

Abstract
Hydropersulfide and hydropolysulfide metabolites are increasingly important reactive sulfur species (RSS) regulating numerous cellular redox dependent functions. Intracellular production of these species is known to occur through RSS interactions or through translational mechanisms involving cysteinyl t-RNA synthetases. However, regulation of these species under cell stress conditions, such as hypoxia, that are known to modulate RSS remain poorly understood. Here we define an important mechanism of increased persulfide and polysulfide production involving cystathionine gamma lyase (CSE) phosphorylation at serine 346 and threonine 355 in a substrate specific manner, under acute hypoxic conditions. Hypoxic phosphorylation of CSE occurs in an AMP kinase dependent manner increasing enzyme activity involving unique inter- and intramolecular interactions within the tetramer. Importantly, both cellular hypoxia and tissue ischemia result in AMP Kinase dependent CSE phosphorylation that regulates blood flow in ischemic tissues. Our findings reveal hypoxia molecular signaling pathways regulating CSE dependent persulfide and polysulfide production impacting tissue and cellular response to stress.
Keywords: Cystathionine gamma lyase, Phosphorylation, Polysulfide, Persulfide, AMP kinase, Ischemia, Molecular modeling
Graphical abstract
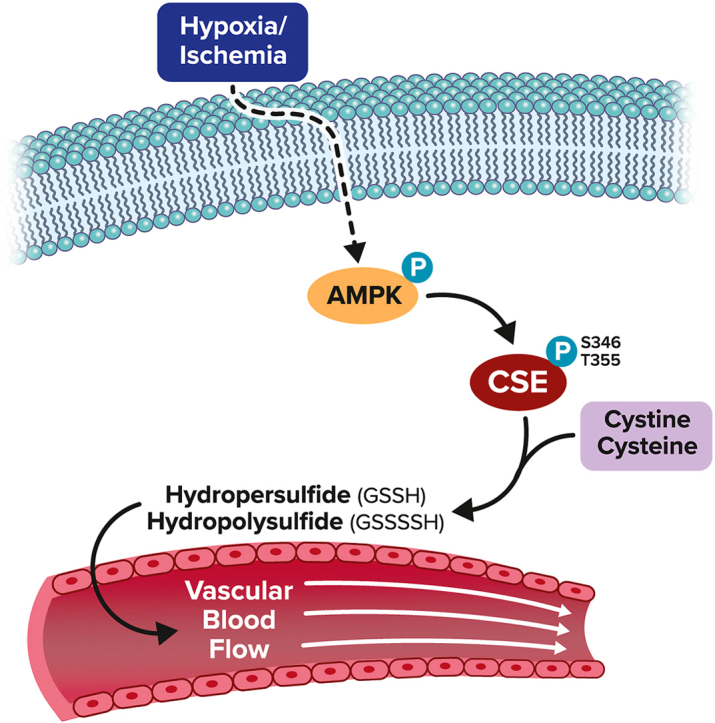
1. Introduction
Reactive sulfur species (RSS) are important for numerous biological functions and pathophysiological roles in cardiovascular, neurological, inflammatory, and immune systems, and in other disease states [[1], [2], [3]]. Hydrogen sulfide (H2S) has received predominant attention among RSS and is produced by enzymes in the trans-sulfuration pathway, cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE/CTH), as well as by 3-mercaptopyruvate sulfurtransferase, which is involved in cysteine catabolism. However, endogenous sulfide may be present in three biochemically distinct forms, including, readily available free sulfide, acid-labile (e.g. iron-sulfur clusters that are localized in active centers of respiratory enzymes), and as bound sulfane sulfur forms (e.g. hydropersulfides and hydropolysulfides) [1,2,4]. Free H2S can be released by acid-labile and bound sulfane sulfur under acidic or reducing conditions, respectively [1,2]. RSS are constantly maintained in mammalian cells and can be influenced either through transcriptional regulation of sulfide synthesis enzymes or via persulfidation popularly called S-sulfhydration of cysteine residues on target proteins [5,6]. Importantly, bound sulfane sulfurs have unique biochemical properties among all RSS as they can act as nucleophiles (reductant) or electrophiles (oxidant) depending on intracellular conditions impacting a wide range of molecules [4]. Our group has previously demonstrated that ischemic and mechanical remodeling of the vasculature involves upregulation of sulfide metabolites that ramify on NO-dependent cytoprotective responses revealing important roles for cardiovascular function [7]. However, specific mechanisms governing hypoxic formation of reactive sulfur species production are poorly understood.
It is now clear that hydropersulfides and polysulfides can be synthesized independently of H2S. Polysulfide release from cysteinyl-tRNA synthetases (CARSs) has been demonstrated [8,9] and both CBS and CSE utilize substrates such as cystine or glutathione disulfide, to catalyze the formation of cysteine hydropersulfide or glutathione hydropersulfide, and other polysulfides [2,4,9,10]. These various intracellular polysulfide/persulfide species together constitute the total concentration of sulfane sulfur inside the cell. Importantly, recent studies show differential metabolism of these species during changes in pH or oxygen (hypoxia or ischemia) that may influence lipid peroxidation and ferroptosis [11,12]. As such, persulfides/polysulfides are now considered critical mediators within cellular redox signaling networks that regulate diverse cellular functions. However, a significant limitation remains with the lack of insight into specific mechanisms regulating the formation of per- and polysulfide compounds under various pathophysiological conditions. In this study, we reveal an important AMP kinase/CSE phosphorylation-dependent mechanism during hypoxia/ischemia leading to specific generation of per/polysulfide with potent pathophysiological effects.
2. Results
2.1. Hypoxia induces polysulfide levels via novel CSE phosphorylation
We previously reported an increase in CSE activity and sulfide metabolites under sustained tissue ischemia [7,13] but the mechanism(s) responsible for these effects remain unknown. Murine aortic endothelial cells were exposed to hypoxia and sulfide metabolites measured. We observed a selective three-fold increase in bound sulfane sulfur consisting of per- and polysulfide pools using the monobromobimane (MBB) RP-HPLC quantitative analytical measurement after 30 min of hypoxia; no changes in free or acid labile sulfide was observed (Fig. 1A). We next utilized and standardized intracellular fluorescent probes to detect free H2S using SF7 (Suppl. Fig. 1A), and per-polysulfide using SSP4 probe (Suppl. Fig. 1B) and confirmed that hypoxia increased per-polysulfides (Fig. 1B) without changing free H2S levels (Suppl. Fig. 1C). Furthermore, response from intracellular labeling with either SF7 or SSP4 mirrored measurement of per- and polysulfide pools via MBB-HPLC. We observed that hypoxia significantly increased CSE activity suggesting a primary role for this enzyme in observed per-polysulfide generation (Fig. 1C). Further, to determine the source of increased per-polysulfides and H2S, we used siRNA against known sulfur metabolite producing enzymes, including CSE, CBS, MPST, CARS1 or CARS2, and then exposed murine endothelial cells to hypoxia and measured per-polysulfides using SSP4 (Fig. 1D), and H2S using SF7 (Suppl. Fig. 1D). Significant decreases in mRNA and protein levels of siRNA treatment for CSE, CBS, MPST CARS1 and CARS2 was confirmed by qRT-PCR and Western blot (Suppl. Fig. 1 E-I and J-N). In CSE siRNA cells, hypoxia dependent increased in SSP4 fluorescence was inhibited >70 % (Fig. 1D); while a 30 % reduction was observed in SF7 intensity. A moderate yet significant reduction in SSP4 intensity was also observed with CARS1 siRNA (Fig. 1D). Interestingly, this inhibition is significantly less compared to inhibition of both per-polysulfides (SSP4), and H2S (SF7), respectively via CSE siRNA. These findings are consistent with previous findings of these enzymes in per-polysulfide production.
Fig. 1.

Hypoxic per-polysulfide formation and CSE phosphorylation. Panel A shows MAECs exposed to either normoxia (21 % oxygen) or hypoxic (1 % oxygen) conditions for 30min and analyzed for various sulfide metabolites, including free, acid-labile sulfide and bound sulfane sulfur (including persulfide and polysulfide) using MBB/HPLC method. Panel B Per-polysulfides levels in MAECs treated with either normoxia or hypoxia for 30min using the fluorescent probe SSP4. Panel C CSE enzyme activity under hypoxia compared to normoxia in MAECs. Panel D Per-polysulfide levels in MAECs transfected with either mock, CSE, CBS, MPST, CARS-1 or CARS-2 siRNAs and then respectively were probed with SSP4 under hypoxic conditions. Panel E LC/MS HCD fragmentation spectrum of trypsin digested human CSE purified from normoxic versus hypoxic treated HEK cells. CSE amino acid fragment 334–364 (LFTLAESLGGFESLAELPAIMTHASVLKNDR) was identified by Protein Discoverer 2.5. (i) HCD spectra of native Human CSE [334−364] peptide. (ii) HCD spectra of singly phosphorylated Human CSE [334−364] at Ser346. (iii) HCD spectra of singly phosphorylated Human CSE [334−364] at T355. Panel F Phospho peptide ratios comparing CSE[334–364] and CSE[365–395] from LC/MS HCD fragmentation spectrum of trypsin digested human CSE purified from hypoxia. Panel G CSE activity of Control (Con), WT, or CSE phosphonegative alanine mutant constructs of S346A or T355A transfected into HEK293 cells. Panel H CSE activity of HEK293 cells transfected with either Control, WT, CSE glutamate (E) phospho-mimetics, S346E or T355E, under hypoxia. All the data are averaged from triplicates from each experiment with at least n = 5. ****P < 0.0001; ***P < 0.0002; **P < 0.003; *P < 0.01.
