

Abstract
Tricyclic tetrahydroquinolines (THQs) have been repeatedly reported as hits across a diverse range of high-throughput screening (HTS) campaigns. The activities of these compounds, however, are likely due to reactive byproducts that interfere with the assay. As a lesser studied class of pan-assay interference compounds, the mechanism by which fused THQs react with protein targets remains largely unknown. During HTS follow-up, we characterized the behavior and stability of several fused tricyclic THQs. We synthesized key analogues to pinpoint the cyclopentene ring double bond as a source of reactivity of fused THQs. We found that these compounds degrade in solution under standard laboratory conditions in days. Importantly, these observations make it likely that fused THQs, which are ubiquitously found within small molecule screening libraries, are unlikely the intact parent compounds. We urge deprioritization of tricylic THQ hits in HTS follow-up and caution against the investment of resources to follow-up on these problematic compounds.
1. Significance
Tricyclic tetrahydroquinolines (THQs) are a family of lesser studied pan-assay interference compounds (PAINS). These compounds are found ubiquitously throughout commercial and academic small molecule screening libraries. Accordingly, they have been identified as hits in high-throughput screening campaigns for diverse protein targets. We demonstrate that fused THQs are reactive when stored in solution under standard laboratory conditions and caution investigators from investing additional resource into validating these nuisance compounds.
2. Introduction
Fused tetrahydroquinolines (THQs) are frequent hitters in hit discovery campaigns. Pan-assay interference compounds (PAINS) have been controversial in the recent literature. While some literature supports these as nuisance compounds, other papers describe PAINS as potentially valuable leads.1−4 There have been descriptions of many different classes of PAINS that vary in their frequency of occurrence as hits in the screening literature. Alkylidene barbiturates and 2-hydroxy-phenyl-hydrazones (Figure 1) are examples of enriched PAINS substructures.5 Fused THQs have been identified in multiple screens (Figure 3), some of which have flagged them as false positives or frequent hitters.4,6 Although these compounds have generally good calculated physicochemical properties, no optimized chemical probes or approved drugs contain the chemotype. Still, some fused THQs have been evaluated in vivo and were found to be very poorly behaved as well as demonstrate suboptimal pharmacokinetic properties.7 The number of papers that selected this scaffold during hit discovery campaigns from multiple chemical libraries supports the idea that fused THQs are frequent hitters. At first glance, these compounds appear to be valid, optimizable hits, with reasonable physicochemical properties. Although micromolar and reproducible activity has been reported for multiple THQ analogues on many protein targets, hit-to-lead optimization programs aimed at improving the initial hits (Supporting Information (SI), Table S1) have resulted in no improvement in potency or no discernible structure–activity relationships (SAR). We stress the importance of flagging and/or removing scaffolds from HTS screening collections that generate aberrant results to break the cycle of hit identification and investment of follow-up chemistry on nonprogressable compounds.
Figure 1.

Structures of common PAINS and fused/tricyclic THQs.
Figure 3.
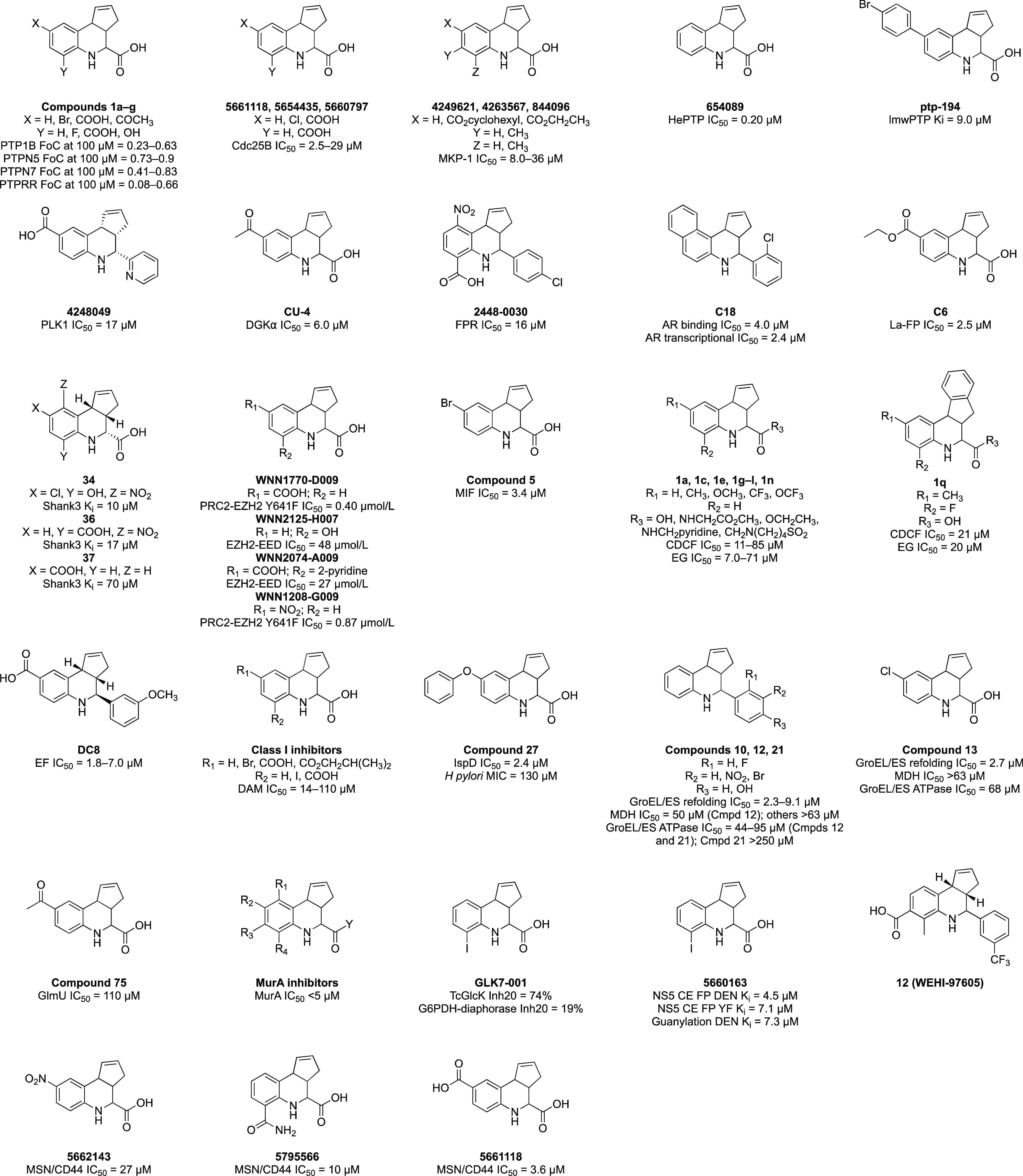
Structures of tricyclic THQs that are exemplified in the literature as screening hits.
2.1. Many Fused THQ Analogues Are Present in Chemical Screening Libraries
The fused/tricyclic THQ scaffold is shown in Figure 1. Analogues have been made with high chemical diversification at the R1–R5 groups with alkyl, aryl, halogen, carboxylic acid, ether, ester, nitro, acyl, amide, and other functional groups. A carboxylic acid is a frequently observed R5 substituent due to its ease of synthesis. This diversification of the fused THQ chemotype has led to a plethora of analogues being included in academic and commercial small molecule screening libraries. Notably, several commercial vendors have screening libraries that include fused THQs available for purchase. A substructure search in a widely used chemical compound database (SciFinder) for the generic structure in Figure 1, where R1–R5 = H yielded over 19 000 compounds bearing the fused THQ core, 15 867 of which were available for purchase by one or more vendor. A deeper dive into the results showed that 1456 of these compounds were available for purchase from more than one vendor. This clearly illustrates that these compounds are very easy to obtain for confirmatory experiments, exacerbating the situation and maximizing the potential expense spent following up on these false leads.
2.2. Few Structures of Protein-Bound Tricyclic THQs Have Been Solved
A search of the PDB for fused THQs yielded only four structures for two proteins. The specific search term queried in the PDB to yield the total answer set of four structures was “tetrahydro-3H-cyclopenta[c]quinoline”. A single peer-reviewed paper described the structure of Shank3 bound to a fused THQ (PDB 3O5N, Figure 2).8 Three additional PDB structures that do not appear in peer reviewed literature include THQs bound to carbonic anhydrase 2:6SYS that contains a tricyclic THQ (Figure 2), and 6SX9 and 6SYB contain THQs with fully saturated cyclopentane rings. The atoms that make hydrogen bonds with the target proteins are highlighted with blue boxes in Figure 2. That both solved structures of bound fused THQs bear electron-withdrawing substituents is coincidental and unlikely to confer added stability to the scaffold. In carbonic anhydrase 2, the primary sulfonamide provides the key pharmacophore for molecular recognition by the enzyme.9−12 The following sections review many papers that have identified fused THQs as hits from high-throughput campaigns (Figure 3). While their activities are modest (in the micromolar range), it is striking that additional cocrystal structures have not been solved.
Figure 2.

Structures of cocrystallized tricyclic THQs. Blue boxes indicate the atoms that make hydrogen bonds with the target proteins.
