

Abstract
Phenylethanolamine N-methyltransferase (PNMT) catalyzes the S-adenosyl-l-methionine (SAM)-dependent methylation of norepinephrine to form epinephrine. Epinephrine is implicated in the regulation of blood pressure, respiration, Alzheimer’s disease, and post-traumatic stress disorder (PTSD). Transition-state (TS) analogues bind their target enzymes orders of magnitude more tightly than their substrates. A synthetic strategy for first-generation TS analogues of human PNMT (hPNMT) permitted structural analysis of hPNMT and revealed potential for second-generation inhibitors [Mahmoodi, N.; et al. J. Am, Chem, Soc. 2020, 142, 14222–14233]. A second-generation TS analogue inhibitor of PNMT was designed, synthesized, and characterized to yield a Ki value of 1.2 nM. PNMT isothermal titration calorimetry (ITC) measurements of inhibitor 4 indicated a negative cooperative binding mechanism driven by large favorable entropic contributions and smaller enthalpic contributions. Cell-based assays with HEK293T cells expressing PNMT revealed a cell permeable, intracellular PNMT inhibitor with an IC50 value of 81 nM. Structural analysis demonstrated inhibitor 4 filling catalytic site regions to recapitulate both norepinephrine and SAM interactions. Conformation of the second-generation inhibitor in the catalytic site of PNMT improves contacts relative to those from the first-generation inhibitors. Inhibitor 4 demonstrates up to 51,000-fold specificity for PNMT relative to DNA and protein methyltransferases. Inhibitor 4 also exhibits a 12,000-fold specificity for PNMT over the α2-adrenoceptor.
Graphicl Abstract

INTRODUCTION
Phenylethanolamine N-methyltransferase (PNMT) catalyzes the conversion of norepinephrine to epinephrine as the final step in the catecholamine biosynthetic pathway (Figure 1). In humans, the expression of PNMT occurs largely in the adrenal gland where epinephrine acts as a hormone for adrenergic receptors.2 The role of epinephrine in the human central nervous system (CNS) is not well understood; however, it is associated with cardiovascular homeostasis, regulation of the circadian cycle, and adrenergic receptor activation.2 Studies suggest that PNMT may also play a role in human disease states including hypertension,3 myocardial infarction,4 Alzheimer’s disease,5 and Parkinson’s diseases.6,7 Catecholamines may also be involved in post-traumatic stress disorder (PTSD). PNMT-knockout mice (Pnmt-KO) induced with PTSD showed a decrease in stress-related behaviors compared to wild-type mice.8 However, when administered with epinephrine, an increase in stress behavior was observed, suggesting that epinephrine may be involved in the persistence of traumatic memories in PTSD, conceivably by enhancing the expression of Nr4a2 and Nr4a3 genes in the hippocampus. Nr4a is a subfamily of orphan nuclear receptor genes, which encode transcription factors Nr4a1, Nr4a2, and Nr4a3, which have been implicated in contextual fear memory formation.8,9 Thus, there is a plausible mechanism suggesting the driving role of epinephrine production as the key to memory persistence in PTSD patient experiences.
Figure 1.

(a) Reaction catalyzed by PNMT converts norepinephrine to epinephrine by transferring a methyl group from S-adenosyl-l-methionine (SAM) to the primary amine, (b) Chemical structures and inhibition constants of known PNMT inhibitors and first-generation transition-state analogues (TSA) inhibitor 3.1 Ki values shown here are measured against PNMT.
