

Abstract
Due to increasing incidence and limited treatments, brain metastases (BM) are an emerging unmet need in modern oncology. Development of effective therapeutics has been hindered by unique challenges. Individual steps of the brain metastatic cascade are driven by distinctive biological processes, suggesting that BM possess intrinsic biological differences compared to primary tumors. Here, we discuss the unique physiology and metabolic constraints specific to BM, as well as emerging treatment strategies that leverage potential vulnerabilities.
INTRODUCTION
Brain metastases (BM) are the most common centra nervous system (CNS) malignancy, and occur in up to 40% of patients in metastatic cancer1. Approximately 200,000 patients are diagnosed with BM annually in the United States. This number will likely increase in the modern era of targeted therapies and immune checkpoint inhibitors (ICI), as patients are living longer with improved extracranial disease control and the risk of developing BM increases with duration of disease2,3. Lung (39–56%), breast (11–19%), and melanoma (6–11%) are among the most common primary tumors to spread to the brain4–6. As progression of BM is the cause of death in 50–75% of patients with BM6, they represent an unmet need in modern oncology and an emerging public health crisis.
Surgical resection followed by radiation to the resection cavity is generally the standard of care for solitary or large (>3 cm) symptomatic lesions. Stereotactic radiosurgery (SRS) is recommended for small (<3 cm) asymptomatic BM and for oligometastatic disease. Hippocampal-sparing whole brain radiation can be considered in more challenging cases such as multiple disseminated BM and leptomeningeal carcinomatosis7. Systemic therapies are generally reserved for patients with an oncogene-addicted BM (e.g. EGFR-mutation, ALK-rearrangement) or those with smaller asymptomatic BM not in urgent need of local control and active concomitant extracranial disease.
Due to improved resolution of commercial MRI’s and recent guidelines that recommend increased screening for BM8, an increasing number of patients present with intracranial lesions that are relatively small and minimally symptomatic. Other patients possess BM in an inoperable location. These cases represent an emerging opportunity for CNS-penetrant systemic therapy, which would reduce the morbidity associated with radiation-induced neurotoxicity or neurosurgical resection. Unfortunately, to date, many agents have limited intracranial efficacy. While targeted therapy and immunotherapy have displayed early promise for BM, the response rates are generally lower than than those observed in ECM9. Often, differential responses for intracranial and extracranial disease are observed, where systemic disease may be adequately controlled with concurrent progression of intracranial tumors.
Arguably the most promising way to revolutionize care for BM likely lies in a better understanding of BM biology in order to identify brain-specific targets. To stimulate future investigation in this field, we have focused this Review on three topics: 1) blood-brain barrier (BBB) physiology, 2) physiologic differences of BM tumor cells compared to extracranial tumors, and 3) mediators of immunosuppression from cell populations specific to the BM microenvironment. We note, however, that our review of these subjects is not meant to be comprehensive, and instead is intended to inspire discussion and collaboration among investigators rather than to present a dogmatic view. Finally, we infuse our thoughts on how these investigations may translate into early-phase trials, biomarkers of treatment response, and precision-based paradigms for BM patients.
BIOLOGY OF BRAIN METASTASES
The brain metastatic cascade
For patients at high risk of developing BM, a preventative strategy may lie in targeting molecular mediators of critical steps of CNS dissemination of solid tumors. To this end, a study using in vivo laser scanning microscopy in a murine model outlined discrete steps of the brain metastatic cascade,10 vascular arrest of tumor cells in intracranial capillaries, extravasation into perivascular space, brain parenchyma invasion, and perivascular growth with angiogenesis (Figure 1). Initial steps appear to be dependent on integrins, which are transmembrane ligands that mediate adhesion between tumor cells and the extracellular environment11–13. Integrin-mediated interaction of tumor cells with the basement membrane modulates vascular arrest through adhesion of tumor cells to the endothelium. However, as 95–99% of brain-arrested cancer cells fail to develop into a macro-metastasis, the metastatic cascade likely involves multiple biological processes, as penetrating the BBB does not necessarily translate into the ability to transition from a micro-metastasis to a macro-metastasis10. Using multimodal correlative microscopy, multiple aspects of the metastatic cascade (e.g. endothelial remodeling, extravasation, BM growth) was found to be dependent on cancer-cell derived matrix metalloprotease 9 (MMP9)14. Additionally, vascular co-option is essential for proliferation of cancer cells of diverse histologies. To our knowledge, clear molecular drivers of vascular co-option have yet to be identified. Another complementary factor to vascular co-option is angiogenesis, as aberrant vascular physiology13,15 and increased VEGF expression are associated with enhanced metastatic potential and higher rates of BM16. However, there have been mixed results with VEGF inhibition in preclinical models and clinical practice17, suggesting that angiogenesis is likely only one, albeit important, step for the brain metastatic cascade. Moving forward, we encourage additional efforts in identifying ‘bottlenecks’ and mediators of these steps within the brain metastatic cascade.
Fig 1. The brain metastatic cascade.
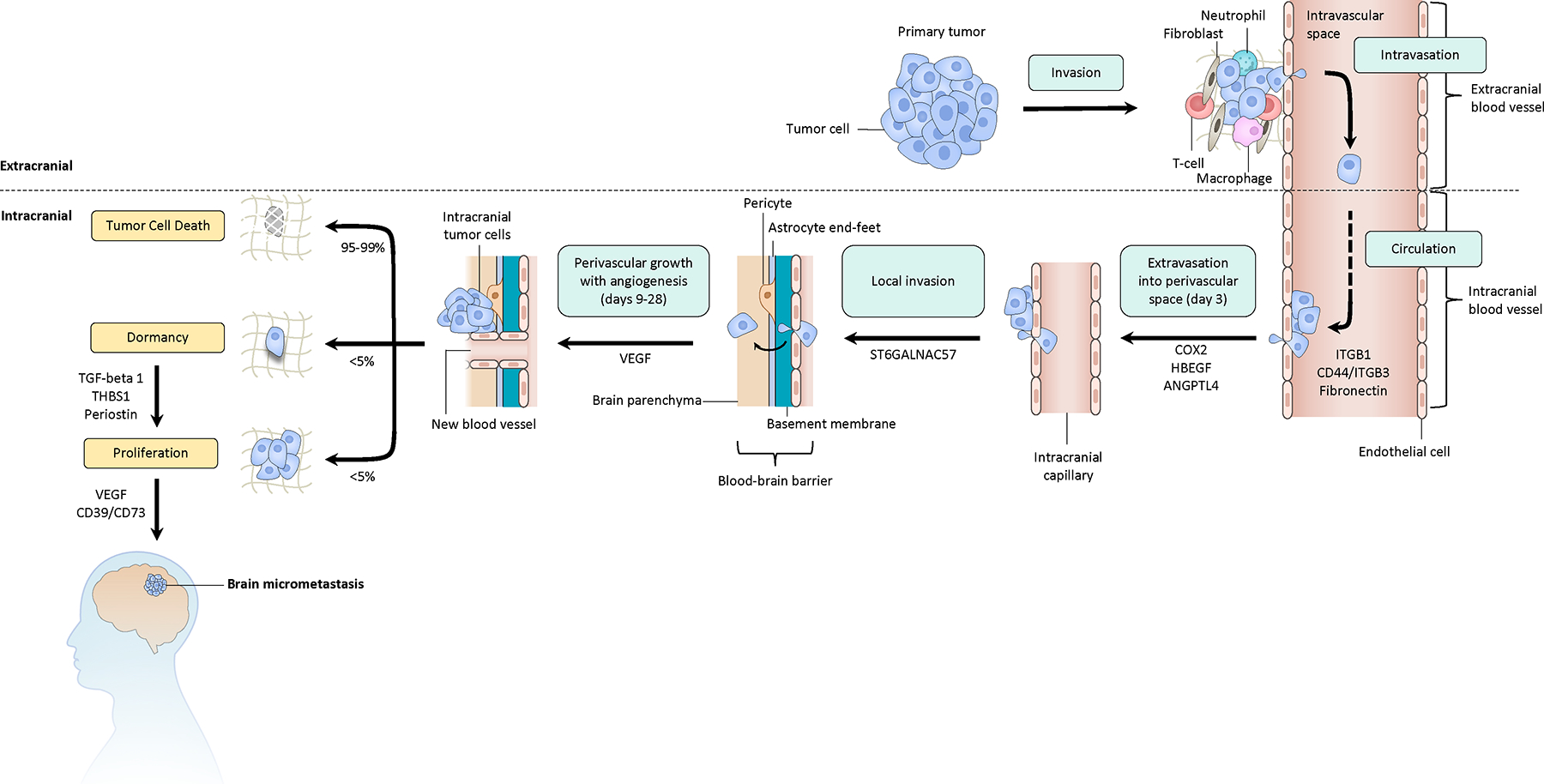
Major steps include: invasion of the tumor cells from the primary site into the extending stroma; intravasation of tumor cells into the peripheral circulation; extravasation of tumor cells into an intracranial perivascular space; local invasion of tumor cells, perivascular growth, and angiogenesis. We note that peri-vascular growth and angiogenesis are two separate steps in the formation of a macrometastasis, and that VEGF is therapeutically relevant are targeting angiogenesis. The majority of tumor cells do not survive following extravasation, with less than 5% lying in a dormant state with the ability to proceed to a proliferative state. This schematic may not recapitulate many nuances of in vivo brain metastasis, as this work was mostly done on cell lines.
Interestingly, a large fraction of cancer cells enter a dormant state after extravasation and rest as single cells along blood vessels without proliferation or regression for long periods of time10. Using an in vivo MR technique that enabled tracking of breast cancer cells in a murine model, three dormant tumor cells for every brain macrometastasis were visualized, serving as a considerable reservoir for tumor cells to awaken18. Release of TGF-beta 1, periostin, and thrombospondin-1 from the basement membrane activate vascular endothelial cells and may drive the transition from dormancy into a proliferative state19,20. Treatment modalities aimed at reducing levels of these proteins may be beneficial in preventing development of macro-metastases.
