

Abstract
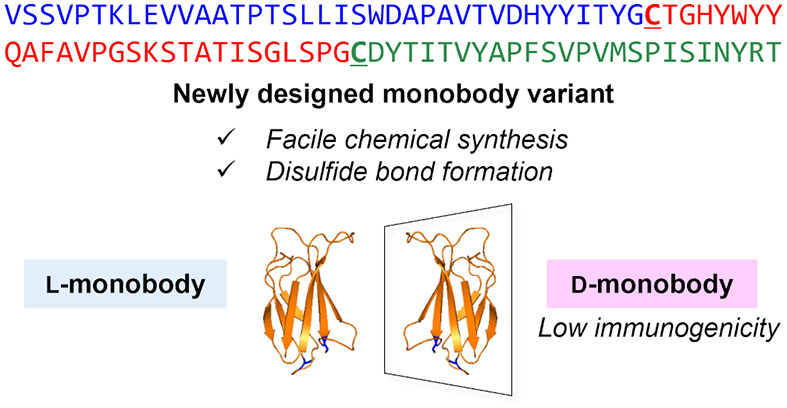
Mirror-image proteins (d-proteins) are promising scaffolds for drug discovery because of their high proteolytic stability and low immunogenic properties. Facile and reproducible processes for the preparation of functional d-proteins are required for their application in therapeutic biologics. In this study, we designed and synthesized a novel monobody variant with two cysteine substitutions that facilitate the synthetic process via sequential native chemical ligations and improve protein stability by disulfide bond formation. The synthetic anti-GFP monobody in this model study exhibited good binding affinity to the target enhanced green fluorescent protein. In vivo administration of the synthetic anti-GFP monobody (l-monobody) to mice induced antidrug antibody (ADA) production, whereas no ADA production was observed following immunization with the mirror-image anti-GFP monobody (d-monobody). These results suggest that the synthetic d-monobody is a non-antibody protein scaffold with low immunogenic properties.
Keywords: Fibronectin type III domain, Immunogenicity, Mirror-image protein, Monobody
A monobody is a non-immunoglobulin protein scaffold derived from the tenth type III domain of human fibronectin (FN3).1 The immunoglobulin domain-like β-sandwich structure of FN3 can hold several complementarity-determining region (CDR)-like variable regions in the sequence. This rigid scaffold facilitates target binding with high affinity and selectivity by restricting the conformational flexibility. Two types of monobodies were designed for screening variable regions (Figure 1): three loops (BC, DE, and FG loops) between β-strands contain variable regions in a loop-only library, and two β-strands (C and D) and two loops (CD and FG) were employed for the diversified positions in a side-and-loop library.1−3 Using this unique protein scaffold for the screening campaign by display technology, monobodies have been used in life science research, including molecular biology and crystallography.3 Additionally, several FN3-based therapeutic candidates have been identified for various target molecules, such as vascular endothelial growth factor receptor 2 (VEGFR2)4 and proprotein convertase subtilisin/kexin type 9.5 For medicinal applications, a monobody would be advantageous over other protein scaffolds because the sequence designed from endogenous fibronectin is less likely to induce an immune response after in vivo administration.6 However, using human-derived sequences for protein therapeutics does not necessarily eliminate the generation of anti-drug antibodies (ADAs), which may impair the therapeutic effects and/or sometimes cause adverse effects.7 Actually, it was reported that the administration of CT-322,8 a pegylated FN3-based protein engineered to bind VEGFR2, caused ADA generation in more than half of the patients.9
Figure 1.

Sequences of monobodies and design of a monobody variant with two cysteine substitutions. (A) Comparison of the variable regions in the side-and-loop and loop-only libraries. The substituted cysteines are underlined. The sequence is based on the anti-GFP monobody GS2.24 (B) Variable regions in the side-and-loop library in the structure of the monobody. Variable regions are colored with cyan, and the arrows indicate the substituted cysteines. (C) Variable regions in the loop-only library in the structure of the monobody.
