

Summary
Fucoxanthin is the most abundant marine carotenoid extracted from seaweed. Our previous study has shown that fucoxanthin inhibited oxidative stress after traumatic brain injury (TBI). However, the effects of fucoxanthin on TBI-induced blood–brain barrier (BBB) destruction have not been well understood. In the present study, we found that fucoxanthin improved neurological dysfunction, reduced brain edema, attenuated cortical lesion volume, and decreased dendrites loss after TBI in vivo. Moreover, fucoxanthin suppressed BBB leakage, preserved tight junction (TJ) and adherens junction (AJ) proteins, and inhibited MMP-9 expression. Furthermore, fucoxanthin alleviated apoptosis and ferroptosis, and activated mitophagy in endothelial cells (ECs) after TBI. However, the protection of fucoxanthin on BBB was attenuated when mitophagy was inhibited. Importantly, fucoxanthin also provided protective effects in bEnd.3 cells after TBI. Taken together, our results suggested that fucoxanthin played a key role in the protection of BBB after TBI through mitophagy.
Subject areas: Cellular physiology, Neuroscience, Molecular neuroscience
Graphical abstract
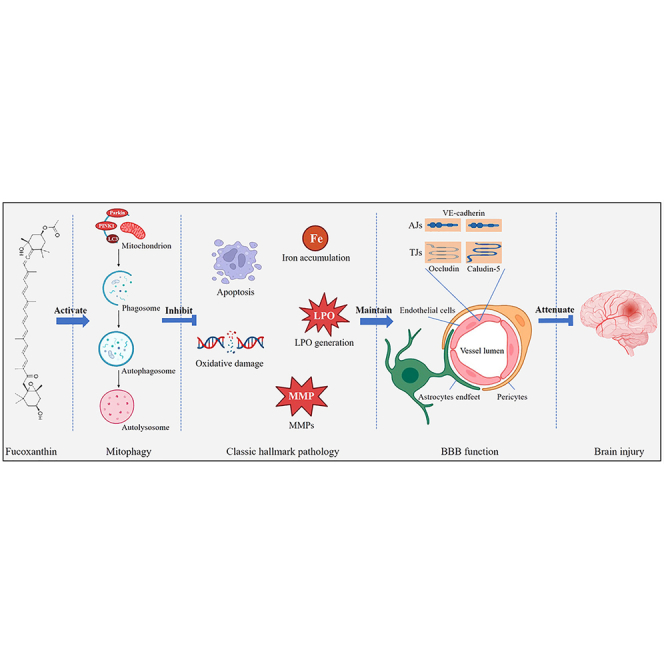
Highlights
-
•
Fucoxanthin suppresses TBI-induced BBB disruption
-
•
Fucoxanthin decreases TBI-induced endothelial cell apoptosis and ferroptosis
-
•
Fucoxanthin activates endothelial cell mitophagy after TBI
-
•
Inhibition of mitophagy reversed the protection of fucoxanthin on BBB after TBI
Cellular physiology; Neuroscience; Molecular neuroscience
Introduction
Traumatic brain injury (TBI) is one of the leading causes of disability and death worldwide. It can be induced by external trauma such as traffic, industrial, falls and military conflicts.1 The pathogenesis of TBI involves a complicated set of events, including the primary damage caused by mechanical forces and the secondary damage triggered by blood–brain barrier (BBB) disruption, synaptic dysfunction, neuronal apoptosis and oxidative damage.2 The current treatments for TBI include surgery, hyperbaric oxygen therapy and nerve dehydration. However, millions of survivors of TBI are often left with mental, physical and intellectual disabilities that reduce the quality of life and pose a heavy financial burden on the public health system.3 Given these realities, it is urgently needed to better understand the pathophysiological mechanisms of TBI and develop effective therapeutic strategies for patients suffering from TBI.
BBB disruption is one of the important pathological mechanisms after TBI, which occurs at the beginning and accompanies the whole process of TBI.4 The destruction of BBB can initiate the release of macrophages, neutrophils and lymphocytes at the injury site, thus aggravating the brain damage.5 BBB is a lipophilic and dynamic barrier which separates the brain from blood flow.6 The functional and structural integrity of BBB is critical for maintaining the homeostasis of brain microenvironment.7 Due to its highly specialized nature, BBB can selectively prevent the exchange of neurotoxic molecules, regulate the trafficking of macromolecules, amino acids, ions and peptides between the brain and blood.8 The integrity of BBB is primarily preserved by endothelial cells (ECs) and basement membrane. It is also regulated by pericytes, oligodendrocytes and immunes cells.9 The damage of BBB after TBI may lead to neurodegenerative and neurological deficits in the brain.10,11 Therefore, repairing of BBB can be the effective therapeutic method for the treatment of TBI.
Fucoxanthin is a marine carotenoid found in the chloroplasts of seaweeds and diatoms.12 It is abundant in nature, accounting for about 10% of the total production of carotenoids.13 Furthermore, fucoxanthin has been suggested to be safe by the European Food Safety Authority and the US Food and Drug Administration.14 Fucoxanthin possesses unique structural features, including allene bond, epoxide and acetyl groups.15 Due to its structural diversity, fucoxanthin has health benefits such as anti-inflammation, antioxidant, anti-obesity and anti-diabetic effects.16 Recently, studies have revealed the effective role of fucoxanthin in the treatment of central nervous system (CNS) diseases. It has been indicated that fucoxanthin inhibited oxidative damage and brain injury after subarachnoid hemorrhage (SAH) by the deacetylation of forkhead transcription factors of the O class (FoxO) and p53 through the activation of sirtuin 1 (SIRT1).17 In addition, our previous study has demonstrated that fucoxanthin suppressed cell death, oxidative stress and apoptosis in TBI via the nuclear factor erythroid 2-related factor 2 (Nrf2)-antioxidant-response element (ARE) and Nrf2-autophagy pathways.18 However, the effects of fucoxanthin in TBI-induced BBB destruction have not been fully explained. In this study, we explored whether fucoxanthin protected BBB against TBI and the underlying mechanisms.
Results
Fucoxanthin improved the neurological outcomes, reduced brain edema and decreased lesion volume after traumatic brain injury
To determine whether fucoxanthin protected mice against TBI, we firstly used the NSS (Table 1), rotarod test, MWM and OFT to evaluate the neurological function of mice after TBI. As shown in Figure 1A, the motor performance of fucoxanthin-treated TBI mice was better than that of vehicle-treated TBI mice. Moreover, the rotarod test showed that the administration of fucoxanthin obviously improved neurological function after TBI (Figure 1B). Furthermore, in the MWM test, fucoxanthin-treated TBI mice spent shorter escape latency (Figure 1C) and less swimming distance (Figure 1D) to find the platform than vehicle-treated TBI mice (Figure 1E). In addition, the OFT showed that the total moving distance of mice was decreased after fucoxanthin treatment (Figures 1F and 1G).
Table 1.
Neurological severity scoring (NSS)
| Items | Description | Points |
|
|---|---|---|---|
| Success/Failure | |||
| Exit circle | Ability and initiative to exit a circle of 30 cm diameter (time limit: 3 min) | 0 | 1 |
| Mono-/hemiparesis | Paresis of upper and/or lower limb of contralateral side | 0 | 1 |
| Straight walk | Alertness, initiative, and motor ability to walk straight, when placed on the floor | 0 | 1 |
| Startle reflex | Innate reflex (flinching in response to a loud hand clap) | 0 | 1 |
| Seeking behavior | Physiological behavior as a sign of “interest” in the environment | 0 | 1 |
| Beam balancing | Ability to balance on a beam 7 mm in width for at least 10 s | 0 | 1 |
| Round stick balancing | Ability to balance on a round stick 5 mm in diameter for at least 10 s | 0 | 1 |
| Beam walk: 3 cm | Ability to cross a beam (length × width, 30 × 3 cm) | 0 | 1 |
| Beam walk: 2 cm | Same task but with increased difficulty (beam width = 2 cm) | 0 | 1 |
| Beam walk: 1 cm | Same task but with increased difficulty (beam width = 1 cm) | 0 | 1 |
| Maximum score | 10 | ||
Figure 1.

Effects of fucoxanthin in TBI
(A and B) Mice were subjected to TBI and received 0.05 mmol/L of fucoxanthin or vehicle i.c.v. injection 30 min after TBI. NSS (A) and Rotarod test (B) were evaluated at 1, 3, 5, 7 and 14 days after TBI and fucoxanthin treatment.
(C and D) Mice were subjected to TBI and received 0.05 mmol/L of fucoxanthin or vehicle i.c.v. injection 30 min after TBI. MWM test was evaluated at 15–21 days after TBI and fucoxanthin treatment.
(E) Representative images of swimming traces in MWM test at 21 days post TBI.
(F) Mice were subjected to TBI and received 0.05 mmol/L of fucoxanthin or vehicle i.c.v. injection 30 min after TBI. OFT was evaluated at 15–21 days after TBI and fucoxanthin treatment.
(G) Representative images of tracks in OFT at 21 days post TBI.
(H–K) Mice were subjected to TBI and received 0.05 mmol/L of fucoxanthin or vehicle i.c.v. injection 30 min after TBI. Brain water content (H), brain tissue loss (I and J), and neuronal spine density (K) were examined at 3 days after TBI and fucoxanthin treatment. n = 3 per group.
Data were presented as mean ± SD; ∗∗∗p < 0.001 versus sham group; #p < 0.05, ##p < 0.01, ###p < 0.001 versus TBI + vehicle group.
We then used brain water content to confirm the neuroprotective effects of fucoxanthin. As shown in Figure 1H, the brain water content was increased at 3 days after TBI. However, the treatment of fucoxanthin decreased the brain water content. Then, we determined whether fucoxanthin affected TBI-induced cortical lesion volume. Figures 1I and 1J showed that TBI induced obvious brain tissue loss (red lines). However, the treatment of fucoxanthin reduced the lesion volume (Figures 1I and 1J). Finally, we used Golgi-Cox staining to examine the effects of fucoxanthin on neuronal dendrites and dendritic spines. As shown in Figure 1K, the neurons lost their dendrites after TBI. However, fucoxanthin protected the neuronal spine density against TBI (Figure 1K).
