

Abstract
Stroke is a debilitating disease. Current effective therapies for stroke recovery are limited to neurorehabilitation. Most stroke recovery occurs in a limited and early time window. Many of the mechanisms of spontaneous recovery after stroke parallel mechanisms of normal learning and memory. While various efforts are in place to identify potential drug targets, an emerging approach is to understand biological correlates between learning and stroke recovery. This review assesses parallels between biological changes at the molecular, structural and functional levels during learning and recovery after stroke, with a focus on drug and cellular targets for therapeutics.
Keywords: Stroke, learning and memory, neurorehabilitation, dendritic spines, axonal sprouting, gene systems, cortical maps, functional motor recovery
Introduction
Stroke causes lasting neurological impairments, making it the leading cause of adult disability. With advancements in early interventions, such as with recanalization and thrombolysis, the death toll resulting from a stroke has declined by 16.7 percent1 from 2006 to 2016. While these efforts increase survival, the proportion of stroke survivors with motor and cognitive impairments is on the rise. Currently, neurorehabilitation is the only therapy for stroke recovery. Neurorehabilitative therapies encompass repetitive training2, specific muscle training3,4, constraint-induced movement5, robot-assisted gait training6 and other forms of reproducible behavioral activity, such as with virtual reality modalities7,8. The idea is that practice makes perfect. Repetitive and task-specific training have been hypothesized to induce forms of plasticity that are beneficial for stroke recovery. However, our understanding of the biological substrates that effect processes beneficial to recovery, or what these processes might be, are limited. Imaging studies in patients are limited to changes in gross anatomical features or macroscopic changes in functional connectivity across brain regions. While these outcomes can be loosely correlated to recovery, relevant changes at the synapse, neuronal circuit, or in gene expression underlying recovery remain elusive. Pre-clinical models, such as rodent models of stroke, have furthered our understanding of these changes, allowing us to dissect mechanisms that lead to motor recovery after stroke. This review addresses biological substrates that have been defined, with an emphasis on parallels between mechanisms underlying normal learning processes in the brain and those of spontaneous biological recovery after stroke.
Are learning-induced changes recapitulated during motor recovery?
The brain undergoes a remarkable degree of plasticity in an age and environmentally dependent manner. Both learning and stroke are marked by temporal periods of heightened plasticity. For learning, this period is present during development and shaped by sensory processing, and has been shown to be critical for language acquisition and development of other cognitive domains9,10. The most sensitive period of the brain to changes in circuitry and physiology from alterations in outside (sensory) input has been termed the critical period9,10. Following stroke, a critical period exists in the subacute phase of stroke in which the most significant change occurs in behavioral recovery and brain circuitry and physiology. This has been demonstrated with the use of neuromodulatory drugs and activity-based therapies11–13.
Evidence from neurorehabilitative training and associative motor recovery suggests that learning processes may shape plasticity responses to output functional motor recovery. In other words, a learning process introduced after stroke, or by the stroke itself, gives rise to functional changes normally associated with learning, which act as substrates for motor recovery after stroke. While a direct causal link between learning-induced biological changes and recovery after stroke is yet to be experimentally determined, several lines of evidence show that biological changes within neuronal populations during learning and recovery after stroke share similar structural changes and gene transcriptional programs14,15. For example, induction of learning-induced biological changes, such as a genetic modulation underlying learning, may be evident during motor recovery after stroke. This review defines such processes that are active during learning as well as recovery after stroke, and hence identifies mechanisms that underlie motor recovery after stroke. Most descriptions on learning in this review pertain to learning of a motor skill, a modality most relevant to neurorehabilitation.
Dendritic spine plasticity during learning and stroke recovery
Cortical plasticity, first described as instability in response to cortical stimulation16, forms the basis of adaptation of the cortex to changes in the environment. Learning is an integral part of adapting to change. Although many studies have shown changes in cortical plasticity during learning, more recent studies have shown the involvement of the motor cortex in storing information that pertains to learned motor movements. A biological readout of this plasticity-related process has been through the quantification of sub-micron volume protrusions on dendrites that carry chemical information for synapses. These protrusions, termed dendritic spines, exhibit changes in numbers through formation of new spines or deletion of old spines, and can cluster on dendrites during learning processes (Figure 1). The concept that the cortex acts as a seat of learning stemmed from observations on increased dendritic spine density in the visual cortex after animals were exposed to enriched conditions17. The plasticity of the cortex during skilled movement such as a unilateral reaching task was first demonstrated in the 1970’s, where training of one limb led to structural changes in cortical layer 5 dendrites that were otherwise absent in the hemisphere contralateral to the trained limb18. Cortical layer V is the output layer of motor cortex to the spinal cord and brain stem movement centers. Since then, with the introduction of better imaging technology, the use of a similar training task has led to in-depth characterization of changes in synapse number, strength and structural changes during motor training. For example, repetitive motor training has been associated with the formation of new dendritic spines on layer 5 dendrites during the acquisition phase of skill learning19. Similarly, clustering of dendritic spines with repeated learning is indicative of strengthened synapses associated with the task19, while destabilization or deletion of spines leads to a loss of the motor memory associated with the task20,21. The direct link between the association of newly formed spines with a new motor skill was recently demonstrated with an elegant use of tools where selective optogenetic silencing of task-associated dendritic spines led to a complete erasure of motor memory connected with the task22. Spatial characterization of spine dynamics has shown that task-associated spines are compartmentalized to specific dendritic branches22. These studies highlight the complexity with which motor learning affects neuronal circuits to the level of synapses on specific branches of neurons, presumably those directly involved in the task. Compartment-specific spine changes have also been observed in layers 2/3 where spines gained in superficial dendrites are lost as training ceases but after the skill has been gained, whereas dendrites in deeper layers retain spines gained during learning even in the absence of training23. This leads to the question of whether the disruption of neuronal circuits dedicated towards a specific motor task such as during a stroke can be remodeled onto different branches in adjacent spared tissue.