We have focused on developing mirror-image monobodies for novel protein therapeutics to overcome this immunogenicity shortcoming. Mirror-image peptides (d-peptides) and proteins (d-proteins) are expected to have favorable pharmacokinetic and safety profiles10 because the sequences consisting entirely of d-amino acids are less susceptible to proteolytic degradation by endogenous peptidases.11 The less efficient processing of d-peptide- or d-protein-based biologics in antigen-presenting cells (APCs) may avoid antigen presentation on major histocompatibility complex (MHC) molecules for T cell recognition, leading to less ADA generation via T cell activation.12
Several studies have explored mirror-image peptide and protein therapeutics that bind several target molecules by mirror-image screening technologies.10,13−16 In this technology, the synthetic mirror-image protein of a target molecule (d-target) is used to screen a phage-display library (mirror-image phage display). When the hit sequences that bind with the d-target are identified, the d-peptide therapeutics are prepared by chemical synthesis, which should exhibit binding with the native target molecule. Recent success in developing a 13 kDa bivalent d-protein antagonist for vascular endothelial growth factor A17−19 suggests that mirror-image monobodies (∼10 kDa) are also promising protein scaffolds with less immunogenicity for drug discovery. We envisioned that establishing the preparation protocols of mirror-image monobodies would extend the scope of mirror-image screening using the synthetic target d-proteins20−22 into an unexplored modality.23 In this study, we investigated the potential of the mirror-image monobody as a suitable less immunogenic non-antibody scaffold in drug discovery.24 Comparative structural and biological analyses were conducted between native and mirror-image monobodies, which were obtained by establishing a facile synthetic process.
For this model study, we selected a monobody, GS2, that binds with green fluorescent protein (GFP).2 This anti-GFP monobody, GS2, was identified by phage-display selection from a side-and-loop library. Our initial attempt to establish the synthetic protocol of the monobody scaffold was conducted for the native sequence of GS2 (Scheme S1). Because the monobody sequence does not contain conserved Cys residues, native chemical ligations (NCLs)25 and a desulfurization strategy were employed for ligations at the Xaa-Ala sites.26 The sequence of GS2 was successfully constructed from four peptide segments; however, temporary modification with a solubilizing auxiliary was also required to improve the low solubility of the intermediate and purification by HPLC after NCLs. We postulated that these steps would hamper the efficient construction of monobody sequences, which would be identified by mirror-image screening for target proteins.27
To overcome the drawbacks of considerable synthetic efforts, we designed a novel monobody variant in which two cysteine substitutions were introduced into the sequence to facilitate the synthetic process by stepwise NCLs. We chose Glu38 in the CD-loop and Val66 in the EF-loop of GS2 for substitution with cysteines, which are located in close proximity to potentially form a disulfide bond (Figure 1). These two residues are also located outside of the variable regions in a side-and-loop library of monobodies and on the opposite side of the variable regions (BC-, DE-, and FG-loops) in a loop-only library of monobodies. A previous study on Centyrin, an FN3-based protein scaffold, revealed that single cysteine mutations at the corresponding residues had relatively small effects on the target binding.28 Therefore, the overall synthetic process can be designed without dependence on the hit bioactive sequences in the variable regions. Additionally, because the upstream residues of Glu38 and Val66 are glycines (Gly37 and Gly65) in the sequence of GS2, substitutions with cysteines enable efficient NCLs at the Gly–Cys junction.29 Thus, this modified monobody is a non-antibody scaffold that combines sufficient target binding and less synthetic effort, which is suitable for a mirror-image screening strategy.
We investigated two synthetic routes for modified GS2 (mGS2) with cysteine substitutions. Our initial attempt to synthesize mGS2 via an N-to-C NCL strategy is depicted in Scheme 1. The N-terminal peptide segment l-1 and middle segment l-2a were constructed by Fmoc-based solid-phase peptide synthesis on N-acyl-N′-methyl-benzimidazolinone (MeNbz)30 and diaminobenzoic acid (Dbz)31 linkers, respectively. To improve the solubility of the segments during NCL and purification, a solubilizing auxiliary sequence of four arginines was appended to the C-terminus of peptides l-1 and l-2a. The C-terminal segment l-3 was also synthesized, which contained a histidine tag at the C-terminus for detection and immobilization. 1,2,4-Triazole-mediated NCL32 between peptides l-1 and l-2a provided the intermediate l-4. NaNO2-mediated activation of the Dbz moiety33 in l-4 followed by NCL in the presence of 4-mercaptophenylacetic acid (MPAA) with the C-terminal segment l-3 afforded the expected full sequence of mGS2 (l-6). However, the chromatographic separation of the product from a trace hydrolysate impurity from l-4 was difficult.
Scheme 1. Synthesis of anti-GFP Monobody via N-to-C Native Chemical Ligations.