We next examined CSE protein for post-translational modifications under normoxia versus hypoxia, wild type human CSE FLAG-tagged construct was expressed in murine endothelial cells, treated under normoxia or hypoxia, purified and trypsin digested for LC/MS HCD (higher-energy C-trap dissociation) fragmentation spectrum (Fig. 1E). A three-fold increase in phospho-peptide abundance ratio in HCD spectra for CSE amino acid fragment [334−364] under hypoxia compared to normoxia was observed. The LC/MS HCD fragmentation spectrum identified hypoxia dependent phosphorylation site at serine 346 (S346) and threonine 355 (T355) (Fig. 1E, inset ii and 1E, inset iii). Moreover, we observed that the abundance ratio of CSE[334–364] peptide fragment was 3 fold higher than CSE[365–395] peptide fragment containing the phospho-inhibitory serine 377 residue under hypoxia compared to normoxia conditions (Fig. 1F). To check if these phosphorylation sites impact CSE activity, we generated phospho-mimetic glutamate or phospho-negative alanine mutants of either S346 or T355 and examined their enzyme activities in transfected HEK cells. A two-fold decrease in cystathionine consumption (i.e. CSE enzyme activity) with the phospho-negative S346A or T355A (Fig. 1G) was observed under hypoxia; whereas the phospho-mimetic S346E or T355E significantly increased CSE enzyme activity under hypoxia (Fig. 1H).
2.2. Substrate influences CSE-mediated per-polysulfide generation under hypoxia
CSE is a promiscuous enzyme where based on its substrate utilization the kinetics can vary with change in the microenvironment [14]. Next, we investigated the role of the identified phosphorylation sites and their role in the subsequent production of per-polysulfide or free H2S levels using different substrates under hypoxia. Wild type and CSE phospho-negative or mimetic constructs were FLAG tagged and used for transient transfection studies in HEK cells and confirmed by FLAG immunoblotting for uniform expression (Suppl. Figs. 2A and B). Hypoxia dependent increases in SSP4 per-polysulfide were completely blunted by either S346A or T355A using cysteine, cystine, or cystathionine substrates (Fig. 2A, B, and C) as well as under normoxic conditions (Suppl. Figs. 3A, B, and C). Free H2S levels were not significantly different with various substrates under hypoxia (Fig. 2D, E, and F) or normoxia (Suppl. Figs. 3D, E, and F).
Fig. 2.

Phospho-negative mutants reduces per-polysulfide levels under hypoxia. HEK293 cells transfected with either Control (Con), wild type CSE (WT), phospho-negative mutant S346A or T355A under hypoxia (1 % oxygen) for 30 min and probed for per-polysulfide (SSP4 fluorophore) or hydrogen sulfide (SF7 fluorophore) signal. Panels A, B, and C show SSP4 signal using cysteine, cystine, or cystathionine as substrates, respectively. Panels D, E, and F show SF7 signal using cysteine, cystine, or cystathionine as substrates, respectively. All data were averaged from triplicates from each experiment with at least n = 5. ****P < 0.0001; **P < 0.003; *P < 0.01.
We next examined the effect of CSE phosphomimetic constructs S346E and T355E under hypoxia with various substrates. A significant 2.5- and 2.75-fold increase, respectively in per-polysulfides was observed with cysteine and cystine (Fig. 3A and B); but only a 1.5-fold increase with cystathionine (Fig. 3C) as a substrate. While S346E or T355E significantly increased SSP4 signal they did not significantly change H2S levels as measured by SF7 under hypoxia (Fig. 3D, E, and F) These data reveal that CSE phospho-mimetic favors hypoxic per-polysulfide production with cystine or cysteine as substrates versus cystathionine. Similar observations of substrate-specific per-polysulfide generation was observed with phosphomimetics S346E or T355E under normoxic conditions (Suppl. Figs. 4A, B, and C) although with substantially reduced SSP4 signal compared to hypoxic conditions. Importantly, no significant changes were noticed with free H2S generation (Suppl. Figs. 4D, E, and F).
Fig. 3.

Phospho-mimetics induces per-polysulfide levels under hypoxia. HEK293 cells transfected with either Control (Con), wild type CSE (WT), phospho-mimetic mutants S346E or T355E under hypoxia (1 % oxygen) for 30 min and probed for per-polysulfide (SSP4 fluorophore) or hydrogen sulfide (SF7 fluorophore) signal. Panels A, B, and C show SSP4 signal using cysteine, cystine, or cystathionine as substrates, respectively. Panels D, E, and F show SF7 signal using cysteine, cystine, or cystathionine as substrates, respectively. All data were averaged from triplicates from each experiment with at least n = 5. ****P < 0.0001; **P < 0.003; *P < 0.01.
2.3. HPE-IAM detection of CSE-mediated glutathione per-polysulfide generation under hypoxia
We further examined specific per/polysulfide species from cystine substrate utilization under hypoxia in mouse aortic endothelial cells transfected with wild type, S346A, T355A, S346E or T355E CSE constructs followed by derivatization with HPE-IAM for per-polysulfide LC/MS analyses [15]. Significant increases in GSSH persulfide and GSSSSH polysulfide with wild type CSE were observed under normoxia (N-WT) compared to hypoxia (H-WT) (Fig. 4A and B, respectively). Both S346A or T355A significantly blunted hypoxic (H-346A or H-355A) GSSH persulfide (Fig. 4A) and GSSSSH polysulfide (Fig. 4B) production compared to hypoxia wild type CSE (H-WT). Conversely, S346E and T355E phospho-mimetics significantly increased hypoxic (H-346E or H-355E) GSSH persulfide and GSSSSH polysulfide above that of wild type CSE (H-WT) (Fig. 4A and B). These data corroborate findings using SSP4 and delineate specific sulfane sulfur species that are formed.
Fig. 4.

Per and polysulfide levels in endothelial cells transfected with CSE phospho mutants. Abundance of per/polysulfide to protein (fold change) was measured by HPE-IAM LC/MS in MAECs transfected with either wild type CSE (WT) under normoxia (N-WT) or hypoxia (H-WT), phospho negative mutants S346A (H-346A), T355A (H-355A); phospho-mimetics mutant S346E (H-346E) or T355E (H-355E) under hypoxia. Panel A reports glutathionine persulfide (GSSH) and panel B illustrates glutathionine polysulfide (GSSSSH). All data are averaged from triplicates from each experiment with at least n = 3. ****P < 0.0001; ***P < 0.0002; **P < 0.003.
2.4. Molecular modeling determines molecular stabilization of CSE via phosphorylation
CSE is arranged as a tetramer with D2 symmetry [16]; so, we questioned whether phosphorylation could modulate CSE activity via allosteric mechanisms. We confirmed these effects of phosphorylation sites S346 or T355 on human CSE tetrameric structure using molecular dynamics simulations (Fig. 5A and B). For reference, each monomer is labeled as hCSEA, hCSEB, hCSEC, and hCSED (Fig. 5A), and the location of S346 shown in purple and T355 in gold (Fig. 5B). As can be seen in Fig. 5C and D, snapshots of the trajectories reveal that phosphorylation of these two residues establishes several new electrostatic interactions within and between each monomer of the CSE tetramer. Phosphorylation of S346 induces novel intra- and inter-molecular electrostatic interactions that include but are not limited to pS346A – E381A, K260B (Fig. 5C, inset i); pS346B – E381B, R257A, K260A (Fig. 5C, inset ii); p346D – T336D, K48A (Fig. 5C, inset iii); and pS346C – S218C, T336C, E345C, E381C, K260D (Fig. 5C, inset iv). We also observed several novel intra- and inter-molecular electrostatic interactions formed by phosphorylation of T355. These include but are not limited to pT355A – H55D, R62D, R119A (Fig. 5D, inset i); pT355B – R119B (Fig. 5D, inset ii); pT355D – Q323D, M354D, H356D, S358D, V359D (Fig. 5D, inset iii); and pT355C – R119C, H356C, R62B, N241B (Fig. 5D, inset iv). We next tested whether phosphorylation modulated interactions with CSE and PLP using the inhibitor propargyl glycine (PAG). PAG treatment of HEK transfected with wild type, S346E, or T355E CSE constructs significantly blunted hypoxia and cystine dependent increases in SSP4 fluorescence (Fig. 5E). Conversely, PAG had no effect on hypoxia and cysteine dependent effects with S346A or T355A (Fig. 5F) suggesting that S346 and T355 phosphorylation preferentially conveys tetramer structural organization enabling PLP binding. These data indicate that S346 and T355 may be unique specific targets for CSE enzyme inhibition that requires further study.
Fig. 5.

