense the extracellular environment to change how it binds and activates PTH1R. This Ca2+ sensing ability is unique among both family A and family B GPCRs. Specifically, we have purified another family B GPCR, glucagon-like peptide 1 receptor (GLP1R) in nanodiscs61 and determined that its ligand binding and activation is not affected by 15 mM Ca2+ (SI Figure S5). Of interest, two family C GPCRs, the calcium sensing receptor (CaSR)62, and the metabotropic glutamate receptor (mGluR)63 are modulated by extracellular calcium. However, the mechanism of the Ca2+ effect on these receptors is distinct from the Ca2+ sensing ability of PTH(1–34). For instance, Ca2+ alone activates the CaSR with an EC50 value of around 4 mM and binds to a distinct and conserved binding site in CaSRs.64 The mechanism of the Ca2+ effect on mGluR is distinct because Ca2+ binds to a specific binding site of 3 acidic residues of mGluR which modulates the effect of glutamate and other ligands.65 In contrast to the built-in calcium sensing ability of PTH(1–34), Ca2+ modulation of mGluR occurs for multiple, small molecule ligands.65 Therefore, our studies underline the uniqueness of a single hormone with dual functions of sensing the extracellular environment and carrying endocrine signals.
In summary, we have highlighted a novel, built-in Ca2+ sensing ability of PTH(1–34), one of two ligands that binds to PTH1R. We probed domains of the ligands that contribute to the Ca2+ sensing ability and found that two residues in the C-terminal domain of PTH(1–34), E19 and E22, decrease the Ca2+ dependent ligand binding. In addition, our studies with chimeric ligands show ligand binding is more complicated and likely influenced by long range or allosteric interactions. Future studies aided by molecular modeling to investigate these allosteric interactions as well as domains of PTH1R that contribute to Ca2+-dependent ligand binding will help elucidate the molecular mechanisms of the ligand binding in PTH1R, facilitating studies to uncover the intricate role of PTH in the bone remodeling process.
Methods
Expression and purification of PTH1R.
PTH1R was overexpressed in HEK293S GnTI− cell lines following previously published nanodisc purification protocols.36 Briefly, PTH1R with the C terminal 1D4 epitope tag was cloned into tetracycline-inducible TetO-pACMV vectors.53, 66 The vectors were transiently transfected into HEK293S GnTI− cells and treated with 1mg/mL genetecin (AmericanBio, Natick, MA) for rounds of selection. The clonal cell line expressing the highest amount of PTH1R was determined by western blot using a mouse 1D4 monoclonal antibody (University of British Colombia). For purification, 10-cm plates of stable cells lines were induced with 2 μg/μL tetracycline and 0.55 mg/mL sodium butyrate for 48 hrs. The cells were harvested, lysed and the membrane fraction separated by sucrose density gradient ultracentrifugation (110,000 × g). The membrane pellet was solubilized with solubilization buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 5 mM CaCl2, 5 mM MgCl2, 2 mM EDTA, 10% glycerol and 0.5% n-Dodecyl-β-D-maltoside (DDM, ACROS Organics)). Membrane scaffold protein (MSP1E3D1)67 and detergent-solubilized 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) were mixed with the solubilized membrane proteins, and Bio-beads were added to initiate nanodisc assembly for 24 hrs. Nanodiscs containing PTH1R were separated from the mixture by single-step affinity purification against 1D4 epitope.
Fluorescence anisotropy.