Reagents and conditions: (a) 1,2,4-triazole, TCEP, 6 M guanidine (pH 7.0); (b) NaNO2, 6 M guanidine, phosphate buffer (pH 3.0); (c) MPAA, TCEP, 6 M guanidine, phosphate buffer (pH 6.2); (d) MPAA, TCEP 6 M guanidine, phosphate buffer (pH 6.5).
Next, we investigated the synthesis of mGS2 via a C-to-N NCL strategy (Scheme 2). Because the N-terminal and C-terminal peptide segments (l-1 and l-3) can be used for the C-to-N NCL strategy, only the middle segment (l-2b) was newly designed, which contains 1,3-thiazolidine-4-carboxylic acid (Thz) for N-terminal temporary protection and a MeNbz linker for C-terminal activation. The resulting peptide segments were assembled in the C-to-N direction. 1,2,4-Triazole-mediated NCL between peptides l-2b and l-3 followed by methoxyamine-mediated deprotection of thiazolidine provided the intermediate l-7. Subsequently, peptide l-7 was conjugated with the N-terminal segment l-1 in the presence of 1,2,4-triazole again to give l-mGS2 (l-6) in 35% overall yield (two steps from the peptide segment l-3). The C-to-N NCL strategy was more straightforward than the N-to-C process because the enhanced solubility of l-7 with a C-terminal histidine tag facilitated the purification processes after NCLs. This simple synthetic process was also used to synthesize the mirror-image d-mGS2 (d-6) using d-amino acids and glycine (37% overall yield from d-3). Our success in the efficient preparation of full-length mGS2 supports the validity of our design and the synthetic process for monobody variants.
Scheme 2. Synthesis of anti-GFP Monobody via C-to-N Native Chemical Ligations.

Reagents and conditions: (a) 1,2,4-triazole, TCEP, 6 M guanidine (pH 7.0); (b) methoxyamine; (c) 2-PDS, 6 M guanidine (pH 8.0).
With the full length of l-mGS2 (l-6) in hand, we investigated the folding conditions to obtain bioactive l-mGS2. There is a reported procedure for purification of an FN3-derived binding protein from E. coli inclusion bodies via refolding by dialysis in acetate buffer (pH 4.5).8 According to the reported protocol8 with some modifications, l-mGS2 was subjected to dialysis procedures under slightly acidic conditions (50 mM acetate buffer, pH 4.5, 5 mM TCEP) to provide the folded protein with substituted cysteines in a reduced form. Of note, when we initially investigated various folding conditions and procedures under neutral pH, l-mGS2 was highly prone to aggregation, and obtaining bioactive l-mGS2 failed. It was reported that FN3 modules and FN3-derived proteins are thermodynamically more stable under acidic conditions compared with neutral conditions.1,34−36 In the current case, the slightly acidic conditions (pH 4.5) would contribute to the formation of stable structure and thus the rapid folding kinetics,34 which presumably prevented the unfavorable self-association. The CD spectrum of folded l-mGS2 suggested the existence of a β-sheet structure, which is a common feature among FN3-derived binding proteins (Figure 2). d-mGS2, which was subjected to identical folding conditions, showed an inverted CD spectrum compared to that of l-mGS2. These results suggested that synthetic d-mGS2 folded correctly to form the mirror-image structure of l-mGS2.
Figure 2.
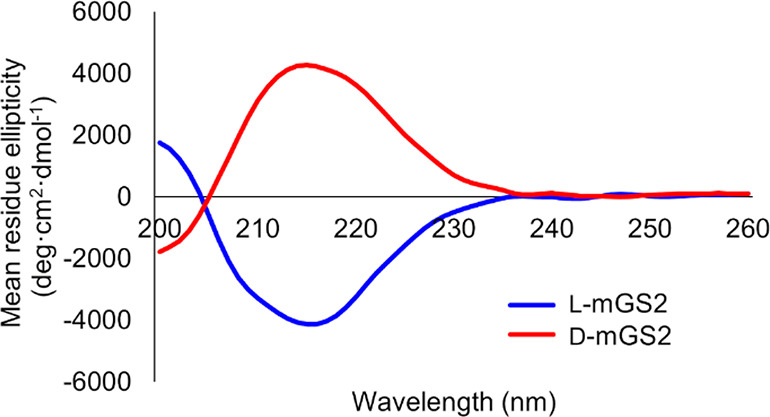
CD spectra of l-mGS2 and d-mGS2.
