

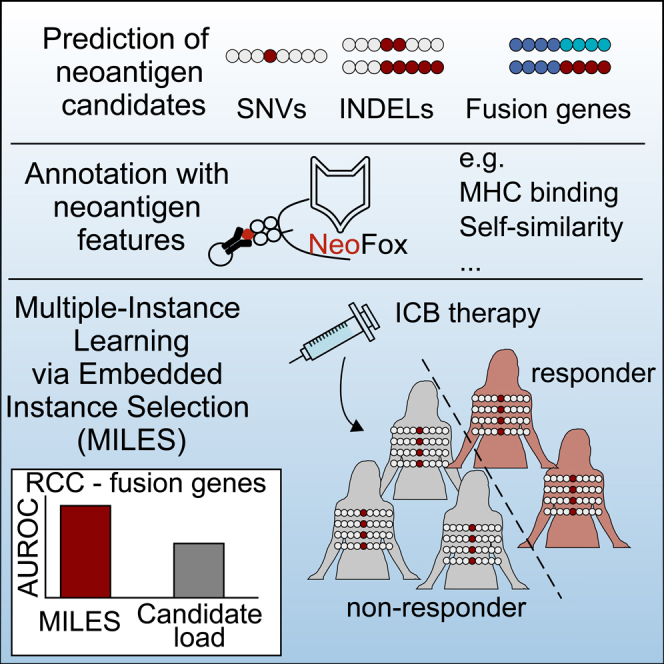
Summary
Previous studies showed that the neoantigen candidate load is an imperfect predictor of immune checkpoint blockade (ICB) efficacy. Further studies provided evidence that the response to ICB is also affected by the qualitative properties of a few or even single candidates, limiting the predictive power based on candidate quantity alone. Here, we predict ICB efficacy based on neoantigen candidates and their neoantigen features in the context of the mutation type, using Multiple-Instance Learning via Embedded Instance Selection (MILES). Multiple instance learning is a type of supervised machine learning that classifies labeled bags that are formed by a set of unlabeled instances. MILES performed better compared with neoantigen candidate load alone for low-abundant fusion genes in renal cell carcinoma. Our findings suggest that MILES is an appropriate method to predict the efficacy of ICB therapy based on neoantigen candidates without requiring direct T cell response information.
Subject areas: Bioinformatics, Immunology, Machine learning
Graphical abstract
Highlights
-
•
Multiple-Instance Learning via Embedded Instance Selection (MILES)
-
•
Prediction of the immune checkpoint blockade (ICB) efficacy
-
•
MILES predicts ICB efficacy with fusion genes in renal cell carcinoma
-
•
MILES might support defining neoantigen features triggering response to ICB
Bioinformatics; Immunology; Machine learning
Introduction
Neoantigens are tumor-specific mutated gene products that are presented in the form of neoepitopes by the major histocompatibility complex (MHC) proteins and recognized by CD8+ or CD4+ T cells. Upon neoepitope recognition, these neoantigen-specific T cells can mediate tumor control in the presence of a favorable tumor microenvironment. Immune checkpoint blockade (ICB) drives tumor control via the functional re-invigoration of neoantigen-specific T cells.1,2,3,4 We previously introduced a concept-based classification of neoantigens, classifying neoantigens that are recognized by such pre-existing re-invigorated T cells and that are predictive for the clinical benefit of ICB therapy as restrained neoantigens.5
Different types of mutation sources can generate neoantigens with diverse molecular characteristics. While neoantigens from single-nucleotide variants (SNVs) usually cause a single amino acid substitution, INDELs (small insertions or deletions) or fusion genes can generate frameshift neoantigens with completely altered amino acid sequences. INDELs can generate immunogenic neoantigens,6,7 and the INDEL burden correlates with the response to ICB in melanoma patients.8,9 Furthermore, a head and neck cancer patient with clinical response to anti-PD-1 therapy harbored only one single immunogenic neoantigen from a fusion gene.10 These observations suggest that neoantigens of all mutation types could act as restrained neoantigens. Previous studies investigating the characteristics of restrained neoantigens from SNVs have shown that clonality,11 the difference in MHC-I binding affinity to the wild-type peptide (differential agretopicity index, DAI),12 and the ratio-based DAI in combination with the sequence similarity to epitopes from known pathogens13 correlated with survival upon ICB therapy. Therefore, the response to cancer immunotherapy is driven not only by neoantigen candidate quantity but also by their quality.14
Further neoantigen features and prioritization methods have been published, and we recently developed a toolbox called NeoFox15 to annotate neoantigen candidates with a variety of neoantigen features. An analysis of how these features characterize restrained neoantigens is still missing—in particular in the context of non-SNV mutation types.
Standardized, unbiased, and systematic immunogenicity screenings of neoantigen candidates providing direct information about neoantigen-specific T cell responses are still limited in their availability for such an in silico analysis. Therefore, we predicted neoantigen candidates from raw whole-exome (WES) and RNA sequencing (RNA-seq) data from five ICB cohorts to examine if the clinical response can be predicted based on the characteristics of neoantigen candidate profiles in the context of the mutation type.
Although traditional supervised machine learning approaches classify labeled instances, multiple instance learning is a special branch that classifies labeled groups (so-called bags) that are formed by a set of instances with unknown labels.16 According to the standard assumption of multiple instance learning, positive bags harbor at least one instance with a hidden positive label and negative bags harbor exclusively negative instances.16,17 For our analysis, patients are referred to as bags, the clinical response to ICB as the bag label, and neoantigen candidates as the instances. Multiple instance learning has been used in the field of cancer immunology for distinguishing tumor from normal samples on their T cell receptor (TCR) sequence profiles18 and for predicting T cell infiltration on neoantigen candidate profiles.19
Here, we used multiple instance learning to predict the clinical response to ICB based on neoantigen candidates of cancer patients in the context of the mutation type. We further identified features that are relevant to predict ICB efficacy and that may characterize restrained neoantigens (i.e., neoantigens that are recognized by ICB reinvigorated T cells).5
Results
Neoantigen candidate loads are heterogeneous in cancer patients
To investigate the characteristics of neoantigen candidates in the context of the mutation type, we identified neoantigen candidates from SNVs, INDELs, and fusion genes in raw WES and RNA-seq data from five melanoma or renal cell carcinoma patient cohorts treated with α-PD-1,20,21,22 α-CTLA-4,23 or α-PD-L120,24 cancer immunotherapy. We then annotated these neoantigen candidates with neoantigen features (Figure 1).
Figure 1.

