

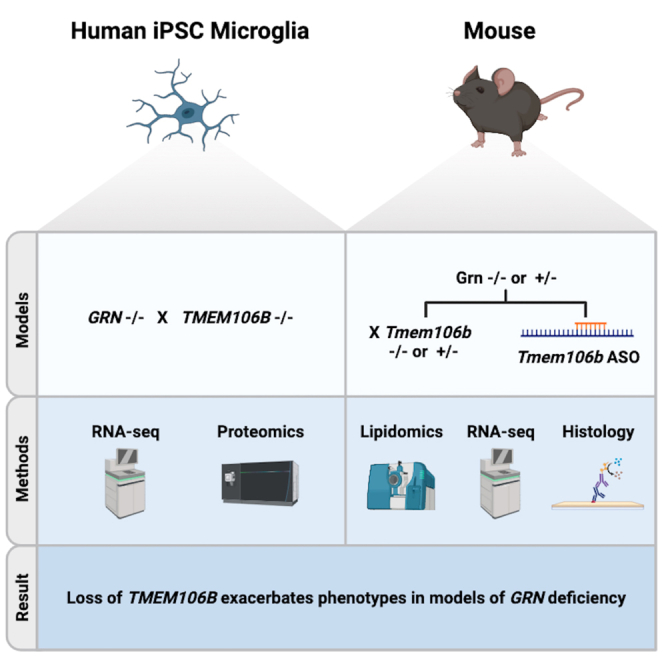
Summary
Heterozygous mutations in the granulin (GRN) gene are a leading cause of frontotemporal lobar degeneration with TDP-43 aggregates (FTLD-TDP). Polymorphisms in TMEM106B have been associated with disease risk in GRN mutation carriers and protective TMEM106B variants associated with reduced levels of TMEM106B, suggesting that lowering TMEM106B might be therapeutic in the context of FTLD. Here, we tested the impact of full deletion and partial reduction of TMEM106B in mouse and iPSC-derived human cell models of GRN deficiency. TMEM106B deletion did not reverse transcriptomic or proteomic profiles in GRN-deficient microglia, with a few exceptions in immune signaling markers. Neither homozygous nor heterozygous Tmem106b deletion normalized disease-associated phenotypes in Grn −/−mice. Furthermore, Tmem106b reduction by antisense oligonucleotide (ASO) was poorly tolerated in Grn −/−mice. These data provide novel insight into TMEM106B and GRN function in microglia cells but do not support lowering TMEM106B levels as a viable therapeutic strategy for treating FTD-GRN.
Subject areas: Molecular medicine, Neuroscience, Cellular neuroscience
Graphical abstract
Highlights
-
•
TMEM106B KO does not rescue GRN KO human microglia omic profile
-
•
Homozygous deletion of Tmem106b induces neuropathology and early mortality in Grn KO mice
-
•
Heterozygous deletion of Tmem106b does not alter phenotypes in Grn KO mice
-
•
Reduction of Tmem106b with antisense oligonucleotides is toxic in Grn KO mice
Molecular medicine; Neuroscience; Cellular neuroscience
Introduction
Genome-wide association studies have identified TMEM106B, a gene encoding a type II lysosomal transmembrane protein, as a bona fide risk factor for frontotemporal lobe dementia (FTD), especially in patients with GRN mutations.1,2,3 Polymorphisms in TMEM106B are associated with FTD risk and age of onset.4 Genetic variants are associated with FTD risk and were initially shown to impact TMEM106B protein levels. TMEM106B non-coding risk variants have been associated with increased levels of TMEM106B mRNA1,5 and protein.6 Compared to the risk variant, the protective isoform of the coding variant T185S results in reduced, unstable TMEM106B protein.7 TMEM106B overexpression in neurons exacerbates lysosomal abnormalities caused by loss of progranulin and can induce neurotoxicity.8,9 Taken together, the data have led to a hypothesis that TMEM106B levels are elevated in disease and reduction of TMEM106B may be therapeutic in FTD-GRN patients.
Progranulin, synthesized primarily in microglia, is a secreted glycoprotein that is taken up by all cell types. Once trafficked to the lysosome, progranulin is processed by cathepsins into granulin peptides, which are important for normal lysosomal function.9,10,11 Patients with homozygous GRN mutations are deficient in granulins10 and exhibit neuronal ceroid lipofuscinosis (NCL), a lysosomal storage disorder.12,13 NCL-related phenotypes have also been reported in FTD patients with heterozygous loss-of-function GRN mutations.14,15,16,17 Studies in Grn −/− mice show changes in lysosomal and immune marker transcripts, which are thought to be derived mainly from microglia.18,19 With age, Grn −/− mice develop a pathological accumulation of lipofuscin, neuroinflammation, and axonal degeneration.19,20,21 Grn −/− mice also display behavior abnormalities such as excessive grooming22 or disinhibition of exploratory behaviors.23 These data suggest that in human disease, loss of progranulin leads first to lysosomal dysfunction in microglia which triggers a cascade of pathological events across cell types, resulting in neurodegeneration and clinical symptoms of FTLD.
Like progranulin, TMEM106B is a lysosomal protein10,24 and is co-expressed with progranulin across cell types. While the function of TMEM106B is not fully understood, it has been described to play a role in lysosomal size and acidification, trafficking, and lysosomal-phagosomal fusion.24 Deletion of Tmem106b and Grn produce opposite phenotypes in mouse25 and transgenic overexpression of Tmem106b worsened lysosomal phenotypes in Grn −/− mice.9 Overexpression of TMEM106B reportedly decreases processing of progranulin into granulins, suggesting a functional interaction of these two proteins at the level of the lysosome.11 Partial deletion of Tmem106b was reported to have little effect on progranulin levels or phenotypes in Grn +/− mice,26 whereas complete deletion of Tmem106b surprisingly resulted in exacerbation of phenotypes and early mortality in Grn −/− mice.27,28,29 Here, we replicate and extend previous findings by testing the impact of both partial and complete deletion of TMEM106B in human and mouse progranulin deficient models, particularly focusing on microglia cell function. Deletion of TMEM106B in human microglia resulted in an omic profile that was distinct from deletion of GRN and led to robust alteration of transcript and protein expression when combined with GRN deletion. Despite robust changes in omics, deletion of the genes had no impact on microglia phagocytic behavior. Deletion of Tmem106b exacerbated phenotypes in Grn −/− mice, while partial reduction of Tmem106b had no benefit on microglial transcriptomics or Grn −/− brain pathology. Furthermore, Tmem106b-targeting antisense oligonucleotides were detrimental in Grn −/− mice. These data suggest that Tmem106b and progranulin have different functional roles in microglia and that when combined, loss of these proteins is detrimental. Thus, reduction of TMEM106B transcript is unlikely to be therapeutic in FTD-GRN patients.
Results
Deletion of TMEM106B does not rescue GRN phenotypes in iPSC-derived human microglia cells
Given the expression of TMEM106B and GRN in human microglia and the observed mouse glia cell phenotypes in Grn −/− mice, we examined whether homozygous deletion of TMEM106B impacts microglia cell phenotypes alone and in combination with homozygous deletion of GRN in iPSC-derived human microglia (iMG). The impact of heterozygous deletion of GRN and TMEM106B may be of interest in the future, but here we selected complete KOs to optimize for detecting small effects. We first characterized the iMG to ensure a high-quality preparation. Immunofluorescent staining revealed robust staining for microglia-specific markers Aif1 and TREM2 (Figure S1A). Transcriptomic and proteomics analysis of the cells confirmed that cells had been differentiated into microglia based on the expression profiles of cell-type-specific markers (Figures S1B–S1D). To investigate the general cellular function in TMEM106B, GRN, and DKO iMG, we measured phagocytosis using a bacteria-phrodo labeled live imaging assay. The assay was first validated using cytoD treatment to inhibit phagocytosis (Figure S2A). No change in phagocytosis was detected in any group relative to control WT iMGs (Figure S2B), suggesting that overall phagocytosis was not affected in TMEM106B, GRN, and DKO compared to isogenic control cells.
We then characterized the TMEM106B, GRN, and DKO iMGs in more detail by transcriptomic profiling (Figures 1A–1D). Based on the differentially expressed genes (Figure 1A; Table S1), the different genotypes showed reproducible expression profiles across their respective biological replicates (Figure 1B; Table S2). As expected, GRN and TMEM106B transcripts were decreased in respective KO cells compared to WT (Figure 1D). When comparing GRN −/− microglia to WT cells, pathway analyses revealed significant (FDR <0.05) elevations in interferon signaling, sphingolipid metabolism, transferrin endocytosis, and decreased cell cycle markers (Figure 1C). Changes to lysosomal transcripts were not a prominent feature of GRN −/− microglia. TMEM106B −/− microglia showed increased expression of pathways related to ECM and collagen formation, and decreased PD1 signaling and RNA translation (Figure 1C). Overall, the GRN −/− transcriptome was not restored in the dKO microglia and distinct differences in pathways related to interleukin and chemokine signaling emerged (Figure 1C). Although a restoration of the GRN −/− transcriptional profile by TMEM106B −/− was not detected, a genotype interaction was detected for a subset of transcripts related to immune signaling (CSF2RA, ETS1, IRAK4, IRF4, and TLR3) or RNA metabolism (PUS7L) (Figure 1D).
Figure 1.

TMEM106B and GRN KO iMG transcriptomic signatures
(A) Volcano plots of differential gene expression profiles from pairwise contrasts of GRN KO vs. WT, TMEM KO vs. WT, double KO vs. WT, and double KO vs. GRN KO. Red indicates transcripts passing the FDR <0.05 & |Log2 FC| > 1 threshold, blue indicates transcripts passing the FDR <0.05 threshold, green represents transcripts passing the |Log2 FC| > 1 threshold, and gray represents the remaining transcripts.
(B) Heatmap of significant (FDR <0.05 & |Log2 FC| > 1) differentially expressed genes presented in the volcano plots.
(C) Enrichment testing results for the top distinct significant (FDR <0.05) pathways for GRN KO vs. WT, TMEM KO vs. WT, and double KO vs. GRN KO. Values for double KO vs. WT are also shown.
(D) Expression of select significant (FDR <0.05 & |Log2 FC| > 1) differentially expressed genes from pairwise contrasts of GRN KO vs. WT, TMEM KO vs. WT, double KO vs. WT, and double KO vs. GRN KO within each genotype. Error bars represent S.E.M. for 3 biological replicates.
We further characterized the TMEM106B, GRN, and DKO iMGs by proteomics (Figures 2A–2D; Table S3), confirming lack of TMEM106B and GRN protein (Figure 2D). Relative to the differentially expressed proteins (Figure 2A; Table S3), the expression profiles of biological replicates were reproducible within genotypes (Figure 2B; Table S4). Compared to WT controls, GRN −/− microglia had significant (padjusted < 0.05) elevation in pathways related to retrograde transport from Golgi to ER and vesicle-mediated transport (Figure 2C). The increased expression of CSF2R and decreased expression of PUS7L proteins were notable (Figure 2D) given their analogous changes at the transcript level and connection to disease.30,31 TMEM106B −/− microglia proteomics revealed altered immune signaling pathways indexed by elevated interleukin and Fc receptor markers, and decreased markers of lipid and carbohydrate metabolism (Figure 2C). Glycosphingolipid metabolism was one of the top significant (FDR <0.05) downregulated pathways for TMEM106B −/− in human iMG (Figure 2C). Of note, the PNPO protein32 showed the greatest decrease in expression, other than TMEM106B itself (Figure 2D). The GRN −/− proteomic profile was apparent in the dKO microglia but with distinct differences in pathways related to increases in ECM and neuroinflammasome and decreased transport of small molecules (Figure 2C). Although a genotype interaction was detected for a small subset of immune-related proteins (TIFAB, TRAF3, IRAK1, CSF2R, and PUS7L) and other proteins (ERO1B, GTR6, and PNPO) that were restored to normal levels (Figure 2D), the collective data indicate that deletion of TMEM106B did not normalize the GRN −/− proteome and resulted in a unique transcriptional and proteomics profile when compared to WT and GRN −/− iPSC-derived human microglia cells.
Figure 2.

TMEM106B and GRN KO iMG proteomic signatures
(A) Volcano plots of differential gene expression profiles from pairwise contrasts of GRN KO vs. WT, TMEM KO vs. WT, double KO vs. WT, and double KO vs. GRN KO. Red indicates proteins passing the FDR <0.05 & |Log2 FC| > 1 threshold, blue indicates proteins passing the FDR <0.05 threshold, green represents proteins passing the |Log2 FC| > 1 threshold, and gray represents the remaining proteins.
(B) Heatmap of significant (FDR <0.05 & |Log2 FC| > 1) differentially expressed proteins presented in the volcano plots.
(C) Enrichment testing results for the top distinct significant (padjusted < 0.05) pathways for GRN KO vs. WT, TMEM KO vs. WT, and double KO vs. GRN KO. Values for double KO vs. WT are also shown.