The bioactivity of synthetic mGS2 proteins was evaluated by surface plasmon resonance (SPR) analysis. Biotin-labeled mGS2 (l-mGS2biotin and d-mGS2biotin) were designed and synthesized for immobilization on the sensor chip (Scheme S2). Enhanced green fluorescent protein (EGFP) at various concentrations was flowed over the sensor chip. Synthetic l-mGS2biotin showed slightly less potent binding toward EGFP (KD = 9.8 ± 4.1 nM) compared with that of biotin-labeled native GS2 (l-GS2biotin; KD = 1.8 ± 1.3 nM) (Figure S1 and Table 1). The binding affinity of these synthetic proteins was comparable to that of recombinant GS2, which was reported previously.2 In contrast, d-mGS2biotin did not bind to EGFP, suggesting that the molecular recognition of synthetic l-mGS2 with EGFP was accomplished stereoselectively.
Table 1. SPR Analysis of Synthetic Monobodies Binding with EGFP.
| Ligand | KD (nM)a |
|---|---|
| l-GS2biotin b | 1.8 ± 1.3 |
| l-mGS2biotin | 9.8 ± 4.1 |
| l-mGS2SS/biotin | 3.7 ± 0.3 |
| d-mGS2biotin | no binding |
KD values were determined from triplicate assays.
KD value of recombinant GS2 was reported to be 3.4 ± 0.2 nM.2
Next, we comparatively investigated the immunogenic properties of folded l-mGS2 and d-mGS2 (Figure 3). BALB/c mice were immunized intraperitoneally with synthetic l- or d-mGS2 three times in combination with a Freund’s adjuvant at days 0, 14, and 28. Plasma samples were collected from mice at days 0, 14, 28, 35 and 44, and the generation of an anti-l-mGS2 antibody or anti-d-mGS2 antibody (ADA against mGS2) was measured by ELISA. Repeated injections caused a gradual increase in the ADA level for l-mGS2-immunized mice, whereas no increase in ADA was observed for d-mGS2-immunized mice. The production of ADA was observed in all l-mGS2-immunized mice on day 44 after the initial administration of the monobody, with some variation in the production levels (Figure S2). In contrast, no ADA generation was observed in all d-mGS2-immunized mice. These results suggest that the mirror-image monobodies are promising protein scaffolds with less immunogenic properties and support previous reports showing that mirror-image peptides and proteins have lower immunogenic properties.12,18,19
Figure 3.
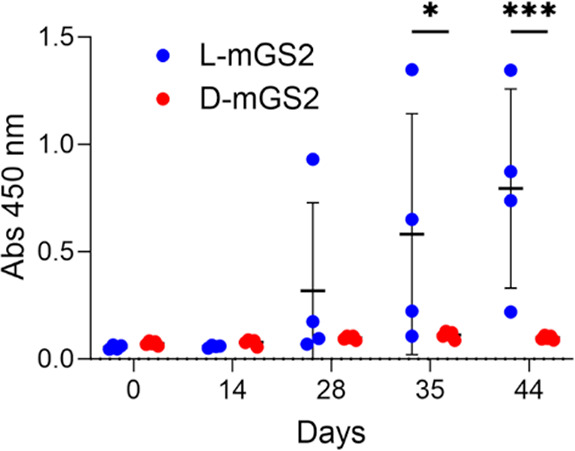
Evaluation of the immunogenicity of l-mGS2 and d-mGS2. Generation of an antidrug antibody (ADA) in mouse sera at days 0, 14, 28, 35, and 44 after injection of l-mGS2 and d-mGS2 was detected by ELISA (l-mGS2: n = 4; d-mGS2: n = 5). Absorbance of 3,3′,5,5′-tetramethylbenzidine (TMB) was measured at 450 nm. Statistical analysis was performed by two-way ANOVA followed by Sidak’s multiple comparisons test. *, p < 0.05, ***, p < 0.001.
