

Visual Abstract
Abstract
The ability to isolate and characterize different hematopoietic stem cell (HSC) or progenitor cell populations opens avenues to understand how hematopoiesis is regulated during development, homeostasis, and regeneration as well as in age-related conditions such as clonal hematopoiesis and leukemogenesis. Significant progress has been made in the past few decades in determining the composition of the cell types that exist in this system, but the most significant advances have come from mouse studies. However, recent breakthroughs have made significant strides that have enhanced the resolution of the human primitive hematopoietic compartment. Therefore, we aim to review this subject not only from a historical perspective but also to discuss the progress made in the characterization of the human postnatal CD34+ HSC–enriched populations. This approach will enable us to shed light on the potential future translational applicability of human HSCs.
Edited by Associate Editor John Crispino, this Review Series focuses on hematopoietic stem cell (HSC) biology, going beyond the work in murine HSC biology and emphasizing development and characterization of human HSCs. Expert authors in the field cover topics ranging from ontogeny to aging and leukemia transformation. We hope that these erudite articles will stimulate research that develops novel strategies to reverse the effects of aging, prevent malignant transformation, and enhance in vitro production of HSCs from pluripotent stem cells.
Introduction
The aptitude to balance between self-renewal, differentiation, and cell fate decisions is the hallmark of long-term hematopoietic stem cells (LT-HSCs). Despite their rarity, slow cycling and multipotent LT-HSCs have the capacity to differentiate into all blood cells at the single-cell level to steadily maintain the adult hematopoietic system. In addition, LT-HSCs have been conceptually defined to have long-term reconstitution capacity (>3-4 months) and the ability to be serially transplantable. This can be accomplished because LT-HSCs, which sit at the top of the hierarchy, are responsible for generating intermediate precursors (eg, short-term HSCs; multipotent cells but without durable and serial reconstitution ability) that in turn give rise to different multipotent progenitor (MPP) populations. These MPPs are known to have limited self-renewal potential but are the actual workhorses of the hematopoietic system. Despite being one of the best-characterized stem cells, many of the biological paradigms have mainly been tested in mice. Given that the generation of blood cells is largely conserved throughout vertebrate evolution, one would think that many of the experimental findings in mice would also be applicable to the human system. However, this has turned out to not be the case entirely, and there is an accumulating body of evidence suggesting that considerable differences exist between the 2 species with many of the important findings in mice not translatable to applications in the human system. For example, SLAM markers that are currently used to identify mouse LT-HSCs are not even expressed and/or useful to delineate human HSC–enriched populations in most postnatal tissues.1 As such, determining the composition and relationship of the cell types that constitute the human stem cell compartment may not only help to identify the cellular and molecular factors that govern healthy and leukemic development but will also facilitate the advancement in the clinical applications of transplantation, gene therapy, stem cell expansion, and tumor-cell purging. In this article, we will critically review the progress in the delineation of the human postnatal CD34+ HSC–enriched population by examining the methodologies and approaches that have been used over the past 3 decades.
Although recent improvements in sorting approaches, in vitro clonal assays, xenotransplantation, and single-cell RNA sequencing (scRNA-seq) have significantly contributed to the progress toward defining human LT-HSCs,2,3 for any given phenotypically defined population, one must be cautious when interpreting such data sets. This is because certain primitive populations could contain cells with stem cell properties that fail to generate any functional readout in the aforementioned assays and are therefore classified as nonstem cells. For example, in vitro clonal assays that are widely used to test HSC-enriched populations have only up to 60% efficiency.2,3 In addition, single-cell transplantation of mouse HSCs and cellular barcoding experiments have revealed that only a few HSCs reconstitute all the cell lineages even within a given phenotypically defined LT-HSC population, with the rest being either restricted or having a biased output. Hence, a combinational approach of single-cell surface phenotype and functional assays should always be used to define new cell populations, especially rare cells, such as HSCs. Furthermore, this is particularly important in the human system because the HSC-enriched population is less well phenotypically characterized than other cell groups. As a result of all these caveats, and given that many of the previously described HSC populations in humans have not been functionally validated for their long-term reconstitution capacity, we have refrained from using the LT-HSC/short-term–HSC nomenclature. Therefore, a more acceptable terminology, HSC-enriched, is used in this review to describe a human hematopoietic cell population that may contain HSCs. In addition, we would also like to take this opportunity to hypothesize the potential roles that some of the cell surface antigens that have been used to enrich human hematopoietic stem and progenitor cells (HSPCs) may have in regulating the biology of these primitive cells (Figure 1). The description of potential molecular regulators of human HSPCs as well as the current human hematopoietic roadmap at the progenitor level will not be covered in this study because they have been comprehensively explored elsewhere.
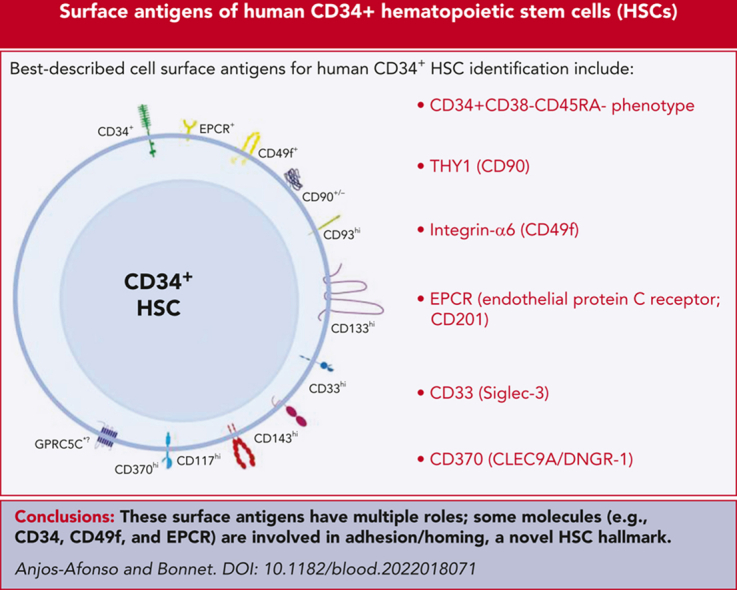
Figure 1.

Hematopoietic stem cell markers and their potential signaling interactions governing their biology. A schematic diagram of potential signaling pathways downstream of key markers expressed on CD34+(CD38/CD45RA)–EPCR+ HSCs (which are also CD49f+ and CD90+/−) upon interacting with their potential ligands in the niche. This highly HSC-enriched population highly expresses CD33, CD93, CD117, CD133, and CD143.30 We hypothesize that these cells may also express GPRC5C and CD370 as well as PAR1 and ATR1 (to convey the EPCR/APC/PAR1 and CD143/Ang-/ATR1 signaling, respectively).
