

Abstract
The latest RNA genomic mutation of SARS-CoV-2 virus, termed the Omicron variant, has generated a stream of highly contagious and antibody-resistant strains, which in turn led to classifying Omicron as a variant of concern. We systematically collected Raman spectra from six Omicron subvariants available in Japan (i.e., BA.1.18, BA.2, BA.4, BA.5, XE, and BA.2.75) and applied machine-learning algorithms to decrypt their structural characteristics at the molecular scale. Unique Raman fingerprints of sulfur-containing amino acid rotamers, RNA purines and pyrimidines, tyrosine phenol ring configurations, and secondary protein structures clearly differentiated the six Omicron subvariants. These spectral characteristics, which were linked to infectiousness, transmissibility, and propensity for immune evasion, revealed evolutionary motifs to be compared with the outputs of genomic studies. The availability of a Raman “metabolomic snapshot”, which was then translated into a barcode to enable a prompt subvariant identification, opened the way to rationalize in real-time SARS-CoV-2 activity and variability. As a proof of concept, we applied the Raman barcode procedure to a nasal swab sample retrieved from a SARS-CoV-2 patient and identified its Omicron subvariant by coupling a commercially available magnetic bead technology with our newly developed Raman analyses.
Keywords: SARS-CoV-2, Omicron subvariants, Raman spectroscopy, principal component analysis, Raman barcode
The ongoing COVID-19 pandemic has prompted an extraordinary response from the worldwide scientific community.1 In a remarkably short span of time, scientists have made breakthroughs in testing,2 vaccinations,3 small-molecule therapeutics,4,5 and plasma treatments.6 Despite the progress, the effort to eradicate SARS-CoV-2 virus globally faces new challenges. The latest one resides in the emergence of multiple highly contagious Omicron subvariants, among which, the strain called BA.5 has so far proved the most contagious one, causing more than 88% of cases in the United States.7 This same strain has so far fully replaced other Omicron strains also in Japan.8 Although the original Omicron strain has represented a relatively mild version of the virus, currently, Omicron continues to drift toward new subvariants potentially outcompeting the previous ones (i.e., the BA.2.75), keeping its antibody escape characteristics9 and surging more quickly than any previous variant in many areas. For all of these combined reasons, Omicron is presently being classified as a “variant of concern”.10
The abundance of mutations is definitely the most striking characteristic of Omicron variants. With a total of about 50 mutations with respect to the wild type (26 of which are unique to the variant and more than 30 specific to amino acids in the spike protein), Omicron is exploiting a significantly enhanced transmissibility. This characteristic can be considered as the combined result of both evading the body’s immune system and forming a stronger grip on the ACE2 receptor protein than previously appeared in other SARS-CoV-2 variants.11−15 Martin et al.16 examined all Omicron mutations and reported that more than a dozen of them could be classified as extremely rare. Some were never seen before, while others abruptly appeared and then disappeared whenever disadvantageous to the virus. Peacock et al.13 also suggested that changes in the spike protein have modified the way Omicron enters cells, with a mechanism of engulfment in endosomes being preferred to the more common one of direct fusion to the cell membrane. Studies on the electrostatic potential of the Omicron variant by Pascarella et al.17 suggested that Omicron possesses an increased positive electrostatic potential level in its spike receptor-binding domain (RBD) interaction and can thus exploit higher affinity for the negatively charged ACE2 on the cell side. Lupala et al.18 also proposed a significantly increased binding affinity of the Omicron RBD toward ACE2 as compared to Delta as an explanation for an increase in infectivity. Those analyses showed that the enhanced ACE2–RBD interface binding is due to both increased hydrogen bonding interaction and enlarging the buried solvent-accessible surface area. Different approaches were also followed to express ACE2–RBD binding affinity, which included considering the binding free energy between the spike receptor-binding domain and ACE2 receptors as a direct link to the infectivity rate of the strain; the more positive the value of binding free energy, the higher the infectivity rate.19,20 These latter studies have clearly shown that understanding the infectivity and transmissibility of SARS-CoV-2 variants necessarily involves monitoring specific virus mutations in the receptor-binding domain and the related changes in binding free energy. Concerning the Omicron variant, two specific mutations have been located that may increase the affinity of the spike protein to ACE2, three that are related to the risk of increasing transmissibility, and an additional set of mutations that may result in evasion of the immune system.21−26 While it is now clear from a genomic viewpoint that Omicron contains key mutations linked to increased infectivity, transmissibility, and immune escape, important chemical aspects of the molecular origin of these characteristics are yet missing.
Raman spectroscopy holds a position of advantage in molecular biology research, which arises from its capability of revealing key characteristics of biological molecules and secondary structures of ordered and intrinsically disordered proteins and peptides.27−30 A Raman spectrum is a partly overlapping collection of signals from biomolecules that compose cellular, bacterial, and viral structures, thus providing a “metabolomic snapshot” of living cells and microbiome. Raman analyses of the molecular distribution and dynamics can significantly advance our knowledge of cell biology and microbiology. Moreover, as a result of instrumental developments that have led to improved speed and sensitivity in spectral acquisition, Raman advances have evolved into two mainstream technologies: (i) data mining, to help decrypting complex and intertwined chemical information contained in the spectral data31 and (ii) bioinformatics, to enable managing the analysis of large sets of biological data.32 Specifically, regarding virology, our previous studies employed data-mining methods to extract molecular characteristics from the Raman spectra of SARS-CoV-2 α and Delta variants/subvariants in comparison with the Japanese ancestral strain, and to translate them into valuable biological information.33,34 In those studies, a “Raman barcode” algorithm was also proposed to track virus evolution and to keep a record of viral spreading. In this study, we target the so far reported six highly contagious Omicron subvariants, BA.1.18, BA.2, BA.4, BA.5, XE, and BA.2.75, by applying the same Raman data-mining method developed in previous analyses.35−37 The Raman characteristics of these subvariants, which we further discuss in relation to their respective infectivity enhancement and antibody escape resistance, are then translated into Raman barcodes to confirm the possibility of unambiguously locating them and decrypting their main characteristics into easily readable information through appropriate apps. As a proof of concept, we shall show here an example of an Omicron subvariant identification from a nasal swab sample retrieved from a Japanese patient upon coupling a commercially available magnetic beads technology with our newly developed Raman barcode approach.
Results and Discussion
Raman Spectra of SARS-CoV-2 Omicron Subvariants
Raman spectroscopic analyses were performed on six Omicron subvariants in order to spectroscopically unfold with high sensitivity and selectivity the fingerprints of their structural differences, metabolomic perturbations, and evolutionary adaptation in response to vaccine. Figure 1 shows normalized Raman spectra collected in two different spectral intervals (i.e., 600–1800 and 2800–3200 cm–1) on (a) BA.1.18, (b) BA.2, (c) BA.4, (d) BA.5, (e) XE, and (f) BA.2.75.
Figure 1.

Normalized and deconvoluted Raman spectra collected in two different spectral intervals (i.e., 600–1800 and 2800–3200 cm–1) on (a) BA.1.18, (b) BA.2, (c) BA.4, (d) BA.5, (e) XE, and (f) BA.2.75 Omicron subvariants; the wavenumbers given in the inset are in units of cm–1. Phe, Tyr, Trp, Met, Arg, Asn, and Gln are abbreviations for phenylalanine, tyrosine, tryptophan, methionine, arginine, asparagine, and glutamine amino acid residues, respectively.
The most immediate and striking finding was that, at a glance, the average spectra of all subvariants appeared to be morphologically very different from each other. Note that this characteristic is in agreement with previous studies of SARS-CoV-2 variants, which similarly showed significant spectral differences from each other (and from those presented in this study).33,34 Such bold spectral differences demonstrate the high sensitivity of the Raman approach to SARS-CoV-2 virus speciation and position the Raman spectroscopic method as a unique tool for virus metabolomic profiling. The Raman spectra in Figure 1 could indeed be regarded as genuine “snapshots” of virion composition at the molecular scale, giving us a chance to link differences in the virus structure to the reported viral characteristics.
Average Raman spectra were deconvoluted into subband components according to the machine-learning algorithm described in the Methods section. Main signals from amino acid residues are labeled in the inset together with the respective wavenumbers (in cm–1), while a complete list of wavenumbers at maximum and related vibrational origins has been given in previous publications.33,38 In the inset of Figure 1a, four spectral intervals, located at 600–750, 750–900, 900–1200, and 1600–1700 cm–1, are labeled as Zones I–IV, respectively. These four zones, which will be analyzed in detail in the next section, contain characteristic signals from C–S bonds, tyrosine ring structures, RNA purines and pyrimidines, and proteins’ secondary structures, respectively. The latter zone, which is referred to as the Amide I vibrational zone, is a composite vibrational mode involving three different bonds, all contained in the so-called amide plane; namely, C–N and C=O stretching and C–H bending.39 Zone IV is important because it allows the relative fractions of secondary protein structures to be located, including α-helix, β-sheet, random coil, and β-turn. An additional spectral zone was observed at high wavenumbers between 2800 and 3100 cm–1. This zone is characteristic of stretching of C–H bonds, belonging to either CH2 or CH3 groups attached to different biological molecules.
One possible way for cross-checking the spectra of different Omicron subvariants in the region 600–1800 cm–1 resides in screening the respective maxima of relative spectral intensity. In a comparison of Figure 1a–f, it immediately appears that spectral maxima occur in different regions for different Omicron subvariants. The band at ∼1005 cm–1, which arises from the symmetric ring breathing of phenylalanine residues,40 was generally observed as a relatively strong signal in the spectra of all subvariants except for BA.1.18 (cf. Figure 1a). However, this band represented the strongest signal only for the BA.4 subvariant (cf. Figure 1c). Kumar et al.41 reported that the Omicron spike protein contains higher amounts of hydrophobic amino acids, phenylalanine and isoleucine, as compared to the Delta variant, while it is lower than Delta in polar amino acids such as asparagine and glutamine. Fingerprints for polar amino acids can be found in the range 1400–1450 cm–1, as scissoring contributions from CH2 vibrations (1426 and 1438 cm–1 for asparagine, and 1427 and 1450 cm–1 for glutamine).42 The main bands related to hydrophobic phenylalanine and polar asparagine/glutamine residues (at ∼1005 and ∼1426 cm–1, respectively) are labeled in Figure 1. The relative intensities of these bands, expressed as the ratio of hydrophobic to polar residues, Rh/p, showed values ranging between ∼2 (for BA.1.18 and BA.2) and 7.5 (for BA.4), while the other subvariants experienced intermediate values. All Omicron subvariants’ values were indeed higher than the value 1.7 found for the Delta variant (data in a previous study),34 although showing with great variability.
In a previous Raman study of the Delta variant,34 we also reported about the prominent spectral contributions assignable to the basic arginine amino acid residue at 617 cm–1 (C–OH bending), 879 cm–1 (C–N torsion), 940 and 982 cm–1 (N–C–N symmetric stretching), 1088 cm–1 (C–N–H2 asymmetric bending), 1102 cm–1 (NH3+ asymmetric rocking), and 1369 cm–1 (COO– symmetric stretching).40,43,44 In ref (34), the N–C–N symmetric stretching band at 940 cm–1 was taken as a Raman fingerprint for the guanidinium group of arginine and indicated as the chemical moiety primarily responsible for the strengthened electrochemical grip on cells of the Delta variant. In the spectrum of the Delta variant,34 the intensity ratio between arginine 940 cm–1 and phenylalanine 1003 cm–1 bands was ∼1, while Figure 1 shows how it drops to <0.2 for all Omicron subvariants. In addition to indicating a significantly altered ratio between signals representing fractions of phenylalanine and arginine residues, these data also suggest a different path to infectivity for the Omicron variants as compared to the Delta one.
An additional feature was that all subvariants except for BA.1.18 (cf. Figure 1a) showed a relatively strong Amide I protein signal. Amide I was the strongest signal in the spectrum only in the case of BA.5 (cf. Figure 1d), but also in the cases of all other subvariants, it yet remained significantly stronger than in BA.1.18. As shown later in more details, the most prominent component among Amide I signals was that related to disordered structures (random coil) for all subvariants, except for BA.1.18 and BA.4. The structure of SARS-CoV-2 virions contains four types of structural proteins, all involved with different functions in infectious virus assembly; namely, spike (S), envelope (E), membrane (M), and nucleocapsid (N) proteins. The Amide I Raman signal collected on virion samples incorporates all four types of proteins with scarce possibility to single out the contributions of the four different types. The presence of misfolding (prion-like) domains was recognized to be lower in Omicron S proteins as compared to α and Delta variants,45 and the S structure of Omicron virions was recently reported to possess a high degree of order,41 as expected for viral strains with high content of phenylalanine residues. Phenylalanine indeed promotes long-range order and thus supports the formation of secondary structures in proteins. On the other hand, when it comes to M protein, which is the most abundant viral structural protein and represents the major driver of virus assembly and membrane budding,46,47 or N protein, which plays an essential role in promoting genome packaging and viral replication upon host infection, the Omicron variant notably incorporates mutations that increase a misfolded protein character.48 Based on the above arguments, a strong intensity of the random coil signal in the Amide I region of the Omicron subvariants should rather be considered as related to M and N rather than S proteins. Accordingly, it could be a fingerprint for enhanced viral replication and budding while being likely unrelated to the binding affinity of the S-protein. This point will be discussed in more detail in the forthcoming sections.
Maxima of the Raman intensity in the spectra of both XE and BA.2.75 occurred in the region between 1200 and 1400 cm–1. This region is representative of a number of different overlapping vibrations for which machine-learning analyses could not locate any univocal assignment to specific molecules. These include CH2 twisting/wagging,49 vibrations from the benzene ring in tryptophan residues,50 and ring breathing modes from RNA bases.51 However, an important feature, peculiar to all Omicron subvariants except for BA.1.18, was the presence of relatively strong signals in the interval 1440–1500 cm–1, which could be imparted to CH2 scissoring and CH2/CH3 deformation vibrational modes (possibly related to palmitic acid molecules).52 These latter signals represent the strongest scatter in the spectrum of BA.2, being also relatively strong for all subvariants except for BA.1.18. These bands were similarly strong in the spectra of both the Delta variant and ancestral Japanese strain.34 The strongest signal for BA.1.18, which is located at ∼1608 cm–1, contains contributions by ring vibrations in tyrosine, tryptophan, and phenylalanine,53 but its quite strong intensity suggests additional contributions by different ring molecules containing C=C bonds. The most striking (and unique) characteristic of the BA.1.18 subvariant among all other subvariants (and other SARS-CoV-2 variants)33,34 consisted in the appearance of a sharp and strong feature at ∼1725 cm–1, paired with another bold signal at ∼3075 cm–1. The former signal, which appears with neighboring signals but with much weaker intensity also in other subvariants, is related to C=O stretching, while the latter signal, which is conspicuously absent in the spectrum of all other subvariants, mainly arises from C=C–H stretching. The presence of a triplet (including one particularly strong subband) in the region 1700–1750 cm–1 indicates that ester and aldehyde groups are embedded in different environments,54 while the band at 3075 cm–1 could be assigned to olefinic C=C–H bonds related to the presence of low-density lipoproteins.55 One possible scenario for explaining the above features is as follows: palmitic acid, which is the most common type of fatty acid, contains 16 carbon atoms and no carbon–carbon double bonds. However, its molecular breakdown upon oxidation (to form carbonyl groups at β carbon sites) generates acetyl coenzyme A (acetyl-CoA), nicotinamide adenine dinucleotide (NAD), and flavin adenine dinucleotide (FAD) molecules. All of those molecules contain C=O and C=C bonds with strong Raman signals at 1608, 1700–1750, and 3075 cm–1,55,56 i.e., the same as those observed here in the BA.1.18 spectrum. Although it is not possible to decrypt from the Raman spectrum, the exact mechanism of β-oxidation in palmitic acid molecules and the stage at which it has occurred during virions’ generation, the fact that all subvariant samples were prepared rigorously in the same way suggests a higher propensity to oxidation and thus to the formation of low-density lipoproteins for palmitoylated structures in the BA.1.18 subvariant. Additional bands in the high-wavenumber zone at 2800–3200 cm–1 represent vibrations of CH, CH2, or CH3 groups as belonging to a variety of different molecules (cf. labels in the inset to Figure 1).50,57−60 The morphology of this spectral area definitely contributes to diversify the subvariants, but any univocal assignment of those high-wavenumber bands to specific molecules is hampered by signal overlapping (e.g., lipids and proteins or lipids and fatty acids).
Raman Analyses in Selected Spectral Zones
Zone I (Figure 2a) shows the Raman spectra of the investigated Omicron subvariants (cf. labels in the inset) in the spectral zone between 600 and 750 cm–1. This wavenumber region is dominated by signals belonging to C–S stretching vibrations. C–S bonds are found in methionine (Met) and cysteine (Cys) in both trans (t) and gauche (g) rotameric configurations.61 Different molecular symmetries for these two S-containing amino acids leads to the observed series of signals at different wavenumbers in Zone I (cf. labels in the inset). In Figure 2b, bands from different rotamers are extracted from the spectrum and located with different colors for better visualization. The examination of C–S band composition revealed remarkable differences among the Omicron subvariants, which thus arise from their fundamental diversity in both the fraction and symmetry of S-containing amino acid residues. C–S stretching bands from methionine residues are found at 645 ± 4 and 655 ± 4 cm–1 (C–S bond stretching vibrations on the CH2 side of molecules in gauche rotameric configuration), 670 ± 3 cm–1 (C–S bond stretching vibrations on the CH2 side of molecules in trans rotameric configuration), 702 ± 4 cm–1 (C–S stretching on the CH3 carboxyl side in gauche configuration), and 717 ± 4 cm–1 (C–S stretching on the CH3 carboxyl side in trans configuration).62,63 Similar to the case of methionine signals, the relative intensities of C–S stretching bands belonging to zwitterionic (monoclinic) cysteine rotamers greatly differed in different Omicron subvariants. C–S stretching bands in cysteine residues were found at around 634 ± 4 and 690 ± 4 cm–1 for gauche and trans rotamers, respectively.64−67 The distinct shifts in wavenumber and bold variations in relative intensity of C–S bond-related Raman bands among different subvariants reflect compositional differences in the fractions of methionine rotamers, which, in turn, is a consequence of the different amino acid sequences to which methionine links. Different molecular symmetry characteristics play a key role in a number of functions during the virus lifetime.68,69 Two parameters (computed from the respective band areal fractions) can be selected for Raman analysis in Zone I: the methionine trans-to-gauche ratio, Rt/g, and the cysteine-to methionine ratio, RC/M. The former parameter showed remarkable variations between minimum and maximum values Rt/g = 0.32 in BA.1.18 and 2.32 in BA.5 subvariants, respectively. These boldly different ratios reflect chiral characteristics peculiar to individual subvariants and could be used as efficient fingerprints in subvariant speciation. Also, the RC/M ratio greatly fluctuated between a minimum of 0.14 in both XE and BA.2.75, and a maximum of 0.66 in BA.1.18 and BA.5. Methionine is a strongly hydrophobic amino acid that is incontrovertibly located at buried sites in the core of globular proteins. Conversely, cysteine is always exposed to the protein surface and its thiol side chain participates as a nucleophile in environmental reactions, being thus susceptible to oxidation and sensing different pH environments at the virion surface.70 The importance of S-containing amino acids on virus production, assembly, and budding will be discussed in detail in the forthcoming sections.