3. Examples of Fused THQs as “Hits” Are Pervasive
Specific examples of screening campaigns that have identified tricyclic THQs as hits have been collected and grouped below, organized by the target protein. This arbitrary organization is for clarity of presentation and simply reflects the biological interests of those executing the campaign. This is not a comprehensive collection of all published data on this ubiquitous scaffold but rather a selection of representative examples from the published literature. The diversity of protein targets captured below supports the premise that the fused THQ scaffold does not yield specific hits for these proteins but that the reported activity is a result of pan-assay interference. Supporting Information, Table S1, includes additional details about the tricyclic THQs hits and screens used to identify them. That so many examples of these compounds as hits exist in the literature is driven by groups seeing other respected scientists publishing and patenting members of this chemical series and interpreting that as support for pushing these compounds forward in their project. A boom in interest in HTS coupled with this phenomenon has produced a steady stream of patents and publications in influential journals featuring fused THQs. As evidence of this, those compounds featured in Figure 3 were all published in the period of 2003–2020.
3.1. Fused THQs Identified in Hit Discovery Efforts for Mammalian Targets
As shown in Figure 3 and summarized below, more than 10 different works have included a tricyclic THQ as a hit. That the targets being pursued were not structurally homologous or involved in related biological functions suggests that the activity was unrelated to a conserved structural fold or motif in the target proteins.
Phosphatases
An HTS campaign using a fragment-based library identified fused THQs as a class of inhibitors of protein tyrosine phosphatases, PTPN5, PTPRR, and PTPN7. Compound 1a with X = Y = H (Figure 3), was computationally docked into PTPRR. Enzymatic inhibition, presented as a fraction of control (FoC) between 8% and 76% was reported at 100 μM.13
An HTS of 10 000 small molecules from the ChemBridge PRIME-Collection compound library identified fused THQ 5661118 with X = COOH and Y = H (Figure 3) as a mixed partial competitive inhibitor of the enzymatic activity of human Cdc25B (IC50 = 2.5 μM). Despite a central role of Cdc25 phosphatases in cell cycle progression, 5661118 failed to elicit cell cycle arrest at ≥30 μM. Presented in the PTPN paper that identified compound 1a is 1c,13 which has the same structure as 5661118. Accordingly, 5661118 demonstrated inhibition of PTP1B with an IC50 = 6.6 μM14 and 0.3 FoC at 100 μM.13
To find inhibitors of mitogen-activated protein kinase phosphatase-1 dual-specificity protein phosphatase (MKP-1), an HTS using the 65 000-member NIH diversity library was performed at 10 μM. A fluorescence-based assay readout identified active compounds (≥50% inhibition at 10 μM) and the assay was employed in dose–response format to select compounds with IC50 < 50 μM, which were then clustered by structural similarity. Cluster 7 included several tricyclic THQs with micromolar potency (4249621, 4263567, and 844096 in Figure 3).15
An inhibitor of hematopoietic protein tyrosine phosphatase (HePTP/PTPN7) was identified using an HTS of 112 000 compounds (from the MLSCN library and ChemBridge DIVERSet) at 20 μM using an assay that employed pNPP as the substrate. Among the active compounds with IC50 < 10 μM was a cluster of tricyclic THQs, including 654089 (IC50 = 0.20 μM, Figure 3).16
Virtual screening of 500 000 compounds was used to identify 34 candidate inhibitors of bovine low molecular weight protein tyrosine phosphatase (lmwPTP). Six of the 34 compounds selected for evaluation were tricyclic THQs. Experimental screening in an enzymatic assay with pNPP as the substrate confirmed nine compounds as competitive inhibitors, including fused THQ ptp-194 (Figure 3) with Ki = 9.0 μM.17
Kinases
A high-throughput time-resolved fluorescence energy transfer (TR-FRET) screen was developed to identify inhibitors of the enzymatic activity of pololike kinase 1 (PLK1). Following interrogation of a 97 000-member library from the NIH repository at 10 μM and concentration–response confirmation, 40 “hit compounds” were identified, seven of which had IC50 < 1 μM and 20 of which had IC50 = 1–5 μM, including the THQ 4248049 (Figure 3) with IC50 = 17 μM.18
Screening the Core Library of 9600 compounds from the Drug Discovery Initiative at the University of Tokyo using a high-throughput ADP-Glo diacylglycerol kinase (DGK) assay identified several putative inhibitors of diacylglycerol kinase α (DGKα). Four compounds demonstrated >20% inhibition at 30–50 μM. Among the hits was CU-4, a fused THQ (Figure 3). CU-4 was found to strongly inhibit the α-, δ-, κ-, and ι- isozymes of DGK at 5 μM and was deprioritized due to a lack of selectivity for DGKα.19
Receptors
Following an in silico screen of 434 000 drug-like molecules from the Chemical Diversity Laboratories collection using a formylpeptide receptor (FPR) homology model, a subset of 4324 compounds were prioritized for screening. Direct binding to human FPR was assessed via the flow cytometry at 67 μM and then in dose–response format, leading to identification of 30 hits from nine chemical families with Ki = 1–32 μM. One of the families contained THQ 2448-0030 (IC50 = 16 μM, Figure 3), which was further characterized as a FPR antagonist.20
An antagonist of the androgen receptor (AR) was identified following a structure-based virtual screen of 200 000 compounds in the Specs database. A total of 32 candidate ligands were evaluated for antagonistic activity using an AR-dependent reporter gene assay at a concentration of 10 μM. Three primary actives were further analyzed in a competitive binding assay using the AR ligand binding domain. A tricyclic THQ, C18 (Figure 3), was found to bind to the AR with IC50 = 4.0 μM and inhibit AR transcriptional activity with IC50 = 2.4 μM. An additional eight tricyclic THQ analogues were prepared, which displayed IC50 = 0.15–2.4 μM as AR antagonists in the reporter gene assay.21
Protein–Protein Interactions (PPIs) or Mediators
Compounds capable of disrupting the interface been the RNA-binding protein La and RNA derived from cyclin D1 mRNA were identified using a high-throughput fluorescence polarization (FP) assay. The ChemBridge DIVERSet library of 50 000 compounds was screened at 100 μM with >80% reduction in signal triggering dose–response follow-up. Six compounds were identified with IC50 = 2.5–50 μM and two, including fused THQ C6 (Figure 3), were selected for orthogonal evaluation. C6 was found to impair complex formation with several RNA partners in electrophoretic mobility shift assays (EMSAs) at 10–20 μM. Additional tricyclic THQs were purchased bearing an isobutyl or butyl ester in place of the ethyl ester of C6 and were found to block La:fD1 interactions at a similar concentration range.22
Shank3 is a central scaffolding protein that interacts with other proteins through a PDZ domain. To find small molecules that bind to the Shank3 domain, a small-molecule FP assay using a high-affinity peptide was performed. The 17 000-member ChemBioNet library was screened at a concentration of 10 μM, yielding 13 hits after removal of those with autofluorescence. In dose–response follow-up, two compounds demonstrated an IC50 < 250 μM, including tricyclic THQ 34 (Figure 3). Next, 21 additional analogues were synthesized but only two demonstrated Ki values <250 μM (17 and 70 μM). One of these fused THQs was 37 (Figure 3), which is also known as 5661118 and was previously identified as a hit in two of the phosphatase screens,13,14 and 36 (Figure 3), that was cocrystallized with the Shank3 PDZ domain (PDB 3O5N).8
The interaction between EED and EZH2 and PRC2 containing EZH2 Y641F, harboring a somatic activating mutation, was targeted via two independent HTS campaigns using libraries of synthetic and natural product-derived compounds from the Chinese National Compound Library. The PRC2-EZH2 Y641F screen employed a homogeneous time-resolved fluorescence (HTRF) assay to screen 250 000 compounds at 10 μM, resulting in 162 hits that demonstrated concentration-dependent inhibition. An FP assay of a different 60 000 compounds at 20 μM resulted in 97 hits for the EZH2-EED interaction. One fused THQ (WNN1770-D009) that was identified as an inhibitor of PRC2-EZH2 Y641F was also found to inhibit WT PRC2-EZH2 and the EZH2-EED interaction, albeit at higher concentrations. Two tricyclic THQs (WNN2125-H007 and WNN2074-A009) were found to be weak EZH2-EED PPI disruptors that also inhibited WT PRC2-EZH2. Finally, WNN1208-G009 was identified as an inhibitor of PRC2-EZH2 Y641F that lacked the inhibitory activity on WT PRC2-EZH2.23
Motility and Transport Proteins
To identify inhibitors of macrophage migration inhibitory factor (MIF), candidate binders from the 11 046-member Coelacanth chemical compound library predicted by computational docking were evaluated for direct binding to MIF by surface plasmon resonance (SPR). Confirmed binders were analyzed for their ability to inhibit MIF tautomerase activity, where compound 5 (Figure 3) demonstrated IC50 = 3.4 μM.24
The ATP binding cassette family C2 (ABCC2) is a multidrug resistance protein family member without known inhibitors, the inhibition of which could reduce acquired chemotherapy resistance. Using a vesicular transport assay and two substrates (EG and CDCF), 432 compounds from the University of Pittsburgh Center for Chemical Methodologies and Library Development library were screened at 80 μM. In total, 96 compounds modulated transport, and 53 remained active when evaluated in dose–response format. Twenty-five of the 96 actives identified in the primary screen were tricyclic THQs. The authors noted that the THQ scaffold has been identified in many other binding assays. Following dose–response studies, 11 fused THQs (1a, 1c, 1e, 1g, 1h, 1i, 1j, 1k, 1l, 1n, and 1q in Figure 3) demonstrated an IC50 < 30 μM in at least one assay.25,26
3.2. Tricyclic THQs Identified in Hit Discovery Campaigns for Nonmammalian Targets
Figure 3 includes tricyclic THQs from manuscripts and a patent that were identified as hits for a diverse range of bacterial, parasitic, and viral proteins. Discovery of these compounds is summarized in the following sections, and additional details are provided in SI, Table S1. The recurrent activity of various fused THQs as hits against a wide range of nonmammalian protein targets is consistent with their pan-assay interference and echoes the conclusion that these compounds are not specific for the protein targets featured in each subsection.
