

Key Points
-
•
The antitumorigenic activity of eftoza is enhanced in combination with venetoclax in preclinical models of AML.
-
•
Eftoza-venetoclax combination is well tolerated in patients with relapsed/refractory AML and is associated with antileukemic responses.
Visual Abstract
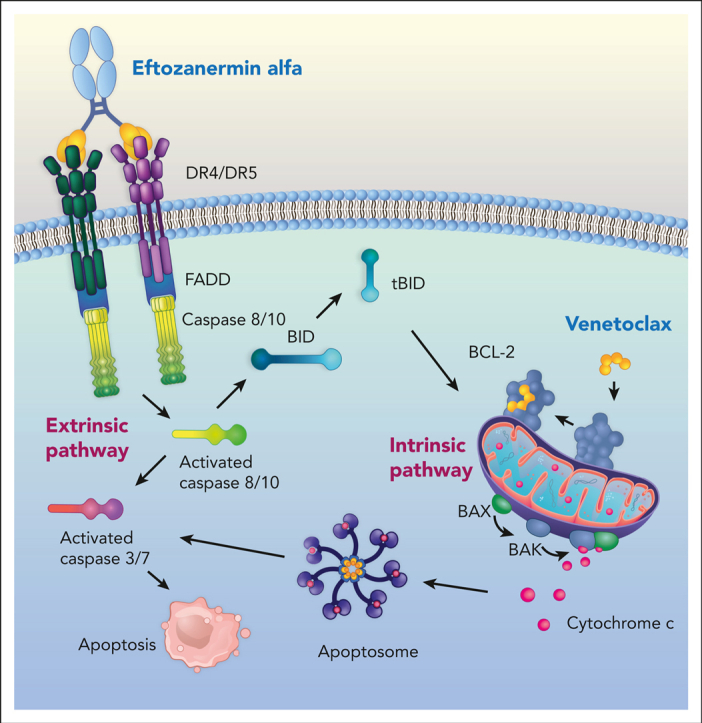
Abstract
Activation of apoptosis in malignant cells is an established strategy for controlling cancer and is potentially curative. To assess the impact of concurrently inducing the extrinsic and intrinsic apoptosis-signaling pathways in acute myeloid leukemia (AML), we evaluated activity of the TRAIL receptor agonistic fusion protein eftozanermin alfa (eftoza; ABBV-621) in combination with the B-cell lymphoma protein-2 selective inhibitor venetoclax in preclinical models and human patients. Simultaneously stimulating intrinsic and extrinsic apoptosis-signaling pathways with venetoclax and eftoza, respectively, enhanced their activities in AML cell lines and patient-derived ex vivo/in vivo models. Eftoza activity alone or plus venetoclax required death receptor 4/5 (DR4/DR5) expression on the plasma membrane but was independent of TP53 or FLT3-ITD status. The safety/tolerability of eftoza as monotherapy and in combination with venetoclax was demonstrated in patients with relapsed/refractory AML in a phase 1 clinical trial. Treatment-related adverse events were reported in 2 of 4 (50%) patients treated with eftoza monotherapy and 18 of 23 (78%) treated with eftoza plus venetoclax. An overall response rate of 30% (7/23; 4 complete responses [CRs], 2 CRs with incomplete hematologic recovery, and 1 morphologic leukemia-free state) was reported in patients who received treatment with eftoza plus venetoclax and 67% (4/6) in patients with myoblasts positive for DR4/DR5 expression; no tumor responses were observed with eftoza monotherapy. These data indicate that combination therapy with eftoza plus venetoclax to simultaneously activate the extrinsic and intrinsic apoptosis-signaling pathways may improve clinical benefit compared with venetoclax monotherapy in relapsed/refractory AML with an acceptable toxicity profile. This trial was registered at www.clinicaltrials.gov as #NCT03082209.
Introduction
Apoptosis is a form of regulated cell death involving distinct biochemical and morphologic changes in response to internal and external stimuli.1 The ability of cancer cells to evade apoptosis is a hallmark of cancer that can lead to tumor progression and drug resistance.1 The 2 molecular apoptotic signaling pathways are the extrinsic or death receptor (DR) signaling pathway and the intrinsic or mitochondrial pathway. Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL), a member of the TNF superfamily of cytokines, preferentially triggers the extrinsic apoptotic pathway by binding as a trimer to 2 closely related cell-surface DRs, TRAIL-R1 (DR4) and TRAIL-R2 (DR5). Cell-surface DR trimerization leads to the formation of a death-inducing signaling complex to recruit and activate caspase-8/10 that, if sufficiently robust, directly activates effector caspase-3 in type I cells to induce apoptosis. In type II cells, in which death-inducing signaling complex–induced caspase-8 activation is insufficient, engagement of the intrinsic/mitochondrial apoptosis-signaling pathway is required to ultimately induce apoptotic cell death.2 First-generation TRAIL receptor agonists were well tolerated in patients with cancer but failed to develop compelling objective responses either as monotherapy or combinations with other therapeutic agents.3 This has been ascribed to biologic properties, including short plasma half-life4,5 and weak higher-order receptor clustering,6,7 which in some cases requires additional Fc-FcγR–mediated crosslinking.8
To overcome the lack of clinical outcome with first-generation TRAIL receptor agonists, eftozanermin alfa (formerly ABBV-621; eftoza) was developed as an agonistic fusion protein consisting of a mutant immunoglobulin G1–Fc linked to 2 single-chain trimers of TRAIL receptor–binding domain monomers, resulting in a total of 6 DR-binding sites per molecule, designed to optimize receptor clustering.9 Eftoza binds selectively to TRAIL receptors with nanomolar affinity to induce optimal receptor clustering in human solid tumor cancer cells that drives on-target apoptosis. This translates into robust antitumorigenic activity in solid tumor models.9 However, the intrinsic apoptosis-signaling pathway that is tightly regulated by the B-cell lymphoma protein-2 (BCL-2) family of proteins is known to play a role in TRAIL receptor agonism resistance. Specifically, overexpression of the BCL-2 family antiapoptotic members can dysregulate the intrinsic apoptotic pathway, promote tumorigenesis, and enable resistance to anticancer therapeutics, including TRAIL.3,9, 10, 11
BCL-2 plays a critical role in maintaining the survival of acute myeloid leukemia (AML) cell lines and primary patient leukemic blasts, progenitor cells, and stem cells.12, 13, 14, 15 Venetoclax is a first-in-class, orally bioavailable BCL-2 selective inhibitor that binds directly to the BCL-2 homology 3–binding groove of BCL-2 to induce caspase-dependent apoptosis16 and demonstrates efficacy in the treatment of patients with chronic lymphocytic leukemia,17 multiple myeloma,18 and AML.19,20 AML is the most common acute leukemia in adults (median age, 67 years21,22) and accounts for the largest number of leukemia-related deaths in the United States (5-year survival rate, 29%19,23). Results from a phase 2 clinical trial of venetoclax in patients with relapsed/refractory (R/R) AML demonstrated an overall response rate (ORR) of 19%.20
Considering that truncated BH3 interacting-domain death agonist, tBID, binds to all antiapoptotic BCL-2 family members because of TRAIL-induced caspase-8 activation,2 we sought to evaluate the extrinsic apoptosis-signaling pathway activity of eftoza in AML initially as a monotherapy, subsequently hypothesizing that simultaneously engaging the intrinsic apoptosis-signaling pathway with venetoclax would lead to more durable antitumorigenic activity than either agent alone. To define the mechanisms of eftoza activity as a monotherapy and in combination with venetoclax, we evaluated preclinical models and analyzed a cohort of patients with R/R AML treated with eftoza alone or in combination with venetoclax within the ambit of a larger phase 1 trial.
Materials and methods
Reagents and cell culture
AML cell lines were purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen (Braunschweig, Germany), American Type Culture Collection (Manassas, VA), Japanese Collection of Research Bioresources Cell Bank (Osaka, Japan), and Coriell Institute for Medical Research (Camden, NJ) (supplemental Table 1, available on the Blood website). All cell lines were cultured in RPMI 1640 (Gibco, Waltham, MA) supplemented with 10% fetal bovine serum (Invitrogen, Waltham, MA), 1% sodium pyruvate, 25 mM 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid, 4.5 g/L glucose, and 1% penicillin/streptomycin (Sigma, Burlington, MA) and maintained in a humidified chamber at 37°C containing 5% CO2. The authenticity of each cell line was verified by short tandem repeat profiling and tested for mycoplasma at the AbbVie Core Cell Line Facility. Eftoza and venetoclax were synthesized by AbbVie Inc (North Chicago, IL) and Apo2L was obtained from PeproTech (Cranbury, NJ). For in vitro studies, venetoclax was dissolved in anhydrous dimethyl sulfoxide as a 10 mM stock and stored at −20 °C. Eftoza was made up in a buffer containing 200 mM L-arginine, 20 mM tris(hydroxymethyl)aminomethane, and 10 mM succinate, at pH 7.2 and stored at −80 °C. For in vivo studies, venetoclax was formulated in 10% ethanol plus 60% Phosal 50 PG (Sigma, MO) plus 30% polyethylene glycol 400. Eftoza was formulated in phosphate-buffered saline.
Generation of engineered cells
Lentiviral transduction of human erythroleukemia (HEL) cells was used to generate cells overexpressing either human DR4 or DR5. CRISPR-Cas9 editing was used to generate BAK1/BAX-deficient OCI-AML5 cells, TP53-deficient MV-4-11 cells, and TP53R248W-overexpressing MV-4-11 cells. Supplemental Methods include additional details.
Detailed methodology on cell viability assays, determination of caspase-3/7 activation, kinetic live-cell imaging of caspase-3/7 activation, and procedures for quantitative flow cytometry can be found in the supplemental Materials and Methods.
Human AML primary samples
Samples from patients with AML were collected after informed consent had been obtained in accordance with the Princess Margaret Cancer Centre University Health Network review committee (Toronto, ON, Canada). Primary AML cells were isolated using Ficoll and treated in culture media containing Iscove's Modified Dulbecco's Medium (Wisent, St. Bruno, QC, Canada) with 2 mM L-glutamine (Gibco), 10% fetal bovine serum (Wisent), 55 μM β-mercaptoethanol (Gibco), 100 μg/mL Primocin (InvivoGen, San Diego, CA) as well as 100 ng/mL stem cell factor, 50 ng/mL FMS-like tyrosine kinase 3 ligand, 40 ng/mL thrombopoietin, 20 ng/mL interleukin-3, 4 ng/mL granulocyte macrophage colony-stimulating factor, and 30 U/mL interleukin-2 (PeproTech). For viability measurements, cells were treated in triplicate for 24 hours, as part of a broad compound screen, before washing and staining with anti-hCD45–BV421 (1:40; clone 2D1, Thermo Fisher Scientific, Waltham, MA) for 20 minutes. The cells were then washed and resuspended in 100 μL Annexin V Buffer (BD Biosciences, Franklin Lakes, NJ) containing Annexin V-FITC (1:50; BD Biosciences) and 7-AAD (1:650; Invitrogen). Flow cytometry was performed using MACSQuant VYB (Miltenyi Biotec, Bergisch Gladbach, Germany) and FlowJo 10 software (BD Biosciences, Ashland, OR) was used to analyze data.
AML tumor cell line, patient-derived, and leukapheresis-based xenograft models
Subcutaneous tumor cell line xenograft studies were conducted in a specific pathogen-free environment in accordance with the internal Institutional Animal Care and Use Committee and accredited by the American Association of Laboratory Animal Science under conditions that meet or exceed standards set by the US Department of Agriculture Animal Welfare Act, Public Health Service policy on humane care and use of animals, and the National Institutes of Health guide on laboratory animal welfare.
Female CB-17 SCID (SKM-1) mice or female SCID-beige (MV-4-11 and OCI-AML2) mice (Charles River Laboratories, Wilmington, MA) were inoculated with either 2 × 106 SKM-1 cells, 5 × 106 MV-4-11 cells, or 1 × 106 OCI-AML2 cells subcutaneously into the right flank. Mice were injected with a 0.1-mL inoculum of 1:1 cell mixture in culture media and adjuvant Matrigel (BD Biosciences, Bedford, MA). When tumors reached ∼200 to 250 mm3, the mice were size-matched into treatment and control groups and treated as indicated. Tumor volume was measured twice per week with electronic calipers and calculated per the formula (L × W2) ÷ 2. All treatment groups consisted of 8 mice per group.
Disseminated xenograft models derived from patients (PDX) with AML were generated in sublethally irradiated NSG mice and treated with human immunoglobulin G1 (5 mg/kg, every other day for 5 doses, intraperitoneally), eftoza (5 mg/kg, every other day × 5, intraperitoneally), venetoclax (50 mg/kg, daily × 21, by mouth), or eftoza and venetoclax in combination at identical monotherapy doses and schedules. The impact on tumor burden was determined in the bone marrow 1 week after end of treatment (n = 5 mice per treatment).
Leukapheresis studies were performed at Champions Oncology Inc (Rockville, MD). NOG-F mice were sublethally irradiated and inoculated with purified cryopreserved primary patient AML isolates obtained via leukapheresis. After tail vein inoculation (up to 2 × 106 cells per mouse), engraftment was monitored in the blood and bone marrow for up to 12 weeks after inoculation. After engraftment was confirmed in surrogate mice (20-200 huCD33+ blasts per μL of peripheral blood), the remaining cohorts were screened and randomized into groups on the basis of blast counts in blood (n = 10) and placed on trial. Mice were treated with eftoza at 3 mg/kg every other day × 5 and venetoclax at 50 mg/kg daily × 21, and the effect on survival (based on animal health observations) was assessed and compared using log-rank statistical analysis.
Statistical analysis
Linear regression and Spearman correlations were used to determine the correlation between 2 variables, the Mantel-Cox test was used to assess significance between Kaplan-Meier survival curves, and either Mann-Whitney U test, Friedman test, or one-way analysis of variance with Tukey's post hoc test was used to determine statistical significance between data sets. Statistical analyses were performed using GraphPad Prism 9.1.0 (GraphPad Software, La Jolla, CA).
Phase 1 trial
A phase 1, first-in-human, open-label, multicenter study was conducted to evaluate eftoza as single agent and in combination with venetoclax in adult patients (≥18 years old) with previously treated solid tumors or hematologic malignancies (#NCT03082209). The study consisted of 2 parts, dose escalation and dose optimization. The dose range of eftoza (1.25-7.5 mg/kg) used in this clinical study was determined from pharmacokinetic (PK)/pharmacodynamic modeling of the response of mice bearing Colo205 tumors treated with single doses of eftoza.9 The results of the dose-escalation portion of the study with eftoza monotherapy in solid tumors have been previously published.24 Patients with an AML diagnosis and with histologically confirmed R/R disease were eligible to enroll in the AML dose-optimization cohort, which we report herein, and received treatment with eftoza as monotherapy or in combination with venetoclax. Key eligibility criteria are in supplemental Table 2. The primary objective of dose optimization was to evaluate the PK profile of eftoza as monotherapy or in combination with venetoclax in patients with R/R AML; secondary objectives were to assess safety and tolerability. Exploratory objectives included evaluation of preliminary antitumor efficacy, and assessment of pharmacodynamic effects and exploratory biomarkers related to drug development.