Molecular dynamics simulations of cystathionine gamma lyase. Three-dimensional structure of CSE as a tetramer and the respective composite monomers generated by AlphaFold. Molecular dynamics simulations of 350 ns were performed with linear constraint solver (LINCS) constraints for all bonds for panel A WT CSE, and panel B CSE with phospho sites S346 (purple), and T355 (gold). Molecular dynamics simulations Phosphorylation of residues S346 and T355 modeled in Pymol using the PyTMs plugin. All were performed using GROMACS 2019 software with the GROMOS 54A7 force field and SPC216 water model. Frames were recorded every 2 ps. Panel C The 300 ns simulation showing extensive intra- and inter-molecular contacts induced by phosphorylation of 346. Each p346 monomer were color coded belongs to and kept the scheme as shown panel A to illustrate which contacts are intra vs inter-molecular contacts. The backbone RMSD was monitored over the production run of each protein to ensure the stability and convergence of the simulated trajectories. p346 leads to a novel interaction between monomer A (p346) and B (K260, R257), monomer B (p346, H217, E345) and monomer A (K260, R257), monomer C (p346, T336) and monomer A (K48), and monomer D (p346, E345, E381) and monomer C (K260). Panel D Intra- and inter-molecular electrostatic interactions formed by phosphorylation of T355. Interactions to pT355A – H55D, R62D, R119A (Fig. 4D, inset i); pT355B – R119B (Fig. 4D, inset ii); pT355D – Q323D, M354D, H356D, S358D, V359D (Fig. 4d, inset iii); and pT355C – R119C, H356C, R62B, N241B (Fig. 4d, inset iv). Panel E HEK293 cells transfected with either Control (Con), wild type CSE (WT), phospho-mimetic mutants S346E or T355E with or without PAG under hypoxia (1 % oxygen) for 30 min and probed for per-polysulfide (SSP4 fluorophore) Panel F HEK293 cells transfected with either Control (Con), wild type CSE (WT), phospho-negative mutants S346A or T355A with or without PAG under hypoxia (1 % oxygen) for 30 min and probed for per-polysulfide (SSP4 fluorophore). All the data are averaged from triplicates from each experiment with at least n = 5. ****P < 0.0001; ***P < 0.0002; *P < 0.01. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
2.5. Hypoxia dependent AMP kinase regulation of CSE phosphorylation and polysulfide formation in endothelial cells
Next, we examined whether conservation of CSE S346 and T355 sequences are similar across different species using the homology search tool OrthologR. Both the CSE phosphorylation sites S346 and T355 are conserved across species, including human, monkey, zebra fish, frog, rat, mouse, C. cerevisiae, D. melanogaster, and C. elegans (Fig. 6A) suggesting a key role for phosphorylation in CSE activity regulation. Assessment of potential protein Serine/Threonine kinases (using Scansite 4) that may phosphorylate CSE at S346 and T355 identified AMP Kinase basophilic Serine/Threonine kinase group motifs (Fig. 6B). Mouse endothelial cells were exposed to hypoxia (0–90 min) and phosphorylation of CSE assessed by immunoblotting with pCSES346 polyclonal antibodies generated by phospho-peptide specific immunization. In parallel, pAMPK was also measured (Fig. 6C). We observed a significant two-fold increase in pCSE at 30, 60 and 90 min, along with a significant increase in pAMPK levels at 30 and 60min (Fig. 6D). We next examined whether AMPK regulates hypoxic CSE phosphorylation and subsequent increases in per-polysulfide using the ATP competitive AMPK inhibitor, dorsomorphin (AMPK-I). Dorsomorphin significantly blunted hypoxia mediated pAMPK and pCSES346 (Fig. 6E and F) that was associated with a significant reduction in hypoxia dependent SSP4 fluorescence (Fig. 6G), as well as SF7 (Suppl. Fig. 5A). Similar effects were observed with HUVECs indicating this pathway is operational in human cells associated with bound sulfide production (Fig. 6H, I and J). We corroborated that hypoxia also increased per-polysulfide SSP4 signal in HUVECs (Suppl. Fig. 5B); however, hypoxia did not significantly increase free H2S SF7 signal (Suppl. Fig. 5C), although AMPK inhibition moderately reduced free H2S SF7 signal (Suppl. Fig. 5D) under hypoxia.
Fig. 6.

Hypoxia induces per-polysulfide via AMPK-mediated phosphorylation of CSE. Panel A Conservation sequence of human CSE at S346 and T355 compared across various species, including human, monkey, Danio rerio (Zebra fish), Xenopus tropicalis (frog), rat, mouse, Saccharomyces cerevisiae, Drosophila melanogaster, and Caenorhabditis elegans. Panel B CSE phospho sites S346 and T355 showing modular signaling domains of protein Serine/Threonine kinases motif groups using Scansite 4. Panel C Representative blots of MAEC treated under hypoxia for 0, 5, 15, 30, 60 and 90 min probed for pCSES346, total CSE, pAMPK, AMPK and GAPDH. Panel D Quantitation of pCSES346 and p-AMPK protein levels, respectively from western blots in Panel A. Panel E Representative blots of MAEC treated with mock or AMPK inhibitor (AMPK-I) Dorsomorphin under hypoxia for 30min. Panel F Quantitation of pCSES346 and pAMPK protein levels, from western blots in Panel E. Panel G MAEC treated with mock or AMPK inhibitor (AMPK-I), Dorsomorphin under hypoxia for 30 min followed by per-polysulfide (SSP4 fluorophore) signal. Panel H Representative blots from HUVEC treated under normoxia or hypoxia or hypoxia + AMPK-I for protein levels of pCSES346, total CSE, pAMPK, AMPK and GAPDH. Panel I quantitation of HUVEC blots for pCSES346 and pAMPK treated with mock or AMPK inhibitor (AMPK-I) Dorsomorphin under hypoxia for 30 min. Panel J HUVECs treated under hypoxia or hypoxia + AMPK-I for 30 min followed by per-polysulfide (SSP4 fluorophore) measurement. Densiometric analysis of p-AMPK and p-CSE346 blots were normalized to total AMPK or total CSE (normalized to GAPDH for protein). All data are averaged from triplicates from each experiment with at least n = 5. ****P < 0.0001; ***P < 0.0002; *P < 0.01.
2.6. AMP kinase isomer inhibition impairs CSE phosphorylation and polysulfide formation in endothelial cells
AMPK is a heterotrimeric protein kinase composed of a catalytic α subunit (α1 or α2) subunit [17,18]. We further investigated the effects of specific AMPK α subunit inhibition on pCSE and per-polysulfide levels using siRNA treatment of mouse aortic endothelial cells. Individual knockdown of AMPKα1 (Fig. 7A) or α2 (Fig. 7B) selectively reduced α1 or α2 mRNA, respectively. We next exposed mouse aortic endothelial cells treated with AMPK siRNAs for α1 or α2 subunits and assessed hypoxic changes by immunoblotting for CSE, AMPK, pAMPK or pCSES346 polyclonal antibodies confirming that siRNA inhibition of AMPK α1 or α2 blunted total and pAMPK while only affecting pCSE346 (Fig. 7C, D and E). Lastly, siRNA inhibition of AMPKα1 or AMPKα2 significantly reduced hypoxia mediated increased per-polysulfide SSP4 fluorescence (Fig. 7F). These data confirm pharmacological inhibitor findings that AMP Kinase activity critically regulates CSE phosphorylation and subsequent sulfane sulfur production.
Fig. 7.

AMPK isomer siRNA reduces hypoxic CSE phosphorylation and per-polysulfide formation. MAEC were treated with si-Con, siAMPKα1 or siAMPKα2 was examined for AMPKα1 and AMPKα2 mRNA expression (Panels A and B). Panel C shows representative blots of pCSES346, total CSE, pAMPK, total AMPK and GAPDH from MAEC treated under hypoxia with siCon, siAMPKα1, or siAMPKα2. Panels D and E respectively show quantitation of pCSES346 and pAMPK protein levels from immunoblots in Panel C. Panel F illustrates SSP4 levels from MAEC transfected with siCon, siAMPKα1, or siAMPKα2 under hypoxia. Densiometric analysis of p-AMPK and p-CSE346 blots were normalized to total AMPK or total CSE (normalized to GAPDH for protein). All data are averaged from triplicates from each experiment with at least n = 5. ****P < 0.0001; ***P < 0.0002; **P < 0.003; *P < 0.01.
2.7. Inhibition of AMP kinase impairs ischemia dependent phosphorylation of CSE, per-polysulfide formation, and blood flow
CSE and H2S regulate vascular dilation and ischemic vascular remodeling involving arteriogenesis and angiogenic properties [7,19]. We examined the effect of tissue ischemia on CSE and AMP kinase phosphorylation using the femoral artery ligation (FAL) model at various time points (0, 3hrs, 24hrs and 5 days) (Fig. 8A). Ischemia increased CSE phosphorylation (pCSES346) rapidly (3 h) in the gastrocnemius muscle that was sustained over 5 days (Fig. 8B). Likewise, ischemic tissue AMP kinase phosphorylation coincided with pCSE beginning at 3 h and lasting over 5 days (Fig. 8B). Next we assessed whether AMPK modulates CSE phosphorylation and activity in vivo, by treating mice with AMPK-I dorsomorphin (10 mg/kg) for 5 days during FAL. Skeletal muscle tissues from non-ischemic (NI), ischemic (Isch) or Isch + AMPK-I groups were probed for pCSE and pAMPK protein levels (Fig. 8C). We found a three-fold increase in pCSES346 and pAMPK protein levels, which was completely blunted with AMPK-I treatment (Fig. 8D). We also evaluated FAL physiological ischemic hindlimb blood flow changes between saline vehicle versus AMPK-I at day 5 (Fig. 8E). Laser speckle blood flow quantification shows that saline vehicle treatment results in a characteristic ∼55–60 % reduction in hind limb blood flow after FAL at day 5 but that AMPK-I treatment resulted in more than a 80 % reduction in hind limb blood flow after FAL (Fig. 8F). Lastly, we examined AMPK-I treatment on FAL plasma per/polysulfide generation using the MBB/HPLC method and found a profound and significant decrease in plasma bound sulfane sulfur with AMPK-I compared to saline vehicle (Fig. 8G).