Figure 1:
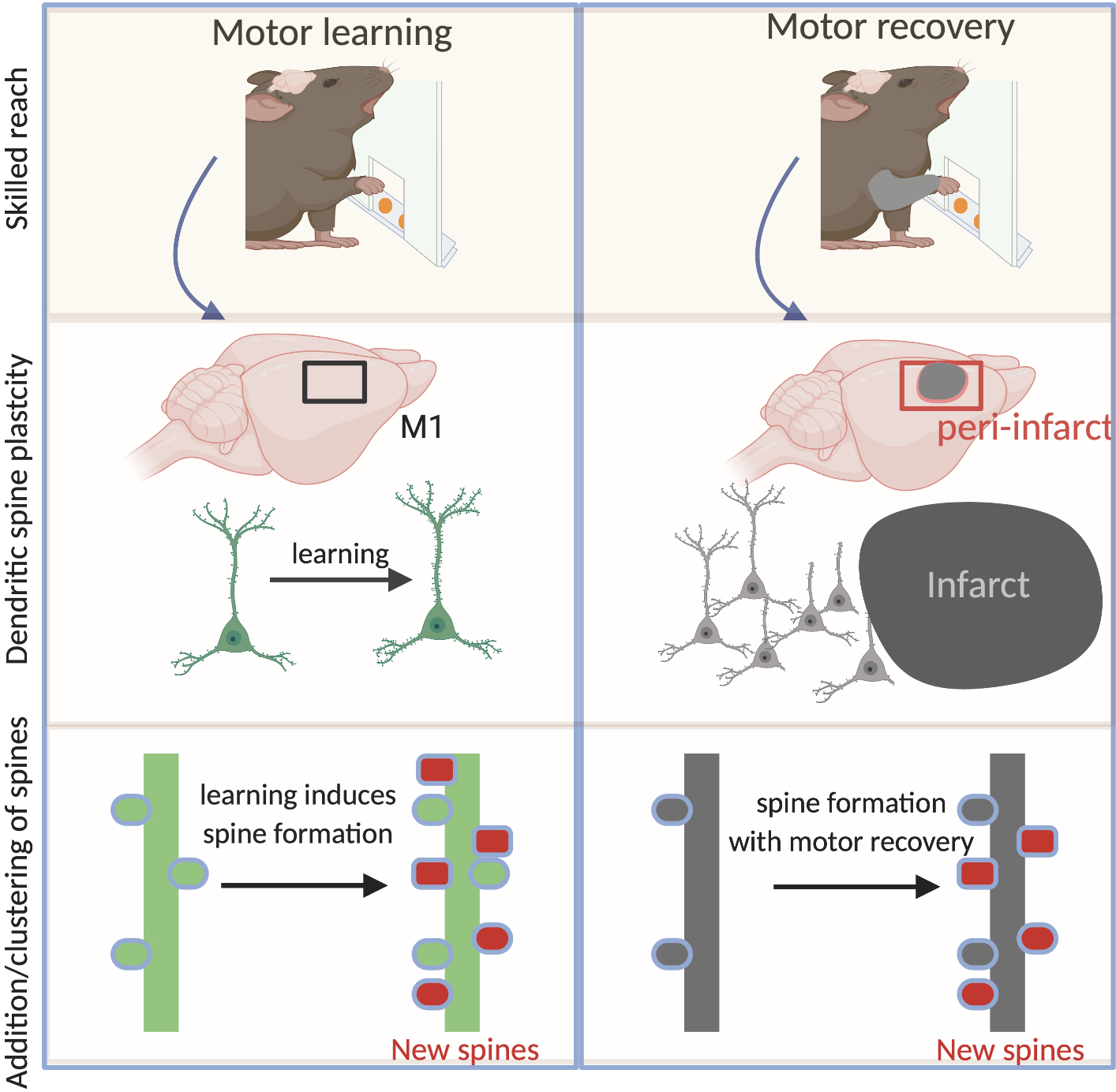
Dendritic spine plasticity- a substrate for motor learning and motor recovery after stroke. Left panel- Motor learning in rodents such as skilled reaching to grab food pellets forces changes on post-synaptic structures on dendrites called spines. These changes occur in motor cortex (M1). Changes include addition of new spines (red) and clustering of spines that lead to strengthened synaptic inputs. Right panel- After stroke, there is loss of synaptic connectivity, particularly near the infarct (peri-infarct tissue). Rehabilitation, spontaneous motor recovery or genetic manipulations such as with CCR5 knockdown induces similar changes on spines as reported with motor learning in the normal brain.
Synaptic dysfunction and spine loss are characteristic of neurodegenerative diseases. Paradoxically, spared tissue adjacent to the infarct in stroke shows a remarkable degree of spine plasticity. For example, dendritic branches from layer 5 neurons in peri-infarct cortex show increased spine formation two weeks after stroke, following the initial loss24,25. This increased plasticity has been partly attributed to increased blood flow, suggesting that a maintenance of tissue perfusion can augment spine formation or retention of spines during tissue stress. Despite the increased turnover of spines in peri-infarct cortex, this amount of plasticity is seemingly insufficient to produce motor recovery. Whether these responses are maladaptive or whether newly formed spines lack sufficient synaptic activation remains an area of further investigation. However, unlike the formation of new spines, preservation of dendritic spines within the first week after stroke is associated with early motor recovery15. The exact cellular mechanisms that lead to spine preservation and subsequent motor recovery are unknown, but presumably preserved spines could be better positioned to receive synaptic input from existing or new synaptic partners.
Similar to learning-induced changes in dendritic plasticity, rehabilitative training after brain injury forces numerous changes in dendritic branching and spine density in the contralesional hemisphere26 and in motor and premotor areas on the same side of the stroke27. Dendritic complexity and increased spine density occur in task-specific neurons in peri-lesion tissue following skilled rehabilitative training28. This broadens the possibility that spontaneous dendritic changes in peri-infarct neurons can be functionally stabilized through repetitive training. A similar output can be produced through frequent intermittent activation of synapses, such as through optogenetic stimulation29. For example, through the use of combinatorial tools to asses stimulation-induced synaptic function in peri-infarct cortex, optogenetic stimulation of thalamocortical axons projecting to peri-infarct somatosensory cortex causes formation of new and stable axonal boutons30. Importantly, these newly formed axonal boutons are responsive to forelimb-evoked calcium transients—indicating that the new synapses stabilized by this activity are functional. These data have been some of the first to directly link optogenetic stimulation with the formation of functional spines during stroke recovery. Given our limited understanding on how synaptic activation of spines through rehabilitation or optogenetic stimulation can augment stroke recovery, the identification of molecular effectors that drive similar changes opens a window for therapeutic potential.
Taken together, learning forces numerous changes in dendritic spines that are causally linked to a learned motor task. Stroke induces a transient loss of spines, whereas neurorehabilitative training increases spine density (Figure1). Changes in spine plasticity are associated with reorganization of molecular pathways. Perturbation of molecular pathways involved in learning and memory influences spine density after stroke. For example, downregulation of a learning and memory receptor, CCR5 (discussed later) prevents initial spine loss after stroke15. This points to the fact that molecular effectors of learning and synaptic plasticity prevent loss of spines and are associated with motor recovery.
Molecular substrates that drive learning and stroke recovery
Both stroke and learning-induced gene expression changes are associated with brain excitability31, synaptogenesis23,32,33 and network connectivity34 (Figure 2). Over the decades, many studies have shed light on molecular pathways that underlie memory formation and consolidation35. GABA and AMPA receptor trafficking play key roles in modulation of cortical excitability and synaptic strength during memory formation both in development and adulthood36–39. Dampening GABA signaling outside the synapse (extrasynaptic GABA signaling), by targeting the alpha5 subunit of the GABA receptor, which mediates inhibition in learning and memory process39, promotes motor recovery after stroke40. Similarly, increasing AMPA receptor signaling also promotes motor recovery11 (for a detailed review, see reference 31). Synaptogenic mechanisms are critical for memory formation, such as axon guidance molecules that mediate neuronal contact and synaptogenesis41. Axon guidance molecules are active during development, where signaling by binding of an axonal guidance receptor on the surface of a cell to its extracellular ligand meditates the migration of the cell or the axon across a region that is enriched in expression of the ligand. Neurons and glia express axon guidance molecules during development and adulthood42,43. In the adult, axon guidance molecules have roles in neurotransmission44. For example, an axon guidance cell-surface receptor-Eph and ligand Ephrin signal in astrocytes and neurons to regulate activity-dependent release of glutamate and subsequent activation of AMPA receptors during LTP and memory storage44. These molecules are also differentially expressed after injury such as stroke45. Ephrin-mediated neuro-glial signaling is active after stroke. Ephrin A5 is highly upregulated in astrocytes following a stroke, where it interacts with neuronal EphA4. Blocking EphA4 in peri-infarct cortex during periods of reduced cortical excitability leads to functional motor recovery and robust axonal sprouting45 (for a detailed review, see reference 46). A recent finding using a knockout model in the mice of Ephrin A5 reports that this molecule may not lead to functional recovery47. The report by de Boer et al uses a knockout model in mice of Ephrin A5 expression and reports no enhancement of functional recovery. The methods of determination of recovery and the data for a lack of effect are not shown. There is an important and critical limitation to this study, and to this overall approach, in the stroke field. Knockdown of a molecule for the entire brain development and life of a mouse, termed “constitutive knockout”, produces compensation in other molecular systems, which assume the role of the knocked out molecular system48,49. Indeed, constitutive knockout for the life of an animal of one axonal growth inhibitor, leads to even compensatory upregulation of other axonal growth inhibitors50. In order to prove that a molecular system has, or does not have, a role in a disease like stroke, the brain cannot have the system knocked out for entire life of the animal (and its brain development), but must have the system knocked out at a discrete interval after the stroke.