Other clinically impactful questions include: what biological factors mediate organ-specific metastasis of cancer? Do these traits arise in the primary tumor, circulating tumor cells, extracranial metastases or the affected organ site? Cancers exhibit clear organ-specific patterns of metastasis; for example, prostate cancer commonly metastasizes to the bone and rarely to the brain, whereas melanoma and lung cancer frequently spread to the CNS. Recent studies, which we present below, suggest that brain-specific metastasic growth results from complex interactions between cancer cells and CNS resident cells that modify the brain TME into a tumor-supportive immunosuppressive niche. Understanding these mechanisms may form the basis for a CNS preventative treatment paradigm for histologies at risk of metastasizing to the CNS that both targets the primary tumor cell and protects the susceptible organ microenvironment.
The blood-brain barrier
Understanding BBB dynamics is critical towards identifying mechanisms of tumor cell extravasation and maximizing on-target effects of candidate therapeutics. In homeostasis, the BBB is a tightly regulated neurovascular unit including endothelial cells, pericytes, and astrocytic end-feet that controls molecular and cellular transport into the CNS21,22. Tumor cells express cell-surface ligands that alter the BBB and facilitate extravasation of cancer cells into the parenchyma. Consequently, the BBB is remodeled into a brain-tumor barrier (BTB), which is characterized by aberrant and dysfunctional pericytes, astrocytic endfeet, and neuronal connections23. The BTB is functionally more permeable than an intact BBB, which permits infiltration of cancer cells, peripheral immune cells, and therapeutics (Figure 2)24,25. Genomic and functional analysis of BM identified the prostaglandin-synthesizing enzyme COX2, heparin-binding epidermal growth factor (HBEGF), and the alpha 2,6-silayltransferase ST6GALNAC5 as mediators of cancer cell passage through the BBB26. ST6GALNAC5 exerted activity through augmenting adhesion to endothelial cells. Another study noted that tumor cells secrete the protease cathepsin S, which degrades junctional adhesion molecules on the BBB and facilitates transmigration27. Interestingly, increased physical activity has been associated with higher BBB integrity and decreased BM formation in murine models28; however discrete molecular mediators that augment BBB integrity are still being explored.
Figure 2: Simplified schematic of BBB and BTB physiology and potential therapeutic strategies.
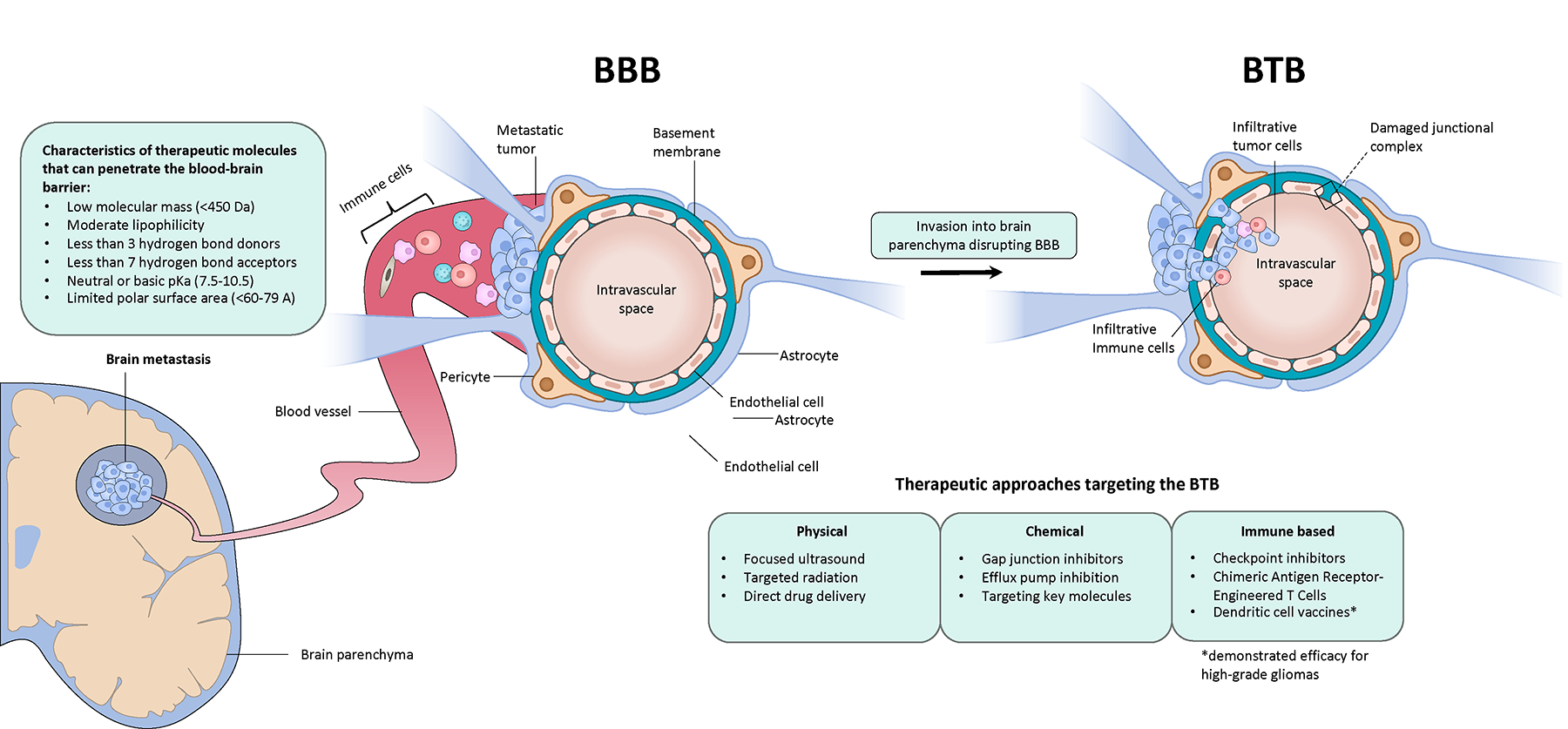
We illustrate biological changes exerted onto the BBB as a result of the brain metastatic cascade. Characteristics of therapeutic molecules that penetrate the BBB are listed. We also note therapeutic strategies augmenting drug efficacy (e.g. increasing immune cell efflux), as well as therapeutically relevant targets within the BTB.
Intracranial efficacy of many systemic therapies is negatively impacted by poor penetration into the CNS. In patients with breast cancer BM, the ratio of trastuzumab levels in the CSF and serum was 1/420 at baseline and rose to only 1/49 – 1/76 following radiation of the BM – which still qualifies as sanctuary site levels29. Another study evaluated drug uptake in a murine BM model through an injection of radiolabeled paclitaxel and doxorubicin and autoradiography of tissue sections30. BM uptake of the drug in question was usually greater than normal brain but only reached cytotoxic concentrations in less than 10% of BM. Given these findings and heterogenous BTB permeability, there is a concerted effort to design brain-permeable compounds. Medicinal properties of successful drugs able to pass the BBB are listed in Figure 231.
Further complicating the issue of CNS drug delivery is considerable heterogeneity in BTB function31–37. Preclinical models of BM have demonstrated differences in permeability within a single BM34,37, across spatially separated BM32, and across different subtypes within the same histology. This heterogeneity results in an uneven and unpredictable distribution of systemic therapies, especially if a patient has multiple BM, and can hamper consistent adequate delivery of a therapeutic agent to BM. However, recent studies indicate that BTB permeability can be modified through select BTB proteins and metabolites, suggesting therapeutic potential in maximizing drug penetration and efficacy. Upregulation of astrocyte-mediated sphingosine 1-phosphate receptor 3 (S1PR3) loosens the BTB through downregulation of tight and adherens junction proteins by secretion of IL-6 and chemokine-ligand 2 (CCL2)33. HER-2 positive breast cancer BM, compared to triple-negative or basal subtypes, possess a less disrupted BTB, which is associated with increased expression of glucose transporter 1 (GLUT1) and breast cancer resistance protein (BCRP)31.
Another set of clinically relevant targets are endothelial drug efflux transporters, which transports substances out of the brain into the blood. Expression of the ATP-binding cassette (ABC) efflux transporters, such as P-glycoprotein, multi-drug resistance (MDR) proteins, and BCRP, have been linked to decreased drug uptake and treatment resistance38,39. Many of these targets have not been evaluated in early-phase trials. Identifying other mediators of drug or effector T cell efflux across the BTB are important considerations to maximize efficacy of immune-based therapies. Other strategies in development include drug-ligand compounds or agents targeting BTB receptor peptides to either trigger receptor-mediated endocytosis across the BTB or prevent efflux of the drug out of the BTB (Figure 2C)22. Moreover, techniques such as drug administration with radiation or focused ultrasound in an attempt to ‘open up’ the BTB are being evaluated22.
Genomic mediators of brain metastases
A logical step towards developing effective systemic therapies for BM is to leverage genomic differences between primary, extracranial, and intracranial metastases to identify molecular drivers of CNS tropism (Figure 3)40. In recent years, increasing availability and size of whole exome (WES) and genome sequencing (WGS) datasets for metastatic tumors have facilitated the identification of candidate variants for functional validation. 41. An analysis of BM of diverse histologies and patient-matched primary tumors and ECM demonstrated near-universal genomic divergence between primary tumor and BM (Figure 3) with more than 50% of BM harboring clinically actionable mutations not detected in the primary tumor. Clinically actionable somatic variants such as CDK, PI3K, and MAPK pathway alterations, were common in BM41,42. Another study in NSCLC patients found that CDK or PI3K pathway alterations or WNT pathway activation were associated with a higher incidence of BM in NSCLC patients42. Furthermore, anatomically distinct BM from the same patient were concordant for 97% of clinically informative driver mutations41. This genomic homogeneity of BM across histologies suggests that unique histology-agnostic physiologic changes are needed for tumors to colonize the CNS.