Figure 2.

(a) Raman spectra of the investigated Omicron subvariants (cf. labels in the inset) in the spectral zone between 600 and 750 cm–1 and (b) bands from different rotamers are extracted from the spectrum and drawn with different colors for better visualization. The values of respective parameters Rt/g and RC/M as computed from the deconvoluted subband areas are listed in the inset. The wavenumbers given in the inset are in units of cm–1 and the abbreviations are defined in the text.
Zone II (Figure 3a) shows the tyrosine doublet, also referred to as the Fermi doublet, for the six Omicron subvariants investigated. The doublet signals appear at 836 ± 5 and 858 ± 3 cm–1, and arise from out-of-plane C–H bending and in-plane ring breathing, respectively. The Fermi doublet is quite sensitive to the surrounding environment, its spectroscopic analysis providing unique information on the chemical environment at the virion/environment interface.71 The intensity ratio, RF = I858/I836, is diagnostic of the H-bonding environment around the tyrosine residues. The RF ratio senses the hydrophobic/hydrophilic balance at the immediate virion surface; the lower the ratio, the more hydrophobic the tyrosine configuration. An increasingly alkaline environment augments tyrosine hydrophobicity and vice versa for an acidic environment. Accordingly, an increasingly low RF value proves surface-protonation conditions pointing to alkaline and more hydrophobic states. According to data in Figure 3b, the Omicron subvariants experienced a surface pH spanning between acidic and neutral values, with BA.1.18 and BA.2.75 having the lowest and the highest surface pH (RF = 2.22 and 1.16, respectively). Despite some minor fluctuations, it clearly appears how the subvariants shifted with time toward an increasing level of surface deprotonation. It should also be noted that a tendency to tyrosine deprotonation, symptomatic of an increased environmental pH, has important implications in virus infectivity, since it implies an enhanced docking ability of the receptor-binding domain while escaping antibody recognition. Moreover, the environmental pH at the virion surface also impacts on the secondary structure of proteins.72 These points will be discussed in more detail in the forthcoming Results and Discussion sections.
Figure 3.

(a) Raman spectra of the investigated Omicron subvariants (cf. labels in the inset) in the spectral zone between 750 and 900 cm–1 and (b) tyrosine doublet, also referred to as the Fermi doublet, is extracted and colored according to the pH scale in the inset together with the respective RF ratios. The wavenumbers given in the inset are in units of cm–1 and the abbreviations are defined in the text.
Zone III (Figure 4a) locates the fingerprint Raman signals of RNA purines and pyrimidines in the spectra of the investigated Omicron subvariants. Spectral fingerprints of cytosine (C) and uracil (U) could be found at 1033 ± 2 and 1057 ± 3 cm–1, these characteristic wavenumbers representing in-plane deformation of C–N–C bonds in heterocyclic aromatic rings.73−75 Despite having the same vibrational origin, these spectral signals differ in wavenumber because they include different functional groups in attachment to the six-membered pyrimidine ring. The same C–N–C vibration also appears in the five-membered imidazole ring of the guanine (G) purine but at a significantly lower wavenumber of 956 ± 6 cm–1, as a consequence of the different ring structure.75,76 A Raman fingerprint for adenine (A) is represented by the cumulative signal from C–N stretching in both five-membered imidazole and six-membered pyrimidine rings found at 1155 ± 7 cm–1.75,77 In Figure 4b, differences in relative intensities of purine and pyrimidine signals are emphasized upon extracting their respective signals from the spectra and by coloring them with different colors, in a comparison among all six subvariants (cf. labels in the inset). Fractional intensities are also given in the inset, which were computed as areal fractions to the sum of all purine and pyrimidine band areas (taken as 100%). Bold differences among “Raman fractions” of RNA bases vividly capture the genome evolution of the Omicron subvariants; if not in terms of absolute fractions as obtained by RNA sequencing, which remain conspicuously the same across Omicron subvariants (Table 1), definitely in terms of base-pairing and association with viral nucleoproteins at preferential RNA sites.78 This point will be discussed in more detail in the Discussion section.
Figure 4.

(a) Raman spectra of the investigated Omicron subvariants (cf. labels in the inset) in the spectral zone between 900 and 1200 cm–1 and (b) fingerprint Raman signals of RNA purines and pyrimidines are extracted from the spectra and colored for better clarity. Fractional intensities are listed in the inset together with the respective Rpl ratios. The wavenumbers given in the inset are in units of cm–1 and the abbreviations are defined in the text.
Table 1. RNA Base Fractions Computed from the Genome Database Show Conspicuous Homogeneity across All Investigated Omicron Subvariants.
| base | BA.1.18 TY38-873 | BA.2 TY40-385 | BA.4 TY41-703 | BA.5 TY41-702 | XE TY41-696 | BA.2.75 TY41-716 |
|---|---|---|---|---|---|---|
| adenine | 29.9% | 29.9% | 29.9% | 29.9% | 29.9% | 32.6% |
| cytosine | 18.3% | 18.3% | 18.3% | 18.3% | 18.3% | 17.8% |
| guanine | 19.6% | 19.6% | 19.6% | 19.6% | 19.6% | 22.5% |
| uracil | 32.1% | 32.2% | 32.2% | 32.2% | 32.2% | 27.1% |
Additional important signals in Zone III were as follows: (i) symmetric ring breathing vibration of phenylalanine (Phe), seen at 1005 ± 3 cm–179 and (ii) phosphodiester linkage (pl) in RNA backbone, detected at ∼1104 ± 2 cm–1.78 As previously mentioned in this section, phenylalanine is a structure-promoting residue, and the intensity of its signal relates to an increased presence of structured proteins.80 On the other hand, the relative intensity of the pl band is proportional to the number of nucleotide units with ordered structures, thus giving the number of nucleotides involved in secondary interactions; the higher the relative intensity, the higher the probability of secondary interactions.81,82 We selected the ratio, Rpl, given by the relative areal intensity of the pl Raman band at ∼1104 ± 2 cm–1 with respect to the total area subtended by fingerprint bands of the four RNA purines/pyrimidines, as a fingerprint parameter characteristic of the “Raman genome”. A comparison of this parameter for different subvariants is given in the next section.
Zone IV (Figure 5a) shows the Amide I region for the six investigated Omicron subvariants (cf. labels in the inset). Deconvoluted Raman signals at 1641 ± 4, 1655 ± 3, 1669 ± 3, and 1682 ± 3/1698 ± 4 cm–1 arise from the same Amide I vibration in different protein secondary structures, namely, α-helix, β-sheet, random coil, and a doublet from two β-turn rotamers (in the literature, referred to as type I and type II), respectively.83 These signals were extracted from the respective subvariant spectra and are replotted in Figure 5b with different colors to emphasize their differences. As seen, the Amide I region distinctly diversified the Omicron subvariants. A feature common to all subvariants, except for BA.1.18 and BA.4, resided in the predominance of the random coil component (i.e., almost undetectable in the spectrum of BA.1.18). The fractional amounts of protein secondary structures were computed from the relative areal intensities in the overall Amide I spectrum, as shown in the inset of Figure 5b. According to Eker et al.,84 a hydrophobic environment (i.e., a higher interface pH at the virions’ surface) leads to a higher propensity to form the β-sheet structure. This was indeed the case of the BA.2.75 subvariant, which showed the lowest value of the Fermi doublet ratio, RF (cf. Figure 3b), and also the highest fraction of the protein β-sheet structure (cf. Figure 5b). This point will be discussed in more detail for all subvariants in the next section.
Figure 5.

(a) Raman spectra of the investigated Omicron subvariants (cf. labels in the inset) in the spectral zone between 1600 and 1750 cm–1, which contain the Amide I region (cf. labels in the inset) and (b) deconvoluted Raman signals from different protein secondary structures are extracted from the respective subvariant spectra and replotted with different colors to emphasize their differences (α-helix, αh; β-sheet, βs; random coil, rc; and β-turn rotamers, βt types I and II). Fractional intensities for different protein secondary structures are listed in the inset together with the respective Rrc ratios. The wavenumbers given in the inset are in units of cm–1 and the abbreviations are defined in the text.
The parameter, Rrc, referred to as the random coil propensity ratio, gives the ratio between the area subtended by the random coil component and the sum of areas from all other secondary structures in the Amide I signal. Therefore, the parameter Rrc describes the degree of disorder in the viral proteins’ structural ensemble (and vice versa RO = 1 – Rrc its degree of order). According to the literature,85 the S-protein in Omicron subvariants is rich in the random coil structure. However, it should be noted that Raman assessments of viral proteins are not capable of distinguishing among S, E, M, and N proteins. Accordingly, a direct link between the observed conformational differences and specific mutations can hardly be inferred. Nevertheless, the present Raman data clearly show that concurrently with an evolutionary trend toward higher pH at the virion surface/environment interface, the conformational state of viral proteins changed, thus also reshaping the overall interaction between virions and host cells.86−88
Testing the Internal Consistency of Spectroscopic Data
In this section, we looked for confirmation of the reliability and soundness of the collected Raman data. Cross-parametric checks were conducted to strengthen the internal consistency and to ensure a sound interpretation of the detected spectroscopic features.
The first spectroscopic confirmation, which concerned the interpretation of RNA-related bands in spectral Zone III, was conducted by screening the phosphodiester linkage ratio, Rpl, across different subvariants. As seen in Figures 1 and 4a, the relative intensity in spectral Zone III varied significantly for different subvariants. Notably, however, the Rpl ratio remained conspicuously constant across all Omicron subvariants (Rpl = 0.24 ± 0.1). This represents an important confirmation since the number of linkages should remain proportional to the number of RNA bases, whether the number of nucleotides involved in secondary interactions is the same. While corroborating the choice of Raman fingerprints for RNA purines and pyrimidines, this evidence also suggests that all of the Omicron subvariants maintain the same degree of cooperative binding of N proteins to RNA. N proteins in the SARS-CoV virus are known to pack up the viral genomic RNA and crucially impact the viability of the virus. Although Raman data cannot clarify the mechanism by which N proteins bind RNA, they suggest that different Omicron subvariants, even those sharing lower sequence homology, similarly construct cooperative binding of N proteins to RNA and share similar RNA-binding ability, in agreement with the published literature.89
A second important confirmation regarding the relationship between the tyrosine ratio RF, representing the degree of alkalinity (and hydrophobic level) of the virion surface, and the protein secondary structure. The plot in Figure 6a shows the relationship between the RF ratio and the fraction, Rβs, of proteins in the β-sheet configuration. The plot is linear and points to higher fractions of the β-sheet structure with increasing pH (cf. arrow in the inset), in agreement with the literature data.84 On the other hand, a nonlinear trend was found for the random coil fraction, Rrc, as a function of tyrosine ratio RF (Figure 6b), with the BA.1.18 subvariant again showing the lowest Rrc value. In this latter case, a steep volumetric increase in random coil configuration was observed up to a saturated value of ∼30% (for tyrosine ratios, RF < 1.65). Several other authors have previously reported conformational shifts toward increasingly disordered protein states upon exposure to increasingly higher pH conditions and/or in alkali-denatured proteins.90−92
Figure 6.
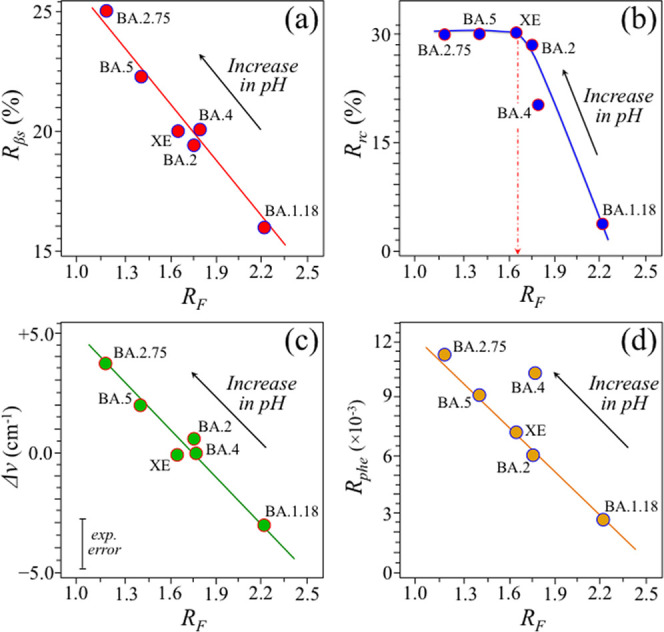
Relationships between the tyrosine ratio RF, representing the degree of alkalinity and hydrophobicity, of the virion surface and (a) β-sheet protein fraction, Rβs, (b) random coil protein fraction, Rrc, (c) band shift for the Trp indole ring deformation band found at 878 cm–1 for neutral pH, and (d) relative areal fraction, Rphe, covered by the symmetric benzene ring breathing vibration of Phe at 1005 ± 3 cm–1 with respect to the cumulative area of Raman scatter in the spectral region between 600 and 1800 cm–1. The consistency among the plots (a–d) represents a cross-parametric check for the evolutional trends of the Omicron subvariants toward increasingly alkaline and hydrophobic surface-protonation conditions.
A crosscheck of the observed alkaline-pH evolutionary trend could be obtained by monitoring the wavenumber shift of the indole ring deformation band in tryptophan (Trp) residues, which appears at 878 cm–1 in neutral pH.93 Similar to the Fermi tyrosine doublet, this Trp band is sensitive to hydrogen bonding and, thus, similar to tyrosine, represents a readout for environmental pH. According to Takeuchi,93 under increasing alkaline conditions (i.e., toward a more substantial non-hydrogen-bonded state), the spectral location of the Trp indole band shifts from 878 cm–1 (spectral location in neutral environment) toward higher wavenumbers, and vice versa for a strong hydrogen-bonded configuration (i.e., in acidic environment). Figure 6c shows a plot of the band shift for the Trp indole ring deformation band (retrieved from subvariants’ spectra in Figure 3) as a function of the RF ratio (cf. also experimental error given in the inset). As seen, this plot obeys a linear trend very similar to that observed for the Rβs(RF) one (shown in Figure 6a), thus confirming an evolutionary shift of the Omicron subvariants toward alkaline (hydrophobic) environmental interfaces.
Finally, a further phenomenological effect accompanying the evolutional hydrophobic shift of the Omicron subvariants was found upon evaluating the relative areal fraction of the symmetric benzene ring breathing vibration of Phe (seen at 1005 ± 3 cm–1) with respect to the entire spectral area between 600 and 1800 cm–1. This parameter, termed Rphe, was also plotted as a function of tyrosine doublet, RF, in Figure 6d. The Rphe(RF) plot was found to be linear, showing a rather precise proportionality dependence with increasing virion/environment interface pH.79 However, unlike the other plots in Figure 6, this latter plot showed an exception for the BA.4 subvariant. Since BA.4 and BA.5 subvariants were reported to share identical S-protein structures with commonly high amounts of Phe residues,94,95 the discrepancy in the BA.4 behavior in the plot of Figure 6d could be related to bond location and balance between surface and bulk Phe residues. Remarkably, in support of this interpretation, a recent study by Hossain et al.96 reported about the mutation, L11F (Leucine → Phenylalanine), in accessory proteins, which is unique to the transmembrane region of the Omicron BA.4 subvariant and possibly impacts on immune modulation. However, as explained in detail in the next section, the Raman spectrum of the BA.4 subvariant was also the one with the highest statistical scatter among all tested subvariants. Therefore, the hypothesis of a spectroscopic link between the nonspike L11F mutation in BA.4 and the exceptionally high relative Raman intensity of its 1005 ± 3 cm–1Phe-ring signal shall require additional confirmations.
Chemometric Analysis and Barcode Identification
Ten locations 10 μm2 in size were selected for each Omicron subvariant sample on which Raman spectra were collected. The obtained 10 spectra per sample were then subjected to principal component analysis (PCA). Figure 7a shows the first, second, and third principal components (PC1, PC2, and PC3, respectively) of PCA analyses of Raman spectra for the six investigated Omicron subvariants. It should be noted that PC1 is mainly influenced by the relative intensity of the spectrum with respect to the point of normalization (e.g., the most intense band of the recorded spectrum); even if accounting for more than 50% of the variance among spectra, this component gives little if any information on the conformation of proteins, amino acid residues, and genetic material. More interesting, from the spectroscopic point of view, is the relationship between PC2 and PC3, directly related to both metabolomics and proteomics of the virions.
Figure 7.