A brief history behind the CD34+CD38–CD45RA– phenotype
Although a very rare HSC population that is negative for CD34 expression has recently been demonstrated to be hierarchically above the CD34+ HSCs4,5 (not discussed in this review), human hematopoiesis is mostly sustained by the latter. Since the first anti-CD34 monoclonal antibody was described, the CD34 antigen has been widely used in different experimental and clinical settings to define human HSPCs because its expression in normal hematopoiesis is mainly confined to primitive cells. This was evidenced by the finding that CD34+ cells purified from the bone marrow (BM), which represent ∼1% to 1.5% of BM mononuclear cells, can reconstitute hematopoiesis in patients undergoing autologous marrow reinfusion after myeloablation, leading to durable donor-derived hematopoietic reconstitution.6 It is now well-established that human HSCs are enriched in the cellular fraction defined as CD34+CD38–/lo. One of the first demonstrations came from a study in 1991 in which the authors attempted to subdivide human BM CD34+ cells using different cell surface antigens known at that time to be differentially expressed by different mature subpopulations, such as CD10, CD7, CD33, CD38, and CD71.7 The authors then reported that human CD34+CD38– marrow cells (∼1% of CD34+ cells)7 lacked differentiation features and appeared highly enriched for blast colony–forming cells, whereas the colonies could be replated up to 5 times.7 Interestingly, sorting for a different subpopulation based on the increasing levels of CD38 expression resulted in a decreasing capacity in blast colony formation,7 thus demonstrating for the first time that the lack of CD38 (cyclic adenosine diphosphate ribose hydrolase) expression is associated with primitive features. Subsequent studies showed that cord blood (CB) CD34+CD38–/lo cells (∼5.5% of CD34+ cells)8 were enriched for long-term culture-initiating cells with an clonogenic output capacity after ∼35 to 60 days when cocultured with BM stroma.8 In this extended long-term culture-initiating cell assay, CD34+CD38+ cells did not generate colony forming units beyond day 40.8 This was further supported when CD34+CD38–/lo cells (∼16% of CD34+ cells)8 were able to engraft in nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice (with a frequency of 1 in ∼600 cells) at 8 weeks after the transplantion, but CD34+CD38+ cells were unable to produce detectable human engraftment at this time point.9 Despite this, human engraftment was detectable at 3 weeks after the transplantation of BM CD34+CD38+ cells (∼40%-50% of CD34+ cells) into a more immunocompromised mouse model (NOD/SCID-β2–macroglobulin–/–)10 or even at later time points, when this population was delivered directly via intrabone and/or with significantly increased cell numbers (>45-fold more cells compared witho CD34+CD38–/lo cells).10,11 All these cumulative results support that CD34+CD38+ cells have very limited self-renewal capacity. Indeed, it is currently well recognized that the CD34+CD38+ population represents hematopoietic progenitor cells that encompass different lymphoid, myeloid, megakaryocytic, and erythroid progenitors12,13 that will not be discussed here. Of note, for a better understanding of the different immunodeficient mouse models that have been developed over the years, we recommend cross checking 2 recent publications.14,15
Differential CD45RA (protein tyrosine phosphatase, receptor type, C, isoform A) expression analysis was also one of the earlier means by which different groups attempted to dissect the heterogeneity of human CD34+ HSPCs. It was first reported that the CD34+CD45RA– population (from BM, CB, and mobilized-PB sources) comprised early precursors (in a colony forming unit assay, these cells differentiated into compact myeloid and erythroid colonies), whereas the CD34+CD45RA+ population contained B lymphoid as well as more committed myeloid progenitor cells.16 Within CB CD34+CD38– cells, the CD45RA+ fraction was showed poor myeloid colony output and no engraftment potential when transplanted into newborn NOD/SCID/IL-2Rγ-null (NOG) mice.17 In the same study, it was shown that the CD45RA+ cells were hierarchically downstream of the CD34+CD38–CD45RA– cell fraction.17 This led to the conclusion that the CD34+CD38–CD45RA+ population (in the BM, it was ∼25%, and in the CB, it was ∼18% of CD34+CD38– cells)17 may define some type of progenitors. Indeed, CD34+CD38–D45RA+ cells were then shown to contain lymphoid-primed multipotent progenitors and multilymphoid progenitors. This cell population was shown to give rise to all lymphoid cell types as well as monocytes, macrophages, and dendritic cells (DCs).18,19
The only way is up with THY1 (CD90)
THY1 or CD90 is another cell surface marker that has been trialed to enrich human HSCs, and its use started around the time when CD38 and CD45RA antigens were described (as mentioned in “A brief history behind the CD34+CD38−CD45RA− phenotype”). Weissman’s group tested whether CD90 could be useful for subfractioning human CD34+ HSPCs. Using fetal BM CD34+ cells, the authors showed that, contrary to the mouse system, CD34+CD90+ cells initiated long-term cocultures at a significantly higher frequency than their CD34+CD90– counterparts. In addition, cultures initiated with CD34+CD90+ cells could be replated, and, importantly, these cells engrafted as well as differentiated in SCID mice.20 The advantages of using CD90 to enrich postnatal human HSC was solidified when it was revealed that CB CD34+CD38–CD90+CD45RA– cells (∼25% of CD34+CD38– cells)17 gave ninefold greater human chimerism than CD34+CD38–CD90–CD45RA– cells (∼50% of CD34+CD38– cells)17 in newborn NOD/SCID-IL-2Rγ-null (NOG) mice 12 weeks after the transplantation.17 Interestingly CD34+CD38–CD90+CD45RA– cells were able to give rise to all primitive CD34+ subpopulations in vivo, whereas CD34+CD38–CD90–CD45RA– cells were incapable to yield CD90+ fraction, thus revealing a hierarchical relationship between these 2 populations.17 Altogether, these findings support the notion that CD34+CD38–CD90+CD45RA– cells represent a population highly enriched in HSCs, whereas the CD90– cells are multipotent progenitors.
Now, the million-dollar question is this, what is the function of the CD90 protein in human HSCs? CD90 is a glycophosphatidylinositol-anchored cell surface protein with a V-like immunoglobulin-domain, with features of extracellular matrix proteins with an integrin-binding site arginine-glycine-aspartate–like tripeptide) and a heparin-binding domain, thus able to bind to integrins and syndecan-4 receptors.21 There are many reports of THY1-associated functions in different cellular settings, most of them describing CD90 as the modulator of integrin signaling (in trans or cis). Many of the integrin-CD90 and syndecan-4–CD90 interaction studies have identified the signaling being induced in the integrin and syndecan-4–expressing cells. Therefore, it is challenging to translate this knowledge into applications in the human HSC context. In addition, because of the lack of a transmembrane domain, it has been difficult to dissect the intracellular signaling induced by this protein. That said, CD90 was shown to interact with αvβ3 integrin, leading to cell signaling in astrocytes.22 It was determined that THY1 protein–lipid raft microdomains (which included the C-terminal Src kinase-binding protein) phosphorylated and inactivated Src, resulting in the activation of p190RhoGAP/RhoA, thus resulting in cytoskeletal alterations and neurite retractions.22 In T cells, CD90 was shown to crosslink, at least in part, with the T-cell receptor complex leading to augmented T-cell activation.23,24 This involves Ca2+ flux, the activation of Lck, Fyn, and Zap-70 protein tyrosine kinases, MAPK, PLCγ, PKC, and PI3K.23,24 This has led to speculation in the field that CD90 may participate in HSC activation; currently, there is no evidence of such function in human HSCs because many of these signaling mediators are not expressed in HSPCs, and one would wonder whether HSCs actually use CD90 to cross talk with an integrin-expressing niche or other mononuclear cells instead. Apart from loss-of-function experiments, it would be very interesting to investigate other potential proteins that are associated with the THY1-lipid raft microdomains in HSCs.
Almost there: the tales of integrin-α6 (CD49f) and EPCR (CD201)
It took more than 15 years for the researchers to adopt the use of CD90/THY1 routinely as a surface antigen to enrich human HSCs. In the meantime, many other contenders, such as CD59, CD117, CD133, CD143, HLA-DR, etc, have been used. In addition, metabolic dyes such as rhodamine 123 (Rho123), BODIPY aminoacetaldehyde (indicative of aldehyde dehydrogenase enzyme activity), and others have also been experimentally explored as HSC enrichment methods. Unfortunately, because of the limited space, these molecules are not discussed in detail in this article, but a brief description of some of them can be seen in Table 1.
Table 1.
