

The chemoresistant population in recurrent osteosarcoma is subclonal at diagnosis, emerges at the time of primary resection due to selective pressure from neoadjuvant chemotherapy, and is characterized by unique oncogenic amplifications.
Abstract
Multiple large-scale genomic profiling efforts have been undertaken in osteosarcoma to define the genomic drivers of tumorigenesis, therapeutic response, and disease recurrence. The spatial and temporal intratumor heterogeneity could also play a role in promoting tumor growth and treatment resistance. We conducted longitudinal whole-genome sequencing of 37 tumor samples from 8 patients with relapsed or refractory osteosarcoma. Each patient had at least one sample from a primary site and a metastatic or relapse site. Subclonal copy-number alterations were identified in all patients except one. In 5 patients, subclones from the primary tumor emerged and dominated at subsequent relapses. MYC gain/amplification was enriched in the treatment-resistant clones in 6 of 7 patients with multiple clones. Amplifications in other potential driver genes, such as CCNE1, RAD21, VEGFA, and IGF1R, were also observed in the resistant copy-number clones. A chromosomal duplication timing analysis revealed that complex genomic rearrangements typically occurred prior to diagnosis, supporting a macroevolutionary model of evolution, where a large number of genomic aberrations are acquired over a short period of time followed by clonal selection, as opposed to ongoing evolution. A mutational signature analysis of recurrent tumors revealed that homologous repair deficiency (HRD)-related SBS3 increases at each time point in patients with recurrent disease, suggesting that HRD continues to be an active mutagenic process after diagnosis. Overall, by examining the clonal relationships between temporally and spatially separated samples from patients with relapsed/refractory osteosarcoma, this study sheds light on the intratumor heterogeneity and potential drivers of treatment resistance in this disease.
Significance:
The chemoresistant population in recurrent osteosarcoma is subclonal at diagnosis, emerges at the time of primary resection due to selective pressure from neoadjuvant chemotherapy, and is characterized by unique oncogenic amplifications.
Graphical Abstract
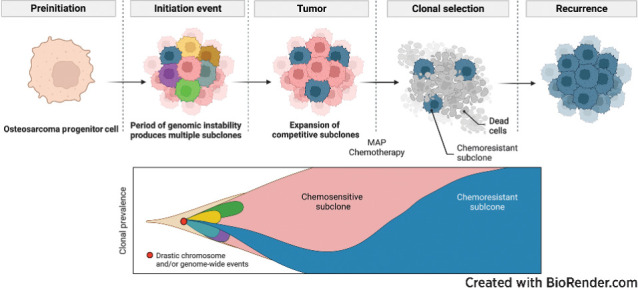
Introduction
Osteosarcoma is an aggressive bone tumor that primarily affects children and young adults. Patients who present with metastatic disease at diagnosis have a poor overall prognosis and those with an inferior response to neoadjuvant chemotherapy have a high risk for recurrence (1–3). Multiple large-scale tumor genomic profiling efforts have been undertaken to describe the genomic underpinnings and identify new potential therapeutic targets for osteosarcoma (4–9). These studies revealed that osteosarcoma, typically characterized by a high degree of chromosomal instability, has a large number of chromosomal deletions, translocations, and amplifications. The common alterations present in osteosarcoma primarily involve tumor suppressor genes (e.g., TP53, RB1, PTEN), whereas targetable activating mutations are rare, making it challenging to link the mutational genotype to a broadly applicable treatment strategy (10, 11). However, recent studies have suggested that targeting focal gene amplifications in consensus driver genes may be an effective strategy for identifying precision-based therapies (4, 12).
Recent genomic studies in osteosarcoma have suggested that metastatic clones do not correspond to the dominant clones present in the primary tumor (13), and osteosarcoma may evolve via parallel evolution (14), with evidence for both monoclonal (15) and polyclonal synchronous seeding of metastases (13, 14). Copy-number alterations (CNA) in consensus driver genes, such as MYC and CDK4 were found to be likely early events (15). Cisplatin-induced mutagenesis has also been highlighted as a potential driver of treatment resistance in recurrent osteosarcoma (15). Despite these initial insights into the clonal heterogeneity of osteosarcoma, the extent to which neoadjuvant chemotherapy affects clonal selection in patients with a poor response to chemotherapy and the degree to which CNAs evolve from diagnosis to relapse remains unclear.
In addition, two competing models of chromosomal evolution in osteosarcoma have been proposed. Evidence exists that implies a dynamic model of genomic instability, which generates a spectrum of chromosomal structures, facilitating the adaptation and selection of diverse phenotypic expressions (16, 17). In osteosarcoma, investigators have inferred a substantial level of copy-number heterogeneity from transcriptional heterogeneity uncovered by single-cell RNA sequencing experiments, supportive of this dynamic model (18, 19). Other studies have emphasized a more static model of tumor development, where early catastrophic events yield complex chromosomal rearrangements, which are maintained with little variation over time (18, 19); supported by comparable structural CNA profiles found in primary, metastatic, and relapse osteosarcoma samples (4, 13, 20–22). This discrepancy poses the question of whether osteosarcoma genomes evolve through continuous cycles of genomic diversification and optimization, or are shaped by early, isolated instability events that dictate long-term tumor development.
To address these open questions about clonal selection, chromosomal evolution, and heterogeneity in osteosarcoma, we performed whole-genome sequencing (WGS) of 37 spatially and temporally separated tumor samples from 8 patients with osteosarcoma who had a poor response to neoadjuvant chemotherapy (<90% necrosis). We describe spatial intermetastatic heterogeneity and temporal clonal evolutionary processes, with a focus on identifying and tracking unique copy-number clones from diagnosis through relapse.
Materials and Methods
Patient consent and tissue processing
This study was approved by the Institutional Review Board (IRB) of the Memorial Sloan Kettering Cancer Center (New York, NY) and conducted in accordance with the Declaration of Helsinki. Informed written consent was obtained from each subject or guardian. Tumor samples and matched normal samples were collected from 10 patients with a pathologically confirmed diagnosis of osteosarcoma, who were identified both retrospectively and prospectively for those who had their tumor banked at diagnosis and at least one other time point. Only patients who consented to an IRB-approved blood and tumor collection protocol were eligible for tumor sequencing. Fresh tumor samples were procured from the operating room in a sterile container. The tissue was processed using scalpels and divided into pea-sized pieces before being stored at −80°C. Frozen tissue samples from several patients were also available through our Precision Pathology Biobanking Center and were acquired using the same protocol. In addition, in several patients, archival tumor specimens in the form of formalin-fixed paraffin-embedded (FFPE) specimens were obtained from both the internal and external pathology departments using the same protocol. Only FFPE samples that were not subjected to harsh decalcification techniques were selected. FFPE samples that had been decalcified using ethylenediaminetetraacetic acid (EDTA) were deemed appropriate for further downstream analysis.
Each frozen tissue sample was submitted to our pathology core, where it was embedded in Tissue-Tek optimum cutting temperature compound and sectioned at 5–10 mm on a Leica Cryostat to create a hematoxylin and eosin–stained slide for review. Each FFPE sample was sectioned using a Leica Microtome. Hematoxylin and eosin slides were evaluated by a trained pathologist to determine tumor content. After pathologic review, tumor samples were isolated via a 21-gauge punch, curl biopsy, or macrodissected from sectioned slides to remove non-neoplastic components. The neoplastic component of each tumor underwent genomic DNA extraction using a Qiagen DNAeasy Blood and Tissue Kit and protocol, whereas the FFPE samples were extracted using a QIAamp DNA FFPE Tissue Kit and protocol.
Peripheral blood mononuclear cells utilized for matched normal sequencing were brought up to 15 mL volume in cold PBS and isolated with the DNeasy Blood & Tissue Kit (QIAGEN, catalog no. 69504) according to the manufacturer's protocol and incubated at 55°C for digestion. DNA was eluted in 0.5X Buffer AE.
WGS and alignment
DNA quantification, library preparation, and WGS were performed using the Integrated Genomics Operation at the Memorial Sloan Kettering Cancer Center (New York, NY). After PicoGreen quantification and quality control using an Agilent BioAnalyzer, 131–500 ng of genomic DNA was sheared using an LE220-plus Focused-ultrasonicator (Covaris, catalog no. 500569), and sequencing libraries were prepared using the KAPA Hyper Prep Kit (Kapa Biosystems KK8504) with modifications. Briefly, libraries were subjected to a 0.5 × size selection using aMPure XP beads (Beckman Coulter, catalog no. A63882) after postligation cleanup. Libraries with <500 ng of genomic DNA were amplified using 5–6 cycles of PCR and pooled equimolar amounts. Libraries containing at least 500 ng of genomic DNA were not amplified. Samples were run on a NovaSeq 6000 in a 150 bp/150 bp paired end run, using the NovaSeq 6000 SBS v1 Kit and an S1, S2, or S4 flow cell (Illumina). The average number of read pairs per normal was 614 million and the average number of read pairs per tumor was 1.3 billion.
WGS pipeline
WGS data were analyzed using the ISABL (23) platform, with methods previously described in detail (24). The additional downstream analyses are described below.