Fucoxanthin attenuated blood–brain barrier damage after traumatic brain injury
BBB damage is a common feature of TBI, which is closely related to the morbidity and mortality in patients with TBI.19 We then investigated whether fucoxanthin attenuated TBI-induced BBB disruption. We firstly assessed the permeability of BBB by EB leakage. The results revealed that TBI increased the permeability of BBB, while the treatment of fucoxanthin by both i.g. administration or i.c.v. injection significantly decreased the BBB permeability (Figure 2A).
Figure 2.

Fucoxanthin attenuated BBB damage after TBI
(A) Representative images and quantitative analysis of the effect of fucoxanthin by 0.05 mmol/L i.c.v. injection and 100 mg/kg i.g. administration on EB leakage at 3 days after TBI.
(B) Western blot assay for the expression of ZO-1 and occludin in the ipsilateral cortex at 3 days after TBI and fucoxanthin i.c.v. injection.
(C) Western blot assay for the expression of claudin-5 and VE-cadherin in the ipsilateral cortex at 3 days after TBI and fucoxanthin i.c.v. injection.
(D) Western blot assay for the expression of MMP-9 in the ipsilateral cortex at 3 days after TBI and fucoxanthin i.c.v. injection. n = 3 per group.
Data were presented as mean ± SD; ∗∗∗p < 0.001 versus sham group; ##p < 0.01, ###p < 0.001 versus TBI + vehicle group. β-actin was used as a loading control.
To explore the mechanisms underlying the protection of fucoxanthin on BBB, we analyzed the expression of tight junction (TJ) proteins (ZO-1, occludin, claudin-5) and adherens junction (AJ) protein (vascular endothelial-cadherin, VE-cadherin). As shown in Figures 2B and 2C, TBI down-regulated the expression of ZO-1, occludin, claudin-5, and VE-cadherin, while fucoxanthin remarkably reversed the expression of these proteins (Figures 2B and 2C).
MMP-9 is a primary proteolytic enzyme that degrades TJ and AJ proteins.10 We then examined the expression of MMP-9 after TBI. We found that TBI increased the expression of MMP-9, which was significantly inhibited by fucoxanthin (Figure 2D). These data suggested that fucoxanthin alleviated BBB leakage by preserving TJ and AJ proteins after TBI, at least partially through the inhibition of MMP-9.
Fucoxanthin suppressed endothelial cell apoptosis after traumatic brain injury
As the primary cell type of BBB, EC survival is responsible for the BBB integrity.20 It has been shown that TBI could induce EC apoptosis, thus destroying BBB.21 To verify the occurrence of EC apoptosis, we firstly used double IF staining of TUNEL and CD31 (an EC marker). The results showed that in the sham group, few apoptotic-positive ECs were detected (Figures 3A and 3B). However, the co-localization of TUNEL-positive and CD31-positive cells were found in the TBI and TBI + vehicle groups (Figures 3A and 3B). Administration of fucoxanthin remarkably decreased the number of TUNEL-positive and CD31-positive cells (Figures 3A and 3B).
Figure 3.

Fucoxanthin suppressed TBI-induced endothelial cell apoptosis
(A and B) TUNEL staining was used to examine the percentage of TUNEL+ and CD31+ cells at 3 days after TBI and fucoxanthin i.c.v. injection.
(C–E) Western blot assay for the expression of Bax and Bcl-2 in the ipsilateral cortex at 3 days after TBI and fucoxanthin i.c.v. injection.
(F and G) Western blot assay for the expression of cleaved caspase-3 in the ipsilateral cortex at 3 days after TBI and fucoxanthin i.c.v. injection. n = 3 per group.
Data were presented as mean ± SD; ∗∗∗p < 0.001 versus sham group; ###p < 0.001 versus TBI + vehicle group. Scale bar of TUNEL staining: 20 μm β-actin was used as a loading control.
To further examine the effects of fucoxanthin on apoptosis, we analyzed several apoptosis markers such as Bax, Bcl-2, and caspase-3. We found that the expression of Bcl-2 (Figures 3C and 3E) was decreased while the expression of Bax (Figures 3C and 3D) and cleaved caspase-3 (Figures 3F and 3G) was increased after TBI. However, the treatment of fucoxanthin evidently reduced TBI-induced apoptosis (Figures 3C–3G).
Fucoxanthin decreased endothelial cells ferroptosis after traumatic brain injury
Ferroptosis can contribute to EC damage in the case of brain injury and inhibition of ferroptosis has been shown to provide neuroprotection on BBB.22 The features of ferroptosis are the iron accumulation-dependent lipid peroxidation (LPO) generation and oxidative damage. To prove the initiation of EC ferroptosis after TBI and fucoxanthin could attenuate TBI-induced EC ferroptosis, we firstly examined the iron accumulation. The Perl’s staining showed that TBI increased iron accumulation in ECs surrounding injury site. However, the treatment of fucoxanthin significantly decreased iron accumulation in ECs (Figures 4A and 4B). We further investigated the potential mechanisms of iron accumulation after TBI. Iron homeostasis is mediated by Tf-TfR internalization for iron uptake, Ft for iron storage and Fpn-1 for iron export in cells. Therefore, we evaluated the expression of TfR, Ft and Fpn-1. The results showed that the levels of TfR and Ft were up-regulated (Figure 4C) and the levels of Fpn-1 were down-regulated in TBI (Figure 4D). Administration of fucoxanthin reversed these effects (Figures 4C and 4D), demonstrating that TBI-caused imbalance of iron homeostasis was restored by fucoxanthin. Then, we assessed the mitochondrial morphology by TEM. As shown in Figure 4E, the shrunken mitochondria with increased bilayer membrane density were observed after TBI, which was considered as the characteristic of ferroptosis. However, fucoxanthin improved the morphology of mitochondria (Figure 4E).
Figure 4.

Fucoxanthin inhibited TBI-induced iron accumulation and mitochondrial damage in endothelial cells
(A and B) Perls’ DAB staining was used to assay iron accumulation at 3 days in endothelial cells after TBI and fucoxanthin i.c.v. injection.
(C) Western blot assay for the expression of TfR and Ft in the ipsilateral cortex at 3 days after TBI and fucoxanthin i.c.v. injection.
(D) Western blot assay for the expression of Fpn-1 in the ipsilateral cortex at 3 days after TBI and fucoxanthin i.c.v. injection.
(E) TEM photomicrographs of mitochondria at 3 days after TBI and fucoxanthin i.c.v. injection. n = 3 per group.
Data were presented as mean ± SD; ∗∗∗p < 0.001 versus sham group; ###p < 0.001 versus TBI + vehicle group. Scale bar: 20 μm β-actin was used as a loading control.
Next, we analyzed the LPO generation by the measurement of MDA and GPx after TBI. We found that the levels of MDA were increased (Figure 5A) and the levels of GPx were decreased (Figure 5B) after TBI. However, the levels of MDA and GPx were reversed after fucoxanthin treatment (Figures 5A and 5B). Subsequently, we examined the oxidative damage by double IF staining of 8-OHdG and CD31, the results revealed that TBI induced oxidative deoxyribonucleic acid (DNA) damage in ECs, while fucoxanthin suppressed TBI-induced oxidative damage (Figure 5C).
Figure 5.

Fucoxanthin inhibited TBI-induced endothelial cell oxidative damage
(A and B) MDA levels (A) and Gpx activity (B) were evaluated by ELISA at 3 days after TBI and fucoxanthin i.c.v. injection.
(C) Representative images of IF staining for 8-OHdG+ and CD31+ cells at 3 days after TBI and fucoxanthin i.c.v. injection. n = 3 per group.
Data were presented as mean ± SD; ∗∗∗p < 0.001 versus sham group; ##p < 0.01, ###p < 0.001 versus TBI + vehicle group. Scale bar: 20 μm.
It has been suggested that ACSL4 and GPX4 were the main regulators of ferroptosis. We then measured the expression of these proteins by IF in ECs. As shown in Figure 6, ACSL4 was up-regulated (Figure 6A) and GPX4 was down-regulated (Figure 6B) in CD31-positive ECs after TBI, while the treatment of fucoxanthin reversed these effects (Figures 6A and 6B). These data indicated that fucoxanthin suppressed ferroptosis after TBI.
Figure 6.

Fucoxanthin decreased the expression of ferroptosis-related proteins in endothelial cells
(A) Representative images of IF staining for ACSL4+ and CD31+ cells at 3 days after TBI and fucoxanthin i.c.v. injection.
(B) Representative images of IF staining for GPX4+ and CD31+ cells at 3 days after TBI and fucoxanthin i.c.v. injection. n = 3 per group.
Data were presented as mean ± SD; ∗∗∗p < 0.001 versus sham group; ###p < 0.001 versus TBI + vehicle group. Scale bar: 20 μm β-actin was used as a loading control.
Fucoxanthin promoted mitophagy after traumatic brain injury
Growing studies have suggested that mitophagy could regulate apoptosis and ferroptosis after TBI.23 Therefore, we wondered whether mitophagy was involved in the protection of fucoxanthin on BBB. We firstly used the double IF staining of LC3 and CD31, the results revealed that TBI increased the co-localization of LC3-positive and CD31-positive cells compared to the sham group, suggesting the formation of autophagosome in ECs (Figure 7A). In addition, the LC3-positive and CD31-positive cells were further increased after fucoxanthin treatment (Figure 7A). The ability of fucoxanthin to induce autophagy was confirmed by TEM. After TBI, the autophagic vacuoles containing cellular organelles were observed. Upon the treatment of fucoxanthin, the numbers of autophagic vacuoles were further increased (Figure 7B). To further prove that mitophagy was activated, we analyzed the expression of mitophagy-related proteins, such as PINK1, Parkin, TOMM20 and LC3. We found that TBI increased the expression of PINK1 (Figures 7C and 7D), Parkin (Figures 7C and 7D), TOMM20 (Figures 7E and 7F) and LC3-II (Figures 7G and 7H). Moreover, the TBI-induced mitophagy was enhanced by fucoxanthin, as proven by further increased expression of PINK1 (Figures 7C and 7D), Parkin (Figures 7C and 7D), TOMM20 (Figures 7E and 7F) and LC3-II (Figures 7G and 7H). These data indicated that fucoxanthin promoted TBI-induced mitophagy in ECs.