Description of some of the most used cell surface antigens to study human HSPC biology for the past 30 y
| Cell surface antigen/gene name | Brief characterization |
Potential and/or hypothetical function(s) in human HSPCs/HSCs | Early reference(s) used to identify/select functional HSPCs/source of human tissue used |
|---|---|---|---|
| CD34 | Sialomucin: type-I integral membrane protein with a short cytoplasmic domain containing 3 protein kinase phosphorylation target sites | Potentially in the rolling process on E- and/or P-selectin–expressing cells (eg, endothelial cells)51; homing; the cytoplasmic domain interacts with CrkL;52 hence, it is hypothesized that it may regulate cell shape, adhesion, and migration | Civin et al53 (BM), Katz et al54 (BM), and Andrews et al55 (BM) |
| CD117/KIT (c-KIT proto-oncogene; receptor tyrosine kinase)/KIT | Type III receptor tyrosine kinase Receptor for SCF Receptor activation leads to PI3K/PDK1/Akt; JAK2; SHP-1/SHP-2; and PLC signaling |
Soluble SCF stimulation leads to PI3K-PDK1-Akt signaling: cell cycle regulation by inducing cell proliferation in mouse HSCs56 Membrane-bound SCF stimulation (from the mesenchymal stroma and endothelial cells): leads to PKI3-actin signaling: cell cycle regulation potentially inducing quiescency and reducing oxidative stress via Akt/nuclear FOXO3A retention in mouse HSCs57,58 Both functions may occur in human HSPCs59 |
Cambareri et al60 (BM) and Ashman et al61 (BM) |
| CD90/THY1 (Thy-1 cell surface antigen)/THY1 | GPI-anchored cell surface protein Lacks transmembrane domain |
No data to support a role in HSPCs Hypothesis that HSPCs may have cross talks with niche cells via integrin binding |
Baum et al20 (FBM) |
| CD133 (prominin-1)/ PROM1 | Pentaspan transmembrane glycoprotein Binds to cholesterol-containing lipid rafts |
Functions of CD133 remain very elusive in many cell types as generally associated with apoptosis, differentiation, metabolism (transferrin update), and autophagy processes No data to support its role in human HSPCs, and too many associated biological processes to hypothesize its role in human HSPCs |
Yin et al62 (BM and CB) |
| CD93/C1qRp (complement component 1Q subcomponent receptor 1)/CD93 | C-type lectin transmembrane receptor Has a highly glycosylated mucin–like domain and a short cytoplasmic tail |
No data to support its role in HSPCs Described to be involved in adhesion, migration, and phagocytosis in other cell types Hypothesis is that it may regulate adhesion/migration |
Danet et al41 (CB) |
| CD143/ACE (angiotensin I converting enzyme)/ACE | Cell surface zinc metallopeptidase that hydrolyzes peptides via the removal of a dipeptide from the C-terminus Substrates include angiotensin I, bradykinin, substance P, Ac-SDKP, etc. |
Could positively control cell proliferation by metabolizing Ac-SDKP and Ang-I Ac-SDKP (released from the N-terminal degradation of thymosin β4 [secreted by endothelial cells]) reduces the proliferation and clonogenicity of human CD34+HLA-DRlo HSPCs63 Ang-II and Ang1–7 (degradation product from Ang-I) promote proliferation of human CD34+ HSPCs via AT1R64,65 |
Jokubaitis et al66 (CB) |
| CD33 (sialic acid–binding immunoglobulin-like lectin 3 [Siglec-3])/CD33 | Cytoplasmic domain with 1 ITIM motif and a second ITIM-like tyrosine residue | Functions of CD33 in HSPCs unknown Hypothesis is that it may regulate inflammatory/immune responses negatively through inhibitory effects on tyrosine kinase–driven signaling pathways44 |
Taussig et al42 (BM and CB) |
| CD49f (integrin subunit α 6)/ITGA6 | Form heterodimers with either integrin β1 (CD29) or integrin β4 (CD104), binding α5-laminins and α3-laminins | May be involved in adhesion to BM mesenchymal stroma cells via α5-laminins interactions26 May interact with Nov/CCN3 (endothelial cells) to control stemness31 |
Notta et al25 (CB) |
| CD201/EPCR (endothelial protein C receptor)/EPCR | Enhances protein C receptor activation Modulator of PARs |
May modulate the type of PAR1 responses in HSCs by regulating APC-like (HSC retention) vs thrombin-like signaling response (HSC mobilization)38 | Fares et al34 (CB) |
| CD370 (C-type lectin domain containing 9A)/CLEC9A | Type II membrane receptor, with a single C-type lectin-like domain Intracellular domain contains a hemi-immunoreceptor tyrosine–based activation signaling motif |
Functions of CD370 in HSPCs unknown; hypothesize that may regulate inflammatory signals | Belluschi et al47 (CB) |
| GPRC5C (G protein-coupled receptor class C group 5-member C)/GPRC5C | G protein-coupled receptor Has short extracellular N-terminus and 7-transmembrane domain motif |
The binding of hyaluronic acid to GPRC5C maintains HSPCs dormant50 | Zhang et al50 (BM, CB) |
The list is roughly organized chronologically in accordance with their first depiction to enrich HSPCs/HSCs. FBM, fetal bone marrow; GPI, glycophosphatidylinositol; SCF, stem cell factor.
Integrin-α6 (CD49f)
To further investigate the underlying biological differences (eg, homing and engraftment) that occurs between CB CD34+CD38–CD45RA–RholoCD90+ HSC–enriched population and CD34+CD38–CD45RA–RholoCD90– MPPs, Notta et al hypothesized that integrins would depict human HSCs because they facilitate niche interactions.25 They compared the cell surface expression of several adhesion molecules between these 2 primitive populations and found that among the few investigated molecules, only integrin-α6 (ITGA6; CD49f) was differentially expressed (twofold).25 From this, the authors investigated whether CD49f could serve to further enrich human HSCs. Based on a series of in vivo experiments, the authors showed credibly that CD34+CD38–CD45RA–RholoCD90+CD49f+ (in CB, it was ∼1.8% of Lin–CD34+CD38– and ∼8% of Lin–CD34+CD38–CD45RA–CD90+)25 cells were highly efficient in generating long-term multilineage grafts (up to 30 weeks), whereas CD34+CD38–CD45RA–RholoCD90+CD49f– MPPs (in CB, it was ∼14% of Lin–CD34+CD38–; ∼56% of Lin–CD34+CD38–CD45RA–CD90– cells)25 provided human chimerism only up to 17 or 18 weeks after thetransplantation into NSG mice.25 Importantly, in vivo limiting dilution assay experiments revealed that the CD34+CD38–CD45RA–RholoCD90+CD49f+ fraction had the highest repopulating cell frequency reported to that point with ∼1 in 10 cells being labeled an HSC.25
Briefly, integrins are type-I transmembrane glycoproteins composed of α and β chains (18 types of α and 8 types of β chains). The different heterodimer combinations have unique affinities for extracellular matrix components, some with specific and others with redundant functions. Integrin-α6 can form heterodimers with either integrin β1 (CD29) or integrin β4 (CD104) and form a receptor for different laminins. Integrin-α6β1 displays strong binding toward α5-laminins (laminin-511/521) and α3-laminin (laminin-332)26 that are expressed by niche cells, such as BM stromal and endothelial cells.27,28 The importance of CD49f-laminin-511/521 interactions in human HSPCs was highlighted in a study by Gu et al., in which authors established that human CD34+ HSPCs, and even more so CD34+CD38– cells, had preferential binding to laminin-511/521 over laminin-111 or -411 in vitro.29 Notably, these interactions were abrogated when cells were treated with the anti-CD49f (GoH3) blocking antibody.29 These results support that the CD34+CD38–CD45RA–RholoCD90+CD49f+ HSC–enriched population could have better adhesion compared with the other CD34+CD38–CD45RA– subpopulations, but this has yet to be directly demonstrated. However, CD49f expression is very low on CD34+CD38–CD45RA– HSPC surfaces,30 and the isolation of the HSC-enriched population is normally performed using the anti-CD49f (GoH3) antibody.25,30 Therefore, it is questionable whether the CD49f–laminin-511/521 interaction is actually crucial for the adhesion of human HSCs in vivo. Interestingly, it was recently demonstrated that CD49f also binds to the matricellular regulator Nov (CCN3; nephroblastoma is overexpressed).31 It was shown that Nov was able to bind to 60% of CB CD34+CD38–CD90+CD45RA– cells, of which 80% were CD49f+,31 and these interactions were abrogated by the same anti-CD49f blocking antibodies.31 Importantly, CD34+CD38–CD90+CD45RA– treated with Nov resulted in a sixfold increase in serially transplantable HSCs that were associated with both metabolic and transcriptional changes (with low oxidative phosphorylation, and MYC and E2F target signatures).31 Because Nov is expressed/secreted by endothelial cells, this interesting data led us to hypothesize that CD49f-Nov interactions may allow human HSCs to maintain stemness in close vicinity to endothelial cells.