Fig. 8.

AMPK inhibition reduces CSE phosphorylation, per-polysulfide and ischemic blood flow. Panel A Representative blots of ischemic gastrocnemius muscle tissues from mice subject to the femoral artery ligation (FAL) 0hrs, 3hrs, 24hrs and 5 days probed for pCSES346, total CSE, pAMPK, AMPK and GAPDH. Panel B Graphic representation of the densitometry quantification depicted in Panel A for pCSE346 and p-AMPK protein expression. Panel C Representative western blots were performed from non-ischemic (NI), ischemic (Isch), and ischemic AMPK-I (Isch + AMPK-I) skeletal muscle (SkM) tissues collected at day 5 post femoral artery ligation. Panel D Graphic representation of the densitometry quantification of pCSE346 and p-AMPK protein expression. Panel E Representative angiogram images of hindlimbs showing blood flow (blue color = lowest flow; red color = highest flow) from saline treated post-ligation at day 5 (upper panel; Sal-D5) and AMPK inhibitor treated post-ligation at day 5 (lower panel; AMPK-I-D5). Panel F Graphic representation of the densitometry quantification depicted in Panel E for ischemic limb blood flow. Panel G Plasma sulfide levels, including free/acid labile pools (F/Al) and bound sulfide of pre-FAL, saline (sal) or AMPK-I treated mice. In all densiometric analysis of western blots, p-AMPK and p-CSE346 are normalized to total AMPK or total CSE that are normalized to GAPDH. All the data were representative of at least n = 5. ****P < 0.0001; **P < 0.003; *P < 0.01. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3. Discussion
The current investigation reveals a unique molecular signaling pathway regulating sulfide metabolism involving hypoxia dependent phosphorylation of CSE and formation of sulfane sulfur. We identified serine 346 (S346) and threonine 355 (T355) as two phosphorylation sites, which enhanced CSE activity under cellular hypoxia and tissue ischemic conditions. Molecular modeling revealed these posttranslational modifications likely stabilize tetramer organization and function due to numerous inter- and intramolecular interactions. This was confirmed by specific phospho-negative mutants that remove these modifications which inhibited hypoxia-mediated increase in CSE activity as well as decreased per-polysulfide generation. Whereas phosphomimetic mutant CSE constructs expressed under hypoxia significantly elevate per-polysulfide generation. Importantly, AMPK inhibition abolished hypoxia/ischemia induced CSE phosphorylation and per-polysulfide generation along with impairment of ischemic vascular responses highlighting a crucial role for cardiovascular and likely other pathophysiological responses.
Previous work has reported an increase in CSE expression and subsequent increase in total sulfide levels under tissue ischemia over prolonged time periods [7]; however, underlying molecular mechanisms have remained unknown. We observed a preferential increase in sulfide as bound sulfane sulfur that includes per-polysulfides. Formation of per-polysulfide biochemical species under hypoxia may be an evolutionarily ideal response from a chemical biology perspective. Preferential formation of polysulfides under hypoxia, as opposed to free H2S, likely reflects cellular responses that ensure reactive sulfide availability under conditions (e.g. low tissue pH) that would favor H2S formation and diffusion. Moreover, polysulfides can readily liberate free sulfide, contribute sulfur to metal complexes, serve as potent nucleophilic and electrophilic redox mediators, and are more biologically stable forms of RSS than H2S alone [4,12,20,21]. Apart from CSE, CBS, and MPST; cysteinyl-tRNA synthetases (CARSs) have been reported to produce per-polysulfides [8,22]. Interestingly, no significant role for formation of per-polysulfides was observed under hypoxia with knockdown of CBS, MPST, or CARS2, with only a moderate role for CARS1. This substantiates earlier observations of CSE-mediated sulfide release under hypoxia [7], and identifies CSE as the predominant enzyme producing per-polysulfides in endothelium under hypoxia.
Dietary modifications, such as methionine restriction (MR) has received considerable attention as they can improve metabolism, including insulin sensitivity and longevity [23,24]. Effects of MR are attributed, to increased CSE/H2S signaling that regulates substrates such as cysteine or cystine [24]. CSE plays a critical role under MR with significant beneficial towards improved metabolism with substrate utilization [23]. Additionally, CSE is known to be promiscuous for its substrate utilization that can change with its microenvironment [6]. With a decrease in cellular cysteine levels, CSE can catalyze β-elimination of cysteine persulfides from cystine, to produce H2S and cysteine [23]. Interestingly, we observed that CSE, does in fact utilize cystine or cysteine as substrates apart from cystathionine to produce per-polysulfides under hypoxic conditions. Moreover, production of these sulfane sulfur metabolites appears more abundant with cystine than cysteine or cystathionine under hypoxia when CSE is phosphorylated. Possible presence of low cystathionine and cysteine levels under hypoxia may also favor utilization of cystine as a substrate thus serving as an alternative pathway to salvage intracellular cysteine and H2S. These results indicate that CSE utilization of cystine or cysteine to generate per-polysulfides under hypoxia (rather than H2S) may be a metabolic and redox pathway to maintain cellular function during stress.
CSE is a PLP-dependent enzyme consisting of 393 amino acid residues and one PLP moiety and is arranged as a tetramer [16]. Pyridoxal phosphate is bound in the active site by Lys212 25. Apart from lysine, PLP is anchored by strong hydrogen bonds between the phosphate moieties, including Gly90, Leu91, Thr211 and Arg62, that can contribute towards stability of its binding with the CSE enzyme [25]. Molecular modeling data demonstrates that phosphorylation of CSE significantly changes the molecular dynamics of CSE via electrostatic interactions, including inter- and intramolecular binding between monomers of CSE tetramer via S346 and T355. Interestingly, a significant number of these novel electrostatic interactions involve amino acids known to be functionally relevant for CSE activity. For instance, Arg62 and Lys212 to form a hydrogen bond with the phosphate moiety of PLP known to be functionally obligatory for CSE activity [25,26]. The unstructured loop containing R119 (residues G115 – Y120) forms a two-turn helix upon CSE binding PLP [25]. Furthermore, a recent study posits that the guanidinium moiety of R119 forms a hydrogen bond with the distal α-carboxylate of cystathionine while maintaining contact with the adjacent monomer via a second hydrogen bond with S242 26. H55 is also a part of a loop (residues D28 – S63) that is disordered in the absence of PLP. There is no electron density in the apo structure for residues T355 – V366, indicating that this region is disordered. Although this region does not directly interact with PLP, it is theorized that it could stabilize one side of the PLP binding pocket. For the apo structure, the T355 – V366 region of the CSE:PLP complex is occupied by the M110 – N118 loop [25]. Finally, N241 plays an important role in the inter-molecular stabilization of PLP in the active site of CSE [26]. Supplement video 1shows non-phosphorylated CSE tetramer molecular interactions over 300 ns, whereas supplement videos 2 and 3illustrate phospho-S346 CSE or phospho-T355 CSE molecular interactions, respectively. Molecular model simulations clearly demonstrate that phospho-S346 engages in horizontal intermolecular monomer interactions, whereas phospho-T355 predominantly engages in vertical intra- and intermolecular monomer interactions. Importantly, phosphorylation of S346 and T355 transition the CSE tetramer from an open to closed conformation, particularly within the PLP binding domain. We posit that phosphorylation of S346 and T355 induce changes in side-chain orientations of numerous functionally relevant residues, thereby altering the function of CSE as observed in our biological experiments. These results highlight the magnitude of conformational changes involved via phosphorylation sites (S346 and T355) towards structural stability of CSE tetramer as indicated by molecular modeling simulations and represent alternative molecular targets for enzyme inhibition beyond interfering with PLP binding.
Prevailing understanding of CSE regulation has predominantly focused on transcriptional regulation via Activating Transcription Factor 4 (ATF4) [27] and specificity protein-1 (Sp1) [28] with Sp1 consensus binding sites present in the core promoter region of the human CSE gene [28]. Additionally, the transcription factor, activating transcription factor 4 (ATF4) regulates CSE under stress conditions [27]. Moreover, regulatory mechanisms governing CSE function and its production of per- and polysulfide generation has remained unknown. We have previously reported CSE regulation of flow-dependent vascular remodeling and endothelial activation preferentially involving altered bound sulfane sulfur levels [29]. 17β-estradiol (E2) can enhance endothelial CSE gene expression and H2S release by promoting phosphorylation of CSE via ERα–Sp1 interaction with binding sites in CSE gene promoter, subsequently improving endothelial function [30,31]. However, this study is predominantly applicable to females and doesn't identify how S56 effects CSE enzyme function. Bibli et al. has also reported that sulfide levels are reduced under disturbed shear stress via phosphorylation of serine 377 thereby inhibiting CSE, due to increased IL-1β 32. However, these mechanisms explain the inhibitory regulation of CSE alone. Our data reveals increased CSE enzyme function under hypoxia via S346 and T355 phosphorylation. Interestingly, our results from LC/MS analysis of CSE posttranslational modification found a significant reduction in phospho peptides CSE[365–395] (containing S377) in comparison to CSE[334–364] (containing S346 and T355). This suggests an inhibition or reduction of phosphorylation of S377, a negative regulator of CSE [32], under hypoxia that requires further study.
