

Abstract
The updated International Consensus Classification (ICC) classification of B-acute lymphoblastic leukemia (B-ALL) and T-acute lymphoblastic leukemia (T-ALL) includes both revisions to subtypes previously outlined in the 2016 WHO classification and several newly described entities. The ICC classification incorporates recent clinical, cytogenetic, and molecular data, with a particular emphasis on whole transcriptome analysis and gene expression (GEX) clustering studies. B-ALL classification is modified to further subclassify BCR::ABL1-positive B-ALL and hypodiploid B-ALL. Additionally, nine new categories of B-ALL are defined, including seven that contain distinguishing gene rearrangements, as well as two new categories that are characterized by a specific single gene mutation. Four provisional entities are also included in the updated B-ALL classification, although definitive identification of these subtypes requires GEX studies. T-ALL classification is also updated to incorporate BCL11B-activating rearrangements into early T-precursor (ETP) ALL taxonomy. Additionally, eight new provisional entities are added to the T-ALL subclassification. The clinical implications of the new entities are discussed, as are practical approaches to the use of different technologies in diagnosis. The enhanced specificity of the new classification will allow for improved risk stratification and optimized treatment plans for patients with ALL.
Keywords: T-lymphoblastic leukemia, T-lymphoblastic lymphoma, B-lymphoblastic leukemia, B-lymphoblastic lymphoma, gene expression clustering
INTRODUCTION
Acute lymphoblastic leukemia (ALL) is an aggressive neoplasm that comprises B- and T- acute lymphoblastic leukemia (B-ALL; T-ALL). The updated International Consensus Classification (ICC) includes both revisions to ALL entities previously described in the 2016 WHO classification, as well as several newly described ALL subtypes (Table 1) [1]. Most entities from the 2016 WHO are retained, but there are several modifications that incorporate findings from recent genomic studies [2]. Many of the newly described entities have characteristic clinical features, and use of the updated classification will assist in risk stratification and treatment selection for these patients.
Table 1.
The International Consensus Classification of ALL
| B-acute lymphoblastic leukemia (B-ALL) |
| B-ALL with recurrent genetic abnormalities |
| B-ALL with t(9;22)(q34.1;q11.2)/BCR::ABL1 |
| with lymphoid only involvement |
| with multilineage involvement |
| B-ALL with t(v;11q23.3)/KMT2A rearranged |
| B-ALL with t(12;21)(p13.2;q22.1)/ETV6::RUNX1 |
| B-ALL, hyperdiploid |
| B-ALL, low hypodiploid |
| B-ALL, near haploid |
| B-ALL with t(5;14)(q31.1;q32.3)/IL3::IGH |
| B-ALL with t(1;19)(q23.3;p13.3)/TCF3::PBX1 |
| B-ALL, BCR::ABL1-like, ABL-1 class rearranged |
| B-ALL, BCR::ABL1-like, JAK-STAT activated |
| B-ALL, BCR::ABL1-like, NOS |
| B-ALL with iAMP21 |
| B-ALL with MYC rearrangement |
| B-ALL with DUX4 rearrangement |
| B-ALL with MEF2D rearrangement |
| B-ALL with ZNF384 rearrangement |
| B-ALL with NUTM1 rearrangement |
| B-ALL with HLF rearrangement |
| B-ALL with UBTF::ATXN7L3/PAN3,CDX2 (“CDX2/UBTF”) |
| B-ALL with IKZF1 N159Y |
| B-ALL with PAX5 P80R |
| Provisional entities (see Table 3): |
| B-ALL, ETV6::RUNX1-like |
| B-ALL, with PAX5 alteration |
| B-ALL, with mutated ZEB2 (p.H1038R)/IGH::CEBPE |
| B-ALL, ZNF384 rearranged-like |
| B-ALL, KMT2A rearranged-like |
| T-acute lymphoblastic leukemia (T-ALL) |
| Early T-cell precursor ALL, BCL11B-activated |
| Early T-cell precursor ALL, NOS |
| T-ALL, NOS |
| Provisional entities (see Table 4) |
| Provisional entity: Natural killer (NK) cell ALL |
B-ACUTE LYMPHOBLASTIC LEUKEMIA/LYMPHOMA
Genomic studies of ALL have identified multiple new entities with distinct driver mutations and commonly distinct gene expression (GEX) profiles evident using clustering algorithms (Figure 1). These discoveries have led to expansions or revisions of the prior 2016 WHO classification.
Figure 1. Gene expression clustering of B-ALL cases.
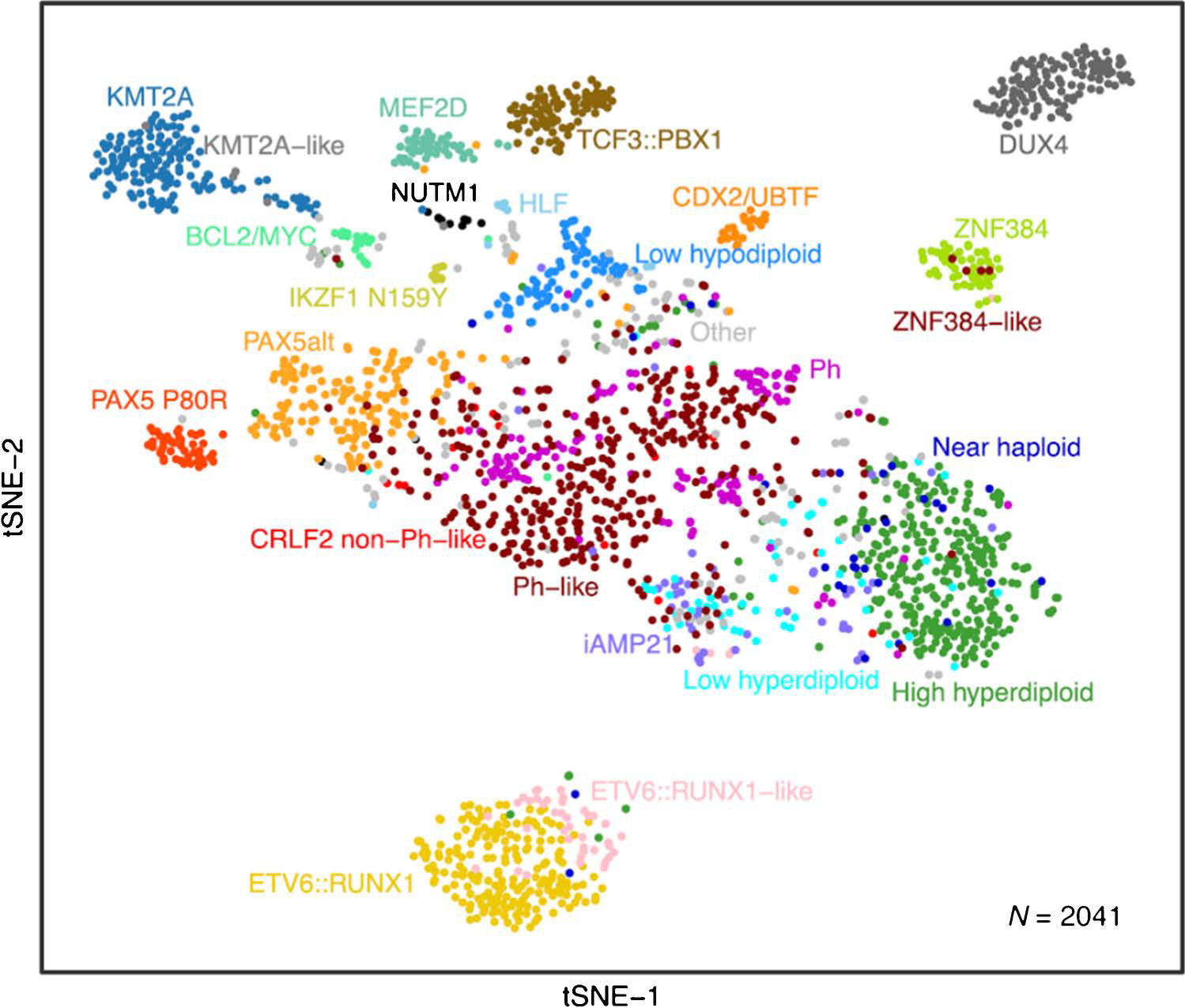
The figure depicts two dimensional clustering using t-distributed stochastic neighbor embedding of whole transcriptome sequencing data from 2041 leukemia samples collected at diagnosis from children or adults with ALL. Cases are color coded by subtype. This approach, and much of the data, were first reported by Gu, et al. [18], and has been updated to include additional CDX2/UBTF cases following recent definition of this subtype.
Revisions to the 2016 WHO classification
B-ALL with t(9;22)(q34.1;q11.2)/BCR::ABL1: lymphoid only and multilineage involvement
The new ICC divides the entity “BCR::ABL1-positive B-ALL” into two subtypes: “B-ALL with t(9;22)(q34.1;q11.2)/BCR::ABL1 with lymphoid only involvement” (BCR::ABL1+ ALL-L), and “B-ALL with t(9;22)(q34.1;q11.2)/BCR::ABL1with multilineage involvement” (BCR::ABL1+ ALL-M). These cases cannot be distinguished by immunophenotyping or differences in the fusion protein (p190 versus p210). The underlying difference between these subtypes reflects the target cell for the transformation event, with a multipotent progenitor serving as the target for BCR::ABL1+ ALL-M, and a later progenitor targeted in BCR::ABL1+ ALL-L. The former thus appears akin to chronic myeloid leukemia presenting in lymphoid blast phase (CML-LBP)[3, 4] and the latter to de novo B-ALL. Although optimal therapy for each has not yet been established, prognosis and treatment may differ, particularly in pediatric patients [3, 5–7].
