

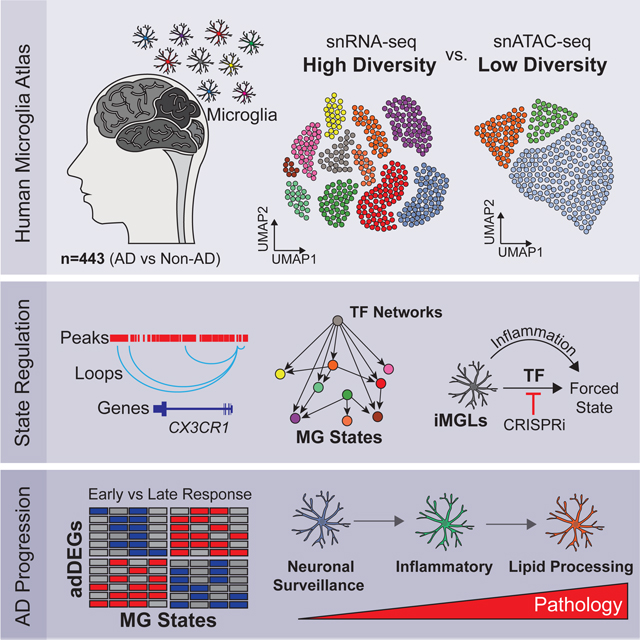
Summary
Altered microglial states affect neuroinflammation, neurodegeneration, and disease, but remain poorly understood. Here, we report 194k single-nucleus microglial transcriptomes and epigenomes across 443 human subjects, and diverse Alzheimer’s disease (AD) pathological phenotypes. We annotate 12 microglial transcriptional states, including AD-dysregulated homeostatic, inflammatory, and lipid-processing states. We identify 1,542 AD-differentially-expressed genes, including both microglia-state-specific and disease-stage-specific alterations. By integrating epigenomic, transcriptomic, and motif information, we infer upstream regulators of microglial cell states, gene-regulatory networks, enhancer-gene links, and transcription factor-driven microglial state transitions. We demonstrate that ectopic expression of our predicted homeostatic-state activators induces homeostatic features in human iPSC-derived microglia-like cells, while inhibiting activators of inflammation can block inflammatory progression. Lastly, we pinpoint the expression of AD-risk genes in microglial states and differential expression of AD-risk genes and their regulators during AD progression. Overall, we provide insights underlying microglial states, including state-specific and AD-stage-specific microglial alterations at unprecedented resolution.
IN BRIEF
Microglia states showing AD-risk-gene expression and AD-progression-associated expression differences were identified from the microglial transcriptome and epigenomes from the 443 human subjects spanning brain regions and diverse clinical and pathological states. Computational framework and functional studies using iPSC-derived microglia defined the diversity of microglial states across disease, the disease-stage changes of gene expression, and the regulatory network that governs microglial state transitions during the progression of AD.
Graphical Abstract
Introduction
Microglia accomplish their diverse roles in the brain by adopting a variety of phenotypic states that are thought to regulate their functional repertoire 1. Several studies have sought to characterize the diversity of microglial states across development, aging, and mouse models of Alzheimer’s disease 2–7. However, findings from mouse models of AD have been difficult to link to humans, as disease-associated microglia states can differ substantially between mouse models and enriched microglial signatures in brains of AD patients 8. Single-cell approaches to profile human microglia isolated from postmortem patient brains have been employed to elucidate cellular states associated with Alzheimer’s disease 9–13. However, due to the relatively low abundance of microglia in the brain, and the shallow depth of reads afforded by single-cell sequencing, the contributions of microglia and its cellular states to AD is still not fully understood.
Here, we analyzed 152,459 microglial transcriptomes across 443 individuals, identifying 12 transcriptional states and their relevance to AD. By integrating gene signatures with snATAC-seq, we unveiled key regulatory networks poised to regulate distinct microglial states. Using stem cell-derived microglia-like cells, we tested transcription factors’ ability to regulate microglial states. We determined AD-stage specific expression changes, linked AD risk genes with microglial states, and outlined potential regulatory pathways in AD prgression. Our data provides the resolution to define the cellular state and disease stage-specific contributions of microglia to AD.
Results
Characterization of Microglia Transcriptional States
To characterize microglia states, we analyzed the transcriptome of 152,459 single microglia nuclei from aged post-mortem brain samples of 217 AD (138 early and 79 late) and 226 control individuals (Table S1, page 1) across 6 brain regions, consisting of prefrontal cortex, hippocampus, mid-temporal cortex, angular gyrus, entorhinal cortex, and thalamus, from ROSMAP study14. We first collected 174,420 immune cells by in silico sorting from brain snRNA-seq datasets using known marker genes (STAR Methods) 11,15,16, which formed into 16 clusters, including 12 clusters of microglia (clusters 0–8 and 10–12, marked by CSF1R, CD74 and C3), one cluster of brain-associated macrophages (BAMs, cluster 9, marked by MRC1, CD163 and LYVE1), one cluster of T cells (cluster 13, marked by CD3E and CD3G), and two clusters of potential doublets with other brain cell types (clusters 14 and 15) (Figure S1A–B). We kept the 12 microglia clusters for in-depth analysis.
We defined these 12 clusters as distinct microglial states based on their molecular signatures and functions (Figure 1A–B and Figure S1C–G, Table S1, pages 2–3). We annotated homeostatic microglia (MG0) with high expression of well-known homeostatic markers P2RY12 and CX3CR1, and neuronal surveillance microglia (MG1) with high expression of neurotransmitter receptors (Figure S1F)17. We found that MG3 showed strongest enrichment to ribosome biogenesis (Figure S1G). We annotated MG4 as a lipid-processing state based on the enrichment of lipid homeostasis and regulation of cholesterol efflux. We annotated MG5 as a phagocytic state due to the significant enrichment of phagocytosis and receptor-mediated endocytosis terms. Of note, MG5 enrichment to phagocytic genes was relatively lower compared to the BAMs (Figure S1C). We annotated MG6 as a stress-related state based on the significant enrichment in response to unfolded protein; MG7 as a glycolytic state because of the enrichment in glycolytic process; MG11 as an antiviral state based on the strong enrichment of viral process and defense response to virus; and MG12 as a cycling state due to the enrichment in DNA replication. We also found three inflammatory states (MG2, MG8 and MG10) showing high expression of several cytokines and cytokine receptors (CCL3, IL1B, IL15, IL4R, IL17R, and IL10RA), and strong functional enrichment in immune responses including cytokine-mediated signaling pathway and regulation of inflammatory response. We further annotated MG2 as inflammatory I (CPEB4/TMEM163/IL4R), MG8 as inflammatory II (LRRK2/SPON1/FOXP1) and MG10 as inflammatory III (IL1B/CCL3/RELB). We next compared these 12 microglial states with a previously published dataset (Olah et al.) 12 and found that MG3 (ribosome biogenesis), MG6 (stress-related), MG10 (inflammatory III), MG11 (antiviral) and MG12 (cycling) states show strongly and significantly alignment across studies, while MG0 (homeostatic) state shows weak but significant alignment (Figure S1H–I, STAR Methods). In addition, comparison with mouse microglial transcriptional states yielded several well-aligned and conserved states across species4 (Figure S1J). We found that all 12 microglial states were robustly detected from each of six brain regions profiled (Figure S2A–B). However, the microglial states showed significant proportional differences across these regions (Figure S2B and S2C).
Figure 1. Microglial transcriptional states in aged human brains.

A. UMAP of 152,459 microglia nuclei with annotated microglial states in snRNA-seq data. B. Heatmap to show 2,228 highly expressed genes in 12 microglial states. The left block with matched colors shows the number of genes in each state. The right shows the representative genes. Z-scores across microglia states are used for the plot. C. Cell fraction distribution in AD pathological groups. Propeller analysis to determine the statistical significance highlighted with ***. D. Immunostaining of human postmortem brain tissue for the pan-macrophage marker IBA1 in cyan and the microglial homeostatic marker P2RY12 in magenta. E. RNAscope in situ hybridization of postmortem prefrontal cortex tissue probed for the microglial marker P2RY12 (red) and counterstained with the nuclei marker DAPI. F. Microglial fraction of P2RY12+ with co-detected MG4 marker, PPARG. G. Microglial fraction of P2RY12+ with co-detected MG2 markers, LRRK2 and FOXP1. E-G. Quantification shown from 8 subjects, averages from microglial fractions of total DAPI nuclei shown for 4 AD and 4 controls. Student’s t-test, ** p-value <0.01. n=91 to 149 cells per group. H. Enrichment analysis of mouse DAM signature across human microglial states. I. Enriched GO terms of highly expressed genes in MG3 vs. all states (upper panel), and MG4 vs. all states (lower panel). J. Co-detection analysis of PPARG and APOE in P2RY12+ microglia from 4 AD subjects with RNAscope. K. APOE and PPARG detection greater than 7 dots reveals discrete APOE+ populations. n= 152 P2RY12+ cells. See also Figure S1–3 and Table S1, pages 1–4.
To evaluate whether the relative proportions of these microglia states change in AD, we tested the statistical significance of cell fractional differences between control, early and late AD individuals in the prefrontal cortex which covered 423 individuals. We found that lipid-processing MG4 and inflammatory MG8 showed significantly-increased fractions in AD, while neuronal surveillance MG1 showed a significantly-decreased fraction in AD (Figure 1C, Figure S2D, STAR Methods). We also performed correlation analysis between the microglial state proportions and multiple pathological variables in PFC to explore the association of those states with specific AD-relevant pathology and confounding factors (Figure S2E, STAR Methods). We found that lipid-processing MG4 microglia showed the most significantly positive correlation with tangles, amyloid, Braak score, and cognitive decline. We found that inflammatory MG8 was more positively correlated with plaques, whereas phagocytic MG5 microglia were more positively correlated with tangles. We also observed that none of the microglial states were dominated by individuals and other covariates including batch, age, sex, or post-mortem interval (PMI) (Figure S2F–K). However, we found that the proportions of several microglial states were significantly different with age (MG5, MG4), PMI (MG1 and MG4), and sex (MG3) (Figure S2K), suggesting the importance of controlling these covariates when we evaluate the proportional changes between phenotypic groups.
We next sought to experimentally validate the enrichment of key transcriptional states in the AD human brain through immunohistochemistry and RNAscope in situ hybridization. Using the pan-macrophage marker IBA1 as a counterstain to label brain-resident macrophages, we performed immunohistochemistry for P2RY12 across 8 AD subjects which revealed high levels of P2RY12 immunoreactivity (Figure 1D and Figure S5). We only observed a small population of IBA1-positive P2RY12-negative cells within the brain parenchyma, perhaps reflecting CNS-infiltrating macrophages (Figure 1D). We reasoned that P2RY12 was therefore a compelling microglia-specific marker for in situ hybridization analysis. We performed RNAscope for P2RY12 in 4 AD and 4 non-AD individuals and found AD subjects had relatively lower fractions of P2RY12-expressing nuclei (Figure 1E).
Since MG4 state showed the relatively higher expression levels of PPARG in snRNA-seq data, we probed for PPARG through RNAscope in AD and non-AD brain tissues. We found a significant increase in the fraction of PPARG+P2RY12+ cells in AD subjects relative to controls (Figure 1F). Additionally, we also observed enrichment of FOXP1+LRRK2+ microglia in these subjects, an expression pattern that marks MG8 inflammatory states (Figure 1G). Collectively, these findings through in situ hybridization corroborates our snRNA cell fraction analysis and points to lipid-processing and inflammatory states as disease-associated microglial states.
We performed enrichment analysis to capture the expression of disease-associated microglia (DAM) genes across our dataset5 (Figure S1D–E). Interestingly, we observed the strongest enrichment of DAM signature genes in MG3, a microglial state that is proportionally unchanged in AD (Figure 1H). Of note, MG3 DAM enrichment is mostly driven by overlap in ribosome protein genes. In addition, we observed significant but relatively lower enrichments of mouse DAM signatures across multiple MG states, including lipid-processing MG4 and inflammatory MG10. Due to the higher prevalence of MG4 microglial fractions in AD brains, we compared the transcriptional programs enriched for MG3 and MG4. Gene ontology analysis for genes enriched in ribosome biogenesis MG3 state over all other microglial states revealed a suite of inflammatory genes enriched for cytokine production and microglial activation pathways (e.g. HLA-A, CD74, C1QB and APOE). For the lipid-processing MG4 state, this analysis uncovered an enrichment of cholesterol and lipid homeostasis genes (e.g. PPARG, APOE, ABCA1 and TREM2) (Figure 1I). High expression of APOE by microglia has been deemed a state marker for both DAM and a broader but similar activated microglia mouse-signature known as microglia neurodegenerative phenotype (MGnD) 18. To determine if higher levels of APOE expression could be distinguished into discrete populations, we performed RNAscope with probes targeting APOE, PPARG and the microglial marker P2RY12 (Figure 1J). We reasoned that levels of PPARG expression could segment high APOE expressing microglia into distinct populations that differentiated ribosome biogenesis MG3 from lipid-processing MG4. We discovered that while the vast majority of PPARG+ microglia expressed high levels of APOE (88% of all PPARG+ nuclei were co-detected with greater than 7 APOE dots), a significant fraction of APOE+ microglia lacked significant levels of PPARG detection (27.6% of all APOE+ nuclei were co-detected with less than 7 PPARG dots) (Figure 1K). Altogether, this indicates that DAM-like signatures in the human AD brain do not encompass a single-state, but rather can be distinguished across many substates.
Distinct Inflammatory States in the Aged Human Brain
Cell proportional analysis also revealed the enrichment of inflammatory MG8 state in AD subjects (Figure 1C). The presence of distinct inflammatory states is intriguing as it could reflect alternative transcriptional programs that are activated by distinct pro-inflammatory stimuli (Figure 2A). Nevertheless, functional categorization of pathways significantly enriched by inflammatory clusters pointed to a pattern of decreasing inflammatory status (MG10 > MG2 > MG8), suggesting that these clusters may instead represent states during the progression of inflammation (Figure 2B).
Figure 2. Microglial inflammatory states capture an inflammatory continuum.

A. UMAP of three inflammatory states of microglia. B. Enrichment of inflammation-related biological processes in three inflammatory states, represented by the -log10(p-value) in heatmap. C. Multiplexed immunostaining of top inflammatory state markers. IBA1 labeled for CNS resident macrophages and derived 3D rendering of cell morphologies, ILB1 for MG10, LRRK2 for MG8 and CPEB4 for MG2 microglia. D. Maximum projection of tile scan from a 2mm biopsy punch of prefrontal cortex tissue after alignment of 4 iterative rounds of RNAscope with inflammatory-state and microglial-specific probes. Inset shows 2 microglia nuclei marked by red arrows, and representative image of detection of microglial-specific probes (right panel). E. Higher magnification of composite image with inflammatory-state probe detection. F. Dendrogram and representative cells for inflammatory state probe detection of heatmap in Figure S4D (upper panel). Cell fraction analysis of 291 microglia for 4 AD subjects. Number of RNAscope dots Z-scored across all cells (lower panel). G. Images from representative microglia nuclei with signatures of discrete inflammatory states. H-J. Transcriptional kinetics of inflammatory state markers in iPS-derived microglia-like cells (iMGLs). I. Treatment with TNFα (40 ng/mL), INFγ (30ng/mL), or LPS (10 ng/mL) for 1 hour and profiled by qPCR. J. Treatment with pre-formed amyloid beta (1–42) fibrils and profiled by RNA-seq; Heatmap of early and late response up-regulated genes. K. Levels of CPEB4, IL1B and LRRK2 expression in response to amyloid treatment. n= 3 biological replicates per group. ANOVA with post-hoc Tukey test. *** p-value <0.001. See also Figure S3–4 and Table S1, pages 4–6.
To determine if these clusters represent distinct populations within the human brain, we immunostained fixed prefrontal cortex brain sections with the macrophage marker IBA1 and used 3D rendering by Imaris to build cellular traces that spatially delineated microglia cell bodies within these sections (Figure 2C, Figure S3). Next, we multiplexed IBA1 staining with additional antibodies against top markers for each cluster (namely, IL1B for MG10, LRRK2 for MG8 and CPEB4 for MG2) (Figure S4A). While IBA1 was highly expressed in all cells, we found instances when the highest expression for each cluster marker was largely in non-overlapping microglia (Figure S4B–C).