EPCR (CD201)
With the addition of anti-CD49f and anti-CD90 stains, it is possible to separate CD34+CD38–CD45RA– cells into 4 subpopulations, CD90+or–CD49f+or–. The CD90+CD49f+ and CD90–CD49f– populations are highly enriched in HSCs and MPPs, respectively;25 and notably, these cell populations have widely been used as references to define stem as well as early progenitors. However, the CD90+CD49f+ population remains heterogeneous25 with a suboptimal purity ranging from 5% to 10%. This is partly due to the difficulties in separating the negative and positive populations, because CD49f is weakly expressed on human CD34+ HSPCs,30 making the separation of these 4 subpopulations difficult and somewhat ambiguous (it has been randomly assigned as the lower/higher 15% to 30% expressing cells). This has affected data reproducibility between different laboratories as well as the interpretation of molecular data (eg, scRNA-seq).
To resolve this, our laboratory members went on to find alternative means to improve human HSC enrichment. While exploring the potential relationship between the different primitive CD34+CD38–CD45RA–CD90+or–CD49+or– subpopulations, we found that the CD90+CD49f+ HSC–enriched fraction (in CB was ∼6%-7% and in BM was ∼2%-4% of CD34+CD38–CD45RA– cells)30 did not comprise all human HSCs because we identified that the CD90–CD49f+ fraction (in CB, it was ∼7%-8%, and in BM, it was ∼1.5%-3% of CD34+CD38–CD45RA– cells)30 was also enriched in HSCs. Importantly, both populations were shown to be interconvertible and gave rise to the whole CD34+ HSPCs in vivo.30 This was not totally surprising because some data had implied that the CD34+CD38–CD45RA–CD90– population may contain cells with self-renewal capacity, albeit at a much lower frequency, with a potential to engraft in secondary NOG mice.17 A close inspection of the seminal work from Dick’s group also revealed that (CD34+CD38–CD45RA–Rholo) CD90–CD49f+ cells were able to sustain long-term engraftment similar to the CD90+CD49f+ cells, although no secondary transplantation experiments were performed to establish their self-renewal properties.25 Our scRNA-seq and functional assays have demonstrated that only a fraction of cells (∼6%-10%) within these 2 populations have long-term repopulating capacity.30 We then identified a primitive human hematopoietic population using endothelial protein C receptor (EPCR)/CD201 as an alternative surface antigen. We demonstrated that the CD34+(CD38/CD45RA)–EPCR+ population (in CB, it was ∼2%-5% and in BM, it was ∼0.1%- 0.3% of CD34+CD38– and ∼0.2%-0.8% and ∼0.03% of CD34+ cells)30 contained all the cells with robust multipotent repopulating capacity within the 2 CD49f+ HSC–enriched populations.30 Although using EPCR as a surface antigen for HSC enrichment was not entirely novel, as it has been previously described in mice32,33 and more recently in unmanipulated and expanded CB HSPCs with UM171,34,35 this newly identified population harbored the highest human HSC frequency identified to date, ∼1 in 3 cells, thus closely matching the purities described in mice.36,37 In addition, our scRNA-seq and in vitro functional studies supported that this newly defined HSC-enriched population appeared to be very homogeneous.30 Altogether, we have defined the most primitive postnatal (CB and BM) CD34+ HSCs that have the potential to sequentially generate CD34+CD90+(CD38/CD45RA)–EPCR– progenitors, then (newly defined) MPPs (CD34+CD90–[CD38/CD45RA]–EPCR–), thus revealing a restructured human HSPC hierarchy at the most primitive level.
From all the cell surface antigens that have been described, the function of EPCR could be the one with a better described role in HSCs. Thanks to the important and detailed works from Lapidot’s group, EPCR was shown to be important in the niche retention and mobilization of mouse HSCs.38 Briefly, EPCR is a transmembrane glycoprotein, and it promotes the activation of protein C (APC) by the thrombin–thrombomodulin complex upon the binding of protein C. EPCR has only a short cytoplasmic tail, hence unlikely to be able to induce cell signaling itself,39,40 but it plays an important role in modulating other mediators localized in the EPCR-lipid rafts, such as protease-activated receptors (PARs).39,40 Indeed, the APC-EPCR activation of PAR1 initiates signaling via β-arrestin 2, resulting in the activation of the PI3K/Akt pathway and Rac1 guanosine triphosphatase and the transactivation of S1P1.39,40 In the context of mouse HSCs, it was described that the adhesion of EPCR+LSK (Lin–Sca-1+c-Kit+) cells to VLA4 (α4β1) integrin was reduced by a blocking anti-EPCR antibody, and this was enhanced upon APC stimulation,38 suggesting a role in HSC retention. Mechanistically, APC–EPCR-PAR1 signaling led to increased eNOS phosphorylation at the negative regulatory site Thr495 and reduced phosphorylation at the positive regulatory site Ser1177, inducing low levels of nitric oxide (NO) production in HSCs.38 The low levels of NO limited Cdc42 activity, which was required for HSC adhesion and BM retention.38 Conversely, treating mice with thrombin (mimicking physiological stress, inflammation, and cytokine-induced mobilization that increases thrombin generation) was shown to induce EPCR shedding (with concomitant increased levels of soluble EPCR in the BM fluid and reduced cell surface EPCR expression on HSCs), which induced HSC egress to the circulation.38 Thrombin-induced PAR1 activation (through the G proteins instead) was evidenced by eNOS phosphorylation shifting at the activating Ser1177 site of PAR1 that led to the NO production.38 Together, we can extrapolate that one of the potential functions of EPCR is to modulate the type of PAR1 responses in human HSCs by regulating APC-like vs thrombin-like signaling responses. Whether similar mechanisms occur in human CD34+(CD38/CD45RA)–EPCR+ HSCs remains to be fully determined. Nonetheless, Sauvageau’s group reported that the knockdown of EPCR in human HSPCs resulted in a ∼10-fold decrease in human engraftment in NSG mice,34 thus supporting an important role of EPCR during regeneration.