AMP-activated protein kinase (AMPK) is a key regulatory molecule for endothelial function, redox homeostasis, and importantly, hypoxia and ischemia/reperfusion injury [33]. AMPK has been reported to mediate myocardial protection from I/R injury [34]. AMPK prevents apoptosis and reduces oxidative stress and inflammation upon I/R injury, which is mediated via AMPK phosphorylation [35]. Specifically, AMPK protects from myocardial I/R injury via phosphorylation of AMPK at Thr172 in ischemic heart tissues [36]. While H2S -mediated induction of AMPK and subsequent pharmacological actions has been previously reported [37], AMPK regulation of CSE and subsequent sulfide metabolite levels have not been known. We demonstrated that AMPK/pAMPK can regulate CSE by increasing its activity via phosphorylation (S346) under hypoxia/ischemia conditions. This increase in CSE phosphorylation subsequently induced per-polysulfide generation, which is significantly blunted upon inhibition of AMPK using dorsomorphin or siRNA. AMPK is a heterotrimeric complex of catalytic α-subunits, regulatory β and γ-subunits. AMPK catalytic α1 and α2 isoforms are activated in cardiac ischemia irrespective of regulatory subunits [38]. AMPK can be activated by LKB1 and CaMKKβ pathways that elicits protective and adaptive mechanisms to various cellular stress conditions in the endothelial cells, including hypoxia [39,40]. However, the molecular regulation and the roles of specific AMPK subunit isoforms is not well defined. Our data demonstrates that under hypoxia, molecular inhibition of either AMPKα1 or AMPKα2 in endothelial cells significantly blunts AMPK activity, hypoxia mediated CSE S346 phosphorylation, and subsequent per-polysulfide production. Importantly, inhibition of AMPK significantly impairs ischemic vascular blood flow in mouse hindlimbs in a model of chronic ischemia that directly involves altered CSE phosphorylation and bound sulfane sulfur production. These results highlight a critical role of AMPK in regulation of CSE phosphorylation and pAMPK in regulating ischemic vascular blood flow. It is conspicuous from our data that CSE S346 phosphorylation remains elevated even at day 5 in ischemic hindlimb, which corroborates with elevated sulfide levels as observed previously and in the current manuscript [7]. This sustained elevation of CSE S346 could be due to stress conditions as an adaptive process. Elevation of per/polysulfide levels endure altered redox environment that could involve both metabolic, and oxygen induced stress. However, future studies are needed to elucidate how both these (or perhaps other) stress conditions may influence in regulating phosphorylation states.
In the family of serine-threonine kinases, AMPK, PKA, PKC, and PKG play key role in catalyzing the phosphorylation of proteins [41,42]. Considering the possibility of CSE being a substrate of these serine-threonine kinases, we performed in silico analysis using Scansite 4.0 tool which detects short protein sequence motifs that are phosphorylated by protein Ser/Thr- or Tyr-kinases. Besides AMPK, Scansite 4.0 analysis didn't reveal a prominent motif for any other kinases. Future studies aiming at how the gain or loss of these kinases affects enzyme function may pave the way to delineating whether these kinases are key modulators of CSE other than AMPK. One of the limitations of our study is that we could not characterize the other phosphorylation site, CSE T355, and its regulation by AMPK or other kinases. We attempted to raise an antibody against pCSE355 but have been currently unsuccessful. We suspect this may be due to locational constraints of T355 residue in protein structure/or antigenicity. Together, these results elucidate molecular mechanisms of increased CSE activity and per-polysulfide formation for tissue and cellular ischemic responses. Implications for this signaling pathway and mechanism are many with immediate relevancy to the regulation of tissue growth, metabolism, adaptation responses, carcinogenesis pathways, and reno-cardiovascular diseases.
4. Methods
4.1. Chemicals and reagents
Sulfane Sulfur Probe 4 (SSP4) and sodium trisulfide (Na2S3) were from Dojindo Molecular Technologies, Inc., MD, USA. Anhydrous sodium sulfide (Na2S) was purchased from Alfa‐Aesar Inc. Diallyl trisulfide (DATS) was procured from Sigma-Aldrich, CA, USA. SF-7 probe was purchased from Cayman Chemicals, MI, USA. β-(4-Hydroxyphenyl)ethyl iodoacetamide (HPE-IAM) obtained from Chem-Impex. The anti-CSE antibody was from Proteintech (IL, USA), anti-CBS, anti-3MST were from Abcam, MA, USA. All secondary fluorophore‐labeled antibodies were obtained from Jackson Immunoresearch Inc (West Grove, PA, USA). Chemicals and tissue culture reagents were obtained from Sigma unless otherwise noted.
4.2. Cell culture and treatments
Human embryonic kidney 293 cells (HEK) were purchased from ATCC, Virginia, USA and Human umbilical vein endothelial cells (HUVECs) were obtained from Lifeline Cell Technology, CA, USA (Cat# FC-0044). Mouse aortic endothelial cells were isolated in our COBRE molecular core and cultured in our laboratory. HUVECs were cultured in VascuLife® Basal Medium (Cat# LM-0002) supplemented with the appropriate LifeFactors® Kit (Cat# LL-0003). All cells were grown in tissue culture flasks under normoxic conditions at 37 °C, 5 % CO2, and 21 % O2. All media was changed every 2–3 days. Cells were transfected with transfection media (Lipofectamine 2000).
For hypoxia treatment, the cells were incubated in the hypoxic chamber (5 % CO2, 37 °C, 1 % O2) for 30min. HUVECs from passages 2–4 was used in the experiments.
4.3. Site-directed mutagenesis of hCSE
Phosphorylation mutants of hCSE of WT, constitutively active phospho negative alanine mutants S346A or T355A; phosphomimetic mutants with glutamate substitution, S346E or T355E. For the generation of hCSE alanine or glutamate mutants, the Site-Directed Mutagenesis kit from Agilent Technologies (Catalog #200522) was utilized. We performed PCR using two synthetic oligonucleotide primers, designed to incorporate the desired mutation. After thermal cycling, DPN I digestion was employed to selectively amplify PCR products containing the mutation. Subsequently, the nicked vector DNA, now carrying the desired mutations, was transformed into competent cells. Plasmids were extracted from the resulting colonies and subjected to Sanger sequencing, thereby confirming the presence of the intended mutants. These mutated plasmids have been transfected into either HEKs, MAECS or HUVECs for various assays as mentioned in the manuscript.
4.4. Measurement of biological pools of H2S
H2S was measured using monobromobimane by RP-HPLC as we previously reported and described in detail elsewhere [43]. Briefly, plasma or tissue samples were collected and stabilized in vials containing 100 mM Tris-HCl buffer (pH 9.5, 0.1 mM DTPA) in a ratio of 1:5 (v/v) and snap-frozen until further analysis. Samples were derivatized with monobromobimane (MBB) and analyzed using a reversed-phase high-performance liquid chromatography (RP-HPLC) system with an Agilent Eclipse XDB-C18 column (5 μm, 80 Å, 4.6 mmx250 mm) for sulfide-dibimane fluorescence detection (excitation: 390 nm; emission: 475 nm).
4.5. Measurement of CSE activity
CSE activity was measured as we previously described [7]. Briefly, samples were incubated with 2 mM cystathionine, 0.25 mM pyridoxal 5′-phosphate in 100 mM Tris-HCl buffer (pH 8.3) for 30 min at 37 °C. 20 % Trichloroacetic acid was added into reaction mixture. After centrifugation, the supernatant was mixed with 2 % ninhydrin reagent and incubated for 5 min at 105 °C then quickly cooled to 4 °C. Samples were then mixed with 97 % ethanol and read at 455 nm using a spectrophotometer (Biotek Inc.). CSE activity was assessed by cystathionine consumption and enzyme activity expressed as fold change calculated from nanomoles of cystathionine consumed per mg of total protein.
4.6. Fluorescent detection of polysulfide metabolite
Polysulfide was detected using a fluorescent probe SSP4 as described earlier [44]. SSP4 was dissolved in DMSO to obtain a 1 mM stock solution. CTAB, a surfactant was dissolved in ethanol to yield a 5 mM stock solution. CTAB and SSP4 (5 μM) were dissolved in HBSS buffer and added to the samples for a total of 100 μl in a black polystyrene, flat-bottomed clear 96-well plate and incubated in the dark at room temperature for 20 min. Samples were measured on a TECAN fluorescence spectrophotometer (emission 525 nm: excitation 485 nm).