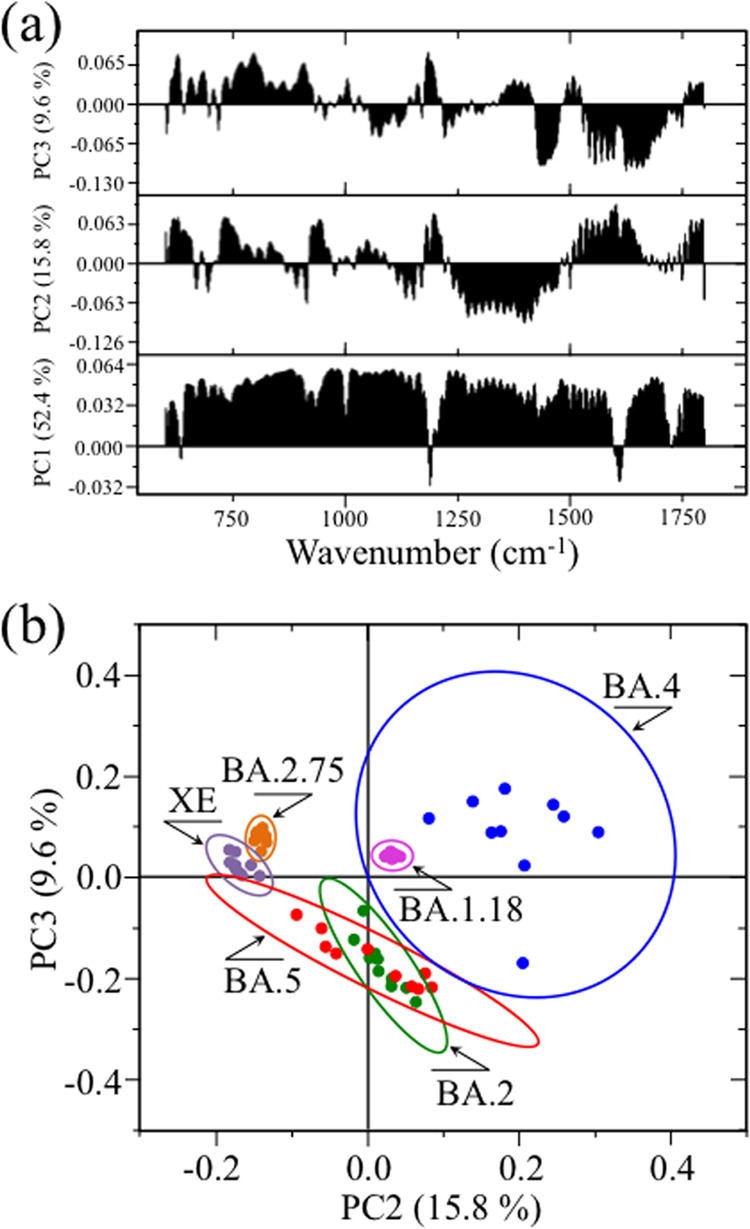
(a) First, second, and third principal components (PC1, PC2, and PC3, respectively) from PCA analysis of Raman spectra at 10 selected locations of different Omicron subvariants and (b) loading vectors of second and third principal components PC2 and PC3 as computed over a PCA analysis of the Raman spectral region 600–1800 cm–1. The plot clearly separated the XE and BA.2.75 subvariants but suffered deficiencies in locating all other Omicron subvariants.
Figure 7b represents the loading vectors of the second and third principal components (PC2 and PC3, respectively) over the spectral region 600–1800 cm–1. This plot shows that PCA analysis succeeded in separating only XE and BA.2.75 subvariants, while it suffered clear deficiencies in locating all other subvariants. In particular, the 10 representative spectra of the BA.4 subvariant showed significantly higher scatter in comparison with other subvariants. At this stage of the Raman investigation, it is not clear what is the reason for the high scatter noticed for the BA.4 subvariant. Since all subvariant samples were prepared exactly in the same way and the Raman measurements were performed with the same instrument under exactly the same measurement conditions, the metabolomic and proteomic scatters found for the BA.4 subvariant in the present untargeted Raman study could actually arise from intrinsic compositional fluctuations of the sample. In summary, the application of the PCA statistical approach, which reduces the dimensionality of the Raman data matrix to two orthogonal variables, conspicuously failed in identifying the majority of the Omicron subvariants.
In previous studies,33,34,97−99 we proposed Raman barcoding as an alternative approach to chemometric PCA analyses in order to overcome speciation deficiencies. The Raman barcode approach, when applied using the algorithm explained in the Methods section, allows a greater depth in capturing structural details than a mere morphological comparison of spectra, as done in PCA analyses. The barcode approach was applied to the average spectra obtained for each Omicron subvariant from the same series of 10 spectra used in PCA analyses. Figure 8 shows sequences of Gaussian–Lorentzian subbands (i.e., the same as those shown in the deconvoluted spectra of Figure 1 and obtained from the machine-learning-based spectral deconvolution described in the Methods section) for the average Raman spectra of each Omicron subvariant sample. The Raman barcodes, given on the right side of Figure 8 were built from the respective subband sequences. As seen, although the barcode approach cannot obviously mitigate the statistical scatter in the Raman spectrum of the BA.4 subvariant, barcodes greatly amplified the differences among the subvariants’ average spectra. Spectral differences arising from different metabolite structures of the Omicron subvariants, once translated into line patterns, made the subvariants distinguishable by the naked eye, thus apparently overcoming PCA deficiencies.
Figure 8.
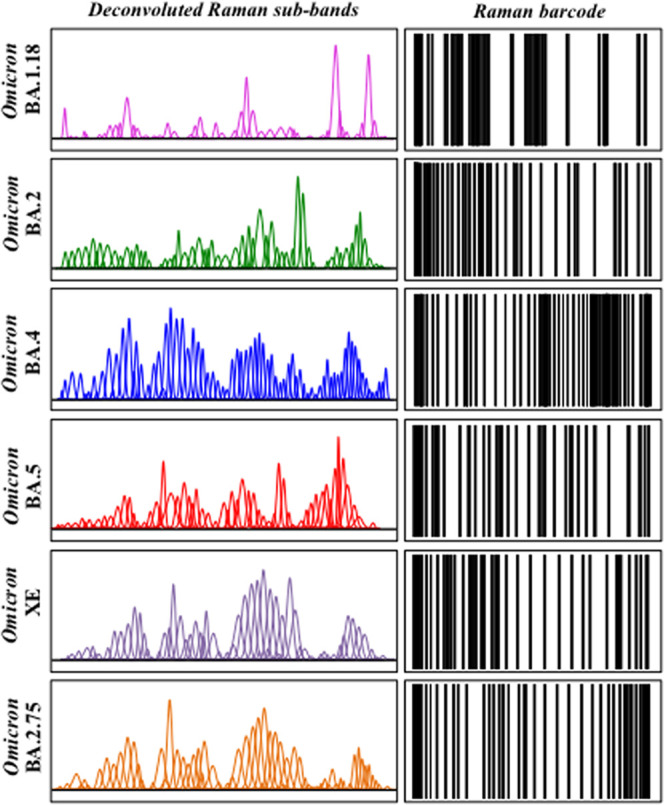
Sequences of Gaussian–Lorentzian subbands from Figure 1 for different Omicron subvariant samples (left side; cf. labels in the inset) and their respective barcodes (right side). The barcode procedure allowed overcoming all PCA deficiencies.
Raman Analysis of the Virion Sample from Infected Human Nasal Swab
Figure 9 gives a schematic draft of virions’ separation from the patient’s swab by means of magnetic beads. In panel (a), the virions are chemically trapped on the beads’ surface (cf. scanning electron micrograph in panel (b)) by electric charge. They are then separated from the swab by magnetic forces and analyzed by Raman spectroscopy (c). Figures 10 and 11 show the results of Raman analysis on isolated swab virions in Zones I (600–750 cm–1) and III (900–1200 cm–1), respectively. These spectral zones were selected because they are considered to be the least affected by Raman signals intrinsic to the magnetic beads and the least impacted by chemical interactions with the beads, respectively. Section (a) of the respective figures gives the results of a comparison between the Raman spectrum library in Figures 2 and 4 with the Raman spectrum of virions separated from the swab. A similar comparison is also given for the related Raman barcodes in the selected spectral areas. The Raman analyses located two subvariants as the closest to the swab virus, namely, the BA.5 and the XE. While in the analysis of S-containing amino acids (Zone I) the two subvariants appear substantially equidistant from the patient’s one, the spectra representing RNA signals (Zone III) point to a slightly closer matching to the BA.5 subvariant (cf. subband analyses in (b), (c), and (d) in each of Figures 10 and 11). Some additional characteristics were also found in the swab spectrum, which are located with light-green subbands and arrows in Figures 10 and 11. In Figure 10, new signals appeared at ∼625 and 729 cm–1, which were not seen in the standard virions’ spectra. These new (weak) signals point to a slight modification of the rotameric structure of cysteine and methionine residues. In support of this interpretation is the appearance of a new signal at 915 cm–1, which could be assigned to stretching of C–COO– carboxylate groups.100 This body of spectral variations points to a scenario in which methionine residues adsorb on the charged bead surface and partly change their rotameric structure while undergoing modifications of its thioether group and terminal structure.
Figure 9.
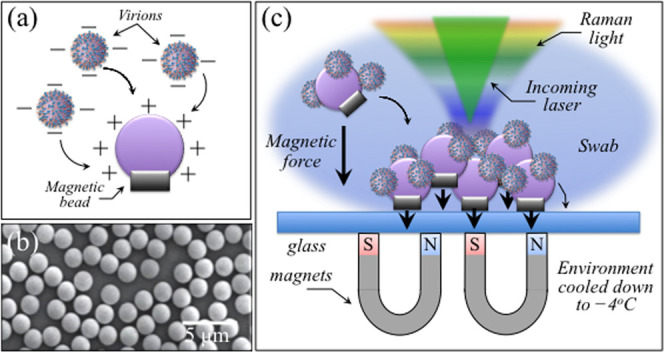
(a) Schematic draft of the electrical charge interaction between virions and magnetic beads, whose scanning electron image is shown in panel (b). In panel (c), the assemblage is shown for Raman measurements on SARS-CoV-2 virions extracted from a human swab and clustered by means of magnetic force. Raman measurements were performed at −4 °C.
Figure 10.

(a) Average Raman spectra of virions extracted from a human swab sample in the spectral zone between 600 and 750 cm–1, in comparison with average (standard) spectra from BA.5 and XE subvariants (the red-shadowed spectral variation refers to the spectral response of virions extracted from the swab nasal sample); the respective Raman barcodes in the same spectral zone are given in the inset. Panels (b), (c), and (d) show deconvoluted Raman subbands from spectra belonging to human nasal swab, standard BA.5 and XE virions, respectively, as shown in panel (a) (cf. labels in the inset). Matching was similarly found with spectra of BA.5 and XE Omicron subvariants. The wavenumbers in the inset to panel (b) are given in units of cm–1 and the abbreviations are defined in the text. Light-green arrows and subbands locate two minor signals that were detected in virions from the swab sample, but not in standard Omicron samples; their possible origins are discussed in the text.
Figure 11.

(a) Average Raman spectra of virions extracted from a human swab sample in the spectral zone between 900 and 1200 cm–1, in comparison with average (standard) spectra from BA.5 and XE subvariants (the red-shadowed spectral variation refers to the spectral response of virions extracted from the swab nasal sample); the respective Raman barcodes in the same spectral zone are given in the inset. Panels (b), (c), and (d) show deconvoluted Raman subbands from spectra belonging to human nasal swab, standard BA.5 and XE virions, respectively, as shown in panel (a) (cf. labels in the inset). Matching was found with spectra of both BA.5 and XE Omicron subvariants, with a slightly closer matching with the spectrum of the former subvariant. The wavenumbers in the inset to panel (b) are given in units of cm–1 and the abbreviations are defined in the text. The light-green arrow and subband locate two minor signals that were detected in virions from the swab sample but not in standard Omicron samples; the possible origin of this signal is discussed in the text.
Two SARS-CoV-2 whole genome sequences were acquired from human infected nasal swab samples through the removal of low-quality variant call sites to avoid false-positive variant calling. We then examined phylogenetic relationships among several SARS-CoV-2 nucleotide sequences, including sequence data from publicly available databases. The ML tree (Figure S1) and Nextclade analyses indicated that genome sequences of the swab samples can be classified into BA.5.2. In the ML tree, the sequences formed a monophyletic clade with sequence data identified as BA.5.2 (bootstrap values 100%).
“Metabolomic Snapshots” of Omicron Subvariants
As shown in the Results section, the systematic collection of Raman spectra revealed at once statistically validated metabolite motifs in a series of six Omicron subvariants. The Raman approach is straightforward and allows characterization of virions’ molecular structures with minimal sample preparation. It also proved capable of providing unexplored patterns of sensitivity in both speciating SARS-CoV-2 Omicron subvariants and unfolding their evolutionary trend. Changes in metabolite motifs not only allowed fast identification of viral strains but also hinted at a direct impact of molecular-scale characteristics on infectivity, production/budding, and capacity of immune evasion. In particular, Raman assessments revealed profound differences in the molecular structure and symmetry of sulfur-containing amino acids, altered hydrophobic interactions of tyrosine residues at the virion–environment interface, markedly different “vibrational fractions” of RNA purines and pyrimidines, and dissimilar protein secondary structures. These aspects are discussed in some detail in the following.
S-Containing Amino Acids and Virus Production
The bold differences in molecular symmetry of S-containing amino acids enabled a swift spectroscopic identification of the six Omicron subvariants and positioned Raman spectroscopy as a fundamental tool in real-time visualization of spatiotemporal viral evolution at the molecular scale. The possibility of quantifying the fractional balance between amino acid residues and their rotamers directly reflects the genomic interplay with the cellular environment in which each specific viral strain developed. The thiol side chain of cysteine indeed participates as a nucleophile in a number of environmentally driven enzymatic reactions.101 Since nucleophilicity measures how rapidly molecules with lone pairs of electrons can react in nucleophilic substitution reactions, larger amounts of cysteine reflect higher propensity to both palmitoylation102 and protein folding (with the subsequent formation of stable intrachain disulfide bonds).103,104 In the study by Lopez et al.,101 the cysteine residues of the coronavirus envelope were found to be conspicuously prone to palmitoylation and were given functional significance upon hinting at their location on the carboxy side of the long hydrophobic domain. The replacement of cysteine residues with alanine residues, which cannot be palmitoylated, did not lead to impairment of viral entry. However, this amino acid substitution significantly reduced the viral yield, namely, the cumulative number of progeny viruses produced by a single cell. It was concluded that cysteine residues and their palmitoylated post-translational modification are functionally important for virus production. In this study, Raman fingerprints of palmitoylation were observed in all subvariants as relatively strong CH2 scissoring and CH2/CH3 deformation signals in the interval 1440–1500 cm–1. An exception to this trend was the BA.1.18 subvariant, for which fingerprints of palmitic acid oxidation to form molecules containing strong C=O and C=C–H spectroscopic features (at ∼1725 and 3075 cm–1, respectively) could be observed. BA.1.18 and BA.5 were the two Omicron subvariants highest in cysteine-to-methionine ratio, RC/M (cf. Figure 2), followed by BA.2. A conspicuously low RC/M value and/or a lack in cysteine palmitoylation in all other subvariants suggest higher protein instability (with no lowered transport), namely, a reduced viral yield (although without reduction in the capacity of entry) as compared to BA.1.18, BA.5, and to a lesser extent to BA.2. The importance of palmitoylation in virus assembly and budding has widely been reported in previously published literature, as a required step to target the protein-to-lipid rafts that allow virions’ assembly and budding.105−107Figure 12a schematically shows how S-palmitoylation occurs through the covalent attachment of 16-C saturated fatty acids to the cysteine residues of membrane proteins via thioester linkages,108 and how such a reversible modification can promote protein trafficking between membrane compartments and protein stability by adding fatty acid chains to the cytosine-rich motif of the S-protein.109,110 According to this mechanism, multiple cysteine clusters in the motif can support the association of proteins to lipid rafts during viral membrane fusion and virion assembly.111−115
Figure 12.
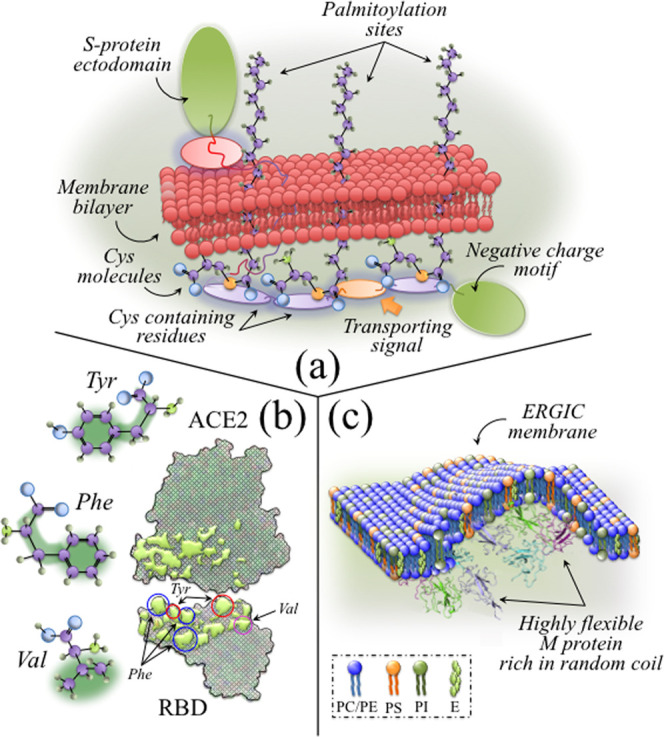
(a) Schematic draft of S-palmitoylation at the covalent attachment of 16-C saturated fatty acids to cysteine residues of membrane proteins via thioester linkages;108 (b) hydrophobic docking sites at the RBD/ACE2 interface (reproduced from ref (124); Val is an abbreviation for valine residue; green areas in the drafts of amino acid residues refer to the hydrophobic areas of the molecules); and (c) schematic draft showing the role of highly flexible and disordered M protein complexes in enhancing membrane bulging (highly flexible M protein drawing is from ref (146)).
Hydrophobic Interactions of the Tyrosine Residue
As explained in the Results section, the intensity ratio of the Fermi doublet at ∼858 and 836 cm–1 is diagnostic of the H-bonding environment around the tyrosine units; the lower the ratio, the higher the environmental pH and the more hydrophobic the surrounding environment.71,116−118 The significant differences detected for different subvariants (cf. Figure 3) revealed that the environment at the surface of the Omicron virions during its temporal evolution has gradually shifted toward alkaline and, thus, more hydrophobic states. It has been reported119 that viral mutations reversing the hydrophobic or hydrophilic performance of residues in the spike receptor-binding domain change the geometric complementarity between antibodies and virions at the binding sites. This in turn leads to breakthroughs in infectivity due to an enhanced ability to escape antibody recognition of the receptor-binding domain.120 In order to understand the importance of surface interaction with environmental water, one should consider that the propensity to form a hydration shell saturates the H-bond formation at hydrophilic groups on the protein surface and greatly affects protein–protein docking.121,122 Because of strong hydrogen bonding interactions in the hydration shell, the molecular dynamics in the immediate surface of proteins (∼1 nm in thickness) is quite different from that in bulk water.121 The hydrophilic groups at the protein surface are normally hydrogen-bonded with the surrounding water molecules; this prevents H-bond donors at the protein surface from bonding with H-bond acceptors of another protein. In other words, the preliminary arrangement of H-bond donor sites, which is in turn decided by the combination of hydrophilic and hydrophobic sites, determines the possible protein–protein docking sites exactly as “a key inside a lock”. The hydrophilic groups at the protein surface represent the hydrogen shell of a protein, and dipole–dipole attractions between CO and NH groups in the protein chains act as pairings of H-bond donors and acceptors.120 At the interface between the receptor-binding domain and ACE2 complex, attraction or repulsion by hydrophobic and hydrophilic groups in water plays the main role in increasing or decreasing binding affinity between the two proteins. According to Pace et al.,123 altering the number of hydrophobic side chains exposed to water indeed represents one of the most crucial driving forces for the docking process. Li et al.124 indicated a hydrophobic-interaction-based mechanism as the main responsible for the enhanced affinity of the SARS-CoV-2 S-protein to Angiotensin-Converting Enzyme 2 (ACE2) as compared to SARS-CoV. Hydrophobic portions in the hydrophilic side chains of the SARS-CoV-2 S-protein, which include tyrosine, phenylalanine, and valine residues, were indicated as the docking sites directly involved with the enhancement of the S-protein/ACE2 affinity. Note that we indeed found an indirect confirmation of this process (with the exception of the BA.4 subvariant) in the plot of phenylalanine relative intensity, which decreased with increasing deprotonation,125 as a function of tyrosine ratio, RF (cf. Figure 6d). Our Raman study is also in agreement with the increased affinity toward ACE2 found by Pascarella et al.17 as a result of an increased positive electrostatic potential level in the spike RBD and by Lupala et al.18 in comparison with the Delta variant.
