

Abstract
Background
Malformations of cortical development (MCDs) have been reported in a subset of patients with pathogenic heterozygous variants in GRIN1 or GRIN2B, genes which encode for subunits of the N-methyl-D-aspartate receptor (NMDAR). The aim of this study was to further define the phenotypic spectrum of NMDAR-related MCDs.
Methods
We report the clinical, radiological and molecular features of 7 new patients and review data on 18 previously reported individuals with NMDAR-related MCDs. Neuropathological findings for two individuals with heterozygous variants in GRIN1 are presented. We report the clinical and neuropathological features of one additional individual with homozygous pathogenic variants in GRIN1.
Results
Heterozygous variants in GRIN1 and GRIN2B were associated with overlapping severe clinical and imaging features, including global developmental delay, epilepsy, diffuse dysgyria, dysmorphic basal ganglia and hippocampi. Neuropathological examination in two fetuses with heterozygous GRIN1 variants suggests that proliferation as well as radial and tangential neuronal migration are impaired. In addition, we show that neuronal migration is also impaired by homozygous GRIN1 variants in an individual with microcephaly with simplified gyral pattern.
Conclusion
These findings expand our understanding of the clinical and imaging features of the ‘NMDARopathy’ spectrum and contribute to our understanding of the likely underlying pathogenic mechanisms leading to MCD in these patients.
INTRODUCTION
N-methyl-D-aspartate receptors (NMDARs) are ligand-gated cation channels mediating excitatory synaptic transmission. The NMDAR is a heteromeric complex typically composed of two GluN1 subunits (encoded by GRIN1) and two GluN2 subunits (encoded by GRIN2A-D). The combination of different subunits influences receptor function.1 2 NMDAR subunits contain a discontinuous ligand-binding domain (S1 and S2), an intracellular re-entrant pore loop (M2) and three transmembrane domains (M1, M3 and M4).3 NMDAR subunits exhibit different spatiotemporal expression patterns. The essential glycine-binding GluN1 subunit is highly and ubiquitously expressed in the fetal and adult brain.2 In contrast, the glutamate-binding subunit GluN2B (encoded by GRIN2B) is highly expressed prenatally with progressive limitation to the forebrain, before being replaced postnatally by GluN2A (encoded by GRIN2A).4
Pathogenic variants in GRIN1 and GRIN2B have been described in individuals with a spectrum of neurological phenotypes. In individuals with heterozygous pathogenic variants, common features include developmental delay, intellectual disability, epilepsy, cortical visual impairment, neurobehavioural problems and movement disorders.5–10 Homozygous variants have only rarely been reported in GRIN1. These include four individuals from two different families with homozygous missense variants, developmental delay and movement disorders but without seizures,9 11 and four individuals from two families with homozygous truncating variants with severe neonatal epileptic encephalopathy.9 12 Homozygous variants in GRIN2B have not been reported yet. To date, 12 patients harbouring de novo heterozygous pathogenic variants in GRIN1 and 6 patients with pathogenic variants in GRIN2B have been reported to have malformations of cortical development (MCDs).13–15 This subset of patients accounts for approximately 10%–15% of all reported individuals with pathogenic variants in either GRIN1 or GRIN2B.16 17 Based on radiological appearance, the MCDs observed in patients with pathogenic variants in GRIN1 or GRIN2B are characterised by variable sulcal depth and/or orientation with normal cortical thickness. This was described as ‘dysgyria’, the generic term for abnormal cerebral cortex, although the malformation seems to closely resemble polymicrogyria.13 18 However, histopathological evidence has not been available from previous patients to confirm this. Associated brain malformations such as dysmorphic basal ganglia, hypoplastic corpus callosum and hippocampal dysplasia have been reported.13 15 The pattern of brain malformations associated with NMDAR-associated MCDs has been noted to resemble that of tubulinopathies.13 15
To provide further insights into the clinical and radiological features of NMDAR-associated MCDs, we report four patients with pathogenic heterozygous variants in GRIN1 and three patients with heterozygous variants in GRIN2B who all presented with complex brain malformations, including dysgyria, and malformations of the hippocampus and basal ganglia. In addition, we present a single patient with a homozygous variant in GRIN1 associated with a simplified gyral pattern (SGP). The neuropathological data, which were available for two fetuses with de novo heterozygous GRIN1 variants and a neonate with the homozygous GRIN1 variant, contribute to our understanding of the underlying pathophysiological mechanisms causing the phenotypic features of NMDAR-associated MCD.
MATERIALS AND METHODS
Patient selection
The patients were identified through the Neuro-MIG Network (European Cooperation in Science and Technology (COST) Action CA16118). Three of the patients (individuals 1, 2 and 7) were part of the Deciphering Developmental Disorders study.19 Clinical data were collected through clinical examination and review of medical records. Imaging data were reviewed by the authors (SB, AEF, NB-B).
Genetic analysis
DNA preparation and genetic investigation were undertaken with informed consent from the parents or guardians of the patients. DNA was derived from the saliva (individuals 1, 2 and 7), blood (individuals 5, 6 and 8) and by amniocentesis (individuals 3 and 4). DNA was extracted using standard protocols. Whole-exome sequencing was performed in individuals 1–4, 6 and 7. Individuals 5 and 8 were tested by next-generation sequencing multigene panels. Sanger sequencing was used in all individuals except for individual 6 to validate the variants and to test the parents. The positions of variants are based on transcripts NM_007327.3 for GRIN1 and NM_000834.3 for GRIN2B. Variants were classified using the American College of Medical Genetics and Genomics and Association for Molecular Pathology criteria.20 21
Electrophysiological analysis
A detailed description of these methods is provided in online supplemental material. In summary, the p.Cys744Tyr and the p.Ala653Thr variants were introduced into a plasmid expression construct containing the human GluN1-1a cDNA (here-after GluN1, based on NM_007327.3) and the p.Met818Thr introduced into human GluN2B cDNA (NM_000834.3) via a QuikChange mutagenesis protocol. The RNAs made from cDNA (cRNA) for wild-type and mutant forms of GluN1 were coinjected with wild-type human GluN2A cRNA and for wild-type and mutant forms of GluN2B with wild-type human GluN1 cRNA into Xenopus laevis oocytes. The expressed receptors were functionally evaluated by two-electrode voltage clamp recordings for dose response to L-glutamate, glycine, Mg2+ and Zn2+ ions and for proton sensitivity.
Neuropathological studies
After complete autopsy, brains were fixed in a 10% formaldehyde-zinc buffer solution. Macroscopic and microscopic examinations were performed according to standardised procedures. Immunohistochemical studies were performed using markers for neuronal progenitors, migrating neurons and interneurons, as well as for cortical layering (online supplemental material). Results were compared with age-matched controls aged 25, 30 and 39 gestational weeks, respectively.
RESULTS
Clinical features of patients with MCD with pathogenic heterozygous variants in GRIN1 and GRIN2B
We identified seven unrelated patients with MCD (individuals 1–7) with de novo heterozygous pathogenic variants in either GRIN1 or GRIN2B. Clinical findings for individuals 1–7 are summarised in table 1. Two individuals with GRIN1 variants (individuals 1 and 2) and three individuals with GRIN2B variants presented with microcephaly, severe developmental delay and epilepsy. The age of seizure onset ranged from the neonatal period to 2 years of age. Seizure types were variable, and were refractory to treatment in two individuals. Individuals 3 and 4 were fetuses with variants in GRIN1 that underwent termination of pregnancy. Autopsy and neuropathological findings from individuals 3 and 4 are described in the following section .