Figure 2:
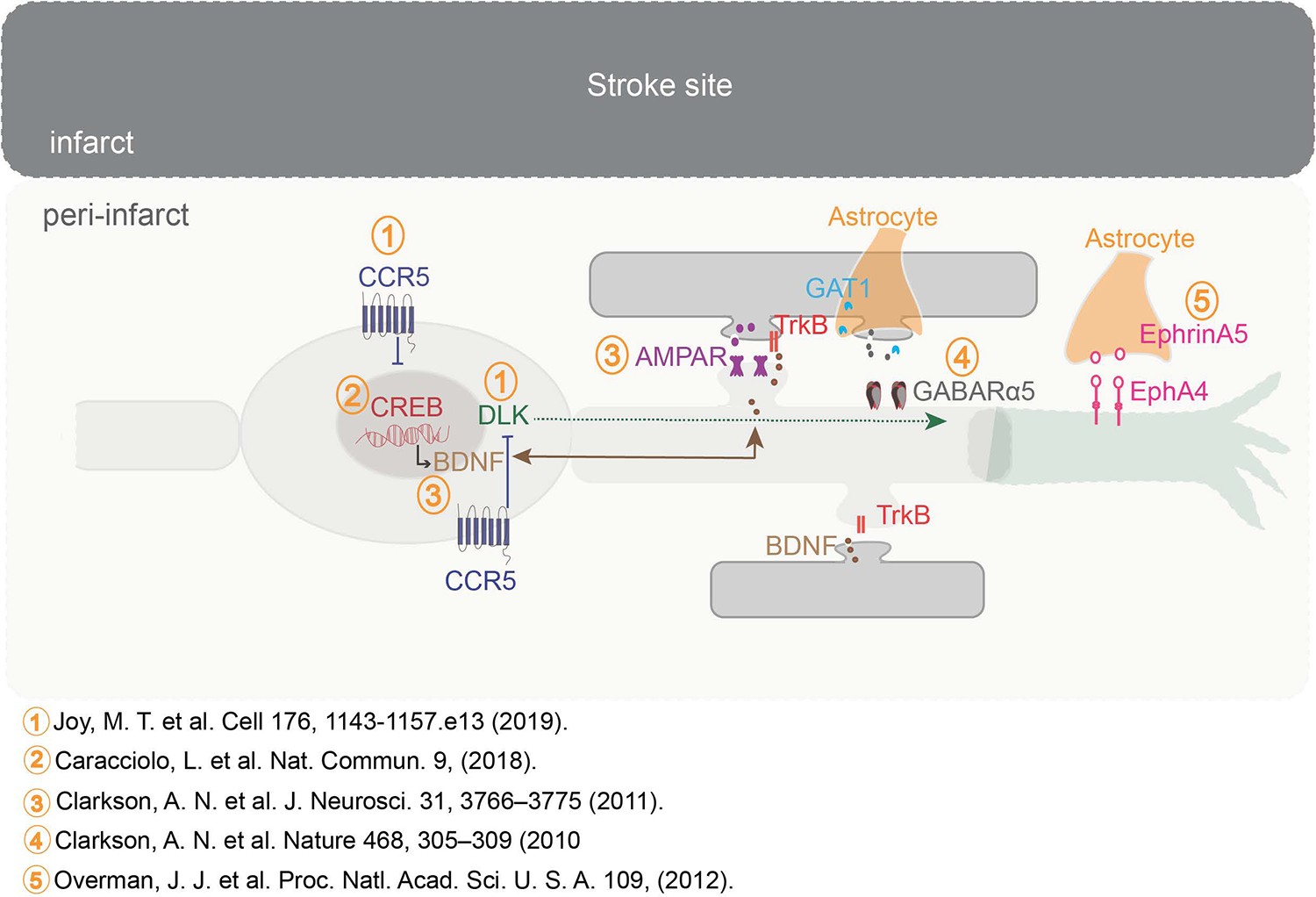
Molecular systems that underlie learning regulate recovery after stroke. The peri-infarct cortex acts a hub for molecular events that regulate recovery after stroke. Transcription factor CREB is critical for memory formation. Increasing CREB signaling after stroke improves recovery of motor function. CREB signaling can lead to signaling pathways that enhance BDNF production. BDNF activates Trk signaling and this pathway in turn is positively modulated by AMPA receptor signaling. Slow and persistent (tonic) GABA signaling through the alpha5 receptor at extra synaptic sites dampens stroke recovery. GABA signaling is persistent due to defects in the GABA transporter (GAT1) expressed by astrocytes that function to transport and clear GABA at extrasynaptic sites. Blocking this pathway enhances motor recovery. CREB signaling is impeded by expression of CCR5 early after stroke. Blocking CCR5 signaling increases CREB signaling and signaling via DLK that acts as a regeneration signal for axonal sprouting. Axonal sprouting is further facilitated through reduced signaling via Ephrin A5 expressed on reactive astrocytes and its receptor EphA4 on neurons, that otherwise serve to collapse arrangement of cytoskeletal proteins.
In these and other studies of functional recovery after stroke, a key issue is alignment between the outcome measures for recovery in the animal model, often the mouse, to those in the human and the establishment of thresholds for meaningful functional gains. Humans have impairments in neurological function as a result of a stroke, which of course affect specific behavioral domains, such as motor, language, attention and sensory domains. In humans, behavioral measures of stroke recovery that do not test impairment, but instead rely on more global measures of disability, are not sensitive to stroke recovery51,52. This limitation in clinical trials with disability outcome measures stems directly from the non-specific—non-domain (motor, language, sensory, etc.)--measures of disability, such as the modified Rankin Scale or the Barthel Index. In mouse models of stroke, behavioral recovery measures that similarly aggregate many functional domains into global scales either do not detect recovery, or over-detect recovery. One such lumped test is the modified Neurological Severity Scale (mNSS), which finds positive behavioral recovery results in nearly every test of a candidate therapy in stroke53. Similarly, just as in human outcome measures in stroke, some commonly used mouse behavioral measures are affected by many different brain functions and brain areas. For example, the Rotorod test, which measures how long a mouse or rat can stay atop a moving, motorized rod, is actually sensitive to spinocerebellar tract damage and repair54, and yet is commonly applied to outcome measures of stroke in the cortex or striatum. Thus, a key issue in this field relates to the use of adequate and aligned behavioral outcome measures in both human and rodent in stroke, which do not test stroke impairments at the general level of disability and which do not lump many functional domains of the nervous system into one common measure. In the studies described in for CREB and CCR5 (below), outcome measures in the mouse are foot placement in gait and individuated use of the forelimb in exploratory behaviors, designed to follow common human behaviors and to narrowly focus on motor control impairments. In these studies, the level of improvement in functional recovery is above the meaningful clinically important difference established in human motor recovery in stroke55,56, which can be extrapolated from the human to the mouse using effect size.