New contenders: CD33 (Siglec-3), CD370 (CLEC9A/DNGR-1), and…
Human HSPCs were long thought of to be devoid of lineage-associated markers, but this has emerged to not be the case. In fact, human HSPCs highly express multiple myeloid antigens, such as CD13 (ANPEP), CD33 (Siglec-3) CD93 (C1qRp), and CD123 (interleukin-3Rα).41,42 Indeed, we have previously shown that most of the in vivo repopulating capacity of Lin–CD34+CD38– HSPCs in NOD/SCID mice was confined in the CD33+ fraction.42 To further enrich human HSCs from the CB CD34+CD38–CD45RA–CD90+CD49f+ population, Eaves’ group has demonstrated that cells with the highest expression of CD33 harbored the highest self-renewal capacity.43 Despite their efforts, the authors did not show further HSC enrichment of their testing population. We later reported that although CD34+(CD38/CD45RA)–EPCR+ HSCs had the highest CD33 expression, a large majority of the downstream CD34+(CD38/CD45RA)–CD90+EPCR– progenitors also had high CD33 expression,30 therefore making CD33 a less powerful antigen for human HSC enrichment.
CD33 is a sialic acid–binding immunoglobulin-like lectin-3 transmembrane receptor, and it can be stimulated by any molecule with sialic acid residues, such as glycoproteins or glycolipids. Intracellularly, the cytoplasmic domain has 1 ITIM motif and a second ITIM-like tyrosine residue. Upon binding to sialic acid, the cytosolic portion of the protein is phosphorylated and acts as a docking site for SHP phosphatases. There is increasing evidence that CD33 has a suppressive endocytic property and antiproliferative/activation functions. It also regulates inflammatory/immune responses negatively through inhibitory effects on tyrosine kinase–driven signaling pathways, features that have been determined in different leukocyte populations.44 Although the function of CD33 in healthy HSPCs remains largely unknown, it is tempting to question whether human HSPCs use CD33 to control inflammatory responses to dampen the NFκB pathway, which can be detrimental to the functions of HSCs.45,46
On a similar line of thought, human HSPCs appear to have retained some features of the innate immune system, perhaps as a surveillance mechanism and/or a way to control inflammatory responses. It is well described that human HSPCs express HLA-DR, which is usually present on professional antigen-presenting cells. Moreover, it was recently reported that human HSPCs also express CD370 (CLEC9A/DNGR-1),47 a protein that was thought to be DC–specific.48 In this interesting study, it was illustrated that CD370 expression was mainly confined in CB CD34+CD38– HSPCs and at the highest level in CD34+CD38–CD45RA–CD90+ cells,47 making CD370 a plausible candidate for further refinement of the HSC population. Unfortunately, no additional functional HSC enrichment was achieved in CD34+CD38–CD45RA–CD90+CD49f+CD370hi cells (reaching an in vivo repopulating frequency of 1 in 13 cells in NSG mice [in CB, it was ∼24% of CD34+CD38–CD45RA–CD90+ and ∼5% of CD34+CD38– cells]).47 Of note, within CD34+CD38–CD45RA–CD90+CD49f+ HSC–enriched cells, a considerable proportion (∼85%) were already shown to be CD370+,47 which might explain why it was not possible to further refine the HSC compartment using an anti-CD370 antibody. Interestingly though, both CD370hi and CD370lo subfractions contained durable repopulating capacity albeit the latter was more sporadic and only restricted to myeloid-lymphoid differentiation.47 Additional functional and transcriptomic studies provided support that the CD370lo subfraction represented the earliest entry point into lymphoid commitment.47 We also observed this feature in the CD34+(CD38/CD45RA)–CD90+EPCR– progenitors.30 It is highly likely that the CD370lo subfraction does not express EPCR. Although these CD370lo cells do express CD49f+, it is known that not all CD49f-expressing cells are EPCR+ and HSCs.30
DNGR-1 is a type II membrane receptor, holding a single C-type lectin-like domain structure and an intracellular domain containing a hemi-immunoreceptor tyrosine–based activation signaling motif. In conventional type-1 DCs, DNGR-1 senses polymeric F-actin that is exposed upon the loss of plasma membrane integrity. Upon the binding of F-actin, it signals through spleen tyrosine kinase and mediates the crosspresentation of dead cell–associated antigens to CD8+ T cells. In addition, it was recently suggested that DNGR-1 may have cross talks with heterologous receptors and dampen heterologous signaling pathways to limit the induction of proinflammatory signals. It was shown in conventional type-1 DCs that the engagement of DNGR-1 by its ligand was accompanied by the phosphorylation of the phosphatase SHP1, which limited the signaling through heterologous receptors such as Dectin-1, resulting in reduced NFκB signaling.49 Clearly, these functions have not been reported in HSPCs; however, it is quite plausible that the latter mechanism could occur in HSPCs; hence, it would be interesting to take this hypothesis further and investigate if human HSCs use DNGR-1 to control NFκB signaling.
One additional surface antigen has been recently described as a potential candidate to enrich for quiescent HSCs: GPRC5C (G protein-coupled receptor class C group 5 member C).50 GPRC5C was found to be differently expressed between CD34+CD38–CD45RA–CD90+CD49f+ HSC–enriched vs CD34+CD38–CD45RA–CD90–CD49f– MPP–enriched cells (in CB and BM). It was then shown that CD34+CD38–GPRC5C+ HSPCs were more confined in the G0 phase of the cell cycle as compared with their negative counterparts (∼95% vs 75%, respectively). Using phenotypic and functional analyses, it was revealed that CD34+GPRC5C+ cells had a ∼twofold enrichment in CD34+CD38–CD45RA–CD90+CD49f+ cells and ∼2.5-fold more repopulating cells as compared with CD34+GPRC5C– (in BM CD34+CD38–CD45RA–CD90+CD49f+, GPRC5C+ made up ∼20% of CD34+CD38– and ∼2%-4% of CD34+)50 cells. However, like many other surface markers, staining with anti-GPRC5C antibody results in a smear-type pattern of staining, making it difficult to reproducibly delineate GPRC5C+ events. Importantly, most CD34+GPRC5C+ cells were CD38+ progenitors, and many MPPs and CD34+CD38–CD45RA+ cells were also found to be positive for GPRC5C expression.50 Despite that, some enrichment of cells with primitive features were attained (as described). Our laboratory members also experienced this (in their observations), thus raising the question of how useful GPRC5C is to further delineate quiescent HSCs from CD34+CD38–CD45RA–CD90+CD49f+ HSC–enriched cells or CD34+(CD38/CD45RA)–EPCR+ HSCs. Nonetheless, it appears that GPRC5C was shown to be activated by hyaluronic acid leading to intracellular calcium signaling.50 Nevertheless, it was unclear from this study how this activation led to gene regulation and subsequent regulation of human HSPC’s quiescence. That said, it would be interesting to determine how human HSCs use GPRC5C to maintain a dormant state.
Concluding remarks
This review highlights several important points. Firstly, despite many of the markers used to identify different HSPC populations that have been characterized for a long time, there is still limited knowledge of their biological roles in human HSPCs. However, many of these surface antigens, from CD34 to CD49f as well as EPCR and, perhaps, GPRC5C appear to be involved, at least in part, in adhesion/homing. Therefore, effective adhesion/homing should also be included as an HSC hallmark in addition to the classical self-renewal, differentiation capacity, and quiescent/dormancy features. Interestingly, some of the surface markers described that are highly expressed on HSCs, such as CD33 and CD370 could be involved in controlling inflammatory signaling and should be further investigated to establish their role in such processes.