To investigate how co-expression of multiple inflammatory state markers can distinguish discrete inflammatory programs in the human brain, we turned to a multiplexing platform through RNAscope technology which allows for many probes to be co-detected through iterative rounds of imaging (STAR Methods). We simultaneously profiled the expression of 9 targets in 4 AD subjects to dissect the relationship of MG8, a weakly inflammatory state that is enriched in AD to other inflammatory states. We further resected prefrontal cortices into a 2mm disc with a biopsy punch so that we could image the entire sample and capture defined tissue borders to aid registration (Figure 2D). Using probes against P2RY12, CX3CR1 and IBA1 we assigned microglia nuclei identity, and subsequently assessed the co-detected levels of MG8 markers LRRK2 and FOXP1, MG2 markers CPEB4 and TMEM163 and MG10 markers CCL3 and RELB (Figure 2E). For cells that expressed more than one inflammatory marker, we found that inflammatory programs were largely overlapping, with MG2 signatures (CPEB4 and TMEM163) bleeding into MG8 and MG10 states in 23% of the inflammatory microglia (Figure 2F). The expression of MG8 and MG10 markers were correlated as demonstrated by the dendrogram depicted from the hierarchical clustering from quantification across all cells (Figure 2F and Figure S4D). In fact, we were able to detect expression of MG8 and MG10 markers in a small fraction of non-overlapping microglial populations, while non-overlapping MG2 states were rarely detected (Figure 2F–G). While it is challenging to extrapolate from these results given the semi-quantitative nature of RNAscope technology, the overlapping programs may be indicative of dynamic transitions in a continuum of inflammation.
We next investigated the transcriptional kinetics of these markers in relation to inflammation. To do so, we derived microglia-like cells (iMGLs) from induced pluripotent stem cells (iPSCs) following established protocols 19 and performed a pulse-and-chase experiment by briefly exposing these cells to distinct pro-inflammatory stimuli, such as Tissue Necrosis Factor-α (TNF-α), Interferon-γ (IFN-γ), bacterial Lipopolysaccharide (LPS) or pre-formed amyloid beta (1–42) fibrils. We then assessed the expression of inflammatory MG2, MG8 and MG10 markers over the course of 96 to 120 hours (Figure 2H and Table S1, page 5). We profiled the expression of IL1B and 6 additional markers of MG10 (including CCL3 and RELB) and observed that presentation of pro-inflammatory stimuli evoked a largely acute response that was completely resolved by 120 hours (Figure S4E–F). We found that markers for MG2 (CPEB4, LIMK2 and FAM129A) and MG8 (LRRK2, GPR155 and CYP27A1) showed sustained activation that were not resolved by the same time frame (Figure 2G and Figure S4E,G–H). To understand if a similar temporal response could be evoked with a more physiological proinflammatory stimulant, we treated iMGLs with pre-formed amyloid beta fibrils, and performed a similar pulse-and-chase experiment at 4 hours, 24 hours and 96 hours post amyloid seeding (Figure 2J). We found a rich transcriptional response that was enriched for inflammatory pathways and clustered by early (upon 4 hours) and late response (upon 24 hours) (Table S1, page 6). Interestingly, amyloid beta fibrils did not evoke CPEB4 transcription activation (MG2 state marker), but recapitulated the early onset of IL1B and the delayed activation of LRRK2 (Figure 2K). To understand if the temporal kinetics of inflammatory programs across these states are captured globally during disease progression, we analyzed the contribution of inflammatory states to transcriptional changes for early and late AD subjects. Indeed, MG10 shows greater transcriptional changes in early disease stages while MG2 and MG8 are more robustly changed in late stages (Figure S4E–H). Collectively, this data suggests that our microglia profiling might be capturing cells along a spectrum of inflammatory responses with amyloid-dependent and independent responses, and not likely bonafide discrete populations.
Prediction of Upstream Transcription Factors of Microglial States
To investigate the upstream regulators that drive distinct microglial states, we leveraged the chromatin accessibility data measured by snATAC-seq using human postmortem brain tissues from a subset of individuals in the same cohort (100 individuals, Table S1, page 7). We obtained 41,832 nuclei of microglia with good quality and captured three epigenomic states including one homeostatic state (HOM) and two activated states (ACT1 and ACT2, Figure 3A, Figure S5A–C, STAR Methods). We found that P2RY12, the homeostatic marker, showed relatively low accessible signals in activated states although ubiquitous accessibility in all microglia(Figure S5B–C). We also observed that CCL3 showed significantly higher accessibility in ACT1, suggesting that the microglia in this epigenomic state could be related to immune response. We also found that SPP1 showed high accessibility in ACT2, consistent with its functions to mediate cell activation and cytokine production 20.
Figure 3. Transcription factors regulate microglial states.

A. UMAP of 41,832 microglia nuclei with annotated epigenetic states in snATAC-seq data. B. Examples of co-accessible links between gene promoters and distal peaks to represent the epigenetic state specific peaks. C. Motif enrichment in the microglial transcriptional state marker genes associated accessible peaks. The -log10(p-value) represents the enrichment. Stars show the TFs that were highly expressed in the corresponding microglial state. D. Heatmap to show the TF enrichment of marker genes in each microglial transcriptional state. Odds ratio represents the enrichment. Stars show the TFs that were highly expressed in the corresponding microglial state. E. TF-target regulatory network in MG0 (homeostatic state). The TFs are combined from gene-set enrichment based and motif based analyses shown in C-D. The pink squared nodes represent the TFs. The light blue circle nodes represent the targets. The size of nodes represents the number of targets. See also Figure S5 and Table S1, pages 7–8.
We noticed that microglia show higher heterogeneity at the transcriptional level than the chromatin accessibility level (Figure 1A, Figure 3A). To evaluate whether this stems from technical bias or unbalanced number of microglia in these two modalities, we analyzed 4,244 microglia nuclei using 10x multiome from human postmortem brains of 19 individuals. We identified nine transcriptomic and two epigenomic clusters using individual modality (Figure S8D), validating our previous observation. We also found that the nine transcriptomic clusters can recapitulate the microglial states we identified in the larger cohort (Figure S5E).
We next called 179,534 accessible peaks and identified 16,800 epigenomic-state-specific peaks (Figure S5F–G, STAR Methods). We annotated these peaks by the number of associated genes and their distance to TSS, and predicted their functions (Figure S5H–I, STAR Methods). We found that homeostatic peaks were enriched near genes acting in regulation of calcineurin-NFAT signaling and lipoprotein particle clearance. Peaks in the activated states were significantly enriched in genes related to cellular response to interleukin, regulations of establishment and maintenance of blood-brain barrier (BBB), and cytotoxic T cell differentiation, consistent with previous findings that activated microglia impair BBB function by producing inflammatory proteins to induce hyperpermeability of BBB in brain diseases21.
To investigate the regulatory machinery of these microglial states, we predicted transcription factors (TFs) through motif enrichment in the peaks of these epigenetically-defined states (Figure S5J). We found that although several TFs (including SPI1, IRF8 and STAT1) showed ubiquitous enrichment in all three states, the enrichment for these factors were considerably stronger in activated states, perhaps reflecting the dynamic balance of microglial TFs that act in both cell fate maintenance and the activation of inflammatory programs. By contrast, several TFs (including ARID5B and ID2) showed specific enrichment in the activated states, suggesting their regulatory potential in certain conditions. We also observed that the peaks from the two activated states were associated with very similar functions and shared many TFs even though the peaks themselves were distinct (Figure S5G–J).
To further refine these gene-regulatory interactions, we linked distal peaks to promoters using the co-accessibility patterns of promoters and distal accessible peaks (STAR Methods). We found 122,658 peak-to-promoter links, implicating 17,109 genes and 46,498 peaks (Table S1, page 8). For example, we found that a peak with strong signal near CX3CR1 in the homeostatic state linked with its promoter, and CCL3 and CSF3R each had peak-to-gene links in a distinct activated state (Figure 3B).
With these co-accessibility, we identified 36 TFs that may modulate microglial states through motif analysis on the distal peaks of microglial state markers (Figure 3C, STAR Methods). To complement this method, we inferred 40 TFs whose known targets were enriched in microglial state makers22 (Figure 3D, STAR Methods). Notably, 7 TFs were identified in both methods, including FOXO3 (MG8), IRF8 (MG2, MG6, MG10), STAT3 (MG3) and RUNX1 (MG7). We also observed that 15 TFs (including SPI1 and CTCF) showed consistent enrichment across all microglial states, suggesting their functions in the maintenance of microglial cell identity and genomic architecture 23,24. Conversely, three TFs exhibited microglial-state-specific enrichment: TFDP1 in MG12 (cycling), aligned with its cell cycle regulation function25, and IRF7 in MG11 (antiviral), reflecting its role in type 1 interferons production for antiviral immunity26.
Several TFs, like ` in inflammatory MG8, were prominently expressed in microglial states, regulating marker genes (Figure 3C–D), aligning with its function in microglial inflammatory activation27. Similarly, HIF1A was highly expressed and significantly regulated the marker genes in MG7 (glycolytic), MG6 (stress) and MG10 (inflammatory), reflecting its functions in glycolytic metabolism, response to inflammation and stress of microglia 28–30. We also conducted single-cell TF activity analysis via DoRothEA31, revealing significant motif/gene-enrichment (Figure S5K).
To further investigate the regulatory mechanisms of TFs, we constructed a TF-target regulatory network (Figure 3E, STAR Methods) for homeostatic state MG0. We found that the homeostatic markers were usually regulated by multiple TFs, suggesting that the TFs may be acting cooperatively, or that transcriptional regulation may be redundant to maintain the robustness of the system. To test whether the TFs collaboratively control gene expression, we evaluated the significance of the shared targets between every pair of TFs (STAR Methods). We found that 13 TFs (including several ETS TF family members, SPI1 and CTCF) significantly shared their targets within the co-regulatory module (Figure S5L). We also observed that the other 14 TFs showed specificity or formed small modules with few TFs, suggesting that these TFs may control the gene expression of a subset of marker genes with specialized functions.
Ectopic Expression of Master Regulators of Homeostatic State in Human Microglia-like Cells
Microglia maintained in vitro adopt increased activated states relative to their in vivo counterparts 32. We postulated that the non-homeostatic nature of iMGLs could be used as a testbed to determine if ectopic expression of master homeostatic TFs is sufficient to drive features of homeostasis in these cells. We began by performing snRNA-seq on iMGLs to determine how in vitro iMGL states compared to in vivo MG states. In addition, we also treated iMGLs with LPS to understand the extent to which these cells can show inflammatory activation states that mirror inflammatory MG2, MG8 and MG10. From this experiment we were able to capture the transcriptome of 18,914 iMGL nuclei that formed 10 clusters, and that was largely segregated by treatment condition (i.e. baseline versus LPS enriched) (Figure 4A and Figure S6A). Computing correlation scores across iMGL and MG states revealed a high degree of similarity between MG12 (a cycling state) and C9, a cell-cycle enriched cluster representing proliferating microglia (Figure 4B and Figure S6A). In addition, we also observed similarities between inflammatory clusters: MG2 and MG10 showed strong similarities to LPS enriched clusters (C0,C8,C3,C6), while MG8 showed some correspondence to iMGLs at baseline (C1 and C4). As predicted, the microglial homeostatic MG0 state was not represented in iMGLs (Figure 4C). Since purinergic signaling via P2RY12 provides a window into the cellular state of microglia (i.e. P2RY12 expression is repressed by activated microglia), we previously leveraged functional ATP uncaging assays to functionally assess homeostatic microglia states in iMGLs bearing sporadic AD risk variants 33. We envisioned that we could use this assay as a functional readout of homeostasis. We began by examining the impact of LPS treatment on P2RY12 levels and determined that LPS dramatically reduces its expression (Figure 4D). Purinergic receptors are potent sensors of ATP, and the binding of ATP to P2RY12 robustly induces downstream intracellular calcium release. As such, we conducted calcium imaging in monocultures of iMGLs at baseline and upon exposure of ATP, with or without LPS pre-treatment (Supplemental Video 1 and Figure S6B). As predicted, we observed that iMGLs pre-treated with LPS showed blunted responses to extracellular ATP compared to baseline conditions (Figure 4F).
Figure 4. TF-driven regulation of microglial state dynamics in iMGLs.

A. UMAP of 18,914 iPSC-derived microglia (iMGL) nuclei, labeled by clusters. LPS enriched clusters shaded in light blue. UMAP with all clusters can be found in Figure S6. B. Enrichment of iMGL marker genes of each cluster in microglial transcriptional states from human post-mortem snRNA-seq data. C. Dotplot to show the expression of marker genes in iMGLs clusters. The size represents the percentage of cells with expression. The color represents the scaled average expression level. D. qPCR analysis of IL1B and P2RY12 expression in iMGLs following 12 hours of 10ng/mL of LPS treatment. Student’s t-test, *** p-value <0.001. n= the mean of 6 biological replicates per group. E. Raster plots for calcium transients in monocultures of iMGLs at baseline or upon ATP uncaging with 405nm stimulation. Peak amplitude change in fluorescent intensity (ΔF/F) is displayed from low (gray) to high (dark blue) in 15 second bins over 33 frames. n= 21 cells. F. TF overexpression screen 8 days post viral infection exposed to neuronal spheroid conditioned media to evoke calcium transients. ANOVA with post-hoc Tukey test. *** p-value <0.001; ** p-value <0.01; * p-value <0.05. n= 80 to 199 cells per group. Red filled bars represent statistically significant changes in contrast to control, while gray-filled bars represent non-significant differences. G. Actin stained iMGLs from RUNX2 and PPARG overexpression and morphology quantification. Image of control sample (iMGLs infected with empty-mCherry lentivirus) in Figure S6H. ANOVA with post-hoc Tukey test. *** p-value <0.001. n= 55 to 76 cells per group. H. Immunostaining of human brain tissue for homeostatic marker TMEM119 and pan-macrophage marker, IBA1. 3D rendering of IBA1 signal was performed on IMARIS and used to generate a mask delineating cell body, shown in yellow. Immunostaining for homeostatic TFs in IBA1 positive cells across the human brain. I. Regression analysis correlating levels of PPARG and RUNX2 in IBA1-positive cells with the homeostatic marker TMEM119. J-K. UMAP from CRISPRi iMGLs treated with LPS carrying sgRNAs targeting FOXO3, HIF1A and FOXP2. K. Enrichment of LPS-treated iMGLs genes for each cluster in microglial transcriptional states from human post-mortem snRNA-seq data. L. Diagram of microglial state transition dynamics. See also Figure S6 and Table S1, page 9.
We selected 25 TFs (Figure 3E) to perform an overexpression screen in iMGLs and assay for increased sensibility to extracellular ATP (i.e. greater homeostatic states) (Table S1, page 9). After 8 days of lentiviral transduction, we observed that 58% of cells were successfully infected based on the expression of a mCherry reporter cassette (Figure S6C). We opted to expose the iMGLs to neuronal conditioned media isolated from forebrain spheroid cultures as we have previously determined that this manipulation induced calcium transients in iMGLs via P2RY12, similarly to ATP uncaging 33. Controls were infected with lentivirus carrying an empty cassette (Figure S6D). We found that ectopic expression of 9 TFs induced significantly higher rates of calcium transients in iMGLs (Figure 4F). Greater calcium dynamics may not necessarily be indicative of homeostasis. To define if TF overexpression was indeed inducing homeostatic features, we next assessed iMGLs through additional approaches. First, we reasoned that microglia morphology could be informative in assessing its cellular state 34. In a dish, iPSC-derived microglia do not resemble the morphology of their homeostatic in vivo counterparts but yet undergo significant morphological changes upon further activation by adopting a more spherical ameboid-like morphology. Secondly, we could determine the relationship between TF expression and homeostatic markers in the human brain. Through these orthogonal approaches, we found that overexpression of RUNX2 and PPARG (2 of the hits from our calcium imaging) reduced the adoption of ameboid-shaped in iMGLs compared to controls (Figure 4G and Figure S6D). In addition, these two TFs were significantly correlated with higher expression of TMEM119 in IBA1-positive cells within the human brain (Figure 4H–I). Co-staining for TMEM119 and P2RY12 confirmed overlapping microglial populations, as TMEM119 is a well-established homeostatic marker 35 (Figure S6D). Through this analysis, we also determined that expression of SMAD3, IRF8 and ELF1 were not significantly correlated to microglial homeostasis in vivo although these were also hits from our calcium imaging screen (Figure 4I and Figure S6F–H).