Returning to molecular mechanisms of stroke recovery in systems involved in the normal synaptic plasticity of learning and memory, recent evidence suggests that biological changes during learning processes and stroke recovery share common molecular pathways outside of the realm of immediate neurotransmitter receptor signaling of glutamate via AMPA receptors or GABA via extrasynaptic GABA receptors. Some examples include neurotrophin signaling via BDNF57, immediate early genes such as Arc58 and growth inhibitors such as Nogo59. While these act as striking examples, our review will focus on the two most recently characterized molecules that have robust effects in both systems-- learning and motor recovery.
CREB
The nuclear transcription factor cAMP response element binding protein (CREB) is a key modulator of learning and memory processes and has been investigated in different model systems that range from aplysia to rodents60,61. CREB is involved in memory formation, storage and retrieval60. At a finer level, CREB expression determines which neurons integrate into a network that stores information pertaining to an event62,63. CREB also regulates learning-induced structural plasticity, such as formation of new spines potentiated through learning64. It is important to note that CREB is involved in learning-dependent structural changes but does not otherwise alter the normal structure of the CNS. In terms of function, CREB downregulation impairs long-term potentiation (LTP)64. The dependency on CREB for learning-induced structural changes indicates that CREB induces a plasticity state in neurons.
Based on its role as a ‘plasticity molecule,’ several studies have shown that CREB upregulation promotes axonal regeneration in vitro as well as in in vivo models of CNS injury65,66. In the context of stroke, CREB upregulation in peri-infarct cortex has substantial effects on motor recovery14. In tasks that test fine motor control of the forelimb, animals with CREB overexpression show enhanced performance such as reduced foot faults, less bias towards use of the impaired limb and improved fine motor control for handling and grabbing, one month after a stroke. Overexpression of CREB through viral transduction of a small population of neurons adjacent to the infarct is sufficient to induce recovery, following a stroke to either the motor cortex or in a larger stroke model inclusive of the motor cortex and striatum. Interestingly, silencing CREB-overexpressing neurons with chemogenetic tools, such as with an inhibitory DREADD (hM4Di)67, reverses its effects on recovery, whereas lifting inhibition in CREB-induced neurons reinstates recovery. In other words, CREB induction drives motor recovery, whereas silencing neurons that selectively upregulate CREB silences the effects on motor recovery. A possibility is that CREB activation within neurons attributes a property of functional dominance, wherein a motor network becomes dependent or is driven by pools of neurons with higher levels of CREB expression. This attribute of functional dominance is present even in the absence of stroke. In healthy animals, neurons with CREB overexpressing neurons in motor cortex, when silenced, show motor deficits similar to animals that have received a stroke to the motor cortex14. A decline in motor performance suggests a selective integration of CREB-overexpressing neurons to a motor network and that this feature is based on a state of increased excitability.
Motor recovery resulting from CREB overexpression after stroke results in widespread gene expression changes14. In healthy animals, CREB overexpression alone causes differential regulation of approximately 200 genes. Being a ubiquitous transcription factor, CREB affects various signaling pathways, including pathways distinct to post-stroke signaling. Prominently, these include fibroblast growth factor (FGF) and suppressor of cytokine signalling-2 (SOCS2) signaling, known to regulate neuronal development and homeostasis,68,69 and have been shown to influence neural repair and axon regeneration. In addition to stroke, a large body of evidence supports a pro-growth role for FGF in other systems of injury68–72. Interestingly, both SOCS2 and FGF have been implicated in signaling during neurogenesis, a process that is upregulated after stroke and can cause functional motor recovery through integration of new neurons into functional cortical circuits in an activity-dependent manner73. These gene expression changes indicate that CREB directly influences recovery after stroke by enabling neurons to engage in larger functional connections in the brain tissue adjacent to the stroke.
CCR5
C-C Chemokine Receptor-5, CCR5, is a G-protein coupled receptor with functions in adaptive immune signaling74. CCR5 is expressed in immune cells such as macrophages, monocytes, T-cells and NK cells. In particular, CCR5 signaling is important for determining localization of CD8 T-cells within lymphoid tissue and subsequent recruitment within inflammatory sites74. CCR5 is also expressed in the CNS, such as in astrocytes, microglia and endothelial cells. Although CCR5 signaling is associated with immune responses, a definitive role for CCR5 in the adult brain was only recently characterized through an extensive screen for potential learning and memory candidate genes75. In a reverse genetic screen of 148 transgenic mouse lines, mice with CCR5 knockdown showed improved performance in various learning and memory tasks, whereas CCR5 overexpression significantly impaired learning and memory. The study clearly constituted a role for CCR5 as a suppressor of cortical plasticity. Although chemokine receptors are not usually envisioned as modulators of synaptic plasticity, a growing body of evidence supports such a role through interactions with neurotransmitters and transcription factors76. In the context of learning and memory, CCR5 knockdown elevates CREB and MAPK signaling75. Furthermore, binding of CCR5 to its ligand RANTES (for ‘regulated upon activation normal T cell expressed and secreted’) biases CCR5 signaling towards activation of the Gαi pathway that inhibits cAMP production77. cAMP production facilities synapse formation during learning and memory60. Production of cAMP following CNS injury promotes a regenerative state78. Given the link between CCR5 and cAMP production, dampening CCR5 could lead to increased cAMP levels required for memory formation. Collectively, G-protein coupled receptor signaling along with interactions with transcription factors such as CREB enable CCR5 to commune plasticity associated-signaling events that range from cAMP generation to synaptogenesis; events that are hallmarks for learning and memory.
We have recently shown that dampening CCR5 signaling within neurons induces substantial motor recovery after stroke15. Knocking down CCR5 function after stroke induces motor recovery in motor tasks that allow quantification of fine motor control of the forelimb. Animals with CCR5 knockdown show improved navigation on a wired grid with fewer foot faults and less bias towards use of the impaired forelimb. CCR5 signaling is active in neurons and glia after stroke79,15. Interestingly, CCR5 is not normally expressed in cortical neurons, but is selectively upregulated following a stroke15. Knockdown of CCR5 in pre-motor cortex following a stroke to the motor cortex induces early and sustained motor recovery. This effect is associated with structural changes differentially regulated by CCR5 knockdown, such as the growth of unique projections to the contralateral pre-motor cortex and preservation of dendritic spines. CCR5 knockdown induces pro-regenerative signaling pathways through upregulation of CREB and dual leucine zipper kinase (DLK) expression. This data indicates a link between the recovery-promoting molecular systems of CCR5 and CREB. DLK, also known as MAP3K12, acts as a prominent axonal regeneration signal in other systems of CNS injury80. DLK activates various transcriptional profiles that pertain to either regeneration or apoptosis. The activation of these opposing pathways is dependent on targets downstream of DLK80. Following a stroke, diminishing CCR5 signaling positively modulates DLK signaling towards switching on a regeneration and/or repair program15. In fact, DLK upregulation is critical for this process. Downregulating DLK function diminishes motor recovery induced through CCR5 knockdown, showing that DLK acts as a signaling hub through which neuronal knockdown of CCR5 restores motor function15.