Single-nucleotide variant filtering
For all 8 patients, single-nucleotide variants (SNV) were called in triplicate by MuTect (25), Strelka (26), and Caveman (27). Only mutations that had a “PASS” flag, were called by at least two mutation callers and observed in less than 2% of the reads of the matched normal sample with 10x coverage were considered for further analysis. It is notoriously difficult to extract high-quality DNA from the osteoid matrix that surrounds viable tumor cells, often leading to sequenced samples with low purity. Across the 8 patients, a mix of samples was prepared as either fresh frozen or FFPE. FFPE samples are thought to be inferior to fresh frozen samples, as the formalin fixation process results in nucleic acid fragmentation, DNA cross-links, and deamination, leading to C>T mutation artifacts. As a result, downstream analysis of FFPE can be challenging when filtering out artifacts from a true positive. Aggressive filtering of low-allele frequency variants has been shown to increase SNV overlap when comparing matched FFPE and fresh frozen samples (28). Given these assumptions, a custom-filtering approach was utilized to maximize the ability to utilize low-purity FFPE samples within a mix of higher-quality FFPE samples and fresh frozen samples for each patient. For fresh frozen samples with an estimated purity of 20%, mutations with a variant allele frequency (VAF) less than 5 were filtered out. If a frozen sample had a purity of less than 20%, no additional filtering was applied, given the potential for filtering out subclonal mutations in an otherwise high-quality frozen tissue sample. For FFPE samples, we filtered out mutations with a VAF less than 20%. These thresholds for FFPE samples were then further purity adjusted, for example, a sample with an estimated purity of 80%, would have mutations below a VAF of 0.16 (0.8 purity * 0.2 VAF filter) initially filtered out. Although this initial filtering for FFPE is strict, the advantage of having multiple samples per patient allows us to utilize the mutation calls in other high-quality samples for a patient to rescue mutations that may have been initially filtered in a low-quality sample. This is accomplished through a pile-up rescue, where all filtered mutations for a patient are combined and then specifically searched in the BAM file that was generated for each sample.
Driver gene analysis
All somatic variants that led to a frameshift insertion, frameshift deletion, in-frame insertion, in-frame deletion, missense, nonsense, nonstop, or splice site/region mutation, or a translation start site were considered. For variants identified as missense or nonsense, we required the variant to be considered a likely functional driver using the LiFD tool (29), which is a two-phase algorithm that pulls from various databases and bioinformatic methods to determine whether a given mutation is likely to be functional. We also considered genes that were significantly mutated in large pediatric cancer and osteosarcoma sequencing studies (4–9, 30). The final list consisted of 639 genes.
Evolutionary analysis
To determine the spatial and temporal dynamics of subclonal diversity within a patient, we first used Treeomics v1.9.2 (31) to derive phylogenies. Treeomics reconstructs the phylogeny of metastatic lesions and maps subclones to their anatomic locations. Treeomics utilizes a Bayesian inference model to account for error-prone sequencing and differing neoplastic cell contents to calculate the likelihood that a specific variant is present or absent. Treeomics then infers a global optimal tree based on mixed-integer linear programming (31).
The HATCHet (32) v1.0.1 algorithm was used to infer allele- and clone-specific copy-number aberrations, clone proportions, and whole-genome duplications (WGD) for each patient. HATCHet was run using the GATK4-CNV custom pipeline, with Battenberg copy-number calls fitted to meet the input requirements for running the tool. Solutions were manually reviewed with the creator of the tool, Simone Zaccaria PhD, to allow for advanced fine-tuning and ensure that the most accurate solutions were selected.
The DeCiFer (33) v1.1.5 algorithm was used to cluster mutations across all samples for each patient, providing descendant cell fractions (DCF) and cancer cell fractions for each cluster. The copy-number input for this algorithm is the output of the HATCHet algorithm. Custom state trees were generated utilizing a maximum copy number between 6 and 8 for each patient (lower maximum copy-number states were selected if the runtime exceeded 48 hours). After clustering of mutations, the CALDER (34) v0.11 algorithm was used to infer evolutionary phylogenies. To run the DeCiFer output through CALDER, the inferred cluster DCF was converted to a read count by multiplying the DCF by 1,000 and then dividing by 2 (because CALDER assumes that all mutations are in heterozygous diploid regions). Therefore, if a mutation has 1,000 reads and an inferred DCF of 40%, the corresponding input will have 200 variant reads and 800 reference reads. Longitudinal constraints were lifted when analyzing OSCE2 and OSCE3, because all analyzed samples were present at diagnosis.
The Palimpest (35) algorithm (version = github commit 4795da2) was used to characterize and visualize mutational signatures (using Cosmic SBS/DBS v3.2) at both clone and sample levels. This algorithm also provided information regarding the timing of duplication and LOH events using previously described methods (36).
Structural variant analysis
Structural variants (SV) were annotated using iAnnotateSV software (37). We used svpluscnv (38) and ShatterSeek (39) to identify regions of chromothripsis. The JaBba tool (40) was used to identify regions with complex rearrangements or amplicon events. The calls from all tools were combined for further downstream analysis.
Data visualizations
Oncoprint was created using the CoMut (41) tool Timescape (https://github.com/shahcompbio/timescape; ref. 42) and Mapscape (https://github.com/shahcompbio/mapscape; ref. 42) were used to visualize temporal and spatial clonal evolution. Tableau Desktop (v2021.4) was used to analyze and visualize the data with charts, bars, and line graphs. Ridgeline graphs were created using R utilizing the ggridges package (https://cran.r-project.org/web/packages/ggridges/vignettes/introduction.html). Sankey plots were created using SankeyMATIC (https://sankeymatic.com/build/). Anatomic cartoons were created using BioRender (https://biorender.com/).
Data availability
WGS generated in this study has been deposited at the European Genomephenome Archive (EGA) under study ID EGAS00001007486 (https://ega-archive.org). All other raw data are available upon request from the corresponding author.
Results
Analysis of SNVs reveals limited driver gene heterogeneity in temporally and spatially distinct osteosarcoma sample
To analyze clonal evolution and intratumoral heterogeneity in osteosarcoma, we performed WGS of tumor tissues from multiple spatially and temporally distinct samples from 8 individuals with relapsed/refractory osteosarcoma. DNA was extracted from 84 samples collected from 10 patients. After initial quality control, we sequenced 62 unique tumor samples from 8 patients with WGS to a target depth of 80x (Supplementary Fig. S1). Of these 8 patients, 4 had localized disease at diagnosis (OSCE4, OSCE5, OSCE6, OSCE9) and 4 had metastatic disease at diagnosis (OSCE1, OSCE2, OSCE3, OSCE10; Fig. 1A), and the age at diagnosis was 11–27 years (4 girls and 4 boys, Fig. 1B). All patients were treated at the Memorial Sloan Kettering Cancer Center and received methotrexate, cisplatin, and doxorubicin (MAP) chemotherapy (subsequent postprocedure treatment, Fig. 1A). Seven of the 8 patients had a poor response to neoadjuvant chemotherapy (<90% necrosis at the time of primary resection, Fig. 1C), while OSCE5 had an upfront resection; therefore, the response to therapy could not be evaluated.
Figure 1.

Characteristics of the patients and samples included in the analysis cohort. A, Oncoprint of sample and patient-level details for each patient. Samples from the same patient are connected by dots and lines at bottom of figure and are in chronological order of time obtained (earliest on left). Bx, biopsy; Rx, resection; sample ending in 0, metastatic site present at diagnosis; sample ending in a number >0, number of relapses. L/R, laterality; d, distal; p, proximal; f, femur; t, tibia; cw, chest wall; H, heart; μL, upper lobe; ll, lower lobe; di, diaphragm; rp, retroperitoneal; l, lobe. B, Age and sex assigned at birth for each patient; patient is on the x-axis, age is on the y-axis, and sex assigned at birth is plotted on the chart as blue for male and pink for female. C, Percent necrosis at time of primary resection for each patient (note OSCE2/OSCE10 both have 45% necrosis). D, Summary of the number of samples per time point for each patient. Darker shades of blue represent higher number of samples at a respective time point. E, Patients are listed on the y-axis and are ordered from longest disease course at the top to shortest at the bottom of the figure. The light blue bars represent length of disease course in months. Events are marked as depicted in the legend with different shapes and colors and plotted along the disease course bar at the time in months that the event occurs. (C, Created with BioRender.com.)
After sequencing was completed, we reviewed the quality of the sequencing data (purity, sequencing coverage) and determined that 37 of the 62 (59%) samples, similar to other studies (43), met our quality control requirements to proceed with further downstream analysis (Supplementary Fig. S1). Of these 37 samples, 17 came from primary sites, with 7 of 8 patients having a pretreatment sample, six of which were biopsies and one (OSCE5), which was a pretreatment primary resection (Fig. 1A and D). Of the 7 patients who did not undergo an upfront resection, all had at least one on-therapy resection sample, and 1 patient (OSCE10) had multiregion sampling from the primary tumor (Fig. 1A and D). The other 20 samples came from metastatic sites, 15 of which were from lung metastases, and seven of the 20 were metastatic sites that were present at diagnosis (Fig. 1A and D). Fresh frozen samples accounted for 15 of the 37 samples selected for downstream analysis, while the remaining 22 were FFPE samples (Fig. 1A). The median purity of fresh frozen samples was 0.75 compared with 0.46 of FFPE samples (Fig. 1A). All 8 patients in this analysis had matched normal blood samples sequenced at a target depth of 40×.
The clinical courses of OSCE2, OSCE3, and OSCE10 were defined as refractory disease with progression on MAP chemotherapy and extremely virulent disease courses, with time from diagnosis to death of 1.08, 1.3, and 1.75 years, respectively (Fig. 1E). OSCE1 and OSCE5 had long protracted relapsing and remitting disease courses, with a time from diagnosis to death of 7 and 6 years, respectively (Fig. 1E). OSCE4 also had a relapsing and remitting disease course but is currently in remission 8 years after diagnosis (Fig. 1E). OSCE6 is alive 4.5 years after diagnosis but has had a recent recurrence (Fig. 1E). OSCE9 is 4 years out from the initial metastatic recurrence (Fig. 1E).