Figure 3: A translational workflow in analyzing patient-derived brain metastasis tissue and identifying new therapeutic targets.
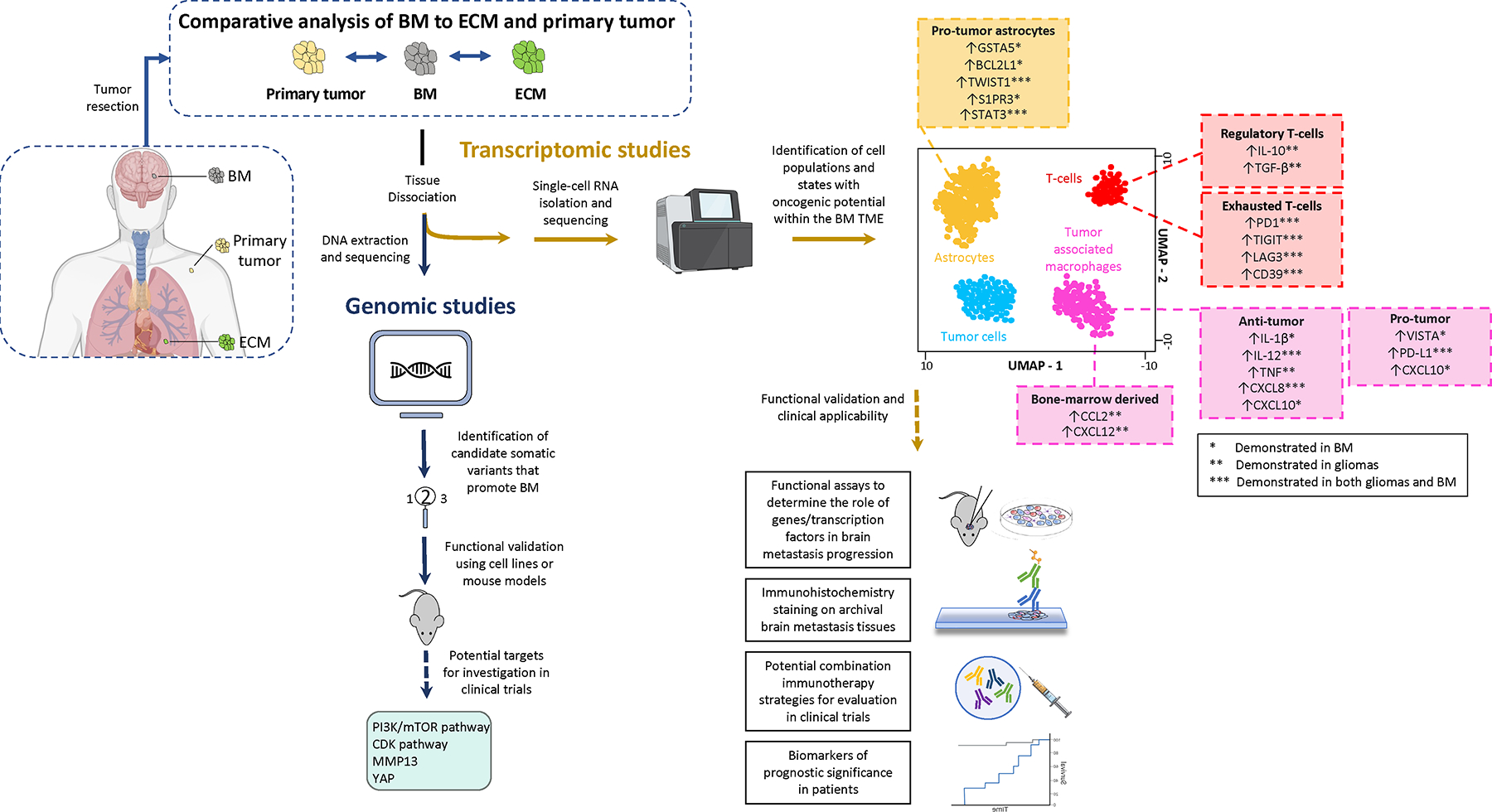
Comparative genomic, transcriptomic, immunogenic, and proteomic analyses of BM to ECM/primary tumor may result in a privileged view of mechanisms of CNS dissemination and therapeutic resistance. BM tissue can be submitted for either genomic or transcriptomic analysis. Following DNA sequencing, comparative analyses between BM and primary tumor WES data may reveal somatic variants associated with increased frequency of BM. Functional validation of these candidate variants have revealed potential targets that are being investigated in trials. Similarly, single-cell expressing profiling has potential to understand TME cell populations or states that play a role in treatment response. While the majority of single-cell profiling efforts in brain tumors have been in gliomas, we anticipate some overlap between immunosuppressive signatures of gliomas to that of BM. These immunosuppressive and tumor-promoting facets of T, glial, and myeloid cell populations may provide new therapeutic opportunities.
Extensive efforts have been devoted to identifying the molecular traits that enable cancer cells to metastasize to the CNS. A WGS analysis of primary and metastatic breast cancer tissues noted that spatially distant metastases often possessed potential driver mutations (e.g. JAK-STAT inactivating mutations) not detected in the primary tumor43. A similar analysis for melanoma metastases revealed increased activation of the PI3K/AKT pathway within BM44. Another study employing a targeted NGS panel from 25,000 patients with metastatic cancers to identify associations between somatic variants and metastatic dissemination45. Metastases, compared to primary tumors, had higher chromosomal instability. Certain somatic alterations (e.g. TP53 mutation, PTEN loss) were associated with increased metastatic burden. Additionally, associations between genomic features and metastasis to specific target organs were detected. For example, lung adenocarcinoma patients with BM had higher frequencies of TP53 mutations, TERT amplification, and EGFR mutations, and a lower frequency of RBM10 mutations.
However, while differences in somatic mutations or copy number aberrations have been reported for diverse genes between BM and ECM, only a few pathways have been functionally validated using pharmaceutical inhibition, or genetic manipulation in brain-tropic cell lines and animal models - namely, the PI3K/mTOR46,47, OXPHOS pathway48, and matrix metalloproteinase family49. Recent clinical studies have shown intracranial efficacy with CNS-penetrant targeted therapies in patients with BM and oncogenic drivers50–56. CNS-penetrant PI3K inhibition in patients with PIK3CA-mutant breast cancer BM has shown promising activity57 and is now being evaluated in clinical trials (NCT04192981; NCT03994796). Similarly, CDK inhibitors have demonstrated activity in BM of diverse histologies that harbor alterations in the CDK pathway40. These findings have inspired the creation of CNS basket trials to evaluate CNS-penetrant targeted therapies for other druggable targets in BM58.
Other analyses suggest that the molecular makeup of the cancer cell, by itself, may not be the only driver of metastasis. A study using WGS data from 2500+ metastatic tumors did not find recurrent cancer-causing mutations associated with either metastasis or organ-specific spread59. Furthermore, there was a high degree of genomic concordance and minimal driver gene heterogeneity between patient-matched spatially distinct metastases. While mutations of metastatic tumors varied widely, commonly implicated genes included: TP53 (52%), CDKN2A (21%), PIK3CA (16%), APC (15%), KRAS (15%), and PTEN (13%). These findings have been corroborated60. WGS analysis of 250 anatomically distinct biopsy pairs collected at different times during a patient’s treatment revealed >90% concordance for a targeted panel of 219 potential oncogenic drivers61. We emphasize, however, that these studies had either a limited number or no BM and some of these findings may not be generalizable for BM physiology. Due to genomic heterogeneity between ECM and BM, we interpret these studies as further evidence that BM possess important biological differences from extracranial tumors. Extracranial organs generally possess similar cellular and metabolic traits with the organ from which the primary tumor originated10,62. Conversely, the brain contains electrically-active cell populations (e.g. astrocytes, microglia, oligodendrocytes) unique to the CNS63. As more than 99% of cancer cells that reach the brain die10, tumors must assume a unique set of physiologic adaptations, different from those seen in extracranial sites, to thrive within the brain parenchyma. Furthermore, as many genomic alterations recurrent within BM (e.g. CDK, PI3K pathway alterations) also occur in ECM41,59, brain colonization probably results from a complex combination of general metastatic pathways, brain-specific vulnerabilities, and cancer-host cell interactions. For example, the molecular makeup of a metastatic tumor can be dependent on its TME. Astrocyte-derived exosomes secrete microRNAs that induce PTEN loss within metastatic tumor cells in the brain64. This loss is reversed when cancer cells leave the brain. These conclusions are supported by an analysis of spatially separated metastases in a breast cancer murine model illustrating unique dynamic organ-specific transcriptional and metabolic signatures, which are dependent on the local TME65.
TME MODULATORS OF BRAIN METASTASES
A growing body of work illustrates synergistic interactions between astrocytes66, macrophages67–69, neutrophils67,70, and natural killer cells71 with cancer cells to potentiate immunosuppression and perpetuate BM growth. With growing availability of single-cell and spatial profiling techniques, there is great potential to identify molecular or transcriptional activation states that can serve as hypotheses for functional validation. As a detailed characterization of each cell population within the BM TME is beyond the scope of this Review, we have focused our attention on highlighting studies of non-malignant cell populations with high translational potential. For a more comprehensive discussion of CNS resident cells in the context of malignancy, we acknowledge expert work by our colleagues72–75. Furthermore, recognizing that BM TME investigation is largely a nascent field, we summarize important unresolved questions.