Moreover, using EPCR as a surface antigen can help identify the purest human HSC population described to date (with a stem cell frequency of ∼1 in 3 cells),5 but, more importantly, anti-EPCR staining clearly demarks a defined subpopulation in CD34+ HSPCs that is mostly composed of CD38lo/−, CD45RA−, and CD49f+, making an easy and reproducible way for purifying human HSCs. However, it remains to be determined whether we could use CD370 and/or GPRC5C markers to go beyond EPCR+ HSCs and attain single-cell transplantation more robustly. Furthermore, it would be interesting to investigate whether the heterogeneity present within the CD34+(CD38/CD45RA)–EPCR+ HSCs can be delineated using an advanced OMICs approach, such as cellular indexing of transcriptomes and epitopes sequencing. A similar approach needs to be applied to examine the human MPP compartment that is currently poorly understood. Indeed, apart from the work by Dick’s group that reported the existence of 2 to 3 MPP subpopulations based on CD71 and BAH1 markers,67 little has been described. More importantly, the scientific community will benefit if research groups could report methodology papers describing the practical details of their recent cell phenotypes to increase reproducibility across various laboratories (eg, appropriate fluorochromes and antibody clones, precise gating strategy/staining pattern for cell populations). Certainly, in some of the recent publications, the authors selected ∼20% to 35% of the high and low antigen-expressing cells as their cell population of interest without specific biological reasoning, which could be problematic for accurately reproducing data between laboratories.
Another major issue over the years has been the use of different immunodeficient mouse models (eg, from SCID to NOD/SCID mice, etc) for performing functional experiments to validate the potency of human HSCs. Hence, readers must take this into consideration when interpreting and comparing past data (in terms of stem cell frequency and differentiation outputs) produced by different laboratories. Currently, NSG mice seem to be the gold standard in vivo model to test human HSPC potential. However, some groups have opted to preferentially analyze the primary engraftment at ∼20 to 28 weeks after the transplantation,25,43,50 and others have performed secondary transplantations30,47 to test the long-term self-renewal capacity of their testing populations. With our long-term experience in this field, we believe that the latter method is the most appropriate. We have indeed observed that using NSG mice allow for the long-term engraftment of human cells, even if the transplantation was done using certain progenitors (eg, CD34+CD90+[CD38/CD45RA]–EPCR– progenitors or CD34+CD90–[CD38/CD45RA]–EPCR– MPPs), as long as substantial cells are being transplanted into the primary mice. However, it is important to note that these progenitor cells are not able to generate successful engraftment in serial transplantation settings (unpublished observations). Therefore, an overall standardization of the different phenotypic analyses and in vivo assays is important for a more consistent, faster, and robust advancement of this field.
Despite the recent improvements in xenotransplantation to assay human blood development, there are certain cell types that are still difficult to evaluate in vivo, for example erythropoietic/megakaryocytic lineages, even in NSGW41 (NSG KitW41/W41) mice.68 High level of human chimerism in these mice is required to attain detectable levels of such cell lineages (unpublished observations). Despite the benefits of the NSGW41 model, it still remains far from perfect, especially when a low number of HSC-enriched cells are transplanted. Furthermore, it is well-known that residual murine macrophages prevent human red blood cell reconstitution in immunodeficient mice. Hence, novel models in which mouse macrophages can be conditionally depleted combined with the expression of human EPO (erythropoietin) and TPO (thrombopoietin) (eg, NSG TgHu(EPO/TPO) LysM-Cre/DTR) may be very useful to properly assay the output of these cell populations. Similarly, making the microenvironment in these mouse models resemble that in human (eg, use of scaffolds seeded with human mesenchymal stroma, endothelial cells, etc)69 when performing future studies will be essential to better understanding human HSC biology.
Finally, the quest for a purer HSC population in humans, particularly determining HSCs across different adult hematopoietic tissues, has been rather slow (please refer to Table 1 for the timeline). There are many reasons for such slow progress, but the increasing difficulties in accessing primary human samples worldwide (eg, due to more restricted ethical regulations) and sustaining/developing specific scientific platforms/models (eg, immunodeficient mice) have not helped speed up the development of this research area. Indeed, many of the recent studies highlighted in this review have been conducted using CB (for being easier to access as compared with adult tissues), therefore it would be imperative to examine whether the current surface antigens could also be useful to delineate similar HSC-enriched populations in different adult hematopoietic tissues. More importantly, the role of these cell surface proteins during the aging process and malignancy needs to be studied. Altogether, these studies would allow not only a better understanding of human HSC biology but will have a higher impact on their use in clinical settings.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Acknowledgments
The authors thank Taylor Hinchly and Syed Mian for proofreading the manuscript.
This work was supported by the Francis Crick Institute, which receives its core funding from Cancer Research UK (CC2027), the UK Medical Research Council (CC2027), the Wellcome Trust (CC2027) (D.B.), and the UK Biotechnology and Biological Sciences Research Council (BB/S017097/) (F.A.-A.).
Authorship
Contribution: F.A.-A. wrote the draft and D.B. provided correction and feedback regarding the final version.
Contributor Information
Fernando Anjos-Afonso, Email: dosanjosafonsof@cardiff.ac.uk.
Dominique Bonnet, Email: dominique.bonnet@crick.ac.uk.
References
- 1.Larochelle A, Savona M, Wiggins M, et al. Human and rhesus macaque hematopoietic stem cells cannot be purified based only on SLAM family markers. Blood. 2011;117(5):1550–1554. doi: 10.1182/blood-2009-03-212803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: a human perspective. Cell Stem Cell. 2012;10(2):120–136. doi: 10.1016/j.stem.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 3.Laurenti E, Gottgens B. From haematopoietic stem cells to complex differentiation landscapes. Nature. 2018;553(7689):418–426. doi: 10.1038/nature25022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhatia M, Bonnet D, Murdoch B, Gan OI, Dick JE. A newly discovered class of human hematopoietic cells with SCID-repopulating activity. Nat Med. 1998;4(9):1038–1045. doi: 10.1038/2023. [DOI] [PubMed] [Google Scholar]
- 5.Anjos-Afonso F, Currie E, Palmer HG, Foster KE, Taussig DC, Bonnet D. CD34(-) cells at the apex of the human hematopoietic stem cell hierarchy have distinctive cellular and molecular signatures. Cell Stem Cell. 2013;13(2):161–174. doi: 10.1016/j.stem.2013.05.025. [DOI] [PubMed] [Google Scholar]
- 6.Link H, Arseniev L, Bahre O, Kadar JG, Diedrich H, Poliwoda H. Transplantation of allogeneic CD34+ blood cells. Blood. 1996;87(11):4903–4909. [PubMed] [Google Scholar]
- 7.Terstappen LW, Huang S, Safford M, Lansdorp PM, Loken MR. Sequential generations of hematopoietic colonies derived from single nonlineage-committed CD34+CD38- progenitor cells. Blood. 1991;77(6):1218–1227. [PubMed] [Google Scholar]