4.7. Identification of phosphorylation by LC-MS/MS
Human CSE from HEK sample cells was performed SDS-PAGE, and then stained with Imperial Protein Stain. The Gel lanes were cut into 2 mm × 2 mm cubes. The cubes underwent trypsin digestion, followed by disulfide bond reduction and cysteine alkylation with dithiothreitol and iodoacetamide, respectively. LC-MS/MS analysis was conducted by a 3000 RSLCnano system coupled via an EasySpray source to an Orbitrap Exploris 480 mass spectrometer as mentioned previously [45]. The digested peptides (5.0 μl) were loaded onto a trap column (PepMap C18, 2 cm × 100 μm, 100 Å) at a flow rate of 20 μl/min using 0.1 % formic acid and separated on an analytical column (EasySpray 50 cm × 75 μm, C18 1.9 μm, 100 Å) with a flow rate of 300 nl/min with a linear gradient of 5–45 % solvent B (100 % ACN, 0.1 % formic acid) over a 120 min gradient. Both precursor and fragment ions were acquired in the Orbitrap mass analyzer. Precursor ions were acquired in m/z range of 375–1500 with a resolution of 120,000 (at m/z 200). Precursor fragmentation was carried out using the higher-energy collisional dissociation method using normalized collision energy (NCE) of 32. The fragment ions were acquired at a resolution of 150,000 (at m/z 200). The scans were arranged in top-speed method with 3 s cycle time between MS and MS/MS. Ion transfer capillary voltage were maintained at 2.1 kV. The raw mass spectrometry data were analyzed using Proteome Discover (version 2.5, Thermo Fisher Scientific) software package with SequestHT using species-specific fasta database and the Percolator peptide validator. Cysteine alkylation was set as a fixed modification. Phosphorylation on serine\threonine\tyrosine residues, and deamidation of asparagine residues were selected as variable modifications.
4.8. Molecular dynamics simulations
The three-dimensional structure of cystathionine gamma lyase used for the molecular dynamics simulations were generated by using AlphaFold to predict the structural elements missing from the previously solved crystal structure (PBD: 3ELP) [46]. Phosphorylation of residues S346 and T355 was modeled in Pymol using the PyTMs plugin [47]. All molecular dynamics simulations were performed using GROMACS 2019 software with the GROMOS 54A7 force field [48] and SPC216 water model. Specifically, each protein was solvated with 72,319 SPC216 explicit water molecules and places in the center of a cubic box of 136 × 136 × 136 Å [47]. Each system was neutralized by adding the appropriate number of Na+ ions. Energy minimization of each structure was followed by a two-step equilibration (i.e., NVT equilibration followed by NPT equilibration). The temperature of each system was controlled through velocity rescaling [49] at 300 K with a time constant of 0.1 ps. The pressure of each system was controlled using the Parrinello-Rahman barostat [50] and set to 1 bar. The particle mesh Ewald algorithm [51] was used to calculate long-range electrostatics while a cutoff of 1.0 nM was used for short-range electrostatics and van der Waals’ interactions. Molecular dynamics simulations of 350 ns were performed for WT, S346-PO4, and T355-PO4 with linear constraint solver (LINCS) constraints for all bonds [52]. Frames were recorded every 2 ps. The backbone RMSD was monitored over the production run of each protein to ensure the stability and convergence of the simulated trajectories.
4.9. Creation of pCSES346 antibody
Polyclonal antibodies to specific phosphorylation site S346 was produced by Genscript, USA Inc. The antibody against the consensus CTH substrate was raised against the following synthetic peptide antigen CAESLGGFE[pSer]LAE. The approach used for phospho antibodies generation was consecutive iterations of subtractive purification using non phosphopeptide resin followed by affinity purification by removing unmodified proteins and/or low sequence complexity epitopes for strong specificity of the phospho-antibody. Briefly, rabbits were immunized with a synthetic phosphorylated peptide (KLH (keyhole limpet hemocyanin) coupled) corresponding to residues surrounding phosphorylation sites. Antibodies are then purified by to isolate the IgG antibody fraction. Affinity chromatography was performed using peptides coupled to SulfoLink resin. Both phosphopeptide-containing resin and the corresponding non-phosphopeptide resin were prepared. Following two rounds of subtractive purification using the non-phosphopeptide resin; protein A eluate was incubated with non-phosphopeptide resin by rotation in a sealed column at room temperature for 1 h to remove antibodies reactive with the non-phospho version of the protein antigen. The column was drained, and the flow-through (containing the desired antibody) was incubated with fresh non-phosphopeptide resin. The flow-through from this second subtractive step was next purified by incubation with phosphopeptide resin. After the phospho-peptide column was washed twice with PBS, phospho-specific antibody (bound to the resin) was eluted with 0.1 m glycine, pH 2.7, and pooled fractions were neutralized with 1 m Tris-HCl, pH 9.5 (∼1–2% of the fraction volume). The eluted phospho-specific antibody was then dialyzed overnight in PBS at 4 °C.
4.10. Western blot analysis
Cells or mouse tissues were homogenized in a lysis solution (Thermofisher Scientific Inc.), containing a protease inhibitor cocktail (Roche, Indianapolis, IN), and phosphatase inhibitor cocktail type I and II (Sigma, Saint Louis, MO). Homogenates were centrifuged at 500×g for 15 min and supernatants were collected. Protein concentrations were analyzed using the Bradford protein assay (BioRad, Hercules, CA). Proteins were separated using 10 % SDS-PAGE (mini or midi Bio-Rad, Hercules, CA) and transferred onto PVDF membranes, and incubated with antibodies against CSE (Cat# 12217-1-AP, Fisher Scientific), total AMPKα (Cat# 5832S), p-AMPKα (Cat#2535S) and α/β-Tubulin (Cat#2148) from Cell Signaling Technology. Chemiluminescent bands were detected and quantified using NIH Image J software.
4.11. Quantitative PCR
Tissue samples were stored in TRIzol reagent (Thermo Fisher Scientific Inc., Waltham, MA, USA) and RNA was isolated as previously reported [53]. RNA concentration was evaluated and purity with a typical OD260 to OD280 ratio of RNA sample was approximately 2.0. cDNA was synthesized using iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA), from 1 μg of total RNA. Quantitative PCR reactions were performed using the universal SYBR Green Supermix (Bio-Rad, CA, USA) on a CFX96 thermal cycler with Bio-Rad CFX Manager software (Bio-Rad, Hercules, CA, USA). The mean threshold cycle (Ct) values were plotted against the logarithm of the cDNA dilution factor. Quantitative PCR primers for genes, including GAPDH, CSE, CBS, AMPKα1, AMPKα2 was used in this study.
4.12. Mouse hindlimb ischemia model and treatment routes
Twelve-week-old male C57BL/6J mice purchased from Jackson Laboratory were used in this study as reported earlier [7]. Mice were housed at the Louisiana State University Health Sciences Center-Shreveport animal resources facility, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. All animal studies were approved by the LSU Institutional Animal Care and Use Committee (LSU IACUC Protocol # P-21-010) and in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. Chronic hindlimb ischemia was induced in 12–16-week-old male C57BL6/J mice, as we have reported previously [7]. After anesthesia with isoflurane (2–3%), the left femoral artery was dissected, separated, ligated and excised distal to profunda femoris artery to create the hindlimb ischemia model. Mice were randomly assigned to different experimental groups by one investigator and were treated and evaluated by a second blinded investigator. AMPK inhibitor, Dorsomorphin (10 mg/kg) was administered i. p. once a day during the length of the study.
4.13. Novadaq SPY imaging analysis
The SPY imaging device (Novadaq Technologies) was used to quantify collateral vessel perfusion, as we have previously described [13]. Briefly, a bolus injection of 30 μl indocyanine green (IC-Green, Akorn Pharmaceutical, Lake Forest, IL) was administered retro-orbitally, and angiograms were captured by an array of light-emitting diodes at a wavelength of 806 nm and recorded for 1 min. Angiograms were taken before ligation on day 0, and post-ligation on day 5 and the percent change in blush rates were calculated as mentioned [13].
4.14. Statistics
All statistical analyses were performed using GraphPad Prism 9 software. Numerical values were shown as mean ± SEM. All data were tested for normality using the Kolmogorov‐Smirnov test, and the data have passed the normality assumption Comparisons of two groups were analyzed using Student's t-test (two tailed) and post-hoc Bonferroni adjustment has been performed. For more than two groups we have performed a two-way ANOVA followed by post-hoc Bonferroni adjustment. We have also performed a post-hoc Tukey's test. P ≤ 0.05 was considered statistically significant. The nonsignificant difference between samples is indicated as “ns”; P values of significant differences are shown in the graphs.
Declaration of competing interest
C.G.K., G.K·K., S.A., and X.S. have a provisional patent on CSE phosphorylation mutants and uses.
Acknowledgements
This work was supported by an Institutional Development Award (IDeA) from the National Institutes of General Medical Sciences of the NIH under grant number P20GM121307 and NHLBI grant HL 149264 to C.G.K; NHBLI HL098435, HL133497, and HL14115 to A.W.O.; NHBLI HL122354, HL145753, HL145753-01S1, and HL145753-03S1 to M.S.B; and P20GM121307 to G.K.K.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2023.102949.