T lymphocytes
The brain has historically been regarded as an immune privileged organ, given the relative paucity of peripheral immune surveillance due to the BBB and lack of conventional lymphatic drainage. As described above, due to pathologic remodeling of the BBB, BM possess variable levels of antigen-presenting dendritic cells as well as CD4+ and CD8+ T-cells, which play key roles in dictating treatment outcomes. CD8+ T cell infiltration within tumors is necessary for ICI response76. Furthermore, the degree of CD3+ or CD8+ lymphocytes and production of inflammatory cytokines correlates with treatment response77,78.
While the bulk of studies evaluating intracranial T-cell dynamics are in in glioblastoma, available data suggests that the BM TME is more immunosuppressive than that of primary tumors or ECM. Several studies comparing patient-matched primary tumors and BM found reduced T cell infiltration and expansion, as well as inhibition of dendritic cell maturation and helper T cell signaling pathways, in BM48,79–81. Similarly, single-cell transcriptomic characterization for patient-matched primary lung cancer, ECM, and BM illustrated a shift towards immunosuppressive T cell phenotypes in metastatic sites82. In metastases, normal myeloid cell populations were replaced with immature dendritic cells, regulatory T cells (Tregs), and exhausted T cells82. Therefore, a logical strategy towards improved treatments for BM are to identify mechanisms that augment T cell entry and cytotoxicity into BM.
However, recent studies present a translational conundrum. As a complicated array of biological factors have been linked to recruitment, trafficking, function, and activation of T cells, it is probable that a multi-target strategy would be needed to fully augment a tumor’s immunogenicity. Interestingly, the presence of extracranial disease can impact intracranial T cell activity. Effector T cell recruitment into BM is augmented by the presence of extracranial tumor lesions via peripheral expansion of effector cells and activation of endothelial cells of the BBB to enhance lymphocyte trafficking into BM83. In addition, dendritic cells and Tregs play a key role in immunosuppression. Intra-tumoral dendritic cells tend to be more immature as the TME inhibits their differentiation. These immature cells can have downstream effects of suppressing T-cell proliferation and recruiting Tregs. Release of regulatory cytokines by Tregs can prevent activation of helper T cells and impair effector T cells. In a pre-clinical model of glioblastoma, targeting glucocorticoid-induced TNFR-related receptor (GITR) on Tregs augments the effect of pembrolizumab by blocking Treg activity and results in a potent anti-tumor effect84. To our knowledge, similar strategies have not yet been tested for BM. Importantly, there is potential in non-invasively monitoring these dynamic changes. A paired single-cell RNA- and T-cell receptor sequencing effort of patient-matched BM and CSF samples identified strong correlations between T-cell clonotypes and phenotypes, suggesting that T-cell dynamics within the BM can be monitored through serial lumbar punctures85.
While enhancing T-cell activation through ICI has demonstrated remarkable intracranial response rates for melanoma and non-small cell lung cancer, most patients with BM eventually progress within the CNS. Current efforts revolve around identifying mechanisms for acquired or innate ICI resistance and then using these putative targets to enhance BM immunogenicity. There is a pressing need to define the molecular factors that mediate T-cell recruitment or shift towards a pro-tumor (Treg) vs. anti-tumor (effector) phenotype. One difficulty in studying these questions is disentangling the roles that distinct TME cell types play in dictating T cell phenotype. While correlative studies using single-cell or spatial techniques have been explored, robust functional valdation is needed. Other strategies in development include using dendritic cell vaccines or chimeric antigen receptor (CAR) T cells to augment T-cell cytotoxic effects against tumor antigens in BM, or using intracranial radiation or targeted therapy in conjunction with ICI to increase T-cell recruitment and limit the effect of Tregs. However, it remains to be seen whether directly augmenting T-cell cytotoxic phenotypes can overcome the immunosuppressive effects of the glial and/or myeloid compartment.
Astrocytes
Astrocytes are the most abundant cell population within the CNS and have diverse homeostatic functions including maintenance of the BBB, neurotransmission, regulation of cerebral blood flow, and synaptic plasticity86,87. In homeostasis, they block the entry of peripheral immune cells into the CNS and mediate the local innate immune response to promote tissue repair. As astrocytes are found only in the brain, metastases that thrive within the brain likely acquire a set of adaptations that allow tumor cells to synergistically interact with an unfamiliar cell type.
Activated astrocytes are prevalent within BM88,89 and play a role in tumor invasion and proliferation. The physiologic factors that mediate the functional shift of astrocytes from the neuro-inflammatory anti-tumor ‘A1’ state to the neuro-protective pro-tumor ‘A2’ state are poorly characterized. In the non-diseased state, astrocytes produce plasmin to promote clearance of tumor cells that enter the brain90. Tumor cells can produce serpins, which are anti-plasminogen activators, to prevent plasmin generation and its metastasis-suppressive effects. Serpin levels in tumors and blood are associated with poor outcomes90. Astrocyte function is also state-dependent; for example metastatic breast cancer cells secrete inflammatory cytokines such as IL-1-beta which ‘activates’ surrounding astrocytes91. Interactions between these activated astrocytes and tumor cells result in an upregulation of stem cell signaling pathways, which ultimately render tumor cells resistant to the deleterious effects of chemotherapy and reactive oxygen species.
In the pro-tumor state, astrocytes facilitate brain invasion through expressing the matrix-degrading enzyme heparinase92. Astrocytes relax endothelial cell junctions through secretion of inflammatory cytokines, such as interferon-alpha and tumor necrosis factor64. Additionally, extravasated cancer cells employ the cadherin-related protein PCDH7 to form connexin 43 (CX43) gap junctions with surrounding astrocytes93. Afterwards, cancer cells exhibit a gap junction-dependent up-regulation of survival genes (GSTA5, BCL2L1, TWIST1) that correlates with chemoresistance94,95, neovascularization, and a shift towards a pro-tumor astrocyte phenotype96,97. Interestingly, in pre-clinical models, brain-permeable gap junction inhibitors reduced intracranial disease burden when given in a therapeutic schedule and augmented the efficacy of chemotherapy93.
Single cell profiling is facilitating the identification of functionally relevant subpopulations of astrocytes and their roles in brain tumors. Available data illustrate substantial region-specific heterogeneity and functional diversity98,99 as astrocytes can be re-programmed from a metastasis-hostile environment to a tumor-promoting immunosuppressive environment33,100. Further complicating this study is that these diverse astrocytic transcriptional signatures do not operate in a vacuum, and interact with microglia, monocytes, and T cells to potentiate these effects. Nonetheless, astrocytes have promising translationally-relevant features. RNA microarray analysis of murine breast cancer BM revealed differential expression of astrocytic sphingosine-1 phosphate receptor 3 (S1PR3) in the neuroinflammatory response of low and high permeability metastases. S1PR3 activation directly resulted in increased BTB permeability to systemic therapy and recruitment of lymphocytes from the peripheral circulation33. Moreover, analysis of single-cell profiling and cell culture models revealed astrocytes within breast cancer BM expressed brain-derived neurotrophic factor and tumor cell tropomyosin kinase receptor B in an estrogen-dependent manner101,102. These factors increased the invasive and tumor-initiating capabilities of breast cancer. This process was inhibited by estrogen depletion, suggesting that astrocytes could be modulated using hormonal therapies. Another study identified an anti-inflammatory subset of astrocytes driven by STAT3 that blunt microglial and tumor-specific T-cells through secreting immune-suppressive cytokines66. Inhibition of STAT3 resulted in impaired viability of tumor cells and reduced outgrowth of BM, suggesting STAT3 may be a therapeutic target. However, as STAT3+ astrocytes are also associated with neurodegenerative disorders103,104, additional investigation is needed to identify mechanisms of BM-specific immunosuppression. Furthermore, as STAT3+ astrocytes are frequently located at BM tumor margins, newer techniques such as spatial transcriptomics may be helpful to identify astrocyte interactions with other resident cell populations that contribute to treatment resistance. These insights will enable rational development of astrocyte-directed therapies for BM.
Tumor-associated macrophages
Tumor-associated macrophages (TAM) for BM consist of microglia and bone-marrow derived macrophages (BMDM). Microglia are macrophages native to the CNS and function as the first line of defense for the innate immune system within the brain parenchyma. BMDM do not usually infiltrate healthy brain but can be recruited to the CNS through mechanisms mediated by CCL2 and CXCL12105,106. BMDM and microglia can comprise up to half of all cells within the TME67,107,108 and illustrate temporal and region-specific transcriptional diversity, dictated by the tissue environment109,110. A thorough mechanistic understanding of TAM-mediated tumor potentiation may identify candidate interactions of therapeutic interest. Here, we summarize available TAM data of therapeutic interest in BM, recognizing that the bulk of TAM exploration in brain tumors, to date, have been in gliomas.
In the initial response to inflammation, microglia repair the damage incurred on the BBB by transmigration of tumor cells, which can protect micrometastases from the tumoricidal effect of systemic therapies. TAM can also transform its phenotype to phagocytose the BBB to facilitate leakage of peripheral immune cells and cytokines into the CNS, which cause widespread inflammation111. After tumor cell extravasation, microglia accumulate at the point of contact between tumor cells and brain parenchyma. Tumor cells use microglial processes in a WNT-dependent fashion to invade and proliferate within the brain parenchyma112. Once macrometastases are established, BM control the recruitment of BMDM to the TME through chemokines such as CCL2 and CX3CL1106,113. BMDM subsequently lose phagocytic activity and acquire tumor-supportive microglial-like states to adapt to the BM TME. In gliomas, this shift to a pro-tumoral TAM state is associated with immunosuppressive gene expression signatures and cytokines (e.g. PD-L1114–116, TGF-beta114,115, IL-10), which have gained traction as potential therapeutic targets. More studies specific for BM are needed.