Next, we tested if we could prevent microglial state dynamics by perturbing master regulators of inflammatory state transitions based on our transcription factor activity analysis. We reasoned that LPS could induce state changes that resembled the progression of weakly inflammatory MG8 to strong inflammatory MG10 states (i.e. C1 to C0 in iMGLs) (Figure 4A). We began by differentiating iMGLs from a dCAS9-KRAB CRISPRi iPSC line and delivering a total of 3 pairs of in vitro transcribed sgRNAs targeting transcription factors predicted by our model to drive inflammatory progression from MG8 into MG10 (FOXO3, HIF1A and FOXP2). 4 Hours after sgRNA delivery, control (transfected iMGLs, but no sgRNAs) or CRISPRi (transfected iMGLs with MG10 TF cocktail) cells were treated with LPS (10ng/ml) for an additional 12 hours and harvested for snRNA-seq (Figure 4J). We found that blocking MG10 TF regulators in the presence of LPS induced new clusters (C0 and C3) and was sufficient to prevent IL1B induction (MG10 state marker) without concomitant gain of homeostatic markers (Figure S6I). Interestingly, this manipulation weakened the execution of the MG10 Inflammatory signature, as well as the enrichment of LPS-induced MG3: Ribosome Biogenesis, MG5: Phagocytic and MG7: Glycolytic signatures, while gaining the enrichment for MG11: Antiviral signature (Figure 4K). By modeling microglial state dynamics with iMGLs, we functionally assess the TF network activity prediction governing microglial state transitionings derived from our postmortem data analysis and provide a roadmap for TF-driven inflammatory state progressions (Figure 4L) (Figure S6J, STAR Methods).
Transcriptional Changes of Microglia States in AD
To investigate the transcriptional changes of microglial states in AD progression, we detected differentially expressed genes (adDEGs) between nonAD and early AD individuals (refers to DEGs in early response), and early AD and late AD individuals (refers to DEGs in late response), in each microglial state, using three methods (single-cell based method: MAST36 and MAST with random effect37, pseudo-bulk based method edgeR38; STAR Methods). We found that the DEGs identified by three methods were significantly overlapped by pairwise comparison (Figure S7A–B), so we defined 1,542 DEGs across 48 comparisons in total which passed the thresholds in at least two methods. We also evaluated the adDEGs biologically by comparing them with previous work9 and found 31 of 48 comparisons show significant enrichment with early, late or end stage DEGs identified in Gerrits et al.9 (Figure S7C, STAR Methods).
We next classified 1,542 adDEGs into three major categories and 51 groups based on their effect size and state-specificity (Figure 5A, Figure S8, STAR Methods, Table S1, pages 10–11). In particular, we found that 473 DEGs in 6 groups showed AD-stage-specific changes in at least two microglial states (Figure 5B), including two groups with up-regulation in AD progression (G1, G2), two groups with down-regulation in early AD and late AD (G25, G26), one group with up-regulation in early AD and down-regulation in late AD (G49), and one group with down-regulation in early AD and up-regulation in late AD (G50). These stage-specific changes of gene expression in AD progression suggest nonlinear and dynamic functions of microglia during the course of AD progression.
Figure 5. Transcriptional changes of microglial states in AD.

A. Barplot showing the number of down-regulated and up-regulated DEGs for each microglial state in early response and late response. B. Transcriptional changes of 473 DEGs in eight groups during AD progression. Red in heatmap represents up-regulated in AD progression, i.e. up-regulated in early AD compared to non-AD and upregulated in late AD compared to early AD, and blue represents down-regulation in AD progression. The numbers in the left block show the number of DEGs per group. The right shows the representative DEGs including known microglia markers, cytokines and their receptors, and top AD risk genes (see more in Figure 6). The heatmap with all DEGs is in Figure S8. C. Gene-gene occurrence network per group of genes in Figure 5B. The edge represents that the two genes are involved in the same Gene Ontology terms. The representative enriched GO terms are shown along with the network. D. Heatmap to show the TF enrichment of DEGs in each group. Odds ratio represents the enrichment. Stars show the TFs that were also differentially expressed and belong to the corresponding group. See also Figure S7–9 and Table S1, pages 10–11.
We next performed Gene Ontology enrichment analysis per group and built a gene-gene network by function sharing (Figure 5C, Figure S9A, STAR Methods, Table S1, pages 12). We found that DEGs in G25, including the homeostatic genes P2RY12 and CX3CR1, were down-regulated in diverse microglial states in early response to AD, but not significantly changed in late response (Figure 5B). These DEGs were significantly enriched in regulations of microglial cell migration, Wnt signaling and neurogenesis, suggesting that the impairment of the microglial homeostatic state may be an early event in AD progression. We also found that DEGs in G2, including APOE, MSR1 and CD74, were up-regulated in late stage but not in early stage and significantly enriched in regulation of chemokine production and amyloid-beta clearance, and cholesterol transport, suggesting that regulatory functions related to lipid accumulation of microglia may be a secondary effect in AD progression.
The DEGs in G26 (including AD risk gene BIN1 and proinflammatory cytokine CCL3), were widely down-regulated in multiple states of late stage of AD progression, and significantly enriched in cellular response to cytokine and insulin stimulus, receptor-mediated endocytosis, and regulation of amyloid-beta formation. We also found that DEGs in G49, including IL1B, CCL4 and VEGFA, were up-regulated in early AD and down-regulated in late AD, and significantly enriched in regulations of cell proliferation, migration and angiogenesis, and cytokine-mediated signaling pathway, consistent with the recent findings on the effects of microglial cytokines in neurodegenerative disease39,40. Additionally, the DEGs in G50, including AD risk genes PICALM, were down-regulated in early stage but up-regulated in late stage, and significantly enriched in regulations of intracellular signal transduction and amyloid-beta formation, and neuron death.
We next inferred the upstream regulators of these DEGs to investigate the transcriptional regulatory mechanisms (STAR Methods). We identified 66 TFs in total, 41 of which showed significant enrichment in 6 groups of earlier identified DEGs that showed AD-stage-specific changes in at least two microglial states (Figure 5D, Figure S9B). Notably, we found that 6 of these TFs also regulated the groups that they were part of, suggesting the importance of these coordinating TFs in the transcriptional regulatory circuits. For example, we observed that RELB, a subunit of NF-κB, showed significant enrichment of regulation of DEGs in G26 which are enriched in cytokine-mediated signaling (Figure 5C), consistent with the crucial role of RELB in neuroinflammation41.
Association of Microglial Transcriptional States with AD Genetics
We next sought to gain insight into the association of microglial states with AD genetics. We tested if microglial states can explain the heritability signal from genome-wide association studies (GWAS) summary statistics 42–44 using MAGMA45 and found inflammatory MG8 and MG2 show the most significant enrichment and MG0 (homeostatic), MG4 (lipid-processing) and MG11(antiviral) also show significant enrichment(Figure 6A, STAR Methods). Remarkably, MG4 and MG8 were increased proportionally in AD individuals, and even though the homeostatic MG0 state showed no significant change in cell proportion (Figure 1C), the homeostatic genes were significantly down-regulated in AD (Figure 5B). This consistency between simultaneous inflammation and downregulation of homeostatic processes highlights the importance of the homeostatic MG0, lipid-processing MG4, and inflammatory MG8 states in AD progression. Additionally, we also found that distinct microglial states show significant association in lupus, ulcerative colitis and triglycerides GWAS signals and weak association in multiple brain disorders (Figure S10A).
Figure 6. Association of microglial transcriptional states with AD genetics.

A. Gene-level enrichment in GWAS signals to associate microglial states with AD genetics using MAGMA. The -log10 (p-value) is shown in the barplot. The red dashed line shows the p-value < 0.05 as a cutoff. The red bars show FDR < 0.05 as a cutoff. B. Expression patterns of top AD risk genes (GWAS p-value < 10e-8) in 12 microglial states. The stars highlight the genes with specifically high expression in the corresponding state. C. Venn diagram showing the significant overlap between DEGs and TWAS genes, evaluated by Fisher’s exact test. D. The representative enriched Gene Ontology biological process of 178 overlapping genes between DEGs and TWAS genes. The -log10(p-value) is shown in the barplot. E. Transcriptional changes of 44 AD risk genes or TWAS genes in the early and late stages of AD in each microglial state. The logFoldChange calculated by edgeR shows the up-/down-regulation in AD progression. The red represents the up-regulation and the blue represents the down-regulation in AD progression. F. Co-expression network of regulators. The regulators include AD risk genes, TWAS genes, and TFs, denoted by nodes. The size of nodes represents the number of categories that one node belongs to. The edge represents the co-expression in at least one comparison (early or late) in each state, weighted by the times of co-expression occurrence. See also Figure S10 and Table S1, pages 12–13.
In addition to the overall enrichment of microglial states in AD GWAS signal, we also found differences in microglial state-specific expression patterns of the top AD risk genes (GWAS Catalog 46, those that are associated with AD variants with p-value < 10e-8, Figure 6B, and p-value < 10e-5, Figure S10B). We found that 35 AD risk genes (with p-value < 10e-8) were highly expressed in at least one microglial state (Figure 6B). For example, TREM2, encoding a lipid receptor, was highly expressed in lipidprocessing MG4, consistent with its function on microglial cholesterol metabolism in AD pathology 47.
To investigate if the 1,542 adDEGs are associated with genetic variants, we identified single-cell eQTLs in microglia, followed by transcriptome wide association study (TWAS) to dissect the associations between genetically regulated gene expression and AD (STAR Methods). We identified 1,036 TWAS genes in microglia (Table S1, page 13) and found that 178 of them were also differentially expressed in AD progression (Figure 6C, Fisher’s exact test, p-value = 1.3e-12), suggesting that the differential expression of these genes may be directly regulated by genetic factors. Gene Ontology enrichment analysis revealed that these 178 genes were significantly enriched in regulation of intracellular signal transduction, cell migration, immune response, and amyloid-beta formation (Figure 6D).
Although the TWAS data associated a group of DEGs directly with AD genetics, AD risk genes uncovered by TWAS could also be differentially expressed due to indirect regulation. To this end, we gauged the differential expression of the top AD risk and TWAS genes in early response and late response for each microglia state, as we previously did for DEGs in Figure 5B, and identified 44 genes (AD risk, TWAS, or both) showing differential expression in single or multiple states (Figure 6F). We found that 17 genes showed the differential expression in the early stage while 34 genes showed the differential expression in the late stage across multiple states. This suggests that the genetic effect on gene expression in AD may be time-sensitive, underscoring the potential significance of stage-specific therapeutics. Interestingly, we observed that only three genes (APOE, BIN1, and PICALM) were both AD risk and TWAS genes, suggesting their potential crucial functions in AD progression.
Lastly, to investigate the potential regulatory relationships between regulators, we built a co-expression network for potential regulators including differential AD risk genes (GWAS), TWAS genes, and upstream TFs of DEGs, by measuring the co-expression between two genes (cor.test p-value < 0.01) in each microglia stage in early and late responses and aggregating the co-expression across all comparisons together (Figure 6F, STAR Methods). We found that seven regulators (APOE, BIN1, RELB, PICALM, THRB, ZBTB16 and HS3ST1) belonged to two categories (GWAS/TWAS, GWAS/TF, or TWAS/TF). Of note is that BIN1 (GWAS/TWAS) and RELB (GWAS/TF) were highly correlated and simultaneously differentially expressed across multiple microglia states (Figure 6E–F), consistent with their functions as key regulators in inflammatory response 48,49. This coexpression network analysis provides insight to dissect the genetic regulatory pathways and possible cooperative mechanisms in microglial response to AD progression, potentially guiding cell-state specific and disease-specific AD therapeutics.
Discussion
Our findings establish that the rich diversity of microglial transcriptional states captured by snRNA-seq profiling is not fully represented by epigenome states defined through snATAC-seq. This conflict between transcriptional programs and chromatin architecture suggests that microglia may retain a relatively permissive chromatin landscape that is crucial to allow dynamic state transitions in response to microenvironment changes. We propose that state transitions can be mediated via the transcriptional activity of master regulator TFs, and define a framework by which the activity of these factors may govern microglial cellular states.
Although we identified a large microglial fraction within the human brain that are characterized by signatures of disease-associated microglia (DAM) states in MG3 ribosome biogenesis, we did not identify enrichment of these cells in AD subjects as previously demonstrated in AD mouse models 5. Notably, unlike the mouse brain, DAM signatures are lowly enriched across all microglial states in our dataset (with the only exception being the homeostatic state MG0). Species-specific differences may account for such divergent observations in disease-state signatures, while the relatively greater diversity of microglial states observed in the human brain could reflect specialized microglial functions in humans pertaining to lipid mobilization and inflammation.
Our data supports the concept that inflammation and lipid metabolism are tightly linked processes, and critically involved in the contribution of microglia to AD pathogenesis 50. According to our state and stage-specific differential analysis, inflammatory processes precede lipid regulation in microglia during disease progression. Through TF regulatory networks we have identified that the lipid-processing MG4 state is closely linked to MG8 inflammatory state. Although we did not empirically determine the relationship between these two clusters, it’s plausible that MG4-MG8 represents an intersection of microglial immunometabolism that is critical to the pathophysiology and progression of AD.
Collectively, our study provides the high resolution required to define the association of microglial cellular states with clinical and pathological variables, disease stages, and AD genetics. Through differential gene expression analysis, our data defines the transcriptional changes associated with microglial states that occur in a disease-stage specific manner, providing a roadmap for the development of therapies aimed at curbing neuroinflammation with disease-stage specificity.
Limitations of the Study
Although many studies have characterized the transcriptional profile of microglia, it is important to note that methodological differences may impact the conservation of disease signatures across studies. These differences include the source of tissue, isolation method and the microglia enrichment strategy. Additional factors such as the low number of nuclei recovered per subject or the sparsity of single-cell data may also affect the data, particularly the DEGs analysis. Lastly, experiments employing cellular reprogramming technologies in this study would be strengthened with the addition of orthogonal experimental approaches.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Manolis Kellis (manoli@mit.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Count matrices, metadata and supplementary data in this study from the ROSMAP cohort are at http://compbio.mit.edu/microglia_states/. ROSMAP data can be requested at https://www.radc.rush.edu. Raw data and processed data for all iMGLs bulk RNA-seq and snRNA-seq in this study are publicly available in GEO database under accession GSE227223.
The codes used in this study are available at http://compbio.mit.edu/scMicroglia/, https://github.com/nasunmit/scMicroglia and Zenodo https://doi.org/10.5281/zenodo.8297901.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS Human post-mortem brain samples from ROSMAP
In this study we obtained human post-mortem brain tissues from the Religious Orders Study and Rush Memory and Aging Project (ROSMAP, each approved by an Institutional Review Board (IRB) of Rush University Medical Center). All participants signed an informed consent, an Anatomic Gift Act for organ donation, and a repository consent to allow the data to be shared 14. We used seven pathological phenotypes for each individual to separate all individuals in this cohort into three groups which we defined as no AD pathology, low AD pathology and high AD pathology by performing a k-means clustering method on the quantitative matrix after z-score normalization. These include pathological phenotypes, e.g., a global AD pathology, molecularly specific amyloid-β and tangles, and global cognition proximate to death. Thus, case status was based on both AD pathology and cognition pathology, and other variables were allowed to freely associate, as previously reported 51–55. We acknowledge that this empirical approach to casesness does not conform to standard diagnostic criteria.
We performed snRNA-seq in the prefrontal cortex for 423 individuals, and four other regions for a subset of 48 individuals, including mid-temporal cortex, angular gyrus, entorhinal cortex, thalamus, and hippocampus for a subset of 65 individuals and an additional 21 individuals (Table S1, page 1). We performed snATAC-seq in the prefrontal cortex for a subset of 92 individuals, and five other regions for a subset of 48 individuals, including mid-temporal cortex, angular gyrus, entorhinal cortex, thalamus, and hippocampus (Table S1, page 7).
Culture of iPSC lines
AICS-0090–391 iPSCs, acquired via the Allen Institute for Cell Science https://www.allencell.org, were maintained on Matrigel-coated plates (Corning; Cat # 354277) with 5% CO2 at 37°C in feeder-free conditions in mTeSR1 media (STEMCELL Technologies; Cat #85850). iPSCs were passaged when 60–80% confluent using ReLeSR (STEMCELL Technologies; Cat# 05872) and reseeded 1:6 onto Matrigel-coated plates.