After filtering and germline subtraction, WGS data identified between 1,684 and 16,215 SNVs per sample (Fig. 1A). Of these, there were between 12 and 181 coding nonsynonymous SNVs per sample (median = 54; Supplementary Fig. S2A). The average number of nonsynonymous SNVs in primary site samples was 42, compared with 73 in metastatic or relapsed samples. SNVs were clustered across all samples for each patient using the DeCiFer (33) algorithm, which determines the DCF of all SNVs for a given cluster in each patient (analogous to the cancer cell fraction but accounting for potential mutation losses; ref. 33). Following previous approaches (33), SNVs were categorized as clonal if they belonged to a cluster in a sample with a DCF ≥ 90%, and subclonal if they belonged to a cluster with a DCF < 90%. The proportion of clonal SNVs ranged from 35.7% to 100% across all samples (median = 68.2%), with relapse samples having the highest proportion (median = 92.7%) of clonal SNVs compared with biopsy, resection, and metastatic samples (median = 66.1%, 64.7%, and 63.6%, respectively; Fig. 1A; Supplementary Fig. S2B). Likely functional driver gene SNVs (29) were identified in 5 of the 8 patients, including genes known to be frequently mutated in osteosarcoma, such as TP53, ATRX, RB1, and CDKN2A (Fig. 1A; refs. 5, 9). These driver genes were clonal (shared) across all samples for each patient, and no new SNVs were likely functional drivers that were unique to any metastatic or recurrent samples (Fig. 1A; Supplementary Fig. S3A).
Because there were no new SNVs in consensus driver genes unique to relapse or metastatic samples, we next examined structural variations and CNAs. SVs in consensus driver genes were shared across all samples for each patient. TP53 SVs involving intron 1 were observed in 5 of 8 patients (OSCE2, OSCE3, OSCE6, OSCE9, and OSCE10; Fig. 1A). A TP53 intron 2 inversion was observed in OSCE4 (Fig. 1A). There was a TP53 SNV in OSCE1 and while there was no TP53 SNV or SV found in OSCE5 (Fig. 1A), there was amplification of MDM2, an important negative regulator of TP53. In OSCE1, there was a deletion event in RB1 and an inversion in ATRX in the pretreatment sample (RdtBx) that was not seen in the primary resection or relapse samples (Fig. 1A). Disruptions in DLG2, a bone tumor supressor gene (44), were observed in 4 patients (OSCE2, OSCE6, OSCE9, and OSCE10; Fig. 1A). Deletion events in DMD, a gene that has been linked to aggressive behavior in human cancers, and is believed to have a potential role as a tumor suppressor, were observed in the three refractory cases (OSCE2, OSCE3, and OSCE9) and were present in all samples for OSCE2 and OSCE3; however, they were only detected in the relapse sample in OSCE10 (Fig. 1A). OSCE9 was found to have a deletion event in PTEN and an in-frame fusion event in ALK (Fig. 1A).
Subclonal somatic CNAs emerge and dominate in recurrent osteosarcoma
A high prevalence of somatic CNAs (SCNA) in osteosarcoma has been reported previously (5, 9). Therefore, we used the HATCHet (32) algorithm to infer both allele- and clone-specific CNAs as well as their relative proportions across multiple samples from the same patient. The average tumor ploidy for each sample ranged from 1.7 in OSCE10 to 3.15 in OSCE3 (Supplementary Fig. S3B). HATCHet (32) identified subclonal CNAs in all but 1 patient (OSCE2; Supplementary Fig. S4A) and WGDs present at diagnosis in 3 of the 8 patients (OSCE1, OSCE2, and OSCE9; Fig. 1A). Among the 7 patients with subclonal CNAs, 6 were identified as having two major copy-number clones (Fig. 2A–E; Supplementary Fig. S4B), and 1 patient (OSCE10) had three distinct copy-number clones (Fig. 2F). In 4 of the 7 patients (OSCE1, OSCE4, OSCE6, and OSCE10), multiple distinct copy-number clones were identified to be simultaneously present in the primary tumor (Fig. 2A, B, D, and F), but only one of these subclones emerged and dominated at subsequent relapses. In 2 of these patients (OSCE1, OSCE6), the emergence of this clone was observed in the primary resection sample when compared with the pretreatment biopsy for each patient (Fig. 2A and D). In OSCE5, a new copy-number clone emerged in the late relapse samples, which was not identified as being present in the pretreatment sample (Fig. 2C). However, when combining the analysis of mutations and CNAs, the dominant SNV-based clone (which shared the dominant copy-number profile of the late emerging copy-number clone) in the relapse samples was found to be subclonal in the pretreatment sample, providing evidence that the dominant copy-number clone in the relapse sample for OSCE5 was present at diagnosis (Supplementary Fig. S5A). In OSCE10, no subclonal copy-number aberrations were identified in the pretreatment sample; however, a subclonal copy-number clone was detected in the primary resection sample that emerged and dominated at relapse in this patient (Fig. 2F). In summary, we found that in most cases, a minor subclone present in the primary tumor emerged and dominated in patients with relapsed disease.
Figure 2.

Subclonal copy-number clones emerge at relapse. A–F, For each patient there is a panel of three figures. The figure on the left is an oncoprint featuring clone-specific CNAs in recurrently altered genes of interest in osteosarcoma. The top figure is a plot of allele-specific CNAs for each clone, with significant events for each clone circled and highlighted (note y-axis scales are unique for each patient). Clone 1, magenta clone; clone 2, teal clone; clone 3 in OSCE10, gray clone. The major allele is plotted above 0 and the minor allele is plotted below 0. The bottom figure in each panel is a TimeScape plot of the prevalence of each clone at different time points throughout a patient's disease course. G, Combined genome-wide CNAs across all patients in the cohort, with recurrently altered genes highlighted.
In two of three refractory cases (OSCE2, OSCE3), there was no subclonal copy-number clone that was identified in the primary that emerged and dominated in metastatic or relapse samples. In OSCE2, only a single major copy-number clone was identified; in OSCE3, two copy-number clones were identified in the pretreatment sample, with the copy-number subclone emerging and dominating in one of the metastatic sites but in mixed proportion in the three other metastatic sites. OSCE9 did not have a pretreatment sample for comparison but did show two copy-number clones in mixed proportions in the primary resection sample and the two relapse sites.
Determinations regarding branched versus parallel evolution (depicted in the TimeScape plots in Fig. 2A–F; Supplementary Fig. S4B) for patients with ≥2 copy-number clones were based on a review of LOH events in the dominant metastatic or recurrent copy-number clone, using the rationale and methods outlined by Watkins and colleagues (45). Branched evolution with the emergence of the treatment resistant copy-number clone from the dominant pretreatment copy-number clone (clone 2 emerging from clone 1) was observed in 3 patients (OSCE4, OSCE9, and OSCE10). Parallel evolution, where the pretreatment and treatment resistant copy-number clones share the same parent clone (clone 1 and clone 2 share the same parent clone) but evolve in parallel, was seen in 4 patients (OSCE1, OSCE3, OSCE5, OSCE6). LOH events were common in tumor suppressor genes such as TP53, RB1, and PTEN and were mostly shared between clones for each patient, with a median of 77.95% of LOH events shared between all clones (range = 18.9%–86.82%; Supplementary Fig. S5B).
Copy-number amplifications in recurrently altered oncogenes in osteosarcoma characterize chemoresistant copy-number clones
Cohort-wide CNAs reflected what has been previously described in osteosarcoma (4), with gains and amplifications seen in VEGFA, MYC, FOXM1, CDK4, AKT1, AURKB, and CCNE1, and loss/deletion events in PTEN, RB1, and TP53 (Fig. 2G). In contrast to previously identified SNV drivers, the relative proportions of alterations across tumor cells changed with time. Clones were classified as chemoresistant if they were present in the primary site and became dominant at relapse, and chemosensitive if it was the dominant clone in the primary site and became subclonal or eliminated in metastatic or relapse sites. When comparing the genomic alterations between copy-number clones for each patient, deletion or LOH events in tumor suppressor driver genes were often clonal in patients found to have ≥2 copy-number clones (Fig. 2A–F; Supplementary Fig. S4A). In the 5 patients with clear emergence of a chemoresistant copy-number clone (clone 2 in OSCE1, OSCE4, OSCE5, OSCE6, and clone 3 in OSCE10), the resistant clone had a higher degree of MYC gain or amplification then the dominant chemosensitive clone at diagnosis (Fig. 2A–F; Supplementary Fig. S5C). In OSCE10, high-level (log2 ≥ 2) MYC-amplified clone 3 emerged at the time of primary resection and dominated at relapse (Fig. 2F). Notably, this treatment-resistant clone was present in four of five multiregion samples from the primary resection, suggesting intratumoral heterogeneity regarding CNAs depending on the site sampled. In addition to MYC, amplification of CCNE1/CCND3 (OSCE1, OSCE6; Fig. 2A and D), KRAS (OSCE2, OSCE4; Supplementary Fig. S4B; Fig. 2B), IGF1R (OSCE6; Fig. 2D), CDK4 (OSCE10; Fig. 2F), VEGFA (OSCE1, OSCE6, OSCE9; Fig. 2A, D, and E), and LOH at HLA (OSCE4; Fig. 2B; Supplementary Fig. S5D) uniquely characterized treatment-resistant or metastatic copy-number clones in this cohort.
Subclonal selection/emergence evident at the time of primary resection
Three patients within our cohort had a pretreatment sample on therapy resection, and at least one relapse sample (OSCE1, OSCE6, OSCE10), which provided an opportunity to analyze the effect of neoadjuvant chemotherapy on subclonal copy-number selection at the time of primary resection and whether this selection is reflective of the dominant clone at the time of relapse. OSCE1 and OSCE6 share a similar pattern, where a copy-number subclone present at diagnosis emerges as the dominant clone at the time of primary resection and continues to dominate at the first and subsequent relapses. Chemoresistant clone 2 was characterized by mid-level (log2 = 1.5–1.99) CCNE1 amplification in OSCE1 (Fig. 2A) and mid-level IGF1R amplification in OSCE6 (Fig. 2D). A slightly different pattern emerged in the refractory case of OSCE10, in which only a single copy-number clone was present at diagnosis. The primary resection sample underwent multiregion sequencing of five spatially separated sites, which revealed the emergence of two new copy-number clones, with clone 2 present in all five samples and clone 3 in four of five samples. Clone 3 then became the dominant clone in the first relapse specimen, characterized by high-level MYC amplification.