BCR::ABL1+ ALL-L and -M may be distinguished by fluorescence-activated cell sorting coupled with fluorescence in situ hybridization (FISH), because in the latter the fusion signal can be detected in cells of non-B lineage. This can be difficult to demonstrate at diagnosis, when blasts greatly predominate, and cell separation techniques may be required. It may be suspected however, particularly after therapy, when the tumor burden estimated by assessing BCR::ABL1 fusion transcripts is higher than that determined by flow cytometry studies [4, 6, 8]. Distinction between BCR::ABL1+ ALL-M and CML-LBP is difficult without a history of prior CML for CML-LBP.
B-ALL, BCR::ABL1-like: ABL1-class rearranged, JAK-STAT activated, and NOS
The entity “B-ALL, BCR::ABL1-like” was introduced in the 2016 WHO to account for cases that have a GEX profile similar to Ph+ B-ALL, but lack the BCR::ABL1 translocation [2]. Further study has shown that these “BCR::ABL1-like” cases contain a variety of genetic lesions. To incorporate these new data, the ICC separates “B-ALL, BCR::ABL1-like” into three subtypes: ABL1-class rearranged, JAK-STAT activated, and not otherwise specified (NOS). Comprehensive identification of the kinase signaling-activating drivers of BCR::ABL1-like B-ALL requires genetic studies, as none of these entities, apart from rearrangement of CRLF2 in the JAK-STAT subgroup, shows distinctive immunophenotypic abnormalities. While BCR::ABL1-like B-ALL overall has a poor prognosis, there is some variability based on the exact genetic lesion; moreover, the recognition of specific targetable lesions has provided some new therapeutic opportunities for these patients.
The identification of ABL1-class fusions in “B-ALL, BCR::ABL1-like: ABL1-class rearranged” is particularly important because this subgroup may respond to tyrosine kinase inhibitors (TKIs) that target ABL1, such as imatinib and dasatinib [9]. The ABL1 class rearrangements include fusions between ABL1, ABL2, CSF1R, or PDGFRB and various partner genes [10]. Rearrangement of these kinases can be detected by FISH, and the specific fusions by commercially available targeted transcriptome sequencing (TS), as described below. While PDGFRB-r lymphoid neoplasms are typically included in “Myeloid/lymphoid neoplasms with PDGFRB rearrangement,” those presenting as B-ALL (e.g. EBF1::PDGFRB) are best classified as BCR::ABL1-like ALL, ABL1-class rearranged [11]. The utility of separately demonstrating the PDGFRB mutation in the myeloid and lymphoid populations is currently unclear in this relatively rare entity, but in clinically ambiguous cases confirmation of the cells that contain the genetic abnormality may be useful.
The second new subclassification of “B-ALL, BCR::ABL1-like” in the ICC includes “B-ALL, BCR::ABL1-like: JAK-STAT activated cases.” The mutations that result in activation of JAK-STAT pathway signaling are diverse. The most common are CRLF2 rearrangements (CRLF2-r) to IGH or P2RY8 that result in CRLF2 overexpression. Less common JAK-STAT activating alterations are JAK2 fusions, truncating and activating rearrangements of EPOR, and less commonly, IL7R or CRLF2 F232C mutations [15]. Additionally, JAK1, JAK2, or JAK3 mutations are identified in approximately half of CRLF2-r cases, and Ras pathway genes (KRAS, NRAS, PTPN11 and NF1) are also commonly mutated in the setting of CRLF2-r [12]. JAK inhibitors are being formally evaluated in trials [13], although to date these have not shown the level of success seen with ABL1-class-directed TKIs, and there is variability in activity according to underlying alterations.
“B-ALL, BCR::ABL1-like, NOS” encompasses heterogeneous alterations of other kinases and cytokine receptors, but importantly, many have been shown to be responsive to TKIs in preclinical models, particularly kinase rearrangements (FLT3, FGFR1, NTRK3, and PTK2B). Ras signaling mutations are also observed (KRAS, NRAS, NF1, PTPN11, BRAF, CBL) but are not specific to BCR::ABL1-like B-ALL [14]. Responsiveness of the B-ALL, BCR::ABL1-like, NOS cases to TKIs is variable depending on the specific mutation and suitability of the inhibitor. In particular, fusions involving NTKR3 may show profound responses to TKIs such as larotrectinib [15].
B-ALL with Hypodiploidy: Low Hypodiploid and Near Haploid ALL
In the 2016 WHO classification, B-ALLs with gross chromosomal abnormalities were subclassified into B-ALL with hyperdiploidy and hypodiploidy. At that time, it was noted that hypodiploid cases were sometimes separated into near-haploid, low hypodiploid, and high hypodiploid B-ALL [16]. In the ICC, however, B-ALL with hypodiploidy is formally separated into two categories: “B-ALL, low hypodiploid” (32–39 chromosomes), and “B-ALL, near haploid” (24–31 chromosomes).
Low hypodiploidy is more common in adults, and is often associated with IKZF2 deletions and TP53 mutations. Approximately one half of TP53 mutations in children with low hypodiploid B-ALL are germline, suggesting that some of these acute leukemias may arise in the setting of Li Fraumeni syndrome [17]. Near haploid B-ALL is more common in children, and is also associated with a poor prognosis [17]. Both subtypes are prone to doubling of the hypodiploid clone (“masked hypodiploidy”), in which the total chromosome number appears hyperdiploid, but the underlying biology and clinical behavior are those of hypodiploid B-ALL [14]. Despite their very different prognoses, near haploid B-ALL and high hyperdiploid B-ALL share common GEX profiles and concomitant mutations, suggesting a common developmental origin.
B-acute lymphoblastic leukemia: New Entities
The ICC includes nine new entities that were not included in the 2016 WHO (Table 2). All are defined by either chromosomal rearrangements or recurrent sequence mutations. Many of these new entities were identified by large scale studies that correlated RNA-seq based GEX clustering with genetic findings [18, 19].
Table 2.
New molecular entities in B-ALL
| Subtype | Frequency | Prognosis | Partner genes | Common colesions |
|---|---|---|---|---|
| B-ALL with DUX4 rearrangement | 5–10%, highest in AYA | Excellent | Enhancers, most commonly IGH | ERG and IKZF1 deletions; CDKN2A |
| B-ALL with ZNF384 rearrangement | 5–10%, higher in AYA | Variable | EP300 (most common and good prognosis), TCF3, TAF15, CREBBP | ETV6; Ras pathway mutations; FLT3 overexpression |
| B-ALL with MEF2D rearrangement | 3–5% | Poor | BCL9, HNRNPUL1 | CDKN2A; Ras pathway mutations |
| B-ALL with MYC rearrangement | 2–5%, higher in adults and AYA | Poor | IGH | May have BCL2 and/or BCL6 rearrangement s |
| B-ALL with NUTM1 rearrangement | 2% or less; rare in adults, mostly in infants lacking KMT2A rearrangements | Good | ACINI, ZNF618, BRD9, IKZF1, CUX1 | |
| B-ALL CDX2/UBTF | <1%; higher in AYA and female | Poor | UBTF::ATXN7L3 | PAX5-ZCCHC7 deletions, PAX5 overexpression in a subset |
| B-ALL with HLF rearrangement | <<1% children | Very poor | TCF3/4 | |
| B-ALL with mutated PAX5 P80R | 2–5% higher in adult | Intermediate, good in adults | Not applicable | JAK and Ras signaling gene mutations; CDKN2A; IL7R. Deletion or LOF mutation of second PAX5 allele. |
| B-ALL with mutated IKZF1 N159Y | <1% all ages | Intermediate | Not applicable | Gain of chromosome 21 |
New ALL entities defined by translocations
These entities are listed by relative overall frequency, although actual frequency can vary considerably by age group. B-ALL with DUX4 rearrangement and B-ALL with ZNF384-rearrangement each comprise 5–10% of cases, while at the other extreme TCF3/4::HLF rearranged B-ALL is very rare.
B-ALL with DUX4-rearrangement
B-ALL with Double homeobox 4 (DUX4)-rearrangement is relatively common in adolescents and young adults (AYA). This entity is associated with an excellent prognosis in both children and adults, even in the presence of otherwise poor risk genetic features such as IKZF1 deletion, and despite elevated levels of MRD early in therapy [18]. IGH enhancers are the most common rearrangement partner of DUX4, resulting in overexpression of a 3’ truncated isoform of DUX4 [20–22]. The IGH::DUX4 translocation is inconsistently detected by TS, and is difficult to detect by FISH due to the repetitive nature of both the DUX4 and IGH loci; however, overexpression of DUX4 RNA or possibly DUX4 protein [23], or the expression of CD371 on flow cytometric analysis [24] may help identify these cases
B-ALL with ZNF384-rearrangement
Rearrangements of the transcription factor zinc finger 384 (ZNF384) defines a subtype of B-ALL that is most common in children and young adults, and has a variable prognosis [25, 26]. Similar to KMT2A-r B-ALL, these cases are characterized by lineage ambiguity [27]. This entity may present as either B-ALL or B/myeloid mixed phenotype acute leukemia (MPAL) [28, 29], and lineage shifts over the course of disease are common. Despite the immunophenotypic variability, the GEX profiles and secondary genetic alterations of ZNF384-r B/myeloid MPAL and B-ALL are indistinguishable.
ZNF384 has numerous fusion partners, though EP300, TCF3 and TAF15 are most common. The prognosis of ZNF384-r B-ALL depends on the fusion partner; EP300 fusions have the best prognosis, whereas TCF3 fusions have the worst prognosis. This neoplasm is also associated with FLT3 overexpression, but not FLT3 mutations. Confirmation of the diagnosis requires either FISH or TS, with careful attention paid to in-frame indels in the last exon [30, 31]. Of note, B-ALL with ZNF362 rearrangements were originally included in this category in the initial report of the updated ICC, but further data suggest that they may be best considered in the “B-ALL, ZNF384-rearranged-like” provisional category (discussed below).