Disulfide bonds play an important role in the folding of peptide-based and protein-based therapeutics and possibly contribute to improving pharmacological properties.37 Gilbreth et al. previously introduced a new disulfide bond between two mutated cysteines of the third fibronectin type III domain of human tenascin-C to improve the thermodynamic stability and resistance to thermolysin-mediated proteolysis.38 In our newly designed monobody scaffold, because the substituted cysteines, Cys38 and Cys66, are adjacent to each other in the folded state, we expected that an intramolecular disulfide bond between these vicinal cysteines would similarly improve the protein stability. Thus, we investigated the formation of an intramolecular disulfide bond in mGS2 and subsequent refolding for preparing the bioactive monobody. l-mGS2 (l-6) was treated with 2,2′-dithiodipyridine (2-PDS)39 under denaturing conditions (pH 8.0) in guanidine buffer to give l-mGS2SS (l-8) with an intramolecular disulfide bond in 44% yield (Scheme 2). The formation of a disulfide bond in l-8 was confirmed by MS measurement after treatment with iodoacetamide (Figure S3). Peptide l-8 was subjected to folding conditions in a mildly acidic buffer (50 mM acetate buffer, pH 4.5) to provide folded l-mGS2SS. The β-sheet structure was verified by CD spectroscopy, with its spectrum identical to that of l-mGS2 (Figure S4). Additionally, the thermal stability of l-mGS2 and l-mGS2SS was evaluated by monitoring changes to the CD signal at 203 nm (Figure S5). l-mGS2SS showed slightly higher thermal stability than l-mGS2. In the SPR analysis, biotin-labeled l-mGS2SS (l-mGS2SS/biotin) exhibited improved binding affinity with EGFP (KD = 3.7 ± 0.3 nM) compared with l-mGS2biotin and comparable to l-GS2biotin (Figure S1 and Table 1). This observation suggests that the formation of the disulfide bond stabilized the bioactive form of the anti-GFP monobody and/or restored the unfavorable effect(s) of two sulfhydryl groups in l-mGS2 on binding with EGFP.
In conclusion, we have established a facile synthetic process for a newly designed monobody variant with two cysteine substitutions via two-step NCLs from three peptide segments. In the synthetic process, the bioactive sequence in the side-and-loop library and loop-only library, which would be identified by phage-display screening, can be included in each peptide segment, providing a general synthetic approach for various monobody proteins. The resulting synthetic l-mGS2 retained the reported structure of the recombinant protein and sufficient binding to EGFP. The immunogenicity assessment suggested that synthetic d-mGS2 showed significantly less ADA generation compared with l-mGS2. Forming an intramolecular disulfide bond in the designed monobody variant (l-mGS2SS) improved the binding affinity and thermal stability. The mirror-image monobody with lower immunogenicity should be an attractive non-antibody scaffold for developing novel therapeutic biologics. The application to mirror-image screening using particular display technologies is ongoing in our laboratory.
Acknowledgments
This work was supported by JSPS KAKENHI, Japan (JP18H02555, JP20K21252, JP22H02747, JP22K19376, JP22KJ1842); Research on Development of New Drugs (JP20ak0101144) from AMED, Japan; The Tokyo Biochemical Research Foundation; Astellas Foundation for Research on Metabolic Disorders; and Takeda Science Foundation.
Glossary
Abbreviations
- ADA
antidrug antibody
- APC
antigen presenting cells
- CDR
complementarity determining region
- Dbz
diaminobenzoic acid
- EGFP
enhanced green fluorescent protein
- FN3
type III domain of human fibronectin
- GFP
green fluorescent protein
- MeNbz
N-acyl-N′-methyl-benzimidazolinone
- MHC
major histocompatibility complex
- MPAA
4-mercaptophenylacetic acid
- NCL
native chemical ligation
- 2-PDS
2,2′-bispyridyl disulfide
- SPR
surface plasmon resonance
- TCEP
tris(2-carboxyethyl)phosphine
- Thz
1,3-thiazolidine-4-carboxylic acid
- TMB
3,3′,5,5′-tetramethylbenzidine
- VEGFR2
vascular endothelial growth factor receptor 2
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.3c00342.