METHOD DETAILS
Nuclei isolation from frozen postmortem brain tissue, snRNA-seq and snATAC-seq
We isolated nuclei from frozen postmortem brain tissue as previously described (Mathys et al., Nature 2019)11 with some modifications. Briefly, we homogenized the brain tissue in 700 μL Homogenization buffer and filtered the homogenate through a 40 μm cell strainer (Corning, NY), added 450 μL Working solution and loaded it as a 25% OptiPrep solution on top of a 30%/40% OptiPrep density gradient (750 μL 30% OptiPrep solution, 300 μL 40% OptiPrep solution). We separated the nuclei by centrifugation using a fixed rotor, tabletop centrifuge (5 minutes, 10000g, 4°C). We collected the nuclei pellet at the 30%/40% interphase, transferred it on a new tube, washed it twice with 1mL ice-cold PBS containing 0.04% BSA (centrifuged 3 minutes, 300g, 4°C) and finally resuspended it in 100 μL PBS containing 0.04% BSA. After counting, we diluted the nuclei to a concentration of 1000 nuclei per μL. We used the isolated nuclei for the droplet-based 10x scRNA-seq assay, targeting 5000 nuclei per brain region and individual, and prepared libraries using the Chromium Single-Cell 3′ Reagent Kits v3 (10x Genomics, Pleasanton, CA) according to the manufacturer’s protocol. We sequenced pooled libraries using the NovaSeq 6000 S2 sequencing kits (100 cycles, Illumina). For the snATAC profiling, we spun down the remaining nuclei, resuspended them in 1x Diluted Nuclei buffer and adjusted their concentration to 2500 nuclei per μL. We proceeded with library construction targeting 5000 nuclei per brain region and individual using the Chromium Single-Cell ATAC Reagent Kit v1 (10x Genomics, Pleasanton, CA) following the manufacturer’s protocol. We sequenced the pooled libraries using the NovaSeq 6000 S2 sequencing kits (100 cycles, Illumina).
snRNA-seq data processing
We mapped the raw reads to human reference genome version GRCh38 and quantified unique molecular identifiers (UMIs) counts for each gene in each cell using CellRanger software v3.0.1 (10x Genomics) 56. We pre-processed this count matrix (gene-cell) using the Seurat R package v.4.0.3 57). We kept the cells with more than 500 UMIs and less than 5% mitochondrial genes, and genes with expression at least in 50 cells for further analysis. We normalized the counts by the total UMI counts for each cell, multiplied by 10,000, and then log-transformed. We used the top 2000 highly variable genes for principal component analysis (PCA) and the top 30 principal components (PCs) as inputs to perform UMAP. We used Harmony for batch correction 58. We used the resolution as 0.5 to identify clusters. We used DoubletFinder to estimate the potential doublets formed by two or more cells based on the by default parameters 59. The cells with high doublet scores (0.2 as cutoff) were removed for further analysis.After generating clusters, one cluster showing high expression of markers of two or more cell types was also treated as doublets and removed for further analysis.
snATAC-seq data processing
We processed snATAC-seq data using the same computational pipeline as Xiong et al. accompanied manuscript. Specifically, we firstly generated raw data of FASTQ for each sample by demultiplexing the reads with cellranger-atac software(v1.1.0) 60, and then mapped the reads to human reference genome version GRCh38 using “cellranger-atac count” to obtain the fragment file for each sample. We processed the snATAC-seq data using ArchR (v1.0.1) 61. We removed the potential doublets using the “filterDoublets” function in ArchR. We kept the cells with TSS enrichment more than 6 and the number of fragments between 1000 to 100,000 for further analysis. We performed Iterative LSI dimension reduction and clustering using the matrix of 500 bp tile-based, with parameters “iterations=4, resolution=0.2, varFeat=50000”. The UMAP was used to visualize cell embedding for all cells. We generated the gene score matrix using ArchR, and annotated the cell type for each cluster based on the gene score of well-known markers in the brain 11. We integrated the clusters that were annotated as microglia/immune cells for further analysis.
In Silico Sorting to enrich immune cells and cell type annotation in snRNA-seq data
For the full datasets with all cell types (2.8 million cells), we first annotated the cell type for each cluster based on three widely-used canonical markers of major cell types in the brain (including excitatory and inhibitory neurons, astrocytes, oligodendrocytes, OPCs, microglia and vascular cells) 11and a list of markers for immune cells 11,62. We also tested the enrichment of a large set of markers 63 in highly expressed genes for each cluster to confirm the annotation based on several marker genes. We next calculated the cell type scores (i.e. astrocyte, oligodendrocyte, microglia, etc) for each cell, which were represented by the average expression of a group of markers for each cell type 63. The cells were then selected as microglia/immune cells for further integrative analysis if and only if (1) the clusters that the cells belong to were annotated as microglia/immune cells; and (2) the cells had the highest score for microglia/immune cells, and 3) the score for microglia/immune cells was 2-fold higher than the second highest score. For the selected microglia/immune cells, we followed the same pipeline to perform dimensional reduction and clustering with the same parameters as full datasets. We used the Wilcoxon rank-sum test in Seurat with customized parameters (min.pct = 0.25, logfc.threshold = 0.25) to identify highly expressed genes for each cluster compared to all cells from other clusters.
Cell proportional analysis
We performed propeller64 analysis to evaluate the statistical significance for compositional differences between phenotypic groups of interest (AD groups, age, and sex group) with the consideration of the confounding variables in the model. We used speckle package in R and adjusted p-value < 0.05 as cutoff for significance.
Microglia epigenetic state annotation
For the cells that were annotated as microglia in snATAC-seq data, we re-performed Iterative LSI dimension reduction and clustering using the parameters “iterations=4, resolution=0.5, varFeat=50000” to identify the subtypes of microglia, which were defined as epigenomic states in further analysis. We removed the clusters with less than 1,000 cells and identified the marker genes based on the gene scores (aggregation of snATAC signals around promoter regions). We used these marker genes to annotate the three epigenetic states as one homeostatic state due to lack of activated genes, and two activated states with high gene scores of activation genes.
Peak calling and differential peak analysis
We used MACS2 to call chromatin accessible peaks based on the pipeline in ArchR 61,65, generated peak matrix (peak X cell matrix) and identified marker peaks for each epigenomic in account for difference in data quality among the cell groups by considering TSS enrichment and the number of unique fragments per cell. We used a Wilcoxon test in ArchR to evaluate if the peak was differential between epigenomic states of microglia (cutoff: p.value < 0.01 and log2FoldChange > 0.5). We performed GREAT analysis to annotate the differential peaks in each state 66 with the setup as basalPlusExt and 100kb for both upstream and downstream genomic regions. We used Homer to perform motif analysis by expanding the peaks to 2000bp with the default parameters 67.
Co-accessibility analysis
We identified the co-accessibility between proximal and distal peaks, as the proxy of peak-to-gene links to look for the correlations between peak accessibility and gene expression. The parameters we used included corCutOff = 0.3, FDR=1e-4, upstream = 50000, and downstream = 50000. The pairs of peaks and genes were used for further analysis to associate distal accessible peaks with microglial transactional state signature genes and the differential expression genes in AD.
Identification of differentially expressed genes in AD progression
We applied three methods (two single-cell based methods, regular MAST and MAST with random effect, and a single-cell based method edgeR) to detect the differentially expressed genes for each microglia transcriptional state between control and low AD pathology individuals, and low AD pathology and high AD pathology individuals. We controlled the covariates for all three methods, including number of expressed genes, percentage of mitochondrial genes, percentage of ribosomal genes, age, sex, PMI, race, batch, brain region, and other dementia related pathology and diagnosis (Lewy body dementia and Parkinson’s disease). The genes with FDR <= 0.05 and absolute coefficient >= 0.1 for MAST and MAST with random effect or absolute logFoldChange >= 0.58 (corresponding to fold change >= 1.5) for edgeR were selected for pairwise comparison between these three methods. We then tested if the DEGs identified by each two methods were significantly overlapped (measured by Fisher’s exact test with all expressed genes as background and adjusted p-value < 0.05 as cutoff) and the percentage of overlapping (measured by Jaccard score). We found the DEGs were highly consistent between three methods. We then kept DEGs which were detected as DEGs in at least two methods for further analysis. We classified these DEGs into three categories based on the cell-state-specificity and effect direction to AD progression, which could be further classified into 51 gene groups as shown in Figure S8.
Comparison with public datasets
We downloaded the marker genes of microglial subtypes described in Olah et al.12 and compared them with the marker genes of microglial states in this study by computing the Jaccard score pairwisely and testing the significance of overlapping with Fisher’s exact test. The Jaccard score and significance were visualized by heatmap shown in Figure S1. We also downloaded the marker genes of microglial subtypes in mouse data provided by Hammond et al.4 and performed a similar analysis to compare human and mouse microglia subtypes. We evaluated the adDEGs by comparing withDEGs identified by Gerrits et al.9. We used Jaccard score to represent the percentage of overlapping genes and Fisher’s exact test to measure the statistical significance (FDR <= 0.05).
Prediction of upstream transcription factors
For enrichment-based analysis, we predicted the upstream transcription factors of genes (marker genes or DEGs) using Enrichr in R based on three libraries including TRANSFAC and JASPAR, ChEA, and ENCODE TF ChIP-seq data 22,68,69. We used adjusted p-value <0.05 as a cut-off to select the significant TFs. For motif-based analysis, we linked the microglial state marker genes to distal peaks using the peak-to-gene links represented by co-accessible peaks and then performed motif analysis by expanding the peaks to 2000bp with the default parameters in Homer 67. We kept the TFs with detected expression (average expression > 1 after log2 transformation) in microglia for further analysis. We also applied DoRothEA to predict differentially activated TFs in each microglial state based on the activity score of TFs31. Fold change > 1.5 and p-value < 0.01 were used as cutoffs to select highly activated TFs for each microglial state in DoRothEA analysis. To compare the TFs identified by enrichment/motif based method and DoRothEA, we calculated the Jaccard score to represent the percentage of overlapping and performed Fisher’s exact test to evaluate the statistical significance (FDR < 0.05 as a cutoff and all expressed TFs as background).
These upstream TFs may or may not be highly and specifically expressed by the microglial states that they regulate. To evaluate the upstream TFs were highly expressed in which microglial state, we checked the upstream TFs for each state and counted which of those TFs had high expression in all states.We then normalized that number by the total number of TFs that are marker genes in each state. We next defined a score by normalizing the sum of the values obtained in the previous step as one, represented as the potential of state-state transition. We performed a permutation test to evaluate whether the score is statistically significant for each pair of states with transition potential. Specifically, we randomly selected the same number of TFs as the enriched TFs for each state from the full database, performed the same analysis as above, and compared the score with the real score to obtain p-values (permutation times: 1000). The result was visualized by the heatmap (Figure S6J), showing the score by color and significance by star. A model of transitions between MG8, MG10 and MG0 is shown in Figure 4L.
Immunohistochemistry staining
Fixed human brain tissue (prefrontal cortex) was sectioned using a vibratome at 40 μm. The sections were blocked for one hour at room temperature (RT) in blocking buffer (PBS containing 5% bovine serum albumin, 1% normal donkey serum and 0.3% Triton X-100). The sections were incubated for 48 hours at 4°C with the primary antibodies in blocking buffer. A list of all primary antibodies can be found in Table S1, page 4. Following primary antibody incubation, the sections were washed three times for 5 minutes each at RT in PBS and then incubated with secondary antibodies (dilution 1:1000) for two hours at RT. The slices were once again washed three times at RT in PBS and incubated in TrueBlack (Biotium; Cat#23007) for 90 seconds. After a final round of washes, the slices were mounted on Superfrost Plus Slides (Fisherbrand; Cat#12–550-15) in Fluromount-G (SouthernBiotech; Cat#0100–01). Primary antibodies were visualized with Alexa-Fluor 405, Alexa-Fluor 488, Alexa-Fluor 555, and Alexa-Fluor 647 antibodies (Molecular Probes), and cell nuclei visualized with Hoechst 33342 (Sigma-Aldrich; Cat#94403) using a confocal microscope (LSM 900; Zeiss) with a 20× or 63× objective. For experiments quantifying fluorescent intensity, images were analyzed with Imaris imaging software (Oxford Instruments) for 3D reconstruction of IBA1 surfaces. Mean intensity values for each channel within IBA surfaces were quantified. For inflammatory state marker expression in multiplexed immunostaining experiments, values for each marker were visualized by creating expression gradients for each channel and superimposed onto IBA-positive surfaces using Imaris visualization tools, as presented in Supplemental Figure S5B. For quantification, this data was z-scored across cells and plotted as a heatmap clustered by hierarchical clustering with Morpheus (https://software.broadinstitute.org/morpheus/).
RNAscope and HiPlex
Tissue blocks from the prefrontal cortex were cored into a 2mm disc with a disposable biopsy punch (UniCore Punch Kit 2.00 mm) and frozen in OCT for cryosectioning at 12um thickness on the cryostat (Leica CM3050S). Sections were directly mounted on Superfrost Plus Slides (Fisherbrand; Cat#12–550-15) and allowed to dry at −20 celsius for 1 to 2 hours. Tissue was then processed following the recommended protocol by ACD Biotechne RNAscope assays. To quench autofluorescence, we applied TrueBlack (Biotium; Cat#23007) for 90 seconds after the final step (DAPI application) of the RNAscope assay procedure, and gently rinsed with PBS prior to coverslipping and imaging. For HiPlex multiplexing, we reapplied DAPI and TrueBlack at the end of every round. 12×12 tile scans with 12z steps were acquired with a confocal microscope (LSM 900; Zeiss) with 63× oil immersion objective. Maximum projection images were manually aligned on Photoshop (Adobe Photoshop 2021) with DAPI nuclei stain and tissue boundaries serving as guides for registration. Microglia were then identified based on probe detection for P2RY12. IBA1 and CX3CR1 probes were weakly detected and were used as confirmation probes. After manually assigning nuclei as microglia (greater than P2RY12 dots with co-detection of IBA1 and CX3CR1), images containing microglia were further processed with QuPath (v0.4.3) for probe quantification. Manual curation of images were performed to ensure accurate probe detection by an investigator blind to the sample group. The distribution of dot numbers varied greatly by probes. For example, while CPEB4 probe detected 0–30+ dots per nuclei, RELB probe detected 0–9+ dots. To infer expression enrichment, the number of dots were Z-scored per probe across all samples.
Microglia induction protocol
Embryoid bodies (EBs) were generated following an adapted iPSC seeding strategy (Marton et al., 2019) with a previously established protocol (Sloan et al., 2018). 60–80% confluent iPSCs were incubated in ReLeSR (STEMCELL Technologies; Cat# 05872) for one minute at room temperature, followed by a 3-minute dry incubation at 37°C. iPSC colonies were then scraped in mTeSR1 media (STEMCELL Technologies; Cat # 85850) and dissociated into a single-cell suspension through pipetting. After centrifugation at 300 g for 5 minutes, the cell pellet was resuspended in 2 mL of mTeSR1 media supplemented with ROCK inhibitor (1:1000) (BioVision; Cat# 2342–5) and counted with a Countess II automated cell counter. 5 × 106 cells were then plated into each well of an AggreWell plate (STEMCELL Technologies; Cat# 34815) for EB induction. After 48 h, 15–30 EBs were moved onto each well of Matrigel-coated plates (Corning; Cat # 354277). Using the STEMdiff Hematopoietic Kit (STEMCELL Technologies; Cat#05310), EBs were differentiated into hematopoietic progenitor cells (HPCs). After collection and centrifugation at 300 g for 5 minutes, nonadherent HPCs were resuspended in 1 mL of microglia differentiation media (MDM) (1:1 DMEM/F12 (Thermo Fisher Scientific; Cat#11330–057): Neurobasal media (Gibco; Cat# 21103049) supplemented with IL-34 (PeproTech; Cat#200–34) and m-CSF (PeproTech; Cat#300–25) as described (McQuade et al., 2018). Cells were plated at 200,000 cells per well in 6-well tissue culture plates and maintained in MDM with 5% CO2 at 37°C for at least two weeks before experiments.
CRISPRi in iMGLs for MG10 TF repression
dCAS9-KRAB-BFP (CRISPRi) iPS line was acquired from Allen Institute for Cell Science (allencell.org). Duplicate sgRNAs per target were synthesized following the protocol from Precision sgRNA Synthesis Kit (ThermoFisher #A29377) from guide RNA sequences for FOXO3, HIF1A and FOXP3 taken from libraries derived by (Horlbeck et al., 2016 eLife). HIF1A_1: GCTGGCCGAAGCGACGAAGA HIF1A_2: GCCTCCTGTCCCCTCAGACG; FOXP2_1: GAAGCCGGGAGACCAGACAC FOXP2_2: GAATCAGCATTTAATCACTA; FOXO3_1: GGAGGGGAAAGGGAAGCGCC FOXO3_2: GTGGCGGCGCGCGAGCTGAC. iMGLs were differentiated from CRISPRi iPSCs following the microglial induction protocol above and a cocktail with mixed sgRNAs delivered to iMGLs via lipofectamine CRISPRMAX (Invitrogen #CMAX00003). Following 4 hours post sgRNA transfection, cells were exposed to 10ng/ml of LPS overnight. Control iMGLs were mock transfected (no sgRNAs in lipofectamine) but also exposed to LPS. Cells were processed for snRNA-seq following our nuclei isolation protocol detailed below.