Identification of neoantigen candidates in immune checkpoint blockade cohorts
Publicly available WES and RNA-seq from patient cohorts who had been treated with ICB therapy were collected. Neoantigen candidates from SNVs and INDELs were identified with an in-house property pipeline25 (“iCaM2”), and candidates from fusion genes were identified with EasyFuse.26 Neoantigen candidates were annotated with neoantigen features using NeoFox.15
The distribution of the neoantigen candidate load per patient varied between mutation types and datasets (Figures 2 and S1 and Table S1). Although the median load of neoantigen candidates derived from SNVs was 208 in the three melanoma datasets (“Hugo”, “Riaz”, “Van Allen”), the median SNV-derived neoantigen candidate load was 39 in the two renal cell carcinoma datasets (“Miao”, “McDermott”) (Figure 2A; Table S1). Neoantigen candidates from INDELs or fusion genes were in general rarer than SNV-derived neoantigen candidates in all datasets. The relative proportion of neoantigen candidates from INDELs or fusion genes per patient with respect to neoantigen candidate load of all mutation types was higher in patients of the renal cell carcinoma datasets (“RCC”) in comparison to the melanoma datasets (“MEL”) (Figures 2B and 2C).
Figure 2.

Overview of neoantigen candidates in the context of the mutation type in cohorts treated with immune checkpoint blockade
(A) The neoantigen candidate load from SNVs, INDELs, or fusion genes per patient in five ICB cohorts.
(B and C) The proportion of neoantigen candidates from each mutation type (SNV, INDEL, or fusion genes) relative to neoantigen candidate load from all mutation types was determined for each patient in the (B) melanoma (“MEL”) cohorts and (C) renal cell carcinoma cohorts (“RCC”). The proportion is shown on the y axis, and indexed patients are shown on the x axis.
(D‒I) Density plots showing the density distribution of (D) MHC-I binding rank, (E) MHC-II binding rank, (F) MHC-I amplitude, (G) amplitude MHC-II, (H) self-similarity MHC-I, and (I) self-similarity MHC-II of neoantigen candidates from SNVs, INDELs, and fusion genes in a combined dataset of all ICB cohorts. See also Figure S1.
Next, we combined neoantigen candidates from the five ICB cohorts and compared the density distribution of selected neoantigen features between SNV-, INDEL-, and fusion-gene-derived candidates (Figures 2D–2I). The distribution of the best-predicted MHC-I and MHC-II binding rank was comparable for SNV-, INDEL-, and fusion-gene-derived neoantigen candidates, indicating that neoantigen candidates from different mutation types shared comparable MHC binding ability (Figures 2D and 2E). INDELs and fusion genes were associated with higher amplitude MHC-II (rank) values in comparison to SNVs (Figures 2G). This suggested that the non-SNV mutation types are more likely to generate predicted MHC-II epitopes with improved MHC-II binding ranks compared with their wild-type counterpart. As expected, the best-predicted MHC-I and MHC-II epitopes of INDEL- and fusion-gene-derived neoantigen candidates were less similar to their wild-type counterpart in comparison to SNV-derived candidates (Figures 2H and 2I).
The neoantigen candidate load is an imperfect predictor of the response to ICB
We systematically evaluated whether the predicted neoantigen candidate load significantly differed between responders and non-responders to ICB for each mutation type and dataset. The neoantigen candidate load was defined with respect to different MHC-I and MHC-II binding affinity thresholds, while considering either all or only expressed neoantigen candidates (Figures 3A–3D and S2A‒S2F).
Figure 3.

Neoantigen candidate load is an imperfect predictor of the response to immune checkpoint blockade
(A‒C) The INDEL-derived neoantigen candidate load was compared between responder and non-responder in the Riaz cohort based on (A) all predicted neoantigen candidates, (B) candidates with MHC-I or MHC-II binding affinity <50 nM, and (C) expressed candidates with MHC-I or MHC-II binding affinity <50 nM.
(D) The neoantigen candidate load was compared between responder and non-responder with respect to the mutation type, MHC binding ability, and RNA expression. The plot represents the resulting p value from each comparison. Comparisons that resulted in a p value <0.05 are shown in purple, whereas non-significant comparisons are shown white. The y axis represents a definition of the neoantigen candidate load. Neoantigen candidates with MHC-I or MHC-II binding affinity lower than the respective threshold (“MHC affinity cutoff”) were used. The column “only expressed” indicates if all neoantigen candidates (“−”) or only neoantigen candidates confirmed in the RNA-seq (“+”) were used. All comparisons were performed in each individual ICB cohort and in a combined dataset of all cohorts (“all”), the melanoma cohorts (“MEL”), or the renal cell carcinoma cohorts (“RCC”). The letters A–C refer to respective subpanel above. Statistical testing was performed with Wilcoxon signed ranked test. P-values were corrected for multiple testing on the same dataset with Benjamini Hochberg method. Statistical tests resulting in p values <0.05 after multiple testing correction were considered as significant. See also Figure S2.
In general, patients responding to ICB therapy harbored significantly higher SNV-derived neoantigen candidate loads compared with non-responding patients when combining all analyzed ICB cohorts independent of the thresholds for MHC binding affinity and expression to define the neoantigen candidate load (p < 0.05) (Figures 3D and S2A‒S2C). Interestingly, the INDEL-derived neoantigen candidates with good MHC-I or MHC-II binding properties correlated with ICB efficacy in one individual melanoma cohort (“Riaz”) (Figures 3A–3D). The fusion-gene-derived neoantigen candidate load generally did not correlate with ICB efficacy (Figures 3D and S2D‒S2F).
These observations support previous findings by other studies27,28,29 that the SNV or SNV-derived neoantigen candidate load alone is an imperfect predictor of the response to ICB.
Multiple-Instance Learning via Embedded Instance Selection to predict the response to ICB on neoantigen candidates
The imperfect correlation between neoantigen candidate load and the response to ICB motivated us to examine whether ICB efficacy can be predicted by considering the qualitative features of neoantigen candidates.14 Therefore, all patients were represented by their predicted neoantigen candidates annotated with selected neoantigen features. We used 29 neoantigen features that were annotated with NeoFox15 such as MHC binding properties or the self-similarity (Table 1).
Table 1.