Table 1.
Clinical and imaging features of novel patients with variants in GRIN1 and GRIN2B
| Individual | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| Gene | GRIN1 | GRIN1 | GRIN1 | GRIN1 | GRIN2B | GRIN2B | GRIN2B | GRIN1 |
| Mutation | c.1658C>T, p.(Ser553Leu) | c.1972G>T, p.(Asp658Tyr) | c.1957G>A, p.(Ala653Thr) | c.2231G>A, p.(Cys744Tyr) | c.2437C>G, p.(Leu813Val) | c.2438T>C, p.(Leu813Pro) | c.2453T>C, p.(Met818Thr) | c.1422C>A, p.(Tyr474*) |
| Inheritance | De novo | De novo | De novo | De novo | De novo | De novo | De novo | Homozygous, inherited |
| Age at last review | 19 months | n/a | 25 GW | 30 GW | 23 months | 2.5 years | 15 months | 6 days |
| Sex | Male | Male | Male | n/a | Male | Male | Male | Female |
| DD | Severe | Severe | n/a | n/a | Severe | Severe | Severe | n/a |
| Age sz onset | Birth/prenatal | 4 months | n/a | n/a | 4 months | 2 years 3 months | 10 weeks | Early postnatal |
| Sz type | n/a | Absences, drops, tonic extensions | n/a | n/a | Epileptic spasms, tonic sz | GTCS with fever | Infantile spasms, rapid and rolling eye movements | Status epilepticus |
| Refractory sz | No | Yes | n/a | n/a | Yes | No | No | Yes |
| Neurology | Hypertonia | Spastic quadriplegia | n/a | n/a | Spastic quadriplegia | Spastic quadriplegia, truncal hypotonia and limb hypertonia, pseudobulbar palsy | Hypertonia | n/a |
| Dysmorphism | Long palpebral fissures, shallow orbits, overlapping fingers | Thin upper lips, anteverted nostrils, high frontal hairline, wide nasal bridge | Prominent philtrum, broad nasal bridge, anteverted nostrils | Marked nasal ridge, prominent philtrum, flat face, camptodactyly | No | No | No | No |
| OFC | 41.5 cm (−5.2 SD) | 0.4–2nd percentile | Low brain weight, OFC −2 SD | Low brain weight | 44 cm (−3.2 SD) | 44.8 cm (−2.2 SD) | 44 cm (9th centile) at 10 months, at 2.5 years 45 cm (−3.96 SD) | Brain weight within normal limits |
| Dysgyria | Bilateral diffuse (A>P) | Bilateral diffuse (A>P) | Bilateral diffuse | Bilateral diffuse | Bilateral diffuse | Bilateral diffuse (peri-Sylvian and parietal lobe most severly affected) | Asymmetric (left > right) | Microcephaly with simplified gyral pattern |
| Other features | Reduced WM, enlarged LV, hypoplastic CC, abnormal myelination in BS, dysmorphic hippocampus | Reduced WM, enlarged LV, hypoplastic CC, dysmorphic BG | Enlarged third ventricle, absent gyration, ACC | Enlarged LV, operculation of Sylvian fissure, enlarged subarachnoid spaces | WM reduced occipitally, colpocephaly, hypoplastic CC, enlarged BG | Reduced WM, enlarged LV, dysmorphic hippocampus | Reduced WM, enlarged left LV, dysmorphic CC and BG | Bilateral symmetric subcortical atrophy with periventricular leucoencephalopathy |
SD is above or below the mean.
ACC, agenesis of corpus callosum; A>P, anterior more severe than posterior; BG, basal ganglia; BS, brainstem; CC, corpus callosum; DD, Developmental delay; GTCS, generalised tonic-clonic seizures; GW, gestational weeks; LV, lateral ventricles; n/a, not applicable; OFC, Occipitofrontal circumference; sz, seizures; WM, white matter.
In total, 25 patients with MCD with heterozygous variants in either GRIN1 or GRIN2B have now been reported in this series and in the wider literature. We reviewed the available clinical information for these 25 patients (online supplemental material tables S1 and S2). Patients with GRIN1 or GRIN2B MCD had overlapping severe clinical phenotypes. Common features in this combined cohort included severe or profound developmental delay or intellectual disability (13 of 13 in GRIN1, 9 of 9 in GRIN2B), epilepsy (13 of 13 in GRIN1, 9 of 9 in GRIN2B), microcephaly (12 of 14 in GRIN1, 5 of 6 in GRIN2B), spastic quadriparesis (9 of 12 in GRIN1, 2 of 3 in GRIN2B) and cortical visual impairment (10 of 11 in GRIN1, 4 of 5 in GRIN2B). Movement disorders were present in three of five individuals with variants in GRIN1, but have not been reported in individuals with variants in GRIN2B. The age of onset of epilepsy was variable across this combined cohort, ranging from the neonatal period to 1 year for GRIN1, and from 10 weeks to 2 years 3 months for GRIN2B. Epilepsy phenotypes were also variable in the combined cohort, including epileptic spasms in 2 of 12 individuals with variants in GRIN1 and 5 of 9 individuals with variants in GRIN2B.
Brain imaging in patients with MCD with pathogenic variants in GRIN1 and GRIN2B
Brain MRI was performed in all seven individuals with heterozygous variants in GRIN1 or GRIN2B in our series between 29 weeks gestation and 3 years of age (table 1, figure 1), except for individual 3 (in which only prenatal ultrasound (US) was performed). All individuals with variants in GRIN1 presented with abnormal gyration. The two living GRIN1 patients (individuals 1 and 2) exhibited bilateral dysgyria, predominantly affecting the frontal regions. Among the GRIN2B patients, individuals 5 and 6 demonstrated diffuse bilateral dysgyria, while individual 7 had bilateral but asymmetric dysgyria predominantly affecting the left hemisphere.
Figure 1.
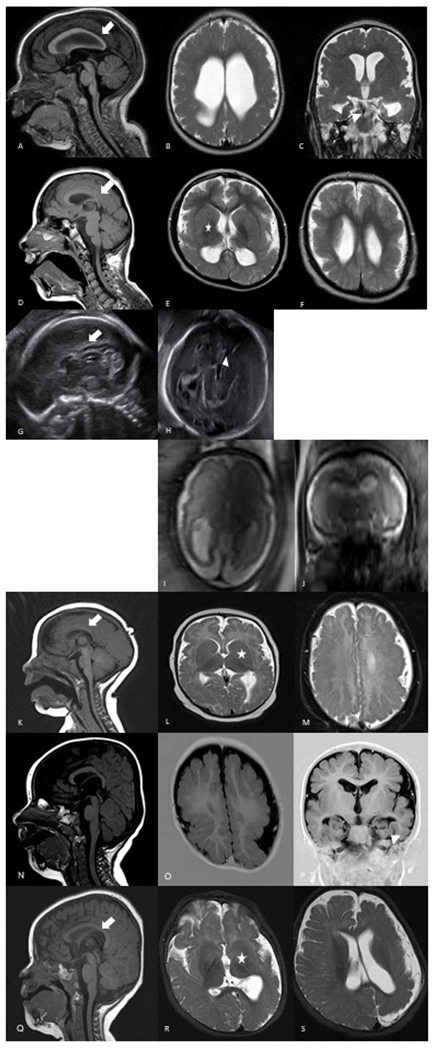
Brain imaging findings of individuals with heterozygous pathogenic variants in GRIN1 and GRIN2B. Individual 1 has diffuse dysgyria, hypoplastic corpus callosum (arrow) and dysmorphic hippocampi (arrowhead) (A–C). Individual 2 has bilateral dysgyria, hypoplastic corpus callosum (arrow) and dysmorphic basal ganglia (asterisk) (D–F). Prenatal ultrasound in individual 3 at 25 gestational weeks shows absent corpus callosum (arrow) and dilated third ventricle (arrowhead) (G–H). Individual 4 has diffuse dysgyria and enlarged lateral ventricles on prenatal MRI at 29 gestational weeks (I–J). Individual 5 presents with bilateral dysgyria, enlarged basal ganglia (asterisk) and hypoplastic corpus callosum (arrow) (K–M). Individual 6 has bilateral symmetric dysgyria and dysmorphic hippocampi (arrowhead) (N–P). Individual 7 has asymmetric dysgyria of the left hemisphere, dysmorphic basal ganglia (asterisk) and corpus callosum (arrow) (Q–S).