Kinetic isotope effect studies indicate a transition state (TS) for PNMT proceeding through an early dissociative SN2 mechanism.9 The rate-limiting step is the transfer of the methyl group from S-adenosyl-l-methionine (SAM) to norepinephrine. The bisubstrate nature of the reaction catalyzed by PNMT led to the development of norepinephrine or SAM substrate analogues as inhibitors. Phenylethylamines and 1,2,3,4-tetrahydroisoquinoline (THIQ) derivatives bearing electron-withdrawing groups are norepinephrine mimics that have been widely investigated for their inhibitory properties (Figure 1).10–12 Despite having Ki values in the nanomolar range (1.6 nM for SK&F 64139 and 120 nM for SK&F 29661),13 most analogues that compete with the binding of norepinephrine exhibit problems of specificity and permeability, prohibiting their further development.14 SAM analogues have been developed as methyltransferase inhibitors, but the wide range of SAM-based methyitransferases poses a challenge to developing selective agents.15–17 A strategy that combines both norepinephrine and SAM mimics has the potential to provide a bisubstrate inhibitor with enhanced specificity. Bisubstrate inhibitors confer a thermodynamic advantage by incorporating two binding moieties within one molecule.18 This approach has provided improved specificity for a PNMT inhibitor relative to the α2-adrenoceptor.19,20 A similar approach has been employed to develop bisubstrate inhibitors of nicotinamide N-methyltransferase (NNMT) and protein N-terminal methyltransferase 1 (NTMT1), with high selectivity for their enzymes.21,22
Transition-state analogues (TSA) mimic the geometry and electrostatic properties of an enzymatic transition state in chemically stable structures and have the potential to bind their cognate active sites orders of magnitude more tightly than substrate analogues.23 TSA are specific for their target protein and have the potential to minimize off-target interactions. Recently, we reported the design, synthesis, and characterization of a chemically unique first-generation TS analogue of human PNMT (inhibitor 3) (Figure 1).1 The crystal structure of PNMT in complex with SK&F 6413924 provided one element of the unique chemical skeleton for inhibitor 3, to give a Ki, value of 12 nM.1 This was among the first TSA inhibitors of methyltransferase enzymes to show affinity in the low nanomolar range.
Crystal structures of PNMT with inhibitor 3 were compared to those of S-adenosyl-l-homocysteine (SAH) and dichloro-THIQ. moieties. The dichloro-THIQ. group of inhibitor 3 is positioned in the norepinephrine binding site where a hydrophobic pocket is formed by Phe182, Tyr222, and Val269 (Figure 2a).1 Compared with the crystal structure with SK&F 64139,22–24 inhibitor 3 binding differs by the position of the dichloro-THIQ.group being approximately 1.5 Å closer to the SAM binding pocket (Figure 2b). Therefore, a TSA with a longer linker, replacing the propyl linker of inhibitor 3 with a butyl group was proposed to place die dichloro-THIQ. deeper into the norepinephrine binding pocket and afford improved binding (Figure 2c).
Figure 2.

Structural comparison of the PNMT binding sites, (a) Binding site of PNMT in the complex with inhibitor 3. (b) Superposition of the binding pocket of SK&F 64139-(magenta) and inhibitor 3-bound (green) structures. The dichloro-THIQ. moiety of inhibitor 3 is closer to the SAM binding pocket than the structures with SK&F 64139. The linker connecting the SAM analogue and dichloro-THIQ. is indicated, (c) Structures of TSA inhibitor 3 and proposed TSA inhibitor 4 with additional methylene in the linker (shown in red).
Clinically relevant inhibitors of PNMT have potential use in PTSD treatment.8,25 The challenge in testing the biological activity of PNMT inhibitors is the lack of a reliable cell-based assay to test the efficacy of newly designed and synthesized PNMT inhibitors. SK&F 64139 and SK&F 29661, the best-known PNMT inhibitors, have been used to evaluate PNMT activity in many published studies.26–28 Studies involving testing in animal models demonstrated that receptor interaction rather than PNMT inhibition was the primary mechanism of physiological action. Cell-based assays provide a biological test of cellular permeability and efficacy of inhibitors. Indeed, issues with permeability of SK&F 2966129,30 have hindered further development of PNMT inhibitors.
Expression of PNMT mRNA is dominant in human adrenal tissue with far lower expression in CNS tissues. Only one progenitor human pheochromocytoma cell line (hPheo1) is available for PNMT expression.31 In our hands, no expression of PNMT was observed in hPheo1 cells when differentiation was induced with dexamethasone. The origin of hPheo1 from progenitor human pheochromocytoma tumor cells is controversial.32
In the absence of human cell lines expressing PNMT, rat pheochromocytoma cell lines (PC12) and mouse pheochromocytoma cell lines (MPC) have been reported for the study of catecholamine synthesis.33,34 However, PC12 cells differ from human adrenal chromaffin cells with little to no expression of PNMT and low levels of epinephrine, an inappropriate model to test inhibitors of PNMT.28,33,35,36 The MPC cells exhibit substantial PNMT expression levels, hence, they have a direct relevance to adrenal chromaffin cells.37,38 In these cell lines, PNMT expression occurs at the mRNA level, but with variable protein expression upon differentiation with dexamethasone.34 When PNMT expression is observed in the MPC cell line, the epinephrine concentration remains low, requiring electrochemical detection (ECD) with high-performance liquid chromatography (HPLC) as a selective and sensitive detection method.39,40 Liquid chromatography and tandem mass spectrometry (LC–MS/MS) have also been used, following sample derivatization steps.41,42 These challenges inspired us to develop a technique for reliable expression of PNMT at the cellular level. Robust PNMT expression in cultured cells provides cellular synthesis of epinephrine, which can be analyzed by enzyme-linked immunosorbent assay (ELISA) protocols and used as an index for PNMT inhibition at the cellular level.