Description of neoantigen features
| Feature | Description | Reference |
|---|---|---|
| rnaExpression | RNA expression | – |
| rnaVariantAlleleFrequency | Variant allele fraction | – |
| Best_rank_MHCI_score | Best predicted MHC-I binding rank per neoantigen candidate | Reynisson et al.30 |
| Best_rank_MHCII_score | Best predicted MHC-II binding rank per neoantigen candidate | Reynisson et al.30 |
| MixMHCpred_best_rank | Best predicted MixMHCpred rank per neoantigen | Bassani-Sternberg et al.31 |
| MixMHC2pred_best_rank | Best predicted MixMHC2pred rank per neoantigen | Racle et al.32 |
| Amplitude_MHCI_affinity | Ratio of the MHC-I affinity score between the best predicted MHC-I neoepitope and its corresponding wild-type peptide | Łuksza et al.,13 Balachandran et al.33 |
| Amplitude_MHCII_rank | Ratio of the MHC-II rank score between the best predicted MHC-II neoepitope and its corresponding wild-type peptide | Adapted from Łuksza et al.,13 Balachandran et al.33 |
| DAI_MHCI_affinity | Difference in the MHC-I affinity score between the best predicted MHC-I neoepitope and its corresponding wild-type peptide | Duan et al.34 |
| PHBR_I | The harmonic mean of best predicted MHC-I binding rank across the MHC-I genotype | Marty et al.35 |
| PHBR_II | The harmonic mean of best predicted MHC-II binding rank across the MHC-II genotype | Marty Pyke et al.36 |
| Generator_rate_MHCI | Number of predicted MHC-I neoepitopes per neoantigen candidate | Rech et al.37 |
| Generator_rate_MHCII | Number of predicted MHC-II neoepitopes per neoantigen candidate | Rech et al.37 |
| Selfsimilarity_MHCI | Similarity to the self-proteome of the best predicted MHC-I neoepitope per neoantigen candidate | Bjerregaard et al.38 |
| Selfsimilarity_MHCII | Similarity to the self-proteome of the best predicted MHC-II neoepitope per neoantigen candidate | Adapted from Bjerregaard et al.38 |
| Dissimilarity_MHCI | Similarity to the self-proteome of the best predicted MHC-I neoepitope per neoantigen candidate | Richman et al.39 |
| Pathogensimiliarity_MHCI_9mer | Similarity of the best predicted MHC-I neoepitope per neoantigen candidate to known pathogens | Łuksza et al.,13 Balachandran et al.33 |
| Pathogensimiliarity_MHCII | Similarity of the best predicted MHC-II neoepitope per neoantigen candidate to known pathogens | Adapted from Łuksza et al.,13 Balachandran et al.33 |
| Hex_alignment_score_MHCI | Similarity of the best predicted MHC-I neoepitope per neoantigen candidate to known pathogens | Chiaro et al.40 |
| Hex_alignment_score_MHCII | Similarity of the best predicted MHC-II neoepitope per neoantigen candidate to known pathogens | Adapted from Chiaro et al.40 |
| IEDB_Immunogenicity_MHCI | IEDB immunogenicity score for the best predicted MHC-I neoepitope | Calis et al.41 |
| IEDB_Immunogenicity_MHCII | IEDB immunogenicity score for the best predicted MHC-II neoepitope | Adapted from Calis et al.41 |
| vaxrank_binding_score | Cumulative MHC I binding score per neoantigen candidate | Rubinsteyn et al.42 |
| vaxrank_total_score | Combination of vaxrank binding score with variant allele expression | Rubinsteyn et al.42 |
| Priority_score | Combinatorial score of MHC-I binding rank and variant allele expression | Bjerregaard et al.43 |
| Recognition_Potential_MHCI_9mer | Combinatorial score of amplitude MHC-I and pathogen similarity | Balachandran et al.33 |
| Neoag_immunogenicity | Machine learning model | Łuksza et al.,13 Smith et al.44 |
| T cell_predictor_score | Machine learning model | Besser et al.45 |
| PRIME_best_rank | Machine learning model | Schmidt et al.46 |
MHC, major histocompatibility complex; DAI, differential agretopicity index; PHBR, Patient Harmonic-mean Best Rank; IEDB, Immune Epitope Database.
Then, we used multiple instance learning to predict ICB efficacy based on the set of annotated and unlabeled neoantigen candidates. Patients are referred to as bags with the response to ICB as their label (Figure 4A). Each bag is a collection of unlabeled instances, i.e., neoantigen candidates with unknown anti-tumoral activity. The multiple instance learning standard assumption meets the biological assumptions that responders (“positive bags”) must harbor at least one true neoantigen (“positive instance”), whereas non-responders (“negative bags”) must harbor only neoantigen candidates that cannot trigger anti-tumoral activity (“negative instances”).17 The MILES (Multiple-Instance Learning via Embedded Instance Selection)47 algorithm was chosen as the algorithm of choice in this study as it performed well in a previous benchmarking study related to cancer detection based on TCR sequences.18
Figure 4.

Multiple-Instance Learning via Embedded Instance Selection to predict the response to immune checkpoint blockade (ICB) using neoantigen candidates
(A) Multiple instance learning was used to distinguish responder from non-responder, given their neoantigen candidate profiles represented by the annotated neoantigen features.
(B) Models were trained and evaluated in a nested cross-validation (CV) loop for multiple hyperparameter sets on the full dataset. The hyperparameter set with the best median area under the receiver operating characteristic curve (AUROC) across the nested CV approach was selected to represent the performance of the learning method.
(C) The median performance of MILES to predict ICB efficacy on a dataset of SNVs, fusion genes, or INDELS only and a dataset combining all mutation types (“all”) in terms of the AUROC for all cohorts (“MEL+RCC”), the melanoma cohorts (“MEL”), and the renal cell carcinoma cohorts (“RCC”).
(D and E) Comparison of the performance of MILES and the neoantigen candidate load alone on a dataset of SNVs, fusion genes, or INDELS only and a dataset combining all mutation types (“all”) in terms of the AUROC for (D) all cohorts (“MEL+RCC”) and (E) the melanoma cohorts (“MEL”).
(F) The previous analysis was repeated on a dataset without patients with stable disease. The median AUROC values of MILES over a nested cross-validation.
(G) Comparison of the performance of MILES and the neoantigen candidate load alone on a dataset of fusion genes for the renal cell carcinoma cohorts (“RCC”). Data are shown over a nested CV in C–G. Median AUROC with interquartile range as error bars is shown in C and F. AUROC values resulting from the nested CV were compared between MILES and the neoantigen candidate load with Wilcoxon signed ranked test in D, E, and G. No multiple testing correction was applied. See also Figures S3–S5.
For a robust performance estimation, the MILES algorithm was trained and evaluated on neoantigen candidates with a nested cross-validation approach across multiple hyperparameter sets48 (Figure 4B). The median area under the receiver operating characteristic curve (AUROC) across the nested cross-validation was used to evaluate the performance of the learning method.
We predicted the response to ICB with MILES on neoantigen candidates from SNVs, INDELs, or fusion genes separately or by a combination of all mutation types in tumor-entity-specific datasets (“MEL”; “RCC”) or in a dataset combining all ICB cohorts (“MEL+RCC”) (Figure 4C; Table S2). The set of hyperparameters with the best performance differed for the learning approaches trained on SNV, INDEL, fusion genes or on all mutation types (Table S2). Training and evaluating the MILES approach on SNV-derived neoantigen candidates from all ICB cohorts (“MEL+RCC”) achieved a median AUROC of 0.62 (Figure 4C; Table S2). We observed an even better performance when evaluating MILES on a dataset restricted to the three melanoma cohorts (“MEL”) for SNV-specific (median AUROC = 0.69) and combined (median AUROC = 0.75) approach (Figure 4C).