We reviewed the available neuroradiology for all 25 patients with heterozygous NMDAR MCD (online supplemental tables S1 and S2). We found that the MRI phenotypes of GRIN1 and GRIN2B patients were very similar (online supplemental material table S3). Common MRI features in the combined cohort included diffuse bilateral dysgyria often with some posterior sparing, reduced white matter, enlarged lateral ventricles, hippocampal dysplasia, hypoplastic corpus callosum, and enlarged or dysmorphic basal ganglia. The brainstem was normal on imaging in all GRIN1 and GRIN2B patients, except for mildly reduced myelination of the brainstem in individual 1 in this study. The cerebellum was abnormal in three individuals with variants in GRIN1, but normal in all individuals with variants in GRIN2B. It was not possible to distinguish GRIN1-related and GRIN2B-related MCDs based on radiological findings alone.
Molecular and electrophysiological findings in patients with MCD with pathogenic variants in GRIN1 and GRIN2B
We identified three GRIN1 and two GRIN2B variants that have not previously been reported. The variants were classified as pathogenic or likely pathogenic (online supplemental material tables S4 and S5). The p.(Ser553Leu) GRIN1 variant in individual 1 is a pathogenic variant previously reported in an unrelated patient with MCD.13 The p.(Met818Thr) GRIN2B variant in individual 7 has previously been reported in a patient without MCD.15 The seven variants were located in the same regions where previous MCD-associated NMDAR variants have clustered (figure 2).
Figure 2.
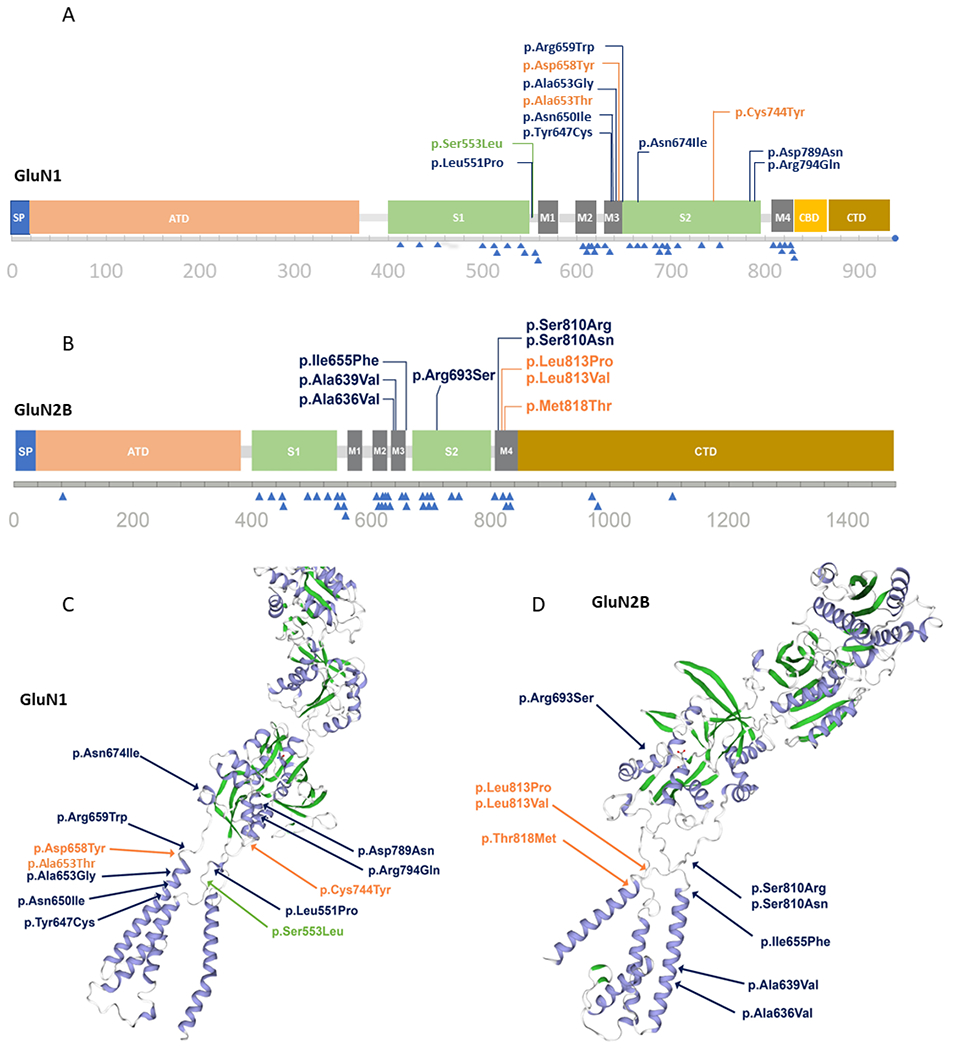
Distribution of variants in a linear model and 3D protein structure for GRIN1 (A, C) and GRIN2B (B, D). In the linear model, clustering of MCD-associated variants (top) and non-MCD associated variants (bottom, blue triangles) is highlighted. Functional domains are highlighted in the 3D model: blue: helices; green: sheets. Reported variants from this cohort are written in orange, variants reported in the literature are blue, and the recurring variants in the literature and our cohort are green. The 3D structure is based on Swiss models NMDZ1_HUMAN glutamate receptor ionotropic, NMDA1 Q05586 and NMDE2_HUMAN Q13224 glutamate receptor ionotropic, and NMDA 2B. 3D, three-dimensional; ATD, amino-terminal domain; CBD, carboxy-terminal domain; MCD, malformation of cortical development; SP, signal peptide.
To investigate the impact of two GRIN1 variants and one GRIN2B variant, functional studies were performed. The full results are given in section 3 of the online supplemental material. Compared with wild-type GluN1 the concentrations that elicit a half-maximal current (EC50) for GluN1-C744Y, GluN1-A653T and GluN2B-M818T were reduced for L-glutamate and for glycine, indicating these agonists have significantly greater potency (ie, produce responses at lower agonist concentrations). Mutant receptors were also less sensitive to inhibition by protons. Expression of the variants GluN1-C744Y and GluN1-A653T subunit protein does not appear to be altered substantially as the expressed levels of both surface and total receptor protein were modest. Overall, these in vitro electrophysiological and biochemical analyses suggest that the three variants cause a gain of function (GOF).