From the above reasoning, one could interpret the evolution of proteins’ surface hydration shell in Omicron subvariants as an attempt by the virus to “design” a surface structure that, by shifting surface-protonation toward alkaline and more hydrophobic conditions, features an increasing docking capacity while escaping the effect of vaccination by altering the “lock” configuration with respect to monoclonal antibodies. Hydrophobic docking sites at the RBD/ACE2 interface are schematically represented in Figure 12b.124,126
Vibrational Fractions of RNA Purines and Pyrimidines
In the Results section, the possibility has been shown to locate fingerprint bands of RNA purines and pyrimidines in the Raman spectrum of Omicron subvariants by exploiting differences in both ring structures and functional groups linked to them. Remarkably, the set of relative intensity values of such fingerprint signals were peculiar to each subvariant (cf. Figure 4), despite the fact that the corresponding base fractions computed from the genome database were conspicuously homogeneous across all subvariants (except for the subvariant BA.2.75 with variations <16%; cf. Table 1). As previously shown for other SARS-CoV-2 variants,33,34 the RNA Raman fractions do not generally match those obtained by genome analyses because of the combined effects of base-pairing, signal cross sections, and differences in viral nucleoproteins associated with preferential RNA sites.127 Such amplified differences in RNA-bases-related Raman signals is indeed a very fortunate circumstance since they enable viral speciation down to the subvariant level without the need of RNA sequencing and within a very short time of a Raman measurement (i.e., in the order of minutes).33,34,128
The interaction between RNA viruses and host innate and adaptive responses represents a quite complex issue whose underlying mechanisms are generally multifactorial and conspicuously hidden in the details of enzymatic chemistry. Based on bioinformatics studies,129,130 it has been suggested that RNA viruses are capable of adapting their genome to inhibit and divert the innate cell defenses beyond the process of merely coding it for developing structural and nonstructural proteins for virion assembly and genome replication. In the stream of these findings, one could postulate that also the RNA genome of Omicron subvariants possibly contains specific genes for escaping the immune response, interceptable in alternative reading frames. With this in mind, we looked at the Raman fractions of different RNA bases in different Omicron subvariants (cf. Table 1 and the RNA-base fractional values in the inset to Figure 4). An interesting finding was that the fractions of uracil detected by Raman spectroscopy in all Omicron subvariants were nearly twice or even higher than that measured in the Japanese ancestral strain (JPN/TY/WK-521)33,34 with the exception of the BA.1.18 subvariant, which showed a comparable value (10.4 vs 11.2%). Note that the same trend of uracil enrichment was observed in Raman studies of two α subvariants (QK002 and QHN001)33 and for the Delta variant (TY11-927).34 In a recent study by Kosuge et al.,128 disproportionate point mutation to uracil was reported for SARS-CoV-2 strains successive to the Wuhan strain. The uracil point mutation was related to apolipoprotein B mRNA editing-enzyme catalytic polypeptides (APOBECs), an enzymatically driven process that increases the release of two main proinflammatory molecules produced by macrophages (i.e., tumor necrosis factor-α and interleukin-6). Genome data in Table 1 clearly show that the genome of all Omicron subvariants is indeed enriched in uracil at the expenses of cytosine. As far as α and Delta variants/subvariants were concerned, also Raman fractional data of RNA bases confirmed the increase in uracil fractions (with respect to the Japanese ancestral strain) as systematically accompanied by a nearly equivalent decrease in cytosine33,34 in agreement with the bioinformatics study in ref (131). However, when comparing the Raman fractional data of the Omicron subvariants, the inverse proportionality between the uracil and cytosine fractions was never preserved, which suggests the presence of multiple base-pairing effects and or other enzymatic processes overlapping the APOBECs’ effect in the Omicron mutation. No other obvious trend could be found with evolving Omicron mutations except for the fraction of guanine base, which remained conspicuously low and constant across all subvariants. Although the above observations do not allow a final clarification of whether Raman assessments could unequivocally locate the contribution of host RNA editing to point mutations and immune reactions, they provide further confirmation on how swiftly the Raman technology could allow genome comparisons to track possible RNA editing effects.
Protein Secondary Structures
The rapid evolution of SARS-CoV-2 variants and their rapid spreading around the world has triggered research on viral protein disorder in search for correlations with infection pathways and in support of practices of rational drug design.132,133 Disordered regions in viral proteins are linked to enzyme catalysis and strongly correlated to viral infectivity and pathogenicity because they provide proteins with the ability to easily and promiscuously bind to host proteins.132 Therefore, understanding the secondary structure of the SARS-CoV-2 virus proteome is considered a key point in evaluating viral infectivity and in designing future treatments for COVID-19.
Protein disorder is generally only indirectly monitored by means of several experimental methods, which include X-ray diffractometry, nuclear magnetic resonance, Raman, and vibrational circular dichroism spectroscopies, as well as hydrodynamic measurements.134 The above methods detect different aspects of protein disorder, which ultimately result in different definitions. In the absence of a commonly agreed definition of protein disorder, one could adopt its thermodynamic definition for polypeptide chains, usually termed the random coil structural state. In this state, the polypeptide experiences all possible degrees of freedom within the conformational space.135 It is thus quite intuitive that disordered proteins could allow for more intramolecular interfaces and interaction partners per unit volume,135,136 while allowing for a flexible regulation with fewer types of linear motives.137 Raman spectroscopy can reveal the secondary structure of intrinsically disordered proteins and peptides by displaying a set of different subbands associated with α-helix, β-sheet, random coil, and two rotameric β-turn structures (cf. Figure 5).29,30 This Raman spectral zone, referred to as the Amide I zone, revealed an evolution in the proteins’ secondary structure toward higher random coil fractions for all subvariants developed after the BA.1.18 one. This structural proteome characteristic is a flag for higher binding propensity and represents one of the important factors leading to enhanced infectivity in all Omicron subvariants post-BA.1.18 (Figures 5 and 6b), which showed significantly higher random coil fraction (28.5–31.0%), with the exception of the BA.4 (i.e., showing an intermediate value of 20.3%). Although incapable of distinguishing among nucleocapsid structural/nonstructural and spike proteins, Raman evaluation of the protein secondary structure revealed an increasing level of structural disorder in the SARS-CoV-2 Omicron subvariants developed after BA.1.18. Following the analyses reported in ref (41), which compared SARS-CoV-2 Delta and Omicron variants, and those given in refs (46−48), which are more general to coronaviruses, M and N proteins (rather than the S one) incorporate the largest fractions of misfolded proteins in SARS-CoV-2. Accordingly, the higher random coil fractions observed in the Omicron subvariants developed after BA.1.18 should mainly be interpreted as a virus structural evolution in nucleocapsid proteins. Such evolution could be the origin of enhanced viral replication and budding propensity rather than being directly related to an enhanced RBD/ACE2 docking, which instead is mainly promoted by the presence of hydrophobic sites in S-protein aromatic residues (cf. Figure 12b).
In a recent review,138 Jackson et al. described in detail the mechanisms of infection by SARS-CoV-2. After cytoplasm entry, the viral replication process included the formation in the perinuclear zone of new membranous structures, referred to as replication organelles and also observed by high-resolution electron microscopy.139 These newly formed structures, which likely originate from the endoplasmic reticulum (ER), incorporate viral replication complexes by sequestering them from cellular molecules. Viral structural proteins are then transferred to the ER–Golgi intermediate compartment (ERGIC), where virus assembly and budding occur.140,141 Although the precise mechanism of budding into the ERGIC lumen is still under debate, the role of M proteins in facilitating viral budding into the ERGIC has been clarified. According to molecular dynamics simulations of M protein complexes, Thrasher Collins et al.142 examined the mechanism by which proteins induced membrane curvature during the budding process. Their study showed how isolated M proteins first created bulges in the cell membrane and then multiple M protein dimers acted together to produce larger amounts of membrane curvature mainly through protein/lipid interactions. M protein/lipid contacts were shown to preferentially occur at phosphatidylinositol (PI) and phosphatidylserine (PS) lipid sites, suggesting that the M proteins dynamically reconfigure the ERGIC membrane to create an optimum lipid environment for curvature to occur. Note that the capacity of PI to induce membrane curvature even at low concentrations (through the action of its five potential H-bonding ring sites) is a well-known characteristic,143,144 supporting the thesis that its affinity for M protein dimers is a main player in initiating the membrane evolution into a curved state. Finally, note that both neutron scattering and X-ray diffraction studies have pointed to the high flexibility of the SARS-CoV-2 nucleocapsid protein as a key factor in its interaction with RNA and in viral replication.145,146 Building upon this understanding, we suggest that a highly disordered M protein structure, as observed here by Raman spectroscopy, might trigger a more efficient membrane bulging by extending with their high flexibility the probability of linking at PI and PS headgroups. A draft showing the role of highly flexible and disordered M protein complexes in enhancing membrane bulging is schematically shown in Figure 12c.
Table 2 summarizes the above metabolomic and proteomic considerations from which it emerges how Raman spectroscopy could contribute to rationalizing the chemistry of SARS-CoV-2 structural evolution, as modulated by the interplay between genomic and founder-effect-driven kinetics. A systematic follow-up of Raman spectroscopic classifications could allow future identifications of the metabolic rewiring of specific strains, as their infective spread proceeds through the evolution of different variants and subvariants.
Table 2. Values (in Brackets) of the Three Main Spectroscopic Parameters Obtained from Raman Characterizations, Namely, RC/M, RF, and Rrc, as Measured for Different Omicron Subvariant Samplesa.
| RC/M | RF | Rrc |
|---|---|---|
| palmitoylation probability | surface hydrophilicity | membrane flexibility |
| BA.1.18 (0.66) | BA.1.18 (2.22) | XE (0.47) |
| BA.5 (0.66) | BA.4 (1.83) | BA.5 (0.44) |
| BA.2 (0.47) | BA.2 (1.76) | BA.2 (0.37) |
| XE (0.14) | XE (1.55) | BA.2.75 (0.35) |
| BA.2.75 (0.14) | BA.5 (1.42) | BA.4 (0.16) |
| BA.4 (0.10) | BA.2.75 (1.16) | BA.1.18 (0.04) |
High values for these spectroscopic ratios represent high palmitoylation probability, high hydrophilicity at the virions’ surface, and high membrane flexibility and bulk capacity, respectively.
Raman Hints of Evolutionary Direction for Omicron Subvariants
The large numbers of Omicron sublineages that continue to emerge during the COVID-19 pandemic represent a potential threat for vaccine breakthrough infections and reinfections. While it is now clear that α and Delta variants developed distinct paths of infectivity and immunological escape profiles as compared to ancestral SARS-CoV-2,147 the evolutionary direction of the Omicron sublineages yet remains a matter of discussion.148−150 Notwithstanding the fact that surveillance and timely sequencing of novel Omicron subvariants remain the main tools to unfold the “strategy” of current and future Omicron sublineages, Raman spectroscopy could contribute to this task by providing an agile and fast approach to analyze sublineage multiplications. The present study provides some evidence in support of this assertion.
So far available clinical data suggest that Omicron subvariants spread faster and evade antibodies more effectively than earlier SARS-CoV-2 variants,151,152 which in turn leads to increased chances of reinfection and infection breakthroughs in the immunized population. Omicron BA.1.18 was reported to replicate ∼70 times faster than the Delta variant in the bronchus, but it was less severe in the human lung.153 Statistical analyses showed an effective reproduction number for BA.2 1.4 times higher than that of BA.1.18, together with antigenicity considerably different from that of BA.1.18.148 The Omicron variants BA.4 and BA.5 were reported to be more transmissible and resistant to immunity generated by previous variants (including BA.1.18) and most monoclonal antibodies.149 Peacock et al.154 reported about an increase in viral copy number in Omicron-infected human nasal airway epithelial cells via the endosomal route. Regarding the origin of the replication efficiency of Omicron, it is known that SARS-CoV viruses can employ two different routes for entering the host cell: membrane fusion and endocytosis.155 In the latter case, which is reported to be the preferred one by the Omicron variant in infecting nasal epithelial cells,154 cysteine residues play a fundamental role. The conservation of cysteine residues is thus fundamental in virus production either as targets for palmitoylation and/or to promote protein–protein interactions through disulfide bonding.101 In comparing previously collected34 Raman spectra of the Delta variant and ancestral Japanese strain with the Omicron ones collected in this study, we notice that the cysteine-to-methionine fractions of the Omicron subvariants BA.1.18, BA.2, and BA.5 are 1.5–2 times higher. They also show Raman fingerprints of palmitoylation, which could provide a rationale for the high viral replication efficiency of these subvariants.
Barozi et al.150 used a combined approach of post molecular dynamics and dynamic residue network analyses to characterize the Omicron sublineages BA.1.18, BA.2, BA.3, and BA.4. Those researchers focused their analyses on the interaction between the spike protein and the human angiotensin-converting enzyme 2, essential for infection and, at the same time, a target for neutralizing antibodies. The results showed that Omicron sublineage mutations altered the protein physicochemical properties and changed the proteins’ conformal flexibility to enhance antibody escape while yet promoting an efficient binding interaction, thus increasing the overall virus transmissibility. Moreover, a clear hint for the antibody escape mechanism was the diverse conformational and dynamic nature of the receptor-binding domain and the greater residue fluctuation of the antigenic hot spots binding to neutralizing antibodies in Omicron sublineages. At the same time, a comparison of computed binding energies and the flexibility of the receptor-binding motif led to the conclusion that the latter factor was indeed the main cause of a favored interaction between the spike receptor-binding domain and the host receptor. In agreement with a number of additional studies,156−160 the computational results in ref (150) showed how the evolutionary Omicron sublineages progressively underwent specific mutations in a trade-off between binding affinity for ACE2 (lower than previous variants) and antibody escape capacity (higher than previous variants). From a different biophysical perspective, the present Raman study supported the hypothesis that the main path in the evolutionary behavior of the Omicron sublineages consisted of a trade-off among binding affinity with the receptor, immune escape attitude, and viral replication/budding. Such a trade-off was made possible mainly by three structural variations, namely, the fraction of cytosine residues, number of hydrophobic RBD sites, and degree of disorder in the M protein structure as landmarks of palmitoylation potential, docking ability, and structural flexibility, respectively. The shift toward alkaline pH, as detected at the surface of Omicron virions and sensed by the tyrosine doublet (cf. Figure 3) was the Raman flag in visualizing the evolutionary development of Omicron subvariants; shifts toward alkaline pH at the virion surface strongly promote the formation of both hydrophobic sites in proteins119,120 and highly flexible random coil structures in M protein.161,162 The shift of the virions’ surface-protonation toward alkaline conditions in successive Omicron subvariants should thus be interpreted as a main factor in their evolutionary development.
According to the above considerations, we selected three parameters derived from the Raman spectra of different Omicron subvariants in order to describe their evolutionary direction as follows: the degree of M protein structural order, RO, related to conformal rigidity and, thus, inversely proportional to the efficiency in virus multiplication and budding; the degree of hydrophilicity, RH, related to environmental pH and to the propensity to form RBD binding hydrophobic sites (and inversely related to the capacity of immune escaping); and the degree of palmitoylation, RP, related to the fraction of cysteine residues and thus representing the propensity for viral multiplication and budding. The parameters RO, RH, and RP were obtained by normalizing the ratios 1 – Rrc, RF, and RC/M, respectively (as defined in the Results section) to fractional percents. Figure 13 shows a triangular graph that summarizes the evolutional characteristics of the investigated Omicron subvariants based on the three selected Raman ratios. The plot shows that for a normalized virological impact of the Omicron variant, the post-BA.1.18 subvariants flipped their infective “strategies” between reproduction/budding and binding enhancements, with BA.4 and XE turning further toward an enhanced immune escape.
Figure 13.
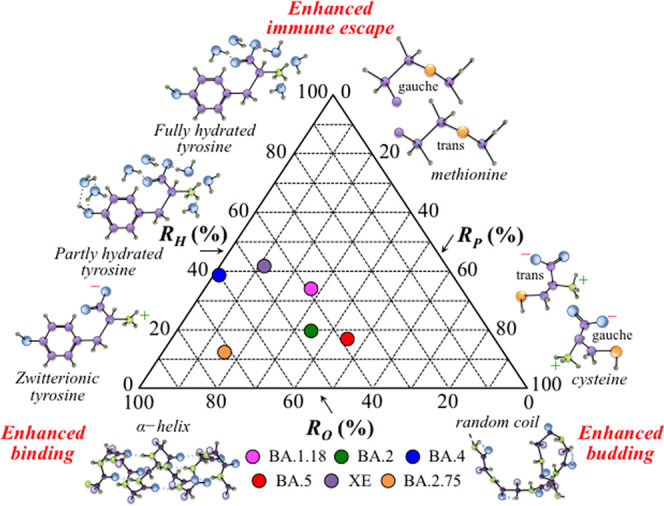
Triangular plot summarizing the evolutional characteristics of the investigated Omicron subvariants based on the three selected Raman ratios, RO, RH, and RP.
COVID-19 Management Using Raman Barcodes
A timely sequencing of positive samples has so far represented the key for discovering possible divergent variants in the SARS-CoV-2 pandemic. Given the possible evolution of Omicron subvariants in patients with chronic infection and immunodeficiency,163 a comprehensive and prompt surveillance of newly evolved variants in COVID-19 patients is key for timely discovering possibly dangerous variants.164,165 In previous studies,33,34 we have proposed the use of a barcode built on the Raman spectrum of different SARS-CoV-2 variants and subvariants in order to achieve an efficient electronic record-keeping and to increase tracking accessibility through apps and user-friendly software. In this study, we have applied the previously developed Raman barcode algorithm to a series of Omicron subvariants and showed that this approach enables one to overcome PCA deficiencies and to successfully speciate different strains belonging to the same variant.