Next, we wanted to directly compare the performance of MILES and using the neoantigen candidate load to predict ICB efficacy. Therefore, we performed an ROC-curve analysis in a nested CV on the neoantigen candidate load as a predictor of ICB efficacy as well (Figures 4D and 4E). This analysis suggested that the MILES approach performed superior to neoantigen candidate load for the mutation-type combined and melanoma-specific dataset (“MEL”, Figure 4E).
As an additional control, we trained the MILES algorithm on datasets with randomized neoantigen candidates but original distribution of neoantigen candidate load (Figure S3). When neoantigen candidates were randomized across patients, MILES performed randomly (e.g., median AUROC = 0.44 for the SNV-specific approach in the “MEL+RCC” cohort).
The MILES algorithm performed randomly on datasets restricted to data from “RCC” cohorts (Figure 4C). The RCC cohorts had a higher fraction of patients with stable disease in comparison to the MEL cohorts (Figures S4A and S4B). We hypothesized that stable disease leading, e.g., to survival benefit may be mediated by neoantigens and re-trained and evaluated MILES on datasets excluding patients with stable disease (Figure 4F; Table S2). Of note, MILES achieved a median AUROC of 0.75 in the fusion-gene-specific approach in RCC cohort, performing superior to the neoantigen candidate load (Figures 4F and 4G). MILES performed randomly on randomized neoantigen candidates in the RCC cohort (Figures S4C and S4D).
Next, we examined which neoantigen features were important to predict ICB efficacy with MILES (Figure 4A). Because the embedding step in the MILES algorithms involves a nonlinear transformation,47 the feature importance could not be estimated internally within the algorithm. Therefore, we repeated the nested cross-validation approach on datasets in which the neoantigen feature of interest was permutated. Then, we approximated feature importance by the delta AUROC of the original learning method and the approach without the feature of interest and considered features achieving a delta AUROC >0.05 as relevant.
We focused the feature importance analysis on MILES approaches that achieved a median AUROC >0.6 as non-random approaches (Figures S5A and S5B). The differential agretopicity index (DAI)34—the difference in MHC binding affinity between the mutated and non-mutated neoepitope candidate—was predicted as important feature in all approaches, suggesting its general relevance (Figures S5A and S5B). Also, the similarity of the best-predicted MHC-II peptide to known pathogenic epitopes in terms of the HEX score40 achieved delta AUROCs higher than 0.05 for the learning approach on SNVs in the MEL and RCC combined dataset (delta AUROC = 0.06) and on all mutation types in the RCC-specific dataset without SD patients (delta AUROC = 0.15). Furthermore, features such as the RNA expression (delta AUROC = 0.06) and PHBR-II36 (delta AUROC = 0.06) and vaxrank42 (delta AUROC = 0.06) were predicted to be relevant in the combined set of neoantigen candidates from all mutation types, specifically in the context of RCC (Figure S5B).
Discussion
Predicting ICB therapy efficacy with neoantigens is still challenging due to lack of appropriate models. We tackled this challenge by predicting—in the context of mutation type and tumor entity—the response to ICB with neoantigen candidate load or with a multiple instance learning approach that relies on neoantigen candidates annotated with neoantigen features.
We identified the SNV-derived neoantigen candidate load as a predictor of the response to ICB in a combined set of all ICB cohorts but not in an individual ICB cohort. This work supported previous findings that the neoantigen candidate load from INDEL mutations correlates with the response to ICB in particular in the context of melanoma.8,9 Furthermore, we observed that the neoantigen candidate load derived from fusion genes was not an indicator for the response to ICB as observed previously.49 The limitations of the mutation or neoantigen candidate load as a predictor of the response to ICB has been extensively studied and discussed in particular in the context of SNVs.27,28,29 It is conceivable that technical shortcomings in the tools used for mutation calling and in the definition of the neoantigen candidate load limit its predictive power in our and other studies. The observation that patients with low neoantigen candidate load also harbor immunogenic neoantigens and can respond to ICB therapy10 suggests that, aside from technical shortcomings, disregarding qualitative traits limits and pursuing solely the neoantigen candidate load would limit predictive power.
Previous studies have used different approaches and prior assumptions to predict the response to ICB based on neoantigen candidates, e.g., based on the best-predicted neoantigen candidate,13,33 the mean across all predicted candidates,12 or by the Cauchy-Schwarz index.50 Here, we predicted the ICB therapy efficacy dependent on neoantigen candidates with multiple instance learning. This approach relies only on the prior assumption that a responder to ICB harbors at least one immunogenic neoantigen, whereas a non-responder lacks immunogenic neoantigens. Evaluating MILES on neoantigen candidate data demonstrated that this approach is able to achieve non-random performance independent of the underlying neoantigen candidate load, as suggested by the evaluation of MILES on randomized data. The MILES approach on the neoantigen candidates from fusion genes improved the prediction of clinical benefit, as compared with that based on the fusion-gene-derived neoantigen candidate load. In particular, predicting the ICB efficacy in RCC by fusion-gene-derived neoantigen candidates was superior to neoantigen candidate load if patients with stable disease were excluded from the analysis.
Previously, we defined neoantigens that are predictive of the clinical outcome of ICB therapy as restrained neoantigens.5 Apart from predicting ICB efficacy, the multiple instance learning approach supports to investigate the features of neoantigen candidates that may contribute to ICB efficacy. We analyzed the relevance of neoantigen features for the learning method and confirmed a previous observation that the DAI of the best-predicted MHC-I neoepitope is a descriptor of neoantigen candidates from all mutation types that may contribute to ICB efficacy.12 Furthermore, the similarity of the best-predicted MHC-II neoepitope to viral epitopes in terms of the HEX algorithm40 appeared to be a relevant neoantigen feature in our analysis. This observation could indicate that at least a subset of the neoantigens in patients responding to ICB may be cross-recognized by heterologous T cells.40,51 However, the external-permutation-based method to estimate feature importance comes with two main limitations: (1) feature importance results might change with permutation and (2) some features might correlate and affect the importance measure of each other. Overcoming these limitations may guide the understanding that qualitative neoantigen features characterize restrained neoantigens in the future.
Here, we showed that multiple instance learning can be used to predict immunotherapy efficacy based on qualitative neoantigen candidate profiles covering multiple mutation types, and we provide the basis for future investigation. In our study, MILES outperformed the neoantigen candidate load only in a few investigated cases. Integrating the potentially complementary neoantigen candidate load and the qualitative multiple instance approach may improve the prediction of the response to ICB in other use cases. A limited set of neoantigen features was integrated into the model approach in this study, mostly targeting the linear sequence of neoantigen candidates and rather focusing on the interaction with MHC molecules.15 Integrating clonality information,11 structural features,52 and novel features that specifically model the interaction between the MHC-bound neoepitope and the TCR repertoire may improve predictions in the future. This could be in particular applicable in the cases for which we retrieved random performance or unimproved performance compared with the neoantigen candidate load in this study. Furthermore, when more data are available, systematic benchmarks may identify the best suitable multiple instance learning algorithm. However, one interesting characteristic of the MILES algorithm used in this study is its internal instance selection approach and its ability to be used for instance classification.47 Therefore, multiple instance learning with instance selection could empower not just prediction of ICB efficacy but also the identification of immunogenic neoantigens in the future.