Figure 7.

Fucoxanthin activated mitophagy in endothelial cells after TBI
(A) Representative images of IF staining for LC3+ and CD31+ at 3 days after TBI and fucoxanthin i.c.v. injection.
(B) TEM photomicrographs of autophagic vacuoles containing cellular material or organelle at 3 days after TBI and fucoxanthin i.c.v. injection.
(C and D) Western blot assay for the expression of PINK1 and Parkin in the ipsilateral cortex at 3 days after TBI and fucoxanthin i.c.v. injection.
(E and F) Western blot assay for the expression of TOMM20 in the ipsilateral cortex at 3 days after TBI and fucoxanthin i.c.v. injection.
(G and H) Western blot assay for the expression of LC3 in the ipsilateral cortex at 3 days after TBI and fucoxanthin i.c.v. injection. n = 3 per group.
Data were presented as mean ± SD; ∗∗∗p < 0.001 versus sham group; ###p < 0.001 versus TBI + vehicle group. Scale bar: 20 μm β-actin was used as a loading control.
To explore whether the protective effects of fucoxanthin on BBB were abated when mitophagy was blocked, we used a mitophagy inhibitor mitochondrial division inhibitor-1 (Mdivi-1). As shown in Figure 8, Mdivi-1 partly reversed the inhibitory effects of fucoxanthin on neurological dysfunction (Figure 8A), brain water content (Figure 8B), EB leakage (Figures 8C and 8D), apoptosis (Figure 8E) and ferroptosis (Figure 8F) in TBI, suggesting that the mitophagy was partly involved in the neuroprotection of fucoxanthin on BBB after TBI.
Figure 8.

Suppression of mitophagy reversed the inhibitory effects of fucoxanthin on neurological deficit, brain edema, BBB damage, apoptosis and ferroptosis
(A) NSS was evaluated at 3 days after TBI, fucoxanthin i.c.v. injection and inhibition of mitophagy.
(B) Brain water content was examined at 3 days after TBI, fucoxanthin i.c.v. injection and inhibition of mitophagy.
(C and D) Representative images (C) and quantitative analysis (D) of the effect of fucoxanthin on EB leakage at 3 days after TBI, fucoxanthin i.c.v. injection and inhibition of mitophagy.
(E) Representative images of IF staining for TUNEL+ and CD31+ cells at 3 days after TBI, fucoxanthin i.c.v. injection and inhibition of mitophagy.
(F) Representative images of IF staining for ACSL4+ and CD31+ cells at 3 days after TBI, fucoxanthin i.c.v. injection and inhibition of mitophagy. n = 3 per group.
Data were presented as mean ± SD; ##p < 0.01, ###p < 0.001 versus TBI + vehicle group; & p < 0.05, && p < 0.01, &&& p < 0.001 versus TBI + fucoxanthin group. Scale bar: 20 μm.
Fucoxanthin protected bEnd.3 cells from traumatic brain injury
The protective effects of fucoxanthin on BBB after TBI were also confirmed in bEnd.3 cells. TB staining and LDH release assay were firstly conducted in cells treated with fucoxanthin. In TB staining, the treatment of fucoxanthin significantly increased the percentage of viable cells (Figure 9A). Similar results were found in LDH release assay (Figure 9B). These results suggested that fucoxanthin provided protection on BBB after TBI in vitro. Furthermore, we examined the role of fucoxanthin on BBB integrity in bEnd.3 cells after TBI. We found that the levels of TEER (Figure 9C) and γ-GT (Figure 9D) were down-regulated after TBI in bEnd.3 cells. However, the treatment of fucoxanthin up-regulated the levels of TEER (Figure 9C) and γ-GT (Figure 9D), indicating that fucoxanthin could improve the integrity and stability of BBB.
Figure 9.

Fucoxanthin protected bEnd.3 cells from TBI
(A and B) bEnd.3 cells were subjected to scratch injury and then treated with 10 μM of fucoxanthin or DMSO for 3 days. The TB staining (A) and LDH release assay (B) were used to evaluate cell viability.
(C) TEER detected the BBB tightness at 3 days in all groups.
(D) Measurement of the levels of γ-GT at 3 days after TBI and fucoxanthin treatment.
(E) Representative images of TUNEL staining for bEnd.3 cells at 3 days in all groups.
(F and G) Western blot assay for the expression of cleaved caspase-3 in bEnd.3 cells at 3 days after TBI and fucoxanthin treatment.
(H and I) Western blot assay for the expression of ACSL4 in bEnd.3 cells at 3 days after TBI and fucoxanthin treatment.
(J and K) Western blot assay for the expression of PINK1 in bEnd.3 cells at 3 days after TBI and fucoxanthin treatment.
(L and M) Western blot assay for the expression of LC3 in bEnd.3 cells at 3 days after TBI and fucoxanthin treatment.
(N and O) The TB staining (N) and LDH release assay (O) were used to evaluate cell viability in bEnd.3 cells at 3 days after TBI, fucoxanthin treatment and inhibition of mitophagy. n = 3 per group.
Data were presented as mean ± SD; ∗∗∗p < 0.001 versus control group; ##p < 0.01, ###p < 0.001 versus TBI + DMSO group; && p < 0.01, &&& p < 0.001 versus TBI + fucoxanthin group. Scale bar: 20 μm β-actin was used as a loading control.
Then, to understand the effects of fucoxanthin on bEnd.3 cell apoptosis, we applied TUNEL staining. Figure 9E showed that compared to the control cells, the TUNEL-positive cells were significantly increased after TBI. When cells were treated with fucoxanthin, the TUNEL-positive cells were decreased (Figure 9E).
We next analyzed the expression of cleaved caspase-3, ACSL4, PINK1 and LC3 by Western blot. We found that compared to the control cells, the expression of cleaved caspase-3 (Figures 9F and 9G), ACSL4 (Figures 9H and 9I), PINK1 (Figures 9J and 9K) and LC3-II (Figures 9L and 9M) was increased in the damaged cells. Fucoxanthin treatment reduced the expression of cleaved caspase-3 (Figures 9F and 9G) and ACSL4 (Figures 9H and 9I) but further increased the expression of PINK1 (Figures 9J and 9K) and LC3-II (Figures 9L and 9M). These results demonstrated that fucoxanthin increased cell viability, maintained BBB integrity, suppressed apoptosis, decreased ferroptosis, and activated mitophagy after TBI in vitro. However, when mitophagy was inhibited by Mdivi-1, the protection of fucoxanthin on cell death was partly attenuated (Figures 9N and 9O), demonstrating that fucoxanthin provided protective effects in bEnd.3 cells partly through the activation of mitophagy after TBI.
Discussion
To the best of our knowledge, this is the first study examining the effects of fucoxanthin on BBB function in TBI. The main findings were as follows: (1) Fucoxanthin improved neurological function, decreased cerebral edema, attenuated lesion volume and reduced dendrites loss after TBI. (2) Fucoxanthin suppressed TBI-induced BBB disruption. (3) Fucoxanthin inhibited apoptosis and ferroptosis in ECs after TBI. (4) Fucoxanthin activated mitophagy in ECs after TBI. (5) Inhibition of mitophagy reversed the protection of fucoxanthin on BBB after TBI.
BBB is a highly selective interface between the blood and brain parenchyma. The damage of BBB can led to brain edema, high intracranial pressure and neurologic deficits after TBI.24 ECs are central to the barrier properties of BBB. These cells have an apical domain and a basolateral domain, and are tightly bound with junctional proteins such as TJs and AJs.25 TJs are located in the apical part of paracellular space and contain cytoplasmic proteins (ZO-1) and transmembrane proteins (occludin, claudin-5).26 TJ proteins are responsible for closing the intercellular cleft, thus preventing free exchange between the CNS and vasculature.10 AJs are located at the basolateral part of paracellular space and compose of cadherin, integrin and their associated proteins.27 VE-cadherin as an AJ protein can regulate TJ adhesion and stability, thus contributing to the maintenance of TJ organization.28 Both TJs and AJs are the molecular basis of BBB damage. For example, in respond to TBI, the destruction of TJ proteins such as ZO-1, occludin, claudin-5, and AJ proteins such as β-catenin were the main factor causing BBB damage. However, the damage of BBB could be reversed by the treatment of basic fibroblast growth factor (bFGF).29 Furthermore, in human BMECs exposed to hypoxia, the treatment of recombinant tissue inhibitor of metalloproteinase-1 (rTIMP1) up-regulated the expression of TJ proteins ZO-1, occludin, claudin-5, and AJ protein VE-cadherin, therefore alleviating TBI-induced loss of junctional proteins.30 In the present study, we examined the changes of TJ proteins (ZO-1, occludin, claudin-5) and AJ protein (VE-cadherin) after TBI. Our results showed that the expression of ZO-1, occludin, claudin-5, and VE-cadherin was down-regulated at 3 days after TBI, while the treatment of fucoxanthin up-regulated the expression of these proteins. Moreover, the protection of BBB by fucoxanthin was further confirmed with EB dye extravasation, suggesting that fucoxanthin suppressed junctional proteins loss and BBB permeability after TBI. In the present study, we found that fucoxanthin could inhibit TBI-induced behavioral changes at least after 20 days after injury. However, we evaluated the protection of fucoxanthin against BBB damage at 3 days after TBI. Because Wei et al. have proposed that BBB damage began to increase at 3 h post-TBI, peaked at 3 days, and decreased gradually while remaining higher than sham injury animals at 7 and 30 days.31 Besides, many studies also examined the BBB damage after TBI at 3 days.22,32,33 Therefore, although fucoxanthin could provide continuous neuroprotection after TBI as shown by behavioral tests, we examined the protective effects of fucoxanthin on BBB damage at its peak time after TBI.