The idea of using a Raman barcode was proposed for the first time in 2016 by Lawson and Rodriguez.166 These researchers proposed a Raman barcode algorithm to recognize counterfeit drug products. The concept behind their Raman barcode identification of drugs was to compare known peaks in the Raman spectra of active pharmaceutical ingredients, which are expected to be in the drug, to the peaks actually observed in the Raman spectrum of the drug. The transformation of the Raman spectra of the drug product and its active pharmaceutical ingredients into respective barcodes was obtained by assigning zero intensity to every spectral frequency except for the frequencies that correspond to Raman peaks. A comparison of the percentage of nonzero-overlap between the expected ingredients’ barcode and the drug product barcode then allowed verifying the identity of active products. The main usefulness of the Lawson and Rodriguez barcode method resides in the possibility to qualitatively identify active products in pharmaceutical products without the need for a comprehensive spectral library for all drug products. Although the so-constructed barcodes are strong indicators of the molecular functional groups present in a given drug substance and thus enable a prompt detection of counterfeits, the Raman relative intensity information is lost in barcoding and hence all spectroscopic insights.
In an effort to substantiate the Raman approach, a method was also proposed to extract a virus sample from the nasal swab of a patient (cf. Figure 9) and to analyze it by Raman spectroscopy in comparison with conventional genomic analyses. The output of Raman analyses was partly consistent with that of genomic analyses; however, the Raman approach could not clearly distinguish between BA.5 and XE subvariants, definitely hinting at the former (in agreement with genomic analyses) only in one of two selected spectral zones (cf. Figures 10 and 11). It should be noted that both Raman and genomic analyses, although performed on the same swab sample, might, in principle, be affected by sample inhomogeneity. Moreover, surface-chemistry interactions between virions and magnetic beads definitely have an impact on the virus structure at the molecular scale and call for additional elaborations of the Raman method. In summary, the present proof-of-concept Raman experiments on a swab sample showed the feasibility of the Raman barcode approach and also its limits and needs for ameliorating the overall analytical procedure.
On the positive side, the Raman barcode approach applied in this study, which was tailored on a machine-learning-construed spectral deconvolution procedure, preserved metabolite information on the virions’ structure, thus linking fundamental features of physical chemistry to the evaluation of virions’ structural characteristics and enabling possible predictions of its evolutionary trends. Once the above limitations could be removed, Raman barcodes will provide a way to decrypt spectroscopic data into easily readable information through appropriate apps in order to allow a prompt classification of on-site collected patients’ specimens without losing metabolomic and proteomic information. Upon automatic linking of specimen barcodes to clouds, a capillary network could be built for real-time tracking and efficient management of COVID-19 pandemics.
Conclusions
Raman spectroscopic analyses of six SARS-CoV-2 Omicron subvariants enabled us to identify putative biomarkers and to provide functional interpretations hypothesizing possible connections between metabolites and the evolutional trends of the virus. Raman analyses revealed the following metabolomic and proteomic characteristics.
-
(i)
The two subvariants BA.1.18 and BA.5 were the richest in cysteine, while all other subvariants showed conspicuously low cysteine fractions. This structural characteristic hints to a lowered protein stability and reduced virus production as compared to BA.1.18 and BA.5.
-
(ii)
The Omicron subvariants evolved toward a surface structure turning surface-protonation conditions into alkaline and more hydrophobic ones; a physicochemical shift likely “designed” to increase RBD/ACE2 binding affinity (through the formation of hydrophobic amino acid sites) while yet featuring immune escape.
-
(iii)
All six Omicron subvariants showed quite different “Raman genomes”, despite their base fractions computed from the genome database being conspicuously homogeneous. Such amplified differences in RNA Raman signals enable viral speciation down to the subvariant level in time frames of minutes without the need of RNA sequencing.
-
(iv)
Significantly high fractions of disordered regions in viral (mainly M) proteins were a landmark for enhanced enzymatic catalysis in all Omicron subvariants and strongly correlated to viral infectivity since providing proteins with the ability to promiscuously bind to host proteins and enhance budding.
Taken together, the above bullet points give novel insight into the function and evolution of the Omicron variant, which is crucial for drug design tackling future Omicron sublineage mutations.
As a proof-of-concept experiment, the Raman method was also applied to a patient’s swab sample to verify its feasibility in actual medical practice. The results of these additional experiments not only gave encouraging results but also showed the need for additional elaboration of the procedure for promptly extracting a measurable virions’ concentration from swab samples.
The availability of virus metabolic profiles by Raman spectroscopy and their encryption into Raman barcodes pave the way to biological interpretations and to a prompt identification of key biochemical pathways that could speed up the development of vaccines and antiviral drugs. Raman metabolomic snapshots of viruses complement the information obtained from genetics by allowing association of infective physiopathological conditions to specific metabolic fingerprints. Structuring a multifaceted metabolomic approach in virology into organized knowledge will likely promote the use of Raman spectroscopy in precision medicine and wellness.
Methods
Preparation of SARS-CoV-2 Subvariants for Raman Analyses
Experiments involving the SARS-CoV-2 virus were conducted in a biosafety Level 3 (BSL-3) biocontainment facility according to BSL-3 work practices. The investigated SARS-CoV-2 viral stocks of the Omicron subvariants were as follows: TY38-873, TY40-385, TY41-703, TY41-702, TY41-686, and TY41-716 (henceforth simply referred to as BA.1.18, BA.2, BA.4, BA.5, XE, and BA.2.75, respectively). All studied subvariants were obtained from the Japanese National Institute of Infectious Diseases after performing detailed genomic characterizations on a series of their biological replicates.
VeroE6/TMPRSS2 cells purchased from JCRB Cell Bank (VeroE6 cells expressing the transmembrane serine protease TMPRSS2) were cultured in growth medium (GM) Dulbecco’s modified Eagle’s minimum essential medium (DMEM) (NACALAI TESQUE, INC., Kyoto, Japan) supplemented with G 418 disulfate aqueous solution (1 mg/mL), penicillin (100 units/mL), streptomycin (100 lg/mL), and 5% fetal bovine serum. The maintenance solution was DMEM supplemented with penicillin, streptomycin, and 0.5% fetal bovine serum. The VeroE6/TMPRSS2 cells were seeded in T-175 flasks (Falcon) and grown in GM upon maintaining them at 37 °C in an atmosphere of 5% CO2.
Viruses were infected at multiplicity of infection (MOI) of 0.01 and propagated in the VeroE6/TMPRSS2 cells at 37 °C for 3–4 days. Then, 90 mL of the culture supernatants was harvested for each variant and centrifuged at 4800 rpm for 20 min. The supernatants for each variant were ultracentrifuged at 24000 rpm, 3 h at 4 °C. After removing the supernatants, the pellets were resuspended in 50 μL of PBS (−), put on glass-bottom dishes (same as above), and air-dried. After fixation with 4% paraformaldehyde for 20 min at room temperature and washing with distilled water twice, the pellets were air-dried again. They were kept in a dry environment at 4 °C until Raman analyses were performed. A virus concentration corresponding to the order of 109 TCID50 was concentrated on each glass bottom (area of 38.5 mm2; 7 mm in diameter).
SARS-CoV-2 particles from COVID-19 donors’ nasopharyngeal swab samples of COVID-19 donors were collected with informed consent. This study was conducted according to the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of University Hospital, Kyoto Prefectural University of Medicine and Kyoto Prefectural Rehabilitation Hospital for Mentally and Physically disabled (Project Identification Code: ERB-C-2587 and RBMR202202). Extraction of the virions from the swab sample was possible upon exploiting a standardized procedure, which employed commercially available magnetic beads (Dynabeads TM Intact Virus Enrichment; Thermo Fisher Scientific, Waltham, MA). The magnetic beads were ∼2 μm in diameter and consisted of polymer-coated Fe2O3; they were optimized for SARS-CoV-2 and used according to the manufacturer’s recommendation.
Viral Whole Genome Sequencing and Data Processing
Human nasopharyngeal swab samples, barcodes 01 (acute phase) and 02 (remission phase), were obtained from a COVID-19 patient whose nasal swab was also used for the Raman analysis. These samples were collected from the patient on different days (September 16, 2022 and 22, 2022). Whole viral genome sequences of SARS-CoV-2 were determined by using the GridION next-generation sequencer (Oxford Nanopore Technologies, U.K.).
Samples were nasopharyngeal swabs collected at UTM (Copan, Italy) and stored at −80 °C. Total viral RNA was extracted using the QIAamp Viral RNA Mini Kit (QIAGEN, Germany). It is an amplicon sequencing analysis by using primers specified by the National Institute of Infectious Diseases. Reverse transcription and MultiplexPCR were performed using the NEBNext ARTIC SARS-CoV-2 Companion kit (New England Biolabs) and Gene Amp PCR system9700 (Thermo Fisher Scientific). Cleanup and measurement of the amount of DNA were performed using the AMPure XP (BECKMAN COULTER) and Qubit 4 Fluorometer (Thermo Fisher Scientific).
Library preparation was performed using the NEBNext RTIC SARS-CoV-2 Companion kit (New England Biolabs) and the Ligation Sequencing kit SQK-LSK109 (Oxford Nanopore Technologies, U.K.).
All of these protocols were performed according to the manual of the National Institute of Infectious Diseases (https://www.niid.go.jp/niid/images/lab-manual/SARS-CoV2_genome_analysis_manual_Nanopore_NEB_ver_1_6_220127.pdf).
For the BAM files generated, we conducted variant calling, filtering out of variants with QUAL < 50, DP < 30, or AD for REF ≥ 30 and AD for ALT ≤ 5, and building of FASTA-formatted consensus sequence, by using BCFtools ver.1.14. In addition, eight whole genome sequence data (GenBank Accession Number MN908947.3 and GISAID Accession Numbers EPI_ISL_7418017, EPI_ISL_7571618, EPI_ISL_9595859, EPI_ISL_14328510, EPI_ISL_13969765, EPI_ISL_13241868, and EPI_ISL_12703378) were retrieved from NCBI (https://www.ncbi.nlm.nih.gov/) and GISAID (https://gisaid.org/) databases.
Real-Time PCR
The analyzed samples were nasopharyngeal swabs collected at the UTM (Copan, Italy). Real-time PCR was performed using the COBAS 6800 system and the COBAS SARS-CoV-2 kit (Roche, Switzerland), and the Ct values of E-gene and ORF-1a/b were calculated.
Phylogenetic Analyses
Multiple sequence alignments of SARS-CoV-2 nucleotide sequences were implemented in MEGA ver. 11.0.13 (Tamura et al., 2021). A phylogenetic tree was constructed based on the maximum-likelihood (ML) method using the Hasegawa–Kishino–Yano substitution model (Hasegawa et al., 1985) with γ-distributed rate variation across sites, estimated by a best-fit substitution model in the MEGA. Nearest-neighbor-interchange (NNI) was applied as the ML heuristic method. Bootstrap analysis was conducted with 1000 replications. Clade classification of each SARS-CoV-2 sequence was conducted in Nextclade v2.14.1 web tool (https://clades.nextstrain.org/).
Raman Spectroscopic Experiments
In situ Raman experiments on the above series of viral strains were conducted with a spectrometer (LabRAM HR800, Horiba/Jobin-Yvon, Kyoto, Japan) set in confocal mode. This specially designed equipment used a holographic notch filter to concurrently provide high-efficiency and high-resolution spectral acquisitions. The wavelength of the incoming laser was 785 nm, with the laser source operating with a power of 10 mW (at the sample surface). The spectrum of a sample consisting of only paraformaldehyde was preliminarily recorded using exactly the same spectroscopic measurement conditions. Under red light, the paraformaldehyde used for fixation appeared only as a broad background, which could easily be eliminated according to a standardized procedure, as explained in the next section. The spectral resolution of ∼1 cm–1 was achieved upon analyzing the Raman scattered light by a double monochromator connected with an air-cooled charge-coupled device (CCD) detector (Andor DV420-OE322; 1024 × 256 pixels); the grating used in the spectrometer had a resolution of 1800 g/mm. The acquisition time of a single spectrum was typically 10 s. However, several consecutive acquisitions were made at the same spot to minimize noise. The laser spot was ∼2 μm as focused on the sample through a 50× optical lens. In collecting average spectra, sets of 10 spectra were collected at different locations over areas of ∼2 mm2 for each strain. Sets of 10 spectra per subvariant were used for statistical analyses, while their respective averages were used for discussing differences among different subvariants. In Raman measurements, agglomerations of virions were first spotted under a microscope into enclaves of paraformaldehyde (typically 50–100 μm in diameter); the agglomerated virions appeared as black or gray “dust particles” with variable focal positions. Then, the focal plane abscissa of the micrometric laser spot was shifted along the z-direction perpendicular to the sample plane in order to maximize the Raman signal. Preliminary x/y-scans were also needed to exactly position the laser spot on the virion agglomerates in order to maximize their Raman signal, with respect to the formaldehyde broad background. The sets of 10 spectra selected for statistical analyses and to compute average spectra for each subvariant were always from different enclaves.
Raman measurements of virions extracted from nasal swab samples collected on magnetic beads were performed by means of the same equipment and under the same measurement conditions mentioned above. The only difference with the above procedure was the measurement temperature (4 °C vs room temperature). The cooling procedure was possible by employing a cooling stage specially designed for the Raman equipment and served to avoid heating of magnetic beads under the laser beam with subsequent disruption of the attached virions.
Treatment and Deconvolution of Raman Spectra
The raw Raman spectra were first subjected to a baseline subtraction procedure. This procedure was preliminarily optimized and standardized by comparing three different methods, namely, polynomial fitting,35 asymmetric least-squares method,36 and penalized spline smoothing based on vector transformation.37 Despite the fact that background removal by polynomial fitting (as determined by a least-squares criterion) is by far the conceptually simplest and most widely used procedure in Raman spectroscopy, the occurrence of background drifts, due to the presence of fluorescence background noise, requires using more complex algorithms. In the present treatment of virus Raman spectra, the overlap by the broad spectrum of paraformaldehyde introduced baseline shifts that differed at different locations or even at the same location for different wavenumber intervals. This required the application of a baseline subtraction procedure insensitive to noise morphology, as given in ref (36) or (37). Both algorithms were suitable and reliable for Raman analyses and gave very close results.
After baseline subtraction as described above, average spectra were deconvoluted into a series of Lorentzian–Gaussian subbands using commercial software (LabSpec 4.02, Horiba/Jobin-Yvon, Kyoto, Japan). In performing this deconvolutive procedure, a machine-learning approach was applied, which employed an in-house-built automatic solver, Sav(ν), exploiting a linear polynomial expression of Lorentzian–Gaussian functions, Vij(Δν, σ, γ), where ν, Δν, σ, and γ represent the Raman frequency, the shift in frequency from the band maximum, ν0, the standard deviation of each band component, and the full width at half-maximum of the Lorentzian component, respectively. An algorithm searching for the minimum value of the difference between the experimental and the fitted average spectrum (cf. subscript “av”) was then set, as follows
| 1 |
where the index i locates each compound in a series of n compounds contributing the overall spectrum, and the index j locates each Lorentzian–Gaussian subband of a series of m compounds in the Raman spectrum of each compound of an n series. Optimization was then launched with a computer program that selected a series of Lorentzian–Gaussian subbands from the deconvoluted spectra of preselected compounds included in a large database of more than 60 key biomolecules in aqueous solution or in solid state according to the chemical and structural peculiarities of virions.28 Upon using spectral subbands of elementary compounds from the library, the automatic algorithm searched for the closest matches to the experimental (average) spectra according to two main criteria: (i) assigning spectral positions (ν0) and full width at half-maximum (σ and γ) values for specific subbands from each elementary compound within ±3 cm–1 (i.e., to account for the experimental error of the spectrometer and to include the possibility of alterations of the molecular structure in the virion structure) and (ii) preserving relative intensities (βij) in the spectra of the elementary compounds. The conditions imposed on band positions, relative intensity, and bandwidths provided a series of mathematical constraints that led to a unique solution. In addition to finding the closest match with the experimental spectrum and identifying the compounds contributing each deconvoluted subband, the above computational procedure enabled locating subbands with single-reference-molecule sourced signal intensity (>90%). Such subbands were taken as spectral fingerprints of the respective molecules.
Chemometric Analysis and Construction of a Raman Barcode
Statistical analyses of the Raman data sets were performed according to principal component analysis (PCA) on 10 selected spectra per subvariant acquired at different locations on each sample. PCA analyses were conducted by means of the Origin software platform (OriginLab Co., Northampton, MA) and displayed by means of a set of “summary indices”, referred to as principal components PC1 and PC2.
Barcodes were generated from deconvoluted Raman spectra using the sequences of Gaussian–Lorentzian subbands. Subband sequences were converted into barcodes by means of an algorithm that assigned to each band a line with thickness equal to 1/50 of the subband full width at half-maximum and a distance from the successive line proportional to the subband area. The Raman barcode could enable efficient electronic record-keeping and increase data accessibility by storing the structural characteristics of different subvariants to be suitably converted into informative text through apps and user-friendly software.
Acknowledgments
The authors thank Messrs. Y. Yamamoto and N. Tomari from Kyoto Municipal Institute of Industrial Technology and Culture for their support in the use of the LabRAM HR800 Raman device. The Strategic Foundational Technology Improvement Support Operation 2019 of the Japanese Government, a Grant-in-aid from Tokuyama Science Foundation 2021, the Japanese Association for Dental Science: Sponsored research 2022-A-2, and the AMED (Grant Number 20he1122006j0001) supported this research.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.3c00312.