Neuropathological findings in GRIN1 patients
Autopsy and neuropathological examination were performed in individuals 3, 4 and 8. Individuals 3 and 4, harbouring de novo heterozygous variants in GRIN1, underwent termination of pregnancy at 25 and 30 weeks gestation, respectively, according to the French law, due to severe brain malformations identified on US imaging. In individual 3, US examination performed at 23 weeks of gestation (WG) revealed agenesis of the corpus callosum and of the septum pellucidum. Severe deformations of the extremities consisting of bilateral camptodactyly and equinovarus talipes were noted. Based on these findings medical termination of pregnancy was proposed and was accepted by both parents. In individual 4, US performed at 24 WG identified mild ventricular dilatation. At 26 WG, control US revealed diffuse dysgyria and MRI performed at 29 WG allowed the detection of absent primary fissures and secondary sulci suggestive of lissencephaly. Neither deformities of the limbs nor polyhydramnios were identified. Both individuals showed mild facial particularities consisting of prominent philtrum, broad nasal bridge with anteverted nostrils and flat face. Macroscopic and histological examinations of all viscera were normal in both cases. Both patients had reduced brain weight for gestational age (<3rd percentile).22 On external macroscopic brain examination of individual 3, the brain surface appeared smooth with a dimple-shaped Sylvian fissure (figure 3A,B). On sagittal and coronal sections, the ventricles were enlarged and the corpus callosum was absent. Histologically, the thin cortical ribbon had a diffuse festooned-like appearance suggestive of polymicrogyria under a thick and irregular molecular layer containing some immature neuronal heterotopias and covered by a persistent transient external granular cell layer (figure 3C). Nodular heterotopias were also noted in the periventricular regions close to the germinative zone of the dorsal telencephalon, to the ganglionic eminences and within the basal ganglia (figure 3D). The infratentorial structures were free of lesions. External macroscopic brain examination in individual 4 revealed diffuse polymicrogyria (figure 3E). The olfactory tracts were absent. The Sylvian fissure was short, vertically oriented and prematurely closed (closure normally occurs between 36 and 38 weeks gestation). The corpus callosum was hypoplastic in its posterior part (figure 3E). The lateral ventricles were moderately dilated. The hippocampi appeared to be malrotated (figure 3F). Histologically, the cortical ribbon was replaced by diffuse and poorly laminated polymicrogyria (figure 3G) covered by a glia limitans which was focally discontinuous with small foci of overmigration. The internal capsule was hypoplastic, the ganglionic eminences had already regressed and the basal ganglia were dysplastic, as were the thalami which were formed by the juxta-position of nodules. Small confetti-like heterotopic nodules were observed throughout the intermediate zone (figure 3H). The cerebellar vermis was hypoplastic but the brainstem and the spinal cord were normal.
Figure 3.
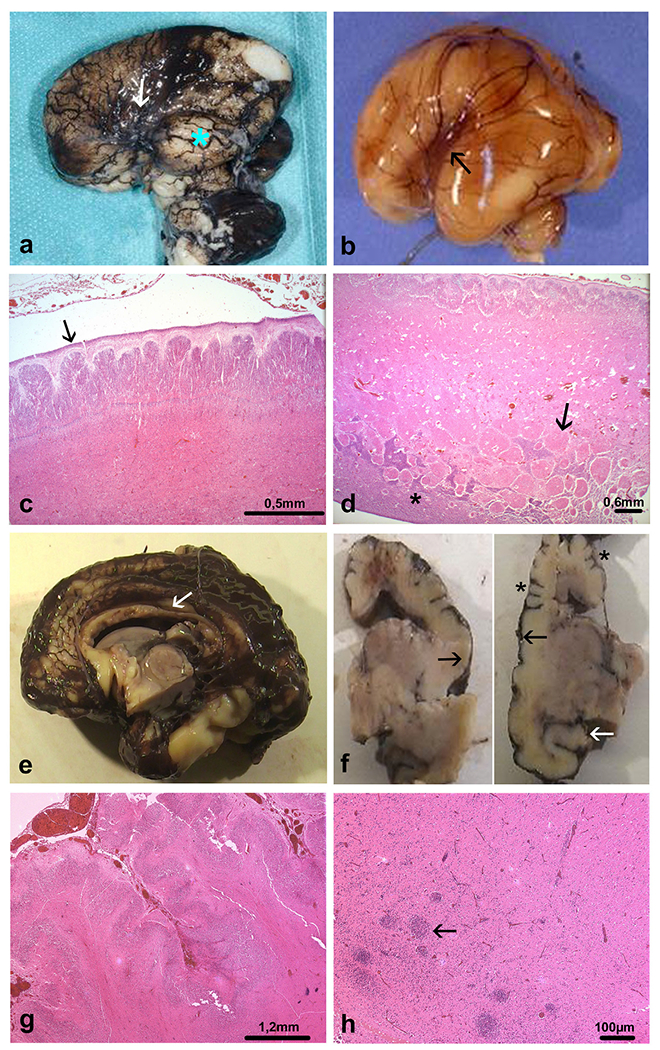
Distinctive brain morphological characteristics from individuals 3 and 4. Individual 3: (A) external view of the left side of the brain, where primary fissures are lacking, with almost indiscernible Sylvian fissure (arrow) and marked temporal lobe hypoplasia (blue asterisk), (B) compared with an age-matched control brain where the Sylvian fissure has a triangular shape (arrow). (C) The brain surface is smooth with a relatively thick transient subpial granular cell layer which should have disappeared from 22 WG (arrow) and diffuse polymicrogyria forming multiple protrusions into the molecular layer (OM ×20). (D) The cerebral mantle is particularly thin (half of the thickness of a normal cortical plate) and the deep intermediate zone contains numerous heterotopic nodules of various sizes (arrow) above the germinative zone of the dorsal telencephalon (asterisk) (OM ×20). Individual 4: (E) median supratentorial sagittal section revealing posterior hypoplasia of the corpus callosum (white arrow), (F) and on coronal sections supernumerary gyri (asterisks), areas of smoothened cortex covering polymicrogyria (black arrows) and malrotated hippocampus (white arrow). (G) Characteristic histological features of unlayered polymicrogyria (H&E, OM ×50), (H) associated with confetti-like nodular heterotopias, measuring about 1.50 μ, distributed throughout the intermediate zone (arrow) (H&E, OM ×50). OM, original magnification; WG, weeks of gestation.
We compared the clinical and neuropathological findings of individuals 3 and 4 with individual 8 with novel homozygous truncating variants (p.Tyr474*) in GRIN1 inherited from asymptomatic heterozygous carrier parents. Shortly after birth, individual 8 developed seizures evolving to status epilepticus. The patient died at 6 days of age. Brain imaging was not performed. Individual 8 had microcephaly with an SGP without tertiary sulci on macroscopic examination (online supplemental material figure S1). The corpus callosum was hypoplastic. Histologically, multiple foci of neuronal depletion with loss of lamination and abnormal persistence of the external transient granular cell layer in some areas were observed. In the hippocampus, the dentate gyrus was dysmorphic and discontinuous. Small germinolytic pseudo-cysts were observed along the ventricular walls (online supplemental material figure S1).
Cortical thickness was measured at 1.97 mm, which does not significantly differ from the cortical thickness measured on several control cortical plates between 30 and 38 WG (1.70–1.90 mm). These data are not in favour of lissencephaly with thin or thick cortex.