(D) Expression of select significant (FDR <0.05 & |Log2 FC| > 1) differentially expressed proteins from pairwise contrasts of GRN KO vs. WT, TMEM KO vs. WT, double KO vs. WT, and double KO vs. GRN KO within each genotype. Error bars represent the S.E.M. for 3 biological replicates.
Homozygous deletion of Tmem106b exacerbates Grn −/− mouse phenotypes
Because microglia interact with neurons and other cell types to impact disease processes, we wanted to test whether deletion of Tmem106b is protective in vivo. We first created a Tmem106b CRISPR knock-out mouse and confirmed loss of Tmem106b protein by western (Figures S3A–S3C). We then crossed Tmem106b −/− mice to the Grn −/− mouse line33 and examined Wt, Tmem −/−, Grn −/−, Tmem106b x Grn double heterozygous (Tmem +/−, Grn +/−) and Tmem106b x Grn double knock-out (Tmem −/−, Grn −/−) mice twice a month starting at ∼10 weeks of age. There were not sufficient numbers of mice with single heterozygous deletions available for this study therefore Tmem +/− and Grn +/− were not included. Behavioral evaluation revealed age-related motor impairment in measures of motor function in dKO mice that was not present in Grn −/− or Tmem −/− mice alone and differed from wild-type controls (Figures 3A–3C). Motor dysfunction manifested as reduced wire hang time, increased limb clasping severity, and increased righting time in the dKO mice was progressively more apparent as mice aged from 2.5 months to 4 months (Figures 3A–3C). Impairments ultimately led to early mortality or euthanasia of the dKO mice at 4 months of age.
Figure 3.

Homozygous deletion of Tmem106b exacerbates Grn KO behavioral and lysosomal phenotypes
(A) Wire hang time was significantly decreased in Tmem −/−, Grn −/− mice and worsened with age. Mixed-effects model (REML) test revealed an overall significant effect of genotype F (4, 79) = 44.12; p < 0.0001, time F (2.611, 141.9) = 11.64; p < 0.0001 and a genotype × time interaction F (12,163) = 3.322; p = 0.0002. Tukey’s multiple comparisons test revealed that at the 3, 3.5, and 4 months time points, the Tmem −/−, Grn −/− group displayed significantly reduced hang times when compared to each of the other 4 genotypes.
(B) Tmem −/−, Grn −/− mice displayed a severe clasping phenotype. REML test on clasping severity revealed an overall effect of genotype F (4, 80) = 49.58; p < 0.0001, time F (2.214, 125.5) = 20.34; p < 0.0001 and a genotype × time interaction F (12,170) = 25.11; p < 0.0001). Tukey’s multiple comparisons test revealed that at the 3.5 and 4 months time points, the Tmem −/−, Grn −/− group displayed significantly increased clasping severity when individually compared to each of the other 4 genotypes.
(C) When placed on their sides Tmem −/−, Grn −/− mice had delayed righting times. REML test on righting latency revealed an overall significant effect for genotype F (4, 82) = 14.06; p < 0.0001, time F (1.602, 94.01) = 10.29; p = 0.0003 and a genotype, time interaction F (12,176) = 11.54; p < 0.0001. Tukey’s multiple comparisons test revealed that at 4 months the Tmem −/−, Grn −/− mice are the only group that displayed a significantly righting phenotype when individually compared to each of the other 4 genotypes. n = 6–15 animals/genotype for behavioral tests.
(D) A significant effect of genotype on LAMP1 staining was detected in whole section analysis (% area) by brain region: forebrain (FB) p = 0.0004, hindbrain (HB) p < 0.0001, and lumbar spinal cord (SC) p < 0.0001 sections by one-way ANOVA. Tmem −/−, Grn −/− mice show significantly increased LAMP1 in HB and SC when compared Grn −/− alone and to all other genotypes. Tmem −/−, Grn −/− differed from all other groups except Grn −/− mice in FB.
(E) An overall effect of genotype was detected for CD68 staining in whole section analysis (% area) by brain region: FB p = 0.0004, HB p < 0.0001 and SC p < 0.0001 using one-way ANOVA. Tmem −/−, Grn −/− mice show significantly increased levels of CD68 in FB, HB, and SC when compared to all other genotypes.
(F) Representative images of CatD staining in FB, HB, and SC showing increases in Tmem −/−, Grn −/− tissues and decreased CatD in Tmem −/− tissue, specifically in spinal cord (see insert).
(G) An overall effect of genotype was detected for CatD staining in whole section analysis (% area) by brain region: FB p = 0.0162, HB p < 0.0001, and SC p < 0.0001 sections by one-way ANOVA. In FB, Grn −/− mice had increased CatD levels compared to Wt mice. Similar to Grn −/− mice, Tmem +/−, Grn +/−, and Tmem −/−, Grn −/− had elevated CatD levels compared to Wt mice. In HB, Grn−/− mice did not show elevated CatD levels. Tmem +/−, Grn +/− differed significantly from Wt mice and Tmem −/−, Grn −/− mice showed significant increases in CatD compared to both Grn −/− and Wt mice. In SC, CatD levels were elevated in Tmem −/−, Grn −/− mice compared to Grn −/− alone and Wt mice, whereas, Tmem −/− mice had significantly decreased levels when compared to the Wt mice. For histology n = 11–16 animals/genotype and 4–5 sections/animal/tissue area/stain. Student’s t test or Tukey’s post-hoc ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, ##p < 0.01 vs. Tmem +/−, Grn +/− and Grn −/− mice. All error bars represent S.E.M.
(H) Representative images of immunofluorescent staining for CatD (green), microglia (Iba1, red), and DAPI (blue).Orange arrow indicate CatD+ve axonal swellings. White arrowhead indicates CatD+ve microglia.
To better understand the mechanisms associated with the loss of Grn and Tmem106b, we first stained for lysosomal markers in the brains and spinal cord of these mice. Immunohistochemistry revealed robust elevations in Lamp1, CD68, and cathepsin D (CatD) staining in dKO mice (Figures 3D–3H, S4A, and S4B.). Most striking was the aberrant distribution of CatD which was elevated in both white and gray matter throughout the spinal cord and hindbrain in dKO compared to Grn −/− alone or to Wt mice. CatD elevations in the forebrain were similar across Grn −/−, Tmem +/−, and dKO mice and all three groups differed from Wt mice (Figures 3F and 3G). In the spinal cord, CatD was elevated in microglia of dKOs (Figures 3F–3H) but was greatly reduced in neurons (Figure S4C) of both Tmem −/− and dKO mice. The significant reduction in CatD staining was confirmed in a separate cohort of 7-month-old Tmem−/− mice (Figure S5A). Moreover, both Tmem −/− and dKO mice displayed CatD filled aberrant axonal swellings consistent with previous studies.28,34
Lysosomal changes in the dKO were associated with histopathology in the forebrain, hindbrain, and spinal cord including decreased solochrome staining, a marker of demyelination (Figure 4B). dKO mice had reduced solochrome staining in both FB and HB. Modest but significant decreases in solochrome staining were also detected in 4-month-old Tmem −/− and Grn −/− mice but only in HB, an effect that was exacerbated in dKO mice compared to the Grn −/− mice alone. Demyelination in dKOs was concomitant with increased amino cupric staining, a marker of axonal degeneration in all 3 tissue regions when compared to Wt mice (Figures 4C and 4D). Given the extent of histopathology, we were surprised not to find a change in ChaT staining in the spinal cord of dKO mice as reported by others28 (Figure S5B). Consistent with the elevation in markers of neurodegeneration in the brain, we detected a robust elevation of plasma NFL in the dKO mice (Figure 4A). A modest but significant elevation in Tmem +/−, Grn+/− mice was also detected (Figure 4A).
Figure 4.

Homozygous deletion of Tmem106b exacerbates neurodegeneration in Grn −/− mice
(A) Plasma NFL levels were elevated in 5 months old Tmem −/−, Grn −/−, and Tmem +/−, Grn +/− mice. A one-way ANOVA revealed an overall effect on genotype p < 0.0001 and post hoc analysis show that that Tmem −/−, Grn −/− and Tmem +/−, Grn +/− have significantly higher levels of plasma NFL when compared to Wt. n = 11–16 animals/genotype.
(B) One-way ANOVA revealed an overall effect of genotype on solochrome staining in FB p < 0.0001 and HB p < 0.0001. Post-hoc analysis revealed significantly reduced solochrome staining in FB and HB of Tmem−/−,Grn−/− mice compared to Wt mice. In HB, solochrome staining was reduced in Grn −/− mice compared to Wts and further reduced in Tmem −/−, Grn −/− mice.
(C) Representative images of dramatic increases in amino cupric silver (AmCuAg) staining in FB, HB, and SC in Tmem −/−, Grn −/− tissues.
(D) Tmem −/−, Grn −/− mice have significantly increased levels of AmCuAg staining in spinal cord and brain. One-way ANOVA reveals an overall effect of genotype on FB p < 0.0001, HB p < 0.0001, SC p < 0.0001. Tmem −/−, Grn −/− mice show significantly increased levels of AmCuAg in brain and spinal cord when compared to all other genotypes. For histology n = 11–16 animals/genotype and 4–5 sections/animal/tissue area/stain. Student’s t test or Tukey’s post-hoc ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. ##p < 0.01 Grn −/− vs. Tmem −/−, Grn −/− All error bars represent S.E.M.
Heterozygous Tmem106b deletion did not alter Grn −/− or Grn +/− phenotypes
Given that we detected a significant elevation in CatD staining (Figures 3F and 3G) and a significant elevation in NFL elevation in 4 months old Tmem +/−, Grn +/− mice, we sought to determine whether partial Tmem106b deletion may have additional biological effects with age. To this end, we characterized 13 months old Grn +/− and −/− mice crossed with Tmem106b +/− mice, germline depletion from birth. For lipid and microglial analyses, all genotypes were included except Tmem −/−, Grn −/− mice due to their early mortality. For histological analyses, Tmem +/−, Grn −/− and Tmem +/−, Grn +/− mice were compared to single KO mice and Wt mice in the same study. There were insufficient mice with single heterozygous deletions available for quantitative analyses of IHC in this study. Brain samples were harvested at 13 months of age when robust neuropathology is present in the Grn −/− mice (Figure S6).
Specifically, microglia were sorted from the thalamus, a region expressing inflammation and neurodegeneration (Figures S6A–S6C). RNA-seq revealed significantly increased (FDR <0.05 & Log2FC > 1) expression of disease-associated microglia (DAM) genes in Grn −/− mice vs. Wt mice, similar to previous findings.18 Among the differentially expressed genes were lysosomal (Gla, Gpnmb, Lyz2, and Atp6v0d2)(Figures 5G–5N.) and lipid-related transcripts (ApoE, Pld3, Lgals3, Gpx3, Smpdl3a, and Pag1) (Figures 5G–5N). Differentially expressed genes showed a similar profile in the Tmem+/− Grn −/− compared to Grn −/− mice. No significant change in microglial transcripts was detected in Grn +/− mice or Tmem +/− Grn +/− mice compared to each other or Wt mice.
Figure 5.

Heterozygous deletion of Tmem106b does not rescue Grn −/− omic profile in 13 months old Grn −/− mice
(A and B) Volcano plots of lipidomic profiles comparing Grn−/− with WT cortex (A) or hippocampus (B). Red dots indicate significantly different lipids.
(C and D) BMP species levels were reduced similarly in Grn −/− and Tmem+/−, Grn−/− mice compared to Wt mice in cortex and hippocampus.
(E and F) Glucosylceramide and glucosylsphingosine levels were elevated in Grn−/− mice but to a lesser extent in Tmem +/−,Grn−/− mice in hippocampus by Mann Whitney U.
(G–N) Bulk RNAseq from isolated thalamic microglia. Select significant (FDR <0.05 & |Log2FC| > 1) differentially expressed genes from pairwise contrasts of Grn −/− vs. Wt and Tmem +/− Grn −/− vs. Grn −/−. N = 5–14 animals/genotype. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. All error bars represent S.E.M.
Because DAM genes have been associated with foamy, lipid-laden microglia in Grn −/− mice,35 we decided to measure lipids in the cortex and hippocampus, using both targeted and non-targeted methods. Compared to Wt mice, both Grn +/− and Grn −/− mice had significantly reduced BMP (Bis(monoacylglycero)phosphate), a glycerophospholipid enriched in lysosomal membranes, which corresponded with elevations in glucosphingolipids and glucoceramides (Figures 5A–5F). Overall, Tmem +/−, Grn +/− mice did not differ from Grn +/− nor did Tmem +/− Grn −/− mice differ from Grn −/− mice with respect to lipid (Figures 5C and 5D) microglia transcripts (Figure 5G–5N), or neuroinflammation and degeneration (Figures 6A, 6B, 6E, and 6F). In addition, 13 months old Tmem+/− Grn+/− did not show changes on any measure, including NFL levels, compared to Wt mice (Figure 6F.) Together these results indicate that heterozygous Tmem106b deletion from birth had limited impact on Grn +/− or Grn −/− phenotypes. However, some region-specific findings were detected. Tmem +/− Grn −/− mice showed partial lowering of GS and GC lipids in the hippocampus (Figures 5E and 5F), a forebrain area where partial reversal in Lamp1 expression was also observed (Figures 6A and 6C). These region-specific findings suggest that Tmem106b may provide varying biological impacts in the CNS depending on cell type36 or its expression level.37
Figure 6.