Bacterial Proteins
A structure-based virtual screen was used to select candidate non-nucleotide inhibitors of the edema factor (EF) of Bacillus anthracis by docking a library of small molecule fragments into the EF active site. The NCI-2000 and ZINC databases were employed in docking studies. The 19 diverse compounds with the best docking scores were evaluated for their ability to reduce cAMP secretion induced by EF in cells by ELISA. Three compounds from orthogonal chemotypes demonstrated inhibition at <10 μM. One of these compounds (DC8, Figure 3) belongs to the tricyclic THQ chemotype.27
A high-throughput fluorescence-based assay was used to screen the 50 000-member ChemBridge PRIME-Collection Library at ∼30 μg/mL (50–200 μM) to identify inhibitors of bacterial DNA adenine methyltransferases (DAM). Fused THQs were one of five major structural classes of inhibitors with 20 of the hits based on the chemotype. Hit expansion yielded another 43 THQs, but these were found to have limited SAR and lack selectivity for DAM. The most potent DAM inhibitors from the tricyclic THQs are summarized in Figure 3 as Class I inhibitors (IC50 = 14–113 μM).28
To find inhibitors of the IspD domain of the IspDF protein from Helicobacter pylori, more than 103 000 small molecules from BASF SE were profiled at 50 μM using a photometric assay. In total, 84 compounds demonstrated IC50 < 10 μM. Twenty-two compounds that had IC50 = 1–3 μM were prioritized for further study, including tricyclic THQ compound 27 (Figure 3). Compound 27 did not show an inhibitory effect when evaluated by an IspF domain assay using 13C NMR and was found to be only a weak inhibitor of H. pylori growth.29
A fluorescence-based HTS of 700 000 small molecules for inhibitors of the bacterial GroEL/GroES-mediated refolding cycle at 10 μM and then in dose–response format identified 21 compounds with IC50 < 10 μM. Tricyclic THQs 10, 12, 13, and 21 (Figure 3) were among the active inhibitors of GroEL/GroES refolding. These compounds were only weakly active or inactive in MDH inhibition and GroEL/GroES ATPase assays. Compounds that were active in the MDH counter screen were flagged as nonselective, while compounds that were active in the ATPase assay were proposed to bind at the ATP-site of these enzymes.30
To identify inhibitors of N-acetylglucosamine-1-phosphate uridyltransferase (GlmU) as potential Myobacterium tuberculosis agents, structure and ligand based computational models were developed and used in a virtual screen of a library of 20 000 compounds from the ChemBridge DIVERSet database. A set of the 125 most active published GlmU inhibitors identified in an HTS of a 216 000-member library by the MLSCN at 30 μM and then confirmed via dose–response follow-up (PubChem Bioassay AID 1376) were used to develop the in silico filters that were applied to the ChemBridge library. Fused THQ compound 75 (Figure 3) was included in these 125 but demonstrated only weak GlmU inhibition. Experimental screening of the candidates from the in silico screen at 100 μM resulted in identification of 15 hits that demonstrated >40% GlmU inhibition, none of which were a fused THQ.31
A patent from Oscient Pharmaceuticals included a series of tricyclic THQs as potent inhibitors of UDP-N-acetylglucosamine enolpyruvyl transferase (MurA) and potential antibacterial agents. These compounds (MurA inhibitors, Figure 3) were evaluated in dose–response fashion in a MurA enzyme assay. The most potent THQ analogues were found to demonstrate inhibition of MurA with an IC50 < 5 μM, but structural details about these analogues were not disclosed.32
Parasitic and Viral Proteins
An HTS of 13 000 small molecules at 20 μM was executed to find inhibitors of Trypanosoma cruzi glucokinase (TcGlcK). The compounds were assembled from the TimTec Diverse Synthetic and Natural Derivatives Libraries. This campaign yielded 25 TcGlcK inhibitors from nine different chemotypes, 13 of which demonstrated TcGlcK IC50 < 20 μM. Among the 25 TcGlcK actives was fused THQ GLK7-001 (Figure 3), which demonstrated 74% TcGlcK inhibition at 20 μM but did not show confirmatory activity in dose–response follow-up using a TcGlcK-coupled assay.33
In a similar approach, a high-throughput FP screen was employed to find GTP-binding inhibitors of the flavivirus NS5 N-terminal capping enzyme. Using an assay for monitoring GTP binding to the capping enzyme in real time, a pilot screen of 46 000 compounds at 12.5 μg/mL was executed with a secondary functional assay used for hit validation. The small molecule library was selected from a compilation of commercially available libraries at the NERCE National Screening Laboratory. One fused THQ (5660163, Figure 3) was among the eight hits identified with Ki < 10 μM.34
3.3. Undisclosed or Unpublished Fused THQs Identified in Screens for Protein–Protein Disruptors
The PAINS concept was derived from a series of assays measuring PPIs.5 Although these assays were performed on undisclosed targets, an effort was made to summarize the frequent hitters, which included tricyclic THQs. The TREAT-AD program35 also ran an HTS for a PPI and identified PAINS, including several fused THQs. Additional details about the fused THQs identified and screens employed are listed in SI, Table S1. This section summarizes fused THQs identified in original PAINS report as well as in a contemporary screen. That tricyclic THQs were a nuisance during PAINS inception and continue to plague the research community reinforces that this is not a solved problem.
Generalized AlphaScreen Assays
Six HTS campaigns were analyzed to identify PAINS within the WEHI 93K HTS library.5 In general terms, these assays were designed for PPIs between a GST-protein target and amphipathic helical ligand (3 separate screens), IL13Rα1-Fc receptor and cytokine IL13, the SH2-binding domain of SOCS2 and gp130, and the SH2-binding domain of SOCS3 and gp130. When screened at a single concentration of 25 or 50 μM, 1–7 tricyclic THQs were identified as primary hits that demonstrated ≥50% inhibition in 2–6 of the AlphaScreen assays. Compound 12 (WEHI-97605) in Figure 3 was an example of the problematic fused THQs for which the authors described follow-up and wasted investment of resources.5
MSN-CD44
Guided by structural studies, the TREAT-AD program developed a TR-FRET-based assay that employed the FERM (four-point-one ezrin radixin moesin) domain of human moesin (MSN) protein and a CD44 peptide to identify inhibitors of the PPI between MSN and CD44.36 This assay was miniaturized for ultra-HTS and a 138 000-member compound library from Emory University was screened at 20 μM. Primary hits were retested in dose–response using the TR-FRET assay, resupplied from commercial vendors, and reconfirmed for activity by TR-FRET. Those 22 hits that reconfirmed were further evaluated in a chromatography-based GST-pulldown assay using cell lysate overexpressing full-length GST-MSN and Venus-Flag-CD44. Three tricyclic THQs (5662143, 5795566, and 5661118 in Figure 3) demonstrated disruption of the PPI with IC50 = 3.6–27 μM.37
4. Fused THQs Are Reactive
The literature has hinted that tricyclic THQs interfere with assays. Like other PAINS, fused THQs appear to be reasonable, albeit small structures with a lipophilic surface that can make hydrophobic interactions and heteroatoms that can engage in hydrogen bonds. Their potential for protein reactivity is masked by these favorable characteristics.4 These discussions simply flag these compounds as potentially problematic but mostly fail to suggest a mechanism for the modest (and nonselective) potency or confirm mechanism(s) by which they might react with a target protein. As the literature supports that these are not advanceable compounds, helping to deconvolute this reactivity has been an aim of ours.
4.1. Mechanism of Reactivity of Fused THQs
While some PAINS structural classes have more obvious and well-described assay interference mechanisms, reasons for the problematic behavior of fused THQs have been left largely undefined. They are not highly colored, excluding photometric interference, sometimes called an “inner filter effect”, as the predominant driver of their problematic behavior. The relevant mechanism of assay interference for fused THQs has been suggested to hinge on the presence of an alkene activated toward nucleophilic attack.5 It has also been proposed that fused THQs could be oxidized in DMSO screening samples to form traces of quinoid-like structures, which would then interfere in an assay and yield false positive results.4,6 Another report discusses that while an original fused THQ sample in solution remained active, the resynthesized compound was inactive. The group hypothesized that the activity was due to traces of the intermediate diaryl imine formed during synthesis, which was stable in a DMSO solution and would find lipophilic protein pockets with which to react. The mildly electrophilic nature was suggested to make it refractory to isolation via reverse phase chromatography.4 Finally, there is a possibility that retrosynthetic degradation may occur in the screening sample, a process that could be promoted by trace amounts of residual acid remaining after purification.4 While many proposed mechanisms could potentially contribute to the observed reactivity of fused THQs, that a reactive structure different from the original fused THQ is present in stock solutions is a uniting theme of this discussion.