A goal of molecular medicine has been to identify pharmaceutical targets for disease that have translational potential. CCR5 fits this description. Maraviroc, an FDA-approved CCR5 antagonist for HIV therapy, when delivered after acute or chronic stroke induces motor recovery, similar to viral genetic manipulations of CCR515. Similarly, stroke patients that carry CCR5Δ32, a mutation that inactivates CCR5, show improved stroke outcomes compared to non-carriers15. The effects of CCR5 in rodent models, its druggable potential and clinical significance in patients makes CCR5 a highly promising target for stroke. Collectively, the role of CCR5 as a transcriptional substrate for memory and learning process parallels its role as a potent genetic target for stroke recovery through induction of shared mechanisms of plasticity.
Learning-associative axonal remodeling and motor recovery
A consequence of gene expression changes during learning result in changes in synaptic strength or remodeling of synaptic inputs. For example, thalamocortical inputs are received by corticospinal neurons in the cortex that control distal and proximal hand function. During motor learning of a skilled reach task, thalamocortical projections onto neurons that project to the distal forelimb involved in grasping show greater firing release probability and amplitude of the signaling events, compared to the proximal limb81. Here, thalamocortical projections show a bias in increased connectivity towards neurons involved in learning. Axonal remodeling that involve growth of new projections have been reported, but these responses as a result of learning are limited, unlike after injury. In Pavlovian eye blink conditioning, a form of motor learning required for timed eyelid closure, skilled performers show structural changes in the cerebellum, such as increased mossy fiber collaterals and de novo axonal connections from the basal pontine nucleus to various regions in the cerebellar nuclei82. In a more relevant motor learning model, where macaques were trained for tool use, new axonal projections form from higher visual centers to the intraparietal regions in the cortex83, suggesting that learning is associated with changes in number and growth of axonal projections. An appreciable body of evidence in humans through longitudinal MRI imaging during learning points towards similar observations on gross structural changes in gray and white matter84,85.
Axonal growth responses are active as early as 7 days after a stroke72. These responses are substantial and have been identified through various anatomical mapping studies15,46,74,87,88. The location, amount and the associated outcome of axonal sprouting vary depending on the size and location of stroke, molecular interventions applied and rehabilitative training. The peri-infarct cortex undergoes a remarkable degree of structural reorganization and growth, the result of time-dependent gene expression changes characteristic to this region. For example, following a focal cortical stroke, the peri-infarct cortex sends out new axonal branches to areas within motor, somatosensory and pre-motor cortices15,46,88. In addition to new axonal outgrowth in local areas, novel growth of bihemispheric projections are also induced15. One could argue that larger sprouting responses have been observed with larger infarcts. In models of large strokes in which more than a quarter of the cortical hemisphere is lost, responses from the contralateral hemisphere to denervated regions in the cervical spinal cord have been reported89. While new projections are formed as a result of the brain’s endogenous response to reorganize after stroke, these axons follow an unconventional trajectory and populate regions of the cortex that are distinct prior to injury. For example, a focal stroke in the somatosensory cortex leads to a loss in connections to the thalamus; however, new axons that sprout take an alternative path into adjacent intracortical regions87. It is unclear if these responses contribute to spontaneous motor recovery, or if these responses are maladaptive.
Genetic manipulations to promote motor recovery or motor training from neurorehabilitation have also shown to induce axonal sprouting responses. Manipulations in EphrinA5, GDF10, or NgR145,88,90 signaling produce new and expansive axonal outgrowth from peri-infarct cortex to adjacent cortical areas, while blockade of CCR515 causes axons to sprout to cortical areas contralateral to the stroke site. These responses were unique in location and were absent in groups with stroke without treatment. Similarly, administration of Nogo-A antibody following a lesion to the primary motor cortex causes sprouting to callosal homotypic pre-motor areas in the contralateral cortex in macaques91. In addition to inter-intra-cortical connections, axonal sprouting resulting from such manipulations after stroke have shown to alter growth responses in descending pathways. Treatment with Nogo-A or inosine induces sprouting of corticospinal axons into the denervated cervical spinal cord 59,90,91,92. Sprouting responses were further intensified when manipulation was paired with skill training59,90. It is worthy to mention that increased sprouting does not positively correlate with motor recovery. A clear demonstration of this is seen when Nogo-A antagonism simultaneously paired with skill training produces a robust sprouting response with null or negative effects on motor recovery59. Interestingly, motor recovery was induced with the introduction of a delay period between NogoA therapy and motor training, but with a less robust axonal sprouting response. These studies suggest that while stroke primes post-stroke neurons to send out new projections, recovery requires complex target selection mechanisms. Non-selective axonal sprouting, or the formation of projections after stroke, may result in detrimental synaptic competition in off-target brain regions.
Cortical map plasticity as a substrate for learning and motor recovery
Growth triggered after loss of axons at the stroke site can lead to changes in topographical brain regions activated during a motor response. These changes in the brain can be mapped using electrical or optical stimulation techniques by pairing the activation of a brain region with the elicited motor response. Characterization of cortical representations during motor behavior, such as a dexterous reach task, have shown the existence of maps with clear boundaries for wrist, digit and trunk movements93. The areas that represent these movements are enlarged during skill training—in other words, the part of the body that is trained shows an enlarged brain representation in its corresponding cortical map. Map reorganization is also dependent on age94. In the aged brain after stroke, where paucity for regenerative potential is pertinent, map reorganization has been shown to be less pronounced or absent.
A substantial body of work has shown that targeted strokes in the motor-sensory areas lead to the displacement or formation of new motor maps in adjoining cortical zones95–99. Map displacement is dependent on the location of the infarct. Map displacement is more prevalent following strokes to the somatosensory cortex where remapping of sensory forelimb function is seen in the motor cortex, although this does not reciprocate in the event of a stroke to the motor cortex95. Maps topographically do not displace following a loss of motor cortex; instead changes are more localized to perilesional cortex such as through an increased state of excitability. These differences in map topography based on lesion location suggest that cortical substrates underlying these maps are segregated based on differences in their cortical output projections or perhaps the intrinsic circuit structure of the particular cortical region (motor vs somatosensory).
A causal relationship between map reorganization and recovery of motor function is yet to be established. While the initial phase of learning induces changes in map topography, these changes normalize over time, making it difficult to assess the importance of topographical changes100. It is possible that map changes are less dynamic following the initial phase of learning. An interesting feature is that map enlargement can be induced through raising cortical excitability by increasing CREB activation after stroke14. However, further studies are required to understand if map enlargement leads to motor recovery. The gross topography of a map lacks resolution to determine changes at the circuit level, such as changes in cortical microcircuits and circuit reorganization during learning and recovery after stroke.