Fluorescence anisotropy experiments measured the binding affinity between PTH1R-ND and the following peptide ligands PTH(1–34), PTH(1–36), PTH(1–14)PTHrP(15–36), PTHrP(1–14)PTH(15–36) and PTH(1–34)E19AE22A. Each of the peptides was labeled with 5/6-fluorescein (FAM) at Lys13 (Neobiolabs, Woburn, MA), previously shown in crystal structures as pointing to the solvent and not perturbing receptor binding.12, 13 The peptides were ordered from Neobiolabs (Woburn, MA), and were weighed and dissolved in anisotropy buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 3mM MgCl2, and 100 μM EDTA). FAM-labeled peptides (~50 nM) were titrated with various concentrations of PTH1R-ND (50nM-1000nM) and the anisotropy recorded using a PTI QuantaMaster C-61 two-channel fluorescence spectrophotometer at 30°C with excitation/emission of 497 nm/518 nm and the slit width of 5 nm. In one experiment, each measurement was averaged over 30 sec with one reading per second. To measure Ca2+-dependent ligand binding, 15 mM CaCl2 was added to the cuvette at each PTH1R-ND concentration and the fluorescence intensity was recorded again to determine the anisotropy. Each titration curve presented in Figure 1 is average of three or more experiments that were carried out using independent preparations of purified PTH1R-ND. CaCl2 solutions were diluted in anisotropy buffer from a 1 M stock solution of CaCl2 dihydrate, minimum 99.0% in ddH2O (Sigma). MgCl2 solutions were diluted in anisotropy buffer from 1 M MgCl2 solution (AmericanBio).
Data Analysis.
The dissociation constant for the equilibrium of the peptide bound receptor dissociating into the free and free receptors is expressed by Eq. (1), which is related to the measured anisotropy values , as described in Eq. (2)36
| (1) |
| (2) |
where is the concentration of PTH1R-ND; is the concentration of peptide; is anisotropy of the bound peptide; and is the anisotropy of the free peptide.
In anisotropy assays, free peptides have low anisotropy due to their small size and fast rotation, while peptides bound to PTH1R-ND have high anisotropy due to the large size of the PTH(1–34)-PTH1R-ND complex and slower rotation. Since the size of molecular entities and photochemical properties of the fluorescence probe determine the values of anisotropy, the anisotropy of free peptides are approximately the same for each peptide being studied, as is the anisotropy of those peptides bound to PTH1R-ND , as shown in the titration curves for the PTH1R-ND concentration at zero and saturated values. Thus, can be determined by fitting the titration curves for each peptide under various conditions.
Flow Cytometry Titrations.
10 cm plates of HEK293S PTH1R stable cells lines were induced with 2 μg/μL tetracycline and 0.55 mg/mL sodium butyrate for 48 hrs. Cells were lifted from the plates with 1X PBS buffer with 2 mM EDTA. Cells were centrifuged and resuspended in Tris buffer with 1% BSA and counted using a hemocytometer. 400,000 cells were aliquoted into eppendorf tubes on ice. Different concentrations of PTH(1–34) were added to each aliquot with or without 15 mM Ca2+. Cells were incubated with PTH(1–34)-FAM for 5 minutes on ice and then sorted using an Accuri C6 Flow cytometer. As a control, uninduced cells were similarly harvested, incubated with the different concentrations of PTH(1–34)-FAM with or without Ca2+, and sorted. For each PTH(1–34)-FAM condition, the mean fluorescence intensity was recorded. To determine the of PTH(1–34) in the presence and absence of 15 mM Ca2+, the data was fitted using the saturation binding equation for one site with total binding measured (equation 3),47 where is the concentration of PTH(1–34), is the mean fluorescence intensity measured, is the maximum fluorescence of specific binding, is the non-specific binding, and the background is the measurement with no labeled ligand present. The value of was obtained from the slope of a linear equation fit to the data from uninduced cells and the background fixed as the mean fluorescence intensity of samples with no PTH(1–34)-FAM added.
| (3) |
Expression and purification of the extracellular domain of PTH1R.
The DNA constructs, pETDuet-1-MBP-PTH1R(ECD)-DsbC and pET15b-DsbC were provided by the Schepartz laboratory (Yale University). pETDuet-1 is a dual expression vector that expresses MBP-PTH1R ECD and the chaperone DsbC under two separate promoters.68 The maltose binding protein (MBP) was fused to the N-terminus of PTH1R ECD to provide additional stability. pET15b-DsbC is a single expression vector that expresses the chaperone DsbC. The His-tagged pET15b-DsbC construct was transformed into E. coli Origami 2 cells, grown to OD600~0.6 and induced for 4 hrs at 37°C with 0.4 mM IPTG. Cells were harvested and lysed by sonication. The clear lysate was incubated with Ni-NTA beads for 1.5 hrs at 4°C. The beads were then washed with 30 mM imidazole and the protein was eluted with 250 mM imidazole. All fractions containing DsbC were pooled and the His-tag was cleaved with biotinylated thrombin to yield pure DsbC. The biotinylated thrombin was removed by passing through streptavidin agarose, collect the supernatant and dialyze again the storage buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5 mM EDTA and 1 mM DTT).