Until recently, single cell profiling of TAM subsets in brain tumors have been hindered by challenges in differentiating between microglia and BMDM and the perturbation of microglial phenotype with conventional tissue dissociation and isolation methods. Discovery of unique markers for microglia and BMDM117–119 (e.g. TMEM119, CX3CR1, and CD49D) have enabled study of these respective populations. In a comparison of TAMs, BM and IDH-wild type gliomas generally possessed a more activated microglial phenotype compared to IDH-mutant gliomas. BM-associated microglia had greater type I interferon signaling and inflammatory nuclear factor-KB (NFKB) signaling, as well as increased expression of CXCL8, a neutrophil attractant. Histopathology review of BM confirmed substantial infiltration of T cells and neutrophils, compared to gliomas67. The functional significance of these transcriptional programs is still being explored. In BM, TAM subsets were differentiated by varying activity of antigen presentation pathways, and expression of pro-inflammatory genes (e.g. hypoxia-inducible factor 1a, IL-1B, VEGFA)120. This transcriptional heterogeneity hints at unique pro-tumoral or anti-tumoral roles performed by different TAM subsets. One of these subpopulations exhibited upregulation of CXCL10, which then promoted an immunosuppressive niche - associated with the molecules VISTA and PD-L1 - and enhanced BM growth. In a transgenic murine model, CXCL10 or VISTA inhibition in combination with anti-PDL1 treatment reduced immune suppression and resulted in BM regression69. These results highlight the dynamic flexibility of CXCL10, as this molecule promotes T-cell recruitment in certain contexts but also immunosuppression through microglial expression of VISTA and PD-L1. Understanding what mediates this plastic phenotypic shift would have high therapeutic potential.
THERAPEUTIC STRATEGIES UNDER DEVELOPMENT
Recent studies have shown intracranial benefit with CNS-penetrant targeted therapies and ICI. These advances have been covered in prior clinical reviews25,121,122. However, the majority of patients with BM exhibit intrinsic or acquired resistance to these therapies. At present, two major categories of BM treatments under investigation include: 1) anti-neoplastic agents that overcome the BBB and 2) targeted or immune-based therapies that interrupt tumor-host cell interactions. A list of select ongoing trials designed specifically for BM patients is listed in Table 1.
Table 1:
Select clinical trials evaluating systemic therapies for patients with BM
| NCT (Phase) | Inclusion / exclusion criteria | Planned Enrollment | Intervention | Study Design | Primary Outcome |
|---|---|---|---|---|---|
| Targeted Therapy | |||||
| NCT04992013 / (2) | CNS basket trial; presence of progressive BM and BRCA1, BRCA2, PARP, DNA repair pathway, or homologous recombination gene alteration within BM | 20 | Niraparib | Single arm open-label | Intracranial clinical benefit rate |
| NCT03994796 / (1/2) | CNS basket trial; presence of progressive BM and CDK pathway, PI3K pathway, or NTRK/ROS1 alteration within BM | 150 | Abemaciclib Paxalisib Entrectinib |
Non-randomized open-label parallel assignment: Arm 1: CDK pathway alteration Arm 2: PI3K pathway alteration Arm 3: NTRK/ROS1 alteration |
Intracranial clinical benefit rate |
| NCT02896335 / 2 | CNS basket trial; presence of progressive BM and CDK pathway alteration within BM | 30 | Palbociclib | Single arm open-label | Intracranial clinical benefit rate |
| NCT03898908 / 2 | BRAF-mutant metastatic melanoma without prior local treatment in the brain | 38 | Encorafenib / binimetinib | Non-randomized open-label parallel assisgnment: Arm 1: asymptomatic BM Arm 2: symptomatic BM |
Intracranial response rate |
| NCT04158947 / 1/2 | HER-2 positive breast cancer BM | 130 | Afatinib and trastuzumab emtansine (T-DM1) | Randomized two-arm study: Arm 1 – T-DM1 + afatinib in dose escalation Arm 2 – T-DM1 + afatinib vs. T-DM1 |
Maximum tolerated dose of afatinib when used in combination with T-DM1; intracranial response rate |
| NCT04752059 / 2 | HER-2 positive breast cancer BM | 15 | Trastuzumab deruxtecan (T-DxD) | Single arm open-label | Intracranial response rate |
| NCT04185883 / 1b/2 | Basket trial for KRAS G12C-mutant solid tumors (progressive BM are allowed) | 1054 | Sotorasib and histology-specific therapeutic | Non-randomized, open-label, multi-arm (for each histology) | Maximum tolerated dose of sotorasib in combination with other therapeutics; extracranial response rate |
| Astrocyte-directed Therapy | |||||
| NCT01904123 / 1 | Progressive melanoma BM or glioma | 8 | WP1066 (STAT3 inhibitor) | Single arm open-label | Maximum tolerated dose; incidence of adverse events |
| NCT02429570 / pilot | Progressive BM of any histology | 30 | Meclofenamate (CX-43 inhibitor) | Single arm open-label | Feasibility |
| Chemotherapy (prevention trial) | |||||
| NCT03190967 / 1/2 | Histology-confirmed HER-2 positive breast cancer BM | 125 | T-DM1 Temozolomide (TMZ) |
Randomized open-label sequential assignment Arm 1 – T-DM1 + TMZ in dose escalation Arm 2 – T-DM1 alone |
Median time to developing new BM; maximum tolerated dose of TMZ when used in combination with T-DM1; |
| Combination immunotherapy regimens | |||||
| NCT04789668 / 1/2 | Progressive BM of any histology | 36 | Bintrafusp alfa + pimasertib | Single arm open-label | Intracranial clinical benefit rate; incidence of dose-limiting toxicities |
| NCT03175432 / 2 | BRAF V600 wild type melanoma BM | 60 | Atezolizumab Bevacizumab Cobimetinib |
Non-randomized open-label parallel assignment Arm 1: atezolizumab, bevacizumab Arm 2: atezolizumab, bevacizumab, cobimetinib |
Intracranial response rate |
| NCT03131908 / 1/2 | Metastatic melanoma with PTEN loss (progressive BM can be enrolled) | 36 | GSK2636771 (PI3K inhibitor) Pembrolizumab |
Single arm open-label | Maximum tolerated dose of GSK2636771 in combination with pembrolizumab; extracranial and intracranial response rate |
| NCT02910700 / 2 | BRAF V600 mutant metastatic melanoma that has progressed on prior PD-1 inhibition (progressive BM can be enrolled) | 51 | Binimetinib Dabrafenib Encorafenib Trametinib Nivolumab |
Non-randomized open-label parallel assignment Arm 1 – nivolumab, dabrafenib Arm 2 – nivolumab, trametinib Arm 3 – nivolumab, encorafenib, binimetinib |
Extracranial and intracranial response rate |
| NCT02886585 / 2 | CNS metastasis of any histology Arm 1 – untreated BM Arm 2 – progressive BM Arm 3 – neoplastic meningitis Arm 4 – 1–4 melanoma BM |
102 | Pembrolizumab Stereotactic radiosurgery |
Non-randomized open-label parallel assignment Arm 1, 2, 3 – pembrolizumab Arm 4 – pembrolizumab + SRS |
Intracranial response rate |
| NCT02816021 / 2 | Metastatic melanoma (progressive BM can be enrolled) Arm 1 – PD-1 naïve Arm 2 – post PD-1 progression |
24 | Azacitidine Pembrolizumab |
Non-randomized open-label parallel assignment | Extracranial and intracranial response rate |
| CAR T-cell therapy | |||||
| NCT03696030 / 1 | CNS metastasis of any histology with HER2-overexpression | 39 | CAR T cell targeting HER2 antigen | Single arm open-label | Treatment-related adverse events |
| Dendritic cell vaccine | |||||
| NCT04348747 / 2 | Triple-negative or HER-2 positive breast cancer BM | 23 | Anti-HER2 / HER3 dendritic cell vaccine | Single arm open-label | Intracranial response rate |
As BM harbor biological differences from primary tumors or ECM, BM genomic characterization and functional studies are instrumental to identify therapies effective for this patient population. Recent studies40,50–52,54–57,123 suggest that targeting oncogenic drivers within BM is a viable therapeutic strategy. Larger multi-institutional studies are now validating this hypothesis58. However, the majority of BM do not possess a genomic alteration that can be targeted with currently available CNS-penetrant targeted therapies41. Pre-clinical studies have identified the PI3K pathway46,124, OXPHOS pathway48, and ST6GALNAC526 are druggable targets in BM. CNS-penetrant inhibitors for these targets are either under development or being evaluated in clinical trials. Furthermore, cancer cells adapt to the BM TME by altering gene and protein expression profiles in situ125,126. Another strategy might be to identify and target the specific vulnerabilities that arise in the process of adapting tumor cells to the BM TME.
While selectively targeting activated oncogenic pathways may result in high initial efficacy, single-agent molecular therapeutics eventually fail in metastatic cancers as tumors will inevitably develop resistance mechanisms. Drugs aimed instead at the BM TME may circumvent this - a prominent example being programmed death ligand (PD-L1), which is expressed on TAM127,128 and mediate CD4+ T-cell suppression129 and Treg expansion114. 130 Combined blockade of CTLA-4 and PD1-PDL1 recently demonstrated increased intracranial efficacy, compared to single-agent ICI130,131. However, many BM patients do not respond even to combination ICI; thusrecent efforts have proposed combining PD1-PDL1 blockade with other CNS TME-targeting agents, such as inhibitors of of TAM receptor tyrosine kinases (RTK - e.g. TYRO3, AXL, MERTK). TAM RTK suppress innate immune responses within the TME. In vivo TAM RTK inhibition with anti-PD1 ICI extended survival in preclinical models of melanoma BM through increasing CD8 T-cell infiltration132. Gap junction and STAT3 inhibitors aimed at tumor astrocytes have shown activity for BM in pre-clinical models66,93. STAT3 inhibitors are now being evaluated in phase I trials for melanoma BM (NCT01904123). As TAM characterization for BM continues to mature, we anticipate study of astrocyte-TAM-tumor interactions will yield potential mechanisms that tumor cells employ to reprogram TAM and astrocytes to an immunosuppressive pro-tumor state. This knowledge can be used to propose immune-based therapies that revert these native cells to an anti-tumor state.