Conclusion
Biological mechanisms underlying learning and memory formation parallel mechanisms of stroke recovery that range from changes at the synapse to motor circuits. Studies on cortical plasticity have identified distinct cellular substrates and molecular systems shared between learning, memory and motor recovery. Synaptic plasticity as viewed in changes in dendritic spine remodeling, axonal remodeling and cortical map function show similar patterns during motor learning and recovery after stroke. Molecular systems that promote neuronal excitability underlying memory formation can enhance recovery after stroke. In fact, these systems drive changes in synapse density and integrate neurons that normally engage in storage of motor memories into a network that accomplishes a motor task that underlies motor recovery. Molecular systems, such as CREB and CCR5, provide pharmacological targets for drugs that promote stroke recovery. However, questions remain in the understanding of links between synaptic plasticity mechanisms and recovery from stroke. While brain reorganization processes have been described at the anatomic levels, organization at the functional level within populations of neurons are areas of further investigation. Furthermore, how does learning such as during neurorehabilitation augment spatiotemporal activity within neuronal assemblies in the motor system to output motor recovery? Is there a state of maladaptiveness reflective at the population level that help understand adaptive and maladaptive structural plasticity? While existing studies discussed here have greatly furthered our understanding on changes in the motor system after a stroke and during functional motor recovery, future studies will dissect mechanistic processes to help understand complex computations within neuronal circuits that underlie recovery of motor function.
Acknowledgements
Supported by the Miriam and Sheldon G Adelson Medical Research Foundation, NIIH grants NS102185, NS085019.
Footnotes
Conflict of Interest
Authors declare no conflict of interests.
References
- 1).Benjamin EJ et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation, 2019; 139, e56–e528. [DOI] [PubMed] [Google Scholar]
- 2).Lang CE, Lohse KR & Birkenmeier RL Dose and timing in neurorehabilitation: prescribing motor therapy after stroke. Curr Opin Neurol. 2015; 28, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Billinger SA et al. Physical activity and exercise recommendations for stroke survivors: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2014; 45, 2532–2553. [DOI] [PubMed] [Google Scholar]
- 4).Harris JE & Eng JJ Strength training improves upper-limb function in individuals with stroke: a meta-analysis. Stroke, 2010; 41, 136–140. [DOI] [PubMed] [Google Scholar]
- 5).Kwakkel G, Veerbeek JM, van Wegen EE & Wolf SL Constraint-induced movement therapy after stroke. Lancet Neurol, 2015; 14, 224–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Duncan PW et al. Body-weight-supported treadmill rehabilitation after stroke. N Engl J Med, 2011; 364, 2026–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).Laver KE et al. Virtual reality for stroke rehabilitation. Cochrane Database Syst Rev; 2017; 11, CD008349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Maier M, Rubio Ballester B, Duff A, Duarte Oller E & Verschure P Effect of Specific Over Nonspecific VR-Based Rehabilitation on Poststroke Motor Recovery: A Systematic Meta-analysis. Neurorehabil Neural Repair, 2018; 33, 112–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).Takesian AE, Bogart LJ, Lichtman JW & Hensch TK Inhibitory circuit gating of auditory critical-period plasticity. Nat Neurosci, 2018; 21, 218–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10).Hartshorne JK, Tenenbaum JB & Pinker S A critical period for second language acquisition: Evidence from 2/3 million English speakers. Cognition, 2018; 177, 263–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Clarkson AN et al. AMPA receptor-induced local brain-derived neurotrophic factor signaling mediates motor recovery after stroke. J Neurosci, 2011; 31, 3766–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Dromerick AW et al. Critical periods after stroke study: translating animal stroke recovery experiments into a clinical trial. Front Hum Neurosci, 2015; 9, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Humm JL, Kozlowski DA, James DC, Gotts JE, Schallert T. Use-dependent exacerbation of brain damage occurs during an early post-lesion vulnerable period. Brain Res, 1998; 783:286–92. [DOI] [PubMed] [Google Scholar]
- 14).Caracciolo L et al. CREB controls cortical circuit plasticity and functional recovery after stroke. Nat Commun, 2018; 9, 2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15).Joy MT et al. CCR5 Is a Therapeutic Target for Recovery after Stroke and Traumatic Brain Injury. Cell, 2019; 176, 1143–1157 e1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).G Brown T, S Sherrington C, On the instability of a cortical point Proc R Soc Lond [Biol], 1912; 85, pp. 250–277. [Google Scholar]
- 17).Globus A, Rosenzweig MR, Bennett EL & Diamond MC Effects of differential experience on dendritic spine counts in rat cerebral cortex. J Comp Physiol Psychol, 1973; 82, 175–181. [DOI] [PubMed] [Google Scholar]
- 18).Greenough WT, Larson JR & Withers GS Effects of unilateral and bilateral training in a reaching task on dendritic branching of neurons in the rat motor-sensory forelimb cortex. Behav Neural Biol, 1985; 44, 301–314. [DOI] [PubMed] [Google Scholar]
- 19).Fu M, Yu X, Lu J & Zuo Y Repetitive motor learning induces coordinated formation of clustered dendritic spines in vivo. Nature, 2012; 483, 92–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).Xu T et al. Rapid formation and selective stabilization of synapses for enduring motor memories. Nature,2009; 462, 915–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21).Yang G, Pan F & Gan WB Stably maintained dendritic spines are associated with lifelong memories. Nature, 2009; 462, 920–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Hayashi-Takagi A et al. Labelling and optical erasure of synaptic memory traces in the motor cortex. Nature, 2015; 525, 333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23).Clark TA, Fu M, Dunn AK, Zuo Y & Jones TA Preferential stabilization of newly formed dendritic spines in motor cortex during manual skill learning predicts performance gains, but not memory endurance. Neurobiol Learn Mem. 2018. Jul;152:50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24).Brown CE, Li P, Boyd JD, Delaney KR & Murphy TH Extensive turnover of dendritic spines and vascular remodeling in cortical tissues recovering from stroke. J Neurosci 27,2007; 4101–4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25).Mostany R et al. Local hemodynamics dictate long-term dendritic plasticity in peri-infarct cortex. J Neurosci, 2010; 30, 14116–14126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26).Takatsuru Y et al. Neuronal circuit remodeling in the contralateral cortical hemisphere during functional recovery from cerebral infarction. J Neurosci, 2009; 29, 10081–10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27).Clark TA et al. Rehabilitative Training Interacts with Ischemia-Instigated Spine Dynamics to Promote a Lasting Population of New Synapses in Peri-Infarct Motor Cortex. J Neurosci. 2019. Oct 23;39(43):8471–8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28).Wang L, Conner JM, Nagahara AH & Tuszynski MH Rehabilitation drives enhancement of neuronal structure in functionally relevant neuronal subsets. Proc Natl Acad Sci U S A, 2016; 113, 2750–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).Cheng MY et al. Optogenetic neuronal stimulation promotes functional recovery after stroke. Proc Natl Acad Sci U S A 111, 2014; 12913–12918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30).Tennant KA, Taylor SL, White ER & Brown CE Optogenetic rewiring of thalamocortical circuits to restore function in the stroke injured brain. Nat Commun; 2017; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Carmichael ST Brain excitability in stroke: the yin and yang of stroke progression. Arch Neurol, 2012; 69, 161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32).Li S, Carmichael ST, Growth-associated gene and protein expression in the region of axonal sprouting in the aged brain after stroke. Neurobiol Dis, 2006; 23, 362–373. [DOI] [PubMed] [Google Scholar]