Limitations of the study
This study comes with certain limitations. The major limitation of our work is the size of the used dataset that leads to large variation while estimating the performance of the neoantigen candidate load or MILES in predicting ICB efficacy with a nested cross-validation. Furthermore, we manually pre-selected neoantigen features used in this study. Neoantigen features such as clonality11 were not considered in our work. The results could be affected by the use of FPKM expression values and the combination of different expression scales when combining mutation types. Moreover, the estimation of the feature importance in MILES models using a permutation-based approach could be imprecise, e.g., due to correlation between features. A direct comparison of mutation types might be imprecise due to uneven occurrence of SNVs, INDELS, and fusion genes in patients.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Hugo dataset | Hugo et al.21 | SRP067938, SRP090294 (WES-seq) and SRP070710 (RNA-seq) |
| Riaz dataset | Riaz et al.22 | SRP095809 (WES-seq) and SRP094781 (RNA-seq) |
| Van Allen dataset | van Allen et al.23 | phs000452.v2.p1 |
| Miao dataset | Miao et al.20 | phs001493.v1.p1 |
| McDermott dataset | McDermott et al.24 | EGAS00001002928 |
| Software and algorithms | ||
| Bwa v0.7.10 | Li and Durbin53 | https://github.com/lh3/bwa |
| Picard v1.110 | Broad Institute | http://broadinstitute.github.io/picard |
| strelka2 v2.0.14 | Kim et al.54 | https://github.com/Illumina/strelka |
| EasyFuse v1.3 | Weber et al.26 | https://github.com/TRON-Bioinformatics/EasyFuse |
| HLA-HD v1.2.0.1 | Kawaguchi et al.55 | https://www.genome.med.kyoto-u.ac.jp/HLA-HD/ |
| STAR v2.4.2a | Dobin et al.56 | https://github.com/alexdobin/STAR |
| Sailfish vBeta-0.7.6 | Patro et al.57 | https://www.cs.cmu.edu/∼ckingsf/software/sailfish/ |
| NeoFox v0.5.3 | Lang et al.15 | https://github.com/TRON-Bioinformatics/neofox |
| Mil | Mil | https://github.com/rosasalberto/mil |
| R v4.1.0 | R Core Team | https://www.r-project.org/ |
| Python v3.7.3 | Python Software Foundation | https://www.python.org/ |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Martin Löwer (Martin.Loewer@TrOn-Mainz.DE).
Materials availability
This study did not generate new unique reagents.
Experimental model and study participant details
Independent datasets from five immune checkpoint blockade trials were collected.20,21,22,23,24
Whole exome sequencing (WES) from tumor and matched normal samples and RNA-seq data of the tumor sample were retrieved from the respective repositories.
Clinical outcome data were collected from the original publications, and only patients with both available WES and RNA-seq were considered in the downstream analysis. The response categories were transformed into a table of binary outcomes. Patients with complete (CR) or partial (PR) response were defined as responders and patients with stable (SD) or progressive (PD) disease as non-responders.
Samples were restricted to ICB-therapy naive samples that were acquired pre-treatment in the Riaz cohort.22 Only patients treated with atezolizumab as a single agent were considered in the analysis in the McDermott cohort.24
Method details
Prediction of neoantigen candidates
Neoantigen candidates were detected using an in-house built standardized pipeline that was described previously.25,58 The pipeline covers the alignment of DNA reads to the reference genome hg19 using bwa (v0.7.10)53 and the removal of duplicated reads with Picard (v1.110) (http://broadinstitute.github.io/picard). An in-house developed proprietary software was used to detect high-confidence single nucleotide variations. INDEL variations were detected with strelka2.54 The detected somatic nonsynonymous mutations were translated into 27mer peptide sequences with the mutation at position 14. Frameshift INDELs were translated until the occurrence of the next stop codon. Unsolvable technical issues arose for two patients; these were excluded from further analysis that included SNV- or INDEL-derived neoantigen candidates.
Neoantigen candidates derived from fusion genes were predicted from RNA-seq data using EasyFuse.26 For the downstream analyses, fusion gene-derived neoantigen candidates had to fulfill the following criteria: (i) an EasyFuse probability score >0.5, (ii) to not be a false-positive fusion gene call from a curated exclusion list of known fusion genes in normal tissue (iii) best break point per fusion gene pair based on the prediction probability of the random forest classifier, (iv) breakpoints must be on the respective exon boundary, (v) frame is not “no_frame” and (vi) exclude neoantigen candidates with “neo_frame” in case “in_frame” neoantigen candidates were predicted for the same fusion gene.
HLA-typing
MHC -I and -II genotypes were detected for each patient with HLA-HD (v1.2.0.1) using the normal WES data.55
Transcript expression analysis
Transcript expression analysis was performed by aligning RNA-seq reads to the hg19 reference genome with STAR (v2.4.2a),56 followed by quantification of transcripts in FPKM (fragments per kilobase of exon model per million reads mapped) with sailfish (vBeta-0.7.6).57
Transcript expression of fusion genes was approximated by the sum of spanning and junction reads.
Annotation of neoantigen candidates
Neoantigen candidates from all mutation types were annotated with published neoantigen features and prioritization algorithms using NeoFox (v0.5.3).15 The predicted neoantigen candidates, MHC-I and -II genotypes of the patient and the tumor type were provided as input.
The wild type counterpart for neoepitope candidates from INDELs or fusion genes was defined by the best hit of the same length in a BLAST (Basic Local Alignment Search Tool) search against the human proteome in NeoFox.
Twenty-nine neoantigen features that were annotated by NeoFox were included in the downstream analyses (Table 1). Features were manually pre-selected to exclude highly coinciding features that derive from the exact same original tool. For instance, we used only the best MHC-I binding rank (Best_rank_MHCI_score), and we neglected the best MHC-I binding affinity score determined by netMHCpan.
Multiple instance learning
The response to ICB was predicted with multiple instance learning on the annotated neoantigen candidates using the MILES (Multiple-Instance Learning via Embedded Instance Selection) algorithm.
MILES was proposed by Chen et al.47 MILES embeds bags into an instance-based feature space using an instance similarity measure between each bag and instances. The dimensionality of this instance-based feature space equals the total number of instances, leading to a high dimensional feature space when the total number of instances in the dataset is high. Not all of the features (instances) may be relevant for classification. Relevant features are selected with a 1-norm support vector machine which is simultaneously used to construct the bag classifier.47
In this work, we used the implemented MILES algorithm from the python library mil (https://github.com/rosasalberto/mil). In order to use the library, we adjusted the function to load the data provided by the package and increased the number of iterations (max_iter) in the LinearSVC of the miles function to 100,000.