B-ALL with MEF2D-rearrangement
Myocyte enhancer factor 2D (MEF2D)-rearrangements are identified in a small subset of B-ALL in children and young adults, and are associated with a relatively poor prognosis. MEF2D is a transcription factor that can rearrange with multiple fusion partners, most often BCL9, resulting in enhanced and/or deregulated function of MEF2D. Deregulation of this transcription factor results in overexpression of HDAC9, suggesting that HDAC inhibitors may have therapeutic potential in this entity [32].
A diagnosis of MEF2D-r B-ALL can be suspected from the immunophenotype, as this subtype characteristically shows expression of cytoplasmic mu chain, low expression of CD10, and moderate expression of CD38 [32, 33]. MEF2D::BCL9 fusion probes are commercially available but custom breakapart probes or transcriptome sequencing are needed for definitive confirmation of all cases.
B-ALL with MYC rearrangement
In everyday clinical practice MYC-rearrangements (MYC-r) are most commonly associated with aggressive mature B-cell lymphomas; however, MYC translocations are also reported in about 4% of adult B-ALL and, rarely, pediatric B-ALL [34–36]. These leukemias are associated with a very poor prognosis [34]. MYC-r B-ALL and MYC-r mature B-cell lymphoma are genetically distinct [37]. Prior work characterizing genetic abnormalities of a small set of IG::MYC positive B-ALL cases showed that the IG::MYC translocation in these acute leukemias results from aberrant VDJ joining in a B cell precursor undergoing VDJ recombination [37]. Accordingly, B-ALLs with MYC-r are characterized by unmutated IGVH genes, since IGVH undergoes somatic hypermutation at a comparatively late stage of B-cell maturation in the germinal center. By contrast, MYC-r mature large B-cell lymphomas show evidence of somatic hypermutation. Additionally, a subset of MYC-r B-ALL with concomitant BCL2 (and/or BCL6) rearrangements have also been described [38] but, unlike high grade lymphomas, there are insufficient data to consider these a separate category of B-ALL.
The clinical presentation of MYC-r mature B-cell lymphomas and MYC-r B-ALL is typically very different, with mass lesions in the former and bone marrow replacement in the latter; however, there is some clinical overlap, and definitive distinction requires laboratory studies. CD34 or TdT expression favors a diagnosis of B-ALL, but not all cases will express these [34, 37]. Moreover, patients with a prior history of mature B-cell lymphoma can sometimes show immunophenotypic features of immaturity (i.e. TdT expression) upon transformation, so a diagnosis of MYC-r B-ALL should be avoided in patients with such a history [39–42]. For this reason, somatic hypermutation analysis may be the most reliable means of recognizing this entity in clinically ambiguous cases.
B-ALL with NUTM1-rearrangement
NUT midline carcinoma family member 1 (NUTM1)-rearranged leukemia is rare, typically affects infants, and has a relatively favorable prognosis [43]. NUTM1-r B-ALL has numerous fusion partners, all of which result in overexpression of “NUT family member 1” (NUT). NUTM1-r B-ALL can be diagnosed using FISH studies directed against NUTM1 [44], and immunohistochemical studies can detect the characteristic NUT overexpression [45].
B-ALL with UBTF::ATXN7L3/PAN3,CDX2 (“CDX2/UBTF”)
B-ALL with UBTF::ATXN7L3/PAN3,CDX2 harbors two alterations in all cases, namely a fusion oncoprotein-encoding rearrangement UBTF::ATXN7L3 and a deletion upstream of FLT3 that results in deregulation of CDX2. This entity is succinctly referred to as “CDX2/UBTF” to acknowledge the two distinct driver mutations. CDX2/UBTF-deregulated B-ALL is rare, tends to affect female adolescents and young adults, and has a poor prognosis [46–48].
B-ALL with HLF-rearrangement
Transcription factor 3 (TCF3)::HLF and Transcription factor 4 (TCF4)::HLF rearranged leukemia are exceptionally rare, and are probably only found in children [49]. B-ALL with either of these rearrangements has a very poor prognosis. A limited series of TCF3::HLF cases showed overexpression of Aurora A kinase in all cases, which may represent a therapeutic target [50], and anti-CD19 therapy and transplant may be efficacious in these patients [51].
New ALL entities with point mutations
Two additional entities with single gene point mutations also generate B-ALLs with distinct patterns in GEX studies. Neither has defining immunophenotypic characteristics, but both are readily detected by next-generation sequencing (NGS).
B-ALL with PAX5 P80R
B-ALL with a PAX5 P80R mutation comprises 2–5% of B-ALL cases, is more common in adults, and has a relatively good prognosis [52, 53]. In general, PAX5 mutations and deletions are common in many B-ALL subtypes, and PAX5 translocations are also reported in a subset of cases [54]. However, PAX5 P80R observed in B-ALL is distinctive as it associated with a distinct GEX profile, and is accompanied by either deletion or inactivating sequence mutation of the wild-type allele, or copy neutral loss of heterozygosity [18]. The product of the PAX5 gene, paired box protein PAX5, is known to play an important role in B-cell differentiation, and also acts a tumor suppressor, with alterations of PAX5 contributing to leukemogenesis in part by resulting in an arrest in B lymphoid maturation [55]. This leukemia tends to have other cooperating lesions, including abnormalities in Ras or JAK-STAT signaling pathways or other kinase pathways, which may suggest additional therapeutic opportunities in these patients [18, 52].
B-ALL with IKZF1 N159Y
B-ALL with a DNA-binding protein Ikaros (IKZF1) N159Y mutation comprises less than 1% of cases, is more common in adults, and has an intermediate prognosis. The IKZF1 N159Y mutation is heterozygous in these cases, and results in abnormal nuclear localization and enhanced cell-cell adhesion [18, 19].
B-acute lymphoblastic leukemia: Provisional entities
Small clusters of cases have been described that have identical GEX profiles as one of the new entities described above, but lack the specific subtype-defining genetic abnormality of the new entity (Table 3). Specific drivers for some but not all of these cases can be defined, but comprehensive identification and definition of these cases requires GEX studies.
Table 3.
Provisional entities in B-ALL
| Subtype | Frequency | Genomics |
|---|---|---|
| ETV6::RUNX1-like | <5%, mostly children; worse prognosis than ETV6::RUNX1 | Fusions or CNAs in ETS family genes including ETV6, FUS and also IKZF1; some cases harbor germline loss-of-function ETV6 mutations |
| PAX5 altered (PAX5alt) | 10% of children and adults | Various mutations (especially compound heterozygosity for R38;R140), intragenic amplifications, and non-kinase fusions (ETV6 most common); CDKN2A co-mutations common |
| ZEB2 H1038R/IGH::CEBPE | < 1% | ZEB2 H1038R or IGH::CEBPE; Frequent NRAS mutations (50%), LMO1 upregulation and downregulation of SMAD1 and BMP2 |
| KMT2A-like | <1% | Some with HOXA fusions |
| ZNF384-like | <1% | Many with alternate ZNF362-r; others unknown |
B-ALL, ETV6::RUNX1-like cases share the same GEX profile and CD27+,CD44 dim/neg phenotype, but lack the defining t(12;21)(p13;q22) [21, 56]. A second provisional entity is the relatively large subtype of B-ALL, with PAX5-alteration (PAX5alt), characterized by a variety of different alterations in PAX5, including point mutations, rearrangements (most commonly ETV6 and NOL4L), and focal intragenic amplification. Not all cases with PAX5 mutations represent this entity, and not all cases with a common GEX pattern can be identified by sequencing, so accurate inclusion in this group requires GEX studies [18, 57]. There are three other very rare and poorly characterized provisional entities that each form a distinct cluster on GEX studies. Cases with co-existing H1038R mutations in ZEB2 and IGH::CEBPE appear to have a poor prognosis [19, 58], and as well as cases with GEX profiles similar to KMT2A-r and ZNF384-r B-ALL, which are designated as “B-ALL, KMT2A-rearranged like” and “B-ALL, ZNF384-rearranged like,” respectively; many, but not all, of the latter have translocations involving ZNF362.
T-ACUTE LYMPHOBLASTIC LEUKEMIA/LYMPHOMA (Table 4)
Table 4.
New entities in T-ALL (other than BCL11B-activated, all are provisional)
| Subtype | Frequency | Partner genes/other rearrangements | Common colesions* |
|---|---|---|---|
| BCL11B-activated | 30% of ETP and T/MPAL, <5% of AML | BETA (14q32 enhancer amplification); ARID1B, CCDC26/MYC; CDK6; STAB1; ETV6; ZEB2; RUNX1; | FLT3-ITD; WT1 |
| TAL1/2-R | 30–40% (TAL2 rare); poor prognosis | TCRA/D; TCRB (TAL2); 1p32 deletion (STIL); intergenic SNV (super enhancer) | CDKN2A, NOTCH1, PTEN, USP7 |
| TLX1-R | 5–10% children; near 30% adult; good prognosis | TCR | PHF6, DNM2, BCL11B, RB1, CDKN1B |
| TLX3-R | 20–25% children <5% adult; good prognosis | TCR; BCL11B; CDK6 | CDKN2A, NOTCH1, FBXW7, PTEN |
| HOXA | 15–25% | HOXA::TCRB/TCRG; KMT2A-R; PICALM::MLLT10; SET::NUP214 | |
| LMO1/2-R | LMO1-R -5% LMO2-R 10% | TCR; cryptic deletion; enhancer/promoter mutations LMO complex with bHLH factors; Extremely high LMO expression | CDKN2A, NOTCH1, FBXW7, PTEN, LEF1 |
| NKX2-R | <5% children | NKX2.1/NKX2.2/NKX2.5::TCR; BCL11B; CDK6 | CDKN2A, NOTCH1 PHF6, LEF1, RPL10 |
| SPI1-R | <5%, children, very poor prognosis | STMN1; TCF7; BCL11B | NRAS,KRAS |
| BHLH, other | <2% |
TCRB::LYL1 TCR::BHLHB1; high LMO expression |
NOTCH1 and CDKN2A mutations are common throughout T ALL, except ETP-ALL
The only specific change made to the classification of T-ALL in the ICC was to include a subtype of ETP-ALL characterized by BCL11B activation, most commonly via rearrangement (see below). Eight provisional entities were also added. Also, although not formally recognized in the ICC, rare cases of BCR::ABL1-positive T-ALL occur.