- 33).Jones TA. Multiple synapse formation in the motor cortex opposite unilateral sensorimotor cortex lesions in adult rats. J Comp Neurol. 1999. Nov 8; 414(1):57–66 [PubMed] [Google Scholar]
- 34).Brown CE, Aminoltejari K, Erb H, Winship IR, Murphy TH, In vivo voltage-sensitive dye imaging in adult mice reveals that somatosensory maps lost to stroke are replaced over weeks by new structural and functional circuits with prolonged modes of activation within both the peri-infarct zone and distant sites. J Neurosci, 2009; 29, 1719–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35).Tonegawa S, Morrissey MD, Kitamura T, The role of engram cells in the systems consolidation of memory. Nat Rev Neurosci, 2018; 19, 485–498. [DOI] [PubMed] [Google Scholar]
- 36).Chittajallu R, Isaac JT, Emergence of cortical inhibition by coordinated sensory-driven plasticity at distinct synaptic loci. Nat Neurosci, 2010; 13, 1240–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37).Diering GH, Huganir RL, The AMPA Receptor Code of Synaptic Plasticity. Neuron, 2018; 100, 314–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38).Yoon JH, Grandelis A, Maddock RJ, Dorsolateral Prefrontal Cortex GABA Concentration in Humans Predicts Working Memory Load Processing Capacity. J Neurosci, 2016; 36, 11788–11794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39).Collinson N et al. , Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the alpha 5 subunit of the GABAA receptor. J Neurosci, 2002; 22, 5572–5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40).Clarkson AN, Huang BS, Macisaac SE, Mody I, Carmichael ST, Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature, 2010; 468, 305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41).Feng G et al. Roles for ephrins in positionally selective synaptogenesis between motor neurons and muscle fibers. Neuron, 2008; 25, 295–306, doi: 10.1016/s0896-6273(00)80895-8. [DOI] [PubMed] [Google Scholar]
- 42).Giger RJ, Hollis ER 2nd, Tuszynski MH, Guidance molecules in axon regeneration. Cold Spring Harb Perspect Biol, 2010; 2, a001867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43).Shen K, Cowan CW, Guidance molecules in synapse formation and plasticity. Cold Spring Harb Perspect Biol 2, 2010; a001842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44).Filosa A et al. Neuron-glia communication via EphA4/ephrin-A3 modulates LTP through glial glutamate transport. Nat Neurosci, 2009; 12, 1285–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45).Overman JJ et al. A role for ephrin-A5 in axonal sprouting, recovery, and activity-dependent plasticity after stroke. Proc Natl Acad Sci U S A, 2012; 109, E2230–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46).Overman JJ & Carmichael ST Plasticity in the injured brain: more than molecules matter. Neuroscientist, 2014; 20, 15–28. [DOI] [PubMed] [Google Scholar]
- 47).de Boer A, Storm A, Rué L, Poppe L, Robberecht W, Lemmens R. Heterozygous Deletion of EphrinA5 Does Not Improve Functional Recovery After Experimental Stroke. Stroke. 2019. Apr;50(4):e101. [DOI] [PubMed] [Google Scholar]
- 48).El-Brolosy MA, Stainier DYR. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet. 2017. Jul 13;13(7):e1006780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49).Tsokas P, Hsieh C, Yao Y, Lesburguères E, Wallace EJC, Tcherepanov A, Jothianandan D, Hartley BR, Pan L, Rivard B, Farese RV, Sajan MP, Bergold PJ, Hernández AI, Cottrell JE, Shouval HZ, Fenton AA, Sacktor TC. Compensation for PKMζ in long-term potentiation and spatial long-term memory in mutant mice. Elife. 2016. May 17;5. pii: e14846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50).Kempf A, Montani L, Petrinovic MM, Schroeter A, Weinmann O, Patrignani A, Schwab ME. Upregulation of axon guidance molecules in the adult central nervous system of Nogo-A knockout mice restricts neuronal growth and regeneration. Eur J Neurosci. 2013. Dec;38(11):3567–79. [DOI] [PubMed] [Google Scholar]
- 51).Cramer SC. Issues important to the design of stroke recovery trials. Lancet Neurol. 2020. Mar;19(3):197–198 [DOI] [PubMed] [Google Scholar]
- 52).Dobkin BH, Carmichael ST. The Specific Requirements of Neural Repair Trials for Stroke. Neurorehabil Neural Repair. 2016. Jun;30(5):470–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53).Hicks A, Schallert T, Jolkkonen J. Cell-based therapies and functional outcome in experimental stroke. Cell Stem Cell. 2009. Aug 7;5(2):139–40. [DOI] [PubMed] [Google Scholar]
- 54).Piao J, Major T, Auyeung G, Policarpio E, Menon J, Droms L, Gutin P, Uryu K, Tchieu J, Soulet D, Tabar V. Human embryonic stem cell-derived oligodendrocyte progenitors remyelinate the brain and rescue behavioral deficits following radiation. Cell Stem Cell. 2015. Feb 5;16(2):198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55).Gladstone DJ, Danells CJ, Black SE. The fugl-meyer assessment of motor recovery after stroke: a critical review of its measurement properties. Neurorehabil Neural Repair. 2002. Sep;16(3):232–40. [DOI] [PubMed] [Google Scholar]
- 56).Lang CE, Edwards DF, Birkenmeier RL, Dromerick AW. Estimating minimal clinically important differences of upper-extremity measures early after stroke. Arch Phys Med Rehabil. 2008. Sep;89(9):1693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57).Kim DY, Quinlan EB, Gramer R & Cramer SC BDNF Val66Met Polymorphism Is Related to Motor System Function After Stroke. Phys Ther, 2016; 96, 533–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58).Kraft AW, Bauer AQ, Culver JP & Lee JM Sensory deprivation after focal ischemia in mice accelerates brain remapping and improves functional recovery through Arc-dependent synaptic plasticity. Sci Transl Med, 2018; 10. [DOI] [PubMed] [Google Scholar]
- 59).Lindau NT et al. Rewiring of the corticospinal tract in the adult rat after unilateral stroke and anti-Nogo-A therapy. Brain, 2014; 137, 739–756, doi: 10.1093/brain/awt336. [DOI] [PubMed] [Google Scholar]
- 60).Kandel ER The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain, 2012; 5, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61).Silva AJ, Kogan JH, Frankland PW, Kida S, CREB and memory. Annu Rev Neurosci, 1998; 21, 127–148. [DOI] [PubMed] [Google Scholar]
- 62).Zhou Y et al. CREB regulates excitability and the allocation of memory to subsets of neurons in the amygdala. Nat Neurosci, 2009; 12, 1438–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63).Yiu AP et al. Neurons are recruited to a memory trace based on relative neuronal excitability immediately before training. Neuron, 2014; 83, 722–735. [DOI] [PubMed] [Google Scholar]
- 64).Middei S et al. CREB selectively controls learning-induced structural remodeling of neurons. Learn Mem, 2012; 19, 330–336. [DOI] [PubMed] [Google Scholar]