Ultimately, given distinct molecular differences and a highly unique TME in BM, compared to ECM, treatments effective for BM may not always be effective for other metastases. In cases where BM harbor a distinct oncogenic driver not detected within the primary tumor or ECM, systemic agents targeting the BM may not result in extracranial responses40,41. As more CNS-specific targets are identified, treating a metastatic cancer patient will likely necessitate a combination of BM-specific drugs, local therapies (e.g. radiation) as well as treatments to target extracranial metastases. However, such combinatorial approaches will require adequate clinical investigation to understand and minimize toxicities. We thus encourage multi-organ TME-based studies to identify shared contributors of metastasis, as well as increased study of BM prevention paradigms, as BM may be easier to treat prophylactically compared to once they have already formed.
BM prevention models are a relatively understudied concept. Prophylactic cranial irradiation was previously recommended for patients with solid tumors at high risk of spreading to the CNS, but has recently fallen out of favor due to neurocognitive sequelae. Therefore, pharmacologic prevention could be attractive to high-risk patients. There is an increasing body of pre-clinical evidence that systemic therapies given at preventative dosing may have higher efficacy than given at therapeutic dosing when macro-metastases have already formed10,133,134. Post-hoc analyses from clinical studies also support this hypothesis. Patients with metastatic RCC treated with sorafenib showed a 75% reduction in development of BM, compared to a 4% clinical response rate on established BM135. Similarly, lapatinib exhibited an intracranial response rate of 6% in patients with HER-2 positive breast cancer BM136, but follow-up analyses noted a significant reduction in the brain as the first site of relapse, which qualifies as a CNS preventative effect137. Recently, pre-clinical efforts have demonstrated preventative efficacy with temozolomide for breast cancer BM138 and PI3K/AKT inhibition for melanoma BM139. A phase 1 trial evaluating intracranial preventative potential of low-dose temozolomide for HER2-positive breast cancer patients is now enrolling (NCT03190967; Table 1).
Pharmacologic prevention is especially appealing due to the poor BBB penetrance of many systemic therapies. Furthermore, in a prevention scenario, it may be possible to lower doses of drugs, and therefore reduce the risk of toxicity, to achieve the desired effect. Targeting essential steps (e.g. vascular co-option) of the brain metastatic cascade may control the outgrowth of micro-metastatic tumor cells and reduce the risk of developing BM. To test candidate therapeutics, we encourage planning of prevention trials enrolling patients at high risk of developing BM (e.g. small cell lung cancer, patients with previously treated BM). A primary prevention trial measures a drug’s ability to prevent development of BM in patients without CNS disease140. These trials, however, can require years of follow-up, and CNS events following the initial development of a BM are ignored. Secondary prevention trials examine the efficacy of an intervention at preventing BM in patients who have been diagnosed with BM and are therefore at a high risk of developing further BM140. This trial design may be a more feasible way of providing initial evidence of a preventative effect. In this scenario, a possible endpoint would be time to development of a new BM.
Finally, another exciting effort is the investigation of non-invasive biomarkers reflective of BM physiology. While precision medicine approaches for BM have demonstrated promising responses, many patients are not able to benefit from this treatment paradigm as molecular or transcriptional analysis of BM is not usually feasible due to the morbidity associated with brain biopsy. One emerging technique to inform the selection of genotype-directed therapies is liquid biopsy. Multiple studies have demonstrated that CSF cell-free tumor DNA (cfDNA) provides a high-fidelity representation of the glioma genome141–143. However, tumor-derived DNA is shed into the CSF for only 40–60% of patients with malignant brain tumors142–144. While the amount of tumor-derived DNA within the CSF correlates with brain tumor burden144, more studies assessing the worth of CSF cfDNA as a prognostic and therapeutic biomarker for BM (e.g. concordance of cfDNA to brain and ECM) are needed. If these issues are optimized, liquid biopsy-based approaches hold promise to increase patient enrollment in future CNS basket trials, particularly those who are poor surgical candidates.
CONCLUSION
Treatment of BM has dramatically improved over the past decade owing to advances in molecular profiling of BM, targeted agents, and immunotherapy. To bolster this progress, we encourage study of diverse facets of BM biology - from individual steps of the brain metastatic cascade to cell populations within the TME – to reveal potential vulnerabilities and prognostic biomarkers (Figure 3. To date, the role of the BM TME and its contribution to the immune response and BM growth are poorly defined. The investigation of the BM TME at the level of single cells to identify candidate gene, protein, or metabolic changes is of great therapeutic interest. Furthermore, burgeoning advances in spatial transcriptomics and proteomics hold promise to transform our understanding of synergistic cross-talk between cell populations in the TME with cancer cells. By understanding the influence of these facets of BM biology on clinical outcomes, we hope our vision of personalized treatment for BM is becoming closer to reality. Clinical applicability of these translational studies will facilitate the development of effective precision-based treatment paradigms and the identification of future targets for BM-directed therapies.
Acknowledgments:
The authors acknowledge helpful feedback by Gromit Persimmon Perrino. PKB is supported by the NCI (1R01CA227156–01, 5R21CA220253–02 and 1R01CA244975–01) and Breast Cancer Research Foundation. AEK is supported by an American Brain Tumor Association Basic Research Fellowship In Honor of Paul Fabbri (BRF1900017), the William G. Kaelin, Jr., MD, Physician-Scientist Award of the Damon Runyon Cancer Research Foundation, American Association for Cancer Research Breast Cancer Research Fellowship, and American Society of Clinical Oncology/Conquer Cancer Young Investigator Award.
Footnotes
Competing Interests Statement: PKB has consulted for Tesaro, Angiochem, Genentech-Roche, ElevateBio, Eli Lilly, SK Life Sciences, Pfizer, Voyager Therapeutics, Sintetica, MPM, Advise Connect Inspire, Kazia,and Dantari, received institutional research funding (to MGH) from Merck, Mirati, Eli Lilly, BMS and Pfizer and has received honoraria from Merck, Medscape, Pfizer, and Genentech-Roche. The remaining authors declare no competing interests.
REFERENCES
- 1.Berghoff AS et al. Predictive molecular markers in metastases to the central nervous system: recent advances and future avenues. Acta Neuropathologica vol. 128 879–891 Preprint at 10.1007/s00401-014-1350-7 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Paterson AHG, Agarwal M, Lees A, Msc JH & Szafran O Brain Metastases in Breast Cancer Patients Receiving Adjuvant Chemotherapy. doi: 10.1002/1097-0142(19820215)49:4. [DOI] [PubMed]
- 3.Sundermeyer ML, Meropol NJ, Rogatko A, Wang H & Cohen SJ Changing patterns of bone and brain metastases in patients with colorectal cancer. Clin Colorectal Cancer 5, 108–113 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Nussbaum ES, Djalilian HR, Cho KH & Hall WA. Brain metastases. Histology, multiplicity, surgery, and survival - PubMed. Cancer 78, 1781–1788 (1996). [PubMed] [Google Scholar]
- 5.Habbous S et al. Incidence and real-world burden of brain metastases from solid tumors and hematologic malignancies in Ontario: a population-based study. Neurooncol Adv 3, 1–14 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barnholtz-Sloan JS et al. Incidence proportions of brain metastases in patients diagnosed (1973 to 2001) in the Metropolitan Detroit Cancer Surveillance System. Journal of Clinical Oncology (2004) doi: 10.1200/JCO.2004.12.149. [DOI] [PubMed] [Google Scholar]
- 7.Westover KD et al. Phase II trial of hippocampal-sparing whole brain irradiation with simultaneous integrated boost for metastatic cancer. Neuro Oncol 22, 1831–1839 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gradishar WJ et al. Clinical practice guidelines in oncology. JNCCN Journal of the National Comprehensive Cancer Network 16, 310–320 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Zhu W et al. Temozolomide for treatment of brain metastases: A review of 21 clinical trials. World J Clin Oncol 5, 19–27 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kienast Y et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med 16, 116–122 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Felding-Habermann B et al. Integrin activation controls metastasis in human breast cancer. Proceedings of the National Academy of Sciences 98, 1853–1858 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malin D et al. αB-crystallin: a novel regulator of breast cancer metastasis to the brain. Clin Cancer Res 20, 56–67 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carbonell WS, Ansorga O, Sibson N & Muschel R The Vascular Basement Membrane as “Soil” in Brain Metastasis. PLoS One 4, e5857 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karreman MA et al. Active Remodeling of Capillary Endothelium via Cancer Cell-Derived MMP9 Promotes Metastatic Brain Colonization. Cancer Res 83, (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Digernes I et al. Brain metastases with poor vascular function are susceptible to pseudoprogression after stereotactic radiation surgery. Adv Radiat Oncol 3, 559–567 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee SK, Huang S, Lu W, Lev DC & Price JE Vascular endothelial growth factor expression promotes the growth of breast cancer brain metastases in nude mice. Clin Exp Metastasis 21, 107–118 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Leenders WPJ et al. Antiangiogenic Therapy of Cerebral Melanoma Metastases Results in Sustained Tumor Progression via Vessel Co-Option. Clinical Cancer Research 10, 6222–6230 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Heyn C et al. In vivo MRI of cancer cell fate at the single-cell level in a mouse model of breast cancer metastasis to the brain. Magn Reson Med 56, 1001–1010 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Ghajar CM et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol 15, 807–817 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Indraccolo S et al. Cross-talk between Tumor and Endothelial Cells Involving the Notch3-Dll4 Interaction Marks Escape from Tumor Dormancy. Cancer Res 69, 1314–1323 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Banks WA From blood-brain barrier to blood-brain interface: new opportunities for CNS drug delivery. Nat Rev Drug Discov 15, 275–292 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Arvanitis CD, Ferraro GB & Jain RK The blood–brain barrier and blood–tumour barrier in brain tumours and metastases. Nature Reviews Cancer vol. 20 26–41 Preprint at 10.1038/s41568-019-0205-x (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dubois LG et al. Gliomas and the vascular fragility of the blood brain barrier. Front Cell Neurosci 8, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dubois LG et al. Gliomas and the vascular fragility of the blood brain barrier. Frontiers in Cellular Neuroscience vol. 8 Preprint at 10.3389/fncel.2014.00418 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Achrol AS et al. Brain metastases. Nature Reviews Disease Primers vol. 5 Preprint at 10.1038/s41572-018-0055-y (2019). [DOI] [PubMed] [Google Scholar]