Maximum-likelihood (ML) tree of SARS-CoV-2 whole genome sequences (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Hardy M. A.; Wright B. A.; Bachman J. L.; Boit T. B.; Haley H. M. S.; Knapp R. R.; Lusi R. F.; Okada T.; Tona V.; Garg N. K.; Sarpong R. Treating a global health crisis with a dose of synthetic chemistry. ACS Cent. Sci. 2020, 6, 1017–1030. 10.1021/acscentsci.0c00637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Marca A.; Capuzzo M.; Paglia T.; Roli L.; Trenti T.; Nelson S. M. Testing for SARS-CoV-2 (COVID-19): A systematic review and clinical guide to molecular and serological in-vitro diagnostic assays. Reprod. BioMed. Online 2020, 41, 483–499. 10.1016/j.rbmo.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deming M. E.; Michael N. L.; Robb M.; Cohen M. S.; Neuzil K. M. Accelerating development of SARS-CoV-2 vaccines - the role for controlled human infection models. N. Engl. J. Med. 2020, 383, e63 10.1056/NEJMp2020076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinner C. D.; Gottlieb R. L.; Criner G. J.; Arribas Lopez J. R.; Cattelan A. M.; Soriano Viladomiu A.; Ogbuagu O.; Malhotra P.; Mullane K. M.; Castagna A.; Chai L. Y. A.; Roestenberg M.; Tsang O. T. Y.; Bernasconi E.; Le Turnier P.; Chang S.-C.; SenGupta D.; Hyland R. H.; Osinusi A. O.; Cao H.; Blair C.; Wang H.; Gaggar A.; Brainard D. M.; McPhail M. J.; Bhagani S.; Ahn M. Y.; Sanyal A. J.; Huhn G.; Marty F. M. Effect of remdesivir vs standard care on clinical status at 11 days in patients with moderate COVID-19. JAMA 2020, 324, 1048–1057. 10.1001/jama.2020.16349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. Fast identification of possible drug treatment of coronavirus disease-19 (COVID-19) through computational drug repurposing study. J. Chem. Inf. Model. 2020, 60, 3277–3286. 10.1021/acs.jcim.0c00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyner M. J.; Wright R. S.; Fairweather D.; Senefeld J. W.; Bruno K. A.; Klassen S. A.; Carter R. E.; Klompas A. M.; Wiggins C. C.; Shepherd J. R. A.; Rea R. F.; Whelan E. R.; Clayburn A. J.; Spiegel M. R.; Johnson P. W.; Lesser E. R.; Baker S. E.; Larson K. F.; Ripoll J. G.; Andersen K. J.; Hodge D. O.; Kunze K. L.; Buras M. R.; Vogt M. N. P.; Herasevich V.; Dennis J. J.; Regimbal R. J.; Bauer P. R.; Blair J. E.; van Buskirk C. M.; Winters J. L.; Stubbs J. R.; Paneth N. S.; Verdun N. C.; Marks P.; Casadevall A. Early safety indicators of COVID-19 convalescent plasma in 5000 patients. J. Clin. Invest. 2020, 130, 4791–4797. 10.1172/JCI140200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://www.yalemedicine.org/news/5-things-to-know-omicron; visited on September 24th 2022.
- https://www.asahi.com/ajw/articles/14682787; visited on September 24th 2022.
- Tuekprakhon A.; Nutalai R.; Dijokaite-Guraliuc A.; Zhou D.; Ginn H. M.; Selvaraj M.; Liu C.; Mentzer A. J.; Supasa P.; Duyvesteyn H. M. E.; Das R.; Skelly D.; Ritter T. G.; Amini A.; Bibi S.; Adele S.; Johnson S. A.; Constantinides B.; Webster H.; Temperton N.; Klenerman P.; Barnes E.; Dunachie S. J.; Crook D.; Pollard A. J.; Lambe T.; Goulder P.; Paterson N. G.; Williams M. A.; Hall D. R.; Fry E. E.; Huo J.; Mongkolsapaya J.; Ren J.; Stuart D. I.; Screaton G. R.; et al. Antibody escape of SARS-CoV-2 Omicron BA.4 and BA.5 from vaccine and BA.1 serum. Cell 2022, 185, 2422–2433. 10.1016/j.cell.2022.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://www.who.int/news/item/26-11-2021-classification-of-omicron-(b.1.1.529)-sars-cov-2-variant-of-concern; visited on September 24th 2022.
- Araf Y.; Akter F.; Tand Y.-d.; Fatemi R.; Parvez Md. S. A.; Zheng C.; Hossain Md. G. Omicron variant of SARS-CoV-2: Genomics, transmissibility, and responses to current COVID-19 vaccines. J. Med. Virol. 2022, 94, 1825–1832. 10.1002/jmv.27588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng B.; Ferreira I. A. T. M.; Abdullahi A.; Saito A.; Kimura I.; Yamasoba D.; Kemp S. A.; Goonawardane N.; Papa G.; Fatihi S.; Rathore S.; Ikeda T.; Toyoda M.; Tan T. S.; Kuramochi J.; Mitsunaga S.; Ueno T.; Charles O. J.; CITIID-NIHR BioResource COVID-19 Collaboration, The Genotype to Phenotype Japan (G2P-Japan) Consortium; Ecuador-COVID-19 Consortium; Smith K. G. C.; Bradley J.; Choi J.; Madissoon E.; Meyer K.; Mlcochova P.; Doffinger R.; Teichmann S. A.; James L.; Lee J. H.; Thukral L.; Sato K.; Gupta R. K. SARS-CoV-2 Omicron spike mediated immune escape, infectivity and cell-cell fusion. Preprint at bioRxiv https://www.biorxiv.org/content/10.1101/2021.12.17.473248v2 (2021).
- Peacock T.; Brown J. C.; Zhou J.; Thakur N.; Newman J.; Kugathasan R.; Sukhova K.; Kaforou M.; Bailey D.; Barclay W. S.. The SARS-CoV-2 variant, Omicron, shows rapid replication in human primary nasal epithelial cultures and efficiently uses the endosomal route of entry. Preprint at bioRxiv https://www.biorxiv.org/content/10.1101/2021.12.31.474653v1 (2022).
- Willett B. J.; Grove J.; MacLean O. A.; Wilkie C.; Logan N.; De Lorenzo G.; Furnon W.; Scott S.; Manali M.; Szemiel A.; Ashraf S.; Vink E.; Harvey W. T.; Davis C.; Orton R.; Hughes J.; Holland P.; Silva V.; Pascall D.; Puxty K.; da Silva Filipe A.; Yebra G.; Shaaban S.; Holden M. T. G.; Pinto R. M.; Gunson R.; Templeton K.; Murcia P. R.; Patel A. H., ; The COVID-19 Genomics UK (COG-UK) Consortium;, Haughney J.; Robertson D. L.; Palmarini M.; Ray S.; Thomson E. C.. The hyper-transmissible SARS-CoV-2 Omicron variant exhibits significant antigenic change, vaccine escape and a switch in cell entry mechanism. Preprint at medRxiv https://www.medrxiv.org/content/10.1101/2022.01.03.21268111v1 (2022).
- Pia L.; Rowland-Jones S. Omicron entry route. Nat. Rev. Immunol. 2022, 22, 144. 10.1038/s41577-022-00681-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D. P.; Lytras S.; Lucaci A. G.; Maier W.; Gruening B.; Shank S. D.; Weaver S.; MacLean O. A.; Orton R. J.; Lemey P.; Boni M. F.; Tegally H.; Harkins G. W.; Scheepers C.; Bhiman J. N.; Everatt J.; Amoako D. G.; San J. E.; Giandhari J.; Sigal A.; NGS-SA Williamson C.; Hsiao N.-y.; von Gottberg A.; De Klerk A.; Shafer R. W.; Robertson D. L.; Wilkinson R. J.; Sewell B. T.; Lessells R.; Nekrutenko A.; Greaney A. J.; Starr T. N.; Bloom J. D.; Murrell B.; Wilkinson E.; Gupta R. K.; de Oliveira T.; Kosakovsky Pond S. L. Selection analysis identifies clusters of unusual mutational changes in Omicron lineage BA.1 that likely impact spike function. Mol. Biol. Evol. 2022, 39, msac061 10.1093/molbev/msac061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascarella S.; Ciccozzi M.; Bianchi M.; Benvenuto D.; Cauda R.; Cassone A. The electrostatic potential of the Omicron variant spike is higher than in delta and delta-plus variants: a hint to higher transmissibility?. J. Med. Virol. 2022, 94, 1277–1280. 10.1002/jmv.27528. [DOI] [PubMed] [Google Scholar]
- Lupala C. S.; Ye Y.; Chen H.; Su X. D.; Liu H. Mutations in RBD of SARS-CoV-2 Omicron variant result in stronger binding to human ACE2 receptor. Biochem. Biophys. Res. Commun. 2022, 590, 34–41. 10.1016/j.bbrc.2021.12.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- K A.; Sharma A.; Kumar D.; Sing S. K.; Gupta G.; Chellappan D. K.; Dua K.; Nagraik R. Molecular aspects of Omicron, vaccine development, and recombinant strain XE: A review. J. Med. Virol. 2022, 94, 4628–4643. 10.1002/jmv.27936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.; Sheersh Boorla V.; Banerjee D.; Chowdhury R.; Cavener V. S.; Nissly R. H.; Gontu A.; Boyle N. R.; Vandegrift K.; Suredran Nair V.; Kuchipudi S. V.; Maranas C. D. Computational prediction of the effect of amino acid changes on the binding affinity between SARS-CoV-2 spike RBD and human ACE2. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2106480118 10.1073/pnas.2106480118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingraham N. E.; Ingbar D. H. The Omicron variant of SARS-CoV-2: understanding the known and living with unknowns. Clin. Transl. Med. 2021, 11, e685 10.1002/ctm2.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahradník J.; Marciano S.; Shemesh M.; Zoler E.; Harari D.; Chiaravalli J.; Meyer B.; Rudich Y.; Li C.; Marton I.; Dym O.; Elad N.; Lewis M. G.; Andersen H.; Gagne M.; Seder R. A.; Douek D. C.; Schreiber G. SARS-CoV-2 variant prediction and antiviral drug design are enabled by RBD in vitro evolution. Nat. Microbiol. 2021, 6, 1188–1198. 10.1038/s41564-021-00954-4. [DOI] [PubMed] [Google Scholar]
- Gong S. Y.; Chatterjee D.; Richard J.; Prevost J.; Tauzin A.; Gasser R.; Bo Y.; Vezina D.; Goyette G.; Gendron-Lepage G.; Medjahed H.; Roger M.; Cote M.; Finzi A. Contribution of single mutations to selected SARS-CoV-2 emerging variants spike antigenicity. Virology 2021, 563, 134–145. 10.1016/j.virol.2021.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benvenuto D.; Angeletti S.; Giovanetti M.; Bianchi M.; Pascarella S.; Cauda R.; Ciccozzi M.; Cassone A. Evolutionary analysis of SARS-CoV-2: how mutation of non-structural protein 6 (NSP6) could affect viral autophagy. J. Infect. 2020, 81, e24–e27. 10.1016/j.jinf.2020.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu P.; Faraone J. N.; Evans J. P.; Zou X.; Zheng Y.-M.; Carlin C.; Bednash J. S.; Lozanski G.; Mallampalli R. K.; Saif L. J.; Oltz E. M.; Mohler P. J.; Gumina R. J.; Liu S.-L. Differential evasion of Delta and Omicron immunity and enhanced fusogenicity of SARS-CoV-2 Omicron BA.4/5 and BA.2.12.1 subvariants. Preprint at. bioRxiv 2022, 10.1101/2022.05.16.492158. [DOI] [Google Scholar]
- Zhang Y.; Zhang T.; Fang Y.; Liu J.; Ye Q.; Ding L. SARS-CoV-2 spike L452R mutation increases Omicron variant fusogenicity and infectivity as well as host glycolysis. Signal Transduction Targeted Ther. 2022, 7, 76. 10.1038/s41392-022-00941-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodo K.; Fujita K.; Sodeoka M. Raman spectroscopy for chemical biology research. J. Am. Chem. Soc. 2022, 144, 19651. 10.1021/jacs.2c05359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezzotti G. Raman spectroscopy in cell biology and microbiology. J. Raman Spectrosc. 2021, 52, 2348–2443. 10.1002/jrs.6204. [DOI] [Google Scholar]
- Tuma R. Raman spectroscopy of proteins: from peptides to large assemblies. J. Raman Spectrosc. 2005, 36, 307–319. 10.1002/jrs.1323. [DOI] [Google Scholar]
- Maiti N. C.; Apetri M. M.; Zagorski M. G.; Carey P. R.; Anderson V. E. Raman spectroscopic characterization of secondary structure in natively unfolded proteins: α-synuclein. J. Am. Chem. Soc. 2004, 126, 2399–2408. 10.1021/ja0356176. [DOI] [PubMed] [Google Scholar]
- Liu Y.-J.; Kyne M.; Wang C.; Yu X.-Y. Data mining in Raman imaging in a cellular biological system. Comp. Struct. Biotechnol. J. 2020, 18, 2920–2930. 10.1016/j.csbj.2020.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Ma X.; Su X.; Huang S.; Xu X.; Zhou X.; Huang W. E.; Knight R. Emerging trends for microbiome analysis: from single-cell functional imaging to microbiome big data. Engineering 2017, 3, 66–70. 10.1016/J.ENG.2017.01.020. [DOI] [Google Scholar]
- Pezzotti G.; Boschetto F.; Ohgitani E.; Fujita Y.; Shin-Ya M.; Adachi T.; Yamamoto T.; Kanamura N.; Marin E.; Zhu W.; Nishimura I.; Mazda O. Raman molecular fingerprints of SARS-CoV-2 British variant and the concept of Raman barcode. Adv. Sci. 2021, 9 (3), 2103287 10.1002/advs.202103287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezzotti G.; Ohgitani E.; Fujita Y.; Imamura H.; Shin-Ya M.; Adachi T.; Yamamoto T.; Kanamura N.; Marin E.; Zhu W.; Nishimura I.; Mazda O. Raman fingerprints of the SARS-CoV-2 Delta variant and mechanisms of its instantaneous inactivation by silicon nitride bioceramics. ACS Infect. Dis. 2022, 8, 1563–1581. 10.1021/acsinfecdis.2c00200. [DOI] [PubMed] [Google Scholar]
- Mazet V.; Carteret C.; Brie D.; Idier J.; Humbert B. Background removal from spectra by designing and minimising a non-quadratic cost function. Chemom. Intell. Lab. Syst. 2005, 76, 121–133. 10.1016/j.chemolab.2004.10.003. [DOI] [Google Scholar]
- He S.; Zhang W.; Liu L.; Huang Y.; He J.; Xie W.; Wu P.; Du C. Baseline correction for Raman spectra using an improved asymmetric least squares method. Anal. Methods 2014, 6, 4402–4407. 10.1039/C4AY00068D. [DOI] [Google Scholar]
- Cai Y.; Yang C.; Xu D.; Gui W. Baseline correction for Raman spectra using penalized spline smoothing based on vector transformation. Anal. Methods 2018, 10, 3525–3533. 10.1039/C8AY00914G. [DOI] [Google Scholar]
- Pezzotti G.; Zhu W.; Adachi T.; Horiguchi S.; Marin E.; Boschetto F.; Ogitani E.; Mazda O. Metabolic machinery encrypted in the Raman spectrum of influenza A virus-inoculated mammalian cells. J. Cell. Physiol. 2020, 235, 5146–5170. 10.1002/jcp.29392. [DOI] [PubMed] [Google Scholar]
- Tuma R. Raman spectroscopy of proteins: From peptides to large assemblies. J. Raman Spectrosc. 2005, 36, 307–319. 10.1002/jrs.1323. [DOI] [Google Scholar]
- Zhu G.; Zhu X.; Fan Q.; Wan X. Raman spectra of amino acids and their aqueous solutions. Spectrochim. Acta, Part A 2011, 78, 1187–1195. 10.1016/j.saa.2010.12.079. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Thambiraja T. S.; Karuppanan K.; Subramaniam G. Omicron and Delta variant of SARS-CoV-2: A comparative computational study of spike protein. J. Med. Virol. 2022, 94, 1641–1649. 10.1002/jmv.27526. [DOI] [PubMed] [Google Scholar]
- Golichenko B. O.; Naseka V. M.; Strelchuk V. V.; Kolomys O. F. Raman study of L-Asparagine and L-Glutamine molecules adsorbed on aluminum films in a wide frequency range. Semicond. Phys., Quantum Electron. Optoelectron. 2017, 20, 297–304. 10.15407/spqeo20.03.297. [DOI] [Google Scholar]
- Hernández B.; Pflüger F.; Derbel N.; De Coninck J.; Ghomi M. Vibrational analysis of amino acids and short peptides in hydrated media. VI. Amino acids with positively charged side chains: L-lysine and L- arginine. J. Phys. Chem. B 2010, 114, 1077–1088. 10.1021/jp909517r. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Rai S. B. Spectroscopic studies of L-arginine molecule. Indian J. Pure Appl. Phys. 2010, 48, 251–255. [Google Scholar]
- Tetz G.; Tetz V. Prion-like domains in spike protein of SARS-CoV-2 differ across its variants and enable changes in affinity to ACE2. Microorganisms 2022, 10, 280. 10.3390/microorganisms10020280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu Y. L.; Teoh K. T.; Lo J.; Chan C. M.; Kien F.; Escriou N.; Tsao S. W.; Nicholls J. M.; Altmeyer R.; Peiris J. S. M.; Bruzzone R.; Nal B. The M, E, and N structural proteins of the severe acute respiratory syndrome coronavirus are required for efficient assembly, trafficking, and release of virus-like particles. J. Virol. 2008, 82, 11318–11330. 10.1128/JVI.01052-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr A. R.; Perlman S.. Coronaviruses: an overview of their replication and pathogenesis. In Coronaviruses, Methods in Molecular Biology; Springer: New York, 2015; Vol. 1282, pp 1–23. 10.1007/978-1-4939-2438-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachrdla M.; Savastano A.; Ibanez de Opakua A.; Cima-Omori M.-S.; Zweckstetter M. Contributions of the N-terminal intrinsically disordered region of the severe acute respiratory syndrome coronavirus 2 nucleocapsid protein to RNA-induced phase separation. Protein Sci. 2022, 31 (9), e4409 10.1002/pro.4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X.; Liu Q.; Benavides-Montano J. A.; Nicola A. V.; Aston D. E.; Rasco B. A.; Aguilar H. C. Detection of receptor-induced glycoprotein conformational changes on enveloped virions by using confocal micro-Raman spectroscopy. J. Virol. 2013, 87, 3130–3142. 10.1128/JVI.03220-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C. R.; Ludwig M.; Asher S. A. Ultraviolet resonance Raman characterization of photochemical transients of phenol, tyrosine, and tryptophan. J. Am. Chem. Soc. 1986, 108, 905–912. 10.1021/ja00265a010. [DOI] [Google Scholar]
- D’Amico F.; Cammisuli F.; Addobbati R.; Rizzardi C.; Gessini A.; Masciovecchio C.; Rossi B.; Pascolo L. Oxidative damage in DNA bases revealed by UV resonant Raman spectroscopy. Analyst 2015, 140, 1477–1485. 10.1039/C4AN02364A. [DOI] [PubMed] [Google Scholar]