- 8.Hao QL, Shah AJ, Thiemann FT, Smogorzewska EM, Crooks GM. A functional comparison of CD34+CD38- cells in cord blood and bone marrow. Blood. 1995;86(10):3745–3753. [PubMed] [Google Scholar]
- 9.Bhatia M, Wang JC, Kapp U, Bonnet D, Dick JE. Purification of primitive human hematopoietic cells capable of repopulating immune-deficient mice. Proc Natl Acad Sci U S A. 1997;94(10):5320–5325. doi: 10.1073/pnas.94.10.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glimm H, Eisterer W, Lee K, et al. Previously undetected human hematopoietic cell populations with short-term repopulating activity selectively engraft NOD/SCID-beta2 microglobulin-null mice. J Clin Invest. 2001;107(2):199–206. doi: 10.1172/JCI11519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taussig DC, Miraki-Moud F, Anjos-Afonso F, et al. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood. 2008;112(3):568–575. doi: 10.1182/blood-2007-10-118331. [DOI] [PubMed] [Google Scholar]
- 12.Hao QL, George AA, Zhu J, et al. Human intrathymic lineage commitment is marked by differential CD7 expression: identification of CD7- lympho-myeloid thymic progenitors. Blood. 2008;111(3):1318–1326. doi: 10.1182/blood-2007-08-106294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manz MG, Miyamoto T, Akashi K, Weissman IL. Prospective isolation of human clonogenic common myeloid progenitors. Proc Natl Acad Sci U S A. 2002;99(18):11872–11877. doi: 10.1073/pnas.172384399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saito Y, Shultz LD, Ishikawa F. Understanding normal and malignant human hematopoiesis using next-generation humanized mice. Trends Immunol. 2020;41(8):706–720. doi: 10.1016/j.it.2020.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mian SA, Anjos-Afonso F, Bonnet D. Advances in human immune system mouse models for studying human hematopoiesis and cancer immunotherapy. Front Immunol. 2020;11 doi: 10.3389/fimmu.2020.619236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fritsch G, Buchinger P, Printz D, et al. Rapid discrimination of early CD34+ myeloid progenitors using CD45-RA analysis. Blood. 1993;81(9):2301–2309. [PubMed] [Google Scholar]
- 17.Majeti R, Park CY, Weissman IL. Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell. 2007;1(6):635–645. doi: 10.1016/j.stem.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doulatov S, Notta F, Eppert K, Nguyen LT, Ohashi PS, Dick JE. Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development. Nat Immunol. 2010;11(7):585–593. doi: 10.1038/ni.1889. [DOI] [PubMed] [Google Scholar]
- 19.Karamitros D, Stoilova B, Aboukhalil Z, et al. Single-cell analysis reveals the continuum of human lympho-myeloid progenitor cells. Nat Immunol. 2018;19(1):85–97. doi: 10.1038/s41590-017-0001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baum CM, Weissman IL, Tsukamoto AS, Buckle AM, Peault B. Isolation of a candidate human hematopoietic stem-cell population. Proc Natl Acad Sci U S A. 1992;89(7):2804–2808. doi: 10.1073/pnas.89.7.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sauzay C, Voutetakis K, Chatziioannou A, Chevet E, Avril T. CD90/Thy-1, a cancer-associated cell surface signaling molecule. Front Cell Dev Biol. 2019;7:66. doi: 10.3389/fcell.2019.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Avalos AM, Valdivia AD, Munoz N, et al. Neuronal Thy-1 induces astrocyte adhesion by engaging syndecan-4 in a cooperative interaction with alphavbeta3 integrin that activates PKCalpha and RhoA. J Cell Sci. 2009;122(Pt 19):3462–3471. doi: 10.1242/jcs.034827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Furlong S, Power Coombs MR, Hoskin DW. Thy-1 stimulation of mouse T cells induces a delayed T cell receptor-like signal that results in Ca2+-independent cytotoxicity. Mol Med Rep. 2017;16(4):5683–5692. doi: 10.3892/mmr.2017.7242. [DOI] [PubMed] [Google Scholar]
- 24.Furlong S, Coombs MRP, Ghassemi-Rad J, Hoskin DW. Thy-1 (CD90) signaling preferentially promotes RORgammat expression and a Th17 response. Front Cell Dev Biol. 2018;6:158. doi: 10.3389/fcell.2018.00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Notta F, Doulatov S, Laurenti E, Poeppl A, Jurisica I, Dick JE. Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science. 2011;333(6039):218–221. doi: 10.1126/science.1201219. [DOI] [PubMed] [Google Scholar]
- 26.Yamada M, Sekiguchi K. Molecular basis of laminin-integrin interactions. Curr Top Membr. 2015;76:197–229. doi: 10.1016/bs.ctm.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 27.Durbeej M, Fecker L, Hjalt T, et al. Expression of laminin alpha 1, alpha 5 and beta 2 chains during embryogenesis of the kidney and vasculature. Matrix Biol. 1996;15(6):397–413. doi: 10.1016/s0945-053x(96)90159-6. [DOI] [PubMed] [Google Scholar]
- 28.Yang Z, Dong P, Fu X, et al. CD49f acts as an inflammation sensor to regulate differentiation, adhesion, and migration of human mesenchymal stem cells. Stem Cells. 2015;33(9):2798–2810. doi: 10.1002/stem.2063. [DOI] [PubMed] [Google Scholar]
- 29.Gu YC, Kortesmaa J, Tryggvason K, et al. Laminin isoform-specific promotion of adhesion and migration of human bone marrow progenitor cells. Blood. 2003;101(3):877–885. doi: 10.1182/blood-2002-03-0796. [DOI] [PubMed] [Google Scholar]
- 30.Anjos-Afonso F, Buettner F, Mian SA, et al. Single cell analyses identify a highly regenerative and homogenous human CD34+ hematopoietic stem cell population. Nat Commun. 2022;13(1):2048. doi: 10.1038/s41467-022-29675-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gupta R, Turati V, Brian D, et al. Nov/CCN3 enhances cord blood engraftment by rapidly recruiting latent human stem cell activity. Cell Stem Cell. 2020;26(4):527–541.e8. doi: 10.1016/j.stem.2020.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kent DG, Copley MR, Benz C, et al. Prospective isolation and molecular characterization of hematopoietic stem cells with durable self-renewal potential. Blood. 2009;113(25):6342–6350. doi: 10.1182/blood-2008-12-192054. [DOI] [PubMed] [Google Scholar]
- 33.Balazs AB, Fabian AJ, Esmon CT, Mulligan RC. Endothelial protein C receptor (CD201) explicitly identifies hematopoietic stem cells in murine bone marrow. Blood. 2006;107(6):2317–2321. doi: 10.1182/blood-2005-06-2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fares I, Chagraoui J, Lehnertz B, et al. EPCR expression marks UM171-expanded CD34(+) cord blood stem cells. Blood. 2017;129(25):3344–3351. doi: 10.1182/blood-2016-11-750729. [DOI] [PubMed] [Google Scholar]
- 35.Tomellini E, Fares I, Lehnertz B, et al. Integrin-alpha3 is a functional marker of ex vivo expanded human long-term hematopoietic stem Cells. Cell Rep. 2019;28(4):1063–1073.e5. doi: 10.1016/j.celrep.2019.06.084. [DOI] [PubMed] [Google Scholar]
- 36.Gulati GS, Zukowska M, Noh JJ, et al. Neogenin-1 distinguishes between myeloid-biased and balanced Hoxb5(+) mouse long-term hematopoietic stem cells. Proc Natl Acad Sci U S A. 2019;116(50):25115–25125. doi: 10.1073/pnas.1911024116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamamoto R, Wilkinson AC, Ooehara J, et al. Large-scale clonal analysis resolves aging of the mouse hematopoietic stem cell compartment. Cell Stem Cell. 2018;22(4):600–607.e4. doi: 10.1016/j.stem.2018.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gur-Cohen S, Itkin T, Chakrabarty S, et al. PAR1 signaling regulates the retention and recruitment of EPCR-expressing bone marrow hematopoietic stem cells. Nat Med. 2015;21(11):1307–1317. doi: 10.1038/nm.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oganesyan V, Oganesyan N, Terzyan S, et al. The crystal structure of the endothelial protein C receptor and a bound phospholipid. J Biol Chem. 2002;277(28):24851–24854. doi: 10.1074/jbc.C200163200. [DOI] [PubMed] [Google Scholar]