Appendix A. Supplementary data
The following are the Supplementary data to this article.Suppl. Figure 1. CSE role in sulfide and per-polysulfide. Panel A Hydrogen sulfide (SF7 fluorophore) signal in HEK293 cells with either Control (Con), 100 μM Na2S or Na2S3 treatments. Panel B Per-polysulfide levels (SSP4) in HEK293 cells with either Control (Con), 100 μM DATS or Na2S3 treatments. Panel C Hydrogen sulfide (SF7) fold change in HEK293 cells treated under normoxia or hypoxia. Panel D Hydrogen sulfide (SF7) levels in MAECs transfected with either mock, CSE, CBS, MPST, CARS-1 or CARS-2 siRNAs and then respectively were probed with SF7 under hypoxic conditions. Panels E-I CSE, CBS, MPST, CARS1 and CARS2 mRNA expressions; Panels J-N Representative western blots and quantitation of protein levels of CSE, CBS, MPST, CARS1 and CARS2, following transfection of HEK293 cells with respective siRNA. All the data were averaged from triplicates from each experiment with at least n = 5. ****P < 0.0001; *P < 0.01.Suppl. Figure 2. CSE expression profiles in HEK cells. Panel A shows blots of Flag, CSE and GAPDH protein levels from HEKs transfected with vehicle (Veh), WT, or phospho-negative mutants, S346A or T355A. Panel B illustrates Flag, CSE, and GAPDH levels from HEK transfected with vehicle (Veh), WT, or phospho-mimetics S346E or T355E. All data are averaged from triplicates from each experiment with at least n = 3. ****P < 0.0001Suppl. Figure 3. Sulfide levels in normoxia with phospho-negative mutants. Panels A-C show SSP4 per/polysulfide signal in HEK cells transfected with either Control (Con), wild type CSE (WT), phospho-negative mutant S346A or T355A under normoxia with cysteine, cystine, or cystathionine as respective substrates. Panels D-F show SF7 hydrogen sulfide signal in HEK cells transfected with either Control (Con), wild type CSE (WT), phospho-negative mutant S346A or T355A under normoxia with cysteine, cystine, or cystathionine as respective substrates. All data were averaged from triplicates from each experiment with at least n = 5. ****P < 0.0001; **P < 0.003; *P < 0.01.Suppl. Figure 4. Sulfide levels in normoxia with phospho-mimetic mutants. Panels A-C show SSP4 per/polysulfide signal in HEK cells transfected with either Control (Con), wild type CSE (WT), phospho-mimetic mutant S346E or T355E under normoxia with cysteine, cystine, or cystathionine as respective substrates. Panels D-F show SF7 hydrogen sulfide signal in HEK cells transfected with either Control (Con), wild type CSE (WT), phospho-mimetic mutant S346E or T355E under normoxia with cysteine, cystine, or cystathionine as respective substrates. All data were averaged from triplicates from each experiment with at least n = 5. ****P < 0.0001; **P < 0.003; *P < 0.01Suppl. Figure 5. AMPK regulation of CSE and per-polysulfide in endothelial cells. Panel A SF7 fold change in MAECs treated with mock or AMPK-I. Panel B SSP4 levels from HUVECs under normoxia vs hypoxia. Panel C SF7 levels from HUVECs under normoxia vs hypoxia. Panel D SF7 levels from HUVECs under hypoxia vs hypoxia + AMPK-I.Suppl. Figure 6. Representative whole blots of pCSES346 (Panel A) and total CSE (Panel A) from MAECs treated under hypoxia for 0, 5, 15, 30, 60 and 90 min.
Data availability
Data will be made available on request.
References
- 1.Kimura H. Hydrogen sulfide and polysulfides as signaling molecules. Proc. Jpn. Acad. B Phys. Biol. Sci. 2015;91:131–159. doi: 10.2183/pjab.91.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kolluru G.K., Shackelford R.E., Shen X., Dominic P., Kevil C.G. Sulfide regulation of cardiovascular function in health and disease. Nat. Rev. Cardiol. 2023;20:109–125. doi: 10.1038/s41569-022-00741-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polhemus D.J., Lefer D.J. Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circ. Res. 2014;114:730–737. doi: 10.1161/circresaha.114.300505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fukuto J.M., Ignarro L.J., Nagy P., Wink D.A., Kevil C.G., Feelisch M., Cortese-Krott M.M., Bianco C.L., Kumagai Y., Hobbs A.J., et al. Biological hydropersulfides and related polysulfides - a new concept and perspective in redox biology. FEBS Lett. 2018;592:2140–2152. doi: 10.1002/1873-3468.13090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paul B.D., Snyder S.H. Protein sulfhydration. Methods Enzymol. 2015;555:79–90. doi: 10.1016/bs.mie.2014.11.021. [DOI] [PubMed] [Google Scholar]
- 6.Banerjee R. Catalytic promiscuity and heme-dependent redox regulation of H2S synthesis. Curr. Opin. Chem. Biol. 2017;37:115–121. doi: 10.1016/j.cbpa.2017.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kolluru G.K., Bir S.C., Yuan S., Shen X., Pardue S., Wang R., Kevil C.G. Cystathionine γ-lyase regulates arteriogenesis through NO-dependent monocyte recruitment. Cardiovasc. Res. 2015;107:590–600. doi: 10.1093/cvr/cvv198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ida T., Sawa T., Ihara H., Tsuchiya Y., Watanabe Y., Kumagai Y., Suematsu M., Motohashi H., Fujii S., Matsunaga T., et al. vol. 111. 2014. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling; pp. 7606–7611. (Proceedings of the National Academy of Sciences of the United States of America). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akaike T., Ida T., Wei F.Y., Nishida M., Kumagai Y., Alam M.M., Ihara H., Sawa T., Matsunaga T., Kasamatsu S., et al. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat. Commun. 2017;8:1177. doi: 10.1038/s41467-017-01311-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moutiez M., Aumercier M., Teissier E., Parmentier B., Tartar A., Sergheraert C. Reduction of a trisulfide derivative of glutathione by glutathione reductase. Biochem. Biophys. Res. Commun. 1994;202:1380–1386. doi: 10.1006/bbrc.1994.2083. [DOI] [PubMed] [Google Scholar]
- 11.Barayeu U., Schilling D., Eid M., Xavier da Silva T.N., Schlicker L., Mitreska N., Zapp C., Gräter F., Miller A.K., Kappl R., et al. Hydropersulfides inhibit lipid peroxidation and ferroptosis by scavenging radicals. Nat. Chem. Biol. 2023;19:28–37. doi: 10.1038/s41589-022-01145-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu Z., Khodade V.S., Chauvin J.-P.R., Rodriguez D., Toscano J.P., Pratt D.A. Hydropersulfides inhibit lipid peroxidation and protect cells from ferroptosis. J. Am. Chem. Soc. 2022;144:15825–15837. doi: 10.1021/jacs.2c06804. [DOI] [PubMed] [Google Scholar]
- 13.Bir S.C., Kolluru G.K., McCarthy P., Shen X., Pardue S., Pattillo C.B., Kevil C.G. Hydrogen sulfide stimulates ischemic vascular remodeling through nitric oxide synthase and nitrite reduction activity regulating hypoxia-inducible factor-1alpha and vascular endothelial growth factor-dependent angiogenesis. J. Am. Heart Assoc. 2012;1 doi: 10.1161/JAHA.112.004093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banerjee R. Catalytic promiscuity and heme-dependent redox regulation of H(2)S synthesis. Curr. Opin. Chem. Biol. 2017;37:115–121. doi: 10.1016/j.cbpa.2017.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kasamatsu S., Ida T., Koga T., Asada K., Motohashi H., Ihara H., Akaike T. High-precision sulfur metabolomics innovated by a new specific probe for trapping reactive sulfur species. Antioxidants Redox Signal. 2021;34:1407–1419. doi: 10.1089/ars.2020.8073. [DOI] [PubMed] [Google Scholar]
- 16.Messerschmidt A., Worbs M., Steegborn C., Wahl M.C., Huber R., Laber B., Clausen T. Determinants of enzymatic specificity in the Cys-Met-metabolism PLP-dependent enzymes family: crystal structure of cystathionine gamma-lyase from yeast and intrafamiliar structure comparison. Biol. Chem. 2003;384:373–386. doi: 10.1515/bc.2003.043. [DOI] [PubMed] [Google Scholar]
- 17.Banko M.R., Allen J.J., Schaffer B.E., Wilker E.W., Tsou P., White J.L., Villén J., Wang B., Kim S.R., Sakamoto K., et al. Chemical genetic screen for AMPKα2 substrates uncovers a network of proteins involved in mitosis. Mol. Cell. 2011;44:878–892. doi: 10.1016/j.molcel.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schaffer B.E., Levin R.S., Hertz N.T., Maures T.J., Schoof M.L., Hollstein P.E., Benayoun B.A., Banko M.R., Shaw R.J., Shokat K.M., Brunet A. Identification of AMPK phosphorylation sites reveals a network of proteins involved in cell invasion and facilitates large-scale substrate prediction. Cell Metabol. 2015;22:907–921. doi: 10.1016/j.cmet.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pardue S., Kolluru G.K., Shen X., Lewis S.E., Saffle C.B., Kelley E.E., Kevil C.G. Hydrogen sulfide stimulates xanthine oxidoreductase conversion to nitrite reductase and formation of NO. Redox Biol. 2020;34 doi: 10.1016/j.redox.2020.101447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu B., Yang X., Yuan Z., Wang B. Prodrugs of sulfide and persulfide species: implications in their different pharmacological activities. Curr. Opin. Chem. Biol. 2023;75 doi: 10.1016/j.cbpa.2023.102329. [DOI] [PubMed] [Google Scholar]