The study was conducted in accordance with the Declaration of Helsinki and International Conference on Harmonization Good Clinical Practice guidelines. The study protocol was approved by an independent ethics committee/institutional review board. All patients provided written informed consent.
Supplemental Materials and Methods include additional details on treatment, safety, PK, and tumor assessments.
Results
Eftoza single-agent activity in AML cells in vitro and in vivo
The effect of eftoza on cell viability was determined in a panel of 23 AML cell lines after 24 hours of treatment in vitro (Figure 1A). Eftoza was active in AML cell lines, inducing cell death with EC50 values <10 nM in 16 of 23 (69.6%) AML cell lines with superior potency compared with that of Apo2L (supplemental Table 3). Cell death after eftoza treatment was apoptosis-based, because a loss in cell viability corresponded to a dose- and time-dependent increase in caspase-3/7 activity (Figure 1B; supplemental Figure 1A-B), and the effect of eftoza on cell viability was abrogated by the pan-caspase inhibitor z-VAD-FMK (Figure 1C). The eftoza-sensitive cell line SKM-1 (EC50 = 0.004 nM) was injected subcutaneously into SCID mice to determine single-agent activity of eftoza in vivo (Figure 1D). Eftoza induced robust single-agent antitumor activity with complete regression of SKM-1 tumors.
Figure 1.

Characterization of eftoza single-agent activity in human AML cell lines. (A) AML cell lines were treated with eftoza for 24 hours and the impact on cell viability was determined using CellTiter-Glo. Half maximal effective concentration (EC50) values were calculated from the resulting dose-response curves using GraphPad Prism. Data represent the mean ± standard error of the mean (SEM; n = ≥3). The TP53 status of AML cell lines (supplemental Table 3) was obtained from the World Health Organization International Agency for Research on Cancer. Red bars depict cell lines expressing TP53 wild-type (TP53wt); green bars depict cell lines harboring mutations affecting the expression of TP53 (TP53mut). (B) OCI-AML2 and SKM-1 cells were treated with eftoza for 24 hours and the impact on caspase-3/7 activity was determined in relation to cell viability using Caspase-Glo 3/7 and CellTiter-Glo, respectively. Data represent the mean ± SEM (n = 3). (C) Dose-response curves of OCI-AML2 and SKM-1 cell lines treated with eftoza for 24 hours alone or pretreated 1 hour with z-VAD-FMK (zvad). Viability was determined using CellTiter-Glo. Data represent the mean ± SEM (n = 3). (D) Mice bearing SKM-1 tumors were treated with eftoza (1 mg/kg, every other day × 5) administered intraperitoneally, and the effect on tumor volumes was determined. Data represent the mean ± SEM of 8 mice per treatment group. (E-F) DR4 (E) and DR5 (F) plasma membrane receptor number was determined in a panel of AML cell lines via quantitative flow cytometry. Data represent the mean ± SEM (n ≥2). (G,H) The plasma membrane expression of DR4, DR5, and total DRs (DR4 + DR5) was measured via flow cytometry in the panel of AML cell lines and the correlation with eftoza EC50 values from panel A was determined. The statistical significance was determined using Spearman correlation. (I) Parental and +huDR4- and +huDR5-overexpressing HEL cells were treated with eftoza for 24 hours and the impact on cell viability was determined using CellTiter-Glo. Data represent the mean ± standard deviation (n = 3). Numbers in brackets indicate the mean DR4 or DR5 plasma membrane receptor numbers (mean ± SEM; n = 3).
To explore the molecular mechanism of eftoza-induced apoptosis in AML cell lines, DR4 and DR5 plasma membrane receptor numbers were determined via quantitative flow cytometry (Figure 1E-H). The majority of AML cell lines (18/23) expressed higher DR5 numbers on the plasma membrane than DR4 (Figure 1E,F) and are reflective of data in solid tumor cell lines.9 The numbers of both DR4 and DR5 on the plasma membrane correlated well with their corresponding DR4 (TNFRSSF10A) and DR5 (TNFRSF10B) gene expression (supplemental Figure 2). The cellular activity of eftoza highly correlated with DR5 (r = −0.7967; P < .0001) and DR4 plus DR5 (r = −0.8358; P < .0001) receptor number on the cell surface but not DR4 receptor number alone (r = −2646; P = .2225) (Figure 1G,H). TNFRSF10B gene expression (r = −0.7253; P = .004), but not TNFRSF10A gene expression (r = 0.1069; P = .6631), correlated with eftoza activity (supplemental Figure 3). Interestingly, the 7 resistant AML cell lines with EC50 values ≥10 nM also had either lower DR5 or lower DR4 + DR5 numbers on the plasma membrane than the cell lines that responded to eftoza (Figure 1F; supplemental Table 3), suggesting that a minimal number of DR4/DR5 on the plasma membrane is required for eftoza activity. To demonstrate that eftoza activity in AML cells can be mediated by either DR4 or DR5 plasma membrane expression, we ectopically expressed human TNFRSF10A or human TNFRSF10B in HEL cells, which possess low levels of both DR4 and DR5 on the plasma membrane and are inherently resistant to eftoza-induced apoptosis. HEL cells overexpressing human DR4 (+huDR4) or DR5 (+huDR5) on the plasma membrane were more sensitive to eftoza than the parental cells (Figure 1I). HEL +huDR4 cells were less sensitive to eftoza than +huDR5 cells, possibly because of lower overexpression of DR4 on the plasma membrane than DR5.
The expression of DRs is transcriptionally upregulated by p53 via intronic binding sites,25,26 a tumor suppressor mutated in up to 30% of patients with AML27, 28, 29 and associated with poor responses to therapy.30,31 All AML cell lines expressing TP53wt (9/9) were sensitive to eftoza (EC50 < 10 nM) and expressed >1750 total DRs (DR4 plus DR5) on the plasma membrane, an estimated threshold for sensitivity (supplemental Figure 4A-B; supplemental Table 3). Although TP53mut AML cell lines were collectively less responsive to eftoza treatment compared with their TP53wt counterparts (supplemental Figure 4A-B), TP53mut AML cell lines that expressed cell-surface DR levels >1750 (7/14) were equally sensitive (supplemental Figure 4C). These data collectively indicate that TP53-independent regulation of DR4 and DR5 protein expression can enable eftoza sensitivity in patient populations with poor outcomes to existing therapies.
Enhanced activity of eftoza in combination with the BCL-2 selective inhibitor venetoclax in AML cell lines
BCL-2 was found to be consistently expressed at higher levels than related antiapoptotic proteins B-cell lymphoma-extra large (BCL-XL) and myeloid cell leukemia sequence 1 (MCL-1) in AML cell lines (supplemental Table 4). We sought to evaluate the impact of venetoclax treatment on activity of eftoza in the panel of AML cell lines in vitro, using the Bliss independence model32 to determine synergistic cell death. Synergistic cell death (Bliss sum >0) was observed between eftoza and venetoclax in 15 of the 23 AML cell lines tested. (Figure 2A; supplemental Figure 5A-B). The synergy of eftoza with venetoclax was apoptosis-based because caspase-3/7 activation was enhanced in a time- and dose-dependent manner after combined treatment of eftoza with venetoclax, compared with either agent alone (Figure 2B,C), and the synergistic effect of eftoza-venetoclax could be abrogated by deletion of BAK and BAX (Figure 2D,E). The eftoza-venetoclax combination partially overcame some of the resistance imparted by BAX deletion on venetoclax or eftoza single-agent activity in OCI-AML5 cells in vitro (supplemental Figure 5C). In vivo, the combination of eftoza and venetoclax induced robust tumor regression and was superior to either compound treatment alone in the MV-4-11 and OCI-AML2 AML CDX tumor models (Figure 2F,G).
Figure 2.

The activity of eftoza is enhanced in combination with the BCL-2 selective inhibitor venetoclax in AML cell lines. (A) AML cell lines were treated with eftoza and venetoclax in combination for 24 hours and cell viability was determined using CellTiter-Glo. Synergy was assessed using the Bliss independence model,32 in which Bliss sums represent the cumulative Bliss scores across the combination matrix. Data represent the mean ± SEM of n = ≥3 independent experiments. Red bars depict cell lines expressing TP53wt; green bars depict cell lines harboring TP53mut. (B-C) Live-cell quantification of caspase-3/7 activation in OCI-AML2 cell line over a time period was measured by adding caspase-3/7 green dye at the beginning of treatment and monitoring the cells every hour using IncuCyte ZOOM. (B) Time- and dose-dependent change in caspase-3/7–positive cells as a percentage of total cells per well. Data represent mean ± SEM; n = 3. (C) Caspase-3/7 activity as measured using the area under the concentration-time curve (AUC) from panel B. (D) Dose-response curves of OCI-AML5 parental and (E) BAK1–/–/BAX–/– OCI-AML5 cells, treated alone or in combination with eftoza and venetoclax for 24 hours. Viability was determined using CellTiter-Glo (mean ± SEM; n = 3). (F) Tumor volume change in mice bearing MV-4-11 tumor cells treated with eftoza (3 mg/kg, q2d × 3, IP), venetoclax (50 mg/kg, qd × 6, po), or eftoza and venetoclax in combination. Data represent the mean ± SEM of 8 mice per treatment group. (G) Tumor volume change in mice bearing OCI-AML2 tumor cells treated with eftoza (3 mg/kg, q7d × 2, IP), venetoclax (25 mg/kg, qd × 14, po), or eftoza and venetoclax in combination. Data represent the mean ± SEM of 8 mice per treatment group. (H-J) The plasma membrane expression of DR4, DR5, total DRs (DR4 + DR5), and total BCL-2 expression were measured using flow cytometry in the panel of AML cell lines, and the Spearman rank correlation with the Bliss sums from panel A was determined. IP, intraperitoneally; po, by mouth; qd, daily; q2d; every other day; q7d, every 7 days.
The protein and gene expressions of DR4, DR5, and BC-L2 were characterized to gain further insight into the underlying mechanisms responsible for combinatorial activity between eftoza and venetoclax. Synergistic cell death in AML cells positively correlated with DR5, DR4 + DR5 plasma membrane receptor number, and total BCL-2 protein levels, but not DR4 plasma membrane receptor number alone, as measured via flow cytometry (Figure 2H-J). Synergistic cell death in AML cells also positively correlated with TNFRSF10B and BCL2, but not TNFRSF10A, gene expression levels (supplemental Figure 6). Interestingly, synergistic cell death was observed between eftoza and venetoclax in 14 of the 23 AML cell lines where the total DR number on the plasma membrane (DR4 + DR5) was >1750 copies per cell (supplemental Table 3).
Genomic aberrations, such as FLT3-ITD, ASXL1, and TP53 are frequently detected in patients with AML33, 34, 35 and associated with less durable responses to venetoclax plus hypomethylating agent–based regimens.20,31,36,37 Synergistic cell death between eftoza and venetoclax was observed in all 4 cell lines harboring ASXL1 mutations (known to confer sensitivity to azacitidine-venetoclax; OCI-AML5, SKM-1, PL-21, and Nomo-1) and all 3 cell lines harboring the FLT3-ITD mutation (MV-4-11, PL-21, and MOLM-13) (supplemental Table 3). Furthermore, in TP53mut AML cell lines in which total DR number was not limiting (>1750 receptors per cell), eftoza-venetoclax synergy was equivalent to that observed in TP53wt AML cells (supplemental Figure 4D-E). The impact of TP53 status was further investigated in engineered MV-4-11 cells (TP53–/– and TP53R248W) compared with the parental cells (TP53wt). Despite a decrease in single-agent activity of both venetoclax and eftoza in the TP53R248W or TP53–/– cells compared with parental TP53wt cells (supplemental Figures 5A and 7A-B), the combinatorial activity was equivalent, independent of TP53 status (supplemental Figure 7C,D).
Combination of eftoza and venetoclax reduces tumor burden and improves survival in patient-derived ex vivo and in vivo models of AML
To further investigate the potential consequence of BCL-2 inhibition on the activity of eftoza in AML, we evaluated the effects of eftoza, venetoclax, or eftoza-venetoclax combination treatment on the viability of ex vivo CD45+ cells from 45 naive patients and 19 patients with R/R AML. Although DR4/DR5 expression was not determined, the eftoza-venetoclax combination significantly enhanced cell death in ex vivo samples from both naive patients and patients with R/R AML when compared with either treatment alone. The median response to eftoza treatment was superior to venetoclax alone compared with the eftoza-venetoclax combination, although this did not meet statistical significance in the samples of patients who were R/R. Importantly, subgroups of patients with AML, who were either R/R to venetoclax (or samples collected while receiving venetoclax-based therapies) or harbored TP53 mutations, were highly sensitive to combination treatment compared with either agent alone (Figure 3A). Subsequently, 4 xenograft models derived from patients with AML who were positive for DR5 plasma membrane expression were treated with either eftoza or venetoclax alone or the eftoza-venetoclax combination, and the effects on tumor burden in the bone marrow were evaluated (Figure 3B). These patient-derived models captured several different genomic aberrations, including FLT3-ITD (AML-23 and AML-31) or TP53 mutations (AML-55) (supplemental Table 5). In xenograft models AML-23, AML-31, and AML-40 derived from patients with AML, single-agent eftoza or venetoclax treatment each had varying and limited effects on reducing tumor burden compared with combination treatment (Figure 3B). In xenograft model AML-55 derived from patients with AML (TP53P222L/R248Q mutations), eftoza monotherapy significantly reduced tumor burden (P < .0001) within the bone marrow in the absence of any venetoclax single-agent activity. The addition of venetoclax to eftoza further reduced evidence of disease in AML-55. Similarly, the combination of eftoza and venetoclax extended the loss in the median tumor burden compared with that of either agent alone in 3 additional AML PDX models, with statistical significance observed over venetoclax alone in AML-40 (P < .05) (Figure 3B). Notably, the percentage of DR5+ cells was highest in AML-55 compared with the other 3 xenograft models derived from patients with AML (Figure 3B).
Figure 3.