- 40.Sen P, Gopalakrishnan R, Kothari H, et al. Factor VIIa bound to endothelial cell protein C receptor activates protease activated receptor-1 and mediates cell signaling and barrier protection. Blood. 2011;117(11):3199–3208. doi: 10.1182/blood-2010-09-310706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Danet GH, Luongo JL, Butler G, et al. C1qRp defines a new human stem cell population with hematopoietic and hepatic potential. Proc Natl Acad Sci U S A. 2002;99(16):10441–10445. doi: 10.1073/pnas.162104799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taussig DC, Pearce DJ, Simpson C, et al. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood. 2005;106(13):4086–4092. doi: 10.1182/blood-2005-03-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knapp D, Hammond CA, Hui T, et al. Single-cell analysis identifies a CD33(+) subset of human cord blood cells with high regenerative potential. Nat Cell Biol. 2018;20(6):710–720. doi: 10.1038/s41556-018-0104-5. [DOI] [PubMed] [Google Scholar]
- 44.Duan S, Paulson JC. Siglecs as immune cell checkpoints in disease. Annu Rev Immunol. 2020;38:365–395. doi: 10.1146/annurev-immunol-102419-035900. [DOI] [PubMed] [Google Scholar]
- 45.Nakagawa MM, Chen H, Rathinam CV. Constitutive activation of NF-kappaB pathway in hematopoietic stem cells causes loss of quiescence and deregulated transcription factor networks. Front Cell Dev Biol. 2018;6:143. doi: 10.3389/fcell.2018.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Talkhoncheh MS, Subramaniam A, Magnusson M, Kumar P, Larsson J, Baudet A. Transient inhibition of NF-kappaB signaling enhances ex vivo propagation of human hematopoietic stem cells. Haematologica. 2018;103(9):1444–1450. doi: 10.3324/haematol.2018.188466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Belluschi S, Calderbank EF, Ciaurro V, et al. Myelo-lymphoid lineage restriction occurs in the human haematopoietic stem cell compartment before lymphoid-primed multipotent progenitors. Nat Commun. 2018;9(1):4100. doi: 10.1038/s41467-018-06442-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Helft J, Anjos-Afonso F, van der Veen AG, Chakravarty P, Bonnet D, Reis e Sousa C. Dendritic cell lineage potential in human early hematopoietic progenitors. Cell Rep. 2017;20(3):529–537. doi: 10.1016/j.celrep.2017.06.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cueto FJ, Del Fresno C, Sancho D. DNGR-1, a dendritic cell-specific sensor of tissue damage that dually modulates immunity and inflammation. Front Immunol. 2019;10:3146. doi: 10.3389/fimmu.2019.03146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang YW, Mess J, Aizarani N, et al. Hyaluronic acid-GPRC5C signalling promotes dormancy in haematopoietic stem cells. Nat Cell Biol. 2022;24(7):1038–1048. doi: 10.1038/s41556-022-00931-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.AbuSamra DB, Aleisa FA, Al-Amoodi AS, et al. Not just a marker: CD34 on human hematopoietic stem/progenitor cells dominates vascular selectin binding along with CD44. Blood Adv. 2017;1(27):2799–2816. doi: 10.1182/bloodadvances.2017004317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Felschow DM, McVeigh ML, Hoehn GT, Civin CI, Fackler MJ. The adapter protein CrkL associates with CD34. Blood. 2001;97(12):3768–3775. doi: 10.1182/blood.v97.12.3768. [DOI] [PubMed] [Google Scholar]
- 53.Civin CI, Banquerigo ML, Strauss LC, Loken MR. Antigenic analysis of hematopoiesis. VI. Flow cytometric characterization of My-10-positive progenitor cells in normal human bone marrow. Exp Hematol. 1987;15(1):10–17. [PubMed] [Google Scholar]
- 54.Katz FE, Tindle R, Sutherland DR, Greaves MF. Identification of a membrane glycoprotein associated with haemopoietic progenitor cells. Leuk Res. 1985;9(2):191–198. doi: 10.1016/0145-2126(85)90082-7. [DOI] [PubMed] [Google Scholar]
- 55.Andrews RG, Singer JW, Bernstein ID. Monoclonal antibody 12-8 recognizes a 115-kd molecule present on both unipotent and multipotent hematopoietic colony-forming cells and their precursors. Blood. 1986;67(3):842–845. [PubMed] [Google Scholar]
- 56.Zou P, Yoshihara H, Hosokawa K, et al. p57(Kip2) and p27(Kip1) cooperate to maintain hematopoietic stem cell quiescence through interactions with Hsc70. Cell Stem Cell. 2011;9(3):247–261. doi: 10.1016/j.stem.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 57.Hao J, Zhou H, Nemes K, et al. Membrane-bound SCF and VCAM-1 synergistically regulate the morphology of hematopoietic stem cells. J Cell Biol. 2021;220:e202010118. doi: 10.1083/jcb.202010118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamazaki S, Iwama A, Takayanagi S, et al. Cytokine signals modulated via lipid rafts mimic niche signals and induce hibernation in hematopoietic stem cells. EMBO J. 2006;25(15):3515–3523. doi: 10.1038/sj.emboj.7601236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bhatia M, Bonnet D, Kapp U, Wang JC, Murdoch B, Dick JE. Quantitative analysis reveals expansion of human hematopoietic repopulating cells after short-term ex vivo culture. J Exp Med. 1997;186(4):619–624. doi: 10.1084/jem.186.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cambareri AC, Ashman LK, Cole SR, Lyons AB. A monoclonal antibody to a human mast cell/myeloid leukaemia-specific antigen binds to normal haemopoietic progenitor cells and inhibits colony formation in vitro. Leuk Res. 1988;12(11-12):929–939. doi: 10.1016/0145-2126(88)90021-5. [DOI] [PubMed] [Google Scholar]
- 61.Ashman LK, Cambareri AC, To LB, Levinsky RJ, Juttner CA. Expression of the YB5.B8 antigen (c-kit proto-oncogene product) in normal human bone marrow. Blood. 1991;78(1):30–37. [PubMed] [Google Scholar]
- 62.Yin AH, Miraglia S, Zanjani ED, et al. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 1997;90(12):5002–5012. [PubMed] [Google Scholar]
- 63.Bonnet D, Lemoine FM, Pontvert-Delucq S, Baillou C, Najman A, Guigon M. Direct and reversible inhibitory effect of the tetrapeptide acetyl-N-Ser-Asp-Lys-Pro (Seraspenide) on the growth of human CD34+ subpopulations in response to growth factors. Blood. 1993;82(11):3307–3314. [PubMed] [Google Scholar]
- 64.Heringer-Walther S, Eckert K, Schumacher SM, et al. Angiotensin-(1-7) stimulates hematopoietic progenitor cells in vitro and in vivo. Haematologica. 2009;94(6):857–860. doi: 10.3324/haematol.2008.000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rodgers KE, Xiong S, Steer R, diZerega GS. Effect of angiotensin II on hematopoietic progenitor cell proliferation. Stem Cells. 2000;18(4):287–294. doi: 10.1634/stemcells.18-4-287. [DOI] [PubMed] [Google Scholar]
- 66.Jokubaitis VJ, Sinka L, Driessen R, et al. Angiotensin-converting enzyme (CD143) marks hematopoietic stem cells in human embryonic, fetal, and adult hematopoietic tissues. Blood. 2008;111(8):4055–4063. doi: 10.1182/blood-2007-05-091710. [DOI] [PubMed] [Google Scholar]
- 67.Notta F, Zandi S, Takayama N, et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science. 2016;351(6269):aab2116. doi: 10.1126/science.aab2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McIntosh BE, Brown ME, Duffin BM, et al. Nonirradiated NOD,B6.SCID Il2rgamma-/- Kit(W41/W41) (NBSGW) mice support multilineage engraftment of human hematopoietic cells. Stem Cell Reports. 2015;4(2):171–180. doi: 10.1016/j.stemcr.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Abarrategi A, Mian SA, Passaro D, Rouault-Pierre K, Grey W, Bonnet D. Modelling the human bone marrow niche in mice: from host bone marrow engraftment to bioengineering approaches. J Exp Med. 2018;215(3):729–743. doi: 10.1084/jem.20172139. [DOI] [PMC free article] [PubMed] [Google Scholar]