Early T cell precursor ALL
Early T cell precursor (ETP) ALL is currently diagnosed by immunophenotype, and the 2016 WHO definition has not changed. A subset of T-ALL cases is phenotypically similar to ETP ALL but with expression of CD5 on ≥75% rather than <75% of blasts, and is referred to as “near-ETP ALL” [11]. Near-ETP ALL has different genetic lesions than ETP ALL, with enrichment for TLX3-rearrangements; however, the full repertoire of driver genomic alterations in each group has not been comprehensively defined. Near-ETP ALL also shows minor differences in clinical presentation and response to therapy, and will continue to be considered separately from ETP ALL [59, 60], although because of the lack of understanding of genetic drivers it is not as yet formally included in the classification.
Both ETP and near-ETP ALL are genetically heterogeneous. Approximately one-third of ETP ALLs harbor alterations, most commonly rearrangements that deregulate the T lineage transcription factor gene BCL11B. ETP ALL with BCL11B alterations is associated with interchromosomal rearrangements in 80% of cases, while the remaining cases have focal amplifications that generate a neoenhancer distal to BCL11B [73]. Both alterations produce leukemias with similar GEX profiles, so this group has been termed “BCL11B activated” (BCL11B-a) to clarify that this subtype includes not only BCL11B rearrangements but also other BCL11B-activating genetic alterations, and to exclude BCL11B-r that do not result in activation of BCL11B, particularly BCL11B::TLX3, which is enriched in near-ETP ALL [61–63].
Identical BCL11B alterations to those seen in the subset of ETP ALL discussed above are also present other cases of acute leukemia. These leukemias share a common GEX profile, indicating a common molecular origin, but exhibit variable expression of cCD3 and cMPO, resulting in variable classification as ETP ALL, T/myeloid MPAL (where it represents one third of cases), and less commonly, AML and acute undifferentiated leukemia. Thus, BCL11B-a leukemia may be considered a molecularly defined subset of acute leukemia irrespective of diagnostic immunophenotype. However, in general, the ICC remains largely lineage-based, so for current purposes we only discuss BCL11B in the context of T cell leukemia, specifically ETP ALL.
T-acute lymphoblastic leukemia: Provisional Entities
The eight new provisional entities are classified based on aberrant activation of different families of transcription factors. These entities have various alterations (e.g. T-cell receptor rearrangements, chimeric fusion oncoproteins, and enhancer mutations) deregulating different drivers; thus comprehensive genomic analysis may be required to classify all cases. However, many have translocations that are detectable with available FISH probes or PCR. The provisional entities are described below, and are arranged roughly in order of frequency; however, it should be noted that their true frequency is somewhat unclear due to the relative paucity of broad data collection across populations and uneven definitions of the entities within the literature.
TAL1-r or TAL2-r T-ALLs (abbreviated “TAL1/2-r”) have intermediate prognosis and together comprise approximately 30–40% of cases. TAL1-r T-ALL are more common than rearrangements of TAL2, which are comparatively rare in T-ALL. This subtype has variable fusion partners, with STIL::TAL1 most common, and has a late cortical thymocyte (CD4+ or CD8+) immunophenotype.
TXL3-r and TLX1-r T-ALLs have a good prognosis. They typically have a similar CD4+CD8+CD1a+ phenotype, but are separable by GEX and differ in relative frequency with age, with TXL3-r more common in children and TLX1-r more common in adults. The subset of cases with BCL11B::TLX3 has a different pathogenesis, with hijacking of BCL11B enhancers resulting in TLX3 deregulation, and so should not be confused with the BCL11B-activated group of ETP-ALL either phenotypically or molecularly.
The “HOXA dysregulated” subtype (15–25% of cases) includes a variety of genetic lesions that all result in dysregulation of the homeobox transcription factor HOXA. This entity includes various rearrangements, including HOXA::TRB/TRG, KMT2A-r, PICALM::MLLT10, and SET::NUP214. These cases tend to have an immature immunophenotype, with some meeting the criteria for ETP. T-ALLs with LMO1-r or LMO2-r (abbreviated LMO1/2-r; approximately 15% of cases) also have an immature immunophenotype. These LMO1/2-r cases are associated with very high expression of LMO proteins. LMO expression is not specific to the HOXA dysregulated subtype, as it is also seen in the very rare (<2%) provisional subtype “BHLH, other”, which includes the genetic lesions TRB::LYL1 and TCR::OLIG2/BHLHB1.
The final two subtypes “SPI1-r” and “NKX2-r” are also rare (<5%), and are predominantly reported in children. The fusion partners for the SPI1-r subtype include STMN1, TCF7, and BCL11B; the blasts characteristically express HLA-DR on immunophenotyping studies; and this subtype has a very poor prognosis. The NKX2-r subtype includes rearrangements between NKX2.1, NKX2.2, NKX2.5 and various partners; this rare subtype is not well characterized.
DIAGNOSTIC APPROACH TO ALL
Unlike most neoplasms, morphology plays little role in the diagnosis of ALL, beyond the simple recognition of blasts that need to be further characterized by lineage, most often by flow cytometry. Morphologic characteristics of either the blasts or the background marrow can rarely be helpful in focusing the work-up. For instance, increased eosinophils raise the possibility of B-ALL with t(5;14), although by itself this is not specific. Leukemic cell morphology resembling Burkitt leukemia/lymphoma can be seen in MYC-rearranged B-ALL, and has also been reported in cases of MEF2D-r ALL [64]. However, ALL subclassification requires the application of a variety of ancillary technologies. Tables 5 and 6 summarize how different technologies can be used to identify specific subtypes of B-ALL and T-ALL, respectively.
Table 5.
B-ALL Diagnostic Considerations and Ancillary Testing.
| Clinical Information |
Infant: B-ALL with KMT2A-r, NUTM1-r History of CML: absent in BCR::ABL1 + ALL-M Splenomegaly: favors chronic mveloid leukemia in ivmphoid blast crisis over BCR::ABL1+ ALL-M History of lymphoma: absent in B-ALL with MYC-r Eosinophilia: B-ALL with t(5:14): B-ALL, BCR::ABL1-like, ABL1 class-r (PDGFRB-r) |
|
| Immunopheno type |
Flow cytometry With few exceptions generally not specific enough to be considered diagnostic, but may focus additional testing. |
CD10: nonspecific, but characteristically dim/negative in B-ALL with KMT2A-r, ZNF384-r, CDX2/UBTF, MEF2D-r (MEF2D- r is also CD38++) Myeloid antigens (CD13/33): nonspecific, but typically positive in BCR::ABL1+ ALL-M and -L; B-ALL NUTM1-r, ZNF384-r, or ZNF384-like CD15:B-ALL with KMT2A-r or NUTM1-r (CD15 expression reported in a subset of cases [78]) CD2: B-ALL with DUX4-r (must be expressed with CD371, as CD2 expression by itself is non-specific) CD34: absent in B-ALL with t(1;19) and MYC-r Surface light chain (dim): non-specific: may be seen in B-ALL with MYC-r Markers that may not be available in all laboratories: Cytoplasmic μ chain: B-ALL with TCF3::PBX1, MEF2D-r, or CDX2/UBTF CRLF2: overexpressed in B-ALL, BCR::ABL1-like, IAK-STAT activated with CRLF-r CD24: absent in B-ALL with KMT2A-r CD371: B-ALL with DUX4-r (coexpressed with CD2) CD27+/CD44 dim or negative: B-ALL with t(12;21)(ETV6::RUNX1) and ETV6;;RUNX1-like CD9: strong expression in B-ALL with t(1;19)/TCF3::PBX1; absent in B-ALL with ETV6::RUNX1 |
|
Immunohistochemistry (IHC) |
TdT IHC: Differentiate between B-ALL (usually +) and aggressive mature B-cell lymphoma (rarely +) NUT IHC: Positive in B-ALL with NUTM1-r (consider NUT IHC in infant B-ALL if KMT2A-r negative) DUX4: Positive in B-ALL with DUX4-r |
|
| Cytogenetics | Karyotype |
Modal chromosome number and/or DNA index: B-ALL, Hyperdiploid (>50 chromosomes) B-ALL, Hypodiploid: Near Haploid (24–31) and Low Hypodiploid (32–39 chromosomes; if low hypodiploid consider germline testing) Translocations (may require confirmation by FISH and/or molecular assays): t(9;22), KMT2A-r, t(5;14); t(1;19) |
| Fluorescence in situ hybridization (FISH) |