Heterozygous deletion of Tmem106b does not rescue histopathology in 13 months old Grn −/− mice
(A) Representative images of CD68, LAMP1, and CatD from forebrain (FB) coronal sections.
(B) Overall effects of genotype observed for CD68 staining in whole section analysis (FB) p < 0.0001, (HB) p < 0.0001 sections by one-way ANOVA. Grn −/− mice had significantly increased levels of CD68 compared to Wt mice. Tmem +/−, Grn −/− mice did not differ from Grn −/− mice, showing significantly increased levels of CD68 in FB and HB compared to Wt mice. Also, Tmem −/− mice displayed significant increase in CD68 in HB compared to Wt mice.
(C) Lamp1 staining is significantly altered between genotypes in FB p < 0.0001 and HB p < 0.0001 by one-way ANOVA. Grn −/− mice show significantly increased levels of Lamp1 in FB and HB when compared to all other genotypes. Lamp1 levels were partially reduced in Tmem +/−, Grn −/− compared to Grn −/− mice. Tmem −/− mice displayed a significant increase in Lamp1 in HB only.
(D) One-way ANOVA reveals an overall genotype effect on cathepsin D in FB p = 0.002 and HB p < 0.0001. Grn −/− and Tmem +/−, Grn −/− mice have significantly increased levels of cathepsin D in FB and HB when compared to Wt and did not differ from each other.
(E) One-way ANOVA reveals an overall genotype effect of AmCuAg staining HB p < 0.0001 but not in FB. Tmem −/− and Grn −/−mice each had significantly increased levels of AmCuAg in HB when compared to Wt. Tmem +/−, Grn −/− mice did not differ significantly from Grn−/− mice. For histology, n = 5–10 animals/genotype and 4–5 sections/animal/tissue area/stain.
(F) An overall effect of genotype was detected in plasma NFL levels by one-way ANOVA p < 0.0001. NFL was significantly increased in Tmem −/− and Grn −/− mice compared to Wt mice. Tmem +/−, Grn −/− NFL levels did not differ from Grn −/− mice. Tmem +/−, Grn +/− NFL levels were similar to Wt mice. N = 8–14 animals/genotype. Student’s test or Tukey post-hoc ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. All error bars represent S.E.M. Figure created with BioRender.com.
Unlike Tmem +/− mice, 13 months old Tmem −/− mice had a clear lipid and neuroinflammation phenotype. Significant elevations in CD68 and Lamp1 staining were detected in the hindbrain (Figures 6A–6C) and a ∼50% reduction in lysophosphatidylethanolamine (LPE) species in the hippocampus (Figures S7A–S7D). Brain lipid changes were associated with demyelination, elevated amino cupric staining (Figure 6E) in the hindbrain, and a large increase in plasma NFL (Figure 6F). Sorted microglia from the thalamus did not reveal many DEGs in Tmem −/− mice, indicating that in vivo inflammation and neurodegeneration may be stemming from other cell types or brain regions. Although we did not dissect the spinal cord for this experiment, the aged Tmem −/− mice may have also had degeneration in this region33 which could contribute to elevated NFL in the plasma.
Antisense oligonucleotide reduction of Tmem106b is safe in Wt and Grn +/− mice but toxic in Grn −/− mice
To eliminate any potential confounds of constitutive deletion of the Tmem106b gene during development, we next tested the impact of reducing Tmem106b transcript in Grn −/− mice using antisense oligonucleotides (ASOs) (Figures 7A–7K). A set of four ASOs directed at mouse Tmem106b transcript were first evaluated for tolerability and potency in adult C57Bl/6J mice (Figures 7A, 7C, 7D, and S8). A non-targeting negative control ASO was included for comparison. In wild-type mice, two weeks following a single intracerebroventricular (icv) injection, we found that the level of mRNA knockdown varied across the ASOs from 40% to 75% (Figure 7C) and the level of protein lowering ranged from 30 to 50% (Figure 7D). All ASOs were well-tolerated in C57Bl/6J mice out to 2 weeks post-dose (Figures 7E and S8A) and had minimal effects on other transcripts including inflammation gene sets (Figures S8B–S8D). To test whether Tmem106b ASO treatment might alter pathology in Grn −/− mice, the same set of ASOs were given to 13 months old Grn −/− and age-matched +/− mice. All ASOs were well-tolerated in the Grn +/− mice (Figure 7H) out to four weeks post-injection. Harvest of tissue at this 4-week time point achieved a maximal lowering of 50% Tmem106b transcript and 25% protein (Figures 7F and 7G). However, the same ASOs given to Grn −/− mice caused seizures and morbidity, and mice were euthanized by 5 days–7 days post-dose (Figure 7K). Maximal Tmem106b transcript lowering of 60% and protein lowering of 40% was detected (Figures 7I and 7J). All mice dosed with the negative control ASO were healthy at the time of harvest. While ASO chemistry and target location were somewhat similar across the 4 ASOs, transcriptional profiling indicated differing levels of selectivity (Figures S8B–S8D). Given that all Tmem106b ASOs showed toxicity with no effect in the negative control group, the observed toxicity is likely on-target. Due to this unexpected toxicity, histological assessments of treatment effects on Grn −/− phenotypes were not possible in this experiment. In a separate study, using electron microscopy, we found that Tmem106b ASO accumulated in lysosomes in multiple cell types in both Wt and Grn −/− mice (Figure S9). While we were unable to discern obvious differences between genotypes, this lysosomal accumulation was similar to our previously reported observation in C57Bl/6J mice and in cultured cells.38 In contrast to genetic deletion, there were no signs of motor dysfunction with Tmem106b ASO treatment. The behavioral manifestation of in vivo toxicity as seizure may be dependent upon the distribution of the Tmem106b ASO, primarily bathing the forebrain and hindbrain but not the spinal cord following an intracerebroventricular (ICV) administration. In summary, we found that partial Tmem106b reduction with ASO was safe in Wt and Grn +/− mice but toxic in Grn −/− mice.
Figure 7.

Partial reduction of Tmem106b with ASO treatment is toxic in Grn −/− mice
(A and B) Study designs for Tmem106b ASO dosing experiments in Wt (A) and Grn +/− and Grn −/− mice (B).
(C and D) Significant knockdown of Tmem106b RNA and protein observed in brains of Wt mice with an overall effect of ASO by one-way ANOVA for mRNA, p < 0.001 and protein, p < 0.0074. When compared to saline, all 4 ASOs significantly reduced Tmem106b RNA and protein levels.
(E) Little to no adverse events observed in Wt mice after ASO dosing. n = 6 Wt mice/group.
(F and G) Significant knockdown of Tmem106b RNA was observed in brains of Grn +/− mice with an overall effect of ASO by one-way ANOVA for RNA p < 0.001 but not for protein. When compared to NC ASO, all 4 Tmem106b ASOs significantly reduced Tmem106b mRNA levels but not protein levels.
(H) Little to no adverse event observed in Grn +/− mice after ASO dosing. n = 4–5 Grn +/− mice/group.
(I and J) Significant knockdown of Tmem106b mRNA and protein was observed in brains of Grn −/− mice with an overall effect of ASO by one-way ANOVA for RNA p < 0.001 and protein p = 0.0088. When compared to NC ASO, all 4 Tmem106b ASOs significantly reduced Tmem106b RNA levels with 3/4 ASOs also significantly reducing protein levels. n = 3–5 Grn −/− mice/group.
(K) At 5–7 days of age many of the Grn −/− mice dosed with Tmem106b targeting ASO developed adverse events including death, seizures, and significant weight loss. Post-hoc Student’s t test ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. All error bars represent S.E.M.
Discussion
It has been proposed that loss of progranulin, primarily synthesized in microglia, leads to compensatory activity of the lysosome, hyperactivity of microglia and release of pro-inflammatory cytokines, and downstream neurodegeneration.18,19,22 Consistent with this theory, we found lysosomal proteins were elevated in 4 months old and to a greater extent in 13 months old Grn −/− mice. Lysosomal changes were associated with neuropathology including inflammation, altered bulk lipids, demyelination, and neurodegeneration in specific regions of the brain including the hindbrain, thalamus, hippocampus, and cortex. Full deletion of Tmem106b exacerbated all of the aforementioned Grn −/− pathologies and brought out new pathologies in the spinal cord. Partial deletion of Tmem106b did not normalize or exacerbate Grn +/− or Grn −/− microglial transcriptomics, lipid, or histopathological phenotypes. Surprisingly, partial Tmem106b reduction with ASO treatment was toxic in Grn −/− mice, likely through an on-target mechanism. Detrimental effects of Tmem106b reduction or deletion may be due to synergistic effects with Grn deletion on phagosomal and lysosomal trafficking in myelinated axons,28,29,34 but here we also found distinct lysosomal and lipid phenotypes that likely contribute to the neurodegeneration in dKO mice.
In Grn −/− mice, we found that CatD was somewhat elevated across the spinal cord and hindbrain, regions where we also detected demyelination and neurodegeneration. The elevation in CatD was exacerbated and diffused in dKO mouse tissue. In distinct contrast to Grn −/− mice, young Tmem106b −/− mice were deficient in CatD in the spinal cord. This loss was particularly evident in neurons but not in microglia. With age, significant reductions in CatD were detected in the hindbrain with trends appearing in the forebrain. When we measured cathepsin D in human TMEM106B −/− cultured microglia, no changes were detected at the RNA or protein level. This would suggest that TMEM106B effects on CatD originate in neurons and glial cell types other than microglia. Feng et al.36 reported that Tmem106b −/− mice have reduced CatD protein levels in oligodendrocytes in the cerebellum and further demonstrated a physical interaction between Tmem106b and CatD. Altered cathepsin expression or activity in oligodendrocytes may underlie the finding that Tmem106b −/− mice are also more susceptible than Wt mice to demyelinating effects of cuprizone.39 Indeed, CatD deficiency has been shown to cause profound defects in proteolipid protein trafficking and myelination in mice40 and can lead to NCL16 and juvenile-onset ataxia41 in people. Given that progranulin and granulin peptides can also regulate the activity of several lysosomal enzymes, including CatD,42,43 the totality of the data point to a functional interaction of TMEM106B and progranulin leading to altered CatD production and activity, which may in turn lead to demyelination and neurodegeneration in the dKO mice. Given high expression of Tmem106B in neurons and glia, future single cell studies across cell types will further reveal the biological function of Tmem106B and its dysfunction in disease.
Lipidomics and transcriptomics analyses revealed robust alterations in lipid biology in both Grn −/− and Tmem106B −/− mice. Interestingly, Grn −/− mice had robust reductions in BMP, a glycerophospholipid enriched in lysosomal membranes, which corresponded with elevations in glycosphingolipids (GS) and glycoceramides (GC). This would make sense given the role of BMP in hydrolyzing these lipids under normal conditions. Elevated GS and GC have been associated with neurodegenerative disease in the context of lysosomal disorders such as NCL and Gaucher.44 Systemic treatment with recombinant progranulin restored lipid phenotypes and alleviated neurodegeneration in Grn −/− mice.45 Foamy lipid-laden microglia have been detected in Grn −/− mice,3 indicating that dysfunctional microglia contribute to altered lipid hydrolysis. Here, we report changes in lipids as a result of TMEM106B deficiency in human iMG and in mouse where we also see elevated markers of neurodegeneration. Lipid production may be an important biomarker of neurodegeneration in disorders where progranulin or Tmem106B is reduced or absent.
Interestingly, we detected region-specific impacts of Tmem106b deletion in mice. Both Tmem106b −/− and Tmem106b −/−, Grn −/− mice had more pronounced neuroinflammation and degeneration in the spinal cord and hindbrain compared to the forebrain. These findings are consistent with other publications of motor neuron and Purkinje cell degeneration in these regions.34,36,46 Moreover, we detected hippocampal-specific changes in lysosomal and lipid markers in Tmem106b −/− mice. While heterozygous deletion of Tmem106b did not exacerbate or normalize BMP or glycosphingolipid levels overall in Grn −/− mice, there were modest but notable reversals in GC and GS levels in the hippocampus of Tmem106b +/−, Grn−/− mice. This finding was interesting given that we also detected partial reversal of Lamp1 levels in the forebrain region containing hippocampus. In addition, we detected a significant reduction in LPEs in the hippocampus, not the cortex, of Tmem106B −/− mice, a lipid phenotype distinct from Grn −/− mice. Similar reports of lysosomal or lipid marker changes in specific brain regions in Tmem106b knock-out mice,25,26,46 suggest a potential for regional or cellular differences in Tmem106b function. Future studies interrogating the expression and role of Tmem106b across brain regions in mouse and TMEM106B in human postmortem tissue would be informative.