- 26.Bos PD et al. Genes that mediate breast cancer metastasis to the brain. Nature 459, 1005–1009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sevenich L et al. Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat Cell Biol 16, 876–888 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolff G, Davidson SJ, Wrobel JK & Toborek M Exercise maintains blood-brain barrier integrity during early stages of brain metastasis formation. Biochem Biophys Res Commun 463, 811–817 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stemmler H & Heinemann V Central Nervous System Metastases in HER-2–Overexpressing Metastatic Breast Cancer: A Treatment Challenge. Oncologist (2008) doi: 10.1634/theoncologist.2008-0052. [DOI] [PubMed] [Google Scholar]
- 30.Lockman PR et al. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin Cancer Res 16, 5664–5678 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yonemori K et al. Disruption of the blood brain barrier by brain metastases of triple-negative and basal-type breast cancer but not HER2/neu-positive breast cancer. Cancer 116, 302–308 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Lockman PR et al. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clinical Cancer Research 16, 5664–5678 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gril B et al. Reactive astrocytic S1P3 signaling modulates the blood–tumor barrier in brain metastases. Nature Communications 2018 9:1 9, 1–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Osswald M et al. Impact of Blood-Brain Barrier Integrity on Tumor Growth and Therapy Response in Brain Metastases. Clin Cancer Res 22, 6078–6087 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Wyatt EA & Davis ME Method of establishing breast cancer brain metastases affects brain uptake and efficacy of targeted, therapeutic nanoparticles. Bioeng Transl Med 4, 30 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heye AK, Culling RD, Valdés Hernández MDC, Thrippleton MJ & Wardlaw JM Assessment of blood-brain barrier disruption using dynamic contrast-enhanced MRI. A systematic review. Neuroimage Clin 6, 262–274 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henry MN, Chen Y, McFadden CD, Simedrea FC & Foster PJ In-vivo longitudinal MRI study: an assessment of melanoma brain metastases in a clinically relevant mouse model. Melanoma Res 25, 127–137 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Sun H, Dai H, Shaik N & Elmquist WF Drug efflux transporters in the CNS. Adv Drug Deliv Rev 55, 83–105 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Fricker G & Miller DS Modulation of drug transporters at the blood-brain barrier. Pharmacology 70, 169–176 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Brastianos PK et al. Palbociclib demonstrates intracranial activity in progressive brain metastases harboring cyclin-dependent kinase pathway alterations. Nat Cancer 2, 498–502 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brastianos PK et al. Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov 5, 1164–1177 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang H et al. Original Article Genes Associated With Increased Brain Metastasis Risk in Non-Small Cell Lung Cancer: Comprehensive Genomic Profiling of 61 Resected Brain Metastases Versus Primary Non-Small Cell Lung Cancer (Guangdong Association Study of Thoracic Oncology. Cancer (2019) doi: 10.1002/cncr.32372. [DOI] [PubMed] [Google Scholar]
- 43.Yates LR et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 32, 169–184.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen G et al. Biology of Human Tumors Molecular Profiling of Patient-Matched Brain and Extracranial Melanoma Metastases Implicates the PI3K Pathway as a Therapeutic Target. Clin Cancer Res 20,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nguyen B et al. Genomic characterization of metastatic patterns from prospective clinical sequencing of 25,000 patients. Cell 185, 563–575.e11 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ippen FM et al. The dual PI3K/mTOR pathway inhibitor GDC-0084 achieves antitumor activity in PIK3CA-mutant breast cancer brain metastases. Clinical Cancer Research (2019) doi: 10.1158/1078-0432.CCR-18-3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shih DJH et al. Genomic characterization of human brain metastases identifies drivers of metastatic lung adenocarcinoma. Nat Genet (2020) doi: 10.1038/s41588-020-0592-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fischer GM et al. Molecular profiling reveals unique immune and metabolic features of melanoma brain metastases. Cancer Discov 9, 628–645 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Massagué J & Obenauf AC Metastatic colonization by circulating tumour cells. Nature 529, 298–306 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Davies MA et al. Dabrafenib plus trametinib in patients with BRAFV600-mutant melanoma brain metastases (COMBI-MB): a multicentre, multicohort, open-label, phase 2 trial. Lancet Oncol 18, 863–873 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gadgeel SM et al. Pooled analysis of CNS response to alectinib in two studies of pretreated patients with ALK-positive non-small-cell lung cancer. Journal of Clinical Oncology 34, 4079–4085 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Solomon BJ et al. Intracranial efficacy of crizotinib versus chemotherapy in patients with advanced ALK-positive non-small-cell lung cancer: Results from PROFILE 1014. Journal of Clinical Oncology 34, 2858–2865 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Bachelot T et al. Lapatinib plus capecitabine in patients with previously untreated brain metastases from HER2-positive metastatic breast cancer (LANDSCAPE): A single-group phase 2 study. Lancet Oncol 14, 64–71 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Shaw AT et al. Crizotinib versus Chemotherapy in Advanced ALK -Positive Lung Cancer. New England Journal of Medicine 368, 2385–2394 (2013). [DOI] [PubMed] [Google Scholar]
- 55.Soria J-C et al. Osimertinib in Untreated EGFR -Mutated Advanced Non–Small-Cell Lung Cancer. New England Journal of Medicine 378, 113–125 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Wu YL et al. CNS efficacy of osimertinib in patients with T790M-positive advanced non–small-cell lung cancer: Data from a randomized Phase III trial (Aura3). in Journal of Clinical Oncology vol. 36 2702–2709 (American Society of Clinical Oncology, 2018). [DOI] [PubMed] [Google Scholar]
- 57.Batalini F et al. Response of Brain Metastases From PIK3CA -Mutant Breast Cancer to Alpelisib. JCO Precis Oncol (2020) doi: 10.1200/po.19.00403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brastianos PK et al. Alliance A071701: Genomically guided treatment trial in brain metastases. Journal of Clinical Oncology (2020) doi: 10.1200/jco.2020.38.15_suppl.tps2573. [DOI] [Google Scholar]
- 59.Priestley P et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019 575:7781 575, 210–216 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reiter JG et al. Minimal functional driver gene heterogeneity among untreated metastases. Science 361, 1033–1037 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van de Haar J et al. Limited evolution of the actionable metastatic cancer genome under therapeutic pressure. Nature Medicine 2021 27:9 27, 1553–1563 (2021). [DOI] [PubMed] [Google Scholar]
- 62.Zhang XHF et al. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell 154, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boire A, Brastianos PK, Garzia L & Valiente M Brain metastasis. Nat Rev Cancer 20, 4–11 (2020). [DOI] [PubMed] [Google Scholar]
- 64.Zhang L et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 2015 527:7576 527, 100–104 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Basnet H et al. Flura-seq identifies organ-specific metabolic adaptations during early metastatic colonization. Elife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Priego N et al. STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis article. Nat Med 24, 1024–1035 (2018). [DOI] [PubMed] [Google Scholar]
- 67.Klemm F et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 181, 1643–1660.e17 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bowman RL et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep 17, 2445–2459 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guldner IH et al. CNS-Native Myeloid Cells Drive Immune Suppression in the Brain Metastatic Niche through Cxcl10. Cell 183, 1234–1248.e25 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang L et al. Blocking immunosuppressive neutrophils deters pY696-EZH2–driven brain metastases. Sci Transl Med 12, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Friebel E et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 181, 1626–1642.e20 (2020). [DOI] [PubMed] [Google Scholar]
- 72.Valiente M et al. The Evolving Landscape of Brain Metastasis. Trends Cancer 4, 176–196 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Quail DF & Joyce JA The Microenvironmental Landscape of Brain Tumors. Cancer Cell 31, 326–341 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Doron H, Pukrop T & Erez N A blazing landscape: Neuroinflammation shapes brain metastasis. Cancer Res 79, 423 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Klemm F et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 181, 1643–1660.e17 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Taggart D et al. Anti–PD-1/anti–CTLA-4 efficacy in melanoma brain metastases depends on extracranial disease and augmentation of CD8+ T cell trafficking. Proc Natl Acad Sci U S A 115, E1540–E1549 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Berghoff AS et al. Density of tumor-infiltrating lymphocytes correlates with extent of brain edema and overall survival time in patients with brain metastases. Oncoimmunology 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zakaria R et al. T-cell densities in brain metastases are associated with patient survival times and diffusion tensor MRI changes. Cancer Res 78, 610–616 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mansfield AS et al. Contraction of T cell richness in lung cancer brain metastases. Sci Rep 8, 2171 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kudo Y et al. Suppressed immune microenvironment and repertoire in brain metastases from patients with resected non-small-cell lung cancer. Annals of Oncology 30, 1521–1530 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Alvarez-Breckenridge C et al. Microenvironmental Landscape of Human Melanoma Brain Metastases in Response to Immune Checkpoint Inhibition. Cancer Immunol Res 10, 996–1012 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim N et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat Commun 11, 1–15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Taggart D et al. Anti–PD-1/anti–CTLA-4 efficacy in melanoma brain metastases depends on extracranial disease and augmentation of CD8+ T cell trafficking. Proc Natl Acad Sci U S A 115, E1540–E1549 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Amoozgar Z et al. Targeting Treg cells with GITR activation alleviates resistance to immunotherapy in murine glioblastomas. Nature Communications 2021 12:1 12, 1–16 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rubio-Perez C et al. Immune cell profiling of the cerebrospinal fluid enables the characterization of the brain metastasis microenvironment. Nat Commun 12, 1–10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Freeman MR & Rowitch DH Evolving Concepts of Gliogenesis: A Look Way Back and Ahead to the Next 25 Years. Neuron 80, 613 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ben Haim L & Rowitch DH Functional diversity of astrocytes in neural circuit regulation. Nature Reviews Neuroscience 2016 18:1 18, 31–41 (2016). [DOI] [PubMed] [Google Scholar]