BCR::ABL1 fusion probe: BCR::ABL1+ALL-M will have BCR:ABL1+ granulocytes and may show a discrepancy between total % blasts and % bCr-ABL1+ cells ETV6::RUNX1 fusion probe: t(12;21)(ETV6::RUNX1); also identifies B-ALL with iAMP21. KMT2A breakapart: B-ALL with KMT2A-r TCF3: B-ALL with t(l;19) - confirm with karyotype/fusion studies If the above FISH studies are negative, then reflex to a BCR::ABL1-like panel to evaluate for rearrangements in: ABL1, ABL2, CSF1R, PDGFRB: B-ALL, BCR::ABL1-like, ABL1 class-r CRLF2, IAK2, EPOR: B-ALL, BCR::ABL1-like, IAK-STAT activated NTRK3: subset of B-ALL, BCR::ABL1-like, NOS (responds to TKIs) MYC breakapart: B-ALL with MYC-r (if +, consider adding BCL2 and BCL6 probes) Additional commercially available probes facilitate detection of B-ALL with: MEF2D::BCL9 fusion: MEF2D-r (subset) ZNF384 breakapart: ZNF384-r HLF breakapart: HLF-r NUTM1 breakapart: NUTM1-r |
|
| Molecular Diagnostics |
Gene panel sequencing Often colloquially referred to as a “next generation sequencing (NGS) panel” |
PAX5:
B-ALL with PAX5 P80R IKZF1: B-ALL with IKZFl N159Y Other targetable kinases and important co-mutations (e.g. TP53, IKZF1) |
| Whole Genome Sequencing: DNA based genomic analysis | Aneuploidy Rearrangements, including those that do not result in a chimeric transcript Mutations |
|
|
Targeted molecular assays: RT-PCR using sequence-specific primers for each partner in a known fusion protein |
BCR::ABL1 (p190 versus p210; may be used for MRD):
BCR::ABL+ ALL-M and -L UBTF::ATXN7L3: CDX2/UBTF-deregulated Identification of mutations that may be cryptic on cytogenetic studies: t(l2;2l)(ETV6::RUNX1) t(l;l9)TCF3::PBX1 Rapid detection of specific mutations in order to guide early therapy. |
|
| Targeted transcriptome sequencing: RNA-Seq using specific primers. For fusion transcript analysis, primers are utilized for one member of the fusion, and anchored multiplex PCR captures the full array of in-frame fusion partners. |
BCR and ABL1: BCR::ABL1+ ALL-M and - L; subset of B-ALL, BCR::ABL1-like KMT2A: B-ALL with KMT2A-r ETV6 and RUNX1: B-ALL with t(12;21)(ETV6::RUNX1) TCF3: B-ALL with TCF3::PBX1, TCF3::HLF, TCF3-ZNF384 MYC: B-ALL with MYC-r (if +, then evaluate for BCL2-r, BCL6-r) ABL1, ABL2, CSF1R, PDGFRB: B-ALL, BCR::ABL1-like, ABL1 class-r CRLF2, IAK2, EPOR: B-ALL, BCR::ABL1-like, IAK-STAT activated FLT3, FGFR1, NTRK3, PTK2B: B-ALL, BCR::ABL1-like. NOS The following fusions may not yet be available on standard gene fusion panels: ZNF384: ZNF384-r UBTF::ATXN7L3: CDX2/UBTF-deregulated Evaluation for selected single gene mutations and gene expression is also possible via targeted transcriptome sequencing, and may be incorporated into panels |
|
| Whole Transcriptome Sequencing: RNA-Seq using random primers to evaluate the full transcriptome | Aneuploidy, partial gains/loss of chromosomes Identification of gene rearrangements that generate fusion proteins Mutations in transcribed sequences Gene expression (B-ALL, BCR::ABL1-like; PAX5alt; ETV6::RUNX1-like) |
|
| Somatic Hypermutation | Negative: B-ALL with MYC-r Positive: MYC-r aggressive B-cell lymphomas | |
| Clonality studies | Recommended at diagnosis to facilitate molecular MRD monitoring *The presence or absence of IGH and/or TCR clones cannot utilized for lineage assignment (B-ALL versus T-ALL). |
|
New Entities in the ICC subclassification are underlined.
Table 6.
T-ALL Diagnostic Considerations and Ancillary Testing.
| Clinical Information |
History of CML: consider T-lymphoblastic transformation (rare) Eosinophilia: consider “myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusions,” including FGFR1-r and FLT3-r |
|
| Immunophenot yping | Flow cytometry |
ETP ALL, NOS and ETP ALL, BCL11B-a: cytoplasmic CD3+*, CD7 +, CD8−, CD1a−, one or more myeloid/stem cell markers (CD34, CD117, HLA-DR, CD13, CD3, CD11b, CD65), CD4+/−, CD2+/−, CD5 (<75% of blasts) “Near-ETP T-ALL.”: ETP immunophenotype with ≥75% of blasts CD5+ *T-ALL characteristically expresses cCD3, but a specific level of expression is not required for a diagnosis of T-ALL Note: Dedicated flow cytometry studies to characterize the neoplastic cells for later MRD monitoring is strongly recommended at diagnosis; however, MRD studies utilize a limited panel of antibodies and do not provide comprehensive immunophenotypic analysis of the blasts. |
| Immunohistochemis try |
BCL11B: Positive in a subset of ETP ALL, BCL11B-a; negative in T-ALL with TXL3::BCL11B (TXL3-r) LMO2: Possible role for immunohistochemistry in the detection of T-ALLwith LMO2-r PU.1: needs validation but might be useful for TALL with SPI1-r |
|
| Cytogenetics | Fluorescence in situ hybridization (FISH) |
BCL11B breakapart:
ETP-ALL with BCL11B-a (subset) Note: FISH studies will not identify BCL11B-r alterations from enhancer amplification downstream of BCL11B that results in looping of the enhancer to BCL11B |
| Molecular Diagnostics | Clonality studies | Recommended at diagnosis to facilitate molecular MRD monitoring *The presence or absence of a TCR and/or IGH clone cannot utilized for lineage assignment (T-ALL versus B-ALL). |
New/provisional Entities in the ICC subclassification are underlined.
Prior to the genomic analyses of ALL conducted in the last decade, most recognized ALL subtypes were amenable to detection by chromosomal karyotyping, FISH, and a small panel of molecular approaches to detect recurrent fusions (e.g. ETV6::RUNX1). These approaches are inadequate, however, to detect rearrangements that are cryptic (e.g. DUX4-r); the full diversity of rearrangements in several subtypes (e.g. MEF2D-r, ZNF384-r, NUTM1-r); sequence mutations that are hallmarks of several subtypes (PAX5 P80R and IKZF1 N159Y, and a subset of PAX5alt with PAX5 R38H/R140H mutations); or, in the most extreme example, the full diversity of sequence and structural variants of BCR::ABL-like B-ALL [65]. Comprehensive identification of all alterations requires genomic sequencing. As a single modality, RNA sequencing can identify most alterations required for diagnosis, risk stratification, and therapeutic targeting; however, if this methodology is not available then alternative approaches may be used to detect many ALL subtypes, particularly those that merit consideration of TKI therapy. The capabilities and limitations of each modality are briefly discussed below.
Immunophenotyping.
Immunophenotyping by either flow cytometry or immunohistochemistry (IHC) can often suggest, and in few cases definitively establish, a diagnosis of a particular subtype of ALL. ETP ALL is defined by flow cytometry, and CRLF2 expression by flow correlates very well with that subset of BCR::ABL1-like ALL with CRLF2 rearrangement. It is important to note, however, that CRLF2 overexpression is not specific for BCR::ABL1-like ALL because the translocation is also found as a secondary event in cases of iAMP21 and hyperdiploid ALL. Also, some cases of Down syndrome with CRLF2-r have a GEX profile that is different from other cases of BCR::ABL1-like ALL.
Other phenotypes correlate with many newly defined entities, although few have high specificity. The most promising surrogate is probably co-expression of CD371 and CD2 in DUX4-r B-ALL. CD19+CD27+CD44-/dim is highly but not perfectly predictive of either ETV6::RUNX1 or ETV6::RUNX1-like ALL, and can help identify cases of the latter when ETV6::RUNX1 is not identified. Many of the less common B-ALL entities, including KMT2A-r, ZNF384-r (and ZNF384-like), MEF2D-r, and CDX2/UBTF B-ALL, are characterized by dim or negative CD10; additional immunophenotypic features, such as CD15 expression in KMT2A-r or cytoplasmic mu with high levels of CD38 in MEF2D-r, can help narrow down possibilities.
Many translocations result in overexpression of proteins that are detectable by IHC, although the relevant data are limited. IHC to detect the N-terminus of DUX4 is sensitive and specific for the DUX4-r subtype of B-ALL [23]. NUT expression, which is also used in the diagnosis of NUT carcinomas, has been reported in cases of NUTM1-r B-ALL [45]. Among T cell cases, BCL11B is expressed in the BCL11B-a subtype of ETP-ALL [63]. Interpretation should be done with caution, however, because other cases of non-ETP T-ALL or T/myeloid MPAL may express BCL11B, so IHC must be used in conjunction with flow cytometry to confirm the diagnosis of ETP. LMO2 IHC can recognize a subset of cases of T-ALL that likely includes the LMO1/2-r subtype [66, 67] although it is not specific because the rare “BHLH, other” subtype has elevated LMO proteins. Other immunophenotypic findings are largely untested, but because there are well-characterized antibodies against PU.1 and TTF1, (the gene products of SPI1 and NKX2.1, respectively) these could potentially be useful to detect cases of these T-ALL subsets.[68, 69]
Cytogenetics.
Conventional karyotyping can detect aneuploidy (hyper- and hypodiploidy) and a subset of recurrent rearrangements. Karyotyping is insufficiently sensitive to identify all recurring rearrangements as many are cryptic, most notably ETV6::RUNX1 and DUX4-r, as well as subsets of the multiple partners of other key genes such as MEF2D, NUTM1 and ZNF384.