Nuclei isolation from iMGLs for snRNA-Seq
Nuclei from iMGLs were isolated using the Nuclei Isolation Kit: Nuclei EZ Prep (Cat#NUC101, Sigma-Aldrich) per manufacturer’s instructions. In brief, microglia were washed with cold PBS and lysed in Nuclei EZ Lysis Buffer with RNase Inhibitor (0.2 U/ul) in well by scraping. Lysate was collected, briefly vortexed and incubated on ice for 5 min. Nuclei were collected by centrifuging at 500 x g for 5 min at 4°C. Following a subsequent wash in lysis buffer, nuclei were resuspended in 200 uL Nuclei EZ storage buffer and counted. Fresh nuclei were diluted to a concentration of 800–1200 cells/uL, and approximately 17,400 cells per channel for an estimated recovery of 10,000 cells per channel. SnRNA-seq libraries were generated from fresh nuclei with the Chromium Next GEM Single Cell 3’ Reagent Kit v3.1 (10X Genomics) in accordance with the manufacturer’s instructions.
Calcium Imaging
Microglia were plated in 48-well tissue-culture treated plates and cultured for 5 days. For ATP uncaging experiments in the presence of an inflammatory stimulus, cells were incubated overnight with LPS (10 ng/mL). The day of imaging, microglia were pre-incubated with 1 mM DMNPE- Caged ATP (Cat# 1049; Fisher Scientific) and 1 mM Fluo-4 AM (Cat#F14201; Thermo Fisher Scientific) at 37°C for 30–45 min. In experiments where iMGLs were exposed to spheroid conditioned media (CM), iMGLs were half-fed with CM for 2 h before recording session. Live-cell imaging was performed using Zeiss LSM900 at 37°C and 5% CO2. All images were acquired at 488 nm. Uncaging of ATP was done at 405 nm for 30 s post-baseline acquisition, followed immediately by post-stimulation image acquisition. Images were first stabilized to account for drift in the x-y direction; we used the ImageJ plugin “Linear Stack Alignment with SIFT”. Calcium traces from motion-corrected time series were manually segmented on ImageJ into individual cells based on threshold intensity, variance, and upper and lower limits for cell size. Image segmentation results were separately inspected for quality control. Fluorescence signal time series (DF/F: change in fluorescence divided by baseline fluorescence) were calculated for each segment. The baseline fluorescence for each cell was determined as the minimum fluorescence signal in the baseline recording epoch. Data was plotted from individual cells in one experiment, although the experiments were repeated at least three times. Heatmaps were generated using GraphPad Prism (GraphPad Software)
Transcriptional Time-point Analysis following inflammatory stimulation
In order to evaluate the temporal transcriptional response of targeted DEGs within the MG2, MG8, and MG10 inflammatory clusters, microglia were plated onto 12-well tissue-culture treated plates and cultured for 5 days. Microglia were stimulated with TNFα (40 ng/mL), INFγ (30ng/mL), or LPS (10 ng/mL) for 1 hr, washed 3 times with D-PBS, and left to incubate in culture media. A subset (n = 3 per condition) of untreated microglia were collected for baseline measurements. At each timepoint (1hr, 6hr, 24hr, 120hr), microglia were pelleted for RNA extraction and subsequent analysis by RealTime PCR (qPCR). RNA extraction of iMGLs was achieved with RNeasy Plus Mini Kit (Cat#74134; Qiagen) and reverse transcription was performed with RNA to cDNA EcoDry Premix (Cat#639549; Takara) according to the manufacturer’s instructions. Gene expression was analyzed with RealTime PCR (CFX96; Bio-Rad) and SsoFast EvaGreen Supermix (Cat#1725202; Bio-Rad). Expression data was normalized to housekeeping gene GAPDH using the 2−ΔΔCt relative quantification method. A list of qPCR primers used in this study can be found. For assessment of amyloid-induced transcriptional changes, iMGLs seeded onto 6-well tissue-culture treated plates were exposed to 2μg of pre-formed amyloid beta (1–42) fibrils (rPeptide #AF-1002–1) per well. Baseline controls were untreated but harvested at the same time point as 4 hours after amyloid seeding. 3 wells per time point were harvested, pelleted and flash-frozen. After all time points were collected, pellets were processed for RNA extraction following RNeasy Plus Mini Kit (Cat#74134; Qiagen) and processed for Bulk RNA-seq at the MIT BioMicro Center.
Homeostatic TF Overexpression: Plasmid cloning, lentivirus production and homeostatic assessment.
To screen the overexpression of transcription factors from MG0, lentivirus constructs were purchased from the DNASU Plasmid Repository (Tempe, AZ). Majority of the 25 TFs (22 of them) utilized were obtained in the lentiviral vector, pLX304. The remaining 3 TFs (SUZ12, JARID2, RUNX2) were obtained in pDONR221 format and subsequently subcloned into pLX304 for lentiviral production and overexpression experiments. An empty pLX304 plasmid was used as a control. A list of all the vectors used in this study can be found in Table S1, page 8. The empty and TF-containing plasmids were then transfected into HEK-293T cell line (Cat#CRL-3216; ATCC) with lentiviral packaging and envelope vectors to generate lentivirus following the previously published protocol 70. Lentiviral supernatant was collected 48 h after transfection and centrifuged for 2 h by ultracentrifugation at 25,000 RPM at 4°C. Pellets were resuspended in DPBS and frozen at −80°C until used. For overexpression experiments, lentivirus was top-loaded and allowed to incubate overnight in microglia that had been re-plated onto multi-well plates and cultured for 8 days. Calcium imaging was performed as described above but cells were pre-incubated with conditioned media from forebrain organoid cultures for 2 hours, a procedure established to yield strong calcium transients in iMGLs in a neuronal-activity dependent manner from our previous study33. For microglial morphology analysis, cells were fixed and permeabilized following standard lab protocols after 10 days of TF overexpression and stained with ActinGreen 488 Readyprobes (Invitrogen #R37110). Imaging was performed with LSM 900 confocal microscope and 3D reconstruction of cell surfaces were achieved through Imaris imaging analyses (Oxford Instruments) for sphericity counts. Control iMGLs were infected with lentivirus containing an empty mCherry backbone.
Gene Ontology enrichment analysis
We used Enrichr in R to perform enrichment analysis for Gene Ontology biological process (adjusted p-value < 0.05 as a cutoff)71,72. We selected the representative terms for visualization using heatmap manually based on the diverse functional categories and the number of genes involved in each term.
Gene-level enrichment in GWAS signal by MAGMA
We applied MAGMA.Celltype, a R package, to test which microglial states can explain the heritability signal from GWAS summary statistics. We tested 14 traits73: AD42, lupus74, ulcerative colitis75, Crohn’s disease75, BMI76, triglycerides77, rheumatoid arthritis78, depressive symptoms79, ever smoked80, schizophrenia81, multiple sclerosis82, type 2 diabetes83, neuroticism79, years of education79. We used p-value < 0.05 or FDR < 0.05 as cutoffs to define the weak or strong significant microglial states associated with GWAS signal.
Expression and changes of AD risk genes
We downloaded GWAS associated genes based on GWAS catalog annotation46 for AD. We defined the GWAS associated genes in AD as AD risk genes. The p-value < 10e-8 and < 10e-5 were used as thresholds to select AD risk genes for analysis.
Microglia sc-eQTLs analysis
For microglia sc-eQTLs analysis, we firstly estimated the average expression for each gene using a poisson-gamma model with a cell-level sequencing depth parameter and an individual-level mean parameter for each gene within each individual. The mean parameter was estimated by an integrative variational inference algorithm 84. We then estimated the confounding factors on the individual-level gene expression matrix in an adaptive way. For each gene, we defined control genes by taking the top most correlated genes located in different chromosomes. We performed principal component analysis (PCA) and used the top 50 PCs as covariates.
We next built knock-off filters 85 for a genotype dosage matrix (0, 1, 2 effect allele count) sampled from a cis-window (1Mb around the gene body) for each gene. We estimated PCs of the sampled genotype matrix by conducting singular value decomposition. Using the first factors that account for the >80% of total genetic variation, we estimated structured confounding factors that capture the dependency structures across individuals and genetic variants. We estimated the knock-off design matrix following scalable approaches that have been previously reported 86–88.
We then performed multivariate analysis for each gene by regressing the pseudobulk profiles on the observed and knock-off genotype matrix, respectively. We performed a Bayesian spike-and-slab regression prior to measure the posterior inclusion probability (PIP) of each variant. The difference between PIP values on the observed variable and the knockoff counterpart were used to construct knockoff statistics. We determined the threshold for a desired false discovery rate level using the R knockoff package 89.
TWAS analysis
We performed transcriptome-wide association studies (TWAS) 90,91 within each LD block independently, and included all the genes whose cis-eQTL variants are located in that block. We used LD blocks previously defined based on the 1000 genomes project 92. For each LD block, we first estimated AD risk variation by projecting AD GWAS summary statistics onto the in-sample genotype matrix 93,94. We also predicted the expression value of each gene directly regulated by cis-regulatory variants based on the results of our eQTL analysis. Since 10–100 genes are located within each LD block, we conducted a multivariate regression analysis to identify the most relevant genes and cell types associated with the predicted AD effects. We also calculated gene-by-gene statistics. If the same gene appears multiple times across different LD blocks, we assign genes to the block showing the strongest effect.
Co-expression network analysis
We calculated the Pearson correlation coefficient between each pair of genes (including AD risk genes, TWAS genes, and upstream transcription factors) across the individuals (early response: individuals with no or low AD pathology, late response: individuals with low or high AD pathology) at the individual level to measure the co-expression (p-value < 0.01 in cor.test in R). We aggregated the correlated gene pairs if they appeared in multiple microglia states and comparisons (early/late) and used the occurrence times as weight of the edge shown in the network. We used Cytoscape (v3.9.1) to visualize the co-expression network 95.
QUANTIFICATION AND STATISTICAL ANALYSES
Statistical analyses for experimental validation were performed in GraphPad Prism using a two-tailed Student’s t test or a one-way ANOVA followed by a post hoc Tukey’s test with *p < 0.05 considered significant. Multiple comparisons were corrected with the Dunnett method as described in the figure legends. Studies were performed blindly and automated whenever possible with IMARIS or ImageJ cell, and multiple investigators confirmed quantification results. Data in graphs are expressed as mean, and error bars represent the standard error of the mean (SEM). For computational data analysis, Wilcoxon rank-sum test was used for calling differentially expressed genes and differentially accessible peaks. Pearson correlation was used for peak-to-gene links and co-expression network analysis. MAST and edgeR were used for identifying differentially expressed genes between phenotypic groups. Fisher’s exact test was used to evaluate the significance of overlapping genes between two gene sets. Multiple comparisons were corrected by fdr.
Supplementary Material
Figure S1. Microglial transcriptional states in aged human brains (Related to Main Figure 1). A. UMAP of 174,420 in silico sorted immune nuclei from human postmortem brain tissues labeled 16 clusters in snRNA-seq data. B. Expression patterns of canonical markers of brain cell types and immune cell types. The size of dots represents the percentage of cells with expression. The color of dots represents the scaled average expression level. C. Average of log2foldchange of 23 MG5 and C9 shared marker genes. The red dashed line shows the threshold (Fold change > 1.5). D. Dotplot to show the signature score of DAM and homeostatic genes in each microglial transcriptional state shown in Figure 1A. E. Heatmap to show the expression of DAM genes in each microglial state. The average expression in each state after log transformation is shown in the heatmap. F. Expression levels of neurotransmitter receptors in 12 microglial states. The average expression in each state after log transformation is shown in the heatmap. G. Representative enriched Gene Ontology biological processes in each microglial transcriptional state. Enrichr in R was used to perform GO enrichment (adjusted p-value < 0.05 as cutoff). H. Heatmap to show the significant overlap of marker genes between Olah et al. and this study. Jaccard score represents the percentage of pairwise overlapping genes. Fisher’s exact test is used to evaluate the significance of overlapping (adjusted p-value < 0.05 as a cutoff). I. Heatmap to show the signature score of marker genes identified in Olah et al. in each microglial state of this study. The signature score is estimated by the average expression level of marker genes. J. Heatmap to show the significant overlap of marker genes between Hammond et al. (mouse) and this study (human). Jaccard score represents the percentage of pairwise overlapping genes. Fisher’s exact test is used to evaluate the significance of overlapping (adjusted p-value < 0.05 as a cutoff).
Figure S2. Cell abundance across multiple variables (Related to Main Figure 1). A. UMAP of microglia nuclei for each brain region. The number of cells in each cell type is shown. B. Distribution of cell fraction across six brain regions in major cell types. C. Statistical testing summary for comparison between regions by propeller in R. D. Statistical testing summary for comparison between nonAD, earlyAD and lateAD by propeller in R. E. Heatmap to show the correlation between cell fraction and quantitative pathological variable in each microglial transcriptional state. The Pearson correlation coefficient (PCC) is calculated and tested by cor.test in R to measure significance. The PCC values are shown in the heatmap. The significant levels are represented by stars (*: p-value < 0.05, **: p-value < 0.01, and ***: p-value < 0.001). F. Distribution of cell fraction across all individuals in each microglial state. G. Distribution of cell fraction across sequencing batches in each microglial state. H-I. Distribution of ages/PMIs of the individuals across 12 microglial states. J. Cell fraction across sex for each microglial state (bottom) and across cell types for male and female individuals (top). K. Statistical testing summary for comparison in age, PMI and sex by propeller in R.
Figure S3. Immunohistochemistry on a subset of postmortem human brain samples from ROSMAP cohort (Related to Main Figures 1, 2 and 4). Immunostaining for IBA1 and P2RY12 across 8 subjects spanning non-AD, to Early AD and Late AD status. ROSMAP subject IDs are displayed in each corresponding image.
Figure S4. Validation of marker genes associated with inflammatory states and transcriptional response across MG10, MG8 and MG2 markers in iMGLs exposed to pro-inflammatory stimulus (Related to Main Figures 2). A. Multiplexed immunostaining to spatially resolve inflammatory states across the human brain. IBA1 was used to label all CNS resident macrophages, ILB1 to label MG10, LRRK2 to label MG8 and CPEB4 to label MG2 microglia (see list of antibodies used in immunohistochemistry in Table S1, page 4). ROSMAP subject ID, 10248033. B. Fluorescent intensity projection onto 3D renderings of IBA1 signal for each channel of multiplex immunostaining shows distinct microglial populations. C. Heatmap for immunostaining quantification showing relative maximum intensity fluorescence for CPEB4, IL1B and LRRK2. D. Quantification of microglia (P2RY12+IBA+CX3CR1+) from RNAscope HiPlex for inflammatory state markers: LRRK2, FOXP1 (MG8 marker), CPEB4, TMEM163 (MG2 marker), CCL3 and RELB (MG10 marker). Number of dots normalized across cells for each marker and plotted as Z-scores and shown from blue (low) to magenta (high). n=291 microglia from 4 AD subjects were quantified. E-H. qPCR analysis for pulse-and-chase experiment after iMGLs exposure to pro-inflammatory stimuli (TNFa, INFy and LPS). E. List of marker genes and their corresponding MG clusters origin. F. MG Cluster 10 genes. G. MG Cluster 2 genes. H. MG Cluster 8 genes. Note that while IL1B is not shown in this supplemental figure, CPEB4 and LRRK2 are duplicated here from main Figure 2G. n= mean of biological triplicate are shown for each timepoint and treatment. Error bars were removed for ease of interpretation.
Figure S5. Microglial epigenetic states defined by snATAC-seq data (Related to Main Figure 3). A. UMAP of 41,832 microglia nuclei in snATAC-seq labeled by the six brain regions. B. Heatmap to show the gene score patterns of 2,805 marker genes across three epigenomic states in microglia. The known marker genes for microglia are listed on the right. The z-scores are shown in the heatmap. C. Gene score distributions of P2RY12, CCL3 and SPP1 in three microglial epigenomic states. CCL3 shows the significantly high score in ACT1 state and SPP1 shows the significantly high score in ACT2 state, highlighted with star labels. The p-value <0.01 as cutoff in the Wilcoxon rank test. D. UMAPs to show the microglia clusters for RNA, ATAC, ATAC colored by RNA, and RNA-ATAC joint analysis, in multiome data from 19 ROSMAP individuals. E. Heatmap to show the significant enrichment of marker genes of multiomic transcriptional clusters of microglia in 12 microglial states. Jaccard score represents the percentage of pairwise overlapping genes. Fisher’s exact test is used to evaluate the significance of overlapping (adjusted p-value < 0.05 as a cutoff). F. The annotation of accessible peaks in microglia. Colors represent the distinct genomic locations including distal, exonic, intronic and promoter. G. Heatmap to show 16,800 marker peaks in three epigenomic states. Z-score values across microglia states are shown in the heatmap. H. GREAT analysis to annotate the epigenomic specific peaks. The panels from left to right include the distribution of peaks based on the number of associated genes per peak, the distribution of peaks based on the distance to TSS, and the distribution of peaks based on the absolute distance to TSS. I. Representative enriched Gene Ontology biological processes of epigenomic state-peak-associated genes. GREAT was used to perform the annotation and GO enrichment (adjusted p-value < 0.05 as cutoff). J. Heatmap to show the TF/motif enrichment of peaks in each epigenomic state. The -log10(p-value) represents the enrichment. Homer was used for motif analysis with the peaks (expanded to 2kb) as input. Only the TFs with expression in microglia (average expression > 1 after log transformation) are shown. K. Barplot to show the percentage of overlapping TFs identified using motif/enrichment based methods and using DoRothEA, a TF activity based method at single cell level. The red bars show the significance (FDR < 0.05). L. The co-regulatory modules of 27 TFs in MG0. Fisher’s exact test is used to evaluate the significance of overlap of targets between each pair of TFs. Adjusted p-value < 0.05 is used as a cutoff and 89 MG0 marker genes as background. The -log10(p-value) is shown in heatmap.
Figure S6. Modulating microglial states in iMGLs by forced expression of master TFs (Related to Main Figure 4). A. UMAP for iMGL single-nuclei sequencing showing all identified clusters (C1-C9). B. Maximum projection of a live imaging time series in iMGLs labeled with the calcium reporter dye, Fluo-4 AM. C. iMGLs 8 days post-infection with lentiviral vector carrying the red fluorescent protein, mCherry. D. Actin staining is shown for a random field of view for each of the TF overexpression conditions to highlight morphological changes. E. Immunostaining of human brain tissue showing colocalization of P2RY12 in magenta with TMEM119 in yellow. ROSMAP Subject ID, 11409232. F. Immunostaining for homeostatic TFs in IBA1 positive cells across the human brain. G. Correlation analysis for SMAD3, IRF8 and ELF1 with TMEM119. No significant effect was detected. H. Actin staining is shown for iMGLs infected with an empty mCherry control lentivirus, and TF overexpression of PPARG and RUNX2. Note that images of PPARG and RUNX2 are duplicated here from those shown in main Figure 4G to ease comparison with control. I. UMAPs for snRNA-seq experiment with CRISPRi iMGLs. Repression of FOXO3, HIF1A and FOXP2 in LPS treated iMGLs can block the induction of IL1B, but no gain in homeostatic marker P2RY12. J. The score shown in the heatmap represents the enrichment of upstream regulators’ expression in each microglial state. The significance (star in the heatmap) is evaluated by the p-value of a permutation test (permutation times:1000). See details in STAR Methods.
Figure S7. Comparisons of DEGs between methods and datasets (Related to Main Figure 5). A. The pairwise comparison of DEGs between MAST, MAST with random effect (MAST.RE) and edgeR. The bar plot shows the Jaccard index to represent the percentage of overlapping genes. The values inside of the bar are FDR after log10 transformation to represent the significance of overlapping. Each panel represents each comparison (nonAD vs earlyAD, or earlyAD vs lateAD, per state). B. Venn diagram to show the number of genes shared or specific in each method. C. Heatmap to show the significant enrichment of DEGs identified in this study with DEGs identified in Gerrits et al.. Jaccard score represents the percentage of pairwise overlapping genes. Fisher’s exact test is used to evaluate the significance of overlapping (adjusted p-value < 0.05 as a cutoff).
Figure S8. Transcriptional changes of microglial transcriptional states in AD (Related to Main Figure 5). Transcriptional changes of 1,542 DEGs in three major categories with 51 gene groups in AD progression. Heatmap shows the change directions. Red represents up-regulated in AD progression, i.e. up-regulated in low AD pathology when compared to no AD pathology and up-regulated in high AD pathology when compared to low AD pathology, and blue represents down-regulation in AD progression. The numbers in the block on the left of heatmap show the number of DEGs included in each gene group.
Figure S9. Biological processes and TF enrichment in DEGs during AD progression (Related to Main Figure 5). A. Heatmap to show the enriched Gene Ontology biological processes in DEGs of 51 gene groups shown in Figure S8. Enrichr in R was used to perform GO enrichment (adjusted p-value < 0.05 as cutoff). B.Heatmap to show the TF enrichment of DEGs in each group shown in Figure S8. Odds ratio represents the enrichment. Enrichr package in R based on three libraries including TRANSFAC and JASPAR, ChEA, and ENCODE TF ChIP-seq data was used to predict upstream TFs (adjusted p-value <0.05 as a cut-off). Only the TFs with expression in microglia (average expression > 1 after log transformation) are shown.
Figure S10. Expression patterns of AD risk genes in 12 microglial transcriptional states (Related to Main Figure 6). A. Heatmap to show gene-level enrichment in GWAS signals to associate microglial states with multiple traits. MAGMA is used to evaluate the significance of enrichment. The -log10 (p-value) is shown in the heatmap. The blue blocks show the p-value < 0.05 as a cutoff. The stars show FDR < 0.05 as a cutoff. B. AD risk genes were obtained from GWAS catalog (GWAS variant p-value < 10e-5). The stars highlight the genes with specially high expression in the corresponding state.
Table S1 Metadata, markers and functions, antibodies, primers, DEGs, TWAS (Related to Figures 1–6).
Supplemental Video 1 (Related to Figure 4). Calcium Dynamics in iMGLs pre-treated with LPS.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Guinea Pig Anti-IBA1 | Synaptic Systems | Cat#822501 |
| Mouse Anti-IL1B | SantaCruz | Cat#sc-32294 |
| Goat Anti-LRRK2 | R&D Systems | Cat#AF6674 |
| Rabbit Anti-CPEB4 | Abcam | Cat#ab224162 |
| Mouse Anti-TMEM119 | NovusBio | Cat#NBP2–76985 |
| Rabbit Anti-RUNX2 | Cell Signaling | Cat#12556S |
| Rabbit Anti-IRF8 | Cell Signaling | Cat# 83413S |
| Rabbit Anti-ELF1 | Santa Cruz | Cat#sc133096 |
| Rabbit Anti-PPARG | Cell Signaling | Cat#2435S |
| Rabbit Anti-SMAD3 | Cell Signaling | Cat#9523S |
| Rabbit Anti-P2RY12 | Atlas Antibodies | Cat# HPA014518 |
| Alexa-Fluor 505 (Donkey anti-Goat) | Thermo Fisher Scientific | Cat# A-48259 |
| Alexa-Fluor 488 (Donkey anti-Mouse) | Thermo Fisher Scientific | Cat# A-21202 |
| Alexa-Fluor 555 (Donkey anti-Rabbit) | Thermo Fisher Scientific | Cat# A-31572 |
| Alexa-Fluor 555 (Donkey anti-Mouse) | Thermo Fisher Scientific | Cat# A-31570 |
| Alexa-Fluor 647 (Donkey Anti-Guinea Pig) | Jackson Immuno Laboratories, Inc. | 706–605-148 |
| DAPI | Thermo Fisher Scientific | Cat# D1306 |
| Biological samples | ||
| See Table S1 for a list of brain samples. | ||
| Chemicals, peptides, and recombinant proteins | ||
| Nuclei Isolation Reagents | Mathys et al.11 | |
| Matrigel | Corning | Cat # 354277 |
| mTeSR1 media | STEMCELL Technologies | Cat# 05872 |
| True black | Biotium | Cat#23007 |
| Fluromount-G | Southern Biotech | Cat#0100–01 |
| Hoechst 33342 | Sigma-Aldrich | Cat#94403 |
| ReLeSR | STEMCELL Technologies | Cat#05872 |
| ROCK inhibitor | BioVision | Cat#2342–25 |
| DMEM/F12 | Thermo Fisher Scientific | Cat#11330–057 |
| Neurobasal media | Gibco | Cat#21103049 |
| IL-34 | PeproTech | Cat#200–34 |
| m-CSF | PeproTech | Cat#300–25 |
| CRISPRMAX | Invitrogen | Cat#CMAX00003 |
| RNase Inhibitor (0.2 U/μL) | Thermo Fisher Scientific | AM2696 |
| LPS (10 ng/mL) | Sigma | L-2654 |
| DMNPE-Caged ATP | Fisher Scientific | Cat#1049 |
| FLUO4-AM | Invitrogen | Cat# F14201 |
| TNFα | PeproTech | 300–01A |
| INFγ | PeproTech | 300–02 |
| EcoDry Premix | Takara | Cat#639549 |
| SsoFast EvaGreen Supermix | Bio-Rad | Cat#1725202 |
| Pre-formed amyloid beta (1–42) fibrils | rPeptide | Cat#AF-1002–1 |
| ActinGreen 488 Readyprobes (Invitrogen #R37110) | Invitrogen | Cat#R37110 |
| Chromium Single-Cell 3′ Reagent Kits v3 | 10x Genomics | https://www.10xgenomics.com |
| STEMdiff Hematopoietic Kit | STEMCELL Technologies | Cat#05310 |
| sgRNA Synthesis Kit | Thermo Fisher Scientific | Cat#A29377 |
| Nuclei EZ Prep | Sigma-Aldrich | Cat#NUC101 |
| Chromium Next GEM Single Cell 3’ Reagent Kit v3.1 | 10x Genomics, Pleasanton, CA) | Cat#PN-1000268 |
| RNeasy Plus Mini Kit | Qiagen | Cat#74134 |
| Critical commercial assays | ||
| RNAscope HiPlex Probe - Hs-FOXP1-T2 | Advanced Cell Diagnostics | 407231-T2 |
| RNAscope HiPlex Probe - Hs-LRRK2-T1 | Advanced Cell Diagnostics | 421531-T1 |
| RNAscope HiPlex Probe - Hs-RELB-T9 | Advanced Cell Diagnostics | 1236361-T9 |
| RNAscope HiPlex Probe - Hs-CCL3-T8 | Advanced Cell Diagnostics | 455331-T8 |
| RNAscope HiPlex Probe - Hs-TMEM163-T10 | Advanced Cell Diagnostics | 1200321-T10 |
| RNAscope HiPlex Probe - Hs-CPEB4-T4 | Advanced Cell Diagnostics | 1063961-T4 |
| RNAscope HiPlex Probe - Hs-P2RY12-T5 | Advanced Cell Diagnostics | 450391-T5 |
| RNAscope HiPlex Probe - Hs-AIF1-T12 | Advanced Cell Diagnostics | 433121-T12 |
| RNAscope HiPlex Probe - Hs-CX3CR1-T6 | Advanced Cell Diagnostics | 411251-T6 |
| RNAscope HiPlex Probe - Hs-PPARG-O1-T4 | Advanced Cell Diagnostics | 864421-T4 |
| RNAscope HiPlex Probe - Hs-APOE-T8 | 433091-T8 | |
| HiPlex12 Reagents Kit (488, 550; 650) Hs V2 | Advanced Cell Diagnostics | 324445 |
| Deposited data | ||
| Count Matrices and Metadata | This paper | http://compbio.mit.edu/microglia_states/ |
| iMGLs bulk RNA-seq and snRNA-seq | This paper | GEO database: GSE227223 |
| Experimental models: Cell lines | ||
| HEK-293T cell line | ATCC | Cat#CRL-3216 |
| CRISPRi iPSC line(dCas9-KRAB) | Allen Institute for Cell Science | Cat#AICS-0090 cl.391 |
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| FOXO3_1 guide RNA: GGAGGGGAAAGGGAAGCGCC |
Horlbeck et al.96 | NA |
| FOXO3_2 guide RNA: GTGGCGGCGCGCGAGCTGAC |
Horlbeck et al.96 | NA |
| HIF1A _1 guide RNA: GCTGGCCGAAGCGACGAAGA |
Horlbeck et al.96 | NA |
| HIF1A _2 guide RNA: GCCTCCTGTCCCCTCAGACG |
Horlbeck et al.96 | NA |
| FOXP2_1 guide RNA: GAAGCCGGGAGACCAGACAC |
Horlbeck et al.96 | NA |
| FOXP2_2 guide RNA: GAATCAGCATTTAATCACTA |
Horlbeck et al.96 | NA |
| CCL3 qPCR FWD: AGTTCTCTGCATCACTTGCTG REV: CGGCTTCGCTTGGTTAGGAA | https://pga.mgh.harvard.edu/primerbank/ | NA |
| PLEK qPCR FWD: AAGAAGGGGAGCGTGTTCAAT REV: TCAGCGGGATCATTCCTTTGG |
https://pga.mgh.harvard.edu/primerbank/ | NA |
| NFKBIZ qPCR FWD: AGAGGCCCCTTTCAAGGTGT REV:TCCATCAGACAACGAATCGGG |
https://pga.mgh.harvard.edu/primerbank/ | NA |
| RELB qPCR FWD:CAGCCTCGTGGGGAAAGAC REV:GCCCAGGTTGTTAAAACTGTGC | https://pga.mgh.harvard.edu/primerbank/ | NA |
| NFKB1 qPCR FWD: AACAGAGAGGATTTCGTTTCCG REV:TTTGACCTGAGGGTAAGACTTCT |
https://pga.mgh.harvard.edu/primerbank/ | NA |
| TNFAIP3 qPCR FWD: TCCTCAGGCTTTGTATTTGAGC REV:TGTGTATCGGTGCATGGTTTTA |
https://pga.mgh.harvard.edu/primerbank/ | NA |
| FAM129A qPCR FWD: GAAGAGGTTTAGTGCCCTCCT REV:CCTTCTCCTGTCGGAAGAATTG |
https://pga.mgh.harvard.edu/primerbank/ | NA |
| LIMK2 qPCR FWD: GGATTCCCTCACCAACTGGTA REV:AGCCACCATAAAAGGCCCTG |
https://pga.mgh.harvard.edu/primerbank/ | NA |
| CPEB4 qPCR FWD: ACATCTAGCGCATCGTCTCTT REV:ACAACAGAGCACCGTTATTAGC |
https://pga.mgh.harvard.edu/primerbank/merbank/ | NA |
| LRRK2 qPCR FWD: GAGCACGCCTCCAAGTTATTT REV:ACTGGCATTATGAACTGTTAGCA |
https://pga.mgh.harvard.edu/primerbank/ | NA |
| GPR155 qPCR FWD: TTGCCTACAGCAGTAACGCAT REV:GGAAGTGCAAATCTGGAGACA |
https://pga.mgh.harvard.edu/primerbank/ | NA |
| CYP27A1 qPCR FWD: CGGCAACGGAGCTTAGAGG |
https://pga.mgh.harvard.edu/primerbank/ | NA |
| IL-1B qPCR FWD: ATGATGGCTTATTACAGTGGCAA REV:GTCGGAGATTCGTAGCTGGA |
https://pga.mgh.harvard.edu/primerbank/ | NA |
| P2RY12 qPCR FWD: CACTGCTCTACACTGTCCTGT REV:AGTGGTCCTGTTCCCAGTTTG |
https://pga.mgh.harvard.edu/primerbank/ | NA |
| Recombinant DNA | ||
| pLX304 SMAD3 | DNASU | HsCD00440766-SMAD3 |
| pLX304 SMARCA4 | DNASU | HsCD00861075-SMARCA4 |
| pLX304 STAT3 | DNASU | HsCD00443857-STAT3 |
| pDONR221 SUZ12 | DNASU | HsCD00832948-SUZ12 |
| pLX304 TCF4 | DNASU | HsCD00438170-TCF4 |
| pLX304 ELF1 | DNASU | HsCD00445051-ELF1 |
| pLX304 ELF2 | DNASU | HsCD00438947-ELF2 |
| pLX304 ETV5 | DNASU | HsCD00435257-ETV5 |
| pLX304 ETV6 | DNASU | HsCD00445584-ETV6 |
| pLX304 FLI1 | DNASU | HsCD00438298-FLI1 |
| pLX304 IRF8 | DNASU | HsCD00440187-IRF8 |
| pLX304 SPI1 | DNASU | HsCD00936456-SPI1 |
| pLX304 ETS2 | DNASU | HsCD00434239-ETS2 |
| pLX304 CEBPD | DNASU | HsCD00852417-CEBPD |
| pLX304 CTBP2 | DNASU | HsCD00435806-CTBP2 |
| pLX304 CTNNB1 | DNASU | HsCD00852530-CTNNB1 |
| pLX304 ESR1 | DNASU | HsCD00443651-ESR1 |
| pLX304 FOXO3 | DNASU | HsCD00446377-FOXO3 |
| pDONR221 JARID2 | DNASU | HsCD00829788-JARID2 |
| pLX304 LMO2 | DNASU | HsCD00438767-LMO2 |
| pLX304 MTF2 | DNASU | HsCD00435376-MTF2 |
| pLX304 NR3C1 | DNASU | HsCD00445225-NR3C1 |
| pLX304 PIAS1 | DNASU | HsCD00443565-PIAS1 |
| pLX304 PPARG | DNASU | HsCD00441631-PPARG |
| pDONR221 RUNX2 | DNASU | HsCD00829534-RUNX2 |
| pLX304 ARNT | DNASU | HsCD00442555-ARNT |
| pLX304 | DNASU | EvNO00877063 |
| pLV-mcherry | Addgene | Addgene #36084 |
| Software and algorithms | ||
| CellRanger software v3.0.1 | 10x Genomics | https://www.10xgenomics.com/ |
| Seurat R package v.4.0.3 | Hao et al.57 | https://satijalab.org/seurat/ |
| propeller | Phipson et al.64 | https://github.com/Oshlack/speckle |
| DoubletFinder v2.0 | McGinnis et al.59 | https://github.com/chris-mcginnis-ucsf/DoubletFinder |
| cellranger-atac software(v1.1.0) | 10x Genomics | https://www.10xgenomics.com/ |
| ArchR (v1.0.1) | Granja et al.61 | https://www.archrproject.com/ |
| MACS2 | Zhang et al.65 | https://github.com/macs3-project/MACS |
| Homer | Heinz et al.67 | http://homer.ucsd.edu/homer/motif/ |
| EnrichR Appyter | Kuleshov et al.69 | https://maayanlab.cloud/Enrichr |
| ComplexHeatmap | Gu et al.97,98 | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| IMARIS | Oxford Instruments | https://imaris.oxinst.com |
| Morpheus | Broad Institute | https://software.broadinstitute.org/morpheus/ |
| Photoshop | Adobe Photoshop 2021 | https://www.adobe.com |
| QuPath (v0.4.3) | QuPath | https://qupath.github.io |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/ |
| MAGMA.Celltype | Skene et al.45 | https://github.com/neurogenomics/MAGMA_Celltyping |
| R knockoff package | https://web.stanford.edu/group/candes/knockoffs/index.html | https://cran.r-project.org/web/packages/knockoff/index.html |
| Cytoscape (v3.9.1) | The Cytoscape Consortium | https://cytoscape.org/ |
| Other | ||
| Superfrost Plus Slides | Fisherbrand | Cat#12–550-15 |
| 40μm cell strainer | Corning | Cat#431750 |
| Confocal microscope | Zeiss | Cat#LSM 900 |
| Punch Kit 2.00 mm | UniCore | Cat# WHAWB100029 |
| Cryostat | Leica | Cat#CM3050s |
| Automated Cell Counter | Invitrogen | Countess II |
| AggreWell 800 | STEMCELL technologies | Cat#34815 |
| Real-Time PCR | Bio-Rad | CFX96 Touch Real-Time PCR Detection System |
HIGHLIGHTS.
Single-nucleus transcriptomes and epigenomes of human microglia
Microglia state-specific and disease-stage-specific profile in Alzheimer’s disease
Chromatin accessibility poorly captured microglia transcriptional state diversity
Transcription factor networks regulate microglial states and their transitions
Acknowledgements
We thank the study participants and staff of the Rush Alzheimer’s Disease Center. This Tsai lab research was made possible through the generous support of the Robert A. and Renee E. Belfer Family Foundation, The JPB Foundation, Eduardo Eurnekian, Joseph P. DiSabato and Nancy E. Sakamoto, Donald A. and Glenda G. Mattes, Lester A. Gimpelson, Paul Slavik, David Emmes, and Thomas Stocky and Avni Shah. This work was also supported in part by NIH grants AG054012, AG058002, AG062377, NS110453, NS115064, AG062335, NS127187, AG074003 (M.K., L.-H.T.); AG067151, MH109978, MH119509, HG008155, DA053631 (M.K.); 1RF1AG054321 (L.-H.T.); P30AG10161, P30AG72975, R01AG15819, R01AG17917, U01AG46152, U01AG61356, and R01AG57473 (D.A.B.); and grants from the Cure Alzheimer’s Foundation CIRCUITS consortium (M.K., L.-H.T). M.B.V. is supported by the Howard Hughes Medical Institute Hanna H. Gray Postdoctoral Fellowship. We thank Amy Grayson and Patricia Purcell for scientific suggestions.
Footnotes
Declaration of interests
L.-H.T. is a member of the Scientific Advisory Board of Cognito Therapeutics, 4M Therapeutics, Cell Signaling Technology, and Souvien Therapeutics, which has no association to this study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- 1.Butovsky O, and Weiner HL (2018). Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 19, 622–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ellwanger DC, Wang S, Brioschi S, Shao Z, Green L, Case R, Yoo D, Weishuhn D, Rathanaswami P, Bradley J, et al. (2021). Prior activation state shapes the microglia response to antihuman TREM2 in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A. 118. 10.1073/pnas.2017742118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, Cao Y, Lee S-H, Haddick PCG, Ngu H, et al. (2018). Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 22, 832–847. [DOI] [PubMed] [Google Scholar]
- 4.Hammond TR., Dufort C., Dissing-Olesen L., Giera S., Young A., Wysoker A., Walker AJ., Gergits F., Segel M., Nemesh J., et al. (2019). Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 50, 253–271.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290.e17. [DOI] [PubMed] [Google Scholar]
- 6.Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M, De Jager PL, Ransohoff RM, Regev A, and Tsai L-H (2017). Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 21, 366–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sala Frigerio C, Wolfs L, Fattorelli N, Thrupp N, Voytyuk I, Schmidt I, Mancuso R, Chen W-T, Woodbury ME, Srivastava G, et al. (2019). The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep. 27, 1293–1306.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Srinivasan K, Friedman BA, Etxeberria A, Huntley MA, van der Brug MP, Foreman O, Paw JS, Modrusan Z, Beach TG, Serrano GE, et al. (2020). Alzheimer’s Patient Microglia Exhibit Enhanced Aging and Unique Transcriptional Activation. Cell Rep. 31, 107843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerrits E, Brouwer N, Kooistra SM, Woodbury ME, Vermeiren Y, Lambourne M, Mulder J, Kummer M, Möller T, Biber K, et al. (2021). Distinct amyloid-β and tau-associated microglia profiles in Alzheimer’s disease. Acta Neuropathol. 141, 681–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grubman A, Choo XY, Chew G, Ouyang JF, Sun G, Croft NP, Rossello FJ, Simmons R, Buckberry S, Landin DV, et al. (2021). Transcriptional signature in microglia associated with Aβ plaque phagocytosis. Nat. Commun. 12, 3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, et al. (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ, Cimpean M, Khairallah A, Coronas-Samano G, Sankowski R, et al. (2020). Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat. Commun. 11, 6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sankowski R, Böttcher C, Masuda T, Geirsdottir L, Sagar, Sindram E, Seredenina T, Muhs A, Scheiwe C, Shah MJ, et al. (2019). Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 22, 2098–2110. [DOI] [PubMed] [Google Scholar]
- 14.Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, and Schneider JA (2018). Religious Orders Study and Rush Memory and Aging Project. J. Alzheimers. Dis. 64, S161–S189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia FJ, Sun N, Lee H, Godlewski B, Mathys H, Galani K, Zhou B, Jiang X, Ng AP, Mantero J, et al. (2022). Single-cell dissection of the human brain vasculature. Nature 603, 893–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun N, Akay LA, Murdock MH, Park Y, Galiana-Melendez F, Bubnys A, Galani K, Mathys H, Jiang X, Ng AP, et al. (2023). Single-nucleus multiregion transcriptomic analysis of brain vasculature in Alzheimer’s disease. Nat. Neurosci. 26, 970–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu H, Leak RK, and Hu X (2016). Neurotransmitter receptors on microglia. Stroke Vasc Neurol 1, 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’Loughlin E, Xu Y, Fanek Z, et al. (2017). The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47, 566–581.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McQuade A., Coburn M., Tu CH., Hasselmann J., Davtyan H., and Blurton-Jones M. (2018). Development and validation of a simplified method to generate human microglia from pluripotent stem cells. Mol. Neurodegener. 13, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang KX, and Denhardt DT (2008). Osteopontin: role in immune regulation and stress responses. Cytokine Growth Factor Rev. 19, 333–345. [DOI] [PubMed] [Google Scholar]
- 21.da Fonseca ACC, Matias D, Garcia C, Amaral R, Geraldo LH, Freitas C, and Lima FRS (2014). The impact of microglial activation on blood-brain barrier in brain diseases. Front. Cell. Neurosci. 8, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith AM, Gibbons HM, Oldfield RL, Bergin PM, Mee EW, Faull RLM, and Dragunow M (2013). The transcription factor PU.1 is critical for viability and function of human brain microglia. Glia 61, 929–942. [DOI] [PubMed] [Google Scholar]
- 24.Kim S, Yu N-K, and Kaang B-K (2015). CTCF as a multifunctional protein in genome regulation and gene expression. Exp. Mol. Med. 47, e166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Helin K, Wu CL, Fattaey AR, Lees JA, Dynlacht BD, Ngwu C, and Harlow E (1993). Heterodimerization of the transcription factors E2F-1 and DP-1 leads to cooperative trans-activation. Genes Dev. 7, 1850–1861. [DOI] [PubMed] [Google Scholar]
- 26.Chen H-W, King K, Tu J, Sanchez M, Luster AD, and Shresta S (2013). The roles of IRF-3 and IRF-7 in innate antiviral immunity against dengue virus. J. Immunol. 191, 4194–4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shang YC, Chong ZZ, Hou J, and Maiese K (2009). The forkhead transcription factor FOXO3a controls microglial inflammatory activation and eventual apoptotic injury through caspase 3. Curr. Neurovasc. Res. 6, 20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.March-Diaz R, Lara-Ureña N, Romero-Molina C, Heras-Garvin A, Ortega-de San Luis C, Alvarez-Vergara MI, Sanchez-Garcia MA, Sanchez-Mejias E, Davila JC, Rosales-Nieves AE, et al. (2021). Hypoxia compromises the mitochondrial metabolism of Alzheimer’s disease microglia via HIF1. Nature Aging 1, 385–399. [DOI] [PubMed] [Google Scholar]
- 29.Lum JJ, Bui T, Gruber M, Gordan JD, DeBerardinis RJ, Covello KL, Simon MC, and Thompson CB (2007). The transcription factor HIF-1alpha plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev. 21, 1037–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H-S, Zhou Y-N, Li L, Li S-F, Long D, Chen X-L, Zhang J-B, Feng L, and Li Y-P (2019). HIF-1α protects against oxidative stress by directly targeting mitochondria. Redox Biol 25, 101109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garcia-Alonso L, Holland CH, Ibrahim MM, Turei D, and Saez-Rodriguez J (2019). Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res. 29, 1363–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Timmerman R, Burm SM, and Bajramovic JJ (2018). An Overview of Methods to Study Microglia. Front. Cell. Neurosci. 12, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Victor MB, Leary N, Luna X, Meharena HS, Scannail AN, Bozzelli PL, Samaan G, Murdock MH, von Maydell D, Effenberger AH, et al. (2022). Lipid accumulation induced by APOE4 impairs microglial surveillance of neuronal-network activity. Cell Stem Cell 29, 1197–1212.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Colombo G., Cubero RJA., Kanari L., Venturino A., Schulz R., Scolamiero M., Agerberg J., Mathys H., Tsai L-H., Chachólski W., et al. (2022). A tool for mapping microglial morphology, morphOMICs, reveals brain-region and sex-dependent phenotypes. Nat. Neurosci. 25, 1379–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, et al. (2014). Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat. Neurosci. 17, 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, et al. (2015). MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zimmerman KD, Espeland MA, and Langefeld CD (2021). A practical solution to pseudoreplication bias in single-cell studies. Nat. Commun. 12, 738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ji Y, Wang X, Kalicki C, Menta BW, Baumgardner M, Koppel SJ, Weidling IW, Perez-Ortiz J, Wilkins HM, and Swerdlow RH (2019). Effects of Microglial Cytokines on Alzheimer’s Disease-Related Phenomena. J. Alzheimers. Dis. 67, 1021–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang W-Y, Tan M-S, Yu J-T, and Tan L (2015). Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med 3, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mockenhaupt K, Gonsiewski A, and Kordula T (2021). RelB and Neuroinflammation. Cells 10. 10.3390/cells10071609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, Sealock J, Karlsson IK, Hägg S, Athanasiu L, et al. (2019). Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 51, 404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, Boland A, Vronskaya M, van der Lee SJ, Amlie-Wolf A, et al. (2019). Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kang S, Gim J, Lee J, Gunasekaran TI, Choi KY, Lee JJ, Seo EH, Ko P-W, Chung JY, Choi S-M, et al. (2021). Potential Novel Genes for Late-Onset Alzheimer’s Disease in East-Asian Descent Identified by APOE-Stratified Genome-Wide Association Study. J. Alzheimers. Dis. 82, 1451–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skene NG, Bryois J, Bakken TE, Breen G, Crowley JJ, Gaspar HA, Giusti-Rodriguez P, Hodge RD, Miller JA, Muñoz-Manchado AB, et al. (2018). Genetic identification of brain cell types underlying schizophrenia. Nat. Genet. 50, 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buniello A., MacArthur JAL., Cerezo M., Harris LW., Hayhurst J., Malangone C., McMahon A., Morales J., Mountjoy E., Sollis E., et al. (2019). The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, Llapashtica C, Wang J, Kim DJ, Xia D, et al. (2020). TREM2 Regulates Microglial Cholesterol Metabolism upon Chronic Phagocytic Challenge. Neuron 105, 837–854.e9. [DOI] [PubMed] [Google Scholar]
- 48.Sudwarts A, Ramesha S, Gao T, Ponnusamy M, Wang S, Hansen M, Kozlova A, Bitarafan S, Kumar P, Beaulieu-Abdelahad D, et al. (2022). BIN1 is a key regulator of proinflammatory and neurodegeneration-related activation in microglia. Mol. Neurodegener. 17, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang M-G, Sun L, Han J, Zheng C, Liang H, Zhu J, and Jin T (2019). Biological characteristics of transcription factor RelB in different immune cell types: implications for the treatment of multiple sclerosis. Mol. Brain 12, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bernier L-P, York EM, and MacVicar BA (2020). Immunometabolism in the Brain: How Metabolism Shapes Microglial Function. Trends Neurosci. 43, 854–869. [DOI] [PubMed] [Google Scholar]
- 51.Bennett DA, Wilson RS, Schneider JA, Evans DA, Beckett LA, Aggarwal NT, Barnes LL, Fox JH, and Bach J (2002). Natural history of mild cognitive impairment in older persons. Neurology 59, 198–205. [DOI] [PubMed] [Google Scholar]
- 52.Bennett DA, Wilson RS, Schneider JA, Evans DA, Aggarwal NT, Arnold SE, Cochran EJ, Berry-Kravis E, and Bienias JL (2003). Apolipoprotein E epsilon4 allele, AD pathology, and the clinical expression of Alzheimer’s disease. Neurology 60, 246–252. [DOI] [PubMed] [Google Scholar]
- 53.Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC, and Wilson RS (2006). Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 66, 1837–1844. [DOI] [PubMed] [Google Scholar]
- 54.Bennett DA, Schneider JA, Wilson RS, Bienias JL, and Arnold SE (2004). Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch. Neurol. 61, 378–384. [DOI] [PubMed] [Google Scholar]
- 55.Bennett DA, Schneider JA, Aggarwal NT, Arvanitakis Z, Shah RC, Kelly JF, Fox JH, Cochran EJ, Arends D, Treinkman AD, et al. (2006). Decision rules guiding the clinical diagnosis of Alzheimer’s disease in two community-based cohort studies compared to standard practice in a clinic-based cohort study. Neuroepidemiology 27, 169–176. [DOI] [PubMed] [Google Scholar]
- 56.Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P-R., and Raychaudhuri S. (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McGinnis CS, Murrow LM, and Gartner ZJ (2019). DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst 8, 329–337.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, McDermott GP, Olsen BN, Mumbach MR, Pierce SE, Corces MR, et al. (2019). Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 37, 925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Granja JM, Corces MR, Pierce SE, Bagdatli ST, Choudhry H, Chang HY, and Greenleaf WJ (2021). ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 53, 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang X, Lan Y, Xu J, Quan F, Zhao E, Deng C, Luo T, Xu L, Liao G, Yan M, et al. (2019). CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 47, D721–D728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang D, Liu S, Warrell J, Won H, Shi X, Navarro FCP, Clarke D, Gu M, Emani P, Yang YT, et al. (2018). Comprehensive functional genomic resource and integrative model for the human brain. Science 362. 10.1126/science.aat8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Phipson B, Sim CB, Porrello ER, Hewitt AW, Powell J, and Oshlack A (2022). propeller: testing for differences in cell type proportions in single cell data. Bioinformatics 38, 4720–4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM, et al. (2021). Gene Set Knowledge Discovery with Enrichr. Curr Protoc 1, e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Victor MB, Richner M, Hermanstyne TO, Ransdell JL, Sobieski C, Deng P-Y, Klyachko VA, Nerbonne JM, and Yoo AS (2014). Generation of human striatal neurons by microRNA-dependent direct conversion of fibroblasts. Neuron 84, 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Michael Cherry J, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.The Gene Ontology Consortium (2019). The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 47, D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh P-R, Anttila V, Xu H, Zang C, Farh K, et al. (2015). Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bentham J., Morris DL., Graham DSC., Pinder CL., Tombleson P., Behrens TW., Martín J., Fairfax BP., Knight JC., Chen L., et al. (2015). Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 47, 1457–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. (2012). Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, Lango Allen H, Lindgren CM, Luan J ‘an, Mägi R., et al. (2010). Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet. 42, 937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, et al. (2010). Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466, 707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, Kochi Y, Ohmura K, Suzuki A, Yoshida S, et al. (2014). Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506, 376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Okbay A, Baselmans BML, De Neve J-E, Turley P, Nivard MG, Fontana MA, Meddens SFW, Linnér RK, Rietveld CA, Derringer J, et al. (2016). Genetic variants associated with subjective well-being, depressive symptoms, and neuroticism identified through genome-wide analyses. Nat. Genet. 48, 624–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thorgeirsson TE, Gudbjartsson DF, Surakka I, Vink JM, Amin N, Geller F, Sulem P, Rafnar T, Esko T, Walter S, et al. (2010). Sequence variants at CHRNB3-CHRNA6 and CYP2A6 affect smoking behavior. Nat. Genet. 42, 448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control Consortium 2, Sawcer S, Hellenthal G, Pirinen M, Spencer CCA, Patsopoulos NA, Moutsianas L, Dilthey A, Su Z, et al. (2011). Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Morris AP., Voight BF., Teslovich TM., Ferreira T., Segrè AV., Steinthorsdottir V., Strawbridge RJ., Khan H., Grallert H., Mahajan A., et al. (2012). Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet. 44, 981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Park YP, and Kellis M (2021). CoCoA-diff: counterfactual inference for single-cell gene expression analysis. Genome Biol. 22, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Barber RF, and Candès EJ (2015). Controlling the false discovery rate via knockoffs. aos 43, 2055–2085. [Google Scholar]