Eftoza in combination with venetoclax reduces tumor burden and improves survival in patient-derived preclinical models of AML. (A) CD45+ cells from patients with naive (n = 45) and R/R (n = 19) primary AML were treated ex vivo with either eftoza (5 nM), venetoclax (1 μM; VEN), or eftoza and venetoclax in combination for 24 hours, and the impact on viability determined from the Annexin V/-7AAD–positive population via flow cytometry compared with the dimethyl sulfoxide-treated control. The resulting data were further segregated to assess treatment responses in samples from patients with AML who were R/R to venetoclax (or collected while on venetoclax-based therapies; n = 7) or harbored TP53 mutations (n = 15). Statistical difference was determined using a Friedman test, in which ∗∗P < .01 and ∗∗∗∗P < .0001 were considered significant. (B) Disseminated AML PDX models were treated with huIgG1 (5 mg/kg, q2d × 5, IP), eftoza (5 mg/kg, q2d × 5, IP), venetoclax (50 mg/kg, qd × 21, po), or eftoza and venetoclax in combination at identical monotherapy doses and schedules, and the impact on tumor burden was determined in the bone marrow a week after the end of treatment (n = 5 mice per treatment). Median response is shown for each treatment (red bar). Flow cytometry histograms depict the percentage of DR4 (red) and DR5 (green) plasma membrane expression of huIgG1-treated AML cells harvested from the bone marrow of inoculated mice at the end of study. Gray represents the isotype negative control. Statistical difference was determined using a one-way analysis of variance with Tukey's post hoc test, for which ∗P < .05, ∗∗P < .01, and ∗∗∗∗P < .0001 were considered significant. (C) Patient-derived AML peripheral blood mononuclear cells were isolated by leukapheresis and injected into whole-body irradiated NOG-F mice. In 8 to 12 weeks, bone marrow engraftment was confirmed, and the mice were treated with vehicle, eftoza (3 mg/kg, q2d × 5, IP), venetoclax (50 mg/kg, qd × 21, po), or eftoza and venetoclax in combination, and the impact on survival (n = 8 mice per group) either after 20- or 28-day treatment was assessed. Statistical difference was determined using the Mantel-Cox test (supplemental Table 6). huIgG1, human immunoglobulin G1; ns, not significant; po, by mouth; qd, daily; q2d, every other day.
Extending these studies to understand the effect of eftoza-venetoclax combination treatment on survival, we leveraged 3 human AML leukapheresis models. In CTG-2229, eftoza treatment alone extended animal survival when compared with vehicle or venetoclax treatment alone, with no added benefit observed from adding venetoclax with eftoza. In the CTG-2226 AML leukapheresis model, venetoclax alone had an overall survival benefit when compared with vehicle treatment, with added benefit observed from addition of eftoza. In the CTG-2238 AML leukapheresis model, in which neither eftoza nor venetoclax monotherapy had an impact on the overall survival, the combination treatment extended the overall survival when compared with vehicle treatment (Figure 3C; supplemental Table 6).
Phase 1 clinical trial of eftoza in patients with AML
To translate aforementioned preclinical findings into the clinical setting, we initiated a first-in-human clinical trial in patients with R/R AML (#NCT03082209). An eftoza dose range from 1.25 to 7.5 mg/kg was used in this clinical study and determined using PK/pharmacodynamic modeling derived from the response of mice bearing Colo205 tumors treated with a single dose of eftoza.9 As of 30 March 2020, a total of 27 patients with R/R AML were enrolled into a dose-optimization cohort and treated with eftoza as monotherapy (n = 4) or in combination with venetoclax (n = 23). Demographics, clinical characteristics, and disposition of patients are summarized in Table 1.
Table 1.
Patient demographics, clinical characteristics, and disposition
| Characteristic | Monotherapy |
Combination |
|||||
|---|---|---|---|---|---|---|---|
| 1.25 mg/kg (n = 1) | 3.75 mg/kg (n = 1) | 7.5 mg/kg (n = 2) | Total (N = 4) | 3.75 mg/kg (n = 10) | 7.5 mg/kg (n = 13) | Total (N = 23) | |
| Age, years, median (range) | 82 (82-82) | 71 (71-71) | 75 (71-78) | 75 (71-78) | 72 (60-79) | 71 (35-82) | 71 (35-82) |
| Sex, n (%) | |||||||
| Female | 1 (100) | 1 (100) | 1 (50) | 3 (75) | 2 (20) | 7 (54) | 9 (39) |
| Male | 0 | 0 | 1 (50) | 1 (25) | 8 (80) | 6 (46) | 14 (61) |
| Time from diagnosis, years, median (range) | 1.4 (1.4-1.4) | 0.3 (0.3-0.3) | 1.1 (0.8-1.3) | 1.1 (0.3-1.4) | 1.1 (0.2-2.7) | 0.8 (0.1-4.6) | 1 (0.1-4.6) |
| Previous therapies, n (%) | |||||||
| 0 | 0 | 0 | 0 | 0 | 1 (10) | 1 (8) | 2 (9) |
| 1 | 0 | 0 | 1 (50) | 1 (25) | 4 (40) | 6 (46) | 10 (43) |
| 2 | 0 | 1 (100) | 0 | 1 (25) | 5 (50) | 2 (15) | 7 (30) |
| 3 | 0 | 0 | 1 (50) | 1 (25) | 0 | 0 | 0 |
| 4 | 1 (100) | 0 | 0 | 1 (25) | 0 | 0 | 0 |
| ≥5 | 0 | 0 | 0 | 0 | 0 | 4 (31) | 4 (17) |
| Study drug discontinuation, n | 1 | 1 | 2 | 4 | 9 | 12 | 21 |
| Primary reason, n (%) | |||||||
| Adverse event | 0 | 0 | 0 | 0 | 2 (22) | 2 (17) | 4 (19) |
| Withdrawal of consent | 0 | 0 | 0 | 0 | 1 (11) | 1 (8) | 2 (9) |
| Progressive disease | 1 (100) | 0 | 0 | 1 (25) | 5 (56) | 6 (50) | 11 (52) |
| Physician decision | 0 | 1 (100) | 1 (50) | 2 (50) | 0 | 1 (8) | 1 (5) |
| Other | 0 | 0 | 1 (50) | 1 (25) | 1 (11) | 2 (17) | 3 (14) |
All-grade treatment-related adverse events (TRAEs) reported in monotherapy cohorts (50%; 2/4) were stomatitis and infusion-related reaction (n = 1; 1.25 mg/kg) as well as neutropenia and hypophosphatemia (n = 1; 3.75 mg/kg); grade ≥3 TRAE of neutropenia was reported by 1 patient. The most common all-grade TRAEs for patients in combination cohorts are summarized in Table 2 and include increased alanine aminotransferase and aspartate aminotransferase (AST; n = 8; 35% each); grade ≥3 TRAEs were increased alanine aminotransferase (n = 5; 22%) and increased AST (n = 4; 17%). In the good laboratory practice repeat-dose toxicology study using nonhuman primates treated with weekly intravenous infusions of eftoza up to 100 mg/kg per week for 4 consecutive weeks, no increase in serum liver enzymes or hepatocellular apoptosis were observed.9 One patient in the 7.5 mg/kg combination cohort experienced an on-treatment death from pneumonia that was also confounded by the progression of underlying AML. Eftoza demonstrated approximately dose-proportional increase in exposure for the dose range from 1.25 to 7.5 mg/kg administered weekly; findings were similar when administered with and without venetoclax. The harmonic mean terminal elimination half-life of eftoza was ∼29 to 34 hours (Table 3). The maximum plasma concentrations achieved with the 7.5 mg/kg dose of eftoza were ∼103 μg/mL and 134 μg/mL, as a monotherapy and in combination with venetoclax, respectively, corresponding to ∼614 nM and 799 nM. Minimal accumulation was detected after multiple weekly doses. Venetoclax exposures were comparable with those observed historically38,39 and did not appear to vary with eftoza dose.
Table 2.
Most common TRAEs (in >10% of patients) and grade ≥3 TRAEs--combination cohorts
| MedDRA preferred term, n (%) | N = 23 |
|
|---|---|---|
| Any grade | Grade ≥3 | |
| Any TRAE | 18 (78.3) | 7 (30.4)∗ |
| Increased ALT | 8 (34.8) | 5 (21.7) |
| Increased AST | 8 (34.8) | 4 (17.4) |
| Fatigue | 5 (21.7) | 0 |
| Nausea | 5 (21.7) | 0 |
| Decreased appetite | 3 (13.0) | 0 |
| Diarrhea | 3 (13.0) | 1 (4.3) |
| Headache | 3 (13.0) | 0 |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; MedDRA, Medical Dictionary for Regulatory Activities.