Fluorescence in situ hybridization.
FISH remains a valuable approach to detect many of the rearrangements in ALL, as well as the chromosome 21 amplification characteristic of iAMP21 B-ALL. Either interphase or metaphase break-apart and/or co-localization assays may be used to detect rearrangements of ABL1-class kinases [70], CRLF2 (with the same caveats mentioned for flow cytometry) [71], Janus kinases [72], MEF2D [32], NUTM1, and ZNF384 [27], as well as less commonly rearranged kinases (e.g. NTRK3 [15]). FISH may be used to detect the majority of rearrangements involving ABL1 and the JAK-STAT pathway, allowing laboratories to detect “actionable” cases of ALL for which a TKI may be useful. Of note, FISH can also detect BCL11B rearrangements [61,73]; however, the presence of a BCL11B rearrangement is nonspecific as BCL11B-r are observed in both T-ALL with BCL11B::TLX3 and ETP ALL, BCL11B-activated, so a combination of immunophenotyping and FISH is required for accurate diagnosis. Moreover, FISH cannot detect the subtype of BCL11B-activated cases that arise from amplification of a neoenhancer distal to BCL11B, rather than rearrangement to an enhancer on another chromosome. Similarly, FISH cannot detect many of the truncating rearrangements that lead to activation of EPOR (due to the focal insertion of EPOR in IGH or other enhancer regions) [73], or rearrangements of DUX4 (due to the repetitive nature of the DUX4 locus on 4q and 10q) [20].
Targeted molecular assays.
Reverse transcription followed by the polymerase chain reaction (RT-PCR) using primers directed against specific fusion partners is useful for the identification of several rearrangements, including BCR::ABL1, TCF3::PBX1, ETV6::RUNX1, and UBTF::ATXN7L3 (this last rearrangement may also be detected by genomic PCR) [46]. This methodology is particularly useful for detection of cryptic rearrangements, verification of the expression and structure of novel fusions identified by other assays (e.g. FISH), or if rapid detection is warranted to guide early therapy. RT-PCR may also be used to detect many of the recurrent rearrangements of other subtypes, such as BCR::ABL1-like B-ALL, but is limited by the great diversity in fusion partners for several kinases.
Transcriptome sequencing.
Both targeted and unbiased TS are powerful approaches to detect many alterations in ALL. In contrast to the targeted molecular assays described above, where both members of a fusion protein must be known, targeted TS uses anchored multiplex PCR to identify the full array of fusion partners for selected genes. These approaches can identify not only fusion partners but also other abnormalities that are transcribed into RNA, as well as GEX levels. Targeted panels are commercially available, and some can be customized to include selected fusions, single gene mutations, and GEX levels. Targeted approaches may be challenged by some genomic abnormalities, including focal (EPOR) or repetitive (DUX4-r) alterations, but can be designed to identify high DUX4 expression.
Unbiased RNA-seq, or whole TS, is similar to targeted TS, but uses random or oligo-dT priming to sequence all or polyadenylated expressed transcripts, and thus more fully detects rearrangements, aneuploidy, sequence mutations, and GEX levels. This method may be required to assign cases to complex, heterogeneous (e.g. PAX5alt and BCR::ABL1-like), and phenocopy subtypes (e.g. ETV6::RUNX1-like) [18]. Whole TS is gaining increasing acceptance with the availability of standardized analytical approaches [74].
Genome sequencing.
Next generation sequencing of targeted, captured DNA may be used to identify single gene mutations in DNA, such as those seen in BALL with PAX5 P80R. This utility of this method is determined in part by the extent of the selected gene panel; importantly, capture-based sequencing has limited ability to detect structural rearrangements as breakpoints commonly fall outside regions of captured DNA.
As a single platform, whole genome sequencing (WGS) is the most comprehensive approach, and has several advantages over whole TS, including the ability to detect all sequence mutations and structural variants/copy number changes, and to identify rearrangements that may not result in expression of a chimeric transcript, which is particularly common in T-ALL. Recent studies have demonstrated the utility of WGS in clinical diagnostics [75]. While not widely used clinically at present, it is expected that, like TS, WGS will become increasingly adopted in future. Despite the power of this approach, WGS cannot fully replace TS, as it cannot provide information about GEX levels, or directly demonstrate the transcriptional consequences of a rearrangement.
Gene expression clustering.
Following the identification of BCR::ABL1-like ALL, the field has grappled with the problem of (1) how to identify all cases of this entity, as the definition is based on leukemic cell GEX; and (2) how to identify all actionable kinase-signaling driver lesions [76]. Several groups have implemented a low-density multiplexed GEX array methodology that identifies the GEX signature of BCR::ABL1-like ALL, and a subset of drivers of this and other ALL subtypes [77]. This method may be used to direct cases for detailed molecular testing. A limitation is the need to standardize the analytical methodology. In the absence of a GEX approach to identify BCR::ABL1-like ALL, a reasonable compromise is to report an alteration (e.g. CRLF2 rearrangement or rearrangement encoding a kinase fusion oncoprotein) as “consistent with BCR::ABL1-like ALL”.
CONCLUSIONS
The ICC classification of ALL is primarily a genomic-based classification of entities that has been updated based on extensive research done over the past decade. It provides a standard language that should be used in large scale clinical trials to better understand the clinical behavior of these entities. It is recognized that application of this classification in practice will be challenging for many labs as techniques needed to define all of the entities are not yet universally available. For now, many cases of ALL will need to be classified without such comprehensive analysis; if a specific entity cannot be defined, then supplemental information in reports should specify the extent to which certain entities have been excluded. However, given the pace of technological development, it is expected that diagnosis of these new subtypes will be more routine well before the time comes for a new classification.
ACKNOWLEDGEMENTS
The authors thank the members of the working group including Elias Jabbour, Ching-Hon Pui, Kathryn Foucar, Nicola Goekbuget, Hartmut Doehner, and Mignon Loh, as well as Daniel Arber for thoughtful discussion.
Additionally, the authors thank Dr. Qingsong Gao (St. Jude Children’s Research Hospital, Memphis, TN), who created Figure 1.
Funding:
A.S.D.: Financial support was received from P30 CA008748 (National Cancer Institute, National Institutes of Health).
C.G.M.: Financial support was received from P30 CA021765 and R35 CA197695 (National Cancer Institute, National Institutes of Health).
M.J.B.: Financial support was received from U10 CA180886 (National Cancer Institute, National Institutes of Health).
Abbreviations:
- ALL
Acute lymphoblastic leukemia
- B-ALL
B-lymphoblastic leukemia
- T-ALL
T-lymphoblastic leukemia
- ICC
International Consensus Classification
- GEX
gene expression
- BCR::ABL1+ ALL-L
BCR::ABL1 B-ALL with lymphoid only involvement
- BCR::ABL1+ ALL-M
BCR::ABL1-positive B-ALL with multilineage involvement
- CML
chronic myeloid leukemia
- LBP
lymphoid blast phase
- FISH
fluorescence in situ hybridization
- NOS
not otherwise specified
- TKIs
tyrosine kinase inhibitors
- TS
transcriptome sequencing
- AYA
adolescents and young adults
- MPAL
mixed phenotype acute leukemia
- CDX2/UBTF
B-ALL with UBTF::ATXN7L3/PAN3,CDX2
- NGS
next-generation sequencing
- PAX5alt
PAX5-altered B-ALL
- ETP
early T precursor
- BCL11B-a
BCL11B-activated
- TAL1/2-r
TAL1-rearranged or TAL2-rearranged
- LMO1/2-r
LMO1-rearranged or LMO2-rearranged
- IHC
immunohistochemistry
- WGS
whole genome sequencing
Footnotes
Conflict of Interest:
Authors A.S.D. and M.J.B. declare no competing interests. Author C.G.M. has received speaker (Illumina and Amgen) and consultant (Faze, Beam) honoraria, and receives research funding from AbbVie and Pfizer.
Compliance with Ethical Standards:
All authors have made substantial contributions to this review, have read and approved the final version submitted, and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
The contents of Tables 1, 2, 3 and 4 are similar to those published in “Arber DA, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemia: Integrating Morphological, Clinical, and Genomic Data, Blood. 2022 Jun 29;blood.2022015850” because the current review expands on the International Consensus Classification first described in the June 2022 manuscript; however, the contents of the current manuscript have not been copyrighted.