4.2. Tricyclic THQs Decompose in Solution
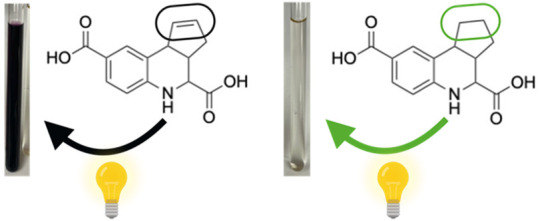
To enable hit confirmation in the TREAT-AD MSN/CD44 screening campaign, fused THQs 5662143, 5795566, and 5661118 (Figure 3) were resynthesized and a few analogues were also prepared. The synthetic scheme for analogues of 5661118 is shown in Scheme 1. Discoloration of solutions prepared for NMR analyses in DMSO-d6 or CDCl3 was noted for both 1 and 5661118 after sitting at ambient temperature for 72 h (Figure 4A, samples (1) and (2) are 1 in DMSO-d6). The same discoloration was observed when a solution of 5661118 in DMSO (10 mM) was prepared for the biological evaluation. A sample of 5661118 (Figure 3) purchased from Aurum Pharmatech (catalogue number GN37789) and 5795566 (Figure 3) purchased from Vitas-M Laboratory (catalogue number STK880452) demonstrated similar discoloration when dissolved in DMSO. Interestingly, when a solution prepared for NMR analysis in DMSO-d6 was kept in the dark at ambient temperature for 72 h, the solution remained clear ((3) in Figure 4A). A color difference when samples were kept in the dark was serendipitously discovered when an NMR sample was stored in a foil lined container. Compared to the benchtop compound ((5) in Figure 4B), this foil-wrapped sample only experienced partial exposure to light at the top of the NMR tube ((6) in Figure 4B). This disparity between solutions kept in the dark versus those exposed to light revealed that one decomposition mechanism was light-catalyzed.
Scheme 1. Synthetic Route to Remake and Diversify Fused THQ 5661118.
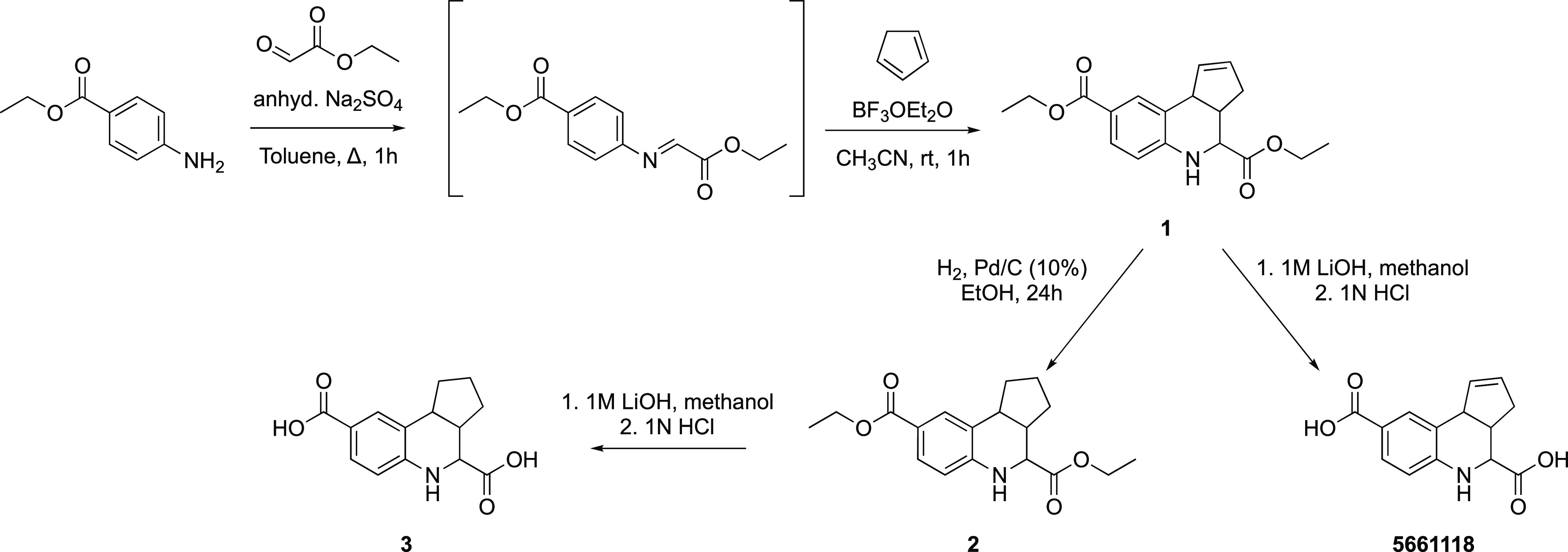
Figure 4.

Fused THQs decompose in solution. (A) Analogue 1 dissolved in DMSO-d6 and kept in the light for 72 h. (1) 25 mg and (2) 5 mg decomposed and changed color (purple), while the sample in DMSO-d6 that was kept in the dark for 72 h remained a clear solution ((3) 5 mg). (B) Analysis of a solution of 1 dissolved in (4) CDCl3 and (5) DMSO-d6 for 48 and 120 h at ambient temperature in the light via thin layer chromatography analysis (eluent: 20% ethyl acetate/80% hexane). Sample (6) was stored in an NMR tube wrapped in foil and exposed to light only from the top for 120 h. (C) The structures of 1 and potential structures of the decomposition product (1a and 1b). (D) LCMS data collected from a discolored solution of 1 (from A), indicating the mass observed for the decomposition material (312 m/z).
Thin layer chromatography (TLC) of the discolored solution of 1 revealed the decomposition of the parent compound into multiple other compounds over time (Figure 4B). Streaking and tailing of diacid 5661118 on the TLC plate precluded a similar analysis of this compound. The separation of 1 and its breakdown products by silica gel column chromatography allowed the isolation of the major decomposition product (SI, Figure S1).
Further analyses of the discolored solutions were carried out via multiple methods. First, the discolored solution ((1) in Figure 4A) was analyzed via mass spectrometry. As shown in Figure 4D, the predominant mass in the solution was not diester 1. Instead, a slightly lower mass of 312 m/z was observed. Potential decomposition products 1a and 1b in Figure 4C were suggested based on the observed mass. While we have elected to draw 1b with specific double bond orientations, it is worth noting that this compound can exist as many different tautomers in which the double bonds within the cyclopentadiene ring are moved around with the five- and adjoining six-membered ring systems (SI, Figure S2). The conjugation of some of these potential alternative structures could confer additional stability versus the form drawn in Figure 4C. The discolored and clear solutions ((2) and (3) in Figure 4A, respectively) were also analyzed via 1H NMR. As shown in Figure 5, the 1H NMR spectrum of the sample kept in the dark for 72 h ((3), Figure 5A) differed from the 1H NMR spectra of the samples kept in the light at ambient temperature ((2) in Figure 5B and (1) in Figure 5C) and new peaks had emerged, especially downfield in the aromatic region (∼6–13 ppm). Despite the additional lower Rf spots observed on the TLC plate in CDCl3 ((4) in Figure 4B), 1H NMR analysis of samples kept in CDCl3 for 120 h in light showed no significant changes in the spectra. An 1H NMR spectrum as well as TLC and LCMS analysis of a small amount of the isolated decomposition product of 1 (SI, Figure S1) revealed that it was a very different chemical structure from the parent fused THQ.
Figure 5.