Experimental procedures for peptide synthesis and biological evaluations, characterization of peptides, and supporting figures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Koide S.; Bailey C. W.; Huang X.; Koide S. The fibronectin type III domain as a scaffold for novel binding proteins. J. Mol. Biol. 1998, 284, 1141–1151. 10.1006/jmbi.1998.2238. [DOI] [PubMed] [Google Scholar]
- Koide A.; Wojcik J.; Gilbreth R. N.; Hoey R. J.; Koide S. Teaching an old scaffold new tricks: monobodies constructed using alternative surfaces of the FN3 scaffold. J. Mol. Biol. 2012, 415, 393–405. 10.1016/j.jmb.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantschel O.; Biancalana M.; Koide S. Monobodies as enabling tools for structural and mechanistic biology. Curr. Opin. Struct. Biol. 2020, 60, 167–174. 10.1016/j.sbi.2020.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker M. H.; Chen Y.; Danehy F.; Dufu K.; Ekstrom J.; Getmanova E.; Gokemeijer J.; Xu L.; Lipovsek D. Antibody mimics based on human fibronectin type three domain engineered for thermostability and high-affinity binding to vascular endothelial growth factor receptor two. Protein Eng. Des. Sel. 2005, 18, 435–444. 10.1093/protein/gzi050. [DOI] [PubMed] [Google Scholar]
- Mitchell T.; Chao G.; Sitkoff D.; Lo F.; Monshizadegan H.; Meyers D.; Low S.; Russo K.; DiBella R.; Denhez F.; Gao M.; Myers J.; Duke G.; Witmer M.; Miao B.; Ho S. P.; Khan J.; Parker R. A. Pharmacologic profile of the adnectin BMS-962476, a small protein biologic alternative to PCSK9 antibodies for low-density lipoprotein lowering. J. Pharmacol. Exp. Ther. 2014, 350, 412–424. 10.1124/jpet.114.214221. [DOI] [PubMed] [Google Scholar]
- Gebauer M.; Skerra A. Engineered protein scaffolds as next generation therapeutics. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 391–415. 10.1146/annurev-pharmtox-010818-021118. [DOI] [PubMed] [Google Scholar]
- Vaisman-Mentesh A.; Gutierrez-Gonzalez M.; DeKosky B. J.; Wine Y. The molecular mechanisms that underlie the immune biology of anti-drug antibody formation following treatment with monoclonal antibodies. Front. Immunol. 2020, 11, 1951–1966. 10.3389/fimmu.2020.01951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamluk R.; Carvajal I. M.; Morse B. A.; Wong H. K; Abramowitz J.; Aslanian S.; Lim A.-C.; Gokemeijer J.; Storek M. J.; Lee J.; Gosselin M.; Wright M. C.; Camphausen R. T.; Wang J.; Chen Y.; Miller K.; Sanders K.; Short S.; Sperinde J.; Prasad G.; Williams S.; Kerbel R. S.; Ebos J.; Mutsaers A.; Mendlein J. D.; Harris A. S.; Furfine E. S. Anti-tumor effect of CT-322 as an adnectin inhibitor of vascular endothelial growth factor receptor-2. mAbs 2010, 2, 199–208. 10.4161/mabs.2.2.11304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolcher A. W.; Sweeney C. J.; Papadopoulos K.; Patnaik A.; Chiorean E. G.; Mita A. C.; Sankhala K.; Furfine E.; Gokemeijer J.; Iacono L.; Eaton C.; Silver B. A.; Mita M. Phase I and pharmacokinetic study of CT-322 (BMS-844203), a targeted adnectin inhibitor of VEGFR-2 based on a domain of human fibronectin. Clin. Cancer Res. 2011, 17, 363–371. 10.1158/1078-0432.CCR-10-1411. [DOI] [PubMed] [Google Scholar]
- Lander A. J.; Jin Y.; Luk L. Y. P. d-Peptide and d-protein technology: recent advances, challenges, and opportunities. ChemBioChem 2023, 24, e202200537 10.1002/cbic.202200537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton R. C. deL.; Milton S. C. F.; Kent S. B. H. Total chemical synthesis of a d-enzyme: the enantiomers of HIV-1 protease show demonstration of reciprocal chiral substrate specificity. Science 1992, 256, 1445–1448. 10.1126/science.1604320. [DOI] [PubMed] [Google Scholar]