MMP-9 has been identified as an important signaling protease that modulated the inflammatory response and contributed to the wound healing process.34 The up-regulation of MMP-9 was correlated with the increase of BBB permeability and decrease of BBB integrity.19 In addition, the induction of MMP-9 after TBI could degrade junctional proteins and lead to the BBB disruption, neuronal apoptosis and infarct volume development.35 Our data demonstrated that the levels of MMP-9 were increased after TBI, while the treatment of fucoxanthin significantly inhibited the levels of MMP-9. However, further studies were needed to explore whether fucoxanthin prevented the activation of MMP-9.
The disruption of junctional proteins by TBI could cause ECs damage, eventually leading to ECs apoptosis.36 Apoptosis is a form of programmed cell death (PCD) that occurs in multicellular organisms.37 Under physiological conditions, apoptosis plays an important role in the development by eliminating unnecessary and unhealthy cells. However, under pathological conditions, accelerated apoptosis can cause neurodegenerative diseases or ischemic damage.37 Apoptosis can be divided into two pathways: the mitochondria-dependent pathway (the intrinsic pathway) and the death receptor-dependent pathway (the extrinsic pathway).38 Bcl-2 and its family members are required for the mitochondria-dependent pathway. Bcl-2, together with Bax, modifies mitochondrial damage, which releases cytochrome c into the cytoplasm and activates cellular caspases, especially the executor caspase-3.32 In our study, we found an increased TUNEL/CD31 double-positive cells after TBI. However, the administration of fucoxanthin reduced TUNEL/CD31 double-positive cells. Combined with the results of decreased bEnd.3 cell death after fucoxanthin treatment, we concluded that fucoxanthin attenuated ECs damage after TBI. Furthermore, our Western blot data also detected a decline of Bcl-2 and activation of Bax and caspase-3 after TBI, while the treatment of fucoxanthin increased the ratio of Bcl-2/Bax and decreased the expression of cleaved caspase-3, indicating that fucoxanthin suppressed cell apoptosis after TBI. However, it was not known whether fucoxanthin inhibited TBI-induced apoptosis in ECs or neurons, further studies were needed to explained it.
Certainly, cells including BMECs would die in the core region of TBI, where tissue primary received physical damage. However, BBB disruption in the penumbra region of TBI was largely due to the transient hyperpermeability of BMECs, but not accompanied with the cell death.11,39,40 Because the transient hyperpermeability was mediated by neuroinflammation and oxidative stress, suppression of the hyperpermeability of BBB was thought to an effective target for TBI therapeutics.41 In the present study, we measured EB leakage and brain water content in the cerebrum and brain hemispheres, respectively, which means that the results mainly reflect BBB disruption in the penumbra region. However, our study lacked consideration of BBB disruption without death BMECs, further studies were needed to examined it.
Growing evidence have indicated that ferroptosis promoted the pathological occurrence and development of TBI. For example, Wang et al. proposed that repetitive mild traumatic brain injury (rmTBI) induced time-dependent alterations in ferroptosis-related markers, such as abnormal iron metabolism, inactivated GPx, decreased GPX4 levels and increased LPO levels. Furthermore, the treatment of mesenchymal stromal cells (MSCs) significantly suppressed rmTBI-mediated ferroptosis.42 Moreover, Zhang et al. found that in a mouse TBI model, the activation of Netrin-1 (NTN1) by NTN1 recombinant decreased the MDA and LPO levels, attenuated the mitochondria shrinkage and up-regulated the GPX4 expression after controlled cortical impact (CCI). However, knockdown of GPX4 counteracted the effects of NTN1 recombinant.43 Ferroptosis is a new non-necrotic and non-apoptotic PCD that kills cells through iron-dependent LPO. The critical pathological mechanisms of ferroptosis have been implicated as iron accumulation, LPO production and oxidative damage.44 Therefore, in the present study, we wondered whether fucoxanthin protected BBB against TBI by the inhibition of ferroptosis. We found that fucoxanthin suppressed iron accumulation in ECs as confirmed by Perls’ DAB staining. LPO produces a wide variety of oxidation product. Among them, MDA is widely studied as biomarkers of LPO. Furthermore, GPx is the antioxidant enzyme that transforms peroxides into innocuous substances.45 Thus, we evaluated the LPO levels by analysis of MDA and GPx. We found that TBI increased the level of MDA and decreased the level of GPx. While the treatment of fucoxanthin down-regulated the MDA level, up-regulated the GPx level and rebalanced the oxidative-antioxidant system. It has been shown that oxidative damage can occur in respond to DNA damage.46 8-OHdG is the modified base of DNA. In nuclear and mitochondrial DNA, 8-OHdG is the result of oxidative DNA damage when deoxyguanosine, a component of DNA, is oxidized.47 In our study, we also found that fucoxanthin attenuated TBI-induced oxidative DNA damage in ECs as evidenced by 8-OHdG/CD31 double staining.
The typical morphological changes of ferroptosis includes a decrease in mitochondrial volume and mitochondrial cristae, and an increase in mitochondrial membrane density.48 Moreover, ferroptosis can be regulated by ACSL4 and GPX4. ACSL4 is an enzyme that facilitates free polyunsaturated fatty acids (PUFAs) to synthesize PUFA-phosphatidylethanolamines, which are unstable and prone to LPO.49 GPX4 is a significant antioxidant enzyme that modulates ferroptosis by protecting cells from LPO. When GPX4 is inhibited, ferroptosis will be triggered as the lethal amount of LPO accumulation.50 Recently, both ACSL4 and GPX4-regulated ferroptosis has been found to participate in the pathological processes of CNS injuries. For example, in a mouse cerebral ischemia-reperfusion (I/R) injury model, the administration of baicalein inhibited the ferroptosis by the inhibition of ACSL4 and activation of GPX4.51 Besides, in a rat SAH model, the treatment of puerarin decreased the expression of ACSL4, increased the expression of GPX4 and alleviated the iron concentration in the ipsilateral hemisphere after SAH.52 In our study, we found that the ferroptosis-related markers such as ACSL4, GPX4 were changed in ECs after TBI. However, these changes were reversed by fucoxanthin, suggesting that fucoxanthin inhibited TBI-induced EC ferroptosis. In the present study, our Western blot data showed that fucoxanthin reduced iron uptake and storage, and promoted iron export after TBI, thus improving iron metabolism disturbances. Besides, our TEM analysis demonstrated that the mitochondrial morphology was changed after TBI, which was maintained by fucoxanthin treatment. These results indicated that fucoxanthin attenuated the iron accumulation and mitochondria damage caused by TBI. However, it is not known whether the accumulation of iron and the damage of mitochondria was in the ECs or in the neurons, further studies were needed to explored it.
Mitophagy is a crucial process of removing undesired and damaged mitochondria to adjust their number and maintain a balanced energy metabolism, which impacts various physiological and pathological courses in the brain.53 Recently, the crosstalk between mitophagy, apoptosis and ferroptosis has been confirmed. Cui et al. reported that in intervertebral disc degeneration (IDD) models, the administration of VO-OHpic suppressed apoptosis and ferroptosis, and activated mitophagy through the Nrf2/HO-1 pathway. While the inhibition of mitophagy by Nrf2-siRNA reversed the preventative effects of VO-OHpic on apoptosis and ferroptosis.54 Moreover, in mouse and neuron TBI models, the administration of human umbilical cord mesenchymal stem cell (HucMSC)-derived exosome inhibited TBI-induced apoptosis and ferroptosis. In addition, HucMSC-derived exosome activated PINK1/Parkin pathway-mediated mitophagy after TBI. However, the protection of HucMSC-derived exosome on apoptosis and ferroptosis was attenuated when mitophagy was inhibited and PINK1 was knockdown.23 Consistent with these results, the present study indicated that mitophagy was activated in ECs after TBI. Suppression of mitophagy by Mdivi-1 reversed the inhibitory effects of fucoxanthin on TBI-induced BBB damage, EC apoptosis and ferroptosis. These data indicated that fucoxanthin prevented TBI-induced BBB disruption by enhancing mitophagy. However, our results were contrary to the findings discovered by Wu et al., they suggested that the treatment of Mdivi-1 could improve TBI-induced injuries to the BBB and relieve cell death by suppressing mitophagy.55,56 The discrepancies may be due to the different TBI model used in these two studies. In our study, we used a CCI model, while in the study conducted by Wu et al., they used a weight-drop model. Depending on different TBI models, mitophagy and cell death can have inhibitory, additive or even synergistic effects. Therefore, in the present study, mitophagy activation might be a potential neuroprotective mechanism of fucoxanthin in TBI model.