Immunohistochemistry revealed less numerous SOX2-positive and PAX6-positive proliferating radial glial cells in the ventricular and outer subventricular zones of individual 3 than observed in the control brain aged 24–25 WG (online supplemental material table S7). MAP2 antibody, which immunolabels radially migrating neurons and pyramidal neurons of layers III and V did not allow identification of any immunoreactive neuron in the micropolygyric cortex of individual 3, these neurons being observed within the periventricular heterotopic nodules (figure 4A). In individuals 4 and 8, very few MAP2-positive neurons were identified in the cortical plate. These neurons were misoriented, dysmorphic and/or immature (figure 4B–D). CTIP2-positive and SATB2-positive neurons were either absent or mislocalised (figure 4E–H). GABAergic interneurons were aberrantly located or almost absent, as were calretininergic interneurons (figure 4I–L). These findings highly suggest that functional NMDARs are essential for proper radial and tangential migration without which neurons cannot reach their final location.
Figure 4.
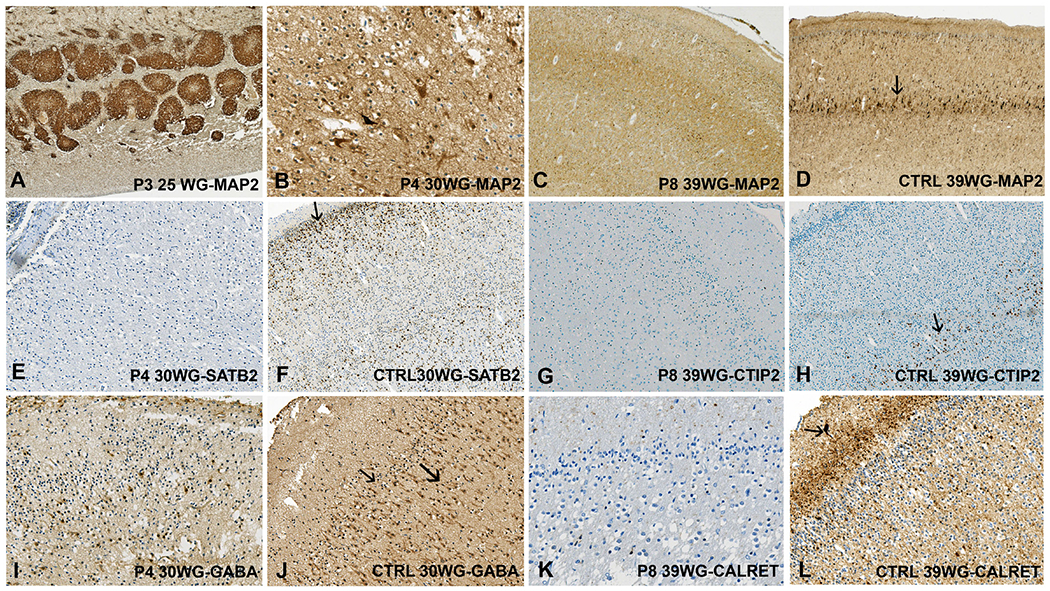
Immunohistochemical findings in individuals 3, 4 and 8. (A) Strong MAP2 immunoreactivity in the periventricular heterotopic nodules in individual 3. (B) Scattered dysmorphic MAP2-positive pyramidal neurons in the cortical plate (individual 4). (C) Decreased density in MAP2-positive haphazardly dispersed pyramidal neurons in the cortical plate (individual 8), (D) compared with the control cortical plate at 39 WG in which the neurons of layers II, III and V are observed, with predominance in layer V (arrow). (E) Almost no SATB2-positive neurons in the cortex of individual 4, (F) contrary to the control brain at 30 WG where SATB2-positive neurons are observed mainly in layer II (arrow) and to a lesser extent in layers III and IV (G) No CTIP2 immunoreactive neurons (individual 8), (H) whereas in the control cortical plate immunoreactive neurons are observed mainly in layer V (arrow). (I) Few GABAergic interneurons dispersed in the micropolygyric cortex (individual 4), (J) by comparison with the control brain where GABA antibody immunolabels interneurons of layers I, II (thin arrow) and III (thick arrow), (K) and with scarce calretininergic interneurons in layer III (individual 8), (L) conversely to the control where calretinin antibody immunolabels the tangential superficial network and Cajal-Retzius cells (arrow) in layer I and interneurons in layers II and III. Dark brown staining: specific; light brown: background staining. Individual 3: 25 WG; individual 4: 30 WG; individual 8: 39 WG. WG, weeks of gestation.
DISCUSSION
We presented seven new patients and reviewed all previously reported patients with NMDAR MCD with de novo heterozygous variants in either GRIN1 or GRIN2B. In addition, we provided the first reports of neuropathological findings in two individuals with heterozygous and one individual with homozygous variants in GRIN1. Our findings expand the current understanding of the phenotypic spectrum of NMDAR-associated MCD and provide insight into the suspected underlying pathophysiological mechanisms.
For the 25 patients with heterozygous variants in either GRIN1 or GRIN2B that have now been reported in this series and in the wider literature, we found a striking similarity in imaging and phenotypic features. The predominant imaging pattern of MCD in both groups was an extensive diffuse, bilateral dysgyria resembling polymicrogyria. Commonly associated brain malformations in both groups include enlarged ventricles, hippocampal dysplasia, hypoplastic corpus callosum, and enlarged or dysmorphic basal ganglia. Hippocampal dysplasia has been reported on MRI in 5 of 15 individuals with variants in GRIN1 and in 5 of 9 patients with variants in GRIN2B. This feature was confirmed by neuropathological examination in individual 4 and in an individual with a homozygous GRIN1 variant (individual 8) in our series. The hippocampal abnormalities are likely due to the high expression of NMDAR in the hippocampus.23 Abnormal basal ganglia were found in 6 of 14 and 8 of 9 of individuals with variants in GRIN1 and GRIN2B, respectively, likely reflecting the importance of glutaminergic signalling passing through the basal ganglia. Both hippocampal dysplasia and dysmorphic basal ganglia are rare in patients with MCD in general. Individuals with variants in tubulin genes, referred to as tubulinopathies, also show complex brain malformations, including dysgyria and dysmorphic basal ganglia. Rarely, hippocampal hypoplasia or dysplasia is present, most frequently in patients with variants in TUBA1A.24 25 Although there appears to be significant overlap of clinical and imaging findings between tubulinopathies and the spectrum observed in individuals with variants in GRIN1 or GRIN2B, tubulinopathies are often associated with abnormalities of the corpus callosum, brainstem and cerebellar hypoplasia or dysplasia, possibly due to the role microtubules play in neuronal migration and axon development.24 In contrast, the cerebellum and brainstem were usually normal in individuals with variants in either GRIN1 or GRIN2B. Prior to genetic testing, variants in GRIN1 and GRIN2B were not suspected based on clinical or imaging features in the individuals presented in this cohort. This might be due to the small number of individuals with NMDAR-related MCD reported previously. During fetal life, the discovery of microcephaly or microlissencephaly with or without corpus callosum agenesis may correspond to a constellation of aetiologies, either environmental or genetic, and there are no specific signs identified to date which could point to GRIN1 and GRIN2B as candidate genes. The homogeneous imaging spectrum observed in individuals with GRIN1-associated and GRIN2B-associated MCD, including diffuse dysgyria, dysmorphic basal ganglia and hippocampal dysplasia without any infratentorial lesions, may therefore be a potential diagnostic clue and should trigger consideration of pathogenic variants in NMDAR based on imaging findings in the future.