- Dintzis H. M.; Symer D. E.; Dintzis R. Z.; Zawadzke L. E.; Berg J. M. A comparison of the immunogenicity of a pair of enantiomeric proteins. Proteins: Struct., Funct. Genet. 1993, 16, 306–308. 10.1002/prot.340160309. [DOI] [PubMed] [Google Scholar]
- Schumacher T. N. M.; Mayr L. M.; Minor D. L. Jr.; Milhollen M. A.; Burgess M. W.; Kim P. S. Identification of d-peptide ligands through mirror-image phage display. Science 1996, 271, 1854–1857. 10.1126/science.271.5257.1854. [DOI] [PubMed] [Google Scholar]
- Chang H.-N.; Liu B.-Y.; Qi Y.-K.; Zhou Y.; Chen Y.-P.; Pan K.-M.; Li W.-W.; Zhou X.-M.; Ma W.-W.; Fu C.-Y.; Qi Y.-M.; Liu L.; Gao Y.-F. Blocking of the PD-1/PD-L1 interaction by a d-peptide antagonist for cancer immunotherapy. Angew. Chem., Int. Ed. 2015, 54, 11760–11764. 10.1002/anie.201506225. [DOI] [PubMed] [Google Scholar]
- Zhou X.; Zuo C.; Li W.; Shi W.; Zhou X.; Wang H.; Chen S.; Du J.; Chen G.; Zhai W.; Zhao W.; Wu Y.; Qi Y.; Liu L.; Gao Y. A novel d-peptide identified by mirror-image phage display blocks TIGIT/PVR for cancer immunotherapy. Angew. Chem., Int. Ed. 2020, 59, 15114–15118. 10.1002/anie.202002783. [DOI] [PubMed] [Google Scholar]
- Malhis M.; Kaniyappan S.; Aillaud I.; Chandupatla R. R.; Ramirez L. M.; Zweckstetter M.; Horn A. H. C.; Mandelkow E.; Sticht H.; Funke S. A. Potent Tau aggregation inhibitor d-peptide selected against Tau-repeat 2 using mirror image phage display. ChemBioChem 2021, 22, 3049–3059. 10.1002/cbic.202100287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal K.; Uppalapati M.; Ault-Riche D.; Kenney J.; Lowitz J.; Sidhu S. S.; Kent S. B. Chemical synthesis and X-ray structure of a heterochiral {d-protein antagonist plus vascular endothelial growth factor} protein complex by racemic crystallography. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 14779–14784. 10.1073/pnas.1210483109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uppalapati M.; Lee D. J.; Mandal K.; Li H.; Miranda L. P.; Lowitz J.; Kenney J.; Adams J. J.; Ault-Riché D.; Kent S. B. H.; Sidhu S. S. A potent d-protein antagonist of VEGF-A is nonimmunogenic, metabolically stable, and longer-circulating in vivo. ACS Chem. Biol. 2016, 11, 1058–1065. 10.1021/acschembio.5b01006. [DOI] [PubMed] [Google Scholar]
- Marinec P. S.; Landgraf K. E.; Uppalapati M.; Chen G.; Xie D.; Jiang Q.; Zhao Y.; Petriello A.; Deshayes K.; Kent S. B. H.; Ault-Riche D.; Sidhu S. S. A non-immunogenic bivalent d-protein potently inhibits retinal vascularization and tumor growth. ACS Chem. Biol. 2021, 16, 548–556. 10.1021/acschembio.1c00017. [DOI] [PubMed] [Google Scholar]
- Noguchi T.; Ishiba H.; Honda K.; Kondoh Y.; Osada H.; Ohno H.; Fujii N.; Oishi S. Synthesis of Grb2 SH2 domain proteins for mirror-image screening systems. Bioconjugate Chem. 2017, 28, 609–619. 10.1021/acs.bioconjchem.6b00692. [DOI] [PubMed] [Google Scholar]
- Shu K.; Noguchi T.; Honda K.; Kondoh Y.; Osada H.; Ohno H.; Fujii N.; Oishi S. Synthesis of the Src SH2 domain and its application in bioassays for mirror-image screening. RSC Adv. 2017, 7, 38725–38732. 10.1039/C7RA07445J. [DOI] [Google Scholar]
- Shu K.; Iwamoto N.; Honda K.; Kondoh Y.; Hirano H.; Osada H.; Ohno H.; Fujii N.; Oishi S. Development of mirror-image screening systems for XIAP BIR3 domain inhibitors. Bioconjugate Chem. 2019, 30, 1395–1404. 10.1021/acs.bioconjchem.9b00154. [DOI] [PubMed] [Google Scholar]
- Agouridas V.; El Mahdi O.; Melnyk O. Chemical protein synthesis in medicinal chemistry. J. Med. Chem. 2020, 63, 15140–15152. 10.1021/acs.jmedchem.0c01082. [DOI] [PubMed] [Google Scholar]
- A similar concept of mirror-image monobody was introduced in a previous conference:; Reichart T.; Kükenshöner T.; Hantschel O. Development of mirror-image monobodies for targeted cancer therapies. J. Pept. Sci. 2018, 24, S67–S68. [Google Scholar]