REFERENCES
- 1.Arber DA, Hasserjian RP, Orazi A, et al. (2022) Classification of myeloid neoplasms/acute leukemia: Global perspectives and the international consensus classification approach. Am J Hematol 97:514–518. 10.1002/ajh.26503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arber DA, Orazi A, Hasserjian R, et al. (2016) The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127:2391–2405. 10.1182/blood-2016-03-643544 [DOI] [PubMed] [Google Scholar]
- 3.Chen Z, Hu S, Wang SA, et al. (2020) Chronic myeloid leukemia presenting in lymphoblastic crisis, a differential diagnosis with Philadelphia-positive B-lymphoblastic leukemia. Leuk Lymphoma 61:2831–2838. 10.1080/10428194.2020.1795160 [DOI] [PubMed] [Google Scholar]
- 4.Hovorkova L, Zaliova M, Venn NC, et al. (2017) Monitoring of childhood ALL using BCR-ABL1 genomic breakpoints identifies a subgroup with CML-like biology. Blood 129:2771–2781. 10.1182/blood-2016-11-749978 [DOI] [PubMed] [Google Scholar]
- 5.Biondi A, Gandemer V, De Lorenzo P, et al. (2018) Imatinib treatment of paediatric Philadelphia chromosome-positive acute lymphoblastic leukaemia (EsPhALL2010): a prospective, intergroup, open-label, single-arm clinical trial. Lancet Haematol 5:e641–e652. 10.1016/S2352-3026(18)30173-X [DOI] [PubMed] [Google Scholar]
- 6.Ware AD, Wake L, Brown P, et al. (2019) B-Lymphoid Blast Phase of Chronic Myeloid Leukemia: A Case Report and Review of the Literature. AJSP Rev Rep 24:191–195 [PMC free article] [PubMed] [Google Scholar]
- 7.Schultz KR, Carroll A, Heerema NA, et al. (2014) Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children’s Oncology Group study AALL0031. Leukemia 28:1467–1471. 10.1038/leu.2014.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cazzaniga G, De Lorenzo P, Alten J, et al. (2018) Predictive value of minimal residual disease in Philadelphia-chromosome-positive acute lymphoblastic leukemia treated with imatinib in the European intergroup study of post-induction treatment of Philadelphia-chromosome-positive acute lymphoblastic leukemia, based on immunoglobulin/T-cell receptor and BCR/ABL1 methodologies. Haematologica 103:107–115. 10.3324/haematol.2017.176917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanasi I, Ba I, Sirvent N, et al. (2019) Efficacy of tyrosine kinase inhibitors in Ph-like acute lymphoblastic leukemia harboring ABL-class rearrangements. Blood 134:1351–1355. 10.1182/blood.2019001244 [DOI] [PubMed] [Google Scholar]
- 10.Roberts KG, Mullighan CG (2015) Genomics in acute lymphoblastic leukaemia: insights and treatment implications. Nat Rev Clin Oncol 12:344–357. 10.1038/nrclinonc.2015.38 [DOI] [PubMed] [Google Scholar]
- 11.Swerdlow SH, Campo E, Harris NL, et al. (2017) WHO classification of tumours of haematopoietic and lymphoid tissues, Revised 4th edition. International Agency for Research on Cancer, Lyon [Google Scholar]
- 12.Reshmi SC, Harvey RC, Roberts KG, et al. (2017) Targetable kinase gene fusions in high-risk B-ALL: a study from the Children’s Oncology Group. Blood 129:3352–3361. 10.1182/blood-2016-12-758979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maese L, Raetz EA (2019) Can Ph-like ALL be effectively targeted? Best Pract Res Clin Haematol 32:101096. 10.1016/j.beha.2019.101096 [DOI] [PubMed] [Google Scholar]
- 14.Roberts KG, Mullighan CG (2020) The Biology of B-Progenitor Acute Lymphoblastic Leukemia. Cold Spring Harb Perspect Med 10:a034835. 10.1101/cshperspect.a034835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roberts KG, Janke LJ, Zhao Y, et al. (2018) ETV6-NTRK3 induces aggressive acute lymphoblastic leukemia highly sensitive to selective TRK inhibition. Blood 132:861–865. 10.1182/blood-2018-05-849554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swerdlow SH, International Agency for Research on Cancer (2008) WHO classification of tumours of haematopoietic and lymphoid tissues, 4. ed. Internat. Agency for Research on Cancer, Lyon [Google Scholar]
- 17.Holmfeldt L, Wei L, Diaz-Flores E, et al. (2013) The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet 45:242–252. 10.1038/ng.2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gu Z, Churchman ML, Roberts KG, et al. (2019) PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet 51:296–307. 10.1038/s41588-018-0315-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li J-F, Dai Y-T, Lilljebjörn H, et al. (2018) Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Proc Natl Acad Sci U S A 115:E11711–E11720. 10.1073/pnas.1814397115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang J, McCastlain K, Yoshihara H, et al. (2016) Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet 48:1481–1489. 10.1038/ng.3691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lilljebjörn H, Henningsson R, Hyrenius-Wittsten A, et al. (2016) Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat Commun 7:11790. 10.1038/ncomms11790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yasuda T, Tsuzuki S, Kawazu M, et al. (2016) Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat Genet 48:569–574. 10.1038/ng.3535 [DOI] [PubMed] [Google Scholar]
- 23.Siegele BJ, Stemmer-Rachamimov AO, Lilljebjorn H, et al. (2022) N-terminus DUX4-immunohistochemistry is a reliable methodology for the diagnosis of DUX4-fused B-lymphoblastic leukemia/lymphoma (N-terminus DUX4 IHC for DUX4-fused B-ALL). Genes Chromosomes Cancer 61:449–458. 10.1002/gcc.23033 [DOI] [PubMed] [Google Scholar]
- 24.Schinnerl D, Mejstrikova E, Schumich A, et al. (2019) CD371 cell surface expression: a unique feature of DUX4-rearranged acute lymphoblastic leukemia. Haematologica 104:e352–e355. 10.3324/haematol.2018.214353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirabayashi S, Butler ER, Ohki K, et al. (2021) Clinical characteristics and outcomes of B-ALL with ZNF384 rearrangements: a retrospective analysis by the Ponte di Legno Childhood ALL Working Group. Leukemia 35:3272–3277. 10.1038/s41375-021-01199-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shago M, Abla O, Hitzler J, et al. (2016) Frequency and outcome of pediatric acute lymphoblastic leukemia with ZNF384 gene rearrangements including a novel translocation resulting in an ARID1B/ZNF384 gene fusion. Pediatr Blood Cancer 63:1915–1921. 10.1002/pbc.26116 [DOI] [PubMed] [Google Scholar]
- 27.Alexander TB, Gu Z, Iacobucci I, et al. (2018) The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 562:373–379. 10.1038/s41586-018-0436-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McGinnis E, Yang D, Au N, et al. (2021) Clinical and laboratory features associated with myeloperoxidase expression in pediatric B-lymphoblastic leukemia. Cytometry B Clin Cytom 100:446–453. 10.1002/cyto.b.21966 [DOI] [PubMed] [Google Scholar]
- 29.Hirabayashi S, Ohki K, Nakabayashi K, et al. (2017) ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica 102:118–129. 10.3324/haematol.2016.151035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Janet NB, Kulkarni U, Arun AK, et al. (2021) Systematic application of fluorescence in situ hybridization and immunophenotype profile for the identification of ZNF384 gene rearrangements in B cell acute lymphoblastic leukemia. Int J Lab Hematol 43:658–663. 10.1111/ijlh.13580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zaliova M, Winkowska L, Stuchly J, et al. (2021) A novel class of ZNF384 aberrations in acute leukemia. Blood Adv 5:4393–4397. 10.1182/bloodadvances.2021005318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gu Z, Churchman M, Roberts K, et al. (2016) Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat Commun 7:13331. 10.1038/ncomms13331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohki K, Kiyokawa N, Saito Y, et al. (2019) Clinical and molecular characteristics of MEF2D fusion-positive B-cell precursor acute lymphoblastic leukemia in childhood, including a novel translocation resulting in MEF2D-HNRNPH1 gene fusion. Haematologica 104:128–137. 10.3324/haematol.2017.186320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu W, Hu S, Konopleva M, et al. (2015) De Novo MYC and BCL2 Double-hit B-Cell Precursor Acute Lymphoblastic Leukemia (BCP-ALL) in Pediatric and Young Adult Patients Associated With Poor Prognosis. Pediatr Hematol Oncol 32:535–547. 10.3109/08880018.2015.1087611 [DOI] [PubMed] [Google Scholar]
- 35.Paietta E, Roberts KG, Wang V, et al. (2021) Molecular classification improves risk assessment in adult BCR-ABL1-negative B-ALL. Blood 138:948–958. 10.1182/blood.2020010144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Navid F, Mosijczuk AD, Head DR, et al. (1999) Acute lymphoblastic leukemia with the (8;14)(q24;q32) translocation and FAB L3 morphology associated with a B-precursor immunophenotype: the Pediatric Oncology Group experience. Leukemia 13:135–141. 10.1038/sj.leu.2401244 [DOI] [PubMed] [Google Scholar]
- 37.Wagener R, López C, Kleinheinz K, et al. (2018) IG-MYC + neoplasms with precursor B-cell phenotype are molecularly distinct from Burkitt lymphomas. Blood 132:2280–2285. 10.1182/blood-2018-03-842088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moench L, Sachs Z, Aasen G, et al. (2016) Double- and triple-hit lymphomas can present with features suggestive of immaturity, including TdT expression, and create diagnostic challenges. Leuk Lymphoma 57:2626–2635. 10.3109/10428194.2016.1143939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ok CY, Medeiros LJ, Thakral B, et al. (2019) High-grade B-cell lymphomas with TdT expression: a diagnostic and classification dilemma. Mod Pathol 32:48–58. 10.1038/s41379-018-0112-9 [DOI] [PubMed] [Google Scholar]