This study reports the design, synthesis, and characterization of a novel TSA inhibitor of PNMT. The interaction of the inhibitors with PNMT is investigated by isothermal titration calorimetry (ITC) and X-ray crystallography. In addition, we report the development of a novel cell-based assay for the evaluation of newly synthesized inhibitors against PNMT and a comparison of the inhibitory specificity for SAM-based DNA and protein methyltransferases. Finally, the specificity of inhibitor 4 for PNMT is compared with its affinity for the α2-adrenoceptor.
RESULTS AND DISCUSSION
Synthesis of TSA inhibitor 4 was designed to repurpose key intermediate 1 developed for the synthesis of inhibitor 3 (Figure 3a).1 Intermediate 1 was converted to fragment B in one step through a Pd-catalyzed hydroformylation reaction.43 Fragment A and fragment C were synthesized as previously described.1 Assembly of the final molecule was achieved by two consecutive reductive animation reactions aided by titanium iso-propoxide.44 Satisfactory results were obtained when the first coupling occurred between the amine group of fragment C and the aldehyde moiety of fragment B to yield compound 2. This was followed by a second reductive animation with fragment A to yield compound 3. A global deprotection step involved trifluoroacetic acid and anisole in water to give TSA inhibitor 4 as a diastereomeric mixture.
Figure 3.
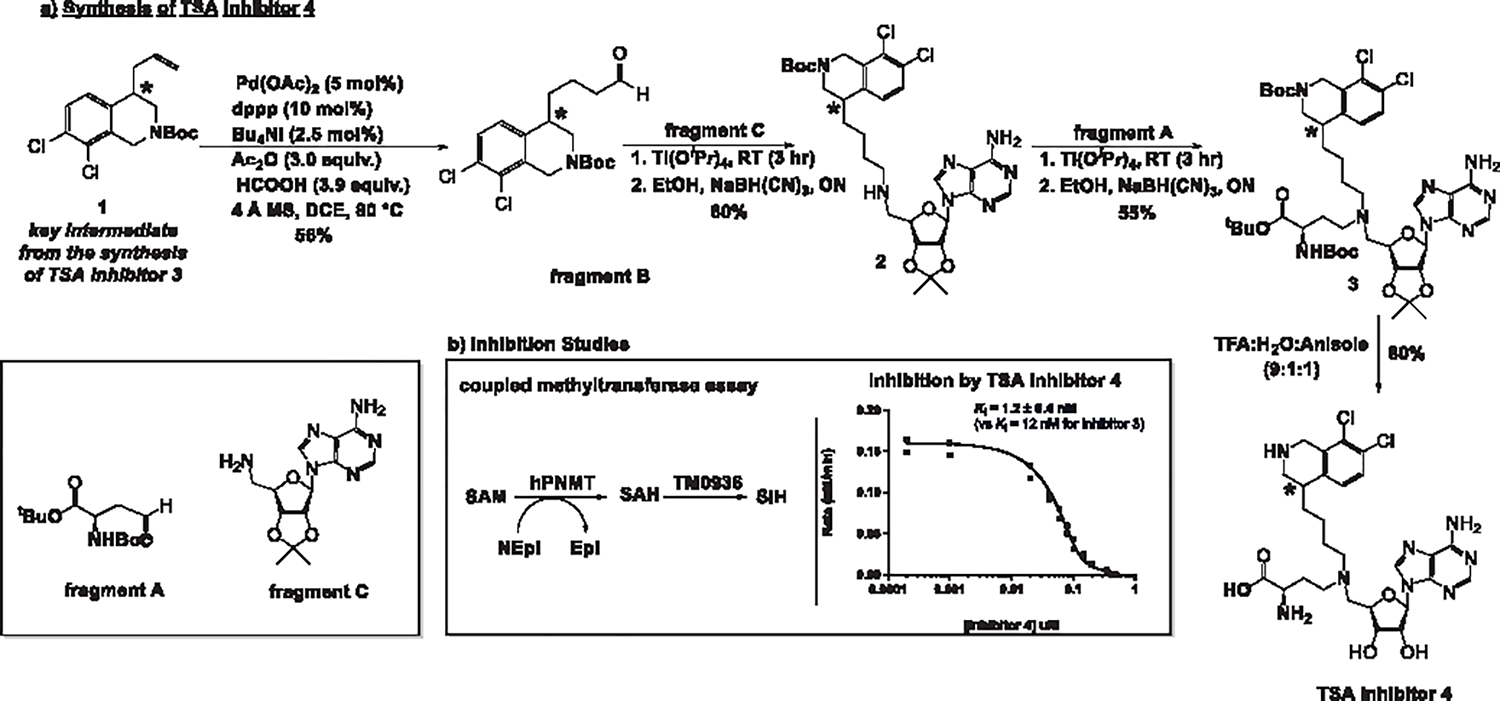
(a) Synthesis of TSA inhibitor 4 (b) Left panel: a coupled PNMT assay uses the deaminase TM0936 to convert SAH, a product of the PNMT reaction into S-inosyl-l-homocysteine (SIH), its inosyl derivative. SAH and SIH show different spectral features that allow for continuous monitoring of the PNMT reaction through a decrease in absorbance at 263 nm.45,46 (b) Right panel: activity profiles as a function of TSA inhibitor 4 concentration. Given the tight-binding nature of inhibitor 4, raw data were fitted to the Morrison equation47 and the inhibition constant was calculated using GraphPad Prism software. Values shown here represent the combined results of two individual trials. The data from both trials are plotted in one graph and the best fit is represented here. * Denotes a stereocenter with unknown stereochemistry (R or S).
Inhibition Assay.