Prior to training, the direction of scaling was harmonized for all neoantigen features, i.e., the scaling was reversed for Best_rank_MHCI_score, Best_rank_MHCII_score, MixMHC2pred_best_rank, MixMHCpred_best_rank, Selfsimilarity_MHCI, Selfsimilarity_MHCII, PRIME_best_rank, PHBR_I, PHBR_II. Missing values were filled with the minimal value of a neoantigen feature across all predicted neoantigen candidate, assuming that a missing value reflects biological irrelevance of the neoantigen candidate of interest.
Quantification and statistical analysis
Candidate load as a predictor of ICB efficacy
We compared the neoantigen candidate load between responder and non-responder to ICB therapy with Wilcoxon signed ranked test in each individual ICB cohort, in tumor entity combined datasets (“MEL”, “RCC”) and in a combined dataset of all cohorts (“all”). Neoantigen candidate load was defined either by neoantigen candidates derived by fusion genes only, INDELs only, SNV only or by all mutation types. Furthermore, the neoantigen candidate load was assessed under multiple MHC binding affinity cutoffs and if the neoantigen candidate load were found in the RNA-seq data. This resulted in many tests on each dataset and p values were corrected for multiple testing with the Benjamini Hochberg method59 in each examined dataset. Statistical tests resulting in p values <0.05 after multiple testing correction were considered as significant.
The number of patients per dataset investigated in this work and the number of predicted neoantigen candidate load (without filtering with respect to MHC binding affinity or RNA expression) are provided in Table S1.
Performance of multiple instance learning
Multiple instance learning models were trained with a plain 10-fold cross-validation on the full dataset to allow a robust estimation of the performance of the learning method across the repeated splits.48
The MILES algorithm comes with the two hyperparameters sigma2 and λ.47 To set the hyperparameters (λ = [0.1,..,1], sigma2 = [50, …,10000000]), an internal 10-fold cross-validation was used. Thus, a 10-fold external cross-validation was used for model validation and within each "fold" another internal 10-fold cross-validation for hyperparameter estimation, amounting to in total 100 runs in a nested cross-validation.
This approach was used to estimate the performance of the learning method in predicting the response to ICB on neoantigen candidates restricted to SNVs, INDELs or fusion genes or on a combined dataset covering neoantigen candidates from SNVs, INDELs and fusion genes. The performance of the learning method was represented by the median AUROC and its interquartile range across the nested cross-validation approach. An AUROC of 0.5 reflects random guessing while an AUROC of 1 reflects a classifier with optimal performance.
To compare the performance of the neoantigen candidate load as a predictor of ICB efficacy to multiple instance learning, ROC-curve analysis was performed as described above for the neoantigen candidate load.
Feature importance
To estimate the importance of each neoantigen feature, the feature of interest was permutated and models were re-trained as described above on that dataset using the best hyper-parameter setting of the original approach. This procedure was repeated 50x and approximated the performance across the 50x nested cross-validations. Then, feature importance was approximated by the delta AUROC of the learning method on the original data and the learning method on the data with permutated feature. Features with delta AUROC ≥0.05 were considered as important in this work.
Acknowledgments
This work was supported by an ERC Advanced Grant to U.S. (ERC-AdG 789256). The authors would like to thank Karen Chu for proof-reading the manuscript and helpful comments. The authors further acknowledge the authors and generators of datasets used in this work and the grants that supported the studies.
Author contributions
Conceptualization: F.L. and M.L.; Methodology: F.L., S.K., and M.L.; Formal Analysis: F.L. and P.S.; Investigation: F.L.; Writing—Original Draft: F.L.; Writing—Review & Editing: F.L., B.S, D.W. S.K., and M.L.; Visualization: F.L.; Supervision: B.S., D.W., S.K., U.S., and M.L.; Project Administration: B.S. and M.L.; Funding Acquisition: B.S., U.S., and M.L.
Declaration of interests
U.S. is the co-founder, shareholder, and CEO at BioNTech.
Published: September 22, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108014.
Supplemental information
Data and code availability
This paper analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table. Analysis code together with results of this study is publicly available at https://github.com/TRON-Bioinformatics/milneo_analysis. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.van Rooij N., van Buuren M.M., Philips D., Velds A., Toebes M., Heemskerk B., van Dijk L.J.A., Behjati S., Hilkmann H., El Atmioui D., et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J. Clin. Oncol. 2013;31:e439–e442. doi: 10.1200/JCO.2012.47.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gubin M.M., Zhang X., Schuster H., Caron E., Ward J.P., Noguchi T., Ivanova Y., Hundal J., Arthur C.D., Krebber W.J., et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–581. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Snyder A., Makarov V., Merghoub T., Yuan J., Zaretsky J.M., Desrichard A., Walsh L.A., Postow M.A., Wong P., Ho T.S., et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2014;371:2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alspach E., Lussier D.M., Miceli A.P., Kizhvatov I., DuPage M., Luoma A.M., Meng W., Lichti C.F., Esaulova E., Vomund A.N., et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature. 2019;574:696–701. doi: 10.1038/s41586-019-1671-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lang F., Schrörs B., Löwer M., Türeci Ö., Sahin U. Identification of neoantigens for individualized therapeutic cancer vaccines. Nat. Rev. Drug Discov. 2022;21:261–282. doi: 10.1038/s41573-021-00387-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roudko V., Bozkus C.C., Orfanelli T., McClain C.B., Carr C., O'Donnell T., Chakraborty L., Samstein R., Huang K.L., Blank S.V., et al. Shared Immunogenic Poly-Epitope Frameshift Mutations in Microsatellite Unstable Tumors. Cell. 2020;183:1634–1649.e17. doi: 10.1016/j.cell.2020.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cimen Bozkus C., Roudko V., Finnigan J.P., Mascarenhas J., Hoffman R., Iancu-Rubin C., Bhardwaj N. Immune Checkpoint Blockade Enhances Shared Neoantigen-Induced T-cell Immunity Directed against Mutated Calreticulin in Myeloproliferative Neoplasms. Cancer Discov. 2019;9:1192–1207. doi: 10.1158/2159-8290.CD-18-1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Litchfield K., Reading J.L., Lim E.L., Xu H., Liu P., Al-Bakir M., Wong Y.N.S., Rowan A., Funt S.A., Merghoub T., et al. Escape from nonsense-mediated decay associates with anti-tumor immunogenicity. Nat. Commun. 2020;11:3800. doi: 10.1038/s41467-020-17526-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turajlic S., Litchfield K., Xu H., Rosenthal R., McGranahan N., Reading J.L., Wong Y.N.S., Rowan A., Kanu N., Al Bakir M., et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype. A pan-cancer analysis. Lancet Oncol. 2017;18:1009–1021. doi: 10.1016/S1470-2045(17)30516-8. [DOI] [PubMed] [Google Scholar]