- 40.Bhavsar S, Liu Y-C, Gibson SE, et al. (2022) Mutational Landscape of TdT+ Large B-cell Lymphomas Supports Their Distinction From B-lymphoblastic Neoplasms: A Multiparameter Study of a Rare and Aggressive Entity. Am J Surg Pathol 46:71–82. 10.1097/PAS.0000000000001750 [DOI] [PubMed] [Google Scholar]
- 41.Nie K, Redmond D, Eng KW, et al. (2021) Mutation landscape, clonal evolution pattern, and potential pathogenic pathways in B-lymphoblastic transformation of follicular lymphoma. Leukemia 35:1203–1208. 10.1038/s41375-020-01014-2 [DOI] [PubMed] [Google Scholar]
- 42.Geyer JT, Subramaniyam S, Jiang Y, et al. (2015) Lymphoblastic transformation of follicular lymphoma: a clinicopathologic and molecular analysis of 7 patients. Hum Pathol 46:260–271. 10.1016/j.humpath.2014.10.021 [DOI] [PubMed] [Google Scholar]
- 43.Boer JM, Valsecchi MG, Hormann FM, et al. (2021) Favorable outcome of NUTM1-rearranged infant and pediatric B cell precursor acute lymphoblastic leukemia in a collaborative international study. Leukemia 35:2978–2982. 10.1038/s41375-021-01333-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hormann FM, Hoogkamer AQ, Beverloo HB, et al. (2019) NUTM1 is a recurrent fusion gene partner in B-cell precursor acute lymphoblastic leukemia associated with increased expression of genes on chromosome band 10p12.31–12.2. Haematologica 104:e455–e459. 10.3324/haematol.2018.206961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pincez T, Landry J-R, Roussy M, et al. (2020) Cryptic recurrent ACIN1-NUTM1 fusions in non-KMT2A-rearranged infant acute lymphoblastic leukemia. Genes Chromosomes Cancer 59:125–130. 10.1002/gcc.22808 [DOI] [PubMed] [Google Scholar]
- 46.Kimura S, Montefiori L, Iacobucci I, et al. (2022) Enhancer retargeting of CDX2 and UBTF::ATXN7L3 define a subtype of high-risk B-progenitor acute lymphoblastic leukemia. Blood 139:3519–3531. 10.1182/blood.2022015444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Passet M, Kim R, Gachet S, et al. (2022) Concurrent CDX2 cis-deregulation and UBTF::ATXN7L3 fusion define a novel high-risk subtype of B-cell ALL. Blood 139:3505–3518. 10.1182/blood.2021014723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yasuda T, Sanada M, Kawazu M, et al. (2022) Two novel high-risk adult B-cell acute lymphoblastic leukemia subtypes with high expression of CDX2 and IDH1/2 mutations. Blood 139:1850–1862. 10.1182/blood.2021011921 [DOI] [PubMed] [Google Scholar]
- 49.Fischer U, Forster M, Rinaldi A, et al. (2015) Genomics and drug profiling of fatal TCF3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat Genet 47:1020–1029. 10.1038/ng.3362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leonard J, Wolf JS, Degnin M, et al. (2021) Aurora A kinase as a target for therapy in TCF3-HLF rearranged acute lymphoblastic leukemia. Haematologica 106:2990–2994. 10.3324/haematol.2021.278692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mouttet B, Vinti L, Ancliff P, et al. (2019) Durable remissions in TCF3-HLF positive acute lymphoblastic leukemia with blinatumomab and stem cell transplantation. Haematologica 104:e244–e247. 10.3324/haematol.2018.210104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Passet M, Boissel N, Sigaux F, et al. (2019) PAX5 P80R mutation identifies a novel subtype of B-cell precursor acute lymphoblastic leukemia with favorable outcome. Blood 133:280–284. 10.1182/blood-2018-10-882142 [DOI] [PubMed] [Google Scholar]
- 53.Nebral K, Denk D, Attarbaschi A, et al. (2009) Incidence and diversity of PAX5 fusion genes in childhood acute lymphoblastic leukemia. Leukemia 23:134–143. 10.1038/leu.2008.306 [DOI] [PubMed] [Google Scholar]
- 54.Mullighan CG, Goorha S, Radtke I, et al. (2007) Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 446:758–764. 10.1038/nature05690 [DOI] [PubMed] [Google Scholar]
- 55.Dang J, Wei L, de Ridder J, et al. (2015) PAX5 is a tumor suppressor in mouse mutagenesis models of acute lymphoblastic leukemia. Blood 125:3609–3617. 10.1182/blood-2015-02-626127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zaliova M, Stuchly J, Winkowska L, et al. (2019) Genomic landscape of pediatric B-other acute lymphoblastic leukemia in a consecutive European cohort. Haematologica 104:1396–1406. 10.3324/haematol.2018.204974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bastian L, Schroeder MP, Eckert C, et al. (2019) PAX5 biallelic genomic alterations define a novel subgroup of B-cell precursor acute lymphoblastic leukemia. Leukemia 33:1895–1909. 10.1038/s41375-019-0430-z [DOI] [PubMed] [Google Scholar]
- 58.Zaliova M, Potuckova E, Lukes J, et al. (2021) Frequency and prognostic impact of ZEB2 H1038 and Q1072 mutations in childhood B-other acute lymphoblastic leukemia. Haematologica 106:886–890. 10.3324/haematol.2020.249094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morita K, Jain N, Kantarjian H, et al. (2021) Outcome of T-cell acute lymphoblastic leukemia/lymphoma: Focus on near-ETP phenotype and differential impact of nelarabine. Am J Hematol 96:589–598. 10.1002/ajh.26144 [DOI] [PubMed] [Google Scholar]
- 60.Liu Y, Easton J, Shao Y, et al. (2017) The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet 49:1211–1218. 10.1038/ng.3909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Di Giacomo D, La Starza R, Gorello P, et al. (2021) 14q32 rearrangements deregulating BCL11B mark a distinct subgroup of T-lymphoid and myeloid immature acute leukemia. Blood 138:773–784. 10.1182/blood.2020010510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Montefiori LE, Bendig S, Gu Z, et al. (2021) Enhancer Hijacking Drives Oncogenic BCL11B Expression in Lineage-Ambiguous Stem Cell Leukemia. Cancer Discov 11:2846–2867. 10.1158/2159-8290.CD-21-0145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fang H, Wang W, El Hussein S, et al. (2021) B-cell lymphoma/leukaemia 11B (BCL11B) expression status helps distinguish early T-cell precursor acute lymphoblastic leukaemia/lymphoma (ETP-ALL/LBL) from other subtypes of T-cell ALL/LBL. Br J Haematol 194:1034–1038. 10.1111/bjh.17681 [DOI] [PubMed] [Google Scholar]
- 64.Sun J, Yu W, Zhang X (2020) MEF2D-rearranged acute lymphoblastic leukemia resembles Burkitt lymphoma/leukemia. Ann Hematol 99:185–188. 10.1007/s00277-019-03857-x [DOI] [PubMed] [Google Scholar]
- 65.Iacobucci I, Kimura S, Mullighan CG (2021) Biologic and Therapeutic Implications of Genomic Alterations in Acute Lymphoblastic Leukemia. J Clin Med 10:3792. 10.3390/jcm10173792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jevremovic D, Roden AC, Ketterling RP, et al. (2016) LMO2 Is a Specific Marker of T-Lymphoblastic Leukemia/Lymphoma. Am J Clin Pathol 145:180–190. 10.1093/ajcp/aqv024 [DOI] [PubMed] [Google Scholar]
- 67.Natkunam Y, Zhao S, Mason DY, et al. (2007) The oncoprotein LMO2 is expressed in normal germinal-center B cells and in human B-cell lymphomas. Blood 109:1636–1642. 10.1182/blood-2006-08-039024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Homminga I, Pieters R, Langerak AW, et al. (2011) Integrated transcript and genome analyses reveal NKX2–1 and MEF2C as potential oncogenes in T cell acute lymphoblastic leukemia. Cancer Cell 19:484–497. 10.1016/j.ccr.2011.02.008 [DOI] [PubMed] [Google Scholar]
- 69.Nasr MR, Rosenthal N, Syrbu S (2010) Expression profiling of transcription factors in B- or T-acute lymphoblastic leukemia/lymphoma and burkitt lymphoma: usefulness of PAX5 immunostaining as pan-Pre-B-cell marker. Am J Clin Pathol 133:41–48. 10.1309/AJCPYP00JNUFWCCY [DOI] [PubMed] [Google Scholar]
- 70.Roberts KG, Li Y, Payne-Turner D, et al. (2014) Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med 371:1005–1015. 10.1056/NEJMoa1403088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mullighan CG, Collins-Underwood JR, Phillips LAA, et al. (2009) Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet 41:1243–1246. 10.1038/ng.469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roberts KG, Yang Y-L, Payne-Turner D, et al. (2017) Oncogenic role and therapeutic targeting of ABL-class and JAK-STAT activating kinase alterations in Ph-like ALL. Blood Adv 1:1657–1671. 10.1182/bloodadvances.2017011296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Iacobucci I, Kimura S, Mullighan CG (2021) Biologic and Therapeutic Implications of Genomic Alterations in Acute Lymphoblastic Leukemia. J Clin Med 10:3792. 10.3390/jcm10173792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schmidt B, Brown LM, Ryland GL, et al. (2022) ALLSorts: an RNA-Seq subtype classifier for B-cell acute lymphoblastic leukemia. Blood Adv 6:4093–4097. 10.1182/bloodadvances.2021005894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rosenquist R, Cuppen E, Buettner R, et al. (2022) Clinical utility of whole-genome sequencing in precision oncology. Semin Cancer Biol 84:32–39. 10.1016/j.semcancer.2021.06.018 [DOI] [PubMed] [Google Scholar]
- 76.Chiaretti S, Messina M, Foà R (2019) BCR/ABL1-like acute lymphoblastic leukemia: How to diagnose and treat? Cancer 125:194–204. 10.1002/cncr.31848 [DOI] [PubMed] [Google Scholar]
- 77.Harvey RC, Kang H, Roberts KG, et al. (2013) Development and Validation Of a Highly Sensitive and Specific Gene Expression Classifier To Prospectively Screen and Identify B-Precursor Acute Lymphoblastic Leukemia (ALL) Patients With a Philadelphia Chromosome-Like (“Ph-like” or “BCR-ABL1-Like”) Signature For Therapeutic Targeting and Clinical Intervention. Blood 122:826. 10.1182/blood.V122.21.826.826 [DOI] [Google Scholar]
- 78.McClure BJ, Pal M, Heatley SL, et al. (2022) Two novel cases of NUTM1-rearranged B-cell acute lymphoblastic leukaemia presenting with high-risk features. Br J Haematol 196:1407–1411. 10.1111/bjh.17995 [DOI] [PubMed] [Google Scholar]