Purification of MBP-PTH1R ECD was carried out as follows. Transformed E. coli Origami 2 cells were grown to OD600~0.6 and induced overnight at 16°C with 0.4 mM IPTG. Cells were harvested and lysed by French press. The lysate was incubated overnight with Ni-NTA beads at 4°C. The beads were then washed with 25 mM imidazole and the protein was eluted with 250 mM imidazole. All fractions containing MBP-PTH1R ECD were pooled and subjected to a disulfide reshuffling reaction in 1 mM each of reduced and oxidized glutathione, 1 mg/ml MBP-PTH1R ECD and 0.4 mg/ml DsbC at 20°C for 20 h. The resultant mixture was incubated with Ni-NTA beads to remove DsbC. The sample was then concentrated and passed twice through a Sephadex 75 gel filtration column. The final pure protein (3.98 mg from a 3 L culture) was stored at −80°C in 50% glycerol.
Fluorescence polarization assays.
Flu-PTH(1–34), which refers to PTH(1–34) with a fluorescein molecule attached to the N-terminal end, was prepared by the Schepartz lab. Direct binding experiments between MBP-PTH1R-ECD and Flu-PTH(1–34) were carried out as follows. First, 16 protein solutions were prepared by 1:1 serial dilutions of MBP-PTH1R ECD in buffer starting with a 200 μM stock. Protein solutions at each concentration in a volume of 18 μL was then transferred in triplicate to a Corning® flat-bottom black polystyrene 384-well plate. Flu-PTH(1–34) (2 μL of a 250 nM stock solution) was then added to each well and incubated at room temperature for 3 hrs. Fluorescence polarization (excitation and emission of 485 nm and 530 nm, respectively) was recorded on an Analyst AD Fluorescence Plate Reader (LJL Biosystems, Sunnyvale, CA). The data were fitted using an equation similar to what was described for the anisotropy assay:
| (4) |
where is the measured fluorescence polarization, is the fluorescence polarization of the free labeled ligand, is the maximum fluorescence polarization of the peptide-protein complex, is the total peptide concentration and is the total protein concentration.69
Cell-based cAMP assays.
HEK293S cells expressing PTH1R were grown and induced in 48-well plates. After ~48 hrs induction, cells were washed with serum-free, Ca2+-free media (Ca2+-free DMEM and 0.2% BSA). Then, cells were treated with 120 μL of cAMP assay buffer (Ca2+-free DMEM containing 200 uM IBMX, 1mg/ml BSA, 35 mM HEPES, pH 7.4), followed by the addition of 60 μL of binding buffer (50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 5 mM KCl, 0.5% FBS and 5% heat-inactivated FBS) containing the peptides (concentrations indicated) and incubated for 30 minutes. Cells were lysed with the lysis buffer (0.1 M HCl and 0.5% Triton X-100), and the cAMP amount under each condition was determined using the Direct cAMP ELISA kit (Enzo Life Sciences). Each point was measured in duplicate in each experiment and each curve represents an average of three experiments.
Supplementary Material
Acknowledgements
The authors would like to thank L. Reagan (Yale University) for use of the PTI QuantaMaster C-61 two-channel fluorescence spectrophotometer, D. Spiegel (Yale University) for use of the Acuri C6 Flow Cytometer and V. Batista (Yale University) for providing the figure of PTH1R in a nanodisc (Scheme 1). JNS received support from the Science, Technology and Research Scholars program (Yale University) and MEB received support from Yale College Dean’s Office. ECY is the recipient of the NSF CAREER Award (MCB-0955407). KJC is the recipient of the NIH Biophysics Training Grant (T32 GM008283–27).
Footnotes
Supplementary Information
Circular dichriosm spectra and cAMP activation assays of all tested hormones, five flow cytometry trials, glucagon-like peptide 1 titrations with and without calcium, PTH(1–34) binding to detergent purified PTH1R with and without calcium (SI Figures S1–S5) and associated methods.
References
- [1].Stevens RC, Cherezov V, Katritch V, Abagyan R, Kuhn P, Rosen H, and Wüthrich K (2013) The GPCR Network: a large-scale collaboration to determine human GPCR structure and function, Nat. Rev. Drug Discovery 12, 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Culhane KJ, Liu Y, Cai Y, and Yan EC (2015) Transmembrane signal transduction by peptide hormones via family BG protein-coupled receptors, Front. Pharmacol. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hollenstein K, de Graaf C, Bortolato A, Wang M-W, Marshall FH, and Stevens RC (2014) Insights into the structure of class B GPCRs, Trends in Pharmacol. Sci. 35, 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pal KM, Karsten: Xu H Eric. (2012) Structure and mechanism for recognition of peptide hormones by Class B G protein coupled receptors, Acta Pharmacol. Sin. 33, 300–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Liang YL, Khoshouei M, Radjainia M, Zhang Y, Glukhova A, Tarrasch J, Thal DM, Furness SGB, Christopoulos G, Coudrat T, Danev R, Baumeister W, Miller LJ, Christopoulos A, Kobilka BK, Wootten D, Skiniotis G, and Sexton PM (2017) Phase-plate cryo-EM structure of a class B GPCR-G-protein complex, Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gardella T, and Jüppner H (2000) Interaction of PTH and PTHrP with their receptors, Rev. Endocr. Metab. Disord. 1, 317–329. [DOI] [PubMed] [Google Scholar]
- [7].Mannstadt M, Jüppner H, and Gardella TJ (1999) Receptors for PTH and PTHrP: their biological importance and functional properties, Am. J. Physiol.-Renal Physiol. 277, F665–F675. [DOI] [PubMed] [Google Scholar]
- [8].Mosekilde L, Søgaard C, Danielsen C, Tørring O, and Nilsson M (1991) The Anabolic Effects of Human Parathyroid Hormone (hPTH) on Rat Vertebral Body Mass Are also Reflected in the Quality of Bone, Assessed by Biomechanical Testing: A Comparison Study between hPTH-(1–34) and hPTH-(1–84)*, Endocrinology 129, 421–428. [DOI] [PubMed] [Google Scholar]
- [9].Parthier C, Reedtz-Runge S, Rudolph R, and Stubbs MT (2009) Passing the baton in class B GPCRs: peptide hormone activation via helix induction?, Trends Biochem. Sci 34, 303–310. [DOI] [PubMed] [Google Scholar]
- [10].Wheatley M, Wootten D, Conner MT, Simms J, Kendrick R, Logan RT, Poyner DR, and Barwell J (2012) Lifting the lid on GPCRs: the role of extracellular loops, Br. J. Pharmacol. 165, 1688–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Castro M, Nikolaev VO, Palm D, Lohse MJ, and Vilardaga J-P (2005) Turn-on switch in parathyroid hormone receptor by a two-step parathyroid hormone binding mechanism, Proc. Natl. Acad. Sci. U. S. A. 102, 16084–16089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Pioszak AA, Parker NR, Gardella TJ, and Xu HE (2009) Structural Basis for Parathyroid Hormone-related Protein Binding to the Parathyroid Hormone Receptor and Design of Conformation-selective Peptides, J. Biol. Chem. 284, 28382–28391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Pioszak AA, and Xu HE (2008) Molecular recognition of parathyroid hormone by its G protein-coupled receptor, Proc. Natl. Acad. Sci. U. S. A. 105, 5034–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Thomas BE, Woznica I, Mierke DF, Wittelsberger A, and Rosenblatt M (2008) Conformational changes in the parathyroid hormone receptor associated with activation by agonist, Mol. Endocrinol. 22, 1154–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cupp ME, Nayak SK, Adem AS, and Thomsen WJ (2013) Parathyroid Hormone (PTH) and PTH-Related Peptide Domains Contributing to Activation of Different PTH Receptor–Mediated Signaling Pathways, J. Pharmacol. Exp. Ther.345, 404–418. [DOI] [PubMed] [Google Scholar]
- [16].Bisello A, Chorev M, Rosenblatt M, Monticelli L, Mierke DF, and Ferrari SL (2002) Selective ligand-induced stabilization of active and desensitized parathyroid hormone type 1 receptor conformations, J. Biol. Chem. 277, 38524–38530. [DOI] [PubMed] [Google Scholar]
- [17].Okazaki M, Ferrandon S, Vilardaga J-P, Bouxsein ML, Potts JT, and Gardella TJ (2008) Prolonged signaling at the parathyroid hormone receptor by peptide ligands targeted to a specific receptor conformation, Proc. Natl. Acad. Sci. U. S. A. 105, 16525–16530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shimizu M, Carter PH, Khatri A, Potts JJT, and Gardella TJ (2001) Enhanced Activity in Parathyroid Hormone-(1–14) and -(1–11): Novel Peptides for Probing Ligand-Receptor Interactions*, Endocrinology 142, 3068–3074. [DOI] [PubMed] [Google Scholar]
- [19].Takasu H, Gardella TJ, Luck MD, Potts JT, and Bringhurst FR (1999) Amino-terminal modifications of human parathyroid hormone (PTH) selectively alter phospholipase C signaling via the type 1 PTH receptor: implications for design of signal-specific PTH ligands, Biochemistry 38, 13453–13460. [DOI] [PubMed] [Google Scholar]
- [20].Goldman ME, McKee RL, Caulfield MP, Reagan JE, Levy JJ, Gay CT, DeHaven PA, Rosenblatt M, and Chorev M (1988) A new highly potent parathyroid hormone antagonist: [D-TRPl2, TYR34] bPTH-(7–34) NH2, Endocrinology 123, 2597–2599. [DOI] [PubMed] [Google Scholar]
- [21].Shimizu N, Dean T, Tsang JC, Khatri A, Potts JT, and Gardella TJ (2005) Novel Parathyroid Hormone (PTH) Antagonists That Bind to the Juxtamembrane Portion of the PTH/PTH-related Protein Receptor, J. Biol. Chem. 280, 1797–1807. [DOI] [PubMed] [Google Scholar]
- [22].Liu Y, Cai Y, Liu W, Li XH, Rhoades E, and Yan EC (2015) Triblock peptide-linker-lipid molecular design improves potency of peptide ligands targeting family B G protein-coupled receptors, Chem. Commun. (Camb) 51, 6157–6160. [DOI] [PubMed] [Google Scholar]
- [23].Shimizu M, Potts JT, and Gardella TJ (2000) Minimization of Parathyroid Hormone: novel amino-terminal parathyroid hormone fragments with enhanced potency in activating the type-1 parathyroid hormone receptor, J. Biol. Chem. 275, 21836–21843. [DOI] [PubMed] [Google Scholar]
- [24].Shimizu N, Guo J, and Gardella TJ (2001) Parathyroid Hormone (PTH)-(1–14) and-(1–11) Analogs Conformationally Constrained by α-Aminoisobutyric Acid Mediate Full Agonist Responses via the Juxtamembrane Region of the PTH-1 Receptor, J. Biol. Chem. 276, 49003–49012. [DOI] [PubMed] [Google Scholar]
- [25].Burr DB (2002) Targeted and nontargeted remodeling, Bone 30, 2–4. [DOI] [PubMed] [Google Scholar]
- [26].Parfitt AM (2002) Targeted and nontargeted bone remodeling: relationship to basic multicellular unit origination and progression, Bone 30, 5–7. [DOI] [PubMed] [Google Scholar]
- [27].Silver I, Murrills R, and Etherington D (1988) Microelectrode studies on the acid microenvironment beneath adherent macrophages and osteoclasts, Exp. Cell Res. 175, 266–276. [DOI] [PubMed] [Google Scholar]