∗One grade 5 TRAE (7.5 mg/kg combination cohort) of pneumonia was reported that was also confounded with the progression of underlying AML.
Table 3.
Preliminary PK parameters of eftoza in AML in cycle 1 after monotherapy or in combination with venetoclax
| PK parameter | 1.25 mg/kg monotherapy (n = 1)∗ | 3.75 mg/kg monotherapy (n = 1)∗ | 3.75 mg/kg combination (n = 10) | 7.5 mg/kg monotherapy (n = 2)∗ | 7.5 mg/kg combination (n = 13) |
|---|---|---|---|---|---|
| Cmax (μg/mL) | 12.9 | 49.3 | 58.9 (62.5, 41) | 98.3, 107 | 134 (139, 27) |
| AUC0-168 (h∗μg/mL) | 352 | 1440 | 1550 (1670, 44) | 2930, 2670 | 3480 (3550, 21) |
| t1/2 (h)† | 30.4 | 31.2 | 34.1 ± 10.1 | 30.3, 20.9 | 28.6 ± 5.93 |
Values showed as geometric mean (arithmetic mean, % CV).
%CV, coefficient of variation percentage; AUC, area under the concentration-time curve; Cmax, maximum plasma concentration; t1/2, terminal phase elimination half-life.
n < 3 presented as individual values.
Showed as harmonic mean ± pseudostandard deviation.
Antileukemic activity was not observed in the 4 patients enrolled in monotherapy cohorts. For the 23 patients in combination cohorts, the complete response (CR) or CR with incomplete blood count recovery (CRi) rate was 26% (n = 6), with 4 CR and 2 CRi. In addition, 1 patient achieved a morphologic leukemia-free state (MLFS). The ORR was 30% (7/23) with inclusion of MLFS. Molecular profiles and responses for patients in combination cohorts are presented in Figure 4. Median duration of response (CRi or better) was 7.8 months (95% confidence interval, 1.2 to not reached). The molecular profile in combination cohorts measured by next-generation sequencing showed aberrations, with IDH1/2 and NRAS/KRAS being the most frequently mutated genes. No genetic aberrations were detected in 2 of 6 patients who reported a CR or CRi. Mutations in IDH2 (n = 1) and ASXL1 (n = 1), respectively, were the only aberration noted in the 2 responders. Duration of responses for a patient with IDH2 mutation was >400 days, with treatment ongoing at time of data cut. The remaining 2 responders had genetic abnormalities in ASXL1, TET2, STAG2, NRAS, WT1, WAC, JAK2, DNMT3A, and NPM1. A FLT3-ITD mutation was documented in 1 patient with morphologic relapse; no mutations in TP53 were observed in enrolled patients.
Figure 4.

Disease response over time in eftoza plus venetoclax combination cohorts. Response per the International Working Group. Disease progression is assessed per European LeukemiaNet. Samples were assessed for DR4/DR5 expression via flow cytometry. DOR, duration of response; MR, morphologic relapse; PD, progressive disease; RD, resistant disease.
In addition, 13 of the 23 patients from combination cohorts had samples available for quantitation of DR4/DR5 via flow cytometry assay; 6 of these patients’ samples (either peripheral blood or bone marrow aspirate) had myeloblasts positive for DR4/DR5 expression. A 67% (4/6) ORR (CR, CRi, or MLFS) was observed among patients with positive DR4/DR5 myeloblasts. CR is ongoing in 1 patient (>400 days; Figure 4).
Discussion
Using preclinical models of AML, we demonstrate that the TRAIL receptor agonistic fusion protein eftoza drives caspase-dependent apoptosis in vitro that translates into antitumorigenic activity in vivo that can be enhanced by concurrently inhibiting BCL-2 with venetoclax. Eftoza single-agent or combinatorial activity depends on DR4 and DR5 expression, 2 factors that are independent predictors of poor prognosis in AML.40 Importantly, the combinatorial activity between eftoza and venetoclax is observed in AML cell lines, patient samples, and patient-derived xenograft models with TP53 and/or FLT3-ITD mutations; genomic aberrations are associated with less durable responses to venetoclax monotherapy or in combination with hypomethylating agents.20,31,34, 35, 36, 37 Eftoza with or without venetoclax was also active in AML cell lines deficient in BAX, a proapoptotic factor that drives resistance to venetoclax-based therapies,41 although this was less efficient than in the parental cell line. Given these observations, the development of new venetoclax-based combination regimens with novel therapeutics such as eftoza has the potential to expand treatment options for patients with AML.
In addition to these preclinical data, we demonstrate that treatment with eftoza was tolerated both as monotherapy and in combination with venetoclax in patients with R/R AML (NCT03082209). Although the combination of eftoza plus venetoclax may be associated with an increase in hepatic toxicity, overall, AEs from the combination arm were aligned with those expected from venetoclax monotherapy. Myelosuppression is a predominant toxicity encountered during venetoclax-based therapy42, 43, 44; however, adverse events of anemia, neutropenia, or thrombocytopenia were not prominent in the eftoza-venetoclax combination arm. Notably, there were no cases of tumor lysis syndrome reported for patients treated with eftoza plus venetoclax but larger studies will be needed to address these adverse events.
Eftoza treatment demonstrated approximately dose-proportional increase in exposure in the weekly 1.25- to 7.5 mg/kg dose range that did not vary with or without coadministration of venetoclax. Although limited activity was demonstrated with eftoza monotherapy in preclinical models of AML, no response was observed in the phase 1 monotherapy cohort. Because of low patient numbers (n = 4), conclusions cannot be drawn. Combination therapy with eftoza and venetoclax showed promising antileukemic activity in patients with R/R AML with a CR/CRi rate of 26% (6/23) and ORR of 30% (7/23) with inclusion of MLFS. As a comparison, venetoclax monotherapy in a similar setting reported 19% CR/CRi rate with median duration of response <2 months.20 Conversely, retrospective analysis of outcomes for 86 patients with R/R AML who were treated with venetoclax combinations (hypomethylating agents [azacitidine or decitabine] or low-dose cytarabine) resulted in responses in 31% of patients (CR, 14%; CRi, 10%; MLFS, 7%).43 Notably, an ORR (CR, CRi, or MLFS) of 67% (4/6) was reported in a subgroup of patients positive for DR4/5 receptor expression.
Collectively, these data highlight that simultaneously targeting extrinsic and intrinsic apoptosis-signaling pathways in AML with eftoza and venetoclax, respectively, improves on the activity of either agent alone. This combination is safe and well tolerated in patients with AML and supports further clinical evaluation of eftoza-venetoclax combination in patients with DR4/DR5-positive AML. Effectively activating the extrinsic pathway with the TRAIL receptor agonist eftoza and the intrinsic pathway with venetoclax may lead to more durable antitumor activity in patients with AML.