Clinical features included severe or profound developmental delay or intellectual disability, early-onset epilepsy, microcephaly, spastic quadriparesis and cortical visual impairment. Microcephaly was more common in patients with NMDAR MCD than in NMDAR patients without MCD, and was present in all seven new patients presented here. Overall, 11 of 13 (84.6%) reported GRIN1 MCD and 6 of 6 reported patients with GRIN2B MCD for which information is available have microcephaly (ranging from −2.0 to −7.1 SD in GRIN1 patients). Six out of 23 (26%) patients without MCD with GRIN1 variants reported by Lemke et al9 and 7 of 56 (12.5%) patients without MCD with GRIN2B missense variants reported by Platzer et al15 had microcephaly. Hyperkinetic and stereotyped movement disorders have been described in patients with and without MCD and variants in NMDAR.8 9 13–15 Interestingly, none of the individuals in our series has been reported to have abnormal movements. However, it is possible that abnormal movements were present but not recognised, limited by limb spasticity or misdiagnosed as seizures.
We provided the first neuropathological reports of three individuals with variants in GRIN1. Individuals 3 and 4 were reported with dysgyria resembling polymicrogyria on brain MRI and de novo heterozygous pathogenic variants in GRIN1. Neuropathological examination confirmed polymicrogyria. Both individuals also had extreme microcephaly, nodular heterotopias in the periventricular and intermediate zones, gaps of the glia limitans and overmigration foci in the meninges, arhinencephaly, dysplastic basal ganglia and hippocampi, as well as cerebellar hypoplasia. In addition, we provide first evidence that homozygous loss-of-function (LOF) variants also cause MCD. Neuropathological examination of individual 8 carrying homozygous GRIN1 variants has revealed microcephaly and an SGP associated with defective lamination of the cortex and malrotated hippocampi.
Taken together, the features observed argue for a crucial role of GRIN1 in the morphological organisation of the different neuroanatomical structures during fetal life. Immunohistochemical findings also strongly argue for decreased neuron production as well as delayed and inappropriate radial and tangential migration resulting in microcephaly with polymicrogyria or simplified pattern. Noteworthy, using NMDAR RNA interference with in utero electroporation in the cortex of rat embryos, Jiang et al26 found severely delayed radial migration, with more than 80% of migrating MAP2-positive projection neurons which could not reach the upper layers of the cortical plate and with 30% of the cells remaining located in the intermediate zone. In addition, it has been shown that developmental disruption of NMDAR has major effects on interneuron localisation leading to inhibitory network deficits, in so far as ionotropic NMDARs are normally expressed in tangentially migrating interneurons.27
It remains uncertain why a subset of patients with NMDAR variants develop severe MCDs. It has already been highlighted that GRIN variants associated with diffuse dysgyria tend to have GOF effects, as has been shown by functional studies for two variants in GRIN1 (p.(Ala653Thr) and p.(Cys744Thr)) and one variant in GRIN2B (p.Met818Thr) presented here, while many non-MCD variants are hypofunctional.9 13 NMDAR variants with GOF effects have also been observed in patients in which no MCDs were reported.7 15 28 29 These observations raise the possibility that a patient’s genetic background or non-genetic events (eg, viral infection or hypoxic-ischaemic injury) may be contributing to the pathogenesis of NMDAR MCDs. Another explanation can be subtle changes of cortical architecture that might be missed on imaging studies, as has been shown in individual 8 with SGP and homozygous LOF variants. Of note, the homozygous variants in individual 8 were truncating variants. These are likely to cause complete LOF of GRIN1, as has been reported in three siblings and one additional individual with a similar clinical course of neonatal epileptic encephalopathy with homozygous truncating variants in GRIN1.9 12 Homozygous truncating variants causing complete absence of the protein might therefore be at the most severe end of clinical and neuropathological features of LOF variants in GRIN1. Thus, the growing evidence for a genotypeȓphenotype correlation suggests that intrinsic properties of specific NMDAR variants need to be considered as well.10 An intermediate imaging phenotype with milder or focal dysgyria has not yet been reported, neither for variants in GRIN1 nor GRIN2B, suggesting different mechanisms of pathogenic variants on protein function. Nevertheless, the SGP in individual 8 demonstrates that MCD can also occur in patients with homozygous LOF variants in GRIN1. Thus, the hypothesis of an all-or-nothing mechanism, resulting in two patient populations with severe MCD and without MCD, is not supported. Finally, the lack of polymicrogyria in the individual with biallelic LOF variants supports the hypothesis that clinical, histological and pathophysiological differences between gain and loss of function GRIN variants exist.
The seven heterozygous NMDAR variants reported in our series were located in the same regions as previously reported MCD-associated variants. MCD-associated GRIN1 variants (figure 1) have clustered in the S1-M1 linker (pre-M1) region, the Lurcher motif in the M3 region and the S2 domain of GluN1.13 MCD-associated GRIN2B variants have clustered in the M3, S2 and M4 domains of GluN2B.15 Interestingly, while non-MCD variants in GRIN1 and GRIN2B have been described, no MCD-associated GRIN1 variants have yet been reported in the GluN1 M4 helix (a region where around half of non-MCD GRIN1 variants have been located7 9) or the pre-M1 region of GluN2B. This may reflect functional differences between these two domains between GluN1 and GluN2 subunits.
In the patients reported to date, the spectrum of NMDAR variants, including recurrent variants, such as the p.(Ser553Leu) GRIN1 variant in individual 1, is different from the variants found in patients without MCD. An exception to this observation has been the recurrent p.(Met818Thr) in GRIN2B, located in the GluN2B M4 helix, which has been reported in both a patient with MCD (individual 7 in this paper) and a previously reported patient without MCD.15 Neuroimaging data from the patient without MCD have not been available for review. The variable brain phenotype associated with the recurrent p.(Met818Thr) variant may indicate that additional modifiers, such as non-genetic events or genetic background, can influence the outcome.
Electrophysiological analysis of the GluN1 p.Cys744Tyr variant seen in individual 4 with morphologically polymicrogyria confirmed that the p.Cys744Tyr variant causes GOF effects. The Cys744 residue forms a disulfide bond with another cysteine in the S2 domain at residue 798.30 Mutation of either Cys744 or Cys798 to alanine has been shown to increase the potency of glutamate and reduce proton inhibition.31 The Cys744-Cys798 disulfide bond has been shown to modulate the behaviour of NMDAR in response to redox environment.30 32 In pathological situations (eg, following stroke or hypoxia) the brain environment becomes more reductive, which favours free thiol over disulfide formation.30 It has been proposed that the Cys744-Cys798 bond is a ‘molecular oxygen sensor’ in the brain, responding to low partial pressure of oxygen (pO2) levels by controlling how much nitric oxide (NO) can inhibit N-methyl-D-aspartate (NMDA) function.30
There is evidence that disruption of NMDA signalling can influence the proliferation, migration and maturation of neuronal progenitor cells and postmitotic neurons.26 33 A higher prevalence of microcephaly in patients with MCD, together with increased potency of the NMDAR during electrophysiological studies and reduced neuronal density on histology, suggests increased NMDAR signalling leading to excitotoxic cell death may contribute to the increased frequency of microcephaly. This also suggests that the impact on proliferation or survival of neurons is more common or more severe in MCD-associated NMDAR variants. Alternatively, it has been noted that MCD-associated NMDAR variants tend to have GOF effects.13 This is consistent with animal models of NMDAR agonist-induced polymicrogyria.34–36
In conclusion, patients with MCD with GRIN1 and GRIN2B variants reported to date have overlapping phenotypic features, consisting of early presentation of global developmental delay, epilepsy and microcephaly. Brain imaging mostly shows severe dysgyria, histologically in line with polymicrogyria in two cases, and often with associated brain malformations including dysmorphic basal ganglia and hippocampi. These findings extend our understanding of the ‘NMDARopathy’ spectrum. Our neuropathological findings for the first time provide evidence that GRIN1 is implicated in the generation, migration and cortical positioning of neurons and interneurons in humans, ultimately leading to MCD.