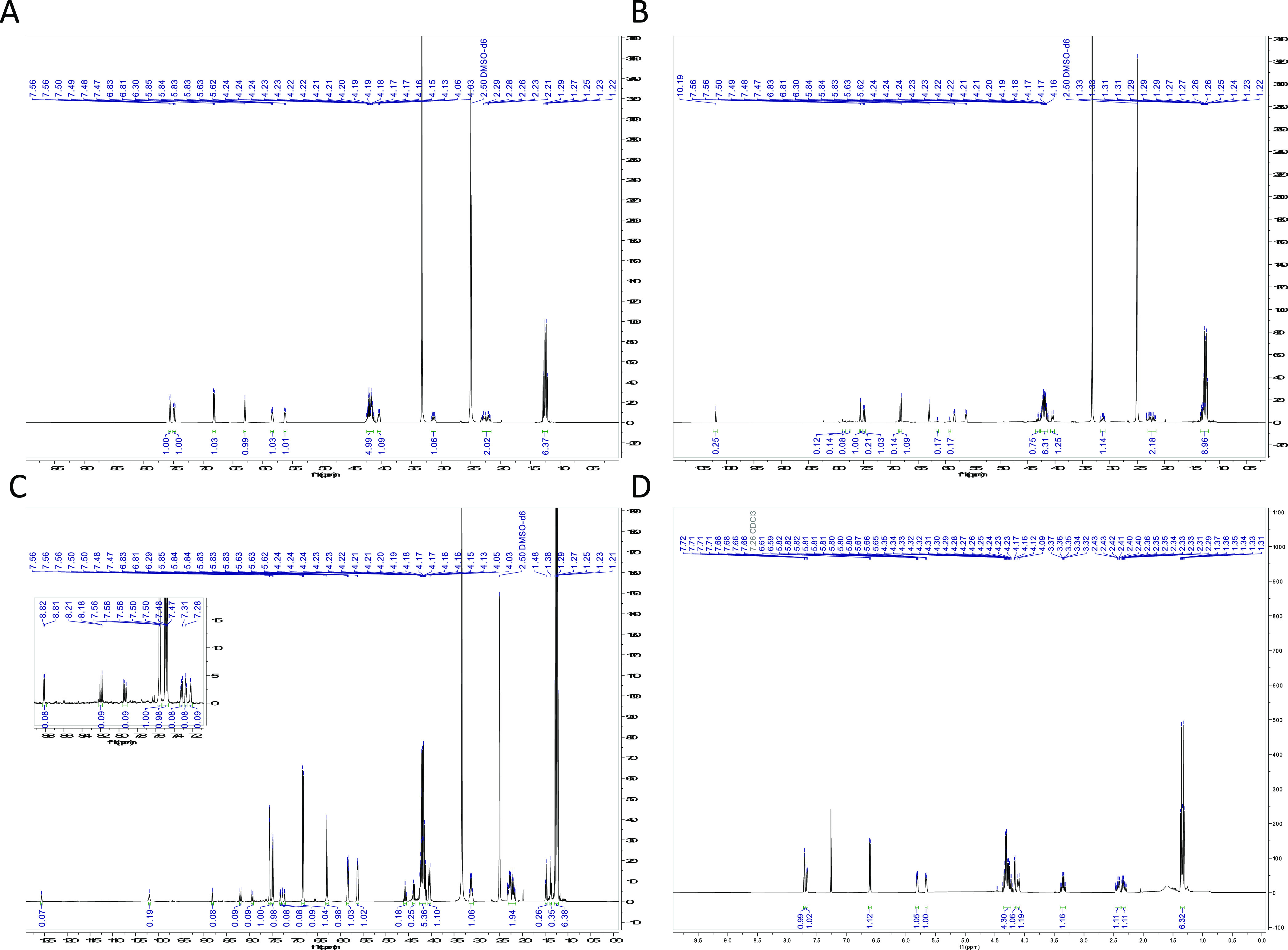
1H NMR spectra of 1 in various states of decomposition. (A) 1H NMR spectrum of 5 mg of 1 in DMSO-d6 after 72 h in the dark. (B) 1H NMR spectrum of 5 mg of 1 in DMSO-d6 after 72 h in the light (benchtop indoors), showing decomposition. (C) 1H NMR spectrum of 25 mg of 1 in DMSO-d6 after 72 h in the light (benchtop indoors), showing decomposition. (D) 1H NMR spectrum of 5 mg of 1 in CDCl3 after 120 h in the light (benchtop indoors).
4.3. A Saturated Analogue Lacks Activity and Reactivity
Structural modifications were made to ascertain the part of the tricyclic THQ system that contributed to its decomposition. The double bond in the cyclopentene ring was reduced to yield THQs 2 and 3 (Scheme 1). No discoloration was observed when these compounds were dissolved in DMSO, even after 5 days at ambient temperature. When evaluated in an MSN/CD44 TR-FRET assay (SI, Figure S3) these saturated analogues were inactive (3, Figure 6). Parent compound 5661118 was evaluated in parallel (Figure 6). By comparing the data for 5661118 and 3, it was concluded that the double bond in the cyclopentene ring was at least partially responsible for the promiscuous activity of 5661118 as well as its propensity to decompose in solution. It is likely that other unsaturated tricyclic THQs also undergo oxidative decomposition in solution, which accounts for their pan-assay interference (Figure 3).
Figure 6.

TR-FRET assay results for (A) 5661118 and (B) saturated analogue 3.
A few variables were considered when analyzing the TR-FRET data. The assay buffer contained 25 mM HEPES at pH 7.5, 0.05% 150 mM NaCl, Tween-20, and 1 mM DTT. Presence of nonionic detergent Tween-20 should have reduced the propensity for both the compound and protein to aggregate. The reducing agent DTT should have suppressed oxidative decomposition of the compound in the assay (but not prior to testing) and limit protein aggregation via preventing disulfide formation. The high Hill coefficient of 3.4 for diacid 5661118 was indicative of a nonstandard dose–response curve, which might be a result of compound aggregation, a nonspecific mechanism of inhibition, or direct interference with the assay readout.38,39 Our evidence-based exercise bolsters the few reports in the literature of fused THQs as PAINS. That a saturated analogue (3) does not manifest the same reactivity as its unsaturated congener (5661118) points to it being a more stable structure. The saturated structure of 3 lacks a double bond that can be oxidized and cannot retrosynthetically degrade to reactive precursors. Compound 3 was also isolated via filtration via Celite, and the contaminating impurities could have been removed. While a clear answer on the mechanism(s) that drive the reactivity of tricyclic THQs has not been defined, at least one part of the scaffold that contributes to its assay interference has been identified and characterized.
5. Perspectives
In reviewing the body of literature around fused THQs, we observed that these compounds are members of multiple large screening libraries employed in HTS and other small molecule screening campaigns. While it might be more widely known that a large HTS will unearth nuisance, nonspecific compounds,4 it is worth noting the fact that small HTS decks (<10 000 compounds) also face this problem. Based on our research, this is true in both physical and virtual screens. Because screening of smaller HTS sets typically yields fewer potential hits, PAINS have a higher probability of being prioritized for follow-up as part of these studies. Overall, the fact that tricyclic THQs are so ubiquitous in academic and commercial screening libraries makes it difficult to purge them from so many compound collections.
When considering the assay readouts employed in these many divergent assays, we found spectrophotometric (absorbance/fluorescence) to be the most popular.5,8,13−15,17,18,20−30,32−34,37 Colorimetric16,31 and chemiluminescent19 readouts were also exemplified. Finally, orthogonal assays, including SPR24 and radiochemical,25,26 have been used in combination with these other methods as part of either filtering or confirmatory experiments. The fact that so many different assays have identified these compounds as hits implies that their discovery is not linked to a specific readout. Although we have not discussed phenotypic screens herein, we believe they are no different from the types of screens described in that if a tricyclic THQ “hits”, it is not worth following up.
Next, we looked at the hits more closely. As shown in Figure 7, certain fused THQs were found by multiple groups in their hit list. Compounds in Figure 7 are labeled by their ChemBridge identifiers and/or CAS registry numbers, and the number of publications that we reviewed that had them shown is listed under each. 5661118 was most frequently identified, including by our group, but also in assays for human phosphatases,13,14 Shank3,8 the PRC2-EZH2 interaction,23 and bacterial DNA adenine methyltransferases (DAM).28 This compound is also covered by multiple commercial patents for a variety of applications, including MIP-1α, RANTES, MASP-2, CNKSR1, and Pin1.6,40−425654172, 5654435, and 4141340 were identified as hits for multiple phosphatases13,14,16 in addition to other targets (DAM for 5654172 and 5654435(28) and MIF for 5654172).24 Compound 5660163 was found to be an active compound for multiple nonhuman targets.33,34 Finally, in our MSN-based study, we found a compound (353484-24-9) that was also exemplified in a targeted screen for the PRC2-EZH2 interaction.23 The repeated inclusion of the same structures as hits from multiple screens supports how common these compounds are in screening sets from different sources, their promiscuity in binding to divergent protein targets, and their exclusion from hit-to-lead optimization campaigns.
Figure 7.
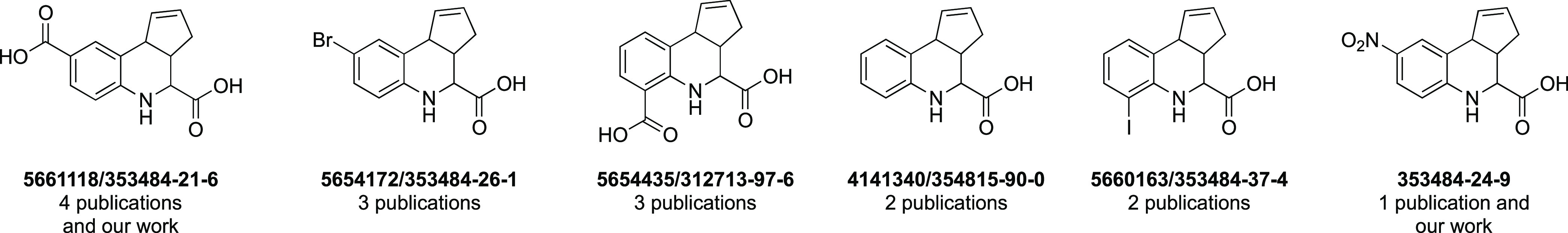
Structures of specific tricyclic THQs that are hits in the multiple discussed screens.