Conflict-of-interest disclosure: E.C. received honoraria or consultation fees from Astellas, Novartis, Nanobiotix, Pfizer, Janssen-Cilag, GLG Pharma, Psioxus Therapeutics, Merck, Medscape, Bristol-Myers Squibb, Gilead Sciences, Seattle Genetics, Pierre Fabre, Boehringer Ingelheim, Cerulean Pharma, EUSA Pharma, Gehrmann Consulting, AstraZeneca, Roche, Guidepoint, Servier, Celgene, AbbVie, Amcure, OncoDNA, and Alkermes; is on a leadership role as director of clinical research for START Madrid and HM Hospitals Group, Madrid and is the founder and president for nonprofit Foundation INTHEOS; is the codirector of Methods in Clinical Cancer Research (MCCR) Workshop, Zeist, Netherlands (joint ECCO-AACR-EORTC-ESMO Workshop on MCCR); has stocks or ownership in START, Oncoart Associated, and International Cancer Consultants; gets direct research funding as project lead from Novartis, AstraZeneca, BeiGene; received institutional financial support for clinical trials from AbbVie, ACEO, Amcure, Amgen, AstraZeneca, Bristol-Myers Squibb, CytomX Therapeutics, GlaxoSmithKline, Genentech/Roche, H3 Biomedicine, Incyte, Janssen, Kura, Lilly, Loxo, Nektar Therapeutics, MacroGenics, Menarini, Merck, Merus, Nanobiotix, Novartis, Pfizer, PharmaMar, Principia, PUMA, Sanofi, Taiho, Tesaro, BeiGene, Transgene, Takeda, Incyte, Inovio, MSD, Psioxus, Seattle Genetics, Mersana, GSK, Daiichi, Nektar, Astellas, ORCA, Boston Therapeutics, Dynavax, Debiopharm, Boehringer Ingelheim, Regeneron, Millennium, Synthon, Spectrum, and Rigontec; is on the scientific board at Psioxus; has memberships for SEOM, EORTC, ESMO, and ASCO; and has a full/part-time employment in HM Hospitals Group and START, Program of Early Phase Clinical Drug Development in Oncology, as a medical oncologist, director clinical research. B.A.C. received institutional research support from AbbVie, Bayer, AstraZeneca, MedImmune, Astellas, Actuate Therapeutics, Pfizer, Dragonfly Therapeutics; and is on the advisory role for Foundation Medicine, Tempus, G1 therapeutics, Seagen. A.S. received research support from Natera; and is an advisory board member of Taiho Oncology. K.I. received grants from AbbVie, during the conduct of the study; grants from Sanofi, Symbio, Bayer, IQVIA, and Otsuka; grants and personal fees from Novartis; personal fees from Takeda, Celgene, Ono, Chugai, Eisai, SRD, MSD, Micron, and Janssen; and personal fees from Kyowa Kirin, outside the submitted work. D.M. received honoraria from Takeda, AbbVie, and Janssen. M.M. and D.A.M. are former employees of AbbVie and stockholders of AbbVie Inc. T.U., S.K.T., C.B., M.S., D.C., F.G.B., S.M.-L., B.C.M., D.S., and D.C.P. are employees of AbbVie and stockholders of AbbVie Inc. The remaining authors declare no competing financial interests.
Acknowledgments
AbbVie and the authors thank all the trial investigators and the patients who participated in this clinical trial. Medical writing support was provided by Mary L. Smith, Aptitude Health, Atlanta, GA, and funded by AbbVie. The authors also thank Rogier Mous (University Medical Center Utrecht), Lot Devriese (University Medical Center Utrecht), John Reagan (Brown University), Adam Olszewski (Brown University), and Marcel Nijland (University Medical Center Groningen) for their invaluable study contributions; and Mark Minden, Dalia Barsyte-Lovejoy, and Genna Luciani (Princess Margaret Cancer Centre) and Dipica Haribhai (AbbVie Precision Medicine Oncology) for their contribution to the ex vivo viability study design and completion. The authors also thank Jason Huska from AbbVie Oncology Discovery for providing the BAX- and BAK/BAX-deficient OCI-AML5 cells.
Venetoclax is being developed in a collaboration between AbbVie and Genentech. AbbVie funded this study and participated in the study design, research, analysis, data collection, interpretation of data, reviewing, and approval of the manuscript. Genentech participated in the review and approval of the manuscript. No honoraria or payments were made for authorship.
Authorship
Contribution: Analysis was initially done by S.K.T., C.B., M.S., D.C., M.M., D.S., D.M., F.G.B., S.M.-L., B.A.C., and D.C.P.; and all authors had access to the data and participated in data collection and interpretation and contributed to the drafting, review, revision, and approval of the manuscript.
Footnotes
Presented in abstract form at the American Association for Cancer Research (AACR) annual meeting, Washington, DC, 1 to 5 April 2017; 12th Meeting on Cell Death, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, 15 to 19 August 2017; 59th annual meeting of the American Society of Hematology, Atlanta, GA, 9 to 12 December 2017; and 61st annual meeting of the American Society of Hematology, Orlando, FL, 7 to 10 December 2019; and in virtual format at the 25th European Hematology Association annual congress, 11 to 21 June 2020 and the 82nd annual meeting of the Japanese Society of Hematology, 10 October to 8 November 2020.
Qualified researchers who engage in rigorous, independent scientific research may request access to anonymized individual and trial-level data after the review and approval of a research proposal, statistical analysis plan, and data sharing agreement. This includes other information (eg, protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. Data requests can be submitted at any time and data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Supplementary Material
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.von Karstedt S, Montinaro A, Walczak H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat Rev Cancer. 2017;17(6):352–366. doi: 10.1038/nrc.2017.28. [DOI] [PubMed] [Google Scholar]
- 3.Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020;17(7):395–417. doi: 10.1038/s41571-020-0341-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelley SK, Harris LA, Xie D, et al. Preclinical studies to predict the disposition of Apo2L/tumor necrosis factor-related apoptosis-inducing ligand in humans: characterization of in vivo efficacy, pharmacokinetics, and safety. J Pharmacol Exp Ther. 2001;299(1):31–38. [PubMed] [Google Scholar]
- 5.Xiang H, Nguyen CB, Kelley SK, Dybdal N, Escandón E. Tissue distribution, stability, and pharmacokinetics of Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand in human colon carcinoma COLO205 tumor-bearing nude mice. Drug Metab Dispos. 2004;32(11):1230–1238. doi: 10.1124/dmd.104.000323. [DOI] [PubMed] [Google Scholar]
- 6.Graves JD, Kordich JJ, Huang TH, et al. Apo2L/TRAIL and the death receptor 5 agonist antibody AMG 655 cooperate to promote receptor clustering and antitumor activity. Cancer Cell. 2014;26(2):177–189. doi: 10.1016/j.ccr.2014.04.028. [DOI] [PubMed] [Google Scholar]
- 7.Tuthill MH, Montinaro A, Zinngrebe J, et al. TRAIL-R2-specific antibodies and recombinant TRAIL can synergise to kill cancer cells. Oncogene. 2015;34(16):2138–2144. doi: 10.1038/onc.2014.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson NS, Yang B, Yang A, et al. An Fcγ receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell. 2011;19(1):101–113. doi: 10.1016/j.ccr.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 9.Phillips DC, Buchanan FG, Cheng D, et al. Hexavalent TRAIL fusion protein eftozanermin alfa optimally clusters apoptosis-inducing TRAIL receptors to induce on-target antitumor activity in solid tumors. Cancer Res. 2021;81(12):3402–3414. doi: 10.1158/0008-5472.CAN-20-2178. [DOI] [PubMed] [Google Scholar]