Timing of copy-number gains reveals large bursts of copy-number gains before diagnosis
The HATCHet algorithm also infers WGD events jointly across all samples for each patient. Of the 5 patients with average ploidy ≥2.5 (OSCE1, OSCE2, OSCE3, OSCE4, OSCE9), WGD was identified in OSCE1, OSCE2, and OSCE9 (Fig. 1A; Supplementary Fig. S2B). The timing of these WGD events as well as other chromosomal duplications can be determined in “molecular time” using previously described methods (35, 36), which compares the number of duplicated versus nonduplicated mutations to estimate the timing of each duplication (Supplementary Fig. S6A–S6E). For the patients who were determined to have WGD events, these appeared to be late events for each respective patient [median point mutation time (pmt) range = 62%–79%; Fig. 3A], compared with the 2 patients with ploidy ≥2.5 without WGD (patients with ploidy <2.5 did not have enough duplication or LOH events to analyze), which appeared to have early synchronous duplications (median pmt range = 16%–25%; Fig. 3A). The 2 patients with the lowest ploidy (OSCE10 and OSCE6) also had the smallest primary tumors (Fig. 3B), whereas all large primary tumors (≥300 cm3) had ploidy ≥2.5 (OSCE2, OSCE9, OSCE3). For the 5 patients with ploidy ≥2.5, we analyzed their earliest primary site samples to assess the natural history of these duplication events in the context of tumorigenesis (Fig. 3C).
Figure 3.

Chromosomal duplication timing analysis reconstructs evolutionary past of genomic instability events. A, Plot of duplication and rearrangement events in molecular time. Left side of figure is plot of duplication events (blue) and LOH events (orange) for select primary site samples. The median duplication time is highlighted for each sample. The plot on the right side of the figure is complex amplicon events (teal) and complex rearrangement events (yellow). Y-axis for both plots is molecular time. The samples are in the same order for each plot. Each plotted event represents an affected chromosomal arm. B, Plot of tumor size (by volume) versus ploidy called by HATCHet. Median size (y-axis) and ploidy (x-axis) values are plotted with dark black lines, with the shaded gray areas representing the range between the lower and upper quartile for each metric. C, Chromosomal duplication timing of five primary site samples. WGD events as called by HATCHet appear to be a late event in our cohort. D–F, Ridgeline plots of the density of duplication events over molecular time for each sample for the selected patients. Notably the highest peak in duplications occurs before diagnosis.
Most of these duplication/LOH events clustered in a single burst of events and were associated with complex rearrangement (chromoplexy, chromothripsis) or complex amplicon (Tyfonas, breakage fusion bridge, double minute; ref. 40) events (Fig. 3A and C). When comparing longitudinal samples from the same patient (Fig. 3D–F), there was no subsequent burst that was greater than the primary site burst. Across the cohort, duplication events appeared to be fixed during tumorigenesis and had decreasing average molecular times when comparing biopsy/resection samples with metastatic/relapse samples (Supplementary Fig. S7A and S7B). LOH events were consistently “late” events, with a median molecular time of 82.16 across the cohort (compared with 21.72 for duplication events; Supplementary Fig. S7C and S7D).
Heterogenous seeding patterns observed in metastatic and recurrent osteosarcoma
Clone-based phylogenies were created to explore the clonal architecture and track the spatial and temporal patterns of evolution. The SNVs across all samples for each patient were clustered using the DeCiFer (33) algorithm. After clustering, each SNV was assigned to a clone with an estimated DCF per sample, and clone-based phylogenetic trees were then constructed using the CALDER (34) algorithm, allowing for the assessment of modes of metastatic seeding and dissemination. At the patient level, there was a heterogeneous mix of dissemination patterns (Figs. 4A–D and 5A–D; Supplementary Fig. S8A–S8H), with complete agreement between clone- and sample-based phylogenies (Supplementary Fig. S3A) regarding whether metastatic dissemination was monophyletic (all metastatic clones shared a common ancestral clone: Figs. 4A, C, and 5E; Supplementary Fig. S8A, S8C, S8E, S8G, and S8H) versus polyphyletic in origin (where ≥2 clones are present from distinct branches of phylogeny whose common ancestor represents the trunk of the primary tumor tree: Figs. 4B, D, and 5B; Supplementary Fig. S8B, S8D, and S8F). Of note, OSCE6 was the only patient among those with a monophyletic origin that had monoclonal seeding (one clone present in the sample, Fig. 4C; Supplementary Fig. S8H). The seeding and dissemination patterns were also assessed for each metastatic sample (Fig. 5E). In 2 patients (OSCE3 and OSCE5), there were examples of different modes of metastatic dissemination among spatially separated metastases from the same resection. In OSCE3 for example, extrapulmonary metastases (RcwM0, RdiM0) demonstrate polyclonal (≥2 clones present in the sample) monophyletic patterns of dissemination, while the pulmonary metastases (RllM0, RulM0) demonstrate monoclonal monophyletic dissemination, with both samples sharing the same ancestral clone (Fig. 5C; Supplementary Fig. S8B).
Figure 4.

SNV-based phylogenies highlighting temporal evolution with clonal mutational signature composition. A–D, Top figure in each panel is a TimeScape plot of the inferred evolutionary phylogeny, highlighting clonal proportions over time. The prevalence of different clones is shown over time on the vertical axis, with the different clones represented by different colors. The horizontal axis represents the time points, which are represented by gray lines. The evolutionary relationships between the clones are shown on the phylogenetic tree and in the TimeScape layout. The bottom right of each panel is a stacked bar plot of the total number of mutations assigned to each clone. Colors represent total number of mutations attributed to each mutational signature, with color legend at bottom of figure. Patient-level metastatic seeding patterns are denoted by the brackets at the top of the page.
Figure 5.

SNV-based phylogenies highlighting spatial evolution and descriptions of metastatic seeding patterns at the sample and patient level. A–D, Spatially and in some cases temporally distinct samples are indicated on the anatomic sites from where the sample originated. The colors represent different clones, and the phylogenetic trees show the evolutionary relationships between these clones. The prevalence of each clone at a particular site is proportional to the colored area of the cellular aggregate representation. E, Sample and patient level dissemination patterns are characterized in these charts. Monoclonal dissemination: single subclone within the primary tumor seeds one or more metastatic lesions; polyclonal dissemination: multiple distinct subclones from the primary tumor seed one or more metastatic lesions; monophyletic origin: all metastatic clones are derived from a recent common ancestor; polyphyletic origin: metastasizing clones are more similar to other subclones within the primary tumor than they are to each other. These descriptions can be considered at the sample level, focused on the clonal make up of a single metastatic site compared with the primary tumor, or taken as a whole, evaluating all spatially or temporally separated samples and how they relate back to the primary tumor. F, Multiregion sequencing was performed on a primary resection sample from OSCE10. Regions D, E, M, O, P were sequenced from the specimen grid depicted. A table of Jaccard similarity indexes based on shared SNVs for these samples is shown in the top left inset. (A–D, Created with BioRender.com.)
Limited heterogeneity after induction chemotherapy
For OSCE10, we obtained a section of the primary tumor sample for multiregion sequencing after the patient had received 10 weeks of induction MAP chemotherapy. The specimen was mapped (Fig. 5F), DNA was extracted from the sections with viable tumor, and then five sections (E, D, M, O, P) with the highest DNA quantity/quality metrics underwent WGS. When examining the clonal architecture of each section (Figs. 4D and 5F), each section had the same four clones (A, D, E, F), with truncal Clone A dominating, except for FRx_M, which did not have clone F, which later became the clone that dominated at relapse. This pattern was also seen with the copy-number clones, where clone 3 was present in all sections except for FRx_M and then dominated at relapse (Fig. 2F). When comparing Jaccard similarity coefficients based on SNV composition across the different sections, adjoining sections shared the highest coefficients (D/E = 0.96, O/P = 0.79, M/P = 0.88; Fig. 5F), whereas nonadjoining sections had similar coefficients, regardless of distance from each other (range = 0.67–0.69; Fig. 5F). As highlighted previously, a single-sample sequencing strategy that sampled from FRx_M would have missed detecting the metastatic subclone that was present in the other four sections.
Most new SNVs in relapsed disease are attributed to homologous repair deficiency–related SBS3 and cisplatin mutational signatures
To further characterize the potential drivers of clonal evolution, mutational signature analysis was performed for each clone (Fig. 4A–D; Supplementary Fig. S8A–S8D). In 3 patients, the largest clone by number of SNVs (OSCE5—Clone C, OSCE6—Clone G, and OSCE10—Clone C) had over half of the SNVs attributed to the DNA-damaging effects of cisplatin chemotherapy (Supplementary Fig. S9A). In 4 patients, the largest clone by number of SNVs (OSCE1—clone H, OSCE3—Clone A, OSCE4—Clone D, and OSCE9—Clone G) had a plurality of SNVs in each clone attributed to single base substitution (SBS) 3, a genomic signature that has been associated with homologous repair deficiency (HRD; Supplementary Fig. S9A). Tumors with this signature are thought to have a BRCAness phenotype and exhibit features similar to those cancers with germline BRCA1 or BRCA2 mutations, even though no mutations in those genes have been identified. In OSCE2 and OSCE3, both refractory cases, the largest number of SNVs was assigned to the truncal clone, clone A, which had a high number of SNVs (OSCE2—1731/2234, OSCE3—2464/4022) associated with HRD-related SBS3 and late replication errors (Sig. 8). In patients with patient-level monophyletic seeding of metastases (OSCE1, OSCE2, OSCE5, OSCE6), where a single ancestral metastatic clone could be identified (OSCE1—Clone D, OSCE2—Clone D, OSCE5—Clone C, OSCE6—Clone C), there were no clear patterns regarding the signature composition identified (Supplementary Fig. S9B). OSCE1—Clone D and OSCE2—Clone D had a plurality of SNVs attributed to HRD-related SBS3, whereas OSCE5—Clone C had the majority of SNVs attributed to cisplatin, and OSCE6 had the majority of its SNVs attributed to reactive oxygen species (ROS) damage (Sig. 18). When looking at doublet base substitution (DBS) signatures at the clonal level, in clones with ≥10 DBS SNVs, cisplatin-associated DBS 5 was the largest contributor of DBS SNVs, accounting for 50% or more of the total SNVs in 17 of 26 clones (Supplementary Fig. S9C).