Here, we compared the effect of partial reduction of Tmem106b by genetic manipulation versus partial reduction using an antisense oligonucleotide approach. Partial reduction was not protective in the Grn −/− mouse model using either approach. Furthermore, it was surprising to detect frank toxicity after partial Tmem106b reduction with ASO treatment given that Tmem106b +/− Grn −/− mice showed no exacerbation of phenotype and no signs of early mortality. Given that Grn −/− mice treated with the negative control ASO were healthy, we suspect that toxicity of Tmem106b ASOs in Grn −/− mice was due to an interaction between Tmem106b deficiency, progranulin deficiency, and ASO treatment. Several possible explanations may be considered. It is plausible that we achieved higher than 50% Tmem106b knockdown earlier in time, e.g., 3 days vs. 7 days post-dose. Similarly, we may have achieved robust knockdown and improper localization of Tmem106b protein to lysosomes in a subpopulation of cells. We have observed that Tmem106b ASO is more potent in microglia than other cell types, likely due to higher uptake into microglia.38 Given that Grn −/− mice already have dysfunctional microglia, ASO toxicity may be related to accumulation in this cell type. However, we have observed that ASOs accumulate in lysosomes of neurons38 as well as in glia and that progranulin deficiency can impair autophagy in neurons18; therefore, ASO accumulation in neurons of Grn −/− mice may further impair autophagy and lead to toxicity. Such an outcome warrants careful investigation of ASO therapeutic safety in lysosomal storage diseases in the future.
While reduction of normal TMEM106B seems unlikely to be therapeutic for FTD-GRN, our preclinical data are helpful in interpreting the role of TMEM106B in FTD and other disorders where lysosomal dysfunction, lipid dysregulation, and immune cell activation are key features.47,48,49,50,51 A recurrent de novo genetic mutation in TMEM106B has been associated with hypomyelinating leukodystrophy.48 Yang et al. found the TMEM106B risk haplotype to be associated with a myelination/lysosomal transcript module in postmortem pTDP43-positive brain tissue.49 Most recently, TMEM106B fibrils have been detected in the normal aging brain and in TDP43-containing postmortem brain,50 implicating a toxic gain of function hypothesis. Insoluble TMEM106B protein was similarly increased in MS plaques compared to controls.51 While TMEM106B aggregates could impact neuronal cell health, aggregation of TMEM106B could result in loss of function of wild-type TMEM106B protein, leading to altered lysosomal activity and lipid formation across cell types, as modeled in mouse KOs in this study. Future studies investigating the impact of TMEM106B snps on TMEM106B biology as well as novel approaches of targeting TMEM106B fibrils for the treatment of neurodegenerative disease are warranted.
Limitations of the study
There were several limitations to the study. Brain and cell-type-specific effects of TMEM106B deletion were not fully evaluated in this experiment. For example, transcriptomics was conducted in sorted microglia from the thalamus but not from other brain regions or cell types. We detected few phenotypes in Grn +/− mice (BMP lipid reductions, altered MG transcriptomics), and we acknowledge that Grn +/− mice do not mimic progranulin haploinsufficiency in people. Improved humanized models of progranulin disease, including human stem cells carrying heterozygous deletions or snps would be useful. In addition, reduction of Tmem106B protein may be beneficial in other TDP43 or neurodegenerative models containing Tmem106B aggregation or fibrillization.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Rabbit Alexa Fluor 488 | Thermofisher | Cat# A21206 |
| Anti-Goat Alexa Fluor 488 | Thermofisher | Cat# A11055 |
| Anti-Tmem106b | Bethyl | Cat # A303-439A |
| Anti-Tmem106b | Cell Signaling | Cat # 93334 |
| Anti-Aif1 human | Wako Fujifilm | Cat# 019-19741 |
| Anti-Trem2 human | R&D Systems | Cat #: AF1828 |
| Anti-Cathepsin D (chromogenic staining) | Abcam | Cat# ab75852 |
| Anti-Cathepsin D (IF staining) | R & D | Cat# AF1029 |
| Anti-Lamp1 | Abcam | Cat# ab25245 |
| Anti-Iba1 | Wako | Cat#019–19741; RRID: AB_839504 |
| Anti-ChaT | Millipore | Cat#AB144P; RRID: AB_2079751 |
| Anti-CD68 | Biorad | Cat#MCA1957T; RRID: AB_2074849 |
| Anti-dMBP | Millipore | Cat#AB5864;RRID: AB_2140351 |
| Anti-NeuN | Synaptic Systems | Cat# 266004 |
| Rabbit polyclonal Anti-ASO antibody (0.5 μg/mL) | Genentech, Inc. | W138- 442 |
| Bacterial strains | ||
| Phrodo Green Zymosan Particles | Thermofisher | Cat #P35365 |
| Chemicals, peptides, and recombinant proteins | ||
| Tandem Mass Tags | ThermoFisher Scientific | Cat # A44522 |
| cOMplete, Mini Protease Inhibitor Cocktail | Sigma-Aldrich | Cat # 11836153001 |
| PhosSTOP, Phosphatase Inhibitor | Sigma-Aldrich | Cat # 4906837001 |
| Dithiothreitol | ThermoFisher Scientific | Cat # A39255 |
| Iodoacetemide | ThermoFisher Scientific | Cat # A39271 |
| Lysyl endopeptidase | FujiFilm Wako Chemicals | Cat # 125-05061 |
| Sequencing Grade Modified Trypsin | Promega | Cat #V5113 |
| hydroxylamine | ThermoFisher Scientific | Cat # 90115 |
| Critical commercial assays | ||
| Mouse neurofilament light | Quanterix | Simoa NF-Light Advantage Kit (#103186) |
| StemDiff hematopoietic kit | Stemcell Technologies | cat #05310 |
| StemDiff microglia differentiation kit | Stemcell Technologies | cat #100-0019; cat #100-0020 |
| Quickstart Bradford Protein Assay Kit | Bio-Rad | Cat # 500202 |
| Deposited data | ||
| iMG Proteomics | MassIVE | MSV000090254 |
| Transcriptomics (Mouse and Human microglia) | GEO | GSE237107 |
| Experimental models: Cell lines | ||
| PGP1 TMEM106B KO iPS line | Synthego | custom |
| PGP1 GRN KO iPS line | Synthego | custom |
| Experimental models: Organisms/strains | ||
| Grn −/− | University of Tokyo | NA |
| TMEM106b −/− | Genentech, Inc. | NA |
| Oligonucleotides | ||
| ETGTATTTCAAATETEA | RICC | ASO 1 |
| ETGTATTTCAAATETEA | RICC | ASO 2 |
| TGACACTATCTCTTEE | RICC | ASO 3 |
| GTATTTCAAATCTEAAAT | RICC | ASO 4 |
| ATTATGTTTATCACEAA | RICC | NC ASO |
| Critical commercial assays | ||
| TMEM106B mouse qPCR probe | Thermofisher Scientific | Cat # 4351370 |
| GAPDH mouse qPCR probe | Applied Biosystems | Cat # 4351309 |
| GRN mouse qPCR probe | Thermofisher | cat# Mm00433848_m1GRN |
| Software and algorithms | ||
| MATLAB for IHC quantification | MathWorks | N/A |
| JMP | https://www.jmp.com/en_us/home.html | |
| Graphpad Prism v9.0 | https://www.graphpad.com/ | |
| In-house mass spectrometry data analysis software | Huttlin et al., 201052 | N/A |
| MASCOT | Matrix Science | N/A |
| R | https://www.r-project.org | |
| MSstats/MSstatsTMT | Huttlin et al., 201052; Huang et al., 202019,53 | http://bioconductor.org/packages/release/bioc/html/MSstatsTMT.html |
| GSNAP | Wu et al., 201654 | https://bioconductor.org/packages/release/bioc/html/gmapR.html |
| HTSeqGenie | Pau & Reeder, 202155 | https://bioconductor.org/packages/release/bioc/html/HTSeqGenie.html |
| Limma | Law et al., 201456 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| ComBat-seq | Zhang et al., 202057 | https://bioconductor.org/packages/release/bioc/html/sva.html |
| Other | ||
| SOLA HRP SPE cartridges | ThermoFisher Scientific | Cat # 60109-001 |
| Sep-pak C18 cartridges | Waters | Cat # WAT054925 |
| Aurora Ultimate column | IonOpticks | Cat # 25075C18 |
| Orbitrap Eclipse Mass Spectrometer | ThermoFisher Scientific | Cat # FSN04-10000 |
| Dionex Ultimate 3000 RSLC nano Proflow | ThermoFisher Scientific | Cat # ULTIM3000RSLCNANO |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Amy Easton (amy.easton@gmail.com).
Materials availability
All unique reagents are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
Data
-
•
Transcriptomic data from mouse and human microglia has been deposited in GEO and will be publicly available (GSE237107 is the reference series).
-
•
Proteomic raw datafiles were deposited into the MassIVE repository with the identifier: Massive: MSV000090254 (reviewer login = MSV000090254_reviewer & password = tmem).
Code
-
•
This study did not generate original code.
Experimental models and study participant details
Mouse lines
Grn −/− mice were generated by M. Nishihara (University of Tokyo, Tokyo, Japan) as described previously.33 Grn +/+ , Grn +/−, and Grn −/− mice were of the genetic background C57BL/6J.
Tmem106B knockout mice were obtained by electroporation-based strategy of C57BL/6J zygotes with 25 ng/μl wild-type Cas9 mRNA (Life Technologies) and 13 ng/μL in vitro-transcribed 2 single-guide RNA into mouse zygotes.58 Tail DNA from resulting offspring was analyzed by PCR and sequencing. Target sequences of sgRNA used to knockout exon 4 and exon 5 are 5′GGGATTTAGAGTAGCCGTTT-3′ PAM: TGG and an CFD algorithm score of 95 and the second sgRNA sequence is 5′-cgactgcccaccgttaaggc-3′ PAM:AGG and an CFD algorithm score of 98. The 4678bp knockout region corresponds to GRCm38/mm10 chr 6: 13,074,630- 13,079,307. Genotyping was carried out using the following primers: Tmem106b_1: TCTGTTGAGATGAAGAGTAGG, Tmem106b_2: CTGAGAACATGAGGAGTGA, and Tmem106b_3:CTTGAGGAGGCTGGTC.The KO band size is 258bp and the wild-type band size is 349bp and the PCR melting temperature is at 55. TMEM106B deletion was verified in brain tissue from 7-month-old mice (Figure S1).
The Tmem106b−/−, Grn−/− cross colony was generated using a heterozygous breeding design to produce littermates with all possible genotypes. Adult C57BL/6J male mice were used for ASO dosing studies (Jackson Laboratory (Bar Harbor, ME: stock #000664). Male and female mice between the ages of 3–6 or 12–13 months of age were used in all other experiments. Mice were housed on a regular light/dark cycle (14:10 h) with ad libitum access to food (LabDiet 5010) and water. All behavioral assessments were conducted during the light phase. All protocols for mouse experiments were approved by the Institutional Animal Care and Use Committee and were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Method details
ASO molecules & synthesis
Oligonucleotides were synthesized as described previously.59 Single-stranded DNA oligonucleotides with complete phosphorothioate backbones and LNA-modified flanks were synthesized on a MerMade 192× synthesizer, MerMade 12 synthesizer (LGC Bioautomation, MI, USA) or an Äkta OligoPilot 100 synthesizer (Cytiva Life Science) utilizing standard phosphoramidite chemistry. For synthesis in small scale, the final 5′-dimethoxytrityl (DMT) group was left on the oligonucleotide for later use as a lipophilic handle during chromatographic purification and for synthesis in larger synthesis scale, the DMT group was removed from the oligonucleotide prior to cleavage and deprotection.
The oligonucleotides synthesized in small scale were cleaved from the solid support on a Hamilton Starlet pipetting robot (Hamilton, Bonaduz, Switzerland) using concentrated aqueous ammonia followed by deprotection in an oven at 65°C for 5–12 h. The oligonucleotides containing 5′-DMT were purified by solid phase extraction in TOP cartridges (Agilent Technologies, Glostrup, Denmark) supported on a Hamilton Starlet pipetting robot (Hamilton, Bonaduz, Switzerland). The full-length oligonucleotide having a 5′-DMT group binds to the column material of the cartridge, allowing for removal of shorter failure sequences. Subsequent removal of the DMT group allowed for elution of the purified oligonucleotide. The collected solution of purified oligonucleotide was evaporated to dryness and subsequently dissolved in PBS. The concentration of oligonucleotide in solution was determined by calculating the Beer-Lambert extinction coefficient and measuring the UV absorbance of the solution at 260 nm. Oligonucleotide identity and purity were validated by reversed-phase ultra-performance liquid chromatography coupled to MS (UPLC-MS).
Cleavage and deprotection of oligonucleotides synthesized in larger synthesis scale (e.g., 20–500 μmol) was performed using a concentrated solution of aqueous ammonia overnight at 65°C. The solid support was removed by filtration and the solution lyophilized overnight. Subsequent purification by ion-exchange high-pressure liquid chromatography (HPLC) chromatography at elevated pH using a gradient of NaCl afforded the purified oligonucleotide. The solution was neutralized and desalted by either size-exclusion chromatography (Äkta Pyrified, Cytiva Life Science) or tangential flow filtration (Äkta CrossFlow, Cytiva Life Science) and the oligonucleotide could be collected after lyophilization. Oligonucleotide identity and purity were validated by reversed-phase UPLC-MS followed by a test for the presence of endotoxins (Endosafe, Charles River Labs). Oligonucleotides with an endotoxin content below 0.2 EU/mg were released for use.