- 10.Danish L, Imig D, Allgöwer F, Scheurich P, Pollak N. Bcl-2-mediated control of TRAIL-induced apoptotic response in the non-small lung cancer cell line NCI-H460 is effective at late caspase processing steps. PLoS One. 2018;13(6):e0198203. doi: 10.1371/journal.pone.0198203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang L, Fang B. Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther. 2005;12(3):228–237. doi: 10.1038/sj.cgt.7700792. [DOI] [PubMed] [Google Scholar]
- 12.Pan R, Hogdal LJ, Benito JM, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014;4(3):362–375. doi: 10.1158/2159-8290.CD-13-0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leverson JD, Phillips DC, Mitten MJ, et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med. 2015;7(279):279ra40. doi: 10.1126/scitranslmed.aaa4642. [DOI] [PubMed] [Google Scholar]
- 14.Lagadinou ED, Sach A, Callahan K, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12(3):329–341. doi: 10.1016/j.stem.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pollyea DA, Stevens BM, Jones CL, et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med. 2018;24(12):1859–1866. doi: 10.1038/s41591-018-0233-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Souers AJ, Leverson JD, Boghaert ER, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19(2):202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 17.Fischer K, Al-Sawaf O, Bahlo J, et al. Venetoclax and obinutuzumab in patients with CLL and coexisting conditions. N Engl J Med. 2019;380(23):2225–2236. doi: 10.1056/NEJMoa1815281. [DOI] [PubMed] [Google Scholar]
- 18.Kaufman JL, Gasparetto C, Schjesvold FH, et al. Targeting BCL-2 with venetoclax and dexamethasone in patients with relapsed/refractory t(11;14) multiple myeloma. Am J Hematol. 2021;96(4):418–427. doi: 10.1002/ajh.26083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guerra VA, DiNardo C, Konopleva M. Venetoclax-based therapies for acute myeloid leukemia. Best Pract Res Clin Haematol. 2019;32(2):145–153. doi: 10.1016/j.beha.2019.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Konopleva M, Pollyea DA, Potluri J, et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 2016;6(10):1106–1117. doi: 10.1158/2159-8290.CD-16-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tallman MS, Wang ES, Altman JK, et al. Acute myeloid leukemia, version 3.2019, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2019;17(6):721–749. doi: 10.6004/jnccn.2019.0028. [DOI] [PubMed] [Google Scholar]
- 22.Daver N, Wei AH, Pollyea DA, Fathi AT, Vyas P, DiNardo CD. New directions for emerging therapies in acute myeloid leukemia: the next chapter. Blood Cancer J. 2020;10(10):107. doi: 10.1038/s41408-020-00376-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rowe JM. Perspectives on current survival and new developments in AML. Best Pract Res Clin Haematol. 2021;34(1):101248. doi: 10.1016/j.beha.2021.101248. [DOI] [PubMed] [Google Scholar]
- 24.LoRusso P, Ratain MJ, Doi T, et al. Eftozanermin alfa (ABBV-621) monotherapy in patients with previously treated solid tumors: findings of a phase 1, first-in-human study. Invest New Drugs. 2022;40(4):762–772. doi: 10.1007/s10637-022-01247-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takimoto R, El-Deiry WS. Wild-type p53 transactivates the KILLER/DR5 gene through an intronic sequence-specific DNA-binding site. Oncogene. 2000;19(14):1735–1743. doi: 10.1038/sj.onc.1203489. [DOI] [PubMed] [Google Scholar]
- 26.Liu X, Yue P, Khuri FR, Sun SY. p53 upregulates death receptor 4 expression through an intronic p53 binding site. Cancer Res. 2004;64(15):5078–5083. doi: 10.1158/0008-5472.CAN-04-1195. [DOI] [PubMed] [Google Scholar]
- 27.Bowen D, Groves MJ, Burnett AK, et al. TP53 gene mutation is frequent in patients with acute myeloid leukemia and complex karyotype, and is associated with very poor prognosis. Leukemia. 2009;23(1):203–206. doi: 10.1038/leu.2008.173. [DOI] [PubMed] [Google Scholar]
- 28.Ok CY, Patel KP, Garcia-Manero G, et al. TP53 mutation characteristics in therapy-related myelodysplastic syndromes and acute myeloid leukemia is similar to de novo diseases. J Hematol Oncol. 2015;8:45. doi: 10.1186/s13045-015-0139-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–2221. doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kadia TM, Jain P, Ravandi F, et al. TP53 mutations in newly diagnosed acute myeloid leukemia: clinicomolecular characteristics, response to therapy, and outcomes. Cancer. 2016;122(22):3484–3491. doi: 10.1002/cncr.30203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hunter AM, Sallman DA. Current status and new treatment approaches in TP53 mutated AML. Best Pract Res Clin Haematol. 2019;32(2):134–144. doi: 10.1016/j.beha.2019.05.004. [DOI] [PubMed] [Google Scholar]
- 32.Borisy AA, Elliott PJ, Hurst NW, et al. Systematic discovery of multicomponent therapeutics. Proc Natl Acad Sci U S A. 2003;100(13):7977–7982. doi: 10.1073/pnas.1337088100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richardson DR, Swoboda DM, Moore DT, et al. Genomic characteristics and prognostic significance of co-mutated ASXL1/SRSF2 acute myeloid leukemia. Am J Hematol. 2021;96(4):462–470. doi: 10.1002/ajh.26110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pasquer H, Tostain M, Kaci N, Roux B, Benajiba L. Descriptive and functional genomics in acute myeloid leukemia (AML): paving the road for a cure. Cancers. 2021;13(4):748. doi: 10.3390/cancers13040748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chyla B, Daver N, Doyle K, et al. Genetic biomarkers of sensitivity and resistance to venetoclax monotherapy in patients with relapsed acute myeloid leukemia. Am J Hematol. 2018;93(8):E202–E205. doi: 10.1002/ajh.25146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Konopleva M, Thirman MJ, Pratz KW, et al. Impact of FLT3 mutation on outcomes after venetoclax and azacitidine for patients with treatment-naïve acute myeloid leukemia. Clin Cancer Res. 2022;28(13):2744–2752. doi: 10.1158/1078-0432.CCR-21-3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pollyea DA, Pratz KW, Wei AH, et al. Outcomes in patients with poor-risk cytogenetics with or without TP53 mutations treated with venetoclax combined with azacitidine. Blood. 2021;138(Supplement 1):224. doi: 10.1158/1078-0432.CCR-22-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Konopleva M, Pollyea DA, Potluri J, et al. A phase 2 study of ABT-199 (GDC-0199) in patients with acute myelogenous leukemia (AML) Blood. 2014;124(21):118. Abstract 118. [Google Scholar]
- 39.Salem AH, Agarwal SK, Dunbar M, Enschede SLH, Humerickhouse RA, Wong SL. Pharmacokinetics of venetoclax, a novel BCL-2 inhibitor, in patients with relapsed or refractory chronic lymphocytic leukemia or non-Hodgkin lymphoma. J Clin Pharmacol. 2017;57(4):484–492. doi: 10.1002/jcph.821. [DOI] [PubMed] [Google Scholar]
- 40.Schmohl JU, Nuebling T, Wild J, et al. Death receptor expression on blasts in AML is associated with unfavorable prognosis. Anticancer Res. 2015;35(7):4043–4052. [PubMed] [Google Scholar]
- 41.Moujalled DM, Brown FC, Chua CC, et al. Acquired mutations in BAX confer resistance to BH3-mimetic therapy in acute myeloid leukemia. Blood. 2023;141(6):634–644. doi: 10.1182/blood.2022016090. [DOI] [PubMed] [Google Scholar]
- 42.Aldoss I, Pullarkat V, Stein AS. Venetoclax-containing regimens in acute myeloid leukemia. Ther Adv Hematol. 2021;12 doi: 10.1177/2040620720986646. 2040620720986646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stahl M, Menghrajani K, Derkach A, et al. Clinical and molecular predictors of response and survival following venetoclax therapy in relapsed/refractory AML. Blood Adv. 2021;5(5):1552–1564. doi: 10.1182/bloodadvances.2020003734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617–629. doi: 10.1056/NEJMoa2012971. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.