Emergence of cisplatin and alkylator signatures helps time the formation of metastases
A simple method for ascertaining whether a given metastasis arose before or after treatment with cisplatin or an alkylator is to find clonal SNVs attributed to the respective signature in a metastatic tumor sample (46). When looking at the dominant clone in the first relapse sample for patients with recurrent disease (OSCE1—Clone D, OSCE4—Clone E/I, OSCE5—Clone C, OSCE6—Clone C/F/G, OSCE9—Clone C/G, OSCE10—Clone C/F), there was a cisplatin signature present in each of these respective clones/clades, indicating that the metastases arose after therapy (Fig. 4A–D; Supplementary Fig. S8C and S8D). In addition, in OSCE1, after the patient received chemotherapy with ifosfamide and etoposide, OSCE1—Clone H and OSCE1—Clone F had mutations attributed to the alkylator signature, SBS11 (Fig. 4A). In the 3 patients with refractory disease (OSCE2, OSCE3, and OSCE10), we can demonstrate which metastatic samples were present at diagnosis and which developed while on therapy. Within OSCE2, Clone F and Clone G were the dominant subclones in RllM0 and RulM0, respectively in OSCE2 (Fig. 5A; Supplementary Fig. S8A). Clone F had 181 of 614 mutations attributed to cisplatin and Clone G had no mutations attributed to cisplatin, evidence Clone F was seeded on therapy as opposed to prior to therapy. In OSCE3, there was no evidence of a cisplatin signature in any of the subclones (Supplementary Fig. S8B), indicating that all metastatic sites had developed prior to initiating therapy. In OSCE10, the metastatic sample (RulM0), which has polyphyletic and polyclonal seeding (Figs. 4D and 5D), showed that the dominant clone/subclone pair of D/G likely seeded on therapy, given that there is a cisplatin signature attributed to approximately half of the mutations (1027/2644) and in all the mutations in subclone G (2727/2727).
Cisplatin-associated hypermutation in a case of refractory osteosarcoma
When evaluating mutational signatures at the sample level, the two most prevalent SBS signatures across all samples were HRD-related SBS3 and cisplatin (Fig. 6A). In the 13 relapse samples, HRD-related SBS3 and cisplatin accounted for more than half of all the SNVs (Fig. 6B). In the 17 primary site samples, the HRD-related SBS3, clock-like (Sig. 5), late replications errors (Sig. 8), ROS damage (Sig. 18), and APOBEC (Sig. 2,13) accounted for 75%–100% of all mutations (Fig. 6C). In six of seven metastatic samples, HRD-related SBS3, clock-like, late replication errors, somatic hypermutation, ROS damage, and APOBEC accounted for all SNVs (Supplementary Fig. S10A). In the metastatic sample thought to have emerged on therapy for OSCE10 (RulM0) and the subsequent relapse sample (LllM1), SNVs attributed to cisplatin accounted for 6,226/7,724 (86%) and 14,276/18,216 (78%) SNVs, respectively (Supplementary Fig. 10A). In the 6 patients with relapsed disease, the number of mutations attributed to HRD-related SBS3 consistently increased from diagnosis to subsequent relapses, with at least 1,000 new SNVs attributed to HRD-related SBS3 when comparing diagnostic and relapse samples (Supplementary Fig. S10B).
Figure 6.

Mutational signature patterns across the cohort. A, Stacked line chart of mutational signature contribution by total number of mutations attributed to each signature. Colors represent the different signatures. B, Stacked bar chart of the relative contribution of HRD-related SBS3 (green) and cisplatin (blue) signature in metastatic and relapse samples. C, Stacked bar chart of the relative contribution of HRD (green), clock-like (pink), ROS damage (red), late replication errors (beige), somatic hypermutation (orange), and APOBEC (teal) in primary site samples. D, Stacked bar chart of the probability that driver gene SNVs were attributed to a mutational signature. Pretreatment samples from patients with driver SNVs were included and the primary resection sample from OSCE9 because no pretreatment sample was available. Colors represent the different signatures.
DBS signatures were also evaluated across the cohort (Supplementary Fig. S10C). DBS signature 5, which is associated with cisplatin, was detected in 21 samples; however, to filter out false positives, a threshold of ≥5 DBS signature 5 SNVs was used to confirm the absence of the signature. Using this filter, DBS signature 5 was present in 15 samples, all metastatic or relapse samples, with complete overlap with the 14 samples that had SBS cisplatin signatures 31 or 35. Only OSCE3_RdiM0 had a DBS cisplatin signature but not an SBS cisplatin signature (Supplementary Fig. S10D).
HRD-related SBS3 and ROS damage linked to driver gene mutagenesis
To identify the mutational processes most likely to be the origin of truncal driver gene SNVs, we calculated the likelihood that each individual SNV was caused by each signature, considering the mutation category and proportion of each mutational signature in the tumor genome (36). To minimize the effect of treatment-related mutagenesis, we limited this analysis to the earliest primary site sample available for patients with truncal driver SNVs. Similar to the observations across all samples, HRD-related SBS3 had the highest probability of attribution in the TP53-driven OSCE1 sample and the RB1-driven OSCE4 samples (OSCE1 probability = 0.49, OSCE4 probability = 0.44; Fig. 6D), and a slightly lower attribution probability than the clock-like signature in OSCE9 (HRD-related SBS3 probability = 0.32, clock-like probability = 0.34; Fig. 6D). Both OSCE3 and OSCE10 had truncal ATRX SNVs, with ROS damage accounting for the highest probability of attribution in both samples (OSCE3 probability = 0.54, OSCE10 probability = 0.42; Fig. 6D).
Discussion
Tumor evolution and clonal heterogeneity have been increasingly recognized as major causes of therapeutic resistance to current antineoplastic therapies (47). These findings extend our understanding of therapeutic resistance in spatially and temporally separated tumor samples from 8 patients with recurrent or refractory osteosarcoma. We found that while clonal driver gene SNVs and structural variants remain largely unchanged over the course of tumor progression, subclonal tumor populations with unique driver gene amplifications are present at diagnosis, emerge after treatment, and persist as the major clone at subsequent relapses.
SCNAs are now increasingly recognized for their prognostic value over SNVs in multiple cancer types (48). Oncogenic CNAs, while heterogeneous across osteosarcoma, represent potential therapeutic targets, given the lack of recurrent targetable SNVs or structural variants (4, 8). Our study revealed that in our 4 patients who had relapsed osteosarcoma with a matched pretreatment sample (OSCE1, OSCE4, OSCE5, OSCE6), there was a subclonal treatment resistant copy-number clone that emerged as the dominant clone in the relapsed setting. Furthermore, in the 2 patients with both a pretreatment sample and on-therapy primary resection (OSCE1, OSCE6), this treatment resistant clone clonally expanded after 10 weeks of neoadjuvant chemotherapy. We believe this finding has important implications for molecular profiling strategies in osteosarcoma, as it suggests that the primary resection sample, and not the pretreatment biopsy, is more reflective of the metastatic potential for a tumor than the pretreatment biopsy, due to the selection pressure of neoadjuvant chemotherapy. Achieving a cure in osteosarcoma requires the extinction of all cancer cells with a successful “first-strike” strategy with MTDs of cisplatin, doxorubicin, and methotrexate (49). For patients for whom this first strike fails (poor necrosis at the time of primary resection), characterizing and targeting the treatment-resistant population of cancer cells using a second-strike strategy may prove to be an effective treatment strategy (49). Our work highlights that molecular profiling of primary resection samples could allow for a more precise “genome-informed” approach (4), aimed at targeting resistant CNAs, to augment MAP chemotherapy.
In contrast to patients with longer relapsing remitting courses (OSCE1, OSCE4, OSCE5, OSCE6, OSCE9), those with more aggressive disease trajectories (OSCE2, OSCE3, OSCE10) displayed unique clonal dynamics (one dominant copy-number clone in OSCE2, two competing copy-number clones in OSCE3, and a resistant clone in OSCE10 that was not identifiable pretreatment), suggesting the presence of more aggressive features at diagnosis for these patients. For instance, deletion events in the DMD gene were unique to these patients and have been associated with poor outcomes in patients with meningioma (50). ATRX nonsense mutations were also truncal for OSCE3 and OSCE10, which has recently been associated with a more aggressive tumor cell phenotype in osteosarcoma (51) and may have contributed to these patients’ refractory disease courses.
The emergence of subclonal CNAs in primary tumors to fully clonal alterations in metastatic or recurrent samples, as demonstrated in our study in 6 of 7 patients with recurrent/refractory disease and pretreatment samples, has been previously described in a subset of adult cancers where analysis of matched primary and metastasis was performed (45). We found MYC gain/amplification to be enriched in the treatment-resistant clone in 6 of 7 patients with more than one clone. Previous studies have shown that MYC amplification is often enriched in metastatic sites (45) and has been previously associated with poor outcomes and increased cell proliferation in osteosarcoma (52–56); however, a recent study questioned its prognostic significance (57). Our study demonstrated that in patients with localized and metastatic disease at diagnosis, MYC amplification is subclonal in pretreatment samples and emerges after 10 weeks of neoadjuvant chemotherapy, highlighting the importance of sample timing when considering the prognostic value of MYC amplification. Furthermore, as we begin to define molecular risk categories within osteosarcoma, our work demonstrates that profiling of posttreatment primary resection samples may reveal previous subclonal amplifications in driver oncogenes; thus, this time point would be more informative when assessing metastatic potential. While multiregion profiling of posttreatment resection from OSCE10 revealed limited heterogeneity among the different sites, there was one site where the metastatic clone was not present, highlighting the potential risk of failing to profile the metastatic clone with single-sample strategies. When considering future sequencing approaches, pooling DNA/RNA extracts from multiple anatomically distinct tumor regions of the primary tumor could be a cost-effective way to improve DNA yield and variant detection, while providing a more complete picture of intratumoral heterogeneity (58).
Our chromosomal duplication timing analysis revealed that gains for the same patient often clustered around the same time point, regardless of whether WGD was present. These bursts of duplications occurred prior to diagnosis, and there were no comparable bursts of duplications in resection, metastatic, or recurrent samples that would reflect ongoing instability. In a pan-cancer cohort, synchronous bursts of copy-number gain were found to occur in 57% of diploid samples and 78% of WGD samples (59). We found that these clustered duplication events were associated with catastrophic complex genomic rearrangement and amplicon events that occurred before diagnosis, such as chromothripsis and tyfonas. In contrast to a previous multi-region osteosarcoma study (15), we demonstrated that tumor ploidy remained consistent across all samples for each patient, which is likely because our copy-number calls were inferred jointly across all samples for each patient, which can improve ploidy estimation in sample sets with wide ranges of purity (32).
These findings of prediagnostic duplication bursts followed by relative genomic stability, combined with our results demonstrating that recurrent osteosarcoma is characterized by the emergence of a preexisting, chemotherapy-resistant subclones, help harmonize a number of seemingly disparate reports within the osteosarcoma literature. Our work suggests that the high degree of SCNAs in osteosarcoma likely stems from an early catastrophic event, yielding a pool of competitive subclones with a shared phylogeny, each originating from a distinct progenitor cell. Over time, those clones with competitive advantages emerge and remain stable, accounting for the observed genomic stability within this complex landscape (22). We propose that a driver CNA detected at relapse that was not apparent at diagnosis is likely due to the rise of a chemoresistant subclone beyond the threshold of bioinformatic detection, rather than the result of ongoing chromosomal instability. Essentially, an amplification like MYC, present in a minority of tumor cells at diagnosis, could be either missed or detected depending on various preanalytical/computational factors. Its apparent emergence at recurrence could be misconstrued as a product of ongoing chromosomal instability. However, if detected at diagnosis and persisting at relapse without the development of any other driver CNA, it would convey a picture of relative genomic stability. Our study uniquely reconciles these viewpoints by tracking the proportion of tumor cells affected by each copy-number clone from diagnosis through recurrence, thereby providing a fresh perspective on the clonal dynamics that underpin SCNAs. However, given the observed stability of SCNA profiles over therapeutic time in patients with multiple recurrences in our study, the potential contribution of epigenetic mechanisms in osteosarcoma progression cannot be disregarded, thus highlighting a promising area for future research.
WGD was confirmed in a subset of patients and was found to be a late event in all three cases. Previous pan-cancer studies have found that WGD events are typically early events in a tumor's molecular time history, but are often preceded by TP53 inactivation (45, 59, 60). Late WGD events have been described in a cohort of patients with hepatocellular carcinoma, and they are typically associated with larger tumors, leading to the conclusion that they may be the last step prior to rapid growth and expansion (36). Our data support a macroevolutionary model of evolution in osteosarcoma (61), with a large number of genomic aberrations acquired over a short period of time secondary to chromosomal instability events, followed by clonal selection, as opposed to ongoing evolution.
Large-scale genomic sequencing studies in osteosarcoma have revealed that there is significant intertumoral heterogeneity in osteosarcoma, with shared mutations typically in tumor suppressor genes rather than in targetable oncogenes (5–8). We observed a heterogeneous mix of metastatic and recurrent seeding patterns in our cohort. We observed only one example of monoclonal, monophyletic dissemination in OSCE6, which is typically a result of a treatment-induced bottle-necking effect. There were three cases of polyclonal/monophyletic dissemination, in which multiple clones were present in the metastatic/recurrent samples, but they all shared a common ancestor, and there were four cases of polyclonal polyphyletic dissemination where multiple distinct clones from the primary tumor seeded a metastatic site. In previous studies, a de novo induced murine model of osteosarcoma demonstrated polyclonal seeding of metastases with ongoing parallel evolution (14), while studies using longitudinal and spatially separated samples have yielded mixed results, demonstrating both polyclonal seeding with parallel evolution (13) and monoclonal monophyletic seeding (15) in a majority of the respective cases from each study. These studies were limited by the lack of pretreatment primary tumors; therefore, the analysis relied on comparing metastatic and recurrent samples to posttreated primaries in many cases.
We demonstrate that while cisplatin and HRD-related SBS3 are active mutagenic processes in osteosarcoma, accounting for most new mutations in relapsed disease, we found no new driver SNVs attributable to these signatures that could account for treatment resistance. The HRD-related SBS3 signature was conserved across all samples in our cohort, consistent with a recent pan-pediatric cancer study that found that 18 of 19 (95%) patients with osteosarcoma had mutations attributed to HRD-related SBS3 (62). The prevalence of the HRD-related SBS3 signature in osteosarcoma is comparable with BRCA1-deficient cancers, suggesting that drugs that target homologous recombination deficient cells, such as PARP inhibitors, may have therapeutic value in osteosarcoma, a concept currently being evaluated in a phase II clinical trial (NCT04417062; ref. 63). A commonly cited limitation of using signature-based assays to assess HRD is that they reflect the HRD state prior to sample acquisition and may not reflect the current state, where HRD may have been restored (64). Our study demonstrated that in osteosarcoma, the number of mutations attributed to HRD-related SBS3 increases at each time point in patients with recurrent disease, suggesting that HRD continues to be an active mutagenic process after diagnosis.
The cisplatin signature was present in all relapse samples and metastatic sites that were thought to have developed during upfront therapy. The extent of cisplatin-induced mutagenesis has been previously described in osteosarcoma, where it was found that cisplatin therapy could potentially increase the mutational burden by 2-fold (15). Although we also found that cisplatin therapy led to large increases in mutational burden in recurrent samples, none of these mutations were likely drivers of treatment resistance, which is consistent with previous studies in patients with platinum-resistant ovarian cancer (65) and osteosarcoma (15). Although we cannot account for CNAs or structural variants induced by cisplatin, recent cell line work in cisplatin-exposed esophageal and liver tumors, found few CNAs or structural variants, suggesting that cisplatin does not contribute significantly to genomic instability (66).
Our study, while offering valuable insights into the genetic heterogeneity and evolution of osteosarcoma, does come with several acknowledged limitations. Primarily, the size and heterogeneity of our patient sample, combined with its descriptive nature, pose challenges to the generalizability of our results. This calls for further validation using larger and more diverse patient cohorts. Our selection bias toward poor responders limits the representation of clonal dynamics in patients showing complete responses to neoadjuvant chemotherapy. Thus, future investigations could gain significant insights from examining the clonal architecture of pretreatment samples from patients who respond fully to therapy. The single spatial sampling used for each time point, apart from the primary resection sample for OSCE10, is another limitation, as it may not fully encapsulate the intricate heterogeneity of the tumor. For instance, while tracking a subclone from the relapse sample back to the pretreatment sample is definitive, not finding a subclone in the pretreatment sample does not necessarily mean it was not present in the pretreatment tumor. This potential sampling bias from single-site sequencing underscores the need for employing multiple spatial sampling or more comprehensive tumor sampling in future research to offer a richer understanding of osteosarcoma genomics. We also acknowledge the inherent limitations associated with the determination of clonality using solver-based inferences from bulk sequencing data, such as the HATCHet tool used in our study. While we have implemented stringent quality control measures to minimize potential artifacts, it remains a possibility that some of the observations could stem from such artifacts. Finally, it is worth mentioning that our study's view on using CNAs to overcome treatment resistance might oversimplify the issue. Osteosarcoma's genetic variability, complexity of mutational patterns, frequent gene amplifications, and the existence of large copy-number gains harboring multiple potential targets all contribute to the challenge of pinpointing specific genetic targets for therapy. Given these complexities, results from preclinical studies targeting CNAs have varied (4, 67). Future studies should take the clonality of CNAs under consideration and prioritize models that were derived from chemoresistant samples when assessing therapies targeting these alterations.
Our findings highlight that the chemoresistant population of tumor cells in osteosarcoma is subclonal at diagnosis and is characterized by unique oncogenic amplifications. As our ability to target these oncogenic amplifications improves, future studies aimed at identifying these oncogenic drivers during upfront therapy may be an effective strategy to eliminate chemoresistant tumor cells and improve survival.