- 65).Gao Y et al. , Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron, 2004; 44, 609–621. [DOI] [PubMed] [Google Scholar]
- 66).Deng K, He H, Qiu J, Lorber B, Bryson JB, Filbin MT. Increased synthesis of spermidine as a result of upregulation of arginase I promotes axonal regeneration in culture and in vivo. J Neurosci. 2009; 29:9545–9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67).Roth BL, DREADDs for Neuroscientists. Neuron, 2016; 89, 683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68).Graham V, Khudyakov J, Ellis P & Pevny L SOX2 functions to maintain neural progenitor identity. Neuron, 2003; 39, 749–765. [DOI] [PubMed] [Google Scholar]
- 69).Dayer AG et al. Expression of FGF-2 in neural progenitor cells enhances their potential for cellular brain repair in the rodent cortex. Brain, 2007; 130, 2962–2976. [DOI] [PubMed] [Google Scholar]
- 70).Yoshimura S et al. FGF-2 regulates neurogenesis and degeneration in the dentate gyrus after traumatic brain injury in mice. J Clin Invest, 2003; 112, 1202–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71).Lee TT, Green BA, Dietrich WD & Yezierski RP Neuroprotective effects of basic fibroblast growth factor following spinal cord contusion injury in the rat. J Neurotrauma, 1999. 16, 347–356. [DOI] [PubMed] [Google Scholar]
- 72).Rabchevsky AG et al. Basic fibroblast growth factor (bFGF) enhances tissue sparing and functional recovery following moderate spinal cord injury. J Neurotrauma, 1999; 16, 817–830. [DOI] [PubMed] [Google Scholar]
- 73).Liang H et al. Region-specific and activity-dependent regulation of SVZ neurogenesis and recovery after stroke. Proc Natl Acad Sci U S A, 2019; 116, 13621–13630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74).Martin-Blondel G, Brassat D, Bauer J, Lassmann H & Liblau RS CCR5 blockade for neuroinflammatory diseases--beyond control of HIV. Nat Rev Neurol, 2016; 12, 95–105. [DOI] [PubMed] [Google Scholar]
- 75).Zhou M et al. CCR5 is a suppressor for cortical plasticity and hippocampal learning and memory. Elife, 2016; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76).Guyon A CXCL12 chemokine and its receptors as major players in the interactions between immune and nervous systems. Front Cell Neurosci 8,2014; 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77).Lorenzen E et al. G protein subtype-specific signaling bias in a series of CCR5 chemokine analogs. Sci Signal, 2018; 11. [DOI] [PubMed] [Google Scholar]
- 78).Hannila SS and Filbin MT The role of cyclic AMP signaling in promoting axonal regeneration after spinal cord injury. Exp Neurol. 2008. Feb; 209(2): 321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79).Galasso JM, Harrison JK & Silverstein FS Excitotoxic brain injury stimulates expression of the chemokine receptor CCR5 in neonatal rats. Am J Pathol, 1998; 153, 1631–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80).Watkins TA et al. , DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc Natl Acad Sci U S A, 2013; 110, 4039–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81).Biane JS, Takashima Y, Scanziani M, Conner JM & Tuszynski MH Thalamocortical Projections onto Behaviorally Relevant Neurons Exhibit Plasticity during Adult Motor Learning. Neuron, 2016; 89, 1173–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82).Boele HJ, Koekkoek SK, De Zeeuw CI & Ruigrok TJ Axonal sprouting and formation of terminals in the adult cerebellum during associative motor learning. J Neurosci, 2013; 33, 17897–17907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83).Hihara S et al. Extension of corticocortical afferents into the anterior bank of the intraparietal sulcus by tool-use training in adult monkeys. Neuropsychologia, 2006; 44, 2636–2646. [DOI] [PubMed] [Google Scholar]
- 84).Zatorre RJ, Fields RD & Johansen-Berg H Plasticity in gray and white: neuroimaging changes in brain structure during learning. Nat Neurosci, 2012; 15, 528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85).Dayan E & Cohen LG Neuroplasticity subserving motor skill learning. Neuron, 2011; 72, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86).Li S et al. An age-related sprouting transcriptome provides molecular control of axonal sprouting after stroke. Nat Neurosci, 2010; 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87).Carmichael ST, Wei L, Rovainen CM & Woolsey TA New patterns of intracortical projections after focal cortical stroke. Neurobiol Dis, 2001; 8, 910–922. [DOI] [PubMed] [Google Scholar]
- 88).Li S et al. GDF10 is a signal for axonal sprouting and functional recovery after stroke. Nat Neurosci, 2015; 18, 1737–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89).Morecraft RJ et al. Frontal and frontoparietal injury differentially affect the ipsilateral corticospinal projection from the nonlesioned hemisphere in monkey (Macaca mulatta). J Comp Neurol, 2016. 524, 380–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90).Wahl AS et al. Neuronal repair. Asynchronous therapy restores motor control by rewiring of the rat corticospinal tract after stroke. Science, 2014; 344, 1250–1255. [DOI] [PubMed] [Google Scholar]
- 91).Hamadjida A et al. Influence of anti-Nogo-A antibody treatment on the reorganization of callosal connectivity of the premotor cortical areas following unilateral lesion of primary motor cortex (M1) in adult macaque monkeys. Exp Brain Res, 2012; 223, 321–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92).Zai L et al. Inosine alters gene expression and axonal projections in neurons contralateral to a cortical infarct and improves skilled use of the impaired limb. J Neurosci, 2009; 29, 8187–8197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93).Tennant KA et al. The organization of the forelimb representation of the C57BL/6 mouse motor cortex as defined by intracortical microstimulation and cytoarchitecture. Cereb Cortex, 2011; 21, 865–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94).Tennant KA et al. Age-dependent reorganization of peri-infarct “premotor” cortex with task-specific rehabilitative training in mice. Neurorehabil Neural Repair, 2015; 29, 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95).Harrison TC, Silasi G, Boyd JD & Murphy TH Displacement of sensory maps and disorganization of motor cortex after targeted stroke in mice. Stroke, 2013; 44, 2300–2306. [DOI] [PubMed] [Google Scholar]
- 96).Brown CE, Aminoltejari K, Erb H, Winship IR & Murphy TH In vivo voltage-sensitive dye imaging in adult mice reveals that somatosensory maps lost to stroke are replaced over weeks by new structural and functional circuits with prolonged modes of activation within both the peri-infarct zone and distant sites. J Neurosci, 2009; 29, 1719–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97).Sawaki L et al. Constraint-induced movement therapy results in increased motor map area in subjects 3 to 9 months after stroke. Neurorehabil Neural Repair, 2008; 22, 505–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98).Kleim JA, Barbay S & Nudo RJ Functional reorganization of the rat motor cortex following motor skill learning. J Neurophysiol, 1998; 80, 3321–3325. [DOI] [PubMed] [Google Scholar]
- 99).Nudo RJ, Milliken GW, Jenkins WM & Merzenich MM Use-dependent alterations of movement representations in primary motor cortex of adult squirrel monkeys. J Neurosci, 1996; 16, 785–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100).Reed A et al. Cortical map plasticity improves learning but is not necessary for improved performance. Neuron, 2011; 70, 121–131. [DOI] [PubMed] [Google Scholar]