- [28].Locklin RM, Khosla S, Turner RT, and Riggs BL (2003) Mediators of the biphasic responses of bone to intermittent and continuously administered parathyroid hormone, J. Cell. Biochem. 89, 180–190. [DOI] [PubMed] [Google Scholar]
- [29].Aslan D, Andersen MD, Gede LB, de Franca TK, Jorgensen SR, Schwarz P, and Jorgensen NR (2012) Mechanisms for the bone anabolic effect of parathyroid hormone treatment in humans, Scand. J. Clin. Lab. Invest. 72, 14–22. [DOI] [PubMed] [Google Scholar]
- [30].Jilka RL (2007) Molecular and cellular mechanisms of the anabolic effect of intermittent PTH, Bone 40, 1434–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Manolagas SC, and Jilka RL (1995) Mechanisms of disease - bone marrow, cytokines, and bone remodeling - emerging insights into the pathophysiology of osteoporosis, N. Engl. J. Med. 332, 305–311. [DOI] [PubMed] [Google Scholar]
- [32].Yang D, Singh R, Diviet P, Guo J, Bouxsein ML, and Bringhurst FR (2007) Contributions of parathyroid hormone (PTH)/PTH-related peptide receptor signaling pathways to the anabolic effect of PTH on bone, Bone 40, 1453–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ohtaki T, Ogi K, Masuda Y, Mitsuoka K, Fujiyoshi Y, Kitada C, Sawada H, Onda H, and Fujino M (1998) Expression, Purification, and Reconstitution of Receptor for Pituitary Adenylate Cyclase-activating Polypeptide large - scale purification of a fucntionally active G protein-coupled receptor produced in SF9 insect cells J. Biol. Chem. 273, 15464–15473. [DOI] [PubMed] [Google Scholar]
- [34].Shimada M, Chen X, Cvrk T, Hilfiker H, Parfenova M, and Segre GV (2002) Purification and characterization of a receptor for human parathyroid hormone and parathyroid hormone-related peptide, J. Biol. Chem. 277, 31774–31780. [DOI] [PubMed] [Google Scholar]
- [35].Gan L, Alexander JM, Wittelsberger A, Thomas B, and Rosenblatt M (2006) Large-scale purification and characterization of human parathyroid hormone-1 receptor stably expressed in HEK293S GnTI− cells, Protein Expression Purif. 47, 296–302. [DOI] [PubMed] [Google Scholar]
- [36].Mitra N, Liu Y, Liu J, Serebryany E, Mooney V, DeVree BT, Sunahara RK, and Yan ECY (2012) Calcium-Dependent Ligand Binding and G-protein Signaling of Family B GPCR Parathyroid Hormone 1 Receptor Purified in Nanodiscs, ACS Chem. Biol. 8, 617–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Michalke K, Huyghe C, Lichière J, Gravière M-E, Siponen M, Sciara G, Lepaul I, Wagner R, Magg C, and Rudolph R (2010) Mammalian G protein-coupled receptor expression in Escherichia coli: II. Refolding and biophysical characterization of mouse cannabinoid receptor 1 and human parathyroid hormone receptor 1, Anal. Biochem. 401, 74–80. [DOI] [PubMed] [Google Scholar]
- [38].Schröder-Tittmann K, Bosse-Doenecke E, Reedtz-Runge S, Ihling C, Sinz A, Tittmann K, and Rudolph R (2010) Recombinant expression, in vitro refolding, and biophysical characterization of the human glucagon-like peptide-1 receptor, Biochemistry 49, 7956–7965. [DOI] [PubMed] [Google Scholar]
- [39].Unson CG (2008) Expression of glucagon receptors in tetracycline-inducible HEK293S GnT1− stable cell lines: An approach toward purification of receptor protein for structural studies, Pept. Sci.90, 287–296. [DOI] [PubMed] [Google Scholar]
- [41].Bayburt TH, and Sligar SG (2010) Membrane protein assembly into Nanodiscs, FEBS Lett. 584, 1721–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Civjan NR, Bayburt TH, Schuler MA, and Sligar SG (2003) Direct solubilization of heterologously expressed membrane proteins by incorporation into nanoscale lipid bilayers, BioTechniques 35, 556–563. [DOI] [PubMed] [Google Scholar]
- [43].Chorev M, Behar V, Yang Q, Rosenblatt M, Mammi S, Maretto S, Pellegrini M, and Peggion E (1995) Conformation of parathyroid hormone antagonists by CD, nmr, and molecular dynamics simulations, Biopolymers 36, 485–495. [DOI] [PubMed] [Google Scholar]
- [44].Rossi AM, and Taylor CW (2011) Analysis of protein-ligand interactions by fluorescence polarization, Nat. Protoc. 6, 365–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Luck MD, Carter PH, and Gardella TJ (1999) The (1–14) fragment of parathyroid hormone (PTH) activates intact and amino-terminally truncated PTH-1 receptors, Mol. Endocrinol. 13, 670–680. [DOI] [PubMed] [Google Scholar]
- [46].Gardella TJ, Luck MD, Wilson AK, Keutmann HT, Nussbaum SR, Potts JT, and Kronenberg HM (1995) Parathyroid Hormone (PTH)-PTH-related Peptide Hybrid Peptides Reveal Functional Interactions between the 1–14 and 15–34 Domains of the Ligand, J. Biol. Chem.270, 6584–6588. [DOI] [PubMed] [Google Scholar]
- [47].Bajaj A, Ćelić A, Ding F-X, Naider F, Becker JM, and Dumont ME (2004) A Fluorescent α-Factor Analogue Exhibits Multiple Steps on Binding to Its G Protein Coupled Receptor in Yeast, Biochemistry 43, 13564–13578. [DOI] [PubMed] [Google Scholar]
- [48].Sklar Larry A., Edwards Bruce S., Graves Steven W., Nolan John P., and Prossnitz ER (2002) Flow Cytometric Analysis of Ligand-Receptor Interactions and Molecular Assemblies, Ann. Rev. Biophys. Biomol. Struct.31, 97–119. [DOI] [PubMed] [Google Scholar]
- [49].Sridharan R, Zuber J, Connelly SM, Mathew E, and Dumont ME (2014) Fluorescent Approaches for Understanding Interactions of Ligands with G Protein Coupled Receptors, Biochim. Biophys. Acta 1838, 15–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Han K-C, Kim JH, Kim K-H, Kim EE, Seo J-H, and Yang EG (2010) Identification of farnesoid X receptor modulators by a fluorescence polarization-based interaction assay, Anal. Biochem. 398, 185–190. [DOI] [PubMed] [Google Scholar]
- [51].Ou J, Tu H, Shan B, Luk A, DeBose-Boyd RA, Bashmakov Y, Goldstein JL, and Brown MS (2001) Unsaturated fatty acids inhibit transcription of the sterol regulatory element-binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the LXR, Proc. Natl. Acad. Sci. U. S. A. 98, 6027–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bledsoe RK, Montana VG, Stanley TB, Delves CJ, Apolito CJ, McKee DD, Consler TG, Parks DJ, Stewart EL, and Willson TM (2002) Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition, Cell 110, 93–105. [DOI] [PubMed] [Google Scholar]
- [53].Reeves PJ, Callewaert N, Contreras R, and Khorana HG (2002) Structure and function in rhodopsin: High-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line, Proc. Natl. Acad. Sci. U. S. A. 99, 13419–13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Reeves PJ, Thurmond RL, and Khorana HG (1996) Structure and function in rhodopsin: High level expression of a synthetic bovine opsin gene and its mutants in stable mammalian cell lines, Proc. Natl. Acad. Sci. U. S. A. 93, 11487–11492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Piserchio A, Shimizu N, Gardella TJ, and Mierke DF (2002) Residue 19 of the Parathyroid Hormone: Structural Consequences, Biochemistry 41, 13217–13223. [DOI] [PubMed] [Google Scholar]