- de Sousa F. F.; Nogueira C. E. S.; Freire P. T. C.; Moreira S. G. C.; Teixeira A. M. R.; de Menezes A. S.; Mendes Filho J.; Saraiva G. D. Conformational change in the C form of palmitic acid investigated by Raman spectroscopy and X-ray diffraction. Spectrochim. Acta, Part A 2016, 161, 162–169. 10.1016/j.saa.2016.02.035. [DOI] [PubMed] [Google Scholar]
- Notingher I.; Verrier S.; Haque S.; Polak J. M.; Hench L. L. Spectroscopic study of human lung epithelial cells (A549) in culture: Living cells versus dead cells. Biopolymers 2003, 72, 230–240. 10.1002/bip.10378. [DOI] [PubMed] [Google Scholar]
- Dritsa V.; Pissaridi K.; Koutoulakis E.; Mamarelis I.; Kotoulas C.; Anastassopoulou J. An infrared spectroscopic study of aortic valve. A possible mechanism of calcification and the role of magnesium salts. In Vivo 2014, 28, 91–98. [PubMed] [Google Scholar]
- Czamara K.; Natorska J.; Kapusta P.; Baranska M.; Kaczor A. Raman microspectroscopy of human aortic valves: investigation of the local and global biochemical changes associated with calcification in aortic stenosis. Analyst 2015, 140, 2164–2170. 10.1039/C4AN01856G. [DOI] [PubMed] [Google Scholar]
- Malini R.; Venkatakrishma K.; Kurien J.; Pai K. M.; Rao L.; Kartha V. B.; Krishna C. M. Discrimination of normal, inflammatory, premalignant, and malignant oral tissue: A Raman spectroscopy study. Biopolymers 2006, 81, 179–193. 10.1002/bip.20398. [DOI] [PubMed] [Google Scholar]
- Koljenović S.; Scut T. B.; Vincent A.; Kros J. M.; Puppels G. J. Detection of meningioma in dura mater by Raman spectroscopy. Anal. Chem. 2005, 77, 7958–7965. 10.1021/ac0512599. [DOI] [PubMed] [Google Scholar]
- Lazar G. Influence of the substrate-electrode applied bias voltage on the properties of sputtered α-C:H thin films. J. Phys. Condens. Matter 2011, 13 (13), 3011–3021. 10.1088/0953-8984/13/13/314. [DOI] [Google Scholar]
- Kline N. J.; Treado P. J. Raman chemical imaging of breast tissue. J. Raman Spectrosc. 1997, 28, 119–124. . [DOI] [Google Scholar]
- Shetty G.; Kendall C.; Shepherd N.; Stone N.; Barr H. Raman spectroscopy: elucidation of biochemical changes in carcinogenesis of oesophagus. Br. J. Cancer 2006, 94, 1460–1464. 10.1038/sj.bjc.6603102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G.; Zhu X.; Fan Q.; Wan X. Raman spectra of amino acids and their aqueous solutions. Spectrochim. Acta, Part A 2011, 78, 1187–1195. 10.1016/j.saa.2010.12.079. [DOI] [PubMed] [Google Scholar]
- Podstawka E.; Ozaki Y.; Proniewicz L. M. Part II: Surface- enhanced Raman spectroscopy investigation of methionine containing heterodipeptides adsorbed on colloidal silver. Appl. Spectrosc. 2004, 58, 581–590. 10.1366/000370204774103417. [DOI] [PubMed] [Google Scholar]
- Cao X.; Fischer G. Conformational and infrared spectral studies of L-methionine and its N-deuterated isotopomer as isolated zwitterions. J. Phys. Chem. A 2002, 106, 41–50. 10.1021/jp012908x. [DOI] [Google Scholar]
- Diaz Fleming G.; Finnerty J. J.; Campos-Vallette M.; Célis F.; Aliaga A. E.; Fredes C.; Koch R. Experimental and theoretical Raman and surface-enhanced Raman scattering study of cysteine. J. Raman Spectrosc. 2009, 40, 632–638. 10.1002/jrs.2175. [DOI] [Google Scholar]
- Parker S. F. Assignment of the vibrational spectrum of L-cysteine. Chem. Phys. 2013, 424, 75–79. 10.1016/j.chemphys.2013.04.020. [DOI] [Google Scholar]
- Bazylewski P.; Divigalpitiya R.; Fanchini G. In situ Raman spectroscopy distinguishes between reversible and irreversible thiol modifications in L-cysteine. RSC Adv. 2017, 7, 2964–2970. 10.1039/C6RA25879D. [DOI] [Google Scholar]
- Hernández B.; Pflüger F.; Adenier A.; Kruglik S. G.; Ghomi M. Side chain flexibility and protonation states of sulfur atom containing amino acids. Phys. Chem. Chem. Phys. 2011, 13, 17284–17294. 10.1039/c1cp21054h. [DOI] [PubMed] [Google Scholar]
- Švančarová P.; Betáková T. Conserved methionine165 of matrix protein contributes to the nuclear import and is essential for influenza A virus replication. Virol. J. 2018, 15 (1), 187 10.1186/s12985-018-1056-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Z.; Liu T.; Offringa D. P.; McInnis J.; Levandowski R. A. Association of influenza virus matrix protein with ribonucleoproteins. J. Virol. 1999, 73, 7467–7473. 10.1128/JVI.73.9.7467-7473.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Requejo R.; Hurd T. R.; Costa N. J.; Murphy M. P. Cysteine residues exposed on protein surfaces are the dominant intramitochondrial thiol and may protect against oxidative damage. FEBS J. 2010, 277, 1465–1480. 10.1111/j.1742-4658.2010.07576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siamwiza M. N.; Lord R. C.; Chen M. C.; Takamatsu T.; Harada I.; Matsuura H.; Shimanouchi T. Interpretation of the doublet at 850 and 830 cm-1 in the Raman spectra of tyrosyl residues in proteins and certain model compounds. Biochemistry 1975, 14, 4870–4876. 10.1021/bi00693a014. [DOI] [PubMed] [Google Scholar]
- Guckeisen T.; Hosseinpour S.; Peukert W. Effect of pH and urea on the protein secondary structure at the water/air interface and in solution. J. Colloid Interface Sci. 2021, 590, 38–49. 10.1016/j.jcis.2021.01.015. [DOI] [PubMed] [Google Scholar]
- Śanchez-Cortés S.; Garcia-Ramos J. V. SERS of cytosine and its methylated derivatives on metal colloids. J. Raman Spectrosc. 1992, 23, 61–66. 10.1002/jrs.1250230108. [DOI] [Google Scholar]
- Mathlouthi M.; Seuvre A. M.; Koenig J. L. F.T.-I.R. and laser-Raman spectra of cytosine and cytidine. Carbohydr. Res. 1986, 146, 1–13. 10.1016/0008-6215(86)85019-4. [DOI] [PubMed] [Google Scholar]
- Madzharova F.; Heiner Z.; Gühlke M.; Kneipp J. Surface- enhanced hyper-Raman spectra of adenine, guanine, cytosine, thymine, and uracil. J. Phys. Chem. C 2016, 120, 15415–15423. 10.1021/acs.jpcc.6b02753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes R. P.; Marques M. P. M.; Valero R.; Tomkinson J.; de Carvalho L. A. E. B. Guanine: a combined study using vibrational spectroscopy and theoretical methods. Int. J. Spectrosc. 2012, 27, 273–292. 10.1155/2012/168286. [DOI] [Google Scholar]
- Lopes R. P.; Valero R.; Tomkinson J.; Marques M. P. M.; Batista de Carvalho L. A. E. Applying vibrational spectroscopy to the study of nucleobases - Adenine as a case study. New J. Chem. 2013, 37, 2691–2699. 10.1039/c3nj00445g. [DOI] [Google Scholar]
- Thomas G. J. Jr.; Hartman K. A. Raman studies of nucleic acids. 8. Estimation of RNA secondary structure from Raman scattering by phosphate-group vibrations. Biochim. Biophys. Acta 1973, 312, 311–322. 10.1016/0005-2787(73)90376-6. [DOI] [PubMed] [Google Scholar]
- Hernández B.; Pflüger F.; Kruglik S. G.; Ghomi M. Characteristic Raman lines of phenylalanine analyzed by a multiconformational approach. J. Raman Spectrosc. 2013, 44, 827–833. 10.1002/jrs.4290. [DOI] [Google Scholar]
- Uversky V. N.; Oldfield C. J.; Midic U.; Xie H.; Xue B.; Vucetic S.; Iakoucheva L. M.; Obradovic Z.; Dunker A. K. Unfoldomics of human diseases: linking protein disorder with diseases. BMC Genomics 2009, 10 (Suppl. 1), S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minchin S.; Lodge J. Understanding biochemistry: structure and function of nucleic acids. Essays Biochem. 2019, 63, 433–456. 10.1042/EBC20180038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino E. J.; Wilkinson K. A.; Coughlan J. L.; Weeks K. M. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE). J. Am. Chem. Soc. 2005, 127, 4223–4231. 10.1021/ja043822v. [DOI] [PubMed] [Google Scholar]
- Sadat A.; Joye I. J. Peak fitting applied to Fourier Transform infrared and Raman spectroscopic analysis of proteins. Appl. Sci. 2020, 10, 5918. 10.3390/app10175918. [DOI] [Google Scholar]
- Eker F.; Cao X.; Nafie L.; Huang Q.; Schweitzer-Stenner R. The structure of alanine based tripeptides in water and dimethyl sulfoxide probed by vibrational spectroscopy. J. Phys. Chem. B 2003, 107, 358–365. 10.1021/jp026958t. [DOI] [Google Scholar]
- Zhang J.; Xiao T.; Cai Y.; Chen B. Structure of SARS-CoV-2 spike protein. Curr. Opin. Virol. 2021, 50, 173–182. 10.1016/j.coviro.2021.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Liu C.; Zhang C.; Wang Y.; Hong Q.; Xu S.; Li Z.; Yang Y.; Huang Z.; Cong Y. Structural basis for SARS-CoV-2 Delta variant recognition of ACE2 receptor and broadly neutralizing antibodies. Nat. Commun. 2022, 13, 871 10.1038/s41467-022-28528-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurkovetskiy L.; Wang X.; Pascal K. E.; Tomkins-Tinch C.; Nyalile T. P.; Wang Y.; Baum A.; Diehl W. E.; Dauphin A.; Carbone C.; Veinotte K.; Egri S. B.; Schaffner S. F.; Lemieux J. E.; Munro J. B.; Rafique A.; Barve A.; Sabeti P. C.; Kyratsous C. A.; Dudkina N. V.; Shen K.; Luban J. Structural and functional analysis of the D614G SARS-CoV-2 spike protein variant. Cell 2020, 183, 739–751. 10.1016/j.cell.2020.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teruel N.; Mailhot O.; Najmanovich R. J. Modelling conformational state dynamics and its role on infection for SARS- CoV-2 Spike protein variants. PLoS Comput. Biol. 2021, 17, e1009286 10.1371/journal.pcbi.1009286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue B.; Blocquel D.; Habchi J.; Uversky A. V.; Kurgan L.; Uversky V. N.; Longhi S. Structural disorder in viral proteins. Chem. Rev. 2014, 114, 6880–6911. 10.1021/cr4005692. [DOI] [PubMed] [Google Scholar]
- Hsu M.-C.; Scheid A.; Choppin P. W. Enhancement of membrane-fusing activity of Sendai virus by exposure of the virus to basic pH is correlated with a conformational change in the fusion protein. Proc. Nat. Acad. Sci. U.S.A. 1982, 79, 5862–5866. 10.1073/pnas.79.19.5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batys P.; Morga M.; Bonarek P.; Sammalkorpi M. pH-induced changes in polypeptide conformation: Force-field comparison with experimental validation. J. Phys. Chem. B 2020, 124, 2961–2972. 10.1021/acs.jpcb.0c01475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H.; Hirai T.; Azuma T.; Hamaguchi K.; Migita S. pH-dependent conformation change of a Bence Jones protein. J. Biochem. 1970, 67, 795–799. 10.1093/oxfordjournals.jbchem.a129311. [DOI] [PubMed] [Google Scholar]
- Takeuchi H. Raman structural markers of tryptophan and histidine side chains in proteins. Biopolymers 2003, 72, 305–317. 10.1002/bip.10440. [DOI] [PubMed] [Google Scholar]
- Tuekprakhon A.; Nutalai R.; Dijokaite-Guraliuc A.; Zhou D.; Ginn H. M.; Selvaraj M.; Liu C.; Mentzer A. J.; Supasa P.; Duyvesteyn H. M. E.; Das R.; Skelly D.; Ritter T. G.; Amini A.; Bibi S.; Adele S.; Johnson S. A.; Constantinides B.; Webster H.; Temperton N.; Klenerman P.; Barnes E.; Dunachie S. J.; Crook D.; Pollard A. J.; Lambe T.; Goulder P.; Paterson N. G.; Williams M. A.; Hall D. R.; Fry E. E.; Huo J.; Mongkolsapaya J.; Ren J.; Stuart D. I.; Screaton G. R.; et al. Antibody escape of SARS-CoV-2 Omicron BA.4 and BA.5 from vaccine and BA.1 serum. Cell 2022, 185, 2422–2433.e13. 10.1016/j.cell.2022.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega J. T.; Serrano M. L.; Pujol F. H.; Rangel H. R. 2020. Role of changes in SARS-CoV- 2 spike protein in the interaction with the human ACE2 receptor: An in-silico analysis. EXCLI J. 2020, 19, 410–417. 10.17179/excli2020-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain A.; Akter S.; Rashid A. A.; Khair S.; Ul Alam A. S. M. R. Unique mutations in SARS-CoV-2 Omicron subvariants’ non-spike proteins: Potential impacts on viral pathogenesis and host immune evasion. Microb. Pathogen. 2022, 170, 105699 10.1016/j.micpath.2022.105699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezzotti G.; Kobara M.; Nakaya T.; Imamura H.; Miyamoto N.; Adachi T.; Yamamoto T.; Kanamura N.; Ohgitani E.; Marin E.; Zhu W.; Nishimura I.; Mazda O.; Nakata T.; Makimura M. Raman spectroscopy of oral Candida species: molecular-scale analyses, chemometrics, and barcode identification. Int. J. Mol. Sci. 2022, 23, 5359. 10.3390/ijms23105359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezzotti G.; Kobara M.; Nakaya T.; Imamura H.; Fujii T.; Miyamoto N.; Adachi T.; Yamamoto T.; Kanamura N.; Ohgitani E.; Marin E.; Zhu W.; Kawai T.; Mazda O.; Nakata T.; Makimura M. Raman metabolomics of Candida auris clades: profiling and barcode identification. Int. J. Mol. Sci. 2022, 23, 11736. 10.3390/ijms231911736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezzotti G.; Zhu W.; Chikaguchi H.; Marin E.; Boschetto F.; Masumura T.; Sato Y.-I.; Nakazaki T. Raman molecular fingerprints of rice nutritional quality and the concept of Raman barcode. Front. Nutr. 2021, 8, 663569 10.3389/fnut.2021.663569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podstawka E.; Ozaki Y.; Proniewicz L. M. Part II: Surface-enhanced Raman spectroscopy investigation of methionine containing heterodipeptides adsorbed on colloid silver. Appl. Spectrosc. 2004, 58, 581–590. 10.1366/000370204774103417. [DOI] [PubMed] [Google Scholar]
- Lopez L. A.; Riffle A. J.; Pike S. L.; Gardner D.; Hogue B. G. Importance of conserved cysteine residues in the Coronavirus envelope protein. J. Virol. 2008, 82, 3000–3010. 10.1128/JVI.01914-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veit M. Palmitoylation of virus proteins. Biol. Cell 2012, 104, 493–515. 10.1111/boc.201200006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouters M. A.; Fan S. W.; Haworth N. L. Disulfides as redox switches: from molecular mechanisms to functional significance. Antioxid. Redox Signaling 2010, 12, 53–91. 10.1089/ars.2009.2510. [DOI] [PubMed] [Google Scholar]
- Arnesano F.; Balatri E.; Banci L.; Bertini I.; Winge D. R. Folding studies of Cox17 reveal an important interplay of cysteine oxidation and copper binding. Structure 2005, 13, 713–722. 10.1016/j.str.2005.02.015. [DOI] [PubMed] [Google Scholar]
- Bhattacharya J.; Peters P. J.; Clapham P. R. Human immunodeficiency virus type 1 envelope glycoproteins that lack cytoplasmic domain cysteines: impact on association with membrane lipid rafts and incorporation onto budding virus particles. J. Virol. 2004, 78, 5500–5506. 10.1128/JVI.78.10.5500-5506.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaedigk-Nitschko K.; Ding M. X.; Levy M. A.; Schlesinger M. J. Site-directed mutations in the Sindbis virus 6K protein reveal sites for fatty acylation and the underacylated protein affects virus release and virion structure. Virology 1990, 175, 282–291. 10.1016/0042-6822(90)90210-I. [DOI] [PubMed] [Google Scholar]
- Yang J.; Lv J.; Wang Y.; Gao S.; Yao Q.; Qu D.; Ye R. Replication of murine coronavirus requires multiple cysteines in the endodomain of spike proteins. Virology 2012, 427, 98–106. 10.1016/j.virol.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D. A.; London E. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 1998, 14, 111–136. 10.1146/annurev.cellbio.14.1.111. [DOI] [PubMed] [Google Scholar]
- Linder M. E.; Deschenes R. J. Palmitoylation: policing protein stability and traffic. Nat. Rev. Mol. Cell Biol. 2007, 8, 74–84. 10.1038/nrm2084. [DOI] [PubMed] [Google Scholar]
- Rocks O.; Peyker A.; Kahms M.; Verveer P. J.; Koerner C.; Lumbierres M.; Kuhlmann J.; Waldmann H.; Wittinghofer A.; Bastiaens P. I. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science 2005, 307 (5716), 1746–1752. 10.1126/science.1105654. [DOI] [PubMed] [Google Scholar]