Supplementary Material
Supplementary Figures
Acknowledgments
We would like to extend our deepest gratitude to the organizations that provided invaluable support for our research. We are immensely grateful for the funding received from the NIH and Memorial Sloan Kettering Cancer Center through the K12 Paul Calabresi Career Development Award for Clinical Oncology. Our research also greatly benefited from the Rally Foundation, which generously provided a Postdoctoral and Clinical Research Fellow Grant and a Young Investigator Award. We further acknowledge the vital support received from the Conquer Cancer Foundation of the American Society of Clinical Oncology with support from the QuadW Foundation through their Young Investigator Award. We would like to thank Hyundai Hope on Wheels for their support via their Young Investigator Award. Lastly, we would like to acknowledge the Olayan Fund for Precision Pediatric Cancer Medicine and the Gold Ribbon Riders, whose support helped underwrite the cost of sequencing and the ISABL platform. The generous contributions from these institutions have been instrumental in facilitating the research presented in this manuscript. The opportunity to conduct this study would not have been possible without their unwavering commitment to advancing the field of pediatric oncology.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclaimer
The Editor-in-Chief of Cancer Research is an author on this article. In keeping with AACR editorial policy, a senior member of the Cancer Research editorial team managed the consideration process for this submission and independently rendered the final decision concerning acceptability.
Authors' Disclosures
M.D. Kinnaman is currently employed by Regeneron Pharmaceuticals and receives equity interest in the company; however, this work was completed prior to the author starting at Regeneron. M.F. Levine reports personal fees from Isabl Inc. outside the submitted work; in addition, M.F. Levine has a patent for managing and accessing experiment data using referential indentifiers pending and a patent for systems and methods for cancer whole-genome and transcriptome sequencing pending. G. Gundem reports other support from ISABL Technologies outside the submitted work. M. Dunigan reports grants from the U.S. government during the conduct of the study. L.H. Wexler reports personal fees from Treeline Biosciences outside the submitted work. J.L. Glade Bender reports grants from NIH during the conduct of the study. W.D. Tap reports personal fees from Eli Lilly, EMD Serono, Mundipharma, C4 Therapeutics, Daiichi Sankyo, Deciphera, Adcendo, Ayala, Kowa, Servier, Bayer, Epizyme, Cogent, Medpacto, Foghorn Therapeutics, Amgen, AmMax Bio, Boehringer Ingelheim, BioAtla, and Inhibrx outside the submitted work; in addition, W.D. Tap has a patent for Companion Diagnostic for CDK4 inhibitors - 14/854,329 pending to MSK/SKI and a patent for Enigma and CDH18 as companion Diagnostics for CDK4 inhibition - SKI2016-021-03 pending to MSK/SKI; in addition, W.D. Tap is a scientific advisory board member of Certis Oncology Solutions, is cofounder of and owns stock in Atropos Therapeutics, and is a scientific advisory board member of Innova Therapeutics. E. Papaemmanuil reports nonfinancial support and other support from Isabl Inc. outside the submitted work; in addition, E. Papaemmanuil has a patent for US20230121103A1 pending. A.L. Kung reports other support from Isabl during the conduct of the study and personal fees from Emendo, Karyopharm, DarwinHealth, and Imago outside the submitted work; in addition, A.L. Kung has a patent for Application Number 17/857,821 pending to Isabl. No disclosures were reported by the other authors.
Authors' Contributions
M.D. Kinnaman: Conceptualization, resources, data curation, software, formal analysis, funding acquisition, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing. S. Zaccaria: Resources, software, formal analysis, supervision, validation, investigation, writing–original draft, writing–review and editing. A. Makohon-Moore: Software, supervision, methodology, project administration, writing–review and editing. B. Arnold: Software, formal analysis, visualization. M.F. Levine: Resources, data curation, software, formal analysis, visualization, methodology. G. Gundem: Resources, software, formal analysis, supervision, visualization, methodology, writing–review and editing. J.E. Arango Ossa: Resources, data curation, software, formal analysis, investigation, visualization, methodology. D. Glodzik: Resources, data curation, software, formal analysis, supervision, investigation, visualization, writing–review and editing. M.I. Rodríguez-Sánchez: Resources, data curation, project administration. N. Bouvier: Resources, data curation, funding acquisition, project administration. S. Li: Resources, data curation, project administration. E. Stockfisch: Resources, data curation, project administration. M. Dunigan: Resources, data curation, project administration. C.C. Cobbs: Resources, supervision, methodology, project administration, writing–review and editing. U.K. Bhanot: Resources, formal analysis, investigation, project administration. D. You: Resources, funding acquisition, project administration. K. Mullen: Software, formal analysis, investigation, visualization, methodology. J.P. Melchor: Conceptualization, resources, funding acquisition, project administration. M.V. Ortiz: Conceptualization, supervision, investigation, methodology, writing–review and editing. T.J. O'Donohue: Investigation, writing–review and editing. E.K. Slotkin: Conceptualization, resources, investigation, writing–review and editing. L.H. Wexler: Conceptualization, investigation, writing–review and editing. F.S. Dela Cruz: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, investigation, methodology, project administration, writing–review and editing. M.R. Hameed: Resources, formal analysis, supervision, investigation, methodology, writing–review and editing. J.L. Glade Bender: Resources, formal analysis, supervision, investigation, writing–review and editing. W.D. Tap: Conceptualization, resources, supervision, writing–review and editing. P.A. Meyers: Conceptualization, resources, data curation, supervision, funding acquisition, writing–review and editing. E. Papaemmanuil: Conceptualization, resources, data curation, software, formal analysis, supervision, investigation, methodology, writing–review and editing. A.L. Kung: Resources, supervision, funding acquisition, investigation, methodology, project administration, writing–review and editing. C.A. Iacobuzio-Donahue: Conceptualization, resources, data curation, software, formal analysis, supervision, funding acquisition, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Kager L, Zoubek A, Pötschger U, Kastner U, Flege S, Kempf-Bielack B, et al. Primary metastatic osteosarcoma: presentation and outcome of patients treated on neoadjuvant cooperative osteosarcoma study group protocols. J Clin Oncol 2003;21:2011–8. [DOI] [PubMed] [Google Scholar]
- 2. Kim MS, Cho WH, Song WS, Lee SY, Jeon DG. Time dependency of prognostic factors in patients with stage II osteosarcomas. Clin Orthop Relat Res 2007;463:157–65. [DOI] [PubMed] [Google Scholar]
- 3. Bielack SS, Kempf-Bielack B, Delling G, Exner GU, Flege S, Helmke K, et al. Prognostic factors in high-grade osteosarcoma of the extremities or trunk: an analysis of 1,702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols. J Clin Oncol 2002;20:776–90. [DOI] [PubMed] [Google Scholar]
- 4. Sayles LC, Breese MR, Koehne AL, Leung SG, Lee AG, Liu H-Y, et al. Genome-informed targeted therapy for osteosarcoma. Cancer Discov 2019;9:46–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Perry JA, Kiezun A, Tonzi P, Van Allen EM, Carter SL, Baca SC, et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci U S A 2014;111:E5564–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Suehara Y, Alex D, Bowman A, Middha S, Zehir A, Chakravarty D, et al. Clinical genomic sequencing of pediatric and adult osteosarcoma reveals distinct molecular subsets with potentially targetable alterations. Clin Cancer Res 2019;25:6346–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Behjati S, Tarpey PS, Haase K, Ye H, Young MD, Alexandrov LB, et al. Recurrent mutation of IGF signalling genes and distinct patterns of genomic rearrangement in osteosarcoma. Nat Commun 2017;8:15936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E, et al. Cell reports report recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep 2014;7:104–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kovac M, Blattmann C, Ribi S, Smida J, Mueller NS, Engert F, et al. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat Commun 2015;6:8940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science 2013;339:1546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meltzer PS, Helman LJ. New horizons in the treatment of osteosarcoma. N Engl J Med 2021;385:2066–76. [DOI] [PubMed] [Google Scholar]
- 12. Persha HE, Kato S, De P, Adashek JJ, Sicklick JK, Subbiah V, et al. Osteosarcoma with cell-cycle and fibroblast growth factor genomic alterations: case report of molecular tumor board combination strategy resulting in long-term exceptional response. J Hematol Oncol 2022;15:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu H, Zhu X, Bao H, Wh Shek T, Huang Z, Wang Y, et al. Genetic and clonal dissection of osteosarcoma progression and lung metastasis. Int J Cancer 2018;143:1134–42. [DOI] [PubMed] [Google Scholar]
- 14. Gambera S, Abarrategi A, González-Camacho F, Morales-Molina Á, Roma J, Alfranca A, et al. Clonal dynamics in osteosarcoma defined by RGB marking. Nat Commun 2018;9:3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brady SW, Ma X, Bahrami A, Satas G, Wu G, Newman S, et al. The clonal evolution of metastatic osteosarcoma as shaped by cisplatin treatment. Mol Cancer Res 2019;17:895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990;61:759–67. [DOI] [PubMed] [Google Scholar]
- 17. Höglund M, Gisselsson D, Mandahl N, Johansson B, Mertens F, Mitelman F, et al. Multivariate analyses of genomic imbalances in solid tumors reveal distinct and converging pathways of karyotypic evolution. Genes Chromosomes Cancer 2001;31:156–71. [DOI] [PubMed] [Google Scholar]