- [56].Shimizu M, Shimizu N, Tsang JC, Petroni BD, Khatri A, Potts JT, and Gardella TJ (2002) Residue 19 of the Parathyroid Hormone (PTH) Modulates Ligand Interaction with the Juxtamembrane Region of the PTH-1 Receptor†, Biochemistry 41, 13224–13233. [DOI] [PubMed] [Google Scholar]
- [57].Gifford JL, Walsh MP, and Vogel HJ (2007) Structures and metal-ion-binding properties of the Ca2+-binding helix–loop–helix EF-hand motifs, Biochem. J. 405, 199–221. [DOI] [PubMed] [Google Scholar]
- [58].Ikura M (1996) Calcium binding and conformational response in EF-hand proteins, Trends in Biochem. Sci. 21, 14–17. [PubMed] [Google Scholar]
- [59].Mahon MJ, and Segre GV (2004) Stimulation by parathyroid hormone of a NHERF-1-assembled complex consisting of the parathyroid hormone I receptor, phospholipase Cβ, and actin increases intracellular calcium in opossum kidney cells, J. Biol. Chem. 279, 23550–23558. [DOI] [PubMed] [Google Scholar]
- [60].Appleton KM, Lee M-H, Alele C, Alele C, Luttrell DK, Peterson YK, Morinelli TA, and Luttrell LM (2013) Chapter Thirteen - Biasing the Parathyroid Hormone Receptor: Relating In Vitro Ligand Efficacy to In Vivo Biological Activity, In Methods in Enzymol. (Conn PM, Ed.), pp 229–262, Academic Press. [DOI] [PubMed] [Google Scholar]
- [61].Cai Y, Liu Y, Culhane KJ, DeVree BT, Yang Y, Sunahara RK, and Yan EC (2017) Purification of family BG protein-coupled receptors using nanodiscs: Application to human glucagon-like peptide-1 receptor, PloS One 12, e0179568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Brown EM, and MacLeod RJ (2001) Extracellular Calcium Sensing and Extracellular Calcium Signaling, Physiol. Rev.81, 239–297. [DOI] [PubMed] [Google Scholar]
- [63].Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, Kumasaka T, Nakanishi S, Jingami H, and Morikawa K (2000) Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor, Nature 407, 971–977. [DOI] [PubMed] [Google Scholar]
- [64].Silve C, Petrel C, Leroy C, Bruel H, Mallet E, Rognan D, and Ruat M (2005) Delineating a Ca2+ Binding Pocket within the Venus Flytrap Module of the Human Calcium-sensing Receptor, J. Biol. Chem. 280, 37917–37923. [DOI] [PubMed] [Google Scholar]
- [65].Jiang JY, Nagaraju M, Meyer RC, Zhang L, Hamelberg D, Hall RA, Brown EM, Conn PJ, and Yang JJ (2014) Extracellular Calcium Modulates Actions of Orthosteric and Allosteric Ligands on Metabotropic Glutamate Receptor 1α, J. Biol. Chem. 289, 1649–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Reeves PJ, Kim J-M, and Khorana HG (2002) Structure and function in rhodopsin: a tetracycline-inducible system in stable mammalian cell lines for high-level expression of opsin mutants, Proc. Natl. Acad. Sci. U. S. A. 99, 13413–13418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Bayburt TH, Grinkova YV, and Sligar SG (2002) Self-assembly of discoidal phospholipid bilayer nanoparticles with membrane scaffold proteins, Nano Lett. 2, 853–856. [Google Scholar]
- [68].Pioszak AA, and Xu HE (2008) Molecular recognition of parathyroid hormone by its G protein-coupled receptor, Proc. Natl. Acad. Sci. U. S. A. 105, 5034–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Denton EV, Craig CJ, Pongratz RL, Appelbaum JS, Doerner AE, Narayanan A, Shulman GI, Cline GW, and Schepartz A (2013) A beta-peptide agonist of the GLP-1 receptor, a class B GPCR, Org. Lett. 15, 5318–5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.