Resources have been invested in diversifying these scaffolds, either through purchase or synthetic efforts. To try to determine whether a hit is truly a hit and to provide confidence that it is a series worth working on, there are two questions worth considering here for the fused THQs. The first question is whether the compounds in question demonstrate selectivity within a target class. The second question is whether analogues within the series show demonstrable and robust structure–activity relationships (SAR) for a single target. For many of the THQ publications, analysis of the data generated when multiple THQ analogues were tested against different enzymes within a family or protein class often demonstrated that no selectivity was observed.13,14,22,23 This points to these compounds being indiscriminate binders. The papers that took advantage of cross-screening within a target class often profiled overlapping targets, such as PTP1B13,14 and PTPN713,16 by groups interested in phosphatase inhibitors. For another phosphatase, several MKP-1 inhibitors that were discovered in the HTS were members of structural classes or shared a pharmacophore with previously reported MKP-1 or other phosphatases. This overlap was viewed favorably as validating the screening assay.15 An alternative viewpoint is that these compounds are promiscuous and bind to many protein targets, making them artifacts rather than assay confirmatory.
In response to the second question, when THQ analogues were prepared and tested against the intended target, they most often did not demonstrate compelling and robust SAR over a broad potency range. Structurally related analogues are either of similar potency or completely inactive.13,14,21,28 This lack of SAR is also captured in Figure 3, where multiple analogues demonstrate similar activities for a single target, and with a few exceptions, the potency across the entire spectrum of compounds hovers between 2 and 20 μM.
Potential mechanisms of fused THQ reactivity have been refuted, and others confirmed. Tricyclic THQs are unstable in solution, and part of their decomposition mechanism is mediated by light. Based on reduction of the double bond in the cyclopentene of fused THQs eliminating decomposition as well as activity of the resulting analogue, it is reasonable to conclude that this position is at least partially responsible for the reactivity of this compound class. It remains a possibility, however, that a reactive imine intermediate that is present when these compounds are synthesized or retrosynthetically formed and present in solution is partially responsible for their reactivity. This further supports the need to remove fused THQs from screening collections. Because vendors will continue to supply the compounds if there are buyers, the onus is on scientists to recognize and deprioritize these PAINS.
PAINS are difficult to confirm as bad actors. Development of methods such as ALARM NMR, an assay developed by Abbott to detect protein-reactive frequent hitters, has enabled pharmaceutical companies to cull their HTS collections. Academic laboratories, however, remain prone to identification of these nonprogressable compounds.4 Assay interference filters that recognized and flag PAINS based on structural motifs have been adopted by some groups, but they are also not perfect because not all potentially reactive structural motifs will be flagged.4,5 Stringent use of these filters can result in reduced chemical diversity but should leave only potentially progressable compounds, and this is a price worth paying. These methods could become more widely adopted to reduce the level of advancement of PAINS in hit-to-lead optimization campaigns. In the absence of such strategies being implemented, efforts such as this one to clean up the literature and increase awareness of classes of PAINS are especially important.
6. Concluding Remarks
It is troublesome that tricyclic THQs are prevalent in the literature as hit compounds for a diverse portfolio of targets. While there are a handful of examples of the confirmation of binding by X-ray crystallography, these structures have been the exception to the rule. Our review and evidence-based experiments solidify the idea that tricyclic THQs are nuisance compounds that cause pan-assay interference in the majority of screens rather than privileged structures worthy of chemical optimization. Their widespread micromolar activities on a broad range of proteins with diverse assay readouts support our assertion that they are unlikely to be valid hits. Notably, unsaturated tricyclic THQs undergo rapid oxidative decomposition in DMSO solution, while the corresponding saturated analogues are stable but lack biological activity. We built upon the already published notion that fused THQs are problematic, and we provided additional data to reinforce the message that these compounds should be avoided.
Fused THQ analogues are present in many screening libraries and readily available for purchase from a plethora of vendors. That the compounds in these screening decks are typically stored as DMSO stock solutions with no special handling to avoid light and in the presence of oxygen could accelerate their decomposition pathways. The compounds thought to be tricyclic THQs in solution have likely decomposed over time, and any observed activity is unlikely to be due to the original compound structures. It is unclear whether routine QC has detected the decomposition of these fused THQs, because so many examples remain in commercial screening libraries. The formation of many different decomposition products coupled with difficulties associated with isolation has complicated the exact assignment of what is being formed. It is clear, however, that the starting fused THQ is not the only small molecule in solution and the heterogeneous stock solution contains reactive species.
In conclusion, we caution the scientific community against investing in resource screening of tricyclic THQs or follow-up of putative hits. By providing this survey of the scientific literature as well as evidence of the decomposition of DMSO solutions of unsaturated THQs, we hope that others will become more aware of this problematic class of pan-assay interfering compounds. It is tragic to continue to watch groups invest time and resources in dead-end hit-to-lead campaigns, and the medicinal chemistry community will benefit everyone if the cycle stops.
Acknowledgments
Our Table of Contents graphic was created with Biorender.com. The authors thank the larger Emory-Sage-SGC TREAT-AD team for their scientific contributions to this work. The Structural Genomics Consortium (SGC) is a registered charity (number 1097737) that receives funds from Bayer AG, Boehringer Ingelheim, the Canada Foundation for Innovation, Eshelman Institute for Innovation, Genentech, Genome Canada through Ontario Genomics Institute, EU/EFPIA/OICR/McGill/KTH/Diamond, Innovative Medicines Initiative 2 Joint Undertaking, Janssen, Merck KGaA (aka EMD in Canada and USA), Pfizer, the São Paulo Research Foundation-FAPESP, and Takeda. Research reported in this publication was supported in part by the NC Biotechnology Center Institutional Support Grant 2018-IDG-1030 and NIH U54AG065187.
Glossary
Abbreviations Used
- AR LBP
AR ligand-binding pocket
- ARR2BP
probasin promoter
- CAS
Chemical Abstracts Service
- CDCF
fluorescent 2′,7′-dichlorofluorescin transport
- CDCl3
deuterated chloroform
- DEN
dengue virus DEN2 strain 16681 AA 1-267
- DMSO-d6
deuterated DMSO
- DTT
dithiothreitol
- EG
radiolabeled β-estradiol 17-(β-d-glucuronide
- ELISA
enzyme-linked immunosorbent assay
- EMSAs
electrophoretic mobility shift assays
- EtOH
ethanol
- FoC
fraction of control
- FP
fluorescence polarization
- h
hours
- GTP
guanosine-5′-triphosphate
- H. pylori
Helicobacter pylori
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HTRF
homogeneous time-resolved fluorescence
- HTS
high-throughput screening
- MDH
malate dehydrogenase
- MIC
minimum inhibitory concentration
- MLSCN
molecular libraries screening center network
- NIH
National Institutes of Health
- NMR
nuclear magnetic resonance
- PAINS
pan-assay interference compounds
- PDB
Protein Data Bank
- pNPP
p-nitrophenyl phosphate
- PPIs
protein–protein interactions
- QC
quality control
- SAR
structure–activity relationships
- SPR
surface plasmon resonance
- THQ(s)
tetrahydroquinoline(s)
- TLC
thin layer chromatography
- TR-FRET
time-resolved fluorescence energy transfer
- WT
wild-type
- YF
yellow fever strain 17D AA 1-268
Biographies
Frances M. Bashore is a Research Assistant Professor at SGC-UNC in the Division of Chemical Biology and Medicinal Chemistry Department in the UNC Eshelman School of Pharmacy. She is currently focusing on novel protein targets implicated in Alzheimer’s disease as part of the Emory-Sage-SGC TREAT-AD team. She is a chemical biologist with keen interest in the design, synthesis, and biological evaluation of proteolysis targeting chimeras.
Joel Annor-Gyamfi was a former postdoctoral research associate as part of the Emory-Sage-SGC TREAT-AD team. As part of this team, he executed medicinal chemistry toward the development of novel chemical probes for protein targets implicated in Alzheimer’s disease, including MSN/CD44. Joel is currently a research scientist at Sterling Pharma Solutions.
Yuhong Du is an Associate Professor in the Emory University School of Medicine and Associate Director of Assay Development and HTS in the Emory Chemical Biology Discovery Center. She employs HTS to identify small molecule leads to enable drug discovery for many different diseases, including Alzheimer’s disease as part of the Emory-Sage-SGC TREAT-AD team.
Vittorio L. Katis is a researcher at the Center of Medicines Discovery (CMD) at the University of Oxford. He supports the structural biology and assay development efforts of the Emory-Sage-SGC TREAT-AD team. He has expertise in protein production and purification as well as biophysical and biochemical assays.
Felix Nwogbo was a former postdoctoral research associate as part of the Emory-Sage-SGC TREAT-AD team. As part of this team, he worked on assay development and evaluation of test compounds, including for MSN/CD44. Felix is currently a business system analyst for Zifo RnD Solutions.
Raymond G. Flax is a doctoral student in the Division of Chemical Biology and Medicinal Chemistry Department in the UNC Eshelman School of Pharmacy and member of the Axtman laboratory. He has deep-rooted interest in neuroscience and plans to use his synthetic chemistry skills to develop high-quality chemical probes for protein targets implicated in neurodegenerative diseases. Raymond obtained his Bachelor’s degree in Chemistry in 2022 from State University of New York College of Environmental Science and Forestry.
Stephen V. Frye is a Fred Eshelman Distinguished Professor and former Director of the Center for Integrative Chemical Biology and Drug Discovery (CICBDD) at the University of North Carolina at Chapel Hill. Prior to joining UNC to create the CICBDD in 2007, Frye was the worldwide vice president of Discovery Medicinal Chemistry (DMC) at GlaxoSmithKline (GSK). His research interests at UNC have centered in the area of chemical biology of chromatin regulation with an emphasis on methyl-lysine reading domains. Stephen led the medicinal chemistry core for the Emory-Sage-SGC TREAT-AD team for an initial period.
Kenneth H. Pearce is a Professor in the Division of Chemical Biology and Medicinal Chemistry Department and Director of the Center for Integrative Chemical Biology and Drug Discovery (CICBDD) in the UNC Eshelman School of Pharmacy. His primary research interests and expertise are fundamentals of protein methods, biochemical, and cell assay development, medium- and high-throughput screening, hit validation and mode-of-action, biophysical methods for characterizing protein–protein and small molecule–protein interactions, and structure–activity relationships for early drug discovery. Before coming to UNC in mid-2015, he spent more than 18 years at GlaxoSmithKline and legacy companies in the Molecular Discovery Research organization. Ken is part of the medicinal chemistry core of the Emory-Sage-SGC TREAT-AD team.
Haian Fu is a Professor and Chair of the Department of Pharmacology and Chemical Biology, Professor in the Department of Hematology and Medical Oncology, and Associate Dean for Innovation and International Strategies in the Emory University School of Medicine. He is also Leader of the Discovery and Developmental Therapeutics Program, Director of the Emory Chemical Biology Discovery Center, and Winship Partner in Research Endowed Chair in the Winship Cancer Institute of Emory University. He leads a research program dedicated to discovery and evaluation of novel therapeutic options for patients in need. These patients include those with Alzheimer’s disease, as he is a member of the Emory-Sage-SGC TREAT-AD team.
Timothy M. Willson is chief scientist of the SGC-UNC, an open-discovery network for protein kinases based at the UNC Eshelman School of Pharmacy. He has more than 25 years of experience in pharmaceutical research with a track record in discovery of first-in-class clinical candidates. He is widely recognized for scientific leadership in chemical biology and was named one of the world’s 400 most influential biomedical researchers. Tim is part of the medicinal chemistry core of the Emory-Sage-SGC TREAT-AD team.
David H. Drewry is a renowned leader in the medicinal chemistry of protein kinases and is one of the principal architects of the research strategy at the SGC-UNC to build an open and collaborative research network to promote target discovery. His research interests include the art and science of medicinal chemistry, kinase inhibitor design, utilization of annotated sets of kinase inhibitors to build understanding of signaling networks and precompetitive chemical biology to facilitate target identification.
Alison D. Axtman is a Research Associate Professor in the Division of Chemical Biology and Medicinal Chemistry Department in the UNC Eshelman School of Pharmacy. She is also a principal investigator in medicinal chemistry at SGC-UNC. Her interests bridge chemistry and biology. She focuses on using high-quality small molecules to explore and impact disease-propagating biological pathways, especially those that cause neurodegenerative diseases. Active projects are aimed at developing preclinical small molecule candidates to interrogate underexplored protein targets in Alzheimer’s disease and amyotrophic sclerosis. She leads the medicinal chemistry core for the Emory-Sage-SGC TREAT-AD team.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c01277.
Author Contributions
F.M.P. and J.A.G. contributed equally to this work. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Baell J.; Walters M. A. Chemistry: Chemical con artists foil drug discovery. Nature 2014, 513 (7519), 481–483. 10.1038/513481a. [DOI] [PubMed] [Google Scholar]
- Capuzzi S. J.; Muratov E. N.; Tropsha A. Phantom PAINS: Problems with the utility of alerts for Pan-Assay INterference CompoundS. J. Chem. Inf Model 2017, 57 (3), 417–427. 10.1021/acs.jcim.6b00465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siramshetty V. B.; Preissner R.; Gohlke B. O. Exploring activity profiles of PAINS and their structural context in target-ligand complexes. J. Chem. Inf Model 2018, 58 (9), 1847–1857. 10.1021/acs.jcim.8b00385. [DOI] [PubMed] [Google Scholar]
- Baell J. B. Observations on screening-based research and some concerning trends in the literature. Future Med. Chem. 2010, 2 (10), 1529–1546. 10.4155/fmc.10.237. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53 (7), 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Ferrins L.; Falk H.; Nikolakopoulos G. PAINS: Relevance to tool compound discovery and fragment-based screening. Aust. J. Chem. 2013, 66 (12), 1483–1494. 10.1071/CH13551. [DOI] [Google Scholar]
- Ramesh C.; Nayak T. K.; Burai R.; Dennis M. K.; Hathaway H. J.; Sklar L. A.; Prossnitz E. R.; Arterburn J. B. Synthesis and Characterization of Iodinated Tetrahydroquinolines Targeting the G Protein-Coupled Estrogen Receptor GPR30. J. Med. Chem. 2010, 53 (3), 1004–1014. 10.1021/jm9011802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saupe J.; Roske Y.; Schillinger C.; Kamdem N.; Radetzki S.; Diehl A.; Oschkinat H.; Krause G.; Heinemann U.; Rademann J. Discovery, structure-activity relationship studies, and crystal structure of nonpeptide inhibitors bound to the Shank3 PDZ domain. ChemMedChem. 2011, 6 (8), 1411–1422. 10.1002/cmdc.201100094. [DOI] [PubMed] [Google Scholar]
- Xuan G. S.; Zhan J. H.; Zhang A. M.; Li W.; Zheng K. Inhibition of carbonic anhydrase II by sulfonamide derivatives. Pharmazie 2021, 76, 412–415. 10.1691/ph.2021.1590. [DOI] [PubMed] [Google Scholar]
- Gokcen T.; Gulcin I.; Ozturk T.; Goren A. C. A class of sulfonamides as carbonic anhydrase I and II inhibitors. J. Enzyme Inhib Med. Chem. 2016, 31 (sup2), 180–188. 10.1080/14756366.2016.1198900. [DOI] [PubMed] [Google Scholar]
- Pagnozzi D.; Pala N.; Biosa G.; Dallocchio R.; Dessì A.; Singh P. K.; Rogolino D.; Di Fiore A.; De Simone G.; Supuran C. T.; Sechi M. Interaction studies between carbonic anhydrase and a sulfonamide inhibitor by experimental and theoretical approaches. ACS Med. Chem. Lett. 2022, 13 (2), 271–277. 10.1021/acsmedchemlett.1c00644. [DOI] [Google Scholar]
- Elkamhawy A.; Woo J.; Nada H.; Angeli A.; Bedair T. M.; Supuran C. T.; Lee K. Identification of novel and potent indole-based benzenesulfonamides as selective human carbonic anhydrase II inhibitors: Design, synthesis, in vitro, and in silico studies. Int. J. Mol. Sci. 2022, 23 (5), 2540. 10.3390/ijms23052540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswaran J.; von Kries J. P.; Marsden B.; Longman E.; Debreczeni J. E.; Ugochukwu E.; Turnbull A.; Lee W. H.; Knapp S.; Barr A. J. Crystal structures and inhibitor identification for PTPN5, PTPRR and PTPN7: a family of human MAPK-specific protein tyrosine phosphatases. Biochem. J. 2006, 395 (3), 483–491. 10.1042/BJ20051931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brisson M.; Nguyen T.; Vogt A.; Yalowich J.; Giorgianni A.; Tobi D.; Bahar I.; Stephenson C. R.; Wipf P.; Lazo J. S. Discovery and characterization of novel small molecule inhibitors of human Cdc25B dual specificity phosphatase. Mol. Pharmacol. 2004, 66 (4), 824–833. 10.1124/mol.104.001784. [DOI] [PubMed] [Google Scholar]
- Johnston P. A.; Foster C. A.; Shun T. Y.; Skoko J. J.; Shinde S.; Wipf P.; Lazo J. S. Development and implementation of a 384-well homogeneous fluorescence intensity high-throughput screening assay to identify mitogen-activated protein kinase phosphatase-1 dual-specificity protein phosphatase inhibitors. Assay Drug Dev Technol. 2007, 5 (3), 319–332. 10.1089/adt.2007.066. [DOI] [PubMed] [Google Scholar]