- 18. Gao R, Davis A, McDonald TO, Sei E, Shi X, Wang Y, et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat Genet 2016;48:1119–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Umbreit NT, Zhang CZ, Lynch LD, Blaine LJ, Cheng AM, Tourdot R, et al. Mechanisms generating cancer genome complexity from a single cell division error. Science 2020;368:eaba0712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kovac M, Ameline B, Ribi S, Kovacova M, Cross W, Barenboim M, et al. The early evolutionary landscape of osteosarcoma provides clues for targeted treatment strategies. J Pathol 2021;254:556–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Negri GL, Grande BM, Delaidelli A, El‐Naggar A, Cochrane D, Lau CC, et al. Integrative genomic analysis of matched primary and metastatic pediatric osteosarcoma. J Pathol 2019;249:319–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rajan S, Zaccaria S, Cannon MV, Cam M, Gross AC, Raphael BJ, et al. Structurally complex osteosarcoma genomes exhibit limited heterogeneity within individual tumors and across evolutionary time. Cancer Res Commun 2023;3:564–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Medina-Martínez JS, Arango-Ossa JE, Levine MF, Zhou Y, Gundem G, Kung AL, et al. Isabl Platform, a digital biobank for processing multimodal patient data. BMC Bioinformatics 2020;21:549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shukla N, Levine MF, Gundem G, Domenico D, Spitzer B, Bouvier N, et al. Feasibility of whole genome and transcriptome profiling in pediatric and young adult cancers. Nat Commun 2022;13:2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Källberg M, et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods 2018;15:591–4. [DOI] [PubMed] [Google Scholar]
- 27. Jones D, Raine KM, Davies H, Tarpey PS, Butler AP, Teague JW, et al. cgpCaVEManWrapper: simple execution of CaVEMan in order to detect somatic single nucleotide variants in NGS data. Curr Protoc Bioinformatics 2016;56:15.10.1–15.10.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Robbe P, Popitsch N, Knight SJL, Antoniou P, Becq J, He M, et al. Clinical whole-genome sequencing from routine formalin-fixed, paraffin-embedded specimens: pilot study for the 100,000 Genomes Project. Genet Med 2018;20:1196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reiter JG, Baretti M, Gerold JM, Makohon-Moore AP, Daud A, Iacobuzio-Donahue CA, et al. An analysis of genetic heterogeneity in untreated cancers. Nat Rev Cancer 2019;19:639–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gröbner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature 2018;555:321–7. [DOI] [PubMed] [Google Scholar]
- 31. Reiter JG, Makohon-Moore AP, Gerold JM, Bozic I, Chatterjee K, Iacobuzio-Donahue CA, et al. Reconstructing metastatic seeding patterns of human cancers. Nat Commun 2017;8:14114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zaccaria S, Raphael BJ. Accurate quantification of copy-number aberrations and whole-genome duplications in multi-sample tumor sequencing data. Nat Commun 2020;11:4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Satas G, Zaccaria S, El-Kebir M, Raphael BJ. DeCiFering the elusive cancer cell fraction in tumor heterogeneity and evolution. Cell Syst 2021;12:1004–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Myers MA, Satas G, Raphael BJ. CALDER: inferring phylogenetic trees from longitudinal tumor samples. Cell Syst 2019;8:514–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shinde J, Bayard Q, Imbeaud S, Hirsch TZ, Liu F, Renault V, et al. Palimpsest: an R package for studying mutational and structural variant signatures along clonal evolution in cancer. Bioinformatics 2018;34:3380–1. [DOI] [PubMed] [Google Scholar]
- 36. Letouzé E, Shinde J, Renault V, Couchy G, Blanc J-F, Tubacher E, et al. Mutational signatures reveal the dynamic interplay of risk factors and cellular processes during liver tumorigenesis. Nat Commun 2017;8:1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shah RH. iAnnotateSV: Annotation of structural variants detected from NGS; 2021. Available from:https://github.com/rhshah/iAnnotateSV.
- 38. Lopez G, Egolf LE, Giorgi FM, Diskin SJ, Margolin AA. Svpluscnv: analysis and visualization of complex structural variation data. Bioinformatics 2021;37:1912–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cortés-Ciriano I, Lee JJ-K, Xi R, Jain D, Jung YL, Yang L, et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat Genet 2020;52:331–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hadi K, Yao X, Behr JM, Deshpande A, Xanthopoulakis C, Tian H, et al. Distinct classes of complex structural variation uncovered across thousands of cancer genome graphs. Cell 2020;183:197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Crowdis J, He MX, Reardon B, van Allen EM. CoMut: visualizing integrated molecular information with comutation plots. Bioinformatics 2020;36:4348–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Smith MA, Nielsen CB, Chan FC, McPherson A, Roth A, Farahani H, et al. E-scape: interactive visualization of single-cell phylogenetics and cancer evolution. Nat Methods 2017;14:549–50. [DOI] [PubMed] [Google Scholar]
- 43. Goswami RS, Luthra R, Singh RR, Patel KP, Routbort MJ, Aldape KD, et al. Identification of factors affecting the success of next-generation sequencing testing in solid tumors. Am J Clin Pathol 2016;145:222–37. [DOI] [PubMed] [Google Scholar]
- 44. Shao YW, Wood GA, Lu J, Tang Q-L, Liu J, Molyneux S, et al. Cross-species genomics identifies DLG2 as a tumor suppressor in osteosarcoma. Oncogene 2019;38:291–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Watkins TBK, Lim EL, Petkovic M, Elizalde S, Birkbak NJ, Wilson GA, et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature 2020;587:126–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Németh E, Krzystanek M, Reiniger L, Ribli D, Pipek O, Sztupinszki Z, et al. The genomic imprint of cancer therapies helps timing the formation of metastases. Int J Cancer 2019;145:694–704. [DOI] [PubMed] [Google Scholar]
- 47. McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 2017;168:613–28. [DOI] [PubMed] [Google Scholar]
- 48. Smith JC, Sheltzer JM. Systematic identification of mutations and copy number alterations associated with cancer patient prognosis. eLife 2018;7:e39217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Reed DR, Metts J, Pressley M, Fridley BL, Hayashi M, Isakoff MS, et al. An evolutionary framework for treating pediatric sarcomas. Cancer 2020;126:2577–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Juratli TA, McCabe D, Nayyar N, Williams EA, Silverman IM, Tummala SS, et al. DMD genomic deletions characterize a subset of progressive/higher-grade meningiomas with poor outcome. Acta Neuropathol 2018;136:779–92. [DOI] [PubMed] [Google Scholar]
- 51. DeWitt SB, Plumlee SH, Brighton HE, Sivaraj D, Martz EJ, Zand M, et al. Loss of ATRX promotes aggressive features of osteosarcoma with increased NF-κB signaling and integrin binding. JCI Insight 2022;7:e151583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ueda T, Healey JH, Huvos AG, Ladanyi M. Amplification of the MYC gene in osteosarcoma secondary to paget's disease of bone. Sarcoma 1997;1:131–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shimizu T, Ishikawa T, Sugihara E, Kuninaka S, Miyamoto T, Mabuchi Y, et al. c-MYC overexpression with loss of Ink4a/Arf transforms bone marrow stromal cells into osteosarcoma accompanied by loss of adipogenesis. Oncogene 2010;29:5687–99. [DOI] [PubMed] [Google Scholar]
- 54. Scionti I, Michelacci F, Pasello M, Hattinger CM, Alberghini M, Manara MC, et al. Clinical impact of the methotrexate resistance-associated genes C-MYC and dihydrofolate reductase (DHFR) in high-grade osteosarcoma. Ann Oncol 2008;19:1500–8. [DOI] [PubMed] [Google Scholar]
- 55. Gamberi G, Benassi MS, Bohling T, Ragazzini P, Molendini L, Sollazzo MR, et al. C-myc and c-fos in human osteosarcoma: prognostic value of mRNA and protein expression. Oncology 1998;55:556–63. [DOI] [PubMed] [Google Scholar]
- 56. Ladanyi M, Park CK, Lewis R, Jhanwar SC, Healey JH, Huvos AG. Sporadic amplification of the MYC gene in human osteosarcomas. Diagn Mol Pathol 1993;2:163–7. [PubMed] [Google Scholar]
- 57. Taylor AM, Sun JM, Yu A, Voicu H, Shen J, Barkauskas DA, et al. Integrated DNA copy number and expression profiling identifies IGF1R as a prognostic biomarker in pediatric osteosarcoma. Int J Mol Sci 2022;23:8036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Litchfield K, Stanislaw S, Spain L, Gallegos LL, Rowan A, Schnidrig D, et al. Representative sequencing: unbiased sampling of solid tumor tissue. Cell Rep 2020;31:107550. [DOI] [PubMed] [Google Scholar]
- 59. Gerstung M, Jolly C, Leshchiner I, Dentro SC, Gonzalez S, Rosebrock D, et al. The evolutionary history of 2,658 cancers. Nature 2020;578:122–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bielski CM, Zehir A, Penson AV, Donoghue MTA, Chatila W, Armenia J, et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat Genet 2018;50:1189–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vendramin R, Litchfield K, Swanton C. Cancer evolution: Darwin and beyond. EMBO J 2021;40:e108389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ma X, Liu Y, Liu Y, Alexandrov LB, Edmonson MN, Gawad C, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018;555:371–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Forrest SJ, Kinnaman MD, Livingston JA, Vo KT, Merriam P, Clinton C, et al. Phase II trial of olaparib in combination with ceralasertib in patients with recurrent osteosarcoma. J Clin Oncol 39: 15s, 2021. (suppl; abstr TPS11575). [Google Scholar]
- 64. Miller RE, Elyashiv O, El-Shakankery KH, Ledermann JA. Ovarian cancer therapy: homologous recombination deficiency as a predictive biomarker of response to PARP inhibitors. Onco Targets Ther 2022;15:1105–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Patch A-M, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015;521:489–94. [DOI] [PubMed] [Google Scholar]
- 66. Boot A, Huang MN, Ng AWT, Ho S-C, Lim JQ, Kawakami Y, et al. In-depth characterization of the cisplatin mutational signature in human cell lines and in esophageal and liver tumors. Genome Res 2018;28:654–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gill J, Gorlick R. Advancing therapy for osteosarcoma. Nat Rev Clin Oncol 2021;18:609–24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
Data Availability Statement
WGS generated in this study has been deposited at the European Genomephenome Archive (EGA) under study ID EGAS00001007486 (https://ega-archive.org). All other raw data are available upon request from the corresponding author.