Limitations of the study
However, there were some limitations in our study. Firstly, multiple cells such as neurons, microglia, astrocytes and pericytes could be affected by fucoxanthin after TBI. Therefore, whether fucoxanthin produced anti-TBI effects via acting on BBB injury or via neurons, microglia, astrocytes or pericytes was explained in our study. Secondly, fucoxanthin owned a variety of properties such as the regulation of oxidative stress. Thus, it should be clarified that whether the protection of fucoxanthin on BBB after TBI was contributed to other properties. Thirdly, the mechanism of fucoxanthin was not fully disclosed. We only showed a series of data regarding the phenotypic consequences of fucoxanthin treatment under TBI. However, how fucoxanthin exerted these effects, such as inducing mitophagy, was not explained. Fourthly, we only used Mdivi-1 to investigate the involvement of mitophagy in the anti-TBI effects of fucoxanthin, more tool drugs or genetic tools should be used to prove that fucoxanthin attenuated endothelial impairments and BBB destruction via activating the PINK1/Parkin pathway-mediated mitophagy. Fifthly, further studies were required to investigate whether the administration of fucoxanthin in different time courses might provide better protection against TBI-induced BBB damage.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-ZO-1 | Cell Signaling Technology | Cat#13663; RRID:AB_2798287 |
| Rabbit anti-occludin | Cell Signaling Technology | Cat#91131; RRID:AB 2934013 |
| Rabbit anti-claudin-5 | Cell Signaling Technology | Cat#49564; RRID:AB 3065250 |
| Rabbit anti-VE-cadherin | Cell Signaling Technology | Cat#2500; RRID:AB 3065251 |
| Rabbit anti-MMP-9 | Cell Signaling Technology | Cat#13667; RRID:AB 2798289 |
| Rabbit anti-Bcl-2 | Cell Signaling Technology | Cat#3498; RRID:AB 1903907 |
| Rabbit anti-Bax | Cell Signaling Technology | Cat#14796; RRID:AB 2716251 |
| Rabbit anti-cleaved caspase-3 | Cell Signaling Technology | Cat#9661; RRID:AB 2341188 |
| Rabbit anti-TfR | Abcam | Cat#ab84036; RRID:AB 10673794 |
| Rabbit anti-Ft | Abcam | Cat#ab75973; RRID:AB 1310222 |
| Rabbit anti-Fpn-1 | Abcam | Cat#ab58695; RRID:AB 2302072 |
| Rabbit anti-PINK1 | Abcam | Cat#ab216144; RRID:AB 2927726 |
| Mouse anti-Parkin | Abcam | Cat#ab77924; RRID:AB 1566559 |
| Rabbit anti-TOMM20 | Cell Signaling Technology | Cat#42406; RRID:AB 2687663 |
| Rabbit anti-LC3 | Novus Biological | Cat#NB600-1384; RRID:AB 669581 |
| Rabbit anti-β-actin | Bioworld Technology | Cat#AP0060; RRID:AB 2797445 |
| Rabbit anti-8-OHdG | Bioss | Cat#bs-1278R; RRID:AB 10856120 |
| Rabbit anti-ACSL4 | Abcam | Cat#ab155282; RRID:AB 2714020 |
| Rabbit anti-GPX4 | Abcam | Cat#ab125066; RRID:AB 10973901 |
| Rabbit anti-CD31 | Abcam | Cat#ab222783; RRID:AB 2905525 |
| Goat anti-rabbit IgG (H + L) secondary antibody, HRP | Bioworld Technology | Cat#BS13278; RRID:AB 2773728 |
| Goat anti-mouse IgG (H + L) secondary antibody, HRP | Bioworld Technology | Cat#BS12478; RRID:AB 2773727 |
| Chemicals, peptides, and recombinant proteins | ||
| Fucoxanthin | Sigma-Aldrich | Cat#F6932 |
| Mdivi-1 | MedChemExpress | Cat#HY-15886 |
| Critical commercial assays | ||
| TB staining kit | Beyotime Biotechnology | Cat#ST798 |
| LDH cytotoxicity assay kit | Beyotime Biotechnology | Cat#C0016 |
| Perls’ DAB staining kit | ChemicalBook | Cat#CB25533488 |
| MDA assay kit | Beyotime Biotechnology | Cat#S0131M |
| GPx assay kit | Beyotime Biotechnology | Cat#S0056 |
| γ-GT kit | Jiancheng Biochemistry | Cat#C017-2-1 |
| TUNEL staining kit | Roche | Cat#11684817910 |
| Experimental models: Cell lines | ||
| bEnd.3 | ATCC | N/A |
| Experimental models: Organisms/strains | ||
| ICR mice | Animal Center of Jinling hospital | N/A |
| Software and algorithms | ||
| Prism | GraphPad | N/A |
| Adobe Photoshop CS5 | Adobe Systems | N/A |
| SPSS 19.0 | SPSS | N/A |
| ImageJ | NIH | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yixing Lin (lyx2022nj@126.com).
Materials availability
This study did not generate new unique reagents.
Experimental model and study participant details
Animals
The study protocol of animals was approved by Institutional Review Board of Jinling Hospital (Ethical approval number: 2022DZGKJDWLS-00153). This study was carried out in accordance with the principles of the Basel Declaration and recommendations of the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 8023, revised 1978). Male ICR mice (28–32 g) were obtained from Animal Center of Jinling hospital. Mice were housed on a 12 h light/dark cycle at 23 ± 1°C with free access to food and water.
Cell lines
The bEnd.3 cells, immortalized mouse brain microvascular endothelial cell (BMEC) line, was purchased from American Type Culture Collection (ATCC) and grown in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Gibco, New York, United States) and streptomycin/penicillin. The bEnd.3 cells were maintained at 37°C on 5% CO2 incubator.
Method details
TBI models
The in vivo (mouse) TBI model was performed using a Controlled Cortical Impact (CCI) model according to our previous study.23 Mice were anesthetized with 3% isoflurane and subsequently placed on the stereotaxic apparatus. The area to be impacted lies on the right frontal skull (2.5 mm lateral to the midline and 0.5 mm anterior to bregma). Then mice were subjected to cortical contusion injury by a 3.0 mm rounded impactor tip (piston velocity: 3.5 m/s; deformation depth: 1.5 mm, dwell time: 150 ms). The sham-injured mice underwent the same procedures, but did not undergo CCI. The in vitro (bEnd.3 cells) TBI model was conducted using a mechanical stretch injury model according to previous studies.22,57 Stretch-induced bEnd.3 cell injury was used to simulate cellular mechanical injury during CCI in the present study. For mechanical stretch injury experiments, bEnd.3 cells were seeded (0.5∗105/cm2) onto BioFlex 6-well culture plates with collagen-coated Silastic membranes (Flexcell International Corp, Burlington, NC, USA). After the cells were grown to confluence, a Cell Injury Controller II system (Virginia Commonwealth University, Richmond, VA, USA) was applied to generate a biaxial stretch on the cells in the plate. A 50 ms burst of nitrogen gas was released to produce a 7.5 mm downward deformation of the Silastic membrane and adherent cells, analogous to the mechanical stress exerted on brain tissue as previously reported.57
Experimental design
The design of our experiment was shown in Figure S1. Fucoxanthin (Sigma-Aldrich, St.Louis, Mo, USA; catalog number: F6932) was single administration in our experiments. The usage and dosage of fucoxanthin used in our in vivo and in vitro studies were according to our previous study.18 In Evans blue (EB) dye extravasation experiment, mice were randomly divided into five groups: sham, TBI, TBI + vehicle, TBI + fucoxanthin intragastric (i.g.) administration (100 mg/kg) and TBI+ fucoxanthin intracerebroventricular (i.c.v.) injection (0.05 mmol/L). For i.g. administration, fucoxanthin was diluted in olive oil (1 mL/kg) immediately before use. For i.c.v. injection, fucoxanthin was prepared in saline with 10% dimethyl sulphoxide (DMSO). Either fucoxanthin or an equal volume of vehicle was administrated 30 min after TBI. In other in vivo studies, mice were randomly divided into four groups: sham, TBI, TBI + vehicle and TBI+ fucoxanthin i.c.v. injection (0.05 mmol/L). For in vitro experiments, bEnd.3 cells were divided into four groups: control, TBI, TBI + DMSO, TBI + fucoxanthin (10 μM). Fucoxanthin was first dissolved in DMSO and then added to cultured media to reach different final concentrations.
Neurological evaluation, brain water content and lesion volume
Neurologic severity score (NSS) was used to evaluate the neurological impairment of mice at 1 day (d), 3days, 5days, 7days and 14days after TBI. The investigators estimate the ability of mouse to perform 10 different tasks which demonstrate physiological behavior, alertness and motor function. One point is given for failing to perform each task, thus 0 = minimum deficit and 10 = maximum deficit (Table 1).58,59 Rotarod test was used to assess the motor coordination and balance of mice at 1 day, 3days, 5days, 7days and 14days after TBI according to a previous study.60 Morris water maze (MWM)61 and open field test (OFT)61 were used to assess the spatial learning, memory and motor function of mice at 15–21 days after TBI. All animals were subjected to behavior training for 3 days before the surgery and mice displaying abnormal behavior were excluded. For rotarod test, mice were placed on a rod with a rotating speed increasing from 5 to 40 rpm within 300 s and the latency to fall was recorded. Three trials were performed, and the mean latency was recorded. For MWM, mice needed to find a hidden platform in a circular aluminum pool that was surrounded by visual cues placed at the same starting point. Trajectory and navigation parameters were recorded by investigators blind to the experimental groups. For OFT, the open field consisted of a square arena (100 × 100 cm) with 25 squares on the ground and a 20 cm wall around it. A camera was attached to a computer above the arena. The brain water content and lesion volume were conducted according to our previous study.18 For brain water content, mouse brain was taken out and placed onto a cooled brain matrix at 3 days following TBI. The cerebellum and stem were taken away and the ipsilateral tissue was weighed to get the wet weight (ww). Then, the hemisphere was dried for 72 h at 80°C and weighed again to get the dry weight (dw). The brain water content equals (ww-dw)/ww × 100%. For lesion volume, all mice were killed at 3 days after TBI. The brains were collected, post-fixed for 12 h and cryoprotected in 15% sucrose in phosphate buffered saline (PBS). Tissue damage was evaluated by morphometric image analysis. The brain sections that obtained at 500 mm intervals spanning the length of the brain were stained with cresyl violet. The areas of the lesion, injured as well as non-injured cortex and hemisphere were estimated using an image analysis system. Area measurements from each tissue section were obtained and summed, and corresponding volumes were calculated. Lesion volume was quantitatively analyzed with Image-Pro Plus system (version 6.0) and was expressed as % (the percentage volume of the non-injured hemisphere).
Terminal deoxynucleotidyl transferase-mediated dUTP nick 3′-end labeling (TUNEL) staining
For TUNEL and CD31 staining, the sections were firstly incubated with labeling solution containing TUNEL (Roche Inc., USA, catalog number: 11684817910) at 37°C for 1 h. Then, CD31 antibody (1:200; Abcam, Cambridge, MA, USA; catalog number: ab222783) and the corresponding secondary antibody were incubated. Subsequently, 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI, Beyotime Biotechnology, Shanghai, China; catalog number: C1002) staining was performed for 2 min to show the locations of nuclei. Finally, the sections were observed under a fluorescence microscope (Olympus IX71, Japan). The TUNEL/CD31 double-positive cells were counted from ten different fields in each group by Image-Pro Plus 6.0 software (Media Cybernetics, USA).
Golgi-Cox staining
The Golgi-Cox staining was performed to investigate the changes in neuronal morphology and dendritic spines after TBI using the FD Rapid Golgi Stain Kit (FD Neuro Technologies, USA). Briefly, mice were deeply anesthetized and intracardially perfused with saline. The brains were removed quickly and placed in a mixture of solution A and B (1:1) for 18 days in the dark at room temperature. Then, the brains were transferred into the solution C for 72 h in the dark at room temperature. All brains were then cut into 100-μm-thick frozen slices and stained according to the manufacturer’s instructions. The images were captured by confocal microscope (LSM880, Zeiss, Germany).
Measurement of BBB permeability and integrity
BBB permeability was measured by EB dye extravasation at 3 days after TBI in vivo. Briefly, 4% EB dye (4 mL/kg) was injected to mice from the femoral vein and allowed to circulate for 2 h. Then, the brain was perfused with saline, weighed and homogenized in a solution containing 1 mL 50% trichloroacetic acid. After centrifugation at 15,000 rpm for 20 min, EB absorbance of the supernatant was determined at 620 nm. The extravasation of EB dye was expressed as micrograms per gram brain tissue according to the standard curve. BBB integrity was measured by Trans-endothelial electrical resistance (TEER) value and γ-GT activity at 3 days after TBI in vitro. TEER value of bEnd.3 cell monolayer was analyzed using Millcell ERS-2 Voltohmmeter (Millipore, USA). Cells were and grown to monolayer at a 24-well plate and subjected to TEER value detection as the protocols suggested by the manufacturer. Calculation formula was TEER value (Ω × cm2) = TEER (Ω) × surface area (cm2). The resistance value (5Ω × cm2) of empty filter was subtracted from each measurement. For the measurement of γ-GT activity, bEnd.3 cells were scraped from the insert membrane, suspended in PBS and centrifuged. The precipitates were dissociated with 1% Triton X-100 and centrifuged again. The γ-GT activity in the supernatant was determined with a γ-GT kit (J Nanjing Jiancheng Biochemistry Co., Nanjing, China; catalog number: C017-2-1).
Measurement of malondialdehyde (MDA) and glutathione peroxidase (GPx)
The injured cerebral cortex samples were homogenized in 2 mL of PBS. After centrifugation at 10,000 g for 25 min, the MDA and GPx in the supernatant were measured using an MDA assay kit (Beyotime Biotechnology, Shanghai, China; catalog number: S0131M) and GPx assay kit (Beyotime Biotechnology, Shanghai, China; catalog number: S0056), and detected by a spectrophotometer according to the manufacturer’s instructions.
Western blot analysis
Western blot analysis was performed according to our previous study.62 Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidene fluoride (PVDF) membranes and incubated with the following primary antibodies for 24 h at 4°C: zonula occludens-1 (ZO-1) (1:1000; Cell Signaling Technology, Danvers, MA, USA; catalog number: 13663), occludin (1:1000; Cell Signaling Technology, Danvers, MA, USA; catalog number: 91131), claudin-5 (1:1000; Cell Signaling Technology, Danvers, MA, USA; catalog number: 49564), vascular endothelial-cadherin (VE-cadherin) (1:1000; Cell Signaling Technology, Danvers, MA, USA; catalog number: 2500), matrix metalloproteinase-9 (MMP-9) (1:1000; Cell Signaling Technology, Danvers, MA, USA; catalog number: 13667), B-cell lymphoma 2 (Bcl-2) (1:1000; Cell Signaling Technology, Danvers, MA, USA; catalog number: 3498), Bcl-2-associated X protein (Bax) (1:5000; Cell Signaling Technology, Danvers, MA, USA; catalog number: 14796), cleaved caspase-3 (1:1000; Cell Signaling Technology, Danvers, MA, USA; catalog number: 9661), transferring receptor (TfR) (1:1000; Abcam, Cambridge, MA, USA; catalog number: ab84036), ferritin (Ft) (1:1000; Abcam, Cambridge, MA, USA; catalog number: ab75973), ferroportin-1 (Fpn-1) (1:1000; Abcam, Cambridge, MA, USA; catalog number: ab58695), phosphatase and tensin homolog induced kinase 1 (PINK1) (1:1000; Abcam, Cambridge, MA, USA; catalog number: ab216144), Parkinson protein 2 E3 ubiquitin-protein ligase (Parkin) (1:1000; Abcam, Cambridge, MA, USA; catalog number: ab77924), translocase of outer mitochondrial membrane 20 (TOMM20) (1:1000; Cell Signaling Technology, Danvers, MA, USA; catalog number: 42406), microtubule-associated protein light chain 3 (LC3) (1:1000; Novus Biological, Littleton, CO, USA; catalog number: NB600-1384) and β-actin (1:5000; Bioworld Technology, Minneapolis, MN, USA; catalog number: AP0060). Subsequently, the membranes were incubated with corresponding secondary antibodies for 2 h at room temperature. Protein signals were visualized using Chemiluminescent HRP Substrate (WBKLS0500, Millipore) with the Tanon Image System (Tanon-1100) (S/N: 15T12NPFLI6–12,107, Tanon, Shanghai, China). All WB bands were analyzed by ImageJ software.
Immunofluorescence (IF) staining
IF staining was performed according to our previous study.23 Briefly, the sections were incubated with the following primary antibodies overnight in a dark place at 4°C: 8-hydroxyguanosine (8-OHdG) (1:100; Bioss, Massachusetts, Boston, USA; catalog number: bs-1278R), ACSL4 (1:200; Abcam, Cambridge, MA, USA; catalog number: ab155282), GPX4 (1:200; Abcam, Cambridge, MA, USA; catalog number: ab125066) and LC3 (1:200; Novus Biological, Littleton, CO, USA; catalog number: NB600-1384). Subsequently, the sections were incubated with anti-CD31 antibody (1:200; Abcam, Cambridge, MA, USA; catalog number: ab222783) under similar conditions. After washing three times with PBS, the sections were incubated with the corresponding secondary antibodies for 1 h in a dark place at room temperature. Finally, the sections were stained with DAPI for 2 min to show the locations of nuclei. We imaged the fluorescently stained cells via Olympus IX71 inverted microscope system and analyzed using Image-Pro Plus 6.0 software (Media Cybernetics, Silver Spring, MD).
Perls’ diaminobenzidine (DAB) staining
The injured cerebral cortex samples were fixed with 4% paraformaldehyde for 48 h and embedded in paraffin. Samples were cut into 4 μm thick slices for using a Perls’ DAB stain kit (ChemicalBook, Beijing, China; catalog number: CB25533488) according to the manufacturer’s instructions. In short, after dewaxing, staining and color development, the sections were mounted in glycerinum. Finally, six pictures (400 ×) from each section were taken and quantitation using Image-Pro Plus system.
Transmission electron microscope (TEM)
TEM was used to identify mitochondrion and autophagosome as previously described.23 Briefly, at 3 days after TBI, mice were killed and perfused with 2.5% buffered glutaraldehyde. The specimens were collected and fixed in glutaraldehyde with a 1% (w/v) solution of osmium tetroxide. The fixed specimens were then embedded, sectioned, double stained with lead citrate and uranyl acetate, observed under a TEM JEM-1011 (JEOL, Japan).
Cell viability analysis
To analyze the cell viability of bEnd.3 cells, we used trypan blue (TB) staining and lactate dehydrogenase (LDH) assay.62 For TB staining, cells were stained by 0.4% TB (Beyotime Biotechnology, Shanghai, China; catalog number: ST798) after treatment. Stained cells were considered as dead while unstained cells were considered as viable. The number of TB-positive cells and total cell number were counted. Survival value equals = (number of stained cells/number of total cells) × 100%. In addition, an LDH cytotoxicity assay kit (Beyotime Biotechnology, Shanghai, China; catalog number: C0016) was used to confirm the results of TB staining. In brief, cells were treated with LDH release agent and the culture medium was centrifuged. The supernatant was further collected to evaluate the activity of LDH. The OD value at 490 nm was analyzed by a spectrophotometer. The percentage of damaged cells (%) = (OD490exosomemple – OD490media)/(OD490maximum – OD490media) × 100%. OD490maximum = cells treated with LDH release agent and OD490media = only media without any cells.
Quantification and statistical analysis
All statistical analysis was performed with SPSS 19.0 (SPSS Inc., Chicago, IL). The Shapiro-Wilk test was used to confirm whether the data fit a normal distribution. Data were summarized as mean ± SD (normally distributed) or median (non-normally distributed). For normally distributed data, one-way analysis of variance (ANOVA) followed by Tukey’s test was used for multiple group comparisons. The least significant difference (LSD) post hoc test was used to determine statistical differences between groups. Statistical differences between the two groups were compared using Student’s t test. For data that did not conform to a normal distribution, comparisons between groups were made using the Mann-Whitney U test. A value of p < 0.05 was considered statistically significant.
Acknowledgments
This study was supported by Grants from the Jiangsu Postdoctoral Research Funding Program (No. 49251) from Zixiang Cong.