- Sergienko E.; Xu J.; Liu W. H.; Dahl R.; Critton D. A.; Su Y.; Brown B. T.; Chan X.; Yang L.; Bobkova E. V.; Vasile S.; Yuan H.; Rascon J.; Colayco S.; Sidique S.; Cosford N. D. P.; Chung T. D. Y.; Mustelin T.; Page R.; Lombroso P. J.; Tautz L. Inhibition of hematopoietic protein tyrosine phosphatase augments and prolongs ERK1/2 and p38 activation. ACS Chem. Biol. 2012, 7 (2), 367–377. 10.1021/cb2004274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal D.; Blobel J.; Pérez Y.; Thormann M.; Pons M. Structure-based discovery of new small molecule inhibitors of low molecular weight protein tyrosine phosphatase. Eur. J. Med. Chem. 2007, 42 (8), 1102–1108. 10.1016/j.ejmech.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Sharlow E. R.; Leimgruber S.; Shun T. Y.; Lazo J. S. Development and implementation of a miniaturized high-throughput time-resolved fluorescence energy transfer assay to identify small molecule inhibitors of polo-like kinase 1. Assay Drug Dev Technol. 2007, 5 (6), 723–735. 10.1089/adt.2007.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K.; Kunii N.; Sakuma M.; Yamaki A.; Mizuno S.; Sato M.; Sakai H.; Kado S.; Kumagai K.; Kojima H.; Okabe T.; Nagano T.; Shirai Y.; Sakane F. A novel diacylglycerol kinase α-selective inhibitor, CU-3, induces cancer cell apoptosis and enhances immune response. J. Lipid Res. 2016, 57 (3), 368–379. 10.1194/jlr.M062794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards B. S.; Bologa C.; Young S. M.; Balakin K. V.; Prossnitz E. R.; Savchuck N. P.; Sklar L. A.; Oprea T. I. Integration of virtual screening with high-throughput flow cytometry to identify novel small molecule formylpeptide receptor antagonists. Mol. Pharmacol. 2005, 68 (5), 1301–1310. 10.1124/mol.105.014068. [DOI] [PubMed] [Google Scholar]
- Tang Q.; Fu W.; Zhang M.; Wang E.; Shan L.; Chai X.; Pang J.; Wang X.; Xu X.; Xu L.; Li D.; Sheng R.; Hou T. Novel androgen receptor antagonist identified by structure-based virtual screening, structural optimization, and biological evaluation. Eur. J. Med. Chem. 2020, 192, 112156 10.1016/j.ejmech.2020.112156. [DOI] [PubMed] [Google Scholar]
- Sommer G.; Fedarovich A.; Kota V.; Rodriguez R.; Smith C. D.; Heise T. Applying a high-throughput fluorescence polarization assay for the discovery of chemical probes blocking La:RNA interactions in vitro and in cells. PLoS One 2017, 12 (3), e0173246 10.1371/journal.pone.0173246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Du D. H.; Wang J.; Cai X. Q.; Deng A. X.; Nosjean O.; Boutin J. A.; Renard P.; Yang D. H.; Luo C.; Wang M.-W. Identification of catalytic and non-catalytic activity inhibitors against PRC2-EZH2 complex through multiple high-throughput screening campaigns. Chem. Biol. Drug Des 2020, 96 (4), 1024–1051. 10.1111/cbdd.13702. [DOI] [PubMed] [Google Scholar]
- Fukunishi Y.; Mikami Y.; Takedomi K.; Yamanouchi M.; Shima H.; Nakamura H. Classification of chemical compounds by protein–compound docking for use in designing a focused library. J. Med. Chem. 2006, 49 (2), 523–533. 10.1021/jm050480a. [DOI] [PubMed] [Google Scholar]
- Wissel G.; Kudryavtsev P.; Ghemtio L.; Tammela P.; Wipf P.; Yliperttula M.; Finel M.; Urtti A.; Kidron H.; Xhaard H. Exploring the structure-activity relationships of ABCC2 modulators using a screening approach. Bioorg. Med. Chem. 2015, 23 (13), 3513–3525. 10.1016/j.bmc.2015.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissel G.; Deng F.; Kudryavtsev P.; Ghemtio L.; Wipf P.; Xhaard H.; Kidron H. A structure-activity relationship study of ABCC2 inhibitors. Eur. J. Pharm. Sci. 2017, 103, 60–69. 10.1016/j.ejps.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D.; Misra M.; Sower L.; Peterson J. W.; Kellogg G. E.; Schein C. H. Novel inhibitors of anthrax edema factor. Bioorg. Med. Chem. 2008, 16 (15), 7225–7233. 10.1016/j.bmc.2008.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashhoon N.; Pruss C.; Carroll M.; Johnson P. H.; Reich N. O. Selective inhibitors of bacterial DNA adenine methyltransferases. J. Biomol Screen 2006, 11 (5), 497–510. 10.1177/1087057106287933. [DOI] [PubMed] [Google Scholar]
- Honold A.; Lettl C.; Schindele F.; Illarionov B.; Haas R.; Witschel M.; Bacher A.; Fischer M. Inhibitors of the bifunctional 2-C-methyl-d-erythritol 4-phosphate cytidylyl transferase/2-C-methyl-d-erythritol-2,4-cyclopyrophosphate synthase (IspDF) of helicobacter pylori. Helv. Chim. Acta 2019, 102 (3), e1800228 10.1002/hlca.201800228. [DOI] [Google Scholar]
- Johnson S. M.; Sharif O.; Mak P. A.; Wang H. T.; Engels I. H.; Brinker A.; Schultz P. G.; Horwich A. L.; Chapman E. A biochemical screen for GroEL/GroES inhibitors. Bioorg. Med. Chem. Lett. 2014, 24 (3), 786–789. 10.1016/j.bmcl.2013.12.100. [DOI] [PubMed] [Google Scholar]
- Mehra R.; Rani C.; Mahajan P.; Vishwakarma R. A.; Khan I. A.; Nargotra A. Computationally guided identification of novel mycobacterium tuberculosis GlmU inhibitory leads, their optimization, and in vitro validation. ACS Comb. Sci. 2016, 18 (2), 100–116. 10.1021/acscombsci.5b00019. [DOI] [PubMed] [Google Scholar]
- Labaudiniere R. F.; Xiang Y.; Jalluri R. K.; Arvan-Ites A. C.. Antibiotic cycloalkyltetrahydroquinoline derivatives. United States PCT/US2004/025937, 2003.
- Mercaldi G. F.; D’Antonio E. L.; Aguessi A.; Rodriguez A.; Cordeiro A. T. Discovery of antichagasic inhibitors by high-throughput screening with Trypanosoma cruzi glucokinase. Bioorg. Med. Chem. Lett. 2019, 29 (15), 1948–1953. 10.1016/j.bmcl.2019.05.037. [DOI] [PubMed] [Google Scholar]
- Geiss B. J.; Stahla-Beek H. J.; Hannah A. M.; Gari H. H.; Henderson B. R.; Saeedi B. J.; Keenan S. M. A high-throughput screening assay for the identification of flavivirus NS5 capping enzyme GTP-binding inhibitors: implications for antiviral drug development. J. Biomol Screen 2011, 16 (8), 852–861. 10.1177/1087057111412183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal K.; Axtman A. D.; Betarbet R.; Brennan P. E.; Carter G. W.; Frye S. V.; Fu H.; Greenwood A. K.; Longo F. M.; Pearce K. H. Jr.; Edwards A. M.; Levey A. I.; Tool and probe development for emerging targets in Alzheimer’s disease. Alzheimers Dementia 2022, 18, e064748 10.1002/alz.064748. [DOI] [Google Scholar]
- Axtman A. D.; Brennan P. E.; Frappier-Brinton T.; Betarbet R.; Carter G. W.; Fu H.; Gileadi O.; Greenwood A. K.; Leal K.; Longo F. M.; Mangravite L. M.; Edwards A. M.; Levey A. I. Open drug discovery in Alzheimer’s disease. Alzheimer’s Dement: Transl Res. Clin Interv 2023, 9, e12394 10.1002/trc2.12394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y.; Bradshaw W. J.; Leisner T. M.; Annor-Gyamfi J. K.; Qian K.; Bashore F. M.; Sikdar A.; Nwogbo F. O.; Ivanov A. A.; Frye S. V.; Gileadi O.; Brennan P. E.; Levey A. I.; Axtman A. D.; Pearce K. H.; Fu H.; Katis V. L.; Emory-Sage-SCG TREAT-AD Center . Development of FERM domain protein–protein interaction inhibitors for MSN and CD44 as a potential therapeutic strategy for Alzheimer’s disease. BioRxiv 2023, DOI: 10.1101/2023.05.22.541727. [DOI] [PubMed]
- Prinz H. Hill coefficients, dose-response curves and allosteric mechanisms. J. Chem. Biol. 2010, 3 (1), 37–44. 10.1007/s12154-009-0029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emelyanova E. V. Graphical approach to compare concentration constants of Hill and Michaelis-Menten equations. J. Biotechnol Biomed 2018, 1, 94–99. 10.14302/issn.2576-6694.jbbs-18-2280. [DOI] [Google Scholar]
- McMaster B.Anti-inflammatory compositions and methods of use. United States WO2003-US16558, 2003.
- Metz M.; Goldstein S. R.; Keshipeddy S. K.; Kwon D. Y.; Lemus R. H.; Vaddela S. B.; Cutshall N. S.; Gage J. L.; Gruswitz F. A.; Khalaf J.; Little T. L.; Nguyen J. H.; Nollert Von Specht P. K.; Tsoung J.; Cicirelli M.. Preparation of heterocyclic and amino acids as MASP-2 inhibitors and methods of use. United States WO2019-US34220, 2019, .
- Kirkpatrick D. L.; Indarte M.; Ihle N. T.. Methods and compositions for inhibiting CNKSR1. United States WO2013-US75505, 2014.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.