- Dawson P. E.; Muir T. W.; Clark-Lewis I.; Kent S. B. H. Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–779. 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- Jin K.; Li X. Advance in native chemical ligation-desulfurization: a powerful strategy for peptide and protein synthesis. Chem.—Eur. J. 2018, 24, 17397–17404. 10.1002/chem.201802067. [DOI] [PubMed] [Google Scholar]
- An earlier study demonstrated the chemical synthesis of a tenth FN3 analogue, which contained a thioester displacement for a peptide bond:; Williams M. J.; Muir T. W.; Ginsberg M. H.; Kent S. B. H. Total chemical synthesis of a folded β-sandwich protein domain: an analog of the tenth fibronectin type 3 module. J. Am. Chem. Soc. 1994, 116, 10797–10798. 10.1021/ja00102a060. [DOI] [Google Scholar]
- Goldberg S. D.; Cardoso R. M. F.; Lin T.; Spinka-Doms T.; Klein D.; Jacobs S. A.; Dudkin V.; Gilliland G.; O’Neil K. T. Engineering a targeted delivery platform using Centyrins. Protein Eng. Des. Sel. 2016, 29, 563–572. 10.1093/protein/gzw054. [DOI] [PubMed] [Google Scholar]
- Hackeng T. M.; Griffin J. H.; Dawson P. E. Protein synthesis by native chemical ligation: expanded scope by using straightforward methodology. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 10068–10073. 10.1073/pnas.96.18.10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Canosa J. B.; Nardone B.; Albericio F.; Dawson P. E. Chemical protein synthesis using a second-generation N-acylurea linker for the preparation of peptide-thioester precursors. J. Am. Chem. Soc. 2015, 137, 7197–7209. 10.1021/jacs.5b03504. [DOI] [PubMed] [Google Scholar]
- Blanco-Canosa J. B.; Dawson P. E. An efficient Fmoc-SPPS approach for the generation of thioester peptide precursors for use in native chemical ligation. Angew. Chem., Int. Ed. 2008, 47, 6851–6855. 10.1002/anie.200705471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K.; Tsuda S.; Nishio H.; Yoshiya T. 1,2,4-Triazole-aided native chemical ligation between peptide-N-acyl-N′-methyl-benzimidazolinone and cysteinyl peptide. Chem. Commun. 2017, 53, 12236–12239. 10.1039/C7CC07817J. [DOI] [PubMed] [Google Scholar]
- Wang J.-X.; Fang G. M.; He Y.; Qu D.-L.; Yu M.; Hong Z.-Y.; Liu L. Peptide o-aminoanilides as crypto-thioesters for protein chemical synthesis. Angew. Chem., Int. Ed. 2015, 54, 2194–2198. 10.1002/anie.201408078. [DOI] [PubMed] [Google Scholar]
- Plaxco K. W.; Spitzfaden C.; Campbell I. D.; Dobson C. M. Rapid refolding for a proline-rich all-β-sheet fibronectin type III module. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 10703–10706. 10.1073/pnas.93.20.10703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke J.; Hamill S. J.; Johnson C. M. Folding and stability of a fibronectin type III domain of human tenascin. J. Mol. Biol. 1997, 270, 771–778. 10.1006/jmbi.1997.1147. [DOI] [PubMed] [Google Scholar]
- Koide A.; Jordan M. R.; Horner S. R.; Batori V.; Koide S. Stabilization of a fibronectin type III domain by the removal of unfavorable electrostatic interaction on the protein surface. Biochemistry 2001, 40, 10326–10333. 10.1021/bi010916y. [DOI] [PubMed] [Google Scholar]
- Gongora-Beńítez M.; Tulla-Puche J.; Albericio F. Multifaced roles of disulfide bonds. Peptides as therapeutics. Chem. Rev. 2014, 114, 901–926. 10.1021/cr400031z. [DOI] [PubMed] [Google Scholar]
- Gilbreth R. N.; Chacko B. M.; Grinberg L.; Swers J. S.; Baca M. Stabilization of the third fibronectin type III domain of human tenascin-C through minimal mutation and rational design. Protein Eng. Des. Sel. 2014, 27, 411–418. 10.1093/protein/gzu024. [DOI] [PubMed] [Google Scholar]
- Maruyama K.; Nagasawa H.; Suzuki A. 2,2′-Bispyridyl disulfide rapidly induces intramolecular disulfide bonds in peptides. Peptides 1999, 20, 881–884. 10.1016/S0196-9781(99)00076-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.