Author contributions
L.Z was responsible for the data collection, animal experiments and article writing; Z.G.H was responsible for the data analysis; W.S.B was responsible for the literature collection; Y.N.P was responsible for the article review; Y.X.L was responsible for the design of the article; Z.X.C was responsible for the cell experiments and funding support. All authors read and approved the final article.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: October 20, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108270.
Contributor Information
Yixing Lin, Email: lyx2022nj@126.com.
Zixiang Cong, Email: congzixiang86@126.com.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Lu W., Wu Z., Zhang C., Gao T., Ling X., Xu M., Wang W., Jin X., Li K., Chen L., et al. Jujuboside A Exhibits an Antiepileptogenic Effect in the Rat Model via Protection against Traumatic Epilepsy-Induced Oxidative Stress and Inflammatory Responses. Evid. Based. Complement. Alternat. Med. 2022;2022 doi: 10.1155/2022/7792791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang X.J., Ling S., Zhou M.L., Deng H.J., Qi M., Liu X.L., Zhen C., Chen Y.X., Fan X.R., Wu Z.Y., et al. Inhibition of TRPA1 Attenuates Oxidative Stress-induced Damage After Traumatic Brain Injury via the ERK/AKT Signaling Pathway. Neuroscience. 2022;494:51–68. doi: 10.1016/j.neuroscience.2022.02.003. [DOI] [PubMed] [Google Scholar]
- 3.Zhu X., Cheng J., Yu J., Liu R., Ma H., Zhao Y. Nicotinamide mononucleotides alleviated neurological impairment via anti-neuroinflammation in traumatic brain injury. Int. J. Med. Sci. 2023;20:307–317. doi: 10.7150/ijms.80942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu A.G., Yong Y.Y., Pan Y.R., Zhang L., Wu J.M., Zhang Y., Tang Y., Wei J., Yu L., Law B.Y.K., et al. Targeting Nrf2-Mediated Oxidative Stress Response in Traumatic Brain Injury: Therapeutic Perspectives of Phytochemicals. Oxid. Med. Cell. Longev. 2022;2022 doi: 10.1155/2022/1015791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalra S., Malik R., Singh G., Bhatia S., Al-Harrasi A., Mohan S., Albratty M., Albarrati A., Tambuwala M.M. Pathogenesis and management of traumatic brain injury (TBI): role of neuroinflammation and anti-inflammatory drugs. Inflammopharmacology. 2022;30:1153–1166. doi: 10.1007/s10787-022-01017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harris W.J., Asselin M.C., Hinz R., Parkes L.M., Allan S., Schiessl I., Boutin H., Dickie B.R. In vivo methods for imaging blood-brain barrier function and dysfunction. Eur. J. Nucl. Med. Mol. Imaging. 2023;50:1051–1083. doi: 10.1007/s00259-022-05997-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aragón-González A., Shaw P.J., Ferraiuolo L. Blood-Brain Barrier Disruption and Its Involvement in Neurodevelopmental and Neurodegenerative Disorders. Int. J. Mol. Sci. 2022;23 doi: 10.3390/ijms232315271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang X., Chen X. The Crosstalk between the Blood-Brain Barrier Dysfunction and Neuroinflammation after General Anaesthesia. Curr. Issues Mol. Biol. 2022;44:5700–5717. doi: 10.3390/cimb44110386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zlokovic B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 10.Zhang L., Bai W., Sun L., Lin Y., Tian M. Targeting Non-Coding RNA for CNS Injuries: Regulation of Blood-Brain Barrier Functions. Neurochem. Res. 2023;48:1997–2016. doi: 10.1007/s11064-023-03892-1. [DOI] [PubMed] [Google Scholar]
- 11.Li W., Cao F., Takase H., Arai K., Lo E.H., Lok J. Blood-Brain Barrier Mechanisms in Stroke and Trauma. Handb. Exp. Pharmacol. 2022;273:267–293. doi: 10.1007/164_2020_426. [DOI] [PubMed] [Google Scholar]
- 12.Nagata K., Takatani N., Beppu F., Abe A., Tominaga E., Fukuhara T., Ozeki M., Hosokawa M. Monocaprin Enhances Bioavailability of Fucoxanthin in Diabetic/Obese KK-A(y) Mice. Mar. Drugs. 2022;20 doi: 10.3390/md20070446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim S.K., Pangestuti R. Biological activities and potential health benefits of fucoxanthin derived from marine brown algae. Adv. Food Nutr. Res. 2011;64:111–128. doi: 10.1016/B978-0-12-387669-0.00009-0. [DOI] [PubMed] [Google Scholar]
- 14.Zhao X., Gao L., Zhao X. Rapid Purification of Fucoxanthin from Phaeodactylum tricornutum. Molecules. 2022;27 doi: 10.3390/molecules27103189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lau T.Y., Kwan H.Y. Fucoxanthin Is a Potential Therapeutic Agent for the Treatment of Breast Cancer. Mar. Drugs. 2022;20 doi: 10.3390/md20060370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bae M., Kim M.B., Lee J.Y. Fucoxanthin Attenuates the Reprogramming of Energy Metabolism during the Activation of Hepatic Stellate Cells. Nutrients. 2022;14 doi: 10.3390/nu14091902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang X.S., Lu Y., Tao T., Wang H., Liu G.J., Liu X.Z., Liu C., Xia D.Y., Hang C.H., Li W. Fucoxanthin Mitigates Subarachnoid Hemorrhage-Induced Oxidative Damage via Sirtuin 1-Dependent Pathway. Mol. Neurobiol. 2020;57:5286–5298. doi: 10.1007/s12035-020-02095-x. [DOI] [PubMed] [Google Scholar]
- 18.Zhang L., Wang H., Fan Y., Gao Y., Li X., Hu Z., Ding K., Wang Y., Wang X. Fucoxanthin provides neuroprotection in models of traumatic brain injury via the Nrf2-ARE and Nrf2-autophagy pathways. Sci. Rep. 2017;7 doi: 10.1038/srep46763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu M., Gong Y., Jiang L., Zhang M., Gu H., Shen H., Dang B. VEGF regulates the blood-brain barrier through MMP-9 in a rat model of traumatic brain injury. Exp. Ther. Med. 2022;24:728. doi: 10.3892/etm.2022.11664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bechinger P., Serrano Sponton L., Grützner V., Musyanovych A., Jussen D., Krenzlin H., Eldahaby D., Riede N., Kempski O., Ringel F., Alessandri B. In-vivo time course of organ uptake and blood-brain-barrier permeation of poly(L-lactide) and poly(perfluorodecyl acrylate) nanoparticles with different surface properties in unharmed and brain-traumatized rats. Front. Neurol. 2023;14 doi: 10.3389/fneur.2023.994877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng H., Di G., Gao C.C., He G., Wang X., Han Y.L., Sun L.A., Zhou M.L., Jiang X. FTY720 Reduces Endothelial Cell Apoptosis and Remodels Neurovascular Unit after Experimental Traumatic Brain Injury. Int. J. Med. Sci. 2021;18:304–313. doi: 10.7150/ijms.49066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fang J., Yuan Q., Du Z., Fei M., Zhang Q., Yang L., Wang M., Yang W., Yu J., Wu G., Hu J. Ferroptosis in brain microvascular endothelial cells mediates blood-brain barrier disruption after traumatic brain injury. Biochem. Biophys. Res. Commun. 2022;619:34–41. doi: 10.1016/j.bbrc.2022.06.040. [DOI] [PubMed] [Google Scholar]
- 23.Zhang L., Lin Y., Bai W., Sun L., Tian M. Human umbilical cord mesenchymal stem cell-derived exosome suppresses programmed cell death in traumatic brain injury via PINK1/Parkin-mediated mitophagy. CNS Neurosci. Ther. 2023;29:2236–2258. doi: 10.1111/cns.14159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daneman R., Prat A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015;7:a020412. doi: 10.1101/cshperspect.a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Candelario-Jalil E., Dijkhuizen R.M., Magnus T. Neuroinflammation, Stroke, Blood-Brain Barrier Dysfunction, and Imaging Modalities. Stroke Vasc. Interv. Neurol. 2022;53:1473–1486. doi: 10.1161/STROKEAHA.122.036946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matter K., Balda M.S. Signalling to and from tight junctions. Nat. Rev. Mol. Cell Biol. 2003;4:225–236. doi: 10.1038/nrm1055. [DOI] [PubMed] [Google Scholar]
- 27.Hawkins B.T., Davis T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 28.Dejana E., Giampietro C. Vascular endothelial-cadherin and vascular stability. Curr. Opin. Hematol. 2012;19:218–223. doi: 10.1097/MOH.0b013e3283523e1c. [DOI] [PubMed] [Google Scholar]
- 29.Chen P., Tang H., Zhang Q., Xu L., Zhou W., Hu X., Deng Y., Zhang L. Basic Fibroblast Growth Factor (bFGF) Protects the Blood-Brain Barrier by Binding of FGFR1 and Activating the ERK Signaling Pathway After Intra-Abdominal Hypertension and Traumatic Brain Injury. Med. Sci. Monit. 2020;26 doi: 10.12659/MSM.922009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang J., Kang Y., Huang L., Wu L., Peng Y. TIMP1 preserves the blood-brain barrier through interacting with CD63/integrin beta 1 complex and regulating downstream FAK/RhoA signaling. Acta Pharm. Sin. B. 2020;10:987–1003. doi: 10.1016/j.apsb.2020.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei X.E., Zhang Y.Z., Li Y.H., Li M.H., Li W.B. Dynamics of rabbit brain edema in focal lesion and perilesion area after traumatic brain injury: a MRI study. J. Neurotrauma. 2012;29:2413–2420. doi: 10.1089/neu.2010.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang L.Y., Song J.X., Cai H., Wang P.P., Yin Q.L., Zhang Y.D., Chen J., Li M., Song J.J., Wang Y.L., et al. Healthy Serum-Derived Exosomes Improve Neurological Outcomes and Protect Blood-Brain Barrier by Inhibiting Endothelial Cell Apoptosis and Reversing Autophagy-Mediated Tight Junction Protein Reduction in Rat Stroke Model. Front. Cell. Neurosci. 2022;16 doi: 10.3389/fncel.2022.841544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu J., He J., Tian X., Luo Y., Zhong J., Zhang H., Li H., Cen B., Jiang T., Sun X. microRNA-9-5p alleviates blood-brain barrier damage and neuroinflammation after traumatic brain injury. J. Neurochem. 2020;153:710–726. doi: 10.1111/jnc.14963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharma C., Dobson G.P., Davenport L.M., Morris J.L., Letson H.L. The role of matrix metalloproteinase-9 and its inhibitor TIMP-1 in burn injury: a systematic review. Int. J. Burns Trauma. 2021;11:275–288. [PMC free article] [PubMed] [Google Scholar]