ASO sequences
Uppercase denote LNA nucleotides, lowercase denote DNA nucleotides lowercase and underlined indicate 2′O Methyl Nucleotides. All backbone linkages are phosphorothioates, All LNA C residues indicated are 5-methyl C.
| RTR ID | Base sequence | Sugar sequence |
|---|---|---|
| ASO 1 | ETGTATTTCAAATETEA | LLDDDDDDDDDDDLLLL |
| ASO 2 | ETGTATTTCAAATETEA | LLLDDDDDDDDDDLDLL |
| ASO 3 | TGACACTATCTCTTEE | LLLDODDDDDDDDDLL |
| ASO 4 | GTATTTCAAATCTEAAAT | LLLLDDDDDDDDDLDLLL |
| NC ASO | ATTATGTTTATCACEAA | LLDDDDDDDDDDDDLLL |
Antisense oligonucleotides have full phosphorothioate backbone.
In base sequence, E indicates 5-methyl-C.
In sugar sequence, L indicates LNA, D indicates DNA, O indicates 2′-O-Me.
Intracerebroventricular (ICV) injections
ICV injections were performed using a free hand surgical method. Animals were anesthetized with isoflurane, the hair above the parietal region of the skull was removed by shaving and skin was cleaned with Cloraprep Swabsticks (BD, NJ: ref #260100) solution. A midline incision of the skin was made between the occiput and forehead to identify bregma. Coordinates for injection were 1–2 mm to the right of the midline and −0.5 mm posterior from bregma. The needle, fitted with polyethylene tubing and cut so that no more than 3 mm of the needle is available to penetrate the brain, was pushed through the skull to a depth of −2.8 mm and 1 min was given to allow for the brain to seal around the needle. Next, a bolus injection of 5 μL was injected at a rate of 1 μL per sec, using a 26-gauge 25 μL Hamilton syringe. One minute after the injection the needle was slowly withdrawn. The incision was closed with tissue glue and buprenorphine was given for up to 3 days to reduce any pain caused by the procedure.
Behavior
Behavior testing was performed at 2.5, 3, 3.5 and 4 months of age. For wire hang testing mice were placed upright on a wire grid, allowed to grasp the wires and then the grid was gently inverted. Hang time was recorded with a maximum score of 60 s. Mice unable to reach the full 60 s could repeat up to 3 times, after which their maximal score we recorded. Clasping was scored while mouse was suspended by its tail for 10 s and on a 0–3 scale. A score of 0 was given if no clasping was observed. If only mild clasping was observed for less than 50% of the time suspended, a score of 1 was given. A score of 2 was given if both hind paws were retracted toward abdomen for greater than 50% and a score of 3 is given when a mouse clasped both hind paws for the entire time that they were suspended. Righting latencies were recording after mouse was scruffed and laid gently on its side and is reflective of the time it took the mouse to stand up onto its feet.
All protocols for mouse experiments were approved by the Institutional Animal Care and Use Committee and were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Plasma collection and NfL measurements
Collection of blood, plasma separation and NfL measurements were performed as described previously.60 To measure NFL levels in the mice, blood was collected via cardiac puncture. Blood was collected in K2 EDTA Micro500 tubes (part# M500-E, SAI Infusion Technologies, Lake Villa, IL, USA), stored on ice until all samples were collected and then spun down at 4 °C for 3 min at maximum speed. The plasma was pipetted into clean tubes and immediately frozen at −80 °C until processed. In vivo plasma concentration of NfL was measured using the Simoa NF-Light Advantage Kit (#103186) at Quanterix, Inc, Billerica, MA, USA. The Simoa assay is a 2-step digital immunoassay, which measures the quantity of NfL in samples using the Simao HD-1 Analyzer and Single Molecule Array (Simoa) technology.
Tissue collection for qPCR and western blots
When brain was being collected for qPCR and western blots, mice were sacrificed with isoflurane overdose followed by rapid decapitation, and the brain was placed in an ice-cold mouse brain slicer matrix (Stoelting) and sectioned into two (2mm) coronal sections (starting at 0.26mm from bregma) weighing approximately 30–40 mg. One piece of tissue was immersed in RNALater solution (ThermoFisher Scientific, cat. AM7021) and stored at 4°C for RNA extraction and the second piece was immediately frozen in dry ice and stored at −80°C for protein extraction.
RNA isolation and qPCR
RNA was isolated using RNeasy Mini Kit (Qiagen, cat. 74116) using QIAcube robotic workstation (Qiagen). Briefly, brain fragment was transferred from the RNALater solution to 1 mL RLT buffer in 1.5 mL Eppendorf tube containing 3mm tungsten carbide bead (Qiagen, cat 69997). Tissue was lysed using TissueLyser II (Qiagen) (3 min at 30Hz) and centrifuged 3 min at 20000g. 550 μL of the lysate (no foam, no sediment) was transferred to 2 mL Eppendorf tube and placed in QIAcube pre-filled with all the required reagents. RNA was purified according to a program “RNeasy Mini - Animal tissues and cells - Large samples (version 2)” with elution with 50 μL of H2O. RNA concentration was measured using NanoDrop 8000 (ThermoFisher Scientific) spectrophotometer and the concentration was adjusted with H2O to 2 ng/μL. Expression levels on sections from the right hemisphere were measured using TMEM106B Mm00510952_m1 (Thermo Fisher Scientific, cat. 4351370) as probe and a GAPDH probe (cat. 4351309, Applied Biosystems) for normalization. The data were analyzed using the QuantStudioTM Real-time PCR Software, and the readouts of the technical duplicates were averaged. Statistical analysis of TMEM106B reduction was analyzed using a one-way ANOVA in Graphpad prism 9.0.
Western blot procedures
Frozen brain samples were homogenized ∼10V of 1% NP40 in PBS with Roche Complete EDTA-free Protease inhibitor cocktail and Roche PhosSTOP phosphatase inhibitor, using a Qiagen TissueLyser II homogenizer for 3 min at 25Hz. Samples were placed on ice for 20 min then centrifuged at 4C for 20 min at 21,000 g. The supernatant was collected and Western blot samples were prepared in 4x LDS loading buffer with 10x Reducing agent. Equal volumes (10μl) of sample were separated on a 10% Bis-Tris polyacrylamide gel in 1 X MOPS buffer and transferred to a nitrocellulose membrane using an iBlot II semidry transfer system. For supplemental data, frozen brain samples were homogenized ∼10V in radioimmunoprecipitation assay buffer (RIPA) [150 mM sodium chloride, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, and 50 mM tris (pH 8)] with Roche Complete EDTA-free Protease inhibitor cocktail and Roche PhosSTOP phosphatase inhibitor, using a Qiagen TissueLyser II homogenizer for 3 min at 25Hz. Samples were placed on ice for 20 min then centrifuged at 4C for 20 min at 16,000 g. The supernatant was collected and protein concentration was measured by the Pierce BCA protein assay kit method (23227) using BSA as a standard, and samples prepared in Laemmli SDS sample buffer (J60015) were subjected to Tris Glycine SDS–polyacrylamide gel electrophoresis and immunoblotted after Semi dry transfer to nitrocellulose membranes using Tris-Glycine Transfer buffer (LC3675) with 15% methanol for 60 min.
Blots were blocked with fluorescent blocking buffer (Rockland) at room temperature for at least 1 h then incubated overnight at 4C with Rabbit anti-Tmem106b (Bethyl A303-439A, 1:1000, or TMEM106B (EZH7Z) cat# 93334, Cell Signaling) and mouse anti-alpha-tubulin (T6199, Millipore, 1:15000). The blots were washed in Tris-buffered Saline with Tween 20 (TBST), and incubated with Donkey anti Rabbit or Mouse secondary antibodies (Licor 680 or 800) for 2 h at room temperature. Blots were then scanned on a Licor Odyssey DLx system and analyzed with either ImageStudioLite (Licor) or EmperioStudio (Licor) software. Samples were normalized to tubulin to control for equal loading.
Tissues collected for immunohistochemistry
Mice were euthanized using 2.5% tribromoethanol (0.5 mL/25 g body weight) and transcardially exsanguinated with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde (PFA) perfusion for fixation. Spinal cords and brains were dissected and post-fixed overnight in 4% PFA, then transferred into PBS and shipped to NeuroScience Associates, Knoxville, TN, USA for sectioning and histology.
Immunohistochemistry and immunofluorescence in mouse tissue
Lumbar spinal cords and hemi brains were sectioned, stained and quantified as described in Pichon et al.61 Tissues were multiply embedded into a gelatin matrix using MultiBrain Technology (NeuroScience Associates) and each MultiBrain block was sectioned coronally at 25 μm for lumbar spinal cords and at 30 μm for hemi brains. A series of 33 sections, equally spaced at 300 μm intervals throughout the entire lumbar cord and a series of 39 coronal cut sections, equally spaced at 360 μm intervals throughout the entire hemi brain, were used for staining. IHC staining was performed using rat anti-Lamp1 (Abcam #ab25245), goat anti-ChAT (choline acetyltransferase; Millipore #AB144P, AB_2079751), rabbit anti-CatD (Abcam #AB75852), rabbit anti-dMBP (Millipore #AB5864, AB_2140351), rabbit anti-Iba1 (Wako #019–19741, AB_839504), rat anti-CD68 (Bio-Rad #MCA1957T, AB_2074849) counterstained with Nissl (0.05% thionine/0.08M acetate buffer, pH 4.5, Amino cupric silver (de Olmos Stain for degenerating cells), or with solochrome stain (for myelin). To stain for myelin, air dried mounted stained slides were carried through the following sequence: 95% ethanol, 95% ethanol/Formaldehyde; 95% ethanol, 70% ethanol, 5 changes of deionized water (dH2O) then Solochrome staining solution, rinsed with running water, differentiated in Potassium Ferricyanide - Sodium Borate, rinsed in running water. The Solochrome stained slides were counterstained with neutral red solution made in an acetate buffer; pH4.5, dH2O rinses, differentiated in 70% ethanol, 95% ethanol, 100% ethanol, 1:1 100% ethanol/xylene, xylene, then coverslipped with Permount as a bonding medium.
Whole slide imaging for spinal cord was performed at 200× magnification using the Nanozoomer (Hamamatsu Corp, San Jose, CA, USA) system. 10–12 images per mouse in the L3-5 regions were analyzed using MATLAB (Mathworks, Natick, MA, USA). Tissue sections, motor neurons, microglia, and degeneration stains were detected using color thresholds and morphological operations in MATLAB. A regional minima and radial symmetry detection with watershed segmentation followed by morphological, shape and size filtering were used to further enumerate DAB positive cells. Image analysis was performed blinded to experimental grouping and genotype. For brain, 4-5 sections were quantified per region, forebrain (FB) sections quantified fell between (bregma −1.22mm to −2.18) coordinates and the hindbrain (HB) sections quantified fell between (bregma −5.52mm to −6.48) coordinates according to the Allen Brain Atlas. For spinal cord 10 sections from lumbar L3-L5 regions were quantified per animal/stain.
For fluorescent Iba1, NeuN, and CatD unstained sheets (prepared as above) were washed in PBS. Sections were then washed in PBST (PBS with 0.1% Triton X-100), then blocked in 5% bovine serum albumin (BSA) and 5% normal goat serum (NGS) in PBS with 0.3% Triton X-100. Microglia were labeled with rabbit anti-Iba1 (Wako 019–19741, 1:1000), in 1% BSA 1%NGS 0.3% PBS-Triton X100 overnight at 4°C. Sheets of sections were washed 3 × 10 min in PBST, followed by incubation for 2 h at room temperature with secondary antibodies (donkey anti-rabbit, -mouse, or -rat IgG conjugated to Alexa 555 or Alexa 647, Thermo Fisher, 1:500). Sheets of sections were mounted onto slides with 0.1% gelatin in PBS and allowed to dry and adhere to the slide at room temperature. Slides were coverslipped with ProLong Gold Antifade Mountant with DAPI (Molecular Probes; Eugene, OR) and dried.
Lipidomics in mouse tissue
BMP species were analyzed on a Luna NH2 column (3μm, 2.0 × 50 mm) with flow rate at 0.5 mL/min. Gradient started with 17% mobile phase A (97/3 ACN/DCM, 4mM ammonium acetate) and 83% mobile phase B (50/50 ACN/H2O, 4mM ammonium acetate, pH = 8). Mobile phase B ramped to 55% over 9 min and then to 70% from 9 min to 9.5 min. This was subsequently increased to 100% in another minute and was maintained for 13 min and then decreased to original condition for equilibration. Glucosylceramide and glucosylsphingosine were analyzed on a HALO HILIC column (2μm, 3.0 × 50 mm). To separate glucosylsphingosine and galactosylsphingosine, mobile phase A (925/50/25 acetonitrile/IPA/H20, 5mM ammonium formate, 0.5% formic acid) and mobile phase B (925/50/25 H2O/IPA/acetonitrile, 5mM ammonium formate, 0.5% formic acid) were ramped from 98/2 to 90/10 in 8 min and then ramped to 60/40 in 6 min. After rapidly ramped to 10/90 in 2 min, mobile phases returned to original condition for equilibration. To separate glucosylceramide from galactosylceramide, an isocratic gradient was used with mobile phase 94/3/3 ACN/H20/methanol, 5mM ammonium formate, 0.5% formic acid. Flow rate is at 0.4 mL/min.
Mice brain was homogenized in DCM:Methanol(1:1, v/v). After centrifuge, 300ul supernatant was transferred into a v-bottom glass tube. 1mL water, 0.75mL DCM and 1.85mL methanol were added to the supernatant to form a single phase. After 30min, isotope labeled internal standards were added to the mixture, followed by 0.9mL DCM and 1mL water. The mixture was centrifuged at 1000x g for 20 min. Phase separation was achieved after centrifuge. The bottom layer was then collected into a clean glass tube, and the upper layer was re-extracted by adding 1.8mL of DCM. The bottom layer was combined and dried under a gentle stream of nitrogen. The residue was reconstituted in DCM:Methanol (1:1), 10mM ammonium acetate for LipidyzerTM Platform analysis.62 BMP, glucosylceramide and glucosylsphingosine were measured using AB Sciex 6500 LC-MS/MS System. The MRM transitions are 773.5/281.2, 865.5/327.2, 728.3/264.2 and 462.3/282.3 for BMP(18:1/18:1), BMP(22:6/22:6), glucosylceramide(d18:1/18:0) and glucosylsphingosine respectively.
FACS sorting microglia and transcriptomics
Dissociated cells were immunostained, FACS sorted, and RNA was isolated as described by Friedman et al. 2018. Dissociated cells were rotated for 20 min at 4°C with DAPI and the following antibodies targeting neurons, astrocytes, and microglia, respectively: Alexa 488-conjugated anti-NeuN (MAB377X, Millipore Sigma) 1:1,000, PE-conjugated anti-GFAP (561483, BDBiosciences) 1:50, and APC-conjugated anti-CD11b (561690, BD Biosciences) 1:250. Stained cells were centrifuged at 2,000 g for 2 min, washed twice, resuspended in Hibernate A, and 40μm filtered. Cell sorting was performed on a BD FACSAria Fusion equipped with 5 lasers (355, 405, 488, 561, and 638 nm). The instrument was set up with a nozzle size of 100 μm at a frequency of 32 kHz and pressure of 20 psi. The “4-Way Purity” sort mode was used for coincident discrimination. DAPI + signal was used as a parental gate to select for singlet, fixed cell bodies. Further gates were chosen based on isotype controls. CD11b + cells were distinct from other cell populations and gated for microglia. CD11b-cells separated into NeuN+ and NeuN- clusters, of which neurons were gated from the NeuN+ population. All final gates were compared across each of the cell-specific markers described above to assure final cell populations were pure. Unsorted cells were also collected for downstream comparison of TMEM106b mRNA levels, see below. After sorting, cells were centrifuged at 2,600 g for 7 min (neurons) or 6,600 g for 7 min. RNA was extracted from the pellet using the RNeasy Plus Micro kit according to the manufacturer’s instructions (74034, Qiagen). cDNA generation was performed using the Smart-Seq V4 Ultra Low Input RNA Kit (Takara), libraries were generated using the Nextera XT DNA Sample Preparation Kit (Illumina), and sequencing was performed on the NovaSeq 6000.
Tissue processing for electron microscopy
Mouse brains were fixed by perfusion fixation in 4% paraformaldehyde buffered with 0.1 M cacodylate buffer. Brain slices (1 mm thick) were infiltrated with 50% dimethyl sulfoxide for freeze protection, then frozen in liquid nitrogen and freeze-substituted in dry acetone supplemented with 0.01% uranyl acetate and 0.5% glutaraldehyde for 72 h at −78C (on dry ice). The freeze-substituted tissues were then warmed up to room temperature, washed in dimethylformamide and embedded in LR White resin (Electron Microscopy Sciences, Hatfield, PA, USA).
Immunogold labeling and BSE-SEM imaging
Semithin sections (500 nm thickness) were cut with the UMC ultramicrotome (Leica Biosystems, Buffalo Grove, IL, USA) using a DIATOME diamond knife for histology (Electron Microscopy Sciences, Hatfield, PA, USA). Sections were transferred to carbon-coated histology glass slides and dried on a warm plate. Labeling was with a primary (in house, GNE) anti-ASO antibody (rabbit) antibody, a secondary (donkey) anti-rabbit-biotinylated antibody (Jackson ImmunoResearch), followed by streptavidin-conjugated to 20nm colloidal gold particles (Abcam). For a negative labeling control, the primary antibody was omitted. Finally, sections were stained with 2% aqueous uranyl acetate for 15 min and 0.1% Reynold’s lead citrate for 1 min to enhance contrast. Sections were thoroughly rinsed with ultrapure water and dried on a heat plate before being transferred to the SEM.
Scanning electron microscopy (SEM) was performed using a Zeiss Gemini SEM 300 equipped with a field emission gun (Carl Zeiss AG, Oberkochen, Germany). For operation of the GeminiSEM 300 microscope the application software SmartSEM (version 6.01) was used (Carl Zeiss AG, Oberkochen, Germany). Imaging was with a backscatter electron detector (BSD1) at 8.5 mm working distance, 60 μm aperture, 5keV acceleration voltage and with operation of the field emission gun in "high current" mode. For the majority of images, a scan speed of "4″, noise reduction by 5x line averaging and an image size of at least 4096 x 3072 (4k x 3k) pixels was chosen. For imaging of ultrastructural detail pixel sizes between 2 and 5 nm were used. The greyscale of the images was inverted to achieve “TEM-like” representations.
iMicroglia differentiation
The PGP1 iPS cell line was obtained from Synthego and the knockout lines were generated using a nucleofection-based RNP approach to introduce Cas9 and sgRNAs against TMEM106b, GRN and TMEM106b/GRN (DKO). PGP1 WT, TKO, GKO and DKO iPSCs derived microglia were generated using the StemDiff hematopoietic kit (stemcell technologies cat #05310) from days 0–12, and subsequently the StemDiff microglia differentiation (stemcell technologies cat #100-0019) and maturation (cat #100-0020) kits from days 13–40+ with some adaptations to increase cell yield.
On day −1 PGP1 iPSCs colonies were seeded in T75 flasks, aiming for an average colony size of 80–150 microns. On day 0, mTeSR medium was changed out for HPSC media plus supplement A. A half media change was performed on day 2. On day 3, the media was changed to HPSC media plus supplement B. From day 5 to day 10, half media changes were performed by collecting half the media volume, centrifuging at 400g for 8 min, aspirating spent medium while leaving HPSCs undisturbed, replacing the medium and returning to the flask. On day 12, 4.5M HPSCs from each line were harvested for microglia differentiation and replated on matrigel coated T225 flasks (add density of cells). From days 12–23 cells were fed every two days with microglia differentiation medium plus supplement 1 and 2, by topping off the media without removing any conditioned media. On day 24, iMicroglia were harvested and replated on matrigel coated flasks (cell density of 1 x 10ˆ5 cells/cm2). On day 36, microglia were plated for maturation onto their final assay plates (density varies based on application) with microglia differentiation medium plus supplements 1, 2 and 3. iMicroglia were allowed to mature for a maximum of 5–6 days before assaying.
Immunohistochemistry & confocal microscopy
iMicroglia were fixed in 4% PFA for 20 min at room temperature. For staining, cells were permeabilized with 0.05% Triton X- in 1x PBS, 5 min incubation at RT, then blocked for 1 h in 0.5% BSA +5% Donkey serum in PBS-T. Following blocking, cells were incubated overnight at 4C with anti-Aif1(Wako Fuji Film) and anti-Trem2 primary antibodies (R&D Systems) diluted in blocking solution at 1:200. The next day, after 4 washes with PBS-T, cells were incubated with secondary antibodies diluted in blocking solution, protected for light at room temperature for 2 h. Nuclei were counterstained with DAPI (Thermofisher), and 1–2 drops/well of 96w of gold antifade (Thermofisher) was used to preserve fluorescence. Cells were imaged on a PerkinElmer high content PHENIX confocal microscope.
iMicroglia phagocytosis assays
iPSC derived microglia lines were seeded at 30,000 cells per well of a 96 well plate coated with 10ug/mL PDL at day 36 of microglia differentiation. At day 40, cells were treated with 10ug/mL pHrodo green zymosan particles (cat #P35365), prepared according to manufacturer’s instructions, and imaged every hour for 48 h on an Incucyte S3 (Sartorius). Phagocytosis was observed as total integrated fluorescent intensity per image.
iMicroglia RNA isolation and transcriptomics
Total RNA was quantified with Qubit RNA HS Assay Kit (Thermo Fisher Scientific) and quality was assessed using RNA ScreenTape on 4200 TapeStation (Agilent Technologies). For sequencing library generation, the Truseq Stranded mRNA kit (Illumina) was used with an input of 100 ng of total RNA. Libraries were quantified with Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific) and the average library size was determined using D1000 ScreenTape on 4200 TapeStation (Agilent Technologies). Libraries were pooled and sequenced on NovaSeq 6000 (Illumina) to generate 30 million single-end 50-base pair reads for each sample. GSE237107 is the reference Series for the publication.
iMicroglia proteomics
Cells were lysed in a denaturing buffer containing 8M urea, 150 mM NaCl, 50 mM HEPES (pH 8.0), one complete-mini (EDTA free) protease inhibitor (Roche), and one phosphatase inhibitor (PHOSstop) tablet (Roche). Lysates were clarified via high-speed centrifugation (18,000 x g, 15 min) to remove insoluble material and protein concentrations were then determined by Bradford assay. 100 μg of lysate per condition were taken forward for subsequent processing. Lysates were reduced (4.1 mM dithiothreitol, 60 min at 37°C) and alkylated (9.1 mM iodoacetamide, 15 min at room temperature). Lysates were diluted 4-fold and subjected to overnight enzymatic digestion at 37°C with a combination of lysyl-endopeptidase (Wako) and sequencing grade trypsin (Promega), both at an enzyme to protein ratio of 1:50, the latter of which was added to the sample 3 h post incubation with the former. Resultant peptides were acidified with 20% TFA to a final concentration of 1%, clarified via high-speed centrifugation (18,000 x g, 10 min) prior to desalting via SOLA HRP solid phase extraction (Thermo).
Each sample was resuspended with 100 μL of 100 mM HEPES pH 8.0 and labeled with a full vial of TMT16plex labeling reagent (Thermo) for 1 h. Here 12 of 16 tags were utilized (TMT-126 to TMT-132N) corresponding to 12 samples. The reaction was quenched by the addition of 5 μL of 5% hydroxylamine (15 min at room temperature), combined and desalted using via C18 Sep-Pak (Waters) solid phase extraction and eluents were dried to completion.
The TMT-labeled sample was redissolved in 0.15% trifluoracetic acid and subjected to centrifugation at 16,000 x g to remove insoluble material with only the supernatant taken forward for further processing. Offline high pH reversed phase liquid chromatography separation was performed using an Agilent 1100 series HPLC system. Peptides were loaded onto an Agilent Zorbax 300 Extend C-18 analytical column (2.1 × 150 mm and 3.5 μm particle size, Part No: 763750-902). Solvent A consisted of 25 mM Ammonium Formate (pH 9.7), while solvent B was 100% acetonitrile. Peptide fractions were collected in 45 s intervals for a total of 63 min with a linear gradient of 15–60% solvent B. In total 96 fractions were collected and every 25th fraction was combined to form a final set of 24 fractions. Samples were dried in a speedvac, desalted using C18 StageTips as previously described and injected for nLC-MS/MS analysis.63
Each of the 24 samples was reconstituted in 2% acetonitrile (ACN)/0.1% formic acid (FA) and analyzed by nLC-MS/MS on a Dionex UltiMate 3000 RSLCnano Proflow system (Thermo Fisher Scientific) connected to an Orbitrap Eclipse mass spectrometer (Thermo Fisher Scientific). Each sample was analyzed three times, once with a 95 min method and twice with a 185 min method. Peptides were loaded onto an Aurora Series 25 cm x 75 μm I.D. column (IonOpticks) and separated at a flow rate of 300 nL/min over a two-stage linear gradient where buffer B (98% ACN, 2% H2O, 0.1% formic acid) increased from 4% to 30% over 68min (95 min method) or 135 min (185 min method) and then from 30% to 75% over 5.9 min (95 min method) or 15 min (185 min method). SPS-MS3 was implemented for improved quantitative accuracy.64,65 For these analyses, a real-time search against a human database was employed using either the capabilities built within the XCalibur method editor or an in-house instrument API program called InSeqAPI.66 For all runs, intact peptides were surveyed in the Orbitrap (250% normalized AGC target, 120,000 resolution) and the top 10 peptides were selected for MS2 fragmentation (150% normalized AGC target; CID, 30 NCE) and analyzed in the ion trap. Quantitative MS3 scans were performed by selection of the 8 most abundant fragment ions from MS2 spectra and fragmentation at high energy (HCD, 40 NCE) to produce reporter mass ions that were analyzed in the Orbitrap (30,000 resolution). Raw datafiles were deposited into the MassIVE repository with the identifier: MSV000090254 (reviewer login = MSV000090254_reviewer & password = tmem).