- 21.Noguchi N., Saito Y., Niki E. Actions of thiols, persulfides, and polysulfides as free radical scavenging antioxidants. Antioxidants Redox Signal. 2023 doi: 10.1089/ars.2022.0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akaike T., Ida T., Wei F.-Y., Nishida M., Kumagai Y., Alam M.M., Ihara H., Sawa T., Matsunaga T., Kasamatsu S., et al. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat. Commun. 2017;8:1177. doi: 10.1038/s41467-017-01311-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeitner T.M., Azcona J.A., Ables G.P., Cooke D., Horowitz M.C., Singh P., Kelly J.M., Cooper A.J.L. Cystine rather than cysteine is the preferred substrate for β-elimination by cystathionine γ-lyase: implications for dietary methionine restriction. GeroScience. 2023 doi: 10.1007/s11357-023-00788-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hine C., Harputlugil E., Zhang Y., Ruckenstuhl C., Lee B.C., Brace L., Longchamp A., Treviño-Villarreal J.H., Mejia P., Ozaki C.K., et al. Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell. 2015;160:132–144. doi: 10.1016/j.cell.2014.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun Q., Collins R., Huang S., Holmberg-Schiavone L., Anand G.S., Tan C.H., van-den-Berg S., Deng L.W., Moore P.K., Karlberg T., Sivaraman J. Structural basis for the inhibition mechanism of human cystathionine gamma-lyase, an enzyme responsible for the production of H(2)S. J. Biol. Chem. 2009;284:3076–3085. doi: 10.1074/jbc.M805459200. [DOI] [PubMed] [Google Scholar]
- 26.Le Corre L., Padovani D. Mechanism-based and computational modeling of hydrogen sulfide biogenesis inhibition: interfacial inhibition. Sci. Rep. 2023;13:7287. doi: 10.1038/s41598-023-34405-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dickhout J.G., Carlisle R.E., Jerome D.E., Mohammed-Ali Z., Jiang H., Yang G., Mani S., Garg S.K., Banerjee R., Kaufman R.J., et al. Integrated stress response modulates cellular redox state via induction of cystathionine γ-lyase: cross-talk between integrated stress response and thiol metabolism. J. Biol. Chem. 2012;287:7603–7614. doi: 10.1074/jbc.M111.304576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang G., Pei Y., Teng H., Cao Q., Wang R. Specificity protein-1 as a critical regulator of human cystathionine gamma-lyase in smooth muscle cells. J. Biol. Chem. 2011;286:26450–26460. doi: 10.1074/jbc.M111.266643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan S., Yurdagul A., Jr., Peretik J.M., Alfaidi M., Al Yafeai Z., Pardue S., Kevil C.G., Orr A.W. Cystathionine γ-lyase modulates flow-dependent vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 2018;38:2126–2136. doi: 10.1161/atvbaha.118.311402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu X., Yan Q., Liu X., Li P., Li X., Chen Y., Simoncini T., Liu J., Zhu D., Fu X. 17β-Estradiol nongenomically induces vascular endothelial H(2)S release by promoting phosphorylation of cystathionine γ-lyase. J. Biol. Chem. 2019;294:15577–15592. doi: 10.1074/jbc.RA119.008597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fu X., Zhou K., Gao Q., Zheng S., Chen H., Li P., Zhang Y., Suo K., Simoncini T., Wang T. 17β-estradiol attenuates atherosclerosis development: the possible role of hydrogen sulfide. Int. J. Cardiol. 2013;167:1061–1063. doi: 10.1016/j.ijcard.2012.10.071. [DOI] [PubMed] [Google Scholar]
- 32.Bibli S.I., Hu J., Sigala F., Wittig I., Heidler J., Zukunft S., Tsilimigras D.I., Randriamboavonjy V., Wittig J., Kojonazarov B., et al. Cystathionine γ lyase sulfhydrates the RNA binding protein human antigen R to preserve endothelial cell function and delay atherogenesis. Circulation. 2019;139:101–114. doi: 10.1161/circulationaha.118.034757. [DOI] [PubMed] [Google Scholar]
- 33.Rodríguez C., Muñoz M., Contreras C., Prieto D. AMPK, metabolism, and vascular function. FEBS J. 2021;288:3746–3771. doi: 10.1111/febs.15863. [DOI] [PubMed] [Google Scholar]
- 34.Russell R.R., 3rd, Li J., Coven D.L., Pypaert M., Zechner C., Palmeri M., Giordano F.J., Mu J., Birnbaum M.J., Young L.H. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J. Clin. Invest. 2004;114:495–503. doi: 10.1172/jci19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paskeh M.D.A., Asadi A., Mirzaei S., Hashemi M., Entezari M., Raesi R., Hushmandi K., Zarrabi A., Ertas Y.N., Aref A.R., et al. Targeting AMPK signaling in ischemic/reperfusion injury: from molecular mechanism to pharmacological interventions. Cell. Signal. 2022;94 doi: 10.1016/j.cellsig.2022.110323. [DOI] [PubMed] [Google Scholar]
- 36.Shibata R., Sato K., Pimentel D.R., Takemura Y., Kihara S., Ohashi K., Funahashi T., Ouchi N., Walsh K. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat. Med. 2005;11:1096–1103. doi: 10.1038/nm1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang M., Tang W., Zhu Y.Z. An update on AMPK in hydrogen sulfide pharmacology. Front. Pharmacol. 2017;8:810. doi: 10.3389/fphar.2017.00810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J., Coven D.L., Miller E.J., Hu X., Young M.E., Carling D., Sinusas A.J., Young L.H. Activation of AMPK alpha- and gamma-isoform complexes in the intact ischemic rat heart. Am. J. Physiol. Heart Circ. Physiol. 2006;291:H1927–H1934. doi: 10.1152/ajpheart.00251.2006. [DOI] [PubMed] [Google Scholar]
- 39.Nagata D., Mogi M., Walsh K. AMP-activated protein kinase (AMPK) signaling in endothelial cells is essential for angiogenesis in response to hypoxic stress. J. Biol. Chem. 2003;278:31000–31006. doi: 10.1074/jbc.M300643200. [DOI] [PubMed] [Google Scholar]
- 40.Zou M.H., Wu Y. AMP-activated protein kinase activation as a strategy for protecting vascular endothelial function. Clin. Exp. Pharmacol. Physiol. 2008;35:535–545. doi: 10.1111/j.1440-1681.2007.04851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Domańska-Janik K. Protein serine/threonine kinases (PKA, PKC and CaMKII) involved in ischemic brain pathology. Acta Neurobiol. Exp. 1996;56:579–585. doi: 10.55782/ane-1996-1163. [DOI] [PubMed] [Google Scholar]
- 42.Liu C., Ke P., Zhang J., Zhang X., Chen X. Protein kinase inhibitor peptide as a tool to specifically inhibit protein kinase A. Front. Physiol. 2020;11 doi: 10.3389/fphys.2020.574030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shen X., Kolluru G.K., Yuan S., Kevil C.G. Measurement of H2S in vivo and in vitro by the monobromobimane method. Methods Enzymol. 2015;554:31–45. doi: 10.1016/bs.mie.2014.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shieh M., Ni X., Xu S., Lindahl S.P., Yang M., Matsunaga T., Flaumenhaft R., Akaike T., Xian M. Shining a light on SSP4: a comprehensive analysis and biological applications for the detection of sulfane sulfurs. Redox Biol. 2022;56 doi: 10.1016/j.redox.2022.102433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Islam M.Z., Shen X., Pardue S., Kevil C.G., Shackelford R.E. The ataxia-telangiectasia mutated gene product regulates the cellular acid-labile sulfide fraction. DNA Repair. 2022;116 doi: 10.1016/j.dnarep.2022.103344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun Q., Collins R., Huang S., Holmberg-Schiavone L., Anand G.S., Tan C.-H., van-den-Berg S., Deng L.-W., Moore P.K., Karlberg T., Sivaraman J. Structural basis for the inhibition mechanism of human cystathionine γ-lyase, an enzyme responsible for the production of H2S. J. Biol. Chem. 2009;284:3076–3085. doi: 10.1074/jbc.M805459200. [DOI] [PubMed] [Google Scholar]
- 47.Warnecke A., Sandalova T., Achour A., Harris R.A. PyTMs: a useful PyMOL plugin for modeling common post-translational modifications. BMC Bioinf. 2014;15:370. doi: 10.1186/s12859-014-0370-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang W., Lin Z., van Gunsteren W.F. Validation of the GROMOS 54A7 force field with respect to β-peptide folding. J. Chem. Theor. Comput. 2011;7:1237–1243. doi: 10.1021/ct100747y. [DOI] [PubMed] [Google Scholar]
- 49.Bussi G., Donadio D., Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126 doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 50.Parrinello M., Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 1981;52:7182–7190. doi: 10.1063/1.328693. [DOI] [Google Scholar]
- 51.Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. doi: 10.1063/1.470117. [DOI] [Google Scholar]
- 52.Hess B., Bekker H., Berendsen H.J.C., Fraaije J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. doi: 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H. [DOI] [Google Scholar]
- 53.Kolluru G.K., Glawe J.D., Pardue S., Kasabali A., Alam S., Rajendran S., Cannon A.L., Abdullah C.S., Traylor J.G., Shackelford R.E., et al. Methamphetamine causes cardiovascular dysfunction via cystathionine gamma lyase and hydrogen sulfide depletion. Redox Biol. 2022;57 doi: 10.1016/j.redox.2022.102480. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.