- Bos E. C.; Heijnen L.; Luytjes W.; Spaan W. J. Mutational analysis of the murine coronavirus spike protein: effect on cell-to-cell fusion. Virology 1995, 214, 453–463. 10.1006/viro.1995.0056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch B. J.; de Haan C. A. M.; Smits S. L.; Rottier P. J. M. Spike protein assembly into the coronavirion: exploring the limits of its sequence requirements. Virology 2005, 334, 306–318. 10.1016/j.virol.2005.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride C. E.; Machamer C. E. Palmitoylation of SARS-CoV S protein is necessary for partitioning into detergent-resistant membranes and cell–cell fusion but not interaction with M protein. Virology 2010, 405, 139–148. 10.1016/j.virol.2010.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit C. M.; Chouljenko V. N.; Iyer A.; Colgrove R.; Farzan M.; Knipe D. M.; Kousoulas K. G. Palmitoylation of the cysteine-rich endodomain of the SARS- coronavirus spike glycoprotein is important for spike-mediated cell fusion. Virology 2007, 360, 264–274. 10.1016/j.virol.2006.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorp E. B.; Boscarino J. A.; Logan H. L.; Goletz J. T.; Gallagher T. M. Palmitoylations on murine coronavirus spike proteins are essential for virion assembly and infectivity. J. Virol. 2006, 80, 1280–1289. 10.1128/JVI.80.3.1280-1289.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt P. G.; Copeland R. A.; Spiro T. G.; Otlewski J.; Laskowski M. Jr.; Prendergast F. G. Tyrosine hydrogen-bonding and environmental effects in proteins probed by ultraviolet resonance Raman spectroscopy. Biochemistry 1988, 27, 5426–5433. 10.1021/bi00415a007. [DOI] [PubMed] [Google Scholar]
- Krafft C.; Hinrichs W.; Orth P.; Saenger W.; Welfle H. Interaction of Tet repressor with operator DNA and with tetracycline studied by infrared and Raman spectroscopy. Biophys. J. 1998, 74, 63–71. 10.1016/S0006-3495(98)77767-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández B.; Coïc Y.-M.; Pflüger F.; Kruglik S. G.; Ghomi M. All characteristic Raman markers of tyrosine and tyrosinate originate from phenol ring fundamental vibrations. J. Raman Spectrosc. 2016, 47, 210–220. 10.1002/jrs.4776. [DOI] [Google Scholar]
- Yang L.; Li J.; Guo S.; Hou C.; Liao C.; Shi L.; Ma X.; Jiang S.; Zheng B.; Fang Y.; Ye L.; He X. SARS-CoV-2 variants, RBD mutations, binding affinity, and antibody escape. Int. J. Mol. Sci. 2021, 22, 12114. 10.3390/ijms222212114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newberry R. W.; Raines R. T. A prevalent intraresidue hydrogen bond stabilizes proteins. Nat. Chem. Biol. 2016, 12, 1084–1088. 10.1038/nchembio.2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Wang L.; Kao Y.-T.; Qiu W.; Yang Y.; Okobiah O.; Zhong D. Mapping hydration dynamics around a protein surface. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 18461–18466. 10.1073/pnas.0707647104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao B.; Jiménez-Ángeles F.; Nguyen T. D.; Olvera de la Cruz M. Water follows polar and nonpolar protein surface domains. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 19274–19281. 10.1073/pnas.1910225116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace C. N.; Shirley B. A.; McNutt M.; Gajiwala K. Forces contributing to the conformational stability of proteins. FASEB J. 1996, 10, 75–83. 10.1096/fasebj.10.1.8566551. [DOI] [PubMed] [Google Scholar]
- Li J.; Ma X.; Guo S.; Hou C.; Shi L.; Zhang H.; Zheng B.; Liao C.; Yang L.; Ye L.; He X. A hydrophobic-interaction-based mechanism triggers docking between the SARS-CoV-2 spike and angiotensin-converting Enzyme 2. Global Challenges 2020, 4, 2000067 10.1002/gch2.202000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández B.; Pflüger F.; Adenier A.; Kruglik G.; Ghomi M. Vibrational analysis of amino acids and short peptides in hydrated media. VIII. Amino acids with aromatic side chains: L-phenylalanine, L-tyrosine, and L-tryptophan. J. Phys. Chem. B 2010, 114, 15319–15330. 10.1021/jp106786j. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Zhang Y.; Wu L.; Niu S.; Song C.; Zhang Z.; Lu G.; Qiao C.; Hu Y.; Yuen K.-Y.; Wang Q.; Zhou H.; Yan J.; Qi J. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894. 10.1016/j.cell.2020.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voicescu M.; Ionescu S.; Nistor C. L. Spectroscopic study of 3 - hydroxyflavone-protein interaction in lipidic bi-layers immobilized on silver nanoparticles. Spectrochim. Acta, Part A 2017, 170, 1–8. 10.1016/j.saa.2016.06.039. [DOI] [PubMed] [Google Scholar]
- Leonardi A. A.; Sciuto E. L.; Lo Faro M. J.; Morganti D.; Midiri A.; Spinella C.; Conoci S.; Irrera A.; Fazio B. Molecular fingerprinting of the Omicron variant genome of SARS-CoV-2 by SERS spectroscopy. Nanomaterials 2022, 12, 2134. 10.3390/nano12132134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belshaw R.; Pybus O. G.; Rambaut A. The evolution of genome compression and genomic novelty in RNA viruses. Genome Res. 2007, 17, 1496–1504. 10.1101/gr.6305707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witteveldt J.; Blundell R.; Maarleveld J. J.; McFadden N.; Evans D. J.; Simmonds P. The influence of viral RNA secondary structure on interactions with innate host cell defenses. Nucleic Acids Res. 2014, 42, 3314–3329. 10.1093/nar/gkt1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosuge M.; Furusawa-Nishii E.; Ito K.; Saito Y.; Ogasawara K. Point mutation bias in SARS-CoV-2 variants results in increased ability to stimulate inflammatory responses. Sci. Rep. 2020, 10, 17766 10.1038/s41598-020-74843-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenchov R.; Zhou Q. A. Intrinsically disordered proteins: Perspective on COVID-19 infection and drug discovery. ACS Infect. Dis. 2022, 8, 422–432. 10.1021/acsinfecdis.2c00031. [DOI] [PubMed] [Google Scholar]
- Walls A. C.; Park Y. J.; Tortorici M. A.; Wall A.; McGuire A. T.; Veesler D. Structure, Function, and Antigenicity of the SARS- CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. 10.1016/S0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- Linding R.; Jensen L. J.; Diella F.; Bork P.; Gibson T. J.; Russell R. B. Protein disorder prediction: implications for structural proteomics. Structure 2003, 11, 1453–1459. 10.1016/j.str.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Gunasekaran K.; Tsai C.; Kumar S.; Zanuy D.; Nussinov R. Extended disordered proteins: targeting function with less scaffold. Trends Biochem. Sci. 2003, 28, 81–85. 10.1016/S0968-0004(03)00003-3. [DOI] [PubMed] [Google Scholar]
- Wright P. E.; Dyson H. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- Jackson C. B.; Farzan M.; Chen B.; Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. 10.1038/s41580-021-00418-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto A. L.; Rai R. K.; Brown J. C.; Griffin P.; Edgar J. R.; Shah A.; Singanayagam A.; Hogg C.; Barclay W. S.; Futter C. E.; Burgoyne T. Ultrastructural insight into SARS-CoV-2 entry and budding in human airway epithelium. Nat. Commun. 2022, 13, 1609 10.1038/s41467-022-29255-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stertz S.; Reichelt M.; Spiegel M.; Kuri T.; Martinez-Sobrido L.; Garcia-Sastre A.; Weber F.; Kochs G. The intracellular sites of early replication and budding of SARS-coronavirus. Virology 2007, 361, 304–315. 10.1016/j.virol.2006.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith C. S.; Tatti K. M.; Ksiazek T. G.; Rollin P. E.; Comer J. A.; Lee W. W.; Rota P. A.; Bankamp B.; Bellini W. J.; Zaki S. R. Ultrastructural characterization of SARS coronavirus. Emerg. Infect. Dis. 2004, 10, 320–326. 10.3201/eid1002.030913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins L. T.; Elkholy T.; Mubin S.; Hill D.; Williams R.; Ezike K.; Singhal A. Elucidation of SARS-CoV-2 budding mechanisms through molecular dynamics simulations of M and E protein complexes. J. Phys. Chem. Lett. 2021, 12, 12249–12255. 10.1021/acs.jpclett.1c02955. [DOI] [PubMed] [Google Scholar]
- Furse S.; Brooks N. J.; Seddon A. M.; Woscholski R.; Templer R. H.; Tate E. W.; Gaffney P. R. J.; Ces O. Lipid Membrane Curvature Induced by Distearoyl Phosphatidylinositol 4- Phosphate. Soft Matter 2012, 8, 3090–3093. 10.1039/c2sm07358g. [DOI] [Google Scholar]
- Mulet X.; Templer R. H.; Woscholski R.; Ces O. Evidence that phosphatidylinositol promotes curved membrane interfaces. Langmuir 2008, 24, 8443–8447. 10.1021/la801114n. [DOI] [PubMed] [Google Scholar]
- Matsuo T. Viewing SARS-CoV-2 nucleocapsid protein in terms of molecular flexibility. Biology 2021, 10, 454. 10.3390/biology10060454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S.; Yang M.; Hong Z.; Zhang L.; Huang Z.; Chen X.; He S.; Zhou Z.; Zhou Z.; Chen Q.; Yan Y.; Zhang C.; Shan H.; Chen S. Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharm. Sin. B 2020, 10, 1228–1238. 10.1016/j.apsb.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavda V. P.; Patel A. B.; Vaghasiya D. D. SARS-CoV-2 variants and vulnerability at the global level. J. Med. Virol. 2022, 94, 2986–3005. 10.1002/jmv.27717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasoba D.; Kimura I.; Nasser H.; Morioka Y.; Nao N.; Ito J.; Uriu K.; Tsuda M.; Zahradnik J.; Shirakawa K.; Suzuki R.; Kishimoto M.; Kosugi Y.; Kobiyama K.; Hara T.; Toyoda M.; Tanaka Y. L.; Butlertanaka E. P.; Shimizu R.; Ito H.; Wang L.; Oda Y.; Orba Y.; Sasaki M.; Nagata K.; Yoshimatsu K.; Asakura H.; Nagashima M.; Sadamasu K.; Yoshimura K.; Kuramochi J.; Seki M.; Fujiki R.; Kaneda A.; Shimada T.; Nakada T.-a.; Sakao S.; Suzuki T.; Ueno T.; Takaori-Kondo A.; Ishii K. J.; Schreiber G.; Sawa H.; Saito A.; Irie T.; Tanaka S.; Matsuno K.; Fukuhara T.; Ikeda T.; Sato K. Virological characteristics of the SARS-CoV-2 Omicron BA.2 spike. Cell 2022, 185, 2103–2115. 10.1016/j.cell.2022.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha L. B.; Foster C.; Rawlinson W.; Tedla N.; Bull R. A. Evolution of the SARS-CoV-2 omicron variants BA.1 to BA.5: Implications for immune escape and transmission. Rev. Med. Virol. 2022, 32, e2381 10.1002/rmv.2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barozi V.; Edkins A. L.; Bishop O. T. Evolutionary progression of collective mutations in Omicron sublineages towards efficient RBD-hACE2: Allosteric communications between and within viral and human proteins. Comput. Struct. Biotechnol. J. 2022, 20, 4562–4578. 10.1016/j.csbj.2022.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planas D.; Saunders N.; Maes P.; Guivel-Benhassine F.; Planchais C.; Buchrieser J.; Bolland W.-H.; Porrot F.; Staropoli I.; Lemoine F.; Pere H.; Veyer D.; Puech J.; Rodary J.; Baele G.; Dellicour S.; Raymenants J.; Gorissen S.; Geenen C.; Vanmechelen B.; Wawina-Bokalanga T.; Marti-Carreras J.; Cuypers L.; Seve A.; Hocqueloux L.; Prazuck T.; Rey F. A.; Simon-Loriere E.; Bruel T.; Mouqyet H.; Andre E.; Schwartz O. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature 2022, 602, 671–675. 10.1038/s41586-021-04389-z. [DOI] [PubMed] [Google Scholar]
- Liu C.; Ginn H. M.; Dejnirattisai W.; Supasa P.; Wang B.; Tuekprakhon A.; Nutalai R.; Zhou D.; Mentzer A. J.; Zhao Y.; Duyvesteyn H. M. E.; Lopez-Camacho C.; Slon-Campos J.; Walter T. S.; Skelly D.; Johnson S. A.; Ritter T. G.; Mason C.; Costa Clemens S. A.; Gomes Naveca F.; Nascimento V.; Nascimento F.; da Costa C. F.; Resende P. C.; Pauvolid-Correa A.; Siqueira M. M.; Dold C.; Temperton N.; Dong T.; Pollard A. J.; Knight J. C.; Crook D.; Lambe T.; Clutterbuck E.; Bibi S.; Flaxman A.; Bittaye M.; Belij-Rammerstorfer S.; Gilbert S. C.; Malik T.; Carroll M. W.; Klenerman P.; Barnes E.; Dunachie S. J.; Baillie V.; Serafin N.; Ditse Z.; Da Silva K.; Paterson N. G.; Williams M. A.; Hall D. R.; Madhi S.; Nunes M. C.; Goulder P.; Fry E. E.; Mongkolsapaya J.; Ren J.; Stuart D. I.; Screaton G. R. Reduced neutralization of SARS-CoV-2 B.1.617 by vaccine and convalescent serum. Cell 2021, 184, 4220–4236.e4213. 10.1016/j.cell.2021.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui K. P. Y.; Ho J. C. W.; Cheung M.-C.; Ng K.-C.; Ching R. H. H.; Lai K.-L.; Kam T. T.; Gu H.; Sit K.-Y.; Hsin M. K. Y.; Au T. W. K.; Poon L. L. M.; Peiris M.; Nicholls J. M.; Chan M. C. W. SARS-CoV-2 Omicron variant replication in human bronchus and lung ex vivo. Nature 2022, 603, 715–720. 10.1038/s41586-022-04479-6. [DOI] [PubMed] [Google Scholar]
- Peacock T. P.; Brown J. C.; Zhou J.; Thakur N.; Newman J.; Kugathasan R.; Sukhova K.; Kaforou M.; Bailey D.; Barclay W. S. The SARS-CoV-2 variant, Omicron, shows rapid replication in human primary nasal epithelial cultures and efficiently uses the endosomal route of entry. BioRxiv 2022, 10.1101/2021.12.31.474653. [DOI] [Google Scholar]
- Pišlar A.; Mitrovic A.; Sabotic J.; Pecar Fonovic U.; Perisic Nanut M.; Jakos T.; Senjor E.; Kos J. The role of cysteine peptidases in coronavirus cell entry and replication: The therapeutic potential of cathepsin inhibitors. PLoS Pathog. 2020, 16, e1009013 10.1371/journal.ppat.1009013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert M.; Bertoglio F.; Steinke S.; Heine P. A.; Ynga-Durand M. A.; Maass H.; Sammartino J. C.; Cassaniti I.; Zuo F.; Du L.; Korn J.; Milosevic M.; Wenzel E. V.; Krstanovic F.; Polten S.; Pribanic-Matesic M.; Brizic I.; Baldanti F.; Hammarstroem L.; Duebel S.; Sustic A.; Marcotte H.; Strengert M.; Protic A.; Piralla A.; Pan-Hammarstroem Q.; Cicin-Sain L.; Hust M. Human serum from SARS-CoV-2- vaccinated and COVID-19 patients shows reduced binding to the RBD of SARS-CoV-2 Omicron variant. BMC Med. 2022, 20 (1), 102 10.1186/s12916-022-02312-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han P.; Li L.; Liu S.; Wang Q.; Zhang D.; Xu Z.; Han P.; Li X.; Peng Q.; Su C.; Huang B.; Li D.; Zhang R.; Tian M.; Fu L.; Gao Y.; Zhao X.; Liu K.; Qi J.; Gao G. F.; Wang P. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell 2022, 185, 630–640.e10. 10.1016/j.cell.2022.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L.; Zhou L.; Mo M.; Liu T.; Wu C.; Gong C.; Lu K.; Gong L.; Zhu W.; Xu Z. SARS-CoV-2 Omicron RBD shows weaker binding affinity than the currently dominant Delta variant to human ACE2. Signal Transduction Targeted Ther. 2022, 7 (1), 8 10.1038/s41392-021-00863-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iketani S.; Liu L.; Guo Y.; Liu L.; Chan J. F.-W.; Huang Y.; Wang M.; Luo Y.; Yu J.; Chu H.; Chik K. K.-H.; Yuen T. T.-T.; Yin M. T.; Sobieszczyk M. E.; Huang Y.; Yuen K.-Y.; Wang H. H.; Sheng Z.; Ho D. D. Antibody evasion properties of SARS-CoV-2 Omicron sublineages. Nature 2022, 604, 553–556. 10.1038/s41586-022-04594-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar S.; Sarkar R. Mutational and phylogenetic analyses of the two lineages of the Omicron variant. J. Med. Virol. 2022, 94, 1777–1779. 10.1002/jmv.27558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J.; Chen J.; Xiong Y. L. Structural and emulsifying properties of soy protein isolate subjected to acid and alkaline pH-shifting processes. J. Agric. Food Chem. 2009, 57, 7576–7583. 10.1021/jf901585n. [DOI] [PubMed] [Google Scholar]
- Figueroa-González J. J.; Lobato-Calleros C.; Vernon-Carter E. J.; Aguirre-Mandujano E.; Alvarez-Ramirez J.; Martinez-Velasco A. Modifying the structure, physicochemical properties, and foaming ability of amaranth protein by dual pH-shifting and ultrasound treatments. LWT 2022, 153, 112561 10.1016/j.lwt.2021.112561. [DOI] [Google Scholar]
- Cele S.; Karim F.; Lustig G.; San J. E.; Hermanus T.; Tegally H.; Snyman J.; Moyo-Gwete T.; Wilkinson E.; Bernstein M.; Khan K.; Hwa S.-H.; Tilles S. W.; Singh L.; Giandhari J.; Mthabela N.; Mazibuko M.; Ganga Y.; Gosnell B. I.; Karim A. S. A.; Hanekom W.; Van Voorhis W. C.; Ndung’u T.; Lessells R. J.; Moore P. L.; Moosa M.-Y. S.; de Oliveira T.; Sigal A. SARS-CoV-2 prolonged infection during advanced HIV disease evolves extensive immune escape. Cell Host Microbe 2022, 30, 154–162.e155. 10.1016/j.chom.2022.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupferschmidt K. Where did ‘weird’ Omicron come from?. Science 2021, 374, 1179. 10.1126/science.acx9738. [DOI] [PubMed] [Google Scholar]
- Xia S.; Wang L.; Zhu Y.; Lu L.; Jiang S. Origin, virological features, immune evasion and intervention of SARS-CoV-2 Omicron sublineages. Signal Transduction Targeted Ther. 2022, 7 (1), 241 10.1038/s41392-022-01105-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson L. S.; Rodriguez J. D. Raman barcode for counterfeit drug product detection. Anal. Chem. 2016, 88, 4706–4713. 10.1021/acs.analchem.5b04636. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.