- 10.Yang W., Lee K.W., Srivastava R.M., Kuo F., Krishna C., Chowell D., Makarov V., Hoen D., Dalin M.G., Wexler L., et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat. Med. 2019;25:767–775. doi: 10.1038/s41591-019-0434-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGranahan N., Furness A.J.S., Rosenthal R., Ramskov S., Lyngaa R., Saini S.K., Jamal-Hanjani M., Wilson G.A., Birkbak N.J., Hiley C.T., et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–1469. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghorani E., Rosenthal R., McGranahan N., Reading J.L., Lynch M., Peggs K.S., Swanton C., Quezada S.A. Differential binding affinity of mutated peptides for MHC class I is a predictor of survival in advanced lung cancer and melanoma. Ann. Oncol. 2018;29:271–279. doi: 10.1093/annonc/mdx687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Łuksza M., Riaz N., Makarov V., Balachandran V.P., Hellmann M.D., Solovyov A., Rizvi N.A., Merghoub T., Levine A.J., Chan T.A., et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature. 2017;551:517–520. doi: 10.1038/nature24473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McGranahan N., Swanton C. Neoantigen quality, not quantity. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aax7918. [DOI] [PubMed] [Google Scholar]
- 15.Lang F., Riesgo-Ferreiro P., Löwer M., Sahin U., Schrörs B. NeoFox. Annotating neoantigen candidates with neoantigen features. Bioinformatics. 2021;37:4246–4247. doi: 10.1093/bioinformatics/btab344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dietterich T.G., Lathrop R.H., Lozano-Pérez T. Solving the multiple instance problem with axis-parallel rectangles. Artif. Intell. 1997;89:31–71. doi: 10.1016/S0004-3702(96)00034-3. [DOI] [Google Scholar]
- 17.Foulds J., Frank E. A review of multi-instance learning assumptions. Knowl. Eng. Rev. 2010;25:1–25. doi: 10.1017/S026988890999035X. [DOI] [Google Scholar]
- 18.Xiong D., Zhang Z., Wang T., Wang X. A comparative study of multiple instance learning methods for cancer detection using T-cell receptor sequences. Comput. Struct. Biotechnol. J. 2021;19:3255–3268. doi: 10.1016/j.csbj.2021.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park S., Wang X., Lim J., Xiao G., Lu T., Wang T. Bayesian multiple instance regression for modeling immunogenic neoantigens. Stat. Methods Med. Res. 2020;29:3032–3047. doi: 10.1177/0962280220914321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miao D., Margolis C.A., Gao W., Voss M.H., Li W., Martini D.J., Norton C., Bossé D., Wankowicz S.M., Cullen D., et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science. 2018;359:801–806. doi: 10.1126/science.aan5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hugo W., Zaretsky J.M., Sun L., Song C., Moreno B.H., Hu-Lieskovan S., Berent-Maoz B., Pang J., Chmielowski B., Cherry G., et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riaz N., Havel J.J., Makarov V., Desrichard A., Urba W.J., Sims J.S., Hodi F.S., Martín-Algarra S., Mandal R., Sharfman W.H., et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell. 2017;171:934–949.e16. doi: 10.1016/j.cell.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Allen E.M., Miao D., Schilling B., Shukla S.A., Blank C., Zimmer L., Sucker A., Hillen U., Foppen M.H.G., Goldinger S.M., et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McDermott D.F., Huseni M.A., Atkins M.B., Motzer R.J., Rini B.I., Escudier B., Fong L., Joseph R.W., Pal S.K., Reeves J.A., et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 2018;24:749–757. doi: 10.1038/s41591-018-0053-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sahin U., Derhovanessian E., Miller M., Kloke B.P., Simon P., Löwer M., Bukur V., Tadmor A.D., Luxemburger U., Schrörs B., et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222–226. doi: 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- 26.Weber D., Ibn-Salem J., Sorn P., Suchan M., Holtsträter C., Lahrmann U., Vogler I., Schmoldt K., Lang F., Schrörs B., et al. Accurate detection of tumor-specific gene fusions reveals strongly immunogenic personal neo-antigens. Nat. Biotechnol. 2022;40:1276–1284. doi: 10.1038/s41587-022-01247-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jardim D.L., Goodman A., de Melo Gagliato D., Kurzrock R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell. 2021;39:154–173. doi: 10.1016/j.ccell.2020.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wood M.A., Weeder B.R., David J.K., Nellore A., Thompson R.F. Burden of tumor mutations, neoepitopes, and other variants are weak predictors of cancer immunotherapy response and overall survival. Genome Med. 2020;12:33. doi: 10.1186/s13073-020-00729-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McGrail D.J., Pilié P.G., Rashid N.U., Voorwerk L., Slagter M., Kok M., Jonasch E., Khasraw M., Heimberger A.B., Lim B., et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann. Oncol. 2021;32:661–672. doi: 10.1016/j.annonc.2021.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reynisson B., Alvarez B., Paul S., Peters B., Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0. Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020;48:W449–W454. doi: 10.1093/nar/gkaa379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bassani-Sternberg M., Chong C., Guillaume P., Solleder M., Pak H., Gannon P.O., Kandalaft L.E., Coukos G., Gfeller D. Deciphering HLA-I motifs across HLA peptidomes improves neo-antigen predictions and identifies allostery regulating HLA specificity. PLoS Comput. Biol. 2017;13 doi: 10.1371/journal.pcbi.1005725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Racle J., Michaux J., Rockinger G.A., Arnaud M., Bobisse S., Chong C., Guillaume P., Coukos G., Harari A., Jandus C., et al. Robust prediction of HLA class II epitopes by deep motif deconvolution of immunopeptidomes. Nat. Biotechnol. 2019;37:1283–1286. doi: 10.1038/s41587-019-0289-6. [DOI] [PubMed] [Google Scholar]