- 88.Fitzgerald DP et al. Reactive glia are recruited by highly proliferative brain metastases of breast cancer and promote tumor cell colonization. Clin Exp Metastasis 25, 799 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Seike T et al. Interaction between lung cancer cells and astrocytes via specific inflammatory cytokines in the microenvironment of brain metastasis. Clin Exp Metastasis 28, 13–25 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Valiente M et al. Serpins promote cancer cell survival and vascular Co-option in brain metastasis. Cell 156, 1002–1016 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xing F et al. Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating Notch signalling in brain. EMBO Mol Med 5, 384–396 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Marchetti Dario, Li John & Shen Ruijun. Astrocytes Contribute to the Brain-metastatic Specificity of Melanoma Cells by Producing Heparanase | Cancer Research. Cancer Research 4767–4770 https://cancerres.aacrjournals.org/content/60/17/4767.long (2000). [PubMed] [Google Scholar]
- 93.Chen Q et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493–498 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kim SW et al. Role of the endothelin axis in astrocyte- and endothelial cell-mediated chemoprotection of cancer cells. Neuro Oncol 16, 1585–1598 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim SJ et al. Astrocytes upregulate survival genes in tumor cells and induce protection from chemotherapy. Neoplasia 13, 286–298 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang L et al. Astrocytes directly influence tumor cell invasion and metastasis in vivo. PLoS One 8, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stoletov K et al. Role of connexins in metastatic breast cancer and melanoma brain colonization. J Cell Sci 126, 904–913 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.John Lin CC et al. Identification of diverse astrocyte populations and their malignant analogs. Nat Neurosci 20, 396–405 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Batiuk MY et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nature Communications 2020 11:1 11, 1–15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Henrik Heiland D et al. Tumor-associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat Commun 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sartorius CA et al. Estrogen promotes the brain metastatic colonization of triple negative breast cancer cells via an astrocyte-mediated paracrine mechanism. Oncogene 2016 35:22 35, 2881–2892 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Contreras-Zárate MJ et al. Estradiol induces BDNF/TrkB signaling in triple-negative breast cancer to promote brain metastases. Oncogene 2019 38:24 38, 4685–4699 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Reichenbach N et al. Inhibition of Stat3-mediated astrogliosis ameliorates pathology in an Alzheimer’s disease model. EMBO Mol Med 11, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Haim L. Ben et al. The JAK/STAT3 Pathway Is a Common Inducer of Astrocyte Reactivity in Alzheimer’s and Huntington’s Diseases. Journal of Neuroscience 35, 2817–2829 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhu X, Fujita M, Snyder LA & Okada H Systemic Delivery of Neutralizing Antibody Targeting CCL2 for Glioma Therapy. J Neurooncol 104, 83 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mercurio L et al. Targeting CXCR4 by a selective peptide antagonist modulates tumor microenvironment and microglia reactivity in a human glioblastoma model. J Exp Clin Cancer Res 35, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Darmanis S et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep 21, 1399–1410 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Venteicher AS et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Böttcher C et al. Human microglia regional heterogeneity and phenotypes determined by multiplexed single-cell mass cytometry. Nat Neurosci 22, 78–90 (2019). [DOI] [PubMed] [Google Scholar]
- 110.Masuda T et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019 566:7744 566, 388–392 (2019). [DOI] [PubMed] [Google Scholar]
- 111.Haruwaka K et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nature Communications 2019 10:1 10, 1–17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pukrop T et al. Microglia promote colonization of brain tissue by breast cancer cells in a Wnt-dependent way. Glia 58, 1477–1489 (2010). [DOI] [PubMed] [Google Scholar]
- 113.Zhu X, Fujita M, Snyder LA & Okada H Systemic Delivery of Neutralizing Antibody Targeting CCL2 for Glioma Therapy. J Neurooncol 104, 83 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Aslan K et al. Heterogeneity of response to immune checkpoint blockade in hypermutated experimental gliomas. Nature Communications 2020 11:1 11, 1–14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ochocka N et al. Single-cell RNA sequencing reveals functional heterogeneity of glioma-associated brain macrophages. Nature Communications 2021 12:1 12, 1–14 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dusoswa SA et al. Glioblastomas exploit truncated O-linked glycans for local and distant immune modulation via the macrophage galactose-type lectin. Proc Natl Acad Sci U S A 117, 3693–3703 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bowman RL et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies Accession Numbers GSE86573 Bowman et al. CellReports 17, 2445–2459 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Satoh J ichi et al. TMEM119 marks a subset of microglia in the human brain. Neuropathology 36, 39–49 (2016). [DOI] [PubMed] [Google Scholar]
- 119.Bennett FC et al. A Combination of Ontogeny and CNS Environment Establishes Microglial Identity. Neuron 98, 1170–1183.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Schulz M et al. Cellular and Molecular Changes of Brain Metastases-Associated Myeloid Cells during Disease Progression and Therapeutic Response. (2020) doi: 10.1016/j.isci.2020.101178. [DOI] [PMC free article] [PubMed]
- 121.Suh JH et al. Current approaches to the management of brain metastases. Nature Reviews Clinical Oncology 2020 17:5 17, 279–299 (2020). [DOI] [PubMed] [Google Scholar]
- 122.Fecci PE et al. The Evolving Modern Management of Brain Metastasis. Clinical Cancer Research 25, 6570–6580 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bachelot T et al. Lapatinib plus capecitabine in patients with previously untreated brain metastases from HER2-positive metastatic breast cancer (LANDSCAPE): A single-group phase 2 study. Lancet Oncol 14, 64–71 (2013). [DOI] [PubMed] [Google Scholar]
- 124.Chen G et al. Molecular profiling of patient-matched brain and extracranial melanoma metastases implicates the PI3K pathway as a therapeutic target. Clinical Cancer Research 20, 5537–5546 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zeng Q et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 2019 573:7775 573, 526–531 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Park ES et al. Cross-species hybridization of microarrays for studying tumor transcriptome of brain metastasis. Proc Natl Acad Sci U S A 108, 17456–17461 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Maas SLN et al. Glioblastoma hijacks microglial gene expression to support tumor growth. J Neuroinflammation 17, 1–18 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Qian J et al. The IFN-γ/PD-L1 axis between T cells and tumor microenvironment: Hints for glioma anti-PD-1/PD-L1 therapy. J Neuroinflammation 15, 1–13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Butte MJ, Keir ME, Phamduy TB, Sharpe AH & Freeman GJ Programmed death-1 ligand 1 interacts specifically with the B7–1 costimulatory molecule to inhibit T cell responses. Immunity 27, 111–122 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Long GV et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: a multicentre randomised phase 2 study. Lancet Oncol 19, 672–681 (2018). [DOI] [PubMed] [Google Scholar]
- 131.Tawbi HA et al. Combined Nivolumab and Ipilimumab in Melanoma Metastatic to the Brain. New England Journal of Medicine 379, 722–730 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Holtzhausen A et al. TAM Family Receptor Kinase Inhibition Reverses MDSC-Mediated Suppression and Augments Anti–PD-1 Therapy in Melanoma. Cancer Immunol Res 7, 1672–1686 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Palmieri D et al. Profound prevention of experimental brain metastases of breast cancer by temozolomide in an MGMT-dependent manner. Clin Cancer Res 20, 2727–2739 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ilhan-Mutlu A et al. Bevacizumab Prevents Brain Metastases Formation in Lung Adenocarcinoma. Mol Cancer Ther 15, 702–710 (2016). [DOI] [PubMed] [Google Scholar]
- 135.Massard C et al. Incidence of brain metastases in renal cell carcinoma treated with sorafenib. Ann Oncol 21, 1027–1031 (2010). [DOI] [PubMed] [Google Scholar]
- 136.Lin NU et al. Multicenter phase II study of lapatinib in patients with brain metastases from HER2-positive breast cancer. Clinical Cancer Research 15, 1452–1459 (2009). [DOI] [PubMed] [Google Scholar]
- 137.Cameron D et al. A phase III randomized comparison of lapatinib plus capecitabine versus capecitabine alone in women with advanced breast cancer that has progressed on trastuzumab: updated efficacy and biomarker analyses. Breast Cancer Res Treat 112, 533–543 (2008). [DOI] [PubMed] [Google Scholar]
- 138.Zimmer AS et al. Temozolomide in secondary prevention of HER2-positive breast cancer brain metastases. Future Oncology 16, 899 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tehranian C et al. The PI3K/Akt/mTOR pathway as a preventive target in melanoma brain metastasis. Neuro Oncol 24, 213–225 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Steeg PS, Camphausen KA & Smith QR Brain metastases as preventive and therapeutic targets. Nat Rev Cancer 11, 352 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Pentsova EI et al. Evaluating cancer of the central nervous system through next-generation sequencing of cerebrospinal fluid. Journal of Clinical Oncology (2016) doi: 10.1200/JCO.2016.66.6487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Miller AM et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 565, 654–658 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang Y et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc Natl Acad Sci U S A (2015) doi: 10.1073/pnas.1511694112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.De Mattos-Arruda L et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nature Communications 2015 6:1 6, 1–6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]