MS/MS spectra were searched using the MASCOT search algorithm (Matrix Science) against a concatenated target-decoy database of human proteins (UniProt DB version 2021_04) containing common contaminant sequences. Search parameters included trypsin cleavage with an allowance of up to 2 missed cleavage events, a precursor ion tolerance of 50 ppm, and a fragment ion tolerance of 0.8 Da. Searches permitted variable modifications of methionine oxidation (+15.9949 Da) with static modifications for cysteine carbamidomethylation (+57.0215) and TMT tags on lysine and peptide N termini (+304.2071). A false discovery rate (FDR) of 2% was used to filter peptide spectra matches (PSMs) on the peptide level, and subsequently 2% on the protein level using linear discrimination. TMT reporter ions produced by the TMT tags were quantified with an in-house software package known as Mojave by calculating the highest peak within 20 ppm of theoretical reporter mass windows and correcting for isotope purities.66
Statistics
Statistical analyses for in vivo data were performed using Graphpad Prism v9.0 and IHC imaging analysis was conducted in MATLAB (Mathworks, Natick, MA). Unless otherwise indicated, in vivo data were analyzed using a one-way ANOVA followed by Dunnett’s post-hoc test for treatment effects. RNA-seq analysis was performed using the GSNAP aligner,54 HTSeqGenie,55 and voom-limma,56 as previously described.67 For the RNA-seq heatmap, gene expression values were batch corrected using ComBat-Seq.57 For proteomics, The R package MSstatsTMT v.2.0.1 was used to preprocess PSM-level quantification before statistical analysis, to have protein quantification and to perform differential abundance analysis.53 MSstatsTMT estimated log2(fold change) and the standard error by linear mixed effect model for each protein. The inference procedure was adjusted by applying an empirical Bayes shrinkage. To test two-sided null hypothesis of no changes in abundance, the model-based test statistics were compared to the Student’s t test distribution with the degrees of freedom appropriate for each protein and each dataset. The resulting p values were adjusted to control the FDR with the method by Benjamini–Hochberg.
Acknowledgments
Merone Roose-Girma and Charles Wong for creating TMEM106B KO mice, Kim Stark for histology and NFL outsourcing support, Shannon Hambro, Alison Huynh, and Sean Flanagan for necropsy support. Jill Yamada and Emmanuel Chua for Veterinary support and Yan Wu and Genentech Antibody Engineering department for generating the anti-ASO antibody.
Author contributions
S.L.D. conducted and designed all mouse experiments including qPCR, analyzed data, and created figures; B.I.L. ran bioinformatics for mouse and human models, created figures, and graphical abstract; A.S.G. sorted microglia, isolated RNA, and harvested tissue; Q.L. ran and analyzed lipidomics, created figures; G.R. generated iMG, generated imaging data and figure; M.E.R. designed and conducted iMG studies; L.P. conducted proteomics; B.F. supported bioinformatics analyses; W.S. provided lipidomics experimental design and analysis support; C.M.R. provided proteomics analysis; H.N. analyzed in vivo staining; O.F. provided imaging and EM support; M.R. conducted EM; Y.J. ran TMEM106B westerns; G.L. conducted IF in KO mouse tissue; D.H. synthesized ASOs; H.N. screened ASOs; D.M. tested stability of ASOs; H.G. generated in vitro potency of ASOs; Ł.J.K. designed ASOs, provided experimental guidance; B.C. generated iMG cell lines and input on experimental design; B.B. provided analytical input for in vivo studies; W.J.M. conducted westerns, provided experimental design input, generated data, figures, and results text for in vivo studies; A.E. and C.C.H. conceptualized experiments, provided analysis input, and wrote the manuscript.
Declaration of interests
The authors have a published patent application WO 2019/154979 A1 related to this work.
Published: October 29, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108362.
Contributor Information
William J. Meilandt, Email: meilandt.william@gene.com.
Amy Easton, Email: amy.easton@gmail.com.
Supplemental information
References
- 1.Van Deerlin V.M., Sleiman P.M.A., Martinez-Lage M., Chen-Plotkin A., Wang L.S., Graff-Radford N.R., Dickson D.W., Rademakers R., Boeve B.F., Grossman M., et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat. Genet. 2010;42:234–239. doi: 10.1038/ng.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finch N., Carrasquillo M.M., Baker M., Rutherford N.J., Coppola G., Dejesus-Hernandez M., Crook R., Hunter T., Ghidoni R., Benussi L., et al. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology. 2011;76:467–474. doi: 10.1212/WNL.0b013e31820a0e3b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van der Zee J., Van Langenhove T., Kleinberger G., Sleegers K., Engelborghs S., Vandenberghe R., Santens P., Van den Broeck M., Joris G., Brys J., et al. TMEM106B is associated with frontotemporal lobar degeneration in a clinically diagnosed patient cohort. Brain. 2011;134:808–815. doi: 10.1093/brain/awr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cruchaga C., Graff C., Chiang H.H., Wang J., Hinrichs A.L., Spiegel N., Bertelsen S., Mayo K., Norton J.B., Morris J.C., Goate A. Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch. Neurol. 2011;68:581–586. doi: 10.1001/archneurol.2010.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen-Plotkin A.S., Unger T.L., Gallagher M.D., Bill E., Kwong L.K., Volpicelli-Daley L., Busch J.I., Akle S., Grossman M., Van Deerlin V., et al. TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways. J. Neurosci. 2012;32:11213–11227. doi: 10.1523/JNEUROSCI.0521-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Busch J.I., Martinez-Lage M., Ashbridge E., Grossman M., Van Deerlin V.M., Hu F., Lee V.M.Y., Trojanowski J.Q., Chen-Plotkin A.S. Expression of TMEM106B, the frontotemporal lobar degeneration-associated protein, in normal and diseased human brain. Acta Neuropathol. Commun. 2013;1:36. doi: 10.1186/2051-5960-1-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nicholson A.M., Finch N.A., Wojtas A., Baker M.C., Perkerson R.B., 3rd, Castanedes-Casey M., Rousseau L., Benussi L., Binetti G., Ghidoni R., et al. TMEM106B p.T185S regulates TMEM106B protein levels: implications for frontotemporal dementia. J. Neurochem. 2013;126:781–791. doi: 10.1111/jnc.12329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Busch J.I., Unger T.L., Jain N., Tyler Skrinak R., Charan R.A., Chen-Plotkin A.S. Increased expression of the frontotemporal dementia risk factor TMEM106B causes C9orf72-dependent alterations in lysosomes. Hum. Mol. Genet. 2016;25:2681–2697. doi: 10.1093/hmg/ddw127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou X., Sun L., Brady O.A., Murphy K.A., Hu F. Elevated TMEM106B levels exaggerate lipofuscin accumulation and lysosomal dysfunction in aged mice with progranulin deficiency. Acta Neuropathol. Commun. 2017;5:9. doi: 10.1186/s40478-017-0412-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Root J., Merino P., Nuckols A., Johnson M., Kukar T. Lysosome dysfunction as a cause of neurodegenerative diseases: Lessons from frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol. Dis. 2021;154 doi: 10.1016/j.nbd.2021.105360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holler C.J., Taylor G., Deng Q., Kukar T. Intracellular Proteolysis of Progranulin Generates Stable, Lysosomal Granulins that Are Haploinsufficient in Patients with Frontotemporal Dementia Caused by GRN Mutations. eNeuro. 2017;4 doi: 10.1523/ENEURO.0100-17.2017. ENEURO.0100-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee C.W., Stankowski J.N., Chew J., Cook C.N., Lam Y.W., Almeida S., Carlomagno Y., Lau K.F., Prudencio M., Gao F.B., et al. The lysosomal protein cathepsin L is a progranulin protease. Mol. Neurodegener. 2017;12:55. doi: 10.1186/s13024-017-0196-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith K.R., Damiano J., Franceschetti S., Carpenter S., Canafoglia L., Morbin M., Rossi G., Pareyson D., Mole S.E., Staropoli J.F., et al. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am. J. Hum. Genet. 2012;90:1102–1107. doi: 10.1016/j.ajhg.2012.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Almeida M.R., Macário M.C., Ramos L., Baldeiras I., Ribeiro M.H., Santana I. Portuguese family with the co-occurrence of frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis phenotypes due to progranulin gene mutation. Neurobiol. Aging. 2016;41:200.e1–200.e5. doi: 10.1016/j.neurobiolaging.2016.02.019. [DOI] [PubMed] [Google Scholar]
- 15.Götzl J.K., Mori K., Damme M., Fellerer K., Tahirovic S., Kleinberger G., Janssens J., van der Zee J., Lang C.M., Kremmer E., et al. Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol. 2014;127:845–860. doi: 10.1007/s00401-014-1262-6. [DOI] [PubMed] [Google Scholar]
- 16.Valdez C., Wong Y.C., Schwake M., Bu G., Wszolek Z.K., Krainc D. Progranulin-mediated deficiency of cathepsin D results in FTD and NCL-like phenotypes in neurons derived from FTD patients. Hum. Mol. Genet. 2017;26:4861–4872. doi: 10.1093/hmg/ddx364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ward M.E., Chen R., Huang H.Y., Ludwig C., Telpoukhovskaia M., Taubes A., Boudin H., Minami S.S., Reichert M., Albrecht P., et al. Individuals with progranulin haploinsufficiency exhibit features of neuronal ceroid lipofuscinosis. Sci. Transl. Med. 2017;9 doi: 10.1126/scitranslmed.aah5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang M.C., Srinivasan K., Friedman B.A., Suto E., Modrusan Z., Lee W.P., Kaminker J.S., Hansen D.V., Sheng M. Progranulin deficiency causes impairment of autophagy and TDP-43 accumulation. J. Exp. Med. 2017;214:2611–2628. doi: 10.1084/jem.20160999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang M., Modeste E., Dammer E., Merino P., Taylor G., Duong D.M., Deng Q., Holler C.J., Gearing M., Dickson D., et al. Network analysis of the progranulin-deficient mouse brain proteome reveals pathogenic mechanisms shared in human frontotemporal dementia caused by GRN mutations. Acta Neuropathol. Commun. 2020;8:163. doi: 10.1186/s40478-020-01037-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahmed Z., Sheng H., Xu Y.F., Lin W.L., Innes A.E., Gass J., Yu X., Wuertzer C.A., Hou H., Chiba S., et al. Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am. J. Pathol. 2010;177:311–324. doi: 10.2353/ajpath.2010.090915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanaka Y., Chambers J.K., Matsuwaki T., Yamanouchi K., Nishihara M. Possible involvement of lysosomal dysfunction in pathological changes of the brain in aged progranulin-deficient mice. Acta Neuropathol. Commun. 2014;2:78. doi: 10.1186/s40478-014-0078-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lui H., Zhang J., Makinson S.R., Cahill M.K., Kelley K.W., Huang H.Y., Shang Y., Oldham M.C., Martens L.H., Gao F., et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell. 2016;165:921–935. doi: 10.1016/j.cell.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krabbe G., Minami S.S., Etchegaray J.I., Taneja P., Djukic B., Davalos D., Le D., Lo I., Zhan L., Reichert M.C., et al. Microglial NFκB-TNFα hyperactivation induces obsessive-compulsive behavior in mouse models of progranulin-deficient frontotemporal dementia. Proc. Natl. Acad. Sci. USA. 2017;114:5029–5034. doi: 10.1073/pnas.1700477114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feng T., Lacrampe A., Hu F. Physiological and pathological functions of TMEM106B: a gene associated with brain aging and multiple brain disorders. Acta Neuropathol. 2021;141:327–339. doi: 10.1007/s00401-020-02246-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klein Z.A., Takahashi H., Ma M., Stagi M., Zhou M., Lam T.T., Strittmatter S.M. Loss of TMEM106B Ameliorates Lysosomal and Frontotemporal Dementia-Related Phenotypes in Progranulin-Deficient Mice. Neuron. 2017;95:281–296.e6. doi: 10.1016/j.neuron.2017.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arrant A.E., Nicholson A.M., Zhou X., Rademakers R., Roberson E.D. Partial Tmem106b reduction does not correct abnormalities due to progranulin haploinsufficiency. Mol. Neurodegener. 