- 35.Ma P., Huang N., Tang J., Zhou Z., Xu J., Chen Y., Zhang M., Huang Q., Cheng Y. The TRPM4 channel inhibitor 9-phenanthrol alleviates cerebral edema after traumatic brain injury in rats. Front. Pharmacol. 2023;14 doi: 10.3389/fphar.2023.1098228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hernandes M.S., Xu Q., Griendling K.K. Role of NADPH Oxidases in Blood-Brain Barrier Disruption and Ischemic Stroke. Antioxidants. 2022;11 doi: 10.3390/antiox11101966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang N., Cao W., He X., Xing Y., Yang N. Long Non-Coding RNAs in Retinal Ganglion Cell Apoptosis. Cell. Mol. Neurobiol. 2023;43:561–574. doi: 10.1007/s10571-022-01210-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang L., Wang H. Long Non-coding RNA in CNS Injuries: A New Target for Therapeutic Intervention. Molecular therapy. Nucleic acids. 2019;17:754–766. doi: 10.1016/j.omtn.2019.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cash A., Theus M.H. Mechanisms of Blood-Brain Barrier Dysfunction in Traumatic Brain Injury. Int. J. Mol. Sci. 2020;21 doi: 10.3390/ijms21093344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alves J.L. Blood-brain barrier and traumatic brain injury. J. Neurosci. Res. 2014;92:141–147. doi: 10.1002/jnr.23300. [DOI] [PubMed] [Google Scholar]
- 41.Bodnar C.N., Watson J.B., Higgins E.K., Quan N., Bachstetter A.D. Inflammatory Regulation of CNS Barriers After Traumatic Brain Injury: A Tale Directed by Interleukin-1. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.688254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang D., Zhang S., Ge X., Yin Z., Li M., Guo M., Hu T., Han Z., Kong X., Li D., et al. Mesenchymal stromal cell treatment attenuates repetitive mild traumatic brain injury-induced persistent cognitive deficits via suppressing ferroptosis. J. Neuroinflammation. 2022;19:185. doi: 10.1186/s12974-022-02550-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y., Lan J., Zhao D., Ruan C., Zhou J., Tan H., Bao Y. Netrin-1 upregulates GPX4 and prevents ferroptosis after traumatic brain injury via the UNC5B/Nrf2 signaling pathway. CNS Neurosci. Ther. 2023;29:216–227. doi: 10.1111/cns.13997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang S., Wei W., Ma N., Qu Y., Liu Q. Molecular mechanisms of ferroptosis and its role in prostate cancer therapy. Crit. Rev. Oncol. Hematol. 2022;176 doi: 10.1016/j.critrevonc.2022.103732. [DOI] [PubMed] [Google Scholar]
- 45.El-Desoky G.E., Wabaidur S.M., AlOthman Z.A., Habila M.A. Regulatory Role of Nano-Curcumin against Tartrazine-Induced Oxidative Stress, Apoptosis-Related Genes Expression, and Genotoxicity in Rats. Molecules. 2020;25 doi: 10.3390/molecules25245801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rafiee A., Ospina M.B., Pitt T.M., Quémerais B. Oxidative stress and DNA damage resulting from welding fumes exposure among professional welders: A systematic review and meta-analysis. Environ. Res. 2022;214 doi: 10.1016/j.envres.2022.114152. [DOI] [PubMed] [Google Scholar]
- 47.Rahman M.F., Billah M.M., Kline R.J., Rahman M.S. Effects of elevated temperature on 8-OHdG expression in the American oyster (Crassostrea virginica): Induction of oxidative stress biomarkers, cellular apoptosis, DNA damage and gammaH2AX signaling pathways. Fish Shellfish Immunol. Rep. 2023;4 doi: 10.1016/j.fsirep.2022.100079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Y., Xiong R., Xiao T., Xiong L., Wu J., Li J., Feng G., Song G., Liu K. SCARA5 induced ferroptosis to effect ESCC proliferation and metastasis by combining with Ferritin light chain. BMC Cancer. 2022;22:1304. doi: 10.1186/s12885-022-10414-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xin S., Schick J.A. PUFAs dictate the balance of power in ferroptosis. Cell Calcium. 2023;110 doi: 10.1016/j.ceca.2023.102703. [DOI] [PubMed] [Google Scholar]
- 50.Ding J., Lu B., Liu L., Zhong Z., Wang N., Li B., Sheng W., He Q. Guilu-Erxian-Glue alleviates Tripterygium wilfordii polyglycoside-induced oligoasthenospermia in rats by resisting ferroptosis via the Keap1/Nrf2/GPX4 signaling pathway. Pharm. Biol. 2023;61:213–227. doi: 10.1080/13880209.2023.2165114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li M., Meng Z., Yu S., Li J., Wang Y., Yang W., Wu H. Baicalein ameliorates cerebral ischemia-reperfusion injury by inhibiting ferroptosis via regulating GPX4/ACSL4/ACSL3 axis. Chem. Biol. Interact. 2022;366 doi: 10.1016/j.cbi.2022.110137. [DOI] [PubMed] [Google Scholar]
- 52.Huang Y., Wu H., Hu Y., Zhou C., Wu J., Wu Y., Wang H., Lenahan C., Huang L., Nie S., et al. Puerarin Attenuates Oxidative Stress and Ferroptosis via AMPK/PGC1alpha/Nrf2 Pathway after Subarachnoid Hemorrhage in Rats. Antioxidants. 2022;11 doi: 10.3390/antiox11071259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu Y., Jiang T., Hua J., Xiong Z., Dai K., Chen H., Li L., Peng J., Peng X., Zheng Z., Xiong W. PINK1/Parkin-mediated mitophagy in cardiovascular disease: From pathogenesis to novel therapy. Int. J. Cardiol. 2022;361:61–69. doi: 10.1016/j.ijcard.2022.05.025. [DOI] [PubMed] [Google Scholar]
- 54.Cui X., Liu X., Kong P., Du T., Li T., Yang G., Zhang W., Jing X., Wang W. PTEN inhibitor VO-OHpic protects endplate chondrocytes against apoptosis and calcification via activating Nrf-2 signaling pathway. Aging. 2023;15:2275–2292. doi: 10.18632/aging.204612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu Q., Xia S.X., Li Q.Q., Gao Y., Shen X., Ma L., Zhang M.Y., Wang T., Li Y.S., Wang Z.F., et al. Mitochondrial division inhibitor 1 (Mdivi-1) offers neuroprotection through diminishing cell death and improving functional outcome in a mouse model of traumatic brain injury. Brain Res. 2016;1630:134–143. doi: 10.1016/j.brainres.2015.11.016. [DOI] [PubMed] [Google Scholar]
- 56.Wu Q., Gao C., Wang H., Zhang X., Li Q., Gu Z., Shi X., Cui Y., Wang T., Chen X., et al. Mdivi-1 alleviates blood-brain barrier disruption and cell death in experimental traumatic brain injury by mitigating autophagy dysfunction and mitophagy activation. Int. J. Biochem. Cell Biol. 2018;94:44–55. doi: 10.1016/j.biocel.2017.11.007. [DOI] [PubMed] [Google Scholar]
- 57.Liu Y.L., Xu Z.M., Yang G.Y., Yang D.X., Ding J., Chen H., Yuan F., Tian H.L. Sesamin alleviates blood-brain barrier disruption in mice with experimental traumatic brain injury. Acta Pharmacol. Sin. 2017;38:1445–1455. doi: 10.1038/aps.2017.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Flierl M.A., Stahel P.F., Beauchamp K.M., Morgan S.J., Smith W.R., Shohami E. Mouse closed head injury model induced by a weight-drop device. Nat. Protoc. 2009;4:1328–1337. doi: 10.1038/nprot.2009.148. [DOI] [PubMed] [Google Scholar]
- 59.Xu J., Wang H., Ding K., Lu X., Li T., Wang J., Wang C., Wang J. Inhibition of cathepsin S produces neuroprotective effects after traumatic brain injury in mice. Mediators Inflamm. 2013;2013 doi: 10.1155/2013/187873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guo R., Wang X., Fang Y., Chen X., Chen K., Huang W., Chen J., Hu J., Liang F., Du J., et al. rhFGF20 promotes angiogenesis and vascular repair following traumatic brain injury by regulating Wnt/beta-catenin pathway. Biomed Pharmacother. 2021;143 doi: 10.1016/j.biopha.2021.112200. [DOI] [PubMed] [Google Scholar]
- 61.Qiu W., Wu Q., Zhang K., Da X., Tang K., Yuan N., Deng L., Wu M., Zhang Y., Quan J., et al. Xiaoyaosan ameliorates depressive-like behavior and susceptibility to glucose intolerance in rat: involvement of LepR-STAT3/PI3K pathway in hypothalamic arcuate nucleus. BMC Complement. Med. Ther. 2023;23:116. doi: 10.1186/s12906-023-03942-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang L., Fei M., Wang H., Zhu Y. Sodium aescinate provides neuroprotection in experimental traumatic brain injury via the Nrf2-ARE pathway. Brain Res. Bull. 2020;157:26–36. doi: 10.1016/j.brainresbull.2020.01.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.