- 33.Balachandran V.P., Łuksza M., Zhao J.N., Makarov V., Moral J.A., Remark R., Herbst B., Askan G., Bhanot U., Senbabaoglu Y., et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 2017;551:512–516. doi: 10.1038/nature24462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duan F., Duitama J., Al Seesi S., Ayres C.M., Corcelli S.A., Pawashe A.P., Blanchard T., McMahon D., Sidney J., Sette A., et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J. Exp. Med. 2014;211:2231–2248. doi: 10.1084/jem.20141308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marty R., Kaabinejadian S., Rossell D., Slifker M.J., van de Haar J., Engin H.B., de Prisco N., Ideker T., Hildebrand W.H., Font-Burgada J., Carter H. MHC-I Genotype Restricts the Oncogenic Mutational Landscape. Cell. 2017;171:1272–1283.e15. doi: 10.1016/j.cell.2017.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marty Pyke R., Thompson W.K., Salem R.M., Font-Burgada J., Zanetti M., Carter H. Evolutionary Pressure against MHC Class II Binding Cancer Mutations. Cell. 2018;175:416–428.e13. doi: 10.1016/j.cell.2018.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rech A.J., Balli D., Mantero A., Ishwaran H., Nathanson K.L., Stanger B.Z., Vonderheide R.H. Tumor Immunity and Survival as a Function of Alternative Neopeptides in Human Cancer. Cancer Immunol. Res. 2018;6:276–287. doi: 10.1158/2326-6066.CIR-17-0559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bjerregaard A.-M., Nielsen M., Jurtz V., Barra C.M., Hadrup S.R., Szallasi Z., Eklund A.C. An Analysis of Natural T Cell Responses to Predicted Tumor Neoepitopes. Front. Immunol. 2017;8:1566. doi: 10.3389/fimmu.2017.01566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richman L.P., Vonderheide R.H., Rech A.J. Neoantigen Dissimilarity to the Self-Proteome Predicts Immunogenicity and Response to Immune Checkpoint Blockade. Cell Syst. 2019;9:375–382.e4. doi: 10.1016/j.cels.2019.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chiaro J., Kasanen H.H.E., Whalley T., Capasso C., Grönholm M., Feola S., Peltonen K., Hamdan F., Hernberg M., Mäkelä S., et al. Viral Molecular Mimicry Influences the Antitumor Immune Response in Murine and Human Melanoma. Cancer Immunol. Res. 2021;9:981–993. doi: 10.1158/2326-6066.CIR-20-0814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Calis J.J.A., Maybeno M., Greenbaum J.A., Weiskopf D., De Silva A.D., Sette A., Keşmir C., Peters B. Properties of MHC Class I Presented Peptides That Enhance Immunogenicity. PLoS Comput. Biol. 2013;9 doi: 10.1371/journal.pcbi.1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rubinsteyn A., Kodysh J., Hodes I., Mondet S., Aksoy B.A., Finnigan J.P., Bhardwaj N., Hammerbacher J. Computational Pipeline for the PGV-001 Neoantigen Vaccine Trial. Front. Immunol. 2018;8:1807. doi: 10.3389/fimmu.2017.01807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bjerregaard A.-M., Nielsen M., Hadrup S.R., Szallasi Z., Eklund A.C. MuPeXI. Prediction of neo-epitopes from tumor sequencing data. Cancer Immunol. Immunother. 2017;66:1123–1130. doi: 10.1007/s00262-017-2001-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith C.C., Chai S., Washington A.R., Lee S.J., Landoni E., Field K., Garness J., Bixby L.M., Selitsky S.R., Parker J.S., et al. Machine-Learning Prediction of Tumor Antigen Immunogenicity in the Selection of Therapeutic Epitopes. Cancer Immunol. Res. 2019;7:1591–1604. doi: 10.1158/2326-6066.CIR-19-0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Besser H., Yunger S., Merhavi-Shoham E., Cohen C.J., Louzoun Y. Level of neo-epitope predecessor and mutation type determine T cell activation of MHC binding peptides. J. Immunother. Cancer. 2019;7:135. doi: 10.1186/s40425-019-0595-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmidt J., Smith A.R., Magnin M., Racle J., Devlin J.R., Bobisse S., Cesbron J., Bonnet V., Carmona S.J., Huber F., et al. Prediction of neo-epitope immunogenicity reveals TCR recognition determinants and provides insight into immunoediting. Cell Rep. Med. 2021;2 doi: 10.1016/j.xcrm.2021.100194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen Y., Bi J., Wang J.Z. MILES. Multiple-instance learning via embedded instance selection. IEEE Trans. Pattern Anal. Mach. Intell. 2006;28:1931–1947. doi: 10.1109/TPAMI.2006.248. [DOI] [PubMed] [Google Scholar]
- 48.Gütlein M., Helma C., Karwath A., Kramer S. A Large-Scale Empirical Evaluation of Cross-Validation and External Test Set Validation in (Q)SAR. Mol. Inform. 2013;32:516–528. doi: 10.1002/minf.201200134. [DOI] [PubMed] [Google Scholar]
- 49.Wei Z., Zhou C., Zhang Z., Guan M., Zhang C., Liu Z., Liu Q. The Landscape of Tumor Fusion Neoantigens. A Pan-Cancer Analysis. iScience. 2019;21:249–260. doi: 10.1016/j.isci.2019.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu T., Wang S., Xu L., Zhou Q., Singla N., Gao J., Manna S., Pop L., Xie Z., Chen M., et al. Tumor neoantigenicity assessment with CSiN score incorporates clonality and immunogenicity to predict immunotherapy outcomes. Sci. Immunol. 2020;5:eaaz3199. doi: 10.1126/sciimmunol.aaz3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leng Q., Tarbe M., Long Q., Wang F. Pre-existing heterologous T-cell immunity and neoantigen immunogenicity. Clin. Transl. Immunology. 2020;9 doi: 10.1002/cti2.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Devlin J.R., Alonso J.A., Ayres C.M., Keller G.L.J., Bobisse S., Vander Kooi C.W., Coukos G., Gfeller D., Harari A., Baker B.M. Structural dissimilarity from self drives neoepitope escape from immune tolerance. Nat. Chem. Biol. 2020;16:1269–1276. doi: 10.1038/s41589-020-0610-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim S., Scheffler K., Halpern A.L., Bekritsky M.A., Noh E., Källberg M., Chen X., Kim Y., Beyter D., Krusche P., Saunders C.T. Strelka2. Fast and accurate calling of germline and somatic variants. Nat. Methods. 2018;15:591–594. doi: 10.1038/s41592-018-0051-x. [DOI] [PubMed] [Google Scholar]
- 55.Kawaguchi S., Higasa K., Shimizu M., Yamada R., Matsuda F. HLA-HD. An accurate HLA typing algorithm for next-generation sequencing data. Hum. Mutat. 2017;38:788–797. doi: 10.1002/humu.23230. [DOI] [PubMed] [Google Scholar]
- 56.Dobin A. STAR. Ultrafast universal RNA-seq aligner. Bioinformatics. 2012;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Patro R., Mount S.M., Kingsford C. Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nat. Biotechnol. 2014;32:462–464. doi: 10.1038/nbt.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hilf N., Kuttruff-Coqui S., Frenzel K., Bukur V., Stevanović S., Gouttefangeas C., Platten M., Tabatabai G., Dutoit V., van der Burg S.H., et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature. 2019;565:240–245. doi: 10.1038/s41586-018-0810-y. [DOI] [PubMed] [Google Scholar]
- 59.Benjamini Y., Hochberg Y. Controlling the False Discovery Rate. A Practical and Powerful Approach to Multiple Testing. J. Roy. Stat. Soc. B. 1995;57:289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table. Analysis code together with results of this study is publicly available at https://github.com/TRON-Bioinformatics/milneo_analysis. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.