Inhibition constants of the PNMT-catalyzed reaction were measured using a coupled-enzyme assay (Figure 3b).45 Briefly, the deamination of SAH by TM0936 yields S-inosyl-l-homocysteine (SIH) and the change in absorbance is monitored at 263 nm.46 Commonly, the assay mixture contains norepinephrine, inhibitor 4, and excess TM0936 and PNMT in phosphate buffer. Upon initiation with SAM at 30 °C, the UV absorbance was monitored at 263 nm (detailed protocol in the Supporting Information). Since the concentration of the inhibitor was less than 10 times the concentration of the enzyme, the results were fitted to the Morrison equation.47 In this manner, the Ki value of the diastereomeric mixture of inhibitor 4 was calculated to be 1.2 ± 0.4 nM (Figure 3b). TSA inhibitor 4 is an order of magnitude more potent than TSA inhibitor 3.1 An increased number of favorable interactions in the norepinephrine binding pocket could account for more favorable binding (see structural interactions below). However, the addition of a methylene bridge also imposes an entropie penalty, canceling some of the enthalpic improvements gained through active site interactions. We investigated the thermodynamic properties of binding by ITC.
Isothermal Titration Calorimetry (ITC) Studies.
The binding of TSA inhibitor 4 (as a diastereomeric mixture) to PNMT was consistent with a two-site binding model with a negative cooperative sequential mechanism (Figure 4a,b, for details see the Methods section). Human PNMT is a homodimer and two active sites are accessible to the ligand. Inhibitor 4 binding is driven by large entropie (−TΔS) values: −9.7 kcal/mol at the first active site and −14.7 kcal/mol at the second active site of the homodimer (Figure 4c). These large entropie contributions are accompanied by a small favorable enthalpic contribution of −1.6 kcal/mol at the first active site and an unfavorable contribution of 4 kcal/mol at the second active site (Figure 4c). Entropie contributions are attributed to the dynamic components, water exclusion, or hydrophobic differences, while enthalpic contributions are attributed to the formation of hydrogen bonds or ionic interactions between the ligand and active site residues. In ITC analysis, inhibitor 4 binds to the first site of the PNMT homodimer with a Kd of 4.9 nM. Binding at the second site gives a Kd of 15 nM, approximately 3-fold less than the first site, a negative cooperative interaction. The longer linker in inhibitor 4 provides a 10-fold improvement in binding when compared to inhibitor 3,1 suggesting that 4 is a closer mimic of the TS. This increased binding affinity results from improved thermodynamic parameters with increased favorable enthalpy and corresