

Abstract
Most cancer treatment strategies target cell proliferation, angiogenesis, migration, and intravasation of tumor cells in an attempt to limit tumor growth and metastasis. An in vitro platform to assess tumor progression and drug sensitivity could provide avenues to enhance our understanding of tumor metastasis as well as precision medicine. We present a microfluidic platform that mimics biological mass transport near the arterial end of a capillary in the tumor microenvironment. A central feature is a quiescent perfused 3D microvascular network created prior to loading tumor cells or patient-derived tumor organoids in an adjacent compartment. The physiological delivery of nutrients and/or drugs to the tumor then occurs through the vascular network. We demonstrate the culture, growth, and treatment of tumor cell lines and patient-derived breast cancer organoids. The platform provides the opportunity to simultaneously and dynamically observe hallmark features of tumor progression including cell proliferation, angiogenesis, cell migration, and tumor cell intravasation. Additionally, primary breast tumor organoids are viable in the device for several weeks and induce robust sprouting angiogenesis. Finally, we demonstrate the feasibility of our platform for drug discovery and personalized medicine by analyzing the response to chemo- and anti-angiogenic therapy. Precision medicine-based cancer treatments can only be realized if individual tumors can be rapidly assessed for therapeutic sensitivity in a clinically relevant timeframe (≲14 days). Our platform indicates that this goal can be achieved and provides compelling opportunities to advance precision medicine for cancer.
Introduction
Cancers with a poor clinical prognosis are strongly correlated with invasiveness of tumor cells, which is manifested by cell proliferation, angiogenesis, migration, and intravasation of tumor cells. These processes enable malignant cells to invade and metastasize. Most treatment strategies target one or more of these hallmarks of tumor progression in an attempt to limit tumor growth and invasion. All these processes require the tumor to communicate with neighbouring microvascular networks, which is dictated by interstitial fluid flow and diffusion of morphogens. Routine in vitro cell culture is unable to recapitulate this complex 3D behaviour of the tumor microenvironment. An in vitro platform which includes a 3D living dynamic vascular network to assess tumor growth, invasiveness, and drug sensitivity could provide avenues to enhance our understanding of tumor progression as well as vital information for effective treatment.
Organ-on-a-chip technologies have been evolving rapidly over the past decade. Our lab and others have created tissues with functional and dynamic microvascular networks.1–3 These tissues are created from human cells, and the microvessels remain functional when implanted in mice.4 The platforms are made up of optically clear polydimethylsiloxane (PDMS) that allow simple and reproducible observation at cellular and subcellular levels without perturbing the tissue microenvironment. The pressure and fluid flow conditions in these platforms are easily controllable. Thus, it becomes feasible to recreate features of the human vascularized tissue microenvironment, and closely observe the cellular behavior under controlled conditions.
A particularly interesting feature of the microcirculation at the capillary level is that net fluid secretion by convection into the interstitial space occurs near the arterial end of the capillary while net absorption of fluid occurs near the venular end. As a result, the former serves as the source of nutrients (e.g., oxygen, glucose) and anti-cancer drugs for a developing tumor. This arrangement impacts all features of tumor progression. For example, tumor-secreted morphogens, such as vascular endothelial growth factor (VEGF) to stimulate angiogenesis, would need to be transported against the direction of convective flow to the arterial end of the capillary. The latter is consistent with the observation that sprouting angiogenesis occurs from the arterial ends of the capillary.5 Similarly, tumor cells which intravasate into blood capillaries need to migrate against interstitial flow. Thus, mass and fluid transport are central features of tumor progression.
A quiescent microvascular network precedes nascent tumors, and thus the temporal order in which a vascular network and tumor are created within an in vitro system is important. For example, simultaneous seeding of tumor and endothelial cells in a single chamber could lead to rapid overgrowth of tumors but undergrowth of vessels thus limiting the possible observation of events such as intravasation. Moreover, the endothelial cells in such assays are exposed to tumor factors before achieving quiescence, which could impact angiogenesis. Previous platforms have investigated specific hallmarks of cancer including tumor growth,2 invasion,6 intravasation or extravasation,7–9 angiogenesis,10 and drug testing.2,11–15 However, the devices in these studies have given only limited considerations of the mass transfer and the temporal order of tissue development.
In this study, we designed a microfluidic platform incorporating the following important new features: 1) a stable quiescent perfused microvascular network which is established initially in the platform; 2) tumor cells and/or organoids sourced from cells lines or primary tumors which are loaded in close proximity to the preformed microvascular network; 3) delivery of nutrients/drugs via the microvascular bed to mimic the in vivo microenvironment; and 4) flow and dimensions of the microfluidic device designed to specifically mimic the in vivo communication between the nascent tumor and the arterial end of the capillary – the source of nutrients and anti-cancer drugs for a developing tumor. We validated the platform using cell lines and treatment conditions with well-established in vivo responses. We demonstrate that this platform facilitates the culture, growth, and treatment of tumor cell lines, as well as patient-derived tumor organoids (PDTO). Furthermore, the platform can be used to easily visualize angiogenesis, intravasation, proliferation, and migration at high spatiotemporal resolution, and quantifies microenvironmental constraints such as distance, flow, and concentration, that allow a tumor to communicate with the arterial end of the capillary.
Material and methods
Cell culture
Normal human lung fibroblasts (NHLFs) were obtained from Lonza (Allendale, NJ), and were cultured up to seven passages in complete fibroblast growth medium (FGM; Lonza). Endothelial colony forming cell-derived endothelial cells (ECFC-ECs) were extracted from cord blood as detailed previously.4,16 The ECFC-ECs were grown up to seven passages in fully supplemented endothelial growth medium (EGM-2; Lonza the breast). Cancer cell lines MDA-MB-231 and MCF-7 and colorectal cancer cell line, Caco-2 (all from ATCC; Manassas, VA) were cultured in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) with 10% FBS and 1× penicillin– streptomycin. Breast cancer associated fibroblasts (CAFs) and normal breast fibroblasts (NBFs) were previously isolated and immortalized,17 and were cultured in DMEM supplemented with 10% FBS (non-heat activated), 1% penicillin–streptomycin, 1% non-essential amino acids, 1% sodium pyruvate, 1% L-glutamine. The colorectal cell line 268 (CRC-268) was generated from an early passage CRC-268 tumor established through engraftment of patient-derived colorectal liver metastasis tissue in NOD-SCID mice developed at the Solid Tumor Tissue Bank and Registry at Washington University in St. Louis.
Cell transduction
To visualize the cells during the long-term culture in the device, the cells were stably transduced using lentiviral transduction particles. The ECFC-ECs were transduced to constitutively express GFP (green fluorescent protein) using pLJM1-EGFP (Addgene plasmid #19319) derived lentiviral transduction particles (Addgene; Cambridge, MA) and polybrene at 10 μg ml−1 (Millipore). The cancer cell lines were transduced with mCherry using pLV-mCherry (Addgene plasmid #36084) or Azurite using pLV-Azurite (Addgene plasmid #36086) derived lentiviral transduction particles and polybrene.
To prepare these lentiviral vectors, HEK293T cells were plated in a 6-well plate and cultured in DMEM with 10% FBS. 1.5 mL of OptiMEM (Gibco) was combined with 45 μL of Lipofectamine-2000 (Invitrogen #11558–019), and kept at rest for 5 min. 9 μg of plasmid DNA (either EGFP, mCherry, or Azurite) was mixed with 4.5 μg of pMDLg-pRRE (Addgene plasmid #12251), 1.8 μg of pRSV-Rev (Addgene plasmid #12253), 2.7 μg pMD2.G (Addgene plasmid #12259), and 1.5 mL of OptiMEM were then combined with the previously described Lipofectamine–OptiMEM mixture and kept at rest for 25 min at room temperature. HEK293T cells had their media replaced with 1 mL of basal DMEM, at which point 500 μL of the plasmid DNA–OptiMEM–Lipofectamine mixture was added. After 24 hours, each well was replaced with fresh medium. After 48 hours, the viral supernatant was collected, centrifuged to remove debris, and frozen at −80 °C for further use.
Patient-derived tumor organoids (PDTO) preparation and labelling
Two patients with newly diagnosed triple negative breast cancer were recruited to participate in this study under an IRB approved protocol at Washington University in St. Louis. Following informed consent, two 14 gauge core biopsies of tumor tissue were collected prior to chemotherapy. The primary patient tumor biopsies were washed with PBS supplemented with 10 mL of 5 μg mL−1 fungizone and 1% penicillin/streptomycin solution. The tissue was then sectioned into smaller pieces, resuspended in 10 mL of DMEM supplemented with 1% L-glutamine, 1% penicillin/streptomycin, collagenase I, and DN-ase I (final concentration 50 KU ml−1), and incubated in cell culture incubator overnight. The tubes were centrifuged at 200 × g for 4 min. They were then resuspended in 3 mL of media and run through a 70 μm filter, to obtain resultant PDTO that were <70 μm in diameter. Afterwards, the tumor cell suspensions were checked for viability by trypan blue staining. Some primary tissue samples were labelled with CellTracker™ CMDil membrane dye to observe the tumor growth following the manufacturer's protocol. Briefly, the tumor cell suspensions were suspended in 2 μM dye solution at about 0.2 million cells per ml. The suspension was maintained at 37 °C for 5 min, and then for an additional 15 minutes at 4 °C. The tumor cells were subsequently washed to remove excess dye. The PDTO of patient 1 were used for tumor progression experiments and the PDTO from patient 2 were used for analysing various cellular types by immunofluorescence staining.
Quantitative RT-PCR
To analyse the angiogenic potential of CAF and NBF, or to characterize epithelial and mesenchymal phenotypes of the tumor cells, qPCR was performed. The qPCR assay was chosen as it can distinguish subtle differences in expression levels and allows one to effectively normalize data to faithfully compare expression in different cell types. Cells were grown to 80% confluence and lysed using TRIzol reagent (ThermoFisher, Waltham, MA). TRIzol–RNA was mixed with chloroform and tubes were centrifuged. RNA in aqueous phase was removed and then purified using the RNeasy Mini Kit (Qiagen) and dissolved in 30 μL of nuclease-free water. RNA quantity and quality were assessed using a NanoDrop ONE or NanoDrop 1000 (ThermoFisher). RNA was then cleared of potential genomic DNA using the RapidOut DNA Removal Kit (ThermoFisher), per manufacturer's instructions. cDNA synthesis was performed using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems), according to the manufacturer guidelines.
The analysis of angiogenic factors produced by the CAF and NBF was performed using a customized TaqMan Array Plates, and the analysis of epithelial and mesenchymal gene expression in tumor cells was performed using SYBR green chemistry, according to manufacturer protocols. Briefly, custom TaqMan Array Plates, which had gene primers and probes pre-printed into specific wells of a 96-well plate were obtained from Applied Biosystems Inc (ABI; Waltham, MA). The list of genes and their corresponding Assay ID numbers are provided in the ESI† (Table S1). The cDNA was diluted in nuclease-free water and combined with TaqMan Universal PCR Master Mix (ABI) and 20 μL was loaded per well, and RT-qPCR was performed using a Mastercycler RealPlex2 (Eppendorf, Hamburg, Germany). Ct values were then analyzed by normalizing all values per gene to NHLF control. All primer sequences for the SYBR green assay were either selected from previously published literature, or, if unavailable in the published literature, designed using PrimerQuest Tool (IDT; Table S2†). Primers were synthesized from IDT (USA) at HPLC purity. Human 18s rRNA was used as an internal control. Thermal cycling was performed using a 7500 Fast Real-Time PCR System (ABI). The efficiencies were calculated using 7500 software V2.0.6 (ABI).
Microfabrication
The process of microfabrication is described in detail else-where.18 In short, a master mold of SU8 on a silicon wafer was prepared using photolithography in a dust free clean room. The microdevice was created by casting polydimethylsiloxane (PDMS), which was prepared by mixing Sylgard® 184 silicone elastomer base and curing agent (both Dow Corning, Midland, MI) in a 10:1 ratio, on the SU-8 master molds. The device was peeled off of the master mold after heat treatment at 60 °C overnight, and then bonded to a flat PDMS sheet using air plasma. The device bonding was cured briefly at 120 °C, and devices were sterilized using UV prior to use in experiments.
Device design
An essential feature of the device design was to have temporal control over loading and thus the development of the vascular and tumor tissues. Therefore, we created a device with distinct tissue chambers for loading these tissues. The device consists of three tissue chambers running parallel to each other (Fig. 2A) and are communicate with each other via microporous walls. The central tissue chamber was designed for development of a perfused microvascular network, and the two side tissue chambers were designed for loading of tumor or other cells and matrix at the same or different time points. The design of the pores that connect the chambers follows the capillary burst principle as described previously.19 The pore geometry (Fig. 2A) was created to retain the gel in the central chamber and create a bubble free interface between the gels in the central and side chambers. The converging portion of the pore away from the central chamber allows filling of the pores with gel while the central chamber is being loaded. The sudden divergence of pore near the side chambers, reduces the contact angle of the advancing gel front, stopping gel leakage into the side chamber. The total length of the pore was 55 μm and the width of the pore converged from 110 to 30 μm. The side chamber width was 200 μm, which resulted in <255 μm distance between a tumor in the side chambers and the closest capillary in the central chamber. The microvascular chamber communicates with a dedicated pair of microfluidic lines, inlet and outlet, that transport nutrients at defined pressures by convection. The hydrostatic pressure gradient between the two microfluidic lines drives interstitial fluid flow through the central tissue chamber during microvascular network development, and then through the lumens of the vessels once anastomosis of the microvascular network with the microfluidic lines is complete. The hydrostatic pressure gradients are maintained using differential levels of media in the reservoir micropipette tips (Fig. 2A). The dedicated microfluidic lines attached to the side tissue chamber serve as a sink for fluid leakage from the tissue compartment.
Fig. 2.
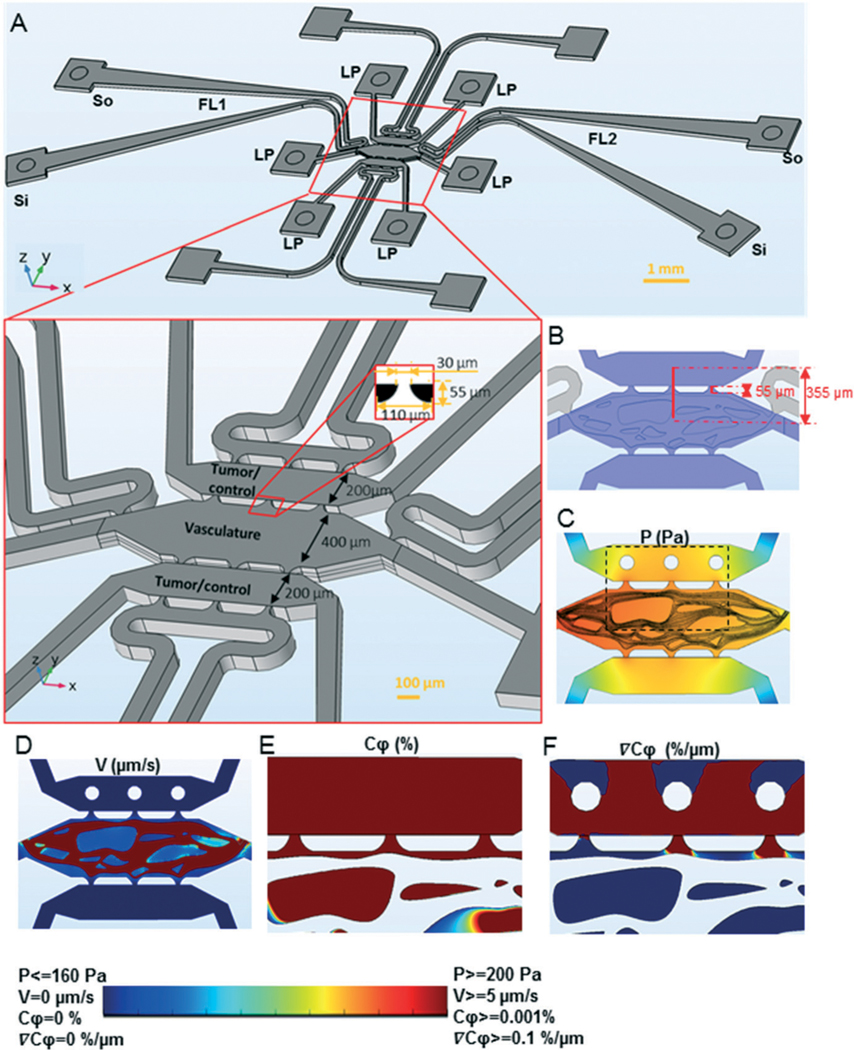
The convection–diffusion model for tumor-vascular communication in the device. A) The device design showing three parallel tissue chambers, each of which is connected to two square tissue loading ports (LP). The tissue chambers are separated by a microporous wall (bottom insert) with a pore design shown in the 2D insert (black is PDMS and white is empty chamber space). The central tissue chamber is for the microvasculature and the two side chambers are for loading tumor or control tissues. The microvascular chamber is attached to two dedicated fluidic lines (FL1 and FL2), which are connected to sources and sinks (so and Si) of their own. The hydrostatic pressure-drop between so and Si drives fluid flow through the fluidic lines, and the hydrostatic pressure drop between the fluidic lines drives flow through the vasculature chamber. Each of the side chambers are attached to a dedicated fluidic line (white), which serves as a sink for excess fluid drainage. B) The device design showing three parallel tissue chambers (blue). The distance between the centers of the top and the central tissue chambers and the length of pore is also indicated. C)–F) A 3D model of microvasculature (central tissue chamber) and tumors (top tissue chamber) was constructed with average pressure drop of 10 mm H2O across the central microvascular and side tissue chambers. The pressure (C) and velocity (D) profiles at the vertical center of the device are shown. The concentration (E) and concentration gradient (F) profiles at the vertical center of the device for an area of the tissue chambers, indicated by the rectangle in C, are shown. The rainbow color scale with the corresponding upper and lower limits of the variables plotted in C–F.
Device loading, tissue growth, and tissue treatments
The microvascular network was created using a 1 : 2 mixture of ECFC-ECs and NHLFs to produce a final concentration of 10 and 20 million cells per ml, respectively. The tissue was prepared in fibrin gel by mixing the cell suspension prepared in bovine fibrinogen (Sigma-Aldrich, St. Louis, MO) and bovine plasma thrombin (Sigma-Aldrich) to produce a final fibrinogen concentration of 10 mg ml−1 and thrombin concentration of 2 U ml−1. The fibrin microtissue was immediately introduced into the device via a tissue loading port of the central tissue chamber (Fig. 2A). The microtissue was maintained for seven days in EGM (Lonza) media maintaining a hydrostatic pressure head of about 10 mm H2O and reversing the direction of flow every day to facilitate the anastomosis of endothelial capillaries with the fluidic lines. This pressure drop was enough to convect the media through the central tissue chamber while maintaining the side tissue chambers dry.
The tumor cell lines or PDTO mixed with growth factor reduced Matrigel™ (Corning) reconstituted in EGM media to yield a final concentration of 2.5 mg ml−1 were loaded into the side tissue chambers of a microvascularized device via loading ports (Fig. 2A). The cell lines were seeded at a concentration of 10 million cells per ml. Each needle biopsy of primary breast cancer was digested as described above providing an approximate organoid cell density of 10 million cells per ml which was adequate to load up to eight tumor chambers. The devices were fed on alternate days by replacing the media in the source attached to the microfluidic lines. In some experiments, the devices were treated with drugs or cytokines by adding them into the highest-pressure source and delivering to the tissue via the microfluidic line. The MDA-MB-231 tumors were grown for two days and VEGF (100 ng ml−1), thrombin (5 U ml−1), or paclitaxel (0–100 μM) treatments were performed for two days. The PDTO were grown for 18 days and paclitaxel (1 μM) treatment was carried out for two days. The CRC-268 tumors were grown for one day and bevacizumab (10 μg ml−1) or TGFβ (50 ng ml−1) treatments were performed for seven days with replenishment of treatment media every alternate day.
Device labelling, imaging, and analysis
The in situ molecular characterization of tissues in the devices was performed by immunofluorescence staining of the tissues as follows. The labelling reagents were introduced via a microfluidic line attached to the central tissue chamber with an average hydrostatic pressure of 25 mm of H2O, while keeping the other side near zero mm H2O. Subsequently, the direction of the interstitial flow was reversed. The flow of reagents was maintained for a day in each direction.3,20 At the completion of an experiment, the microtissues were first fixed with 10% formaldehyde and then labelled with mAbs and nuclear stains. The microtissues were imaged under wide field fluorescence using an IX 83 motorized inverted microscope (Olympus, Tokyo, Japan) or FV1200 Fluoview biological confocal laser scanning microscope (Olympus) connected to computers with MetaMorph Advanced (version 7.8.2.0) or FV10ASW image acquisition and analysis software (both Olympus). The vessel and tumor areas were identified from their respective fluorescent areas in an image using ImageJ1.47V. The growth of tumors in a device was assessed by normalizing the tumor area with day 2 tumor area. The relative tumor growth was defined as the ratio of growth under a test condition to the growth under control condition. The relative vessel growth was calculated as the ratio of vessel area under a test to the area under control condition. The tumor migration was calculated by normalizing the tumor area in the central microvascular chamber with tumor area in the side tumor chamber.
Vessel permeability assay
The barrier function of the vessel was characterized by performing permeability measurements as detailed previously.21,22 Briefly, fluorescently tagged dextran with MW = 70 kDa prepared at 50 μg ml−1 concentration was perfused through the microvessel network. The time lapse images were acquired using an IX 83 motorized inverted microscope. The image analysis was performed using ImageJ 1.47V. The permeability of the vessels was calculated using following equation:21
where is permeability (cm s−1), is fluorescent intensity of filled vessel at , is the rate of change in intensity (s−1), and is the radius of the vessel.
Mathematical model of convection–diffusion in the microfluidic device
The computer aided design (CAD) file used for microfabrication of the device was used to create the boundaries of the device for a mathematical model capable of simulating mass and momentum transport using COMSOL Multiphysics® 5.2a software. The structures of the microvessels were manually drawn, matching approximately the 2D projection image of an actual microvascular network in the device. Three hypothetical tumors with 50 μm radius were created in one of the side tissue chambers. The 2D CAD geometry of the device was extruded to create a height in the third dimension of 100 μm. The microvessel and tumor height were set at 25 μm, which generated rectangular cross sections for the microvessels and the tumors. The vertical centers of the tumors and vessels were aligned with the vertical center of the device. The entire 3D geometry was then imported into COMSOL software as an object geometry. The microvasculature and non-microvascular part consisting of surrounding stroma were modelled as two separate domains. The laminar flow module was used to find pressure and flow through the lumens of the microvessels and the two fluidic lines connected with it. The no-slip boundary condition was applied for all surfaces except the microfluidic entrance and exit. The hydrostatic pressure heads in the source and sink drove the flow in the microfluidic lines and microvessels. The permeability of the microvessel walls were modeled using Darcy's law. The hydraulic conductivity of the microvessel wall was assumed to be equal to the previously reported value for human umbilical cord endothelial cell in vitro cultures (3.5 × 10−7 cm s−1 cm-H2O−1).23 The flow through porous media module was used for the nonmicrovessel (extracellular matrix) portion. The flow entered the domain normally from the microvascular wall, and the loading tips of tumor tissue served as outlets. To find the concentration profiles, the transport of diluted species module was coupled with the fluid flow modules with concentration boundary condition at the tumor surface. The pressure, velocity, and concentration fields were used to interpret the magnitude and pattern of convection and diffusion.
We also performed simulations on a hypothetical tissue in 2D. A microvessel at the bottom boundary of the tissue leaked fluid flow, which was absorbed by the microvessel at the top. The tumors were set at finite concentration (source of pro-angiogenic factors). The interstitial fluid velocity through the tissue was varied between 0.01–5 μm s−1.
The following values for physical constants were used in the simulations: diffusion coefficient of dextran, 7 × 10−11 m2 s−1; porosity and hydraulic permeability of fibrin gel, 0.99 and 1.5 × 10−13 m2, respectively;18 porosity and hydraulic permeability of Matrigel™, 0.99 and 1.5 × 10−16 m2, respectively;24 the dynamic viscosity at 25 °C and density of water, 0.89 cP and 1000 kg m−3, respectively; the diffusion coefficient of VEGF, 10−11 m2 s−1.
Analytical solution of convection-diffusion model
The convective transport directed towards tumor and diffusive transport in the opposite direction (Fig. 1A and B) was modelled and analytically solved to derive device design parameters. One dimensional steady state convection-diffusion equation for incompressible 1D flow is as follows,
| (1) |
Fig. 1.

A convection–diffusion model for tumor-capillary communication. A) The arterial end of capillaries leak fluid which feeds (oxygen, glucose) tumors. The fluid is then reabsorbed into the capillaries near the venular end or lymphatics. Pro-angiogenic factors secreted from the tumor must diffuse against the direction of flow to reach the nutrient-rich arterial end of the capillary. The direction of fluid flow is indicated by arrows, and the tumor-secreted factor gradient is indicated by the filled green triangle. B) A one-dimensional mathematical model was created for the region of interest (dotted box in A) – the arterial end of the capillary. The arrows indicate the direction of flow and the green color gradient indicates the concentration of tumor-derived factors. C) and D) The analytical solution of the model (eqn (2) and (3)) was plotted for various Pe values, including the biologically relevant range of Pe (0.1–10) and Pe beyond which critical values of and (the green-dotted line) cannot be maintained. E) 2D model of A) was constructed, in which the capillary near the arterial end is at the bottom of the image (red rectangle), and the venules and lymphatics at the top (blue rectangle). At low Pe (= 0.2), the tumor-secreted factors can diffuse against the direction of flow (top panel) and reach the arterial end of the capillary, but at high Pe (= 100) the concentration of tumor-secreted factors against the direction of flow is virtually negligible. The color scale indicates values of .
The negative sign of the advective term indicates flow in the negative direction, which is due to flow from the micro-vessel towards the tumor. The concentration of tumor factors () is assumed to be constant in the tumor mass (, e.g., VEGF). Here, is the diffusion coefficient, is the interstitial fluid velocity, represents the distance that the microvessel could be placed from the tumor to achieve a zero concentration for . was assumed to be zero, due to the comparatively rapid luminal flow. Eqn (1) can be analytically solved using the above boundary conditions to yield following equations:
| (2) |
| (3) |
where is the non-dimensional concentration and is the non-dimensional concentration gradient (fractional change per μm distance); is the Peclet number; is the dimensionless distance of the vessel wall from tumor. At the vessel wall, was calculated by assuming capillary internal radius of 3.5 μm and wall thickness of 1.5 μm, as .
Statistics
Statistical analysis was performed using one-way ANOVA and Tukey test for multiple comparisons or Studen’s t-test. All data are presented as the mean ± standard deviation. Results were considered statistically significant for p < 0.05.
Results
Design constraints based on biologically relevant mass transfer requirements
The capillaries at the arterial end of the vessel are at a sufficiently high hydrostatic pressure (∼30 mmHg) to cause leakage of fluid and nutrients into the surrounding tissue. Thus, any tumor secreted factors, such as VEGF, would have to diffuse against this convective flow to reach the capillaries and activate angiogenesis (Fig. 1A). The goal of our microfluidic device design was to simulate these in vivo biological mass transfer features of the tumor microenvironment (TME). We utilized the fractional concentration () and spatial concentration gradient () of a tumor-secreted factor and the ratio of volume of tissue to the surface area of a capillary nourishing the tissue (V/S) as the primary characteristic features of the problem, and then determined the constraints on three scaling parameters to design the microfluidic device: 1) Pecleťs number (Pe); 2) dimensionless distance between tumor and capillary (X); and 3) the mean residence time of molecules in fluid flow that nourishes the tissue.
The interstitial fluid velocity in small tumors (<4 mm) has been reported to be <0.5 μm s−1.25,26 Considering the maximum distance of a capillary from any cell in vivo to be 200 μm,27 this interstitial fluid velocity corresponds to Pe = 10 for a morphogen such as VEGF. Our analytical model (eqn (2)) suggests that at Pe = 10 and X = 0.975 (i.e., d = 200 μm), the corresponding value for is 0.001%. That is, if a tumor is to induce angiogenesis from the arterial end of a capillary, then becomes the minimum concentration of a tumorsecreted factor such as VEGF. Eqn (2) demonstrates that depends on two dimensionless parameters Pe and X, and to maintain above the critical value, the value of one dimensionless parameter dictates the upper bound on the second dimensionless parameter. For example, if , then the maximum allowable Pe is 8.3 to maintain . However, beyond Pe > 25, cannot be maintained above the critical value regardless of the value of X (Fig. 1C). We confirmed this effect in 2D using COMSOL simulations. As shown in Fig. 1E, the tumor factors observed towards the arterial end of capillary at a low Pe (0.2) are virtually eliminated at high Pe (= 100). A biologically relevant lower limit of X is 0.5, which can be found for a typical cell of radius 5 μm placed 5 μm away from the capillary center, or x = 5 μm and d = 10 μm. The biological lower limit of Pe is 0.1 for molecules like VEGF, which can be calculated from the reported lower level interstitial flow velocity (0.1 μm s−1 (ref. 26)) and lower limit of d (= 10 μm).
We have previously shown that an average concentration gradient of 0.1% per μm for VEGF is sufficient to induce and bias angiogenesis towards the higher concentration of VEGF.3 We used this as the minimum critical limit of required to bias angiogenesis toward the tumors. The analytical model (eqn (3)) shows that decreases with increasing Pe and increasing X. Further, for Pe > 13 or X > 0.995, cannot be maintained above the critical value (Fig. 1D). X = 0.995 corresponds to d = 1000 μm; in other words, this result suggests that even for almost zero flow (Pe ≪ 0.1), a tumor needs to be <1000 μm from a capillary to direct angiogenesis.
The V/S ratio for a typical capillary (outer diameter 10 μm) in vivo feeding a cylindrical tissue of 200 μm radius is approximately 4000 μm. The residence time, , is an average time a fluid molecule (i.e., water) spends in a tissue before exiting the tissue, and it depends on the volumetric flow rate () and volume of the tissue (V; μm3) with the simple relationship . The range of τ can be found using the interstitial fluid velocities in small tumor tissues ().25,26 for this scenario is in the range of 2.2 to 11 h. Together, these data suggest the following design constraints on the scaling parameters for the microfluidic device: , and to maintain a biologically relevant concentration, concentration gradient, and feeding rate of tumors from the capillary.
Device design for biologically relevant mass transfer between tumor and microvascular tissues
We designed the microfluidic device with three tissue chambers comprised of a central microvascular chamber and two side implantation chambers (Fig. 2A). The width of the tissue chambers was chosen such that the distance between the center of the implantation chambers and the microvascular chamber was 355 μm (X = 0.986) maintaining the constraint on X. To estimate the interstitial flow and concentrations, we constructed a 3D model of the device with a microvascular network (Fig. 2B–F). The microvascular chamber was set at a positive pressure relative to the implantation chambers (Fig. 2C) consistent with the simulation of the arterial end of a capillary (Fig. 1). We chose an average pressure drop of 10 mm H2O between the microvascular chamber and the side tissue chambers because the volume average interstitial fluid velocity in the microvascular chamber was 0.1 μm s−1, which yields Pe = 3.5 (for VEGF and d = 355 μm), well within the critical limits (0.1 < Pe < 13). The interstitial velocity in the pore region is high (= 0.6 μm s−1), offering the maximum resistance to mass transfer (Fig. 2C). Yet due to the short pore length (55 μm; Fig. 2B) designed for the inter-tissue communication, Pe < 3.5 is maintained within the design limits. The simulations demonstrate that the and at these experimental conditions (Fig. 2E and F).
The volumetric flow in the device was calculated at the pores between the microvascular chamber and tumor chamber, and was 10800 μm3 s−1. The dimensions of the device were such that the tissue volume in the device (approximately 108 μm3 or 100 nl) produces a residence time of 2.6 h, which is within the design constraints. Thus, the device geometry and experimental conditions chosen for device design mimic a biologically relevant simulation of the tumor microenvironment between the arterial end of a capillary and a nascent tumor.
Quiescent, perfused, and isolated microvasculature development in the device
An essential feature of the device design was to develop a perfused microvascular network while keeping the adjacent tissue chambers dry for loading of tumor tissue later. To demonstrate this aspect, fluorescently labelled dextran was delivered to a device, in which the central tissue chamber was loaded with fibrin gel. The media flowed across the device but did not leak into the side tissue chambers (Fig. S1†). When fluorescently tagged endothelial cells and fibroblasts were loaded in the device, a microvascular network formed within 7 days (Fig. 3A). Confocal microscopy showed microvessels with clear lumens (Video S1† and Fig. 3C) ellipsoidal in shape (Fig. 3C), which were supported by the stromal cells (Fig. 3C). The microvessels anastomosed to the microfluidic lines (Fig. S2†) using a previously published strategy.2 To confirm perfusion and interconnectivity of the microvessels, 70 kDa TRITC–dextran was introduced into the microfluidic channel, which was then convected into the microfluidic network (Fig. 3D). Vessel perfusion was also confirmed by flowing microbeads through the lumen (Video S2†). The permeability of the vasculature in the device was 8 ± 1 × 10−7 cm s−1, which closely matches the reported permeability of in vivo microvessels (1.5 ± 0.5 × 10−7 cm s−1).28
Fig. 3.
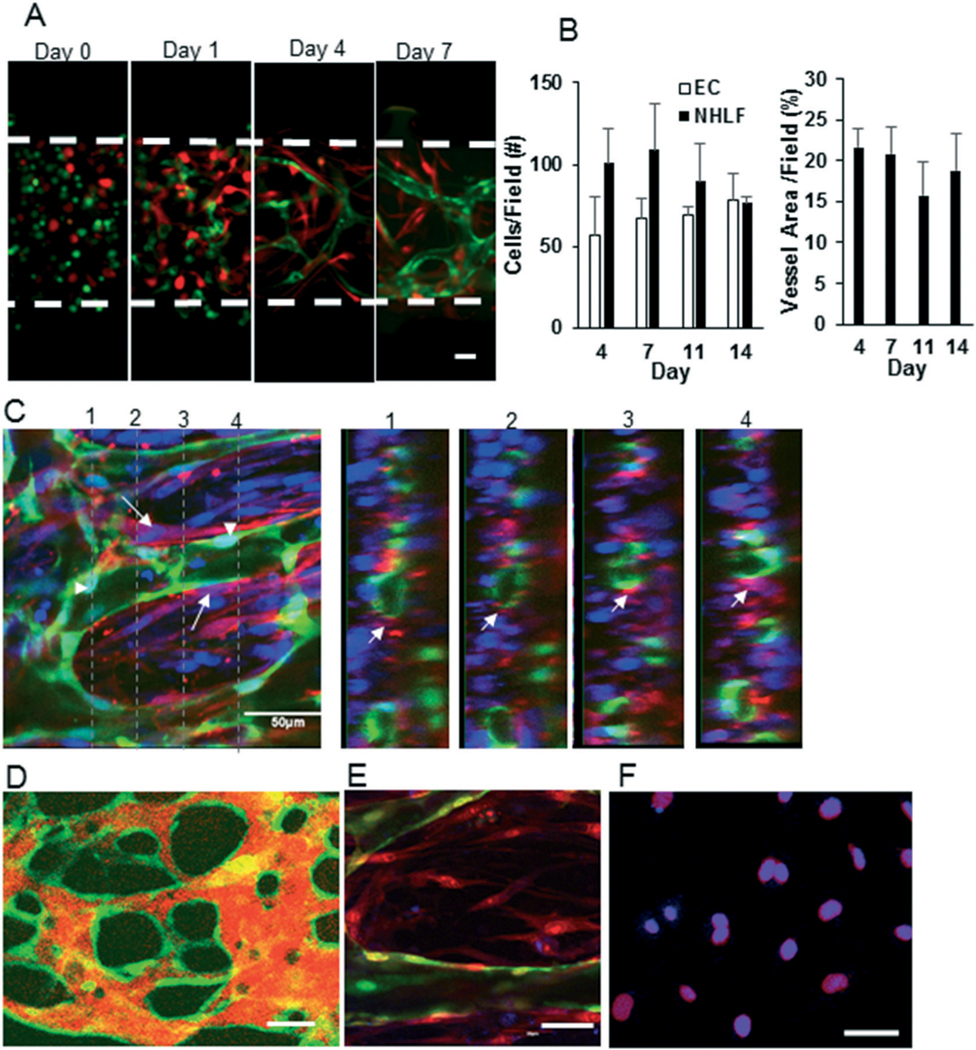
Development of perfused quiescent microvascular network in the device. A) The endothelial cells (green) and fibroblasts (red) were cultured for seven days in the microvascular chamber. The cells begin to self-assemble into a vascular network by day 1 and a full-formed network is present by day 7. B) The number of cells and the area of the vessel network are constant between days 4–14. C) A microvascular network developed using GFP transduced endothelial cells was stained with α-SMA (red), which stains fibroblasts, and DAPI (blue). A confocal Z-stack of the central chamber of the device was obtained. The dotted lines on the left panel indicate the plane at which the Z-stack is shown in the right panels. The arrow shows stromal cell coverage of the capillary. D) Fluorescently labelled dextran (orange) was perfused through microvasculature and a confocal slice of microvasculature (green) was obtained for vessels in the central chamber of the device. E) The microvasculature (green) was developed for seven days and then stained for Ki 67 (blue) and nuclei (propidium iodide; red). The Ki 67 was absent on endothelial cells (green) in the device. F) The endothelial cells were grown in 2D and stained with Ki67 (blue) and nuclear marker DRAQ5 (red) as a positive control for proliferation. The scale bar in A, C, D–F indicates 50 μm.
To find a time window within which the vessels are relatively quiescent, the microvessels in the device were tracked over 14 days. The total number of endothelial cells or fibroblasts and total vessel area did not significantly change from day 4–14 (Fig. 3B). Additionally, endothelial cells at day 7 in the device demonstrated minimal expression of Ki67, a marker of cell proliferation, indicating the cells are in a quiescent state (Fig. 3E). Endothelial cells cultured in 2D (Fig. 3F) served as a positive control. These data confirm that the microvasculature in the device achieves relative quiescence within seven days of culture in the device. Therefore, we chose day 6–8 as the time window for implantation of tumor cells and matrix into the adjacent tissue chambers.
Culture, growth, and chemotherapy treatment of tumors derived from cell lines and PDTO
We first wanted to demonstrate that the microfluidic device would support the growth of tumors. We initially tested breast and colorectal cancer cell lines. Assessment of cell growth in the device 6 days after implantation demonstrated significant growth of CRC-268 and MDA-MB-231 cells (>400%), and slower growth of Caco-2 and MCF-7 cells (<300% and <200%, respectively) (Fig. 4A–D). The more aggressive MDA-MB-231 and CRC-268 tumor cells lines invaded the surrounding stroma and spread, while the less aggressive tumor cell lines formed rounded tumor spheroids with less invasion of surrounding stroma (Fig. 4A and C). To demonstrate the potential for our device to support patient-derived tumors, PDTO labelled with a cell membrane dye were implanted into the device. Over a period of 20 days, the tumor mass of fluorescently labelled cells increased by 400% (Fig. 4E and F). The PDTO were composed of immune cells, endothelial cells, fibroblasts, and epithelial cancer cells as they stained positively for their respective molecular markers, CD45, CD31, αSMA, and EpCAM (Fig. S3†), respectively. The EpCAM positive cancer cells stained positively for Ki67 (Fig. S3†), demonstrating that the cells were proliferating in the device. Additionally, we recorded time lapse videos of the PDTO in the device. The tumors remained viable with notable dynamic movement (Video S3 and Fig. S4†).
Fig. 4.

Various types of tumors grow in the pre-vascularized device. A, C and E) Fluorescently tagged colorectal, breast cancer cell lines, or PDTO were implanted in the device and the growth of the tumors was tracked by using fluorescent area. B, D and F) The growth of tumors with respect to the tumor area on day 2 is presented. The red line indicates base line (100% growth on day 2). The scale bar in A, C and E indicates 50 μm.
To demonstrate the potential of the device to assess drug efficacy, we implanted the breast cancer cell line MDA-MB231 cells or PDTO in the device, and infused paclitaxel, a chemotherapy drug used routinely to treat breast cancer, through the vascular network. We adjusted dosing based absorption of this drug in PDMS using our previously published model,29 which indicated that paclitaxel should be minimally absorbed by PDMS (<0.1%) under our experimental conditions. Tumor size and vascular density both decreased following exposure to paclitaxel (Fig. 5A–C). The IC50 of the MDAMB-231 cells was significantly higher for those cells cultured in the device (7.3 μM) compared to the IC50 (1.0 μM) for the cells cultured in 2D (in culture wells). The relative vessel growth (%) remained unaffected up to the 1 μM drug concentration, beyond which the vessels significantly degraded (Fig. 5A and C); thus, we chose 1 μM to treat the biopsy tumors. The relative vessel growth in the PTDO devices remained unaffected (Fig. 5E), as expected. In contrast, the patient tumor growth was reduced by <50% (Fig. 5D and S5†), demonstrating that PTDO response to chemotherapy can be assessed in the device.
Fig. 5.

The dose response for chemotherapy drug paclitaxel in the device. A) MDA-MB-231 tumor cells (red) were implanted in devices, which had a fully developed microvascular network (green) grown for 7 days. The tumor cells were grown for two days, and then the devices were treated with paclitaxel at indicated concentrations (μM). The images of paclitaxel treated devices were obtained at predetermined time points as indicated. B) The paclitaxel dose–response curve of MDA-MB-231 tumors in devices (3D) and in 2D cell culture was constructed using relative growth (%) of tumor with respect to untreated control tumors. C) The paclitaxel dose–response curve for vessels in MDA-MB-231 devices. D) and E) PDTO devices were grown for 20 days and then treated with 1 μM paclitaxel for two days. The relative growth (%) was found for (D) tumor or (E) vessel network. The scale bar is 100 μm.
Angiogenic potential of cell lines and PDTO in the device
We next wanted to examine the potential of the device to assess tumor angiogenesis. To analyze angiogenesis in the device, we examined the response to a cancer cell line; a CAF, and a PDTO. This choice was based on reports that apart from the cancer cells, CAFs also play important role in tumor progression.30,31 We initially seeded the tissue chambers adjacent to the vascular network with either CRC-268 or breast CAF. After 6 days, a profound angiogenic response was observed for breast CAF and CRC-268 tumors relative to the ECM-only control (Fig. 6A and D). More specifically, the breast CAF and CRC-268 tumors had more than 600% relative vessel growth compared to the ECM-only control (100%). We next tested the angiogenic potential of the PDTO. The PDTO were highly angiogenic in the device (Fig. 6A and D) with more than 300% growth of the angiogenic vessels into the tumor chambers compared to the ECM-only control (100%) after 6 days. To demonstrate long term culture of PDTO, the devices were maintained for 22 days and the vascular network continued to expand and was stable (Fig. 6B).
Fig. 6.

The angiogenic response in the microfluidic device. A) and B) A fibrin-gel-control (no-cells), PDTO, CRC-268 tumor cells, breast CAF, and (C) CAF and NBF cells in fibrin were implanted in the side tissue chambers of the device, which had a fully grown vascular network (green; seven days old) in the central tissue chamber. The devices were maintained for additional (A and C) six or (B) twenty-two days to observe angiogenesis. The microporous wall between the tissue chambers is indicated by white-dotted lines. The scale bar indicates 100 μm. D) The relative vessel growth in the side tissue chambers was calculated at day 6. The red line indicates baseline (100% growth of vessels in gel controls). E) The CRC-268 implanted devices were treated with bevacizumab or control. The relative growth in the side tissue chambers was calculated. F) The CAF and NBF were analyzed by qPCR for angiogenic factors. The data was normalized with respect to the expression in NHLFs.
To further demonstrate the biological utility of our device, we examined the angiogenic response of breast CAF vs. normal breast fibroblasts (NBF) in a single device, with each implanted in two different side tissue chambers. For this setting, the angiogenesis was mainly directed towards the CAF chamber (Fig. 6C). We next examined the expression of 9 different angiogenic factors by qPCR, and found that the proangiogenic factors VEGFA and TGFβ1 were significantly elevated in the CAF relative to the NBF (Fig. 6F).
Finally, we investigated the potential for the device to assess the impact of an anti-angiogenic drug bevacizumab on angiogenesis by CRC-268. Bevacizumab is minimally absorbed by PDMS (<0.1%) under our experimental conditions.29 Bevacizumab treatment resulted in a significant decrease in angiogenic growth by >40% relative to untreated cells (Fig. 6E and S6†).
Tumor invasion and intravasation in the device
We next assessed the potential of the platform to quantify invasion of tumors, which was assessed by measuring the area of tumor spread into the microvascular chamber. We augmented cancer migration by delivering TGFβ (50 ng ml−1) through the vascular network. TGFβ is a well-known inducer of epithelial to mesenchymal transition (EMT).32,33 TGFβ treatment of CRC-268 within the device results in the increased expression of several mesenchymal markers (Fig. 7A). In addition, TGFβ caused increased invasion and migration of the CRC-268 cells into the microvascular chamber of the device by more than 2-fold compared to untreated cells (Fig. 7B). Specifically, about 15% of the untreated cells migrated into the microvascular chamber and about 30% of the TGFβ-treated-tumor migrated into the microvascular chamber (Fig. 7C).
Fig. 7.

The mesenchymal type tumors invade the stroma in the vascular chamber more efficiently. A) CRC-268 cells treated with 50 ng ml−1 TGFβ and the untreated cells were analyzed by qPCR and the data is normalized with respect to the expression in Caco-2 cells. B) Representative devices showing vasculature (green) and CRC-268 cells (red; top panels) or only the cancer cells (bottom panels). The microporous wall between the tissue chambers is indicated by the dotted lines. The scale bar indicates 50 μm. C) The migration (%) of tumor cells into the central tissue chamber.
Finally, we wanted to demonstrate the utility of the platform to observe intravasation of the cancer cells at a single cell level. The devices were fixed with formalin at the end of the experiment and then imaged using high resolution confocal imaging. The entire volume of each of the device was imaged by taking multiple Z-stacks. Tumor intravasation was observed (<5 cells per device Fig. 8A–D), but was a rare event relative to the high number of tumor cells that invaded (hundreds of cells per device) the microvascular chamber. Enhancing the permeability of the vessels by treating with VEGF or thrombin did not significantly increase the rate of intravasation (Fig. 8D).
Fig. 8.
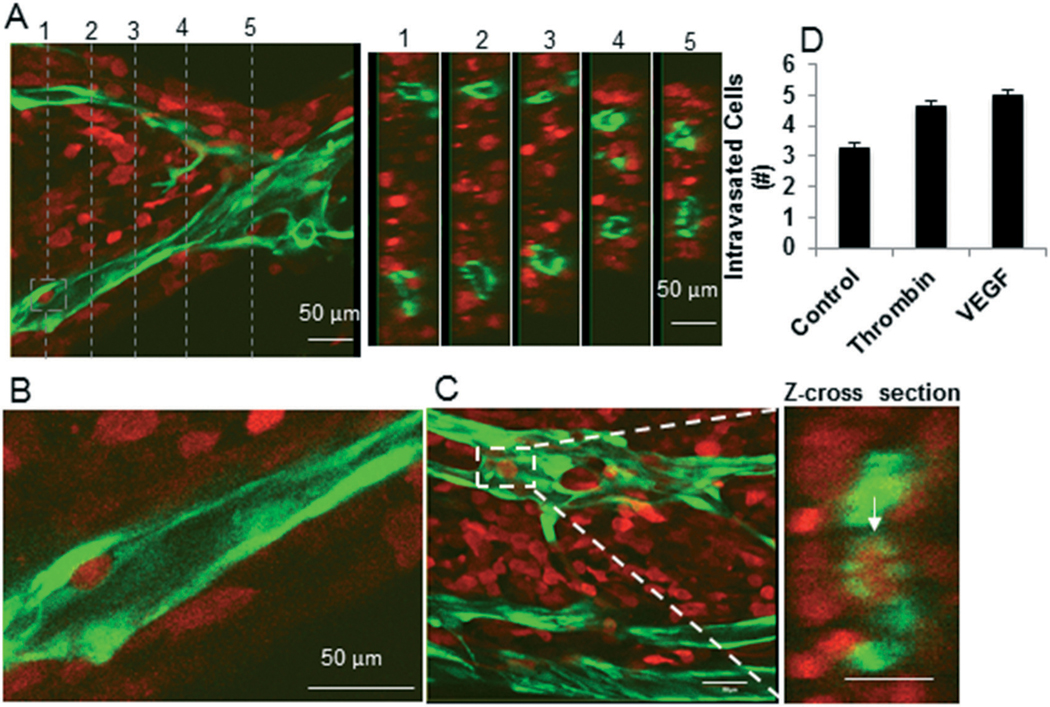
Intravasation of breast cancer cells in microfluidic platform. MDA-MB-231 cells (red) were implanted in a pre-vascularized (green) device and the tumors were grown for four days. A) A section of device with vessels top view and cross-sectional views (right) at five different locations (indicated by lines in the left panel) show no intravasated tumor cells. B) A tumor cell in the process of intravasating into the vessel. C) An intravasated tumor cell (white-rectangle) is shown from top view (left panel). The cross-sectional view at the center of the rectangle in the left panel shows an intravasated tumor cell (right panel). Scale bars in A–C indicate 50 μm. D) The MDA-MB-231 tumors grown for two days devices were treated as indicated for two days, and the number of intravasated tumor cells were counted by reconstructing 3D images of the microvasculature.
Discussion
Precision medicine-based cancer treatments can only be realized if individual patient tumors can be rapidly assessed for sensitivity to specific therapies in a high-throughput fashion. The organ-on-a-chip approach is ideally suited for this task due to the following advantages: 1) human specific stroma; 2) small size of the system (<500 nl); 3) small amount of tissue required for analysis; 4) prospect of high resolution optical measurements; 5) short time (1–2 weeks) of development; and 6) inexpensive fabrication. Recently, tumor microsystems have been developed to investigate specific hallmarks of cancer including tumor growth,2 invasion,6 intravasation or extravasation,7–9 angiogenesis,10 and drug testing.2,11–15 These systems underscore the potential impact of tumor-on-a-chip technologies, but have not yet achieved the level of sophistication of an in vivo tumor microenvironment (TME), where all of the hallmarks of these processes can occur simultaneously or in succession. In this study, we designed a biologically relevant mimic of the TME that includes tumor growth, sprouting angiogenesis, intravasation, and physiologic drug testing capabilities for both cell line and primary tumor biopsy-derived cancers.
Our results demonstrate the ability to culture a wide range of tumor cell lines, including both aggressive (MDA-MB-231, CRC-268) and less aggressive (MCF-7 and Caco-2) lines. More importantly, we were also able to demonstrate growth, angiogenesis, and response to drug for breast patient-derived tumor organoids (PDTO). The processing of the organoids preserves the in vivo architecture of the extracellular matrix, as well as the heterogeneous populations of cells that includes stromal cells and leukocytes. The number of organoids obtained by this method may be less than that obtained by technologies that completely dissociate the tissue and then reform tumor spheroids (e.g. hanging droplet technology); however, the latter method severely compromises the ECM and microenvironment architecture. The primary tissue in our devices was viable for up to three weeks, providing ample opportunity to investigate important responses such as drug sensitivity. This result provides an exciting opportunity for personalized or patient-specific treatment of cancer. Patient-derived tumors implanted in xenograft (PDX) mouse models have advanced our understanding of inter-subject tumor heterogeneity, not only in terms of tumor invasiveness, but also in terms of sensitivity to treatments.34–37 Nonetheless, the use of PDX models in personalized cancer medicine is extremely limited because not all tumors will develop a viable PDX (<10% in some cases), and those that are successful take a relatively long (3–6 months) time to establish.34–36 Interestingly, fibroblasts of human origin facilitate viability of PDX, underscoring the importance of human stromal cells for growth of the tumors.31
Our device design is based on biologic mass transport of morphogens consistent with the in vivo TME. In an in vivo tissue, the capillaries near the arterial end leak fluid, due to a positive Starling's force, and most fluid is reabsorbed by the portion of the capillaries near the venular end, which has a negative Starling's force compared to the interstitial tissue. This delicate distribution of Starling's forces along the length of in vivo capillary is achieved due to a relatively constant osmotic pressure across the capillary wall and a progressively decreasing hydrostatic pressure within the capillary. We chose to model the arterial end of the capillary as this region provides nutrients to the tissues via the microvessels, which impacts tumor growth and drug delivery. In this region, tumor-derived growth factors have to diffuse against the direction of interstitial flow to reach the near-arterial-capillaries to activate angiogenesis. Our theoretical results demonstrate that in order to transport biologically relevant concentrations of a morphogen, the tumor has to be located sufficiently close to the vasculature (d < 1000 μm); the interstitial flow has to be sufficiently small (Pe < 13); and the volume of the tissue small enough to maintain τ < 11 h. We designed our device consistent with these constraints, and demonstrated experimentally that, under these conditions, tumor-derived proangiogenic factors diffuse against interstitial flow to achieve a concentration (gradient) high enough to activate and direct sprouting angiogenesis from the arterial end of a capillary.
Microvascular networks of vessels have been generated by coating endothelial cells on artificially created microchannels. In these assays, endothelial cells attach to the surface of the preformed tube, undermining the normal process of vessel formation resulting in large diameter (>100 μm) endothelial cell-lined tubes.15,38–41 Our approach creates microvessel networks following the developmental process of vasculogenesis,1,3,42 where endothelial cells form vascular lumens in the presence of stromal cells to develop a network of microvessels. These microvessels anastomose with in vivo microvasculature and become perfused with blood when implanted in mice.4 This system provides a more physiologic alternative as the entire microvessel network can respond dynamically to changes in the microenvironment (e.g., presence of drug or tumor-derived morphogen) including both regression (pruning) and growth (angiogenesis) (Fig. 6 and S6†). The permeability of vessels in our device (8 ± 1 × 10−7 cm s−1) falls within the range of permeabilities reported for in vitro microvessels created in microfluidic platforms (4 × 10−7–40 × 10−7 cm s−1) using different types of stromal cells,2,15,43 suggesting that the type of mural cell impacts the permeability.
In vivo microvessels are generally quiescent and are activated to sprout by morphogens released by tumors or wounded tissue. Once activated, the vessels sprout following directional cues such as concentration gradients of morphogens. Our device design allows for the initial creation of a perfused, quiescent microvascular network supported by stromal cells in the central tissue chamber. The microvessels are relatively quiescent (no visible growth and minimal cell division) after 7 days, and thus are more representative of mature in vivo like microvessels. Once the quiescent vascular network is formed, cancer cells or tumor organoids can be implanted adjacent to the microvessel network. This design feature is an important advancement over previous designs,2 in which the seeding of tumor and endothelial cells occurs simultaneously. In this approach, the presence of the tumor during vasculogenesis can potentially impact the phenotype of the microvessel. Our device design, with implantation of cancer cells after mature vessel formation, more accurately represents true angiogenesis and intravasation of tumor cells from a quiescent microvessel network.
We demonstrate sprouting angiogenesis towards tumor cell lines, PDTO, and CAF. This result demonstrates the flexibility of our design to load a range of cell types and organoids in either of the adjacent tissue chambers. In addition, we also demonstrate the feasibility of assessing antiangiogenic therapies in the device (Fig. S6†). Thus, our platform provides the compelling possibility of examining simultaneously the impact of chemotherapeutics (e.g., paclitaxel) and anti-angiogenics (e.g., bevacizumab), which are used together in modern clinical approaches finally, tumor progression (e.g., proliferation, migration, and intravasation) can impact clinical decision-making. Our platform not only assesses the angiogenic potential of a tumor, but also the rate of migration, growth, and intravasation. Thus, the device design has the potential to provide a clinical index of tumor progression, as well as to observe and understand how these processes interact in a cooperative fashion. While this initial study establishes the proof of principle, a more detailed analysis of tumor progression and drug testing, including a larger selection of drugs, and comparison to PDX models and patient clinical indices, of PDTO in the device is needed to further this technology for clinical applications.
In conclusion, we present a tumor-on-a-chip device design that mimics biological mass transport near the arterial end of a capillary in the TME, with the important feature that a quiescent perfused 3D microvascular network is created prior to loading tumor cells or PDTO in adjacent tissue chambers. The result is a platform that can be used to simultaneously observe hallmark features of tumor progression including cell proliferation, cell migration, angiogenesis, and tumor cell intravasation. The platform can also be used to mimic the physiological delivery of drugs to the tumor through the vascular network to assess efficacy. This, combined with our observation that primary tumor organoids are viable for several weeks, provides compelling opportunities to advance precision medicine treatments for cancer.
Supplementary Material
Acknowledgements
This work was supported by grants from the National Institutes of Health (UH3 TR00048, R01 CA170879, R21 CA223836). We would like to thank Mr. Arnold Tao and Ms. Sandra Lam (Washington University in St. Louis) for technical assistance.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c8lc00596f
Conflicts of interest
SCG has equity in Kino Biosciences, a startup company whose core technology involves perfused human micro-vessels. There are no conflicts to declare for all other authors.
References
- 1.Moya ML, Hsu YH, Lee AP, Hughes CC and George SC, Tissue Eng, Part C, 2013, 19, 730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sobrino A, Phan DT, Datta R, Wang X, Hachey SJ, Romero-Lopez M, Gratton E, Lee AP, George SC and Hughes CC, Sci. Rep, 2016, 6, 31589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shirure VS, Lezia A, Tao A, Alonzo LF and George SC, Angiogenesis, 2017, 20, 493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen X, Aledia AS, Popson SA, Him L, Hughes CC and George SC, Tissue Eng, Part A, 2010, 16, 585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang HU, Chen ZF and Anderson DJ, Cell, 1998, 93, 741–753. [DOI] [PubMed] [Google Scholar]
- 6.Truong D, Puleo J, Llave A, Mouneimne G, Kamm RD and Nikkhah M, Sci. Rep, 2016, 6, 34094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang R, Zheng W, Liu W, Zhang W, Long Y and Jiang X, Sci. Rep, 2015, 5, 17768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeon JS, Bersini S, Gilardi M, Dubini G, Charest JL, Moretti M and Kamm RD, Proc. Natl. Acad. Sci. U. S. A, 2015, 112, 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zervantonakis IK, Hughes-Alford SK, Charest JL, Condeelis JS, Gertler FB and Kamm RD, Proc. Natl. Acad. Sci. U. S. A, 2012, 109, 13515–13520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee H, Park W, Ryu H and Jeon NL, Biomicrofluidics, 2014, 8, 054102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fan Y, Nguyen DT, Akay Y, Xu F and Akay M, Sci. Rep, 2016, 6, 25062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuo CT, Chiang CL, Chang CH, Liu HK, Huang GS, Huang RY, Lee H, Huang CS and Wo AM, Biomaterials, 2014, 35, 1562–1571. [DOI] [PubMed] [Google Scholar]
- 13.Mi S, Du Z, Xu Y, Wu Z, Qian X, Zhang M and Sun W, Sci. Rep, 2016, 6, 35544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pradhan S, Smith AM, Garson CJ, Hassani I, Seeto WJ, Pant K, Arnold RD, Prabhakarpandian B and Lipke EA, Sci. Rep, 2018, 8, 3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang Y, Soroush F, Sheffield JB, Wang B, Prabhakarpandian B and Kiani MF, Sci. Rep, 2017, 7, 9359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Melero-Martin JM, Khan ZA, Picard A, Wu X, Paruchuri S and Bischoff J, Blood, 2007, 109, 4761–4768. [DOI] [PubMed] [Google Scholar]
- 17.Alspach E, Flanagan KC, Luo X, Ruhland MK, Huang H, Pazolli E, Donlin MJ, Marsh T, Piwnica-Worms D, Monahan J, Novack DV, McAllister SS and Stewart SA, Cancer Discovery, 2014, 4, 716–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu YH, Moya ML, Abiri P, Hughes CC, George SC and Lee AP, Lab Chip, 2013, 13, 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cho H, Kim HY, Kang JY and Kim TS, J. Colloid Interface Sci, 2007, 306, 379–385. [DOI] [PubMed] [Google Scholar]
- 20.Alonzo LF, Moya ML, Shirure VS and George SC, Lab Chip, 2015, 15, 3521–3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huxley VH, Curry FE and Adamson RH, Am. J. Physiol, 1987, 252, H188–H197. [DOI] [PubMed] [Google Scholar]
- 22.Yuan SY and Rigor RR, in Regulation of Endothelial Barrier Function, Morgan & Claypool Life Sciences, 2010, ch. Methods for Measuring Permeability. [PubMed] [Google Scholar]
- 23.Chang YS, Munn LL, Hillsley MV, Dull RO, Yuan J, Lakshminarayanan S, Gardner TW, Jain RK and Tarbell JM, Microvasc. Res, 2000, 59, 265–277. [DOI] [PubMed] [Google Scholar]
- 24.McCarty WJ and Johnson M, Biorheology, 2007, 44, 303–317. [PMC free article] [PubMed] [Google Scholar]
- 25.Dafni H, Israely T, Bhujwalla ZM, Benjamin LE and Neeman M, Cancer Res, 2002, 62, 6731–6739. [PubMed] [Google Scholar]
- 26.Rutkowski JM and Swartz MA, Trends Cell Biol, 2007, 17, 44–50. [DOI] [PubMed] [Google Scholar]
- 27.Jain RK, Au P, Tam J, Duda DG and Fukumura D, Nat. Biotechnol, 2005, 23, 821–823. [DOI] [PubMed] [Google Scholar]
- 28.Yuan W, Lv Y, Zeng M and Fu BM, Microvasc. Res, 2009, 77, 166–173. [DOI] [PubMed] [Google Scholar]
- 29.Shirure VS and George SC, Lab Chip, 2017, 17, 681–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalluri R, Nat. Rev. Cancer, 2016, 16, 582–598. [DOI] [PubMed] [Google Scholar]
- 31.Kuperwasser C, Chavarria T, Wu M, Magrane G, Gray JW, Carey L, Richardson A and Weinberg RA, Proc. Natl. Acad. Sci. U. S. A, 2004, 101, 4966–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu J, Lamouille S and Derynck R, Cell Res, 2009, 19, 156–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fuxe J and Karlsson MC, Semin. Cancer Biol, 2012, 22, 455–461. [DOI] [PubMed] [Google Scholar]
- 34.Zhang X, Claerhout S, Prat A, Dobrolecki LE, Petrovic I, Lai Q, Landis MD, Wiechmann L, Schiff R, Giuliano M, Wong H, Fuqua SW, Contreras A, Gutierrez C, Huang J, Mao S, Pavlick AC, Froehlich AM, Wu MF, Tsimelzon A, Hilsenbeck SG, Chen ES, Zuloaga P, Shaw CA, Rimawi MF, Perou CM, Mills GB, Chang JC and Lewis MT, Cancer Res, 2013, 73, 4885–4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marangoni E, Vincent-Salomon A, Auger N, Degeorges A, Assayag F, de Cremoux P, de Plater L, Guyader C, De Pinieux G, Judde JG, Rebucci M, Tran-Perennou C, Sastre-Garau X, Sigal-Zafrani B, Delattre O, Dieras V and Poupon MF, Clin. Cancer Res, 2007, 13, 3989–3998. [DOI] [PubMed] [Google Scholar]
- 36.DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MT, Factor R, Matsen C, Milash BA, Nelson E, Neumayer L, Randall RL, Stijleman IJ, Welm BE and Welm AL, Nat. Med, 2011, 17, 1514–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bergamaschi A, Hjortland GO, Triulzi T, Sorlie T, Johnsen H, Ree AH, Russnes HG, Tronnes S, Maelandsmo GM, Fodstad O, Borresen-Dale AL and Engebraaten O, Mol. Oncol, 2009, 3, 469–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bischel LL, Young EW, Mader BR and Beebe DJ, Biomaterials, 2013, 34, 1471–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen DH, Stapleton SC, Yang MT, Cha SS, Choi CK, Galie PA and Chen CS, Proc. Natl. Acad. Sci. U. S. A, 2013, 110, 6712–6717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Verbridge SS, Chakrabarti A, DelNero P, Kwee B, Varner JD, Stroock AD and Fischbach C, J. Biomed. Mater. Res, Part A, 2013, 101, 2948–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng Y, Chen J, Craven M, Choi NW, Totorica S, Diaz-Santana A, Kermani P, Hempstead B, FischbachTeschl C, Lopez JA and Stroock AD, Proc. Natl. Acad. Sci. U. S. A, 2012, 109, 9342–9347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Risau W, Sariola H, Zerwes HG, Sasse J, Ekblom P, Kemler R and Doetschman T, Development, 1988, 102, 471–478. [DOI] [PubMed] [Google Scholar]
- 43.Alimperti S, Mirabella T, Bajaj V, Polacheck W, Pirone DM, Duffield J, Eyckmans J, Assoian RK and Chen CS, Proc. Natl. Acad. Sci. U. S. A, 2017, 114, 8758–8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.