2018;13:32. doi: 10.1186/s13024-018-0264-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou X., Brooks M., Jiang P., Koga S., Zuberi A.R., Baker M.C., Parsons T.M., Castanedes-Casey M., Phillips V., Librero A.L., et al. Loss of Tmem106b exacerbates FTLD pathologies and causes motor deficits in progranulin-deficient mice. EMBO Rep. 2020;21 doi: 10.15252/embr.202050197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feng T., Mai S., Roscoe J.M., Sheng R.R., Ullah M., Zhang J., Katz I.I., Yu H., Xiong W., Hu F. Loss of TMEM106B and PGRN leads to severe lysosomal abnormalities and neurodegeneration in mice. EMBO Rep. 2020;21 doi: 10.15252/embr.202050219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Werner G., Damme M., Schludi M., Gnörich J., Wind K., Fellerer K., Wefers B., Wurst W., Edbauer D., Brendel M., et al. Loss of TMEM106B potentiates lysosomal and FTLD-like pathology in progranulin-deficient mice. EMBO Rep. 2020;21 doi: 10.15252/embr.202050241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chitu V., Biundo F., Shlager G.G.L., Park E.S., Wang P., Gulinello M.E., Gokhan Ş., Ketchum H.C., Saha K., DeTure M.A., et al. Microglial Homeostasis Requires Balanced CSF-1/CSF-2 Receptor Signaling. Cell Rep. 2020;30:3004–3019.e5. doi: 10.1016/j.celrep.2020.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaheen R., Tasak M., Maddirevula S., Abdel-Salam G.M.H., Sayed I.S.M., Alazami A.M., Al-Sheddi T., Alobeid E., Phizicky E.M., Alkuraya F.S. PUS7 mutations impair pseudouridylation in humans and cause intellectual disability and microcephaly. Hum. Genet. 2019;138:231–239. doi: 10.1007/s00439-019-01980-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mills P.B., Surtees R.A.H., Champion M.P., Beesley C.E., Dalton N., Scambler P.J., Heales S.J.R., Briddon A., Scheimberg I., Hoffmann G.F., et al. Neonatal epileptic encephalopathy caused by mutations in the PNPO gene encoding pyridox(am)ine 5′-phosphate oxidase. Hum. Mol. Genet. 2005;14:1077–1086. doi: 10.1093/hmg/ddi120. [DOI] [PubMed] [Google Scholar]
- 33.Kayasuga Y., Chiba S., Suzuki M., Kikusui T., Matsuwaki T., Yamanouchi K., Kotaki H., Horai R., Iwakura Y., Nishihara M. Alteration of behavioural phenotype in mice by targeted disruption of the progranulin gene. Behav. Brain Res. 2007;185:110–118. doi: 10.1016/j.bbr.2007.07.020. [DOI] [PubMed] [Google Scholar]
- 34.Lüningschrör P., Werner G., Stroobants S., Kakuta S., Dombert B., Sinske D., Wanner R., Lüllmann-Rauch R., Wefers B., Wurst W., et al. The FTLD Risk Factor TMEM106B Regulates the Transport of Lysosomes at the Axon Initial Segment of Motoneurons. Cell Rep. 2020;30:3506–3519.e6. doi: 10.1016/j.celrep.2020.02.060. [DOI] [PubMed] [Google Scholar]
- 35.Marschallinger J., Iram T., Zardeneta M., Lee S.E., Lehallier B., Haney M.S., Pluvinage J.V., Mathur V., Hahn O., Morgens D.W., et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 2020;23:194–208. doi: 10.1038/s41593-019-0566-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feng T., Luan L., Katz I.I., Ullah M., Van Deerlin V.M., Trojanowski J.Q., Lee E.B., Hu F. TMEM106B deficiency impairs cerebellar myelination and synaptic integrity with Purkinje cell loss. Acta Neuropathol. Commun. 2022;10:33. doi: 10.1186/s40478-022-01334-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu Y., Sun J.Y., Zhang Y., Zhang H., Gao S., Wang T., Han Z., Wang L., Sun B.L., Liu G. rs1990622 variant associates with Alzheimer’s disease and regulates TMEM106B expression in human brain tissues. BMC Med. 2021;19:11. doi: 10.1186/s12916-020-01883-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Byrnes A.E., Dominguez S.L., Yen C.W., Laufer B.I., Foreman O., Reichelt M., Lin H., Sagolla M., Hötzel K., Ngu H., et al. Lipid nanoparticle delivery changes antisense oligonucleotide activity and cellular distribution in the brain. Mol. Ther. Nucleic Acids. 2023;32:773–793. doi: 10.1016/j.omtn.2023.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou X., Nicholson A.M., Ren Y., Brooks M., Jiang P., Zuberi A., Phuoc H.N., Perkerson R.B., Matchett B., Parsons T.M., et al. Loss of TMEM106B leads to myelination deficits: implications for frontotemporal dementia treatment strategies. Brain. 2020;143:1905–1919. doi: 10.1093/brain/awaa141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo D.Z., Xiao L., Liu Y.J., Shen C., Lou H.F., Lv Y., Pan S.Y. Cathepsin D deficiency delays central nervous system myelination by inhibiting proteolipid protein trafficking from late endosome/lysosome to plasma membrane. Exp. Mol. Med. 2018;50:e457. doi: 10.1038/emm.2017.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hersheson J., Burke D., Clayton R., Anderson G., Jacques T.S., Mills P., Wood N.W., Gissen P., Clayton P., Fearnley J., et al. Cathepsin D deficiency causes juvenile-onset ataxia and distinctive muscle pathology. Neurology. 2014;83:1873–1875. doi: 10.1212/WNL.0000000000000981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beel S., Moisse M., Damme M., De Muynck L., Robberecht W., Van Den Bosch L., Saftig P., Van Damme P. Progranulin functions as a cathepsin D chaperone to stimulate axonal outgrowth in vivo. Hum. Mol. Genet. 2017;26:2850–2863. doi: 10.1093/hmg/ddx162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou X., Paushter D.H., Feng T., Pardon C.M., Mendoza C.S., Hu F. Regulation of cathepsin D activity by the FTLD protein progranulin. Acta Neuropathol. 2017;134:151–153. doi: 10.1007/s00401-017-1719-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schulze H., Sandhoff K. Lysosomal lipid storage diseases. Cold Spring Harbor Perspect. Biol. 2011;3 doi: 10.1101/cshperspect.a004804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Logan T., Simon M.J., Rana A., Cherf G.M., Srivastava A., Davis S.S., Low R.L.Y., Chiu C.L., Fang M., Huang F., et al. Rescue of a lysosomal storage disorder caused by Grn loss of function with a brain penetrant progranulin biologic. Cell. 2021;184:4651–4668.e25. doi: 10.1016/j.cell.2021.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stroobants S., D'Hooge R., Damme M. Aged Tmem106B knockout mice display gait deficits in coincidence with Purkinje cell loss and only limited signs of non-motor dysfunction. Brain Pathol. 2021;31:223–238. doi: 10.1111/bpa.12903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kundu S.T., Grzeskowiak C.L., Fradette J.J., Gibson L.A., Rodriguez L.B., Creighton C.J., Scott K.L., Gibbons D.L. TMEM106B drives lung cancer metastasis by inducing TFEB-dependent lysosome synthesis and secretion of cathepsins. Nat. Commun. 2018;9:2731. doi: 10.1038/s41467-018-05013-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simons C., Dyment D., Bent S.J., Crawford J., D'Hooghe M., Kohlschütter A., Venkateswaran S., Helman G., Poll-The B.T., Makowski C.C., et al. A recurrent de novo mutation in TMEM106B causes hypomyelinating leukodystrophy. Brain. 2017;140:3105–3111. doi: 10.1093/brain/awx314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang H.S., White C.C., Klein H.U., Yu L., Gaiteri C., Ma Y., Felsky D., Mostafavi S., Petyuk V.A., Sperling R.A., et al. Genetics of Gene Expression in the Aging Human Brain Reveal TDP-43 Proteinopathy Pathophysiology. Neuron. 2020;107:496–508.e6. doi: 10.1016/j.neuron.2020.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang A., Xiang X., Wang J., Lee C., Arakhamia T., Simjanoska M., Wang C., Carlomagno Y., Zhang G., Dhingra S., et al. Homotypic fibrillization of TMEM106B across diverse neurodegenerative diseases. Cell. 2022;185:1346–1355.e15. doi: 10.1016/j.cell.2022.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shafit-Zagardo B., Sidoli S., Goldman J.E., DuBois J.C., Corboy J.R., Strittmatter S.M., Guzik H., Edema U., Arackal A.G., Botbol Y.M., et al. TMEM106B puncta is increased in Multiple Sclerosis plaques, and reduced protein in mice causes delayed lipid clearance after CNS injury. Cells. 2023;12:1734. doi: 10.3390/cells12131734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huttlin E.L., Jedrychowski M.P., Elias J.E., Goswami T., Rad R., Beausoleil S.A., Villén J., Haas W., Sowa M.E., Gygi S.P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang T., Choi M., Tzouros M., Golling S., Pandya N.J., Banfai B., Dunkley T., Vitek O. MSstatsTMT: Statistical Detection of Differentially Abundant Proteins in Experiments with Isobaric Labeling and Multiple Mixtures. Mol. Cell. Proteomics. 2020;19:1706–1723. doi: 10.1074/mcp.RA120.002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu T.D., Reeder J., Lawrence M., Becker G., Brauer M.J. GMAP and GSNAP for Genomic Sequence Alignment: Enhancements to Speed, Accuracy, and Functionality. Methods Mol. Biol. 2016;1418:283–334. doi: 10.1007/978-1-4939-3578-9_15. [DOI] [PubMed] [Google Scholar]
- 55.Pau G., Reeder J. Bioconductor version: Release (3.12) 2021. HTSeqGenie: A NGS Analysis Pipeline. [Google Scholar]
- 56.Law C.W., Chen Y., Shi W., Smyth G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Y., Parmigiani G., Johnson W.E. ComBat-seq: batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform. 2020;2:lqaa078. doi: 10.1093/nargab/lqaa078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Modzelewski A.J., Chen S., Willis B.J., Lloyd K.C.K., Wood J.A., He L. Efficient mouse genome engineering by CRISPR-EZ technology. Nat. Protoc. 2018;13:1253–1274. doi: 10.1038/nprot.2018.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Easton A., Jensen M.L., Wang C., Hagedorn P.H., Li Y., Weed M., Meredith J.E., Guss V., Jones K., Gill M., et al. Identification and characterization of a MAPT-targeting locked nucleic acid antisense oligonucleotide therapeutic for tauopathies. Mol. Ther. Nucleic Acids. 2022;29:625–642. doi: 10.1016/j.omtn.2022.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dominguez S., Varfolomeev E., Brendza R., Stark K., Tea J., Imperio J., Ngu H., Earr T., Foreman O., Webster J.D., et al. Genetic inactivation of RIP1 kinase does not ameliorate disease in a mouse model of ALS. Cell Death Differ. 2021;28:915–931. doi: 10.1038/s41418-020-00625-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Le Pichon C.E., Dominguez S.L., Solanoy H., Ngu H., Lewin-Koh N., Chen M., Eastham-Anderson J., Watts R., Scearce-Levie K. EGFR inhibitor erlotinib delays disease progression but does not extend survival in the SOD1 mouse model of ALS. PLoS One. 2013;8 doi: 10.1371/journal.pone.0062342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cao Z., Schmitt T.C., Varma V., Sloper D., Beger R.D., Sun J. Evaluation of the Performance of Lipidyzer Platform and Its Application in the Lipidomics Analysis in Mouse Heart and Liver. J. Proteome Res. 2020;19:2742–2749. doi: 10.1021/acs.jproteome.9b00289. [DOI] [PubMed] [Google Scholar]
- 63.Rappsilber J., Mann M., Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007;2:1896–1906. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- 64.Ting L., Rad R., Gygi S.P., Haas W. MS3 Eliminates Ratio Distortion in Isobaric Multiplexed Quantitative Proteomics. Nat. Methods. 2011;8:937–940. doi: 10.1038/nmeth.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McAlister G.C., Nusinow D.P., Jedrychowski M.P., Wühr M., Huttlin E.L., Erickson B.K., Rad R., Haas W., Gygi S.P. MultiNotch MS3 Enables Accurate, Sensitive, and Multiplexed Detection of Differential Expression across Cancer Cell Line Proteomes. Anal. Chem. 2014;86:7150–7158. doi: 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhuang G., Yu K., Jiang Z., Chung A., Yao J., Ha C., Toy K., Soriano R., Haley B., Blackwood E., et al. Phosphoproteomic Analysis Implicates the MTORC2-FoxO1 Axis in VEGF Signaling and Feedback Activation of Receptor Tyrosine Kinases. Sci. Signal. 2013;6:ra25. doi: 10.1126/scisignal.2003572. [DOI] [PubMed] [Google Scholar]
- 67.Friedman B.A., Srinivasan K., Ayalon G., Meilandt W.J., Lin H., Huntley M.A., Cao Y., Lee S.H., Haddick P.C.G., Ngu H., et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer's Disease Not Evident in Mouse Models. Cell Rep. 2018;22:832–847. doi: 10.1016/j.celrep.2017.12.066. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data
-
•
Transcriptomic data from mouse and human microglia has been deposited in GEO and will be publicly available (GSE237107 is the reference series).
-
•
Proteomic raw datafiles were deposited into the MassIVE repository with the identifier: Massive: MSV000090254 (reviewer login = MSV000090254_reviewer & password = tmem).
Code
-
•
This study did not generate original code.