Supplementary Material
Acknowledgements
The authors would like to thank the patients, families and scientists who contributed to this work, and Rebecca Bui and Dan Teuscher for technical assistance.
Funding
The DDD study presents independent research commissioned by the Health Innovation Challenge Fund (grant number HICF-1009-003). This study makes use of DECIPHER (http://decipher.sanger.ac.uk), which is funded by Wellcome (see Nature PMID: 25533962 or www.ddduk.org/access.html for full acknowledgement). ACJ was funded by an FWO Senior Clinical Investigator Fellowship. SB received funding from the Scientific Fund Willy Gepts. DB is supported by NIHR Research Professorship (RP-2016- 07-011). SFT received funding from the NIH-NINDS (NS111619). YH is supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the National Institutes of Health (R01HD082373) and the National Institute of Mental Health (MH127404). SJM is supported by a grant from the National Institute on Aging (R21AG072142). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Competing interests
SFT is principal investigator on research grants from Biogen and Janssen to Emory; a member of the Scientific Advisory Board for Eumentis, Sage Therapeutics, GRIN2B Foundation and CureGRIN Foundation; co-founder of NeurOp and Agrithera; and coinventor on Emory-owned intellectual property.
Footnotes
Patient consent for publication Parental/guardian consent obtained.
Ethics approval This study involves human participants and was approved by the ethical committee of UZ Brussel (B.U.N.143201214360). The DDD study was approved by the Cambridge South Research Ethics Committee (10/H0305/83). Participants gave informed consent to participate in the study before taking part.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. Anonymised data from this study will be shared by request from any qualified investigator.
REFERENCES
- 1.Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol 2001;11:327–35. [DOI] [PubMed] [Google Scholar]
- 2.Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 2010;62:405–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sobolevsky AI, Beck C, Wollmuth LP. Molecular rearrangements of the extracellular vestibule in NMDAR channels during gating. Neuron 2002;33:75–85. [DOI] [PubMed] [Google Scholar]
- 4.McKay S, Ryan TJ, McQueen J, Indersmitten T, Marwick KFM, Hasel P, Kopanitsa MV, Baxter PS, Martel M-A, Kind PC, Wyllie DJA, O’Dell TJ, Grant SGN, Hardingham GE, Komiyama NH. The developmental shift of NMDA receptor composition proceeds independently of GluN2 subunit-specific GluN2 C-terminal sequences. Cell Rep 2018;25:841–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Endele S, Rosenberger G, Geider K, Popp B, Tamer C, Stefanova I, Milh M, Kortüm F, Fritsch A, Pientka FK, Hellenbroich Y, Kalscheuer VM, Kohlhase J, Moog U, Rappold G, Rauch A, Ropers H-H, von Spiczak S, Tönnies H, Villeneuve N, Villard L, Zabel B, Zenker M, Laube B, Reis A, Wieczorek D, Van Maldergem L, Kutsche K. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet 2010;42:1021–6. [DOI] [PubMed] [Google Scholar]
- 6.Hamdan FF, Gauthier J, Araki Y, Lin D-T, Yoshizawa Y, Higashi K, Park A-R, Spiegelman D, Dobrzeniecka S, Piton A, Tomitori H, Daoud H, Massicotte C, Henrion E, Diallo O, Shekarabi M, Marineau C, Shevell M, Maranda B, Mitchell G, Nadeau A, D’Anjou G, Vanasse M, Srour M, Lafrenière RG, Drapeau P, Lacaille JC, Kim E, Lee J-R, Igarashi K, Huganir RL, Rouleau GA, Michaud JL, S2D Group. Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am J Hum Genet 2011;88:306–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lemke JR, Hendrickx R, Geider K, Laube B, Schwake M, Harvey RJ, James VM, Pepler A, Steiner I, Hörtnagel K, Neidhardt J, Ruf S, Wolff M, Bartholdi D, Caraballo R, Platzer K, Suls A, De Jonghe P, Biskup S, Weckhuysen S. GRIN2B mutations in West syndrome and intellectual disability with focal epilepsy. Ann Neurol 2014;75:147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohba C, Shiina M, Tohyama J, Haginoya K, Lerman-Sagie T, Okamoto N, Blumkin L, Lev D, Mukaida S, Nozaki F, Uematsu M, Onuma A, Kodera H, Nakashima M, Tsurusaki Y, Miyake N, Tanaka F, Kato M, Ogata K, Saitsu H, Matsumoto N. GRIN1 mutations cause encephalopathy with infantile-onset epilepsy, and hyperkinetic and stereotyped movement disorders. Epilepsia 2015;56:841–8. [DOI] [PubMed] [Google Scholar]
- 9.Lemke JR, Geider K, Helbig KL, Heyne HO, Schütz H, Hentschel J, Courage C, Depienne C, Nava C, Heron D, Møller RS, Hjalgrim H, Lal D, Neubauer BA, Nürnberg P, Thiele H, Kurlemann G, Arnold GL, Bhambhani V, Bartholdi D, Pedurupillay CRJ, Misceo D, Frengen E, Strømme P, Dlugos DJ, Doherty ES, Bijlsma EK, Ruivenkamp CA, Hoffer MJV, Goldstein A, Rajan DS, Narayanan V, Ramsey K, Belnap N, Schrauwen I, Richholt R, Koeleman BPC, Sá J, Mendonça C, de Kovel CGF, Weckhuysen S, Hardies K, De Jonghe P, De Meirleir L, Milh M, Badens C, Lebrun M, Busa T, Francannet C, Piton A, Riesch E, Biskup S, Vogt H, Dorn T, Helbig I, Michaud JL, Laube B, Syrbe S. Delineating the GRIN1 phenotypic spectrum: a distinct genetic NMDA receptor encephalopathy. Neurology 2016;86:2171–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.XiangWei W, Jiang Y, Yuan H. De Novo Mutations and Rare Variants Occurring in NMDA Receptors. Curr Opin Physiol 2018;2:27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rossi M, Chatron N, Labalme A, Ville D, Carneiro M, Edery P, des Portes V, Lemke JR, Sanlaville D, Lesca G. Novel homozygous missense variant of GRIN1 in two sibs with intellectual disability and autistic features without epilepsy. Eur J Hum Genet 2017;25:376–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blakes AJM, English J, Banka S, Basu H. A homozygous GRIN1 null variant causes a more severe phenotype of early infantile epileptic encephalopathy. Am J Med Genet A 2022;188:595–599. [DOI] [PubMed] [Google Scholar]