- 86.Wang Y, and Blei DM (2019). The Blessings of Multiple Causes. J. Am. Stat. Assoc. 114, 1574–1596. [Google Scholar]
- 87.Zhu Z, Fan Y, Kong Y, Lv J, and Sun F (2021). DeepLINK: Deep learning inference using knockoffs with applications to genomics. Proc. Natl. Acad. Sci. U. S. A. 118. 10.1073/pnas.2104683118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jiang T, Li Y, and Motsinger-Reif AA (2021). Knockoff boosted tree for model-free variable selection. Bioinformatics 37, 976–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Candes E, Fan Y, Janson L, and Lv J (2018). Panning for gold:’model-X’knockoffs for high dimensional controlled variable selection. Journal of the Royal. [Google Scholar]
- 90.Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BWJH, Jansen R, de Geus EJC, Boomsma DI, Wright FA, et al. (2016). Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48, 245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ, Eyler AE, Denny JC, GTEx Consortium, Nicolae DL, et al. (2015). A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 47, 1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Berisa T, and Pickrell JK (2016). Approximately independent linkage disequilibrium blocks in human populations. Bioinformatics 32, 283–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang G, Sarkar A, Carbonetto P, and Stephens M (2020). A simple new approach to variable selection in regression, with application to genetic fine-mapping. bioRxiv, 501114. 10.1101/501114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Park Y, Sarkar AK, He L, Davila-Velderrain J, De Jager PL, and Kellis M (2017). A Bayesian approach to mediation analysis predicts 206 causal target genes in Alzheimer’s disease. bioRxiv, 219428. 10.1101/219428. [DOI] [Google Scholar]
- 95.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Horlbeck MA, Gilbert LA, Villalta JE, Adamson B, Pak RA, Chen Y, Fields AP, Park CY, Corn JE, Kampmann M, et al. (2016). Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. Elife 5. 10.7554/eLife.19760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- 98.G Z. (2022). Complex heatmap visualization. Imeta 1. 10.1002/imt2.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Microglial transcriptional states in aged human brains (Related to Main Figure 1). A. UMAP of 174,420 in silico sorted immune nuclei from human postmortem brain tissues labeled 16 clusters in snRNA-seq data. B. Expression patterns of canonical markers of brain cell types and immune cell types. The size of dots represents the percentage of cells with expression. The color of dots represents the scaled average expression level. C. Average of log2foldchange of 23 MG5 and C9 shared marker genes. The red dashed line shows the threshold (Fold change > 1.5). D. Dotplot to show the signature score of DAM and homeostatic genes in each microglial transcriptional state shown in Figure 1A. E. Heatmap to show the expression of DAM genes in each microglial state. The average expression in each state after log transformation is shown in the heatmap. F. Expression levels of neurotransmitter receptors in 12 microglial states. The average expression in each state after log transformation is shown in the heatmap. G. Representative enriched Gene Ontology biological processes in each microglial transcriptional state. Enrichr in R was used to perform GO enrichment (adjusted p-value < 0.05 as cutoff). H. Heatmap to show the significant overlap of marker genes between Olah et al. and this study. Jaccard score represents the percentage of pairwise overlapping genes. Fisher’s exact test is used to evaluate the significance of overlapping (adjusted p-value < 0.05 as a cutoff). I. Heatmap to show the signature score of marker genes identified in Olah et al. in each microglial state of this study. The signature score is estimated by the average expression level of marker genes. J. Heatmap to show the significant overlap of marker genes between Hammond et al. (mouse) and this study (human). Jaccard score represents the percentage of pairwise overlapping genes. Fisher’s exact test is used to evaluate the significance of overlapping (adjusted p-value < 0.05 as a cutoff).
Figure S2. Cell abundance across multiple variables (Related to Main Figure 1). A. UMAP of microglia nuclei for each brain region. The number of cells in each cell type is shown. B. Distribution of cell fraction across six brain regions in major cell types. C. Statistical testing summary for comparison between regions by propeller in R. D. Statistical testing summary for comparison between nonAD, earlyAD and lateAD by propeller in R. E. Heatmap to show the correlation between cell fraction and quantitative pathological variable in each microglial transcriptional state. The Pearson correlation coefficient (PCC) is calculated and tested by cor.test in R to measure significance. The PCC values are shown in the heatmap. The significant levels are represented by stars (*: p-value < 0.05, **: p-value < 0.01, and ***: p-value < 0.001). F. Distribution of cell fraction across all individuals in each microglial state. G. Distribution of cell fraction across sequencing batches in each microglial state. H-I. Distribution of ages/PMIs of the individuals across 12 microglial states. J. Cell fraction across sex for each microglial state (bottom) and across cell types for male and female individuals (top). K. Statistical testing summary for comparison in age, PMI and sex by propeller in R.
Figure S3. Immunohistochemistry on a subset of postmortem human brain samples from ROSMAP cohort (Related to Main Figures 1, 2 and 4). Immunostaining for IBA1 and P2RY12 across 8 subjects spanning non-AD, to Early AD and Late AD status. ROSMAP subject IDs are displayed in each corresponding image.
Figure S4. Validation of marker genes associated with inflammatory states and transcriptional response across MG10, MG8 and MG2 markers in iMGLs exposed to pro-inflammatory stimulus (Related to Main Figures 2). A. Multiplexed immunostaining to spatially resolve inflammatory states across the human brain. IBA1 was used to label all CNS resident macrophages, ILB1 to label MG10, LRRK2 to label MG8 and CPEB4 to label MG2 microglia (see list of antibodies used in immunohistochemistry in Table S1, page 4). ROSMAP subject ID, 10248033. B. Fluorescent intensity projection onto 3D renderings of IBA1 signal for each channel of multiplex immunostaining shows distinct microglial populations. C. Heatmap for immunostaining quantification showing relative maximum intensity fluorescence for CPEB4, IL1B and LRRK2. D. Quantification of microglia (P2RY12+IBA+CX3CR1+) from RNAscope HiPlex for inflammatory state markers: LRRK2, FOXP1 (MG8 marker), CPEB4, TMEM163 (MG2 marker), CCL3 and RELB (MG10 marker). Number of dots normalized across cells for each marker and plotted as Z-scores and shown from blue (low) to magenta (high). n=291 microglia from 4 AD subjects were quantified. E-H. qPCR analysis for pulse-and-chase experiment after iMGLs exposure to pro-inflammatory stimuli (TNFa, INFy and LPS). E. List of marker genes and their corresponding MG clusters origin. F. MG Cluster 10 genes. G. MG Cluster 2 genes. H. MG Cluster 8 genes. Note that while IL1B is not shown in this supplemental figure, CPEB4 and LRRK2 are duplicated here from main Figure 2G. n= mean of biological triplicate are shown for each timepoint and treatment. Error bars were removed for ease of interpretation.
Figure S5. Microglial epigenetic states defined by snATAC-seq data (Related to Main Figure 3). A. UMAP of 41,832 microglia nuclei in snATAC-seq labeled by the six brain regions. B. Heatmap to show the gene score patterns of 2,805 marker genes across three epigenomic states in microglia. The known marker genes for microglia are listed on the right. The z-scores are shown in the heatmap. C. Gene score distributions of P2RY12, CCL3 and SPP1 in three microglial epigenomic states. CCL3 shows the significantly high score in ACT1 state and SPP1 shows the significantly high score in ACT2 state, highlighted with star labels. The p-value <0.01 as cutoff in the Wilcoxon rank test. D. UMAPs to show the microglia clusters for RNA, ATAC, ATAC colored by RNA, and RNA-ATAC joint analysis, in multiome data from 19 ROSMAP individuals. E. Heatmap to show the significant enrichment of marker genes of multiomic transcriptional clusters of microglia in 12 microglial states. Jaccard score represents the percentage of pairwise overlapping genes. Fisher’s exact test is used to evaluate the significance of overlapping (adjusted p-value < 0.05 as a cutoff). F. The annotation of accessible peaks in microglia. Colors represent the distinct genomic locations including distal, exonic, intronic and promoter. G. Heatmap to show 16,800 marker peaks in three epigenomic states. Z-score values across microglia states are shown in the heatmap. H. GREAT analysis to annotate the epigenomic specific peaks. The panels from left to right include the distribution of peaks based on the number of associated genes per peak, the distribution of peaks based on the distance to TSS, and the distribution of peaks based on the absolute distance to TSS. I. Representative enriched Gene Ontology biological processes of epigenomic state-peak-associated genes. GREAT was used to perform the annotation and GO enrichment (adjusted p-value < 0.05 as cutoff). J. Heatmap to show the TF/motif enrichment of peaks in each epigenomic state. The -log10(p-value) represents the enrichment. Homer was used for motif analysis with the peaks (expanded to 2kb) as input. Only the TFs with expression in microglia (average expression > 1 after log transformation) are shown. K. Barplot to show the percentage of overlapping TFs identified using motif/enrichment based methods and using DoRothEA, a TF activity based method at single cell level. The red bars show the significance (FDR < 0.05). L. The co-regulatory modules of 27 TFs in MG0. Fisher’s exact test is used to evaluate the significance of overlap of targets between each pair of TFs. Adjusted p-value < 0.05 is used as a cutoff and 89 MG0 marker genes as background. The -log10(p-value) is shown in heatmap.
Figure S6. Modulating microglial states in iMGLs by forced expression of master TFs (Related to Main Figure 4). A. UMAP for iMGL single-nuclei sequencing showing all identified clusters (C1-C9). B. Maximum projection of a live imaging time series in iMGLs labeled with the calcium reporter dye, Fluo-4 AM. C. iMGLs 8 days post-infection with lentiviral vector carrying the red fluorescent protein, mCherry. D. Actin staining is shown for a random field of view for each of the TF overexpression conditions to highlight morphological changes. E. Immunostaining of human brain tissue showing colocalization of P2RY12 in magenta with TMEM119 in yellow. ROSMAP Subject ID, 11409232. F. Immunostaining for homeostatic TFs in IBA1 positive cells across the human brain. G. Correlation analysis for SMAD3, IRF8 and ELF1 with TMEM119. No significant effect was detected. H. Actin staining is shown for iMGLs infected with an empty mCherry control lentivirus, and TF overexpression of PPARG and RUNX2. Note that images of PPARG and RUNX2 are duplicated here from those shown in main Figure 4G to ease comparison with control. I. UMAPs for snRNA-seq experiment with CRISPRi iMGLs. Repression of FOXO3, HIF1A and FOXP2 in LPS treated iMGLs can block the induction of IL1B, but no gain in homeostatic marker P2RY12. J. The score shown in the heatmap represents the enrichment of upstream regulators’ expression in each microglial state. The significance (star in the heatmap) is evaluated by the p-value of a permutation test (permutation times:1000). See details in STAR Methods.
Figure S7. Comparisons of DEGs between methods and datasets (Related to Main Figure 5). A. The pairwise comparison of DEGs between MAST, MAST with random effect (MAST.RE) and edgeR. The bar plot shows the Jaccard index to represent the percentage of overlapping genes. The values inside of the bar are FDR after log10 transformation to represent the significance of overlapping. Each panel represents each comparison (nonAD vs earlyAD, or earlyAD vs lateAD, per state). B. Venn diagram to show the number of genes shared or specific in each method. C. Heatmap to show the significant enrichment of DEGs identified in this study with DEGs identified in Gerrits et al.. Jaccard score represents the percentage of pairwise overlapping genes. Fisher’s exact test is used to evaluate the significance of overlapping (adjusted p-value < 0.05 as a cutoff).
Figure S8. Transcriptional changes of microglial transcriptional states in AD (Related to Main Figure 5). Transcriptional changes of 1,542 DEGs in three major categories with 51 gene groups in AD progression. Heatmap shows the change directions. Red represents up-regulated in AD progression, i.e. up-regulated in low AD pathology when compared to no AD pathology and up-regulated in high AD pathology when compared to low AD pathology, and blue represents down-regulation in AD progression. The numbers in the block on the left of heatmap show the number of DEGs included in each gene group.
Figure S9. Biological processes and TF enrichment in DEGs during AD progression (Related to Main Figure 5). A. Heatmap to show the enriched Gene Ontology biological processes in DEGs of 51 gene groups shown in Figure S8. Enrichr in R was used to perform GO enrichment (adjusted p-value < 0.05 as cutoff). B.Heatmap to show the TF enrichment of DEGs in each group shown in Figure S8. Odds ratio represents the enrichment. Enrichr package in R based on three libraries including TRANSFAC and JASPAR, ChEA, and ENCODE TF ChIP-seq data was used to predict upstream TFs (adjusted p-value <0.05 as a cut-off). Only the TFs with expression in microglia (average expression > 1 after log transformation) are shown.
Figure S10. Expression patterns of AD risk genes in 12 microglial transcriptional states (Related to Main Figure 6). A. Heatmap to show gene-level enrichment in GWAS signals to associate microglial states with multiple traits. MAGMA is used to evaluate the significance of enrichment. The -log10 (p-value) is shown in the heatmap. The blue blocks show the p-value < 0.05 as a cutoff. The stars show FDR < 0.05 as a cutoff. B. AD risk genes were obtained from GWAS catalog (GWAS variant p-value < 10e-5). The stars highlight the genes with specially high expression in the corresponding state.
Table S1 Metadata, markers and functions, antibodies, primers, DEGs, TWAS (Related to Figures 1–6).
Supplemental Video 1 (Related to Figure 4). Calcium Dynamics in iMGLs pre-treated with LPS.
Data Availability Statement
Count matrices, metadata and supplementary data in this study from the ROSMAP cohort are at http://compbio.mit.edu/microglia_states/. ROSMAP data can be requested at https://www.radc.rush.edu. Raw data and processed data for all iMGLs bulk RNA-seq and snRNA-seq in this study are publicly available in GEO database under accession GSE227223.
The codes used in this study are available at http://compbio.mit.edu/scMicroglia/, https://github.com/nasunmit/scMicroglia and Zenodo https://doi.org/10.5281/zenodo.8297901.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.