- 13.Fry AE, Fawcett KA, Zelnik N, Yuan H, Thompson BAN, Shemer-Meiri L, Cushion TD, Mugalaasi H, Sims D, Stoodley N, Chung S-K, Rees MI, Patel CV, Brueton LA, Layet V, Giuliano F, Kerr MP, Banne E, Meiner V, Lerman-Sagie T, Helbig KL, Kofman LH, Knight KM, Chen W, Kannan V, Hu C, Kusumoto H, Zhang J, Swanger SA, Shaulsky GH, Mirzaa GM, Muir AM, Mefford HC, Dobyns WB, Mackenzie AB, Mullins JGL, Lemke JR, Bahi-Buisson N, Traynelis SF, Iago HF, Pilz DT. De novo mutations in GRIN1 cause extensive bilateral polymicrogyria. Brain 2018;141:698–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishimura N, Kumaki T, Murakami H, Enomoto Y, Katsumata K, Toyoshima K, Kurosawa K. Arthrogryposis multiplex congenita with polymicrogyria and infantile encephalopathy caused by a novel GRIN1 variant. Hum Genome Var 2020;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Platzer K, Yuan H, Schütz H, Winschel A, Chen W, Hu C, Kusumoto H, Heyne HO, Helbig KL, Tang S, Willing MC, Tinkle BT, Adams DJ, Depienne C, Keren B, Mignot C, Frengen E, Strømme P, Biskup S, Döcker D, Strom TM, Mefford HC, Myers CT, Muir AM, LaCroix A, Sadleir L, Scheffer IE, Brilstra E, van Haelst MM, van der Smagt JJ, Bok LA, Møller RS, Jensen UB, Millichap JJ, Berg AT, Goldberg EM, De Bie I, Fox S, Major P, Jones JR, Zackai EH, Abou Jamra R, Rolfs A, Leventer RJ, Lawson JA, Roscioli T, Jansen FE, Ranza E, Korff CM, Lehesjoki A-E, Courage C, Linnankivi T, Smith DR, Stanley C, Mintz M, McKnight D, Decker A, Tan W-H, Tarnopolsky MA, Brady LI, Wolff M, Dondit L, Pedro HF, Parisotto SE, Jones KL, Patel AD, Franz DN, Vanzo R, Marco E, Ranells JD, Di Donato N, Dobyns WB, Laube B, Traynelis SF, Lemke JR. GRIN2B encephalopathy: novel findings on phenotype, variant clustering, functional consequences and treatment aspects. J Med Genet 2017;54:460–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Platzer K, Lemke JR. GRIN2B-Related Neurodevelopmental Disorder. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mirzaa G, Amemiya A, eds. GeneReviews®. Seattle (WA: University of Washington, Seattle, 1993. [Google Scholar]
- 17.Platzer K, Lemke JR. GRIN1-Related Neurodevelopmental Disorder. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mirzaa G, Amemiya A, eds. GeneReviews®. Seattle (WA: University of Washington, Seattle, 1993. [Google Scholar]
- 18.Severino M, Geraldo AF, Utz N, Tortora D, Pogledic I, Klonowski W, Triulzi F, Arrigoni F, Mankad K, Leventer RJ, Mancini GMS, Barkovich JA, Lequin MH, Rossi A. Definitions and classification of malformations of cortical development: practical guidelines. Brain 2020;143:2874–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deciphering developmental disorders study. large-scale discovery of novel genetic causes of developmental disorders. Nature 2015;519:223–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ellard S, Baple EL, Berry I, Forrester N, Turnbull C, Owens M, Eccles DM, Abbs S, Scott R, Deans ZC, Lester T, Campbell J, Newman WG, ACGS MDJ. Best practice guidelines for variant classification 2019, 20192019. Available: https://www.leedsth.nhs.uk/assets/Genetics-Laboratory/86fa75f316/ACGS-variant-classification-guidelines-2019.pdf
- 21.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet Med 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guihard-Costa A-M, Ménez F, Delezoide A-L. Organ weights in human fetuses after formalin fixation: standards by gestational age and body weight. Pediatr Dev Pathol 2002;5:559–78. [DOI] [PubMed] [Google Scholar]
- 23.Moriyoshi K, Masu M, Ishii T, Shigemoto R, Mizuno N, Nakanishi S. Molecular cloning and characterization of the rat NMDA receptor. Nature 1991;354:31–7. [DOI] [PubMed] [Google Scholar]
- 24.Oegema R, Cushion TD, Phelps IG, Chung S-K, Dempsey JC, Collins S, Mullins JGL, Dudding T, Gill H, Green AJ, Dobyns WB, Ishak GE, Rees MI, Doherty D. Recognizable cerebellar dysplasia associated with mutations in multiple tubulin genes. Hum Mol Genet 2015;24:5313–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hebebrand M, Hüffmeier U, Trollmann R, Hehr U, Uebe S, Ekici AB, Kraus C, Krumbiegel M, Reis A, Thiel CT, Popp B. The mutational and phenotypic spectrum of TUBA1A-associated tubulinopathy. Orphanet J Rare Dis 2019;14:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang H, Jiang W, Zou J, Wang B, Yu M, Pan Y, Lin Y, Mao Y, Wang Y. The GluN2B subunit of N-methy-D-asparate receptor regulates the radial migration of cortical neurons in vivo. Brain Res 2015;1610:20–32. [DOI] [PubMed] [Google Scholar]
- 27.Soria JM, Valdeolmillos M. Receptor-activated calcium signals in tangentially migrating cortical cells. Cereb Cortex 2002;12:831–9. [DOI] [PubMed] [Google Scholar]
- 28.Swanger SA, Chen W, Wells G, Burger PB, Tankovic A, Bhattacharya S, Strong KL, Hu C, Kusumoto H, Zhang J, Adams DR, Millichap JJ, Petrovski S, Traynelis SF, Yuan H. Mechanistic insight into NMDA receptor dysregulation by rare variants in the GluN2A and GluN2B agonist binding domains. Am J Hum Genet 2016;99:1261–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mullier B, Wolff C, Sands ZA, Ghisdal P, Muglia P, Kaminski RM, André VM. GRIN2B gain of function mutations are sensitive to radiprodil, a negative allosteric modulator of GluN2B-containing NMDA receptors. Neuropharmacology 2017;123:322–31. [DOI] [PubMed] [Google Scholar]
- 30.Takahashi H, Shin Y, Cho S-J, Zago WM, Nakamura T, Gu Z, Ma Y, Furukawa H, Liddington R, Zhang D, Tong G, Chen H-SV, Lipton SA. Hypoxia enhances S-nitrosylation-mediated NMDA receptor inhibition via a thiol oxygen sensor motif. Neuron 2007;53:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sullivan JM, Traynelis SF, Chen HS, Escobar W, Heinemann SF, Lipton SA. Identification of two cysteine residues that are required for redox modulation of the NMDA subtype of glutamate receptor. Neuron 1994;13:929–36. [DOI] [PubMed] [Google Scholar]
- 32.Brimecombe JC, Boeckman FA, Aizenman E. Functional consequences of NR2 subunit composition in single recombinant N-methyl-D-aspartate receptors. Proc Natl Acad Sci U S A 1997;94:11019–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bell S, Maussion G, Jefri M, Peng H, Theroux J-F, Silveira H, Soubannier V, Wu H, Hu P, Galat E, Torres-Platas SG, Boudreau-Pinsonneault C, O’Leary LA, Galat V, Turecki G, Durcan TM, Fon EA, Mechawar N, Ernst C. Disruption of GRIN2B impairs differentiation in human neurons. Stem Cell Reports 2018;11:183–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marret S, Mukendi R, Gadisseux JF, Gressens P, Evrard P. Effect of ibotenate on brain development: an excitotoxic mouse model of microgyria and posthypoxic-like lesions. J Neuropathol Exp Neurol 1995;54:358–70. [DOI] [PubMed] [Google Scholar]
- 35.Takano T, Sawai C, Takeuchi Y. Radial and tangential neuronal migration disorder in ibotenate-induced cortical lesions in hamsters: immunohistochemical study of Reelin, vimentin, and calretinin. J Child Neurol 2004;19:107–15. [DOI] [PubMed] [Google Scholar]
- 36.Takano T, Matsui K. Increased expression of GAP43 in interneurons in a rat model of experimental polymicrogyria. J Child Neurol 2015;30:716–28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. Anonymised data from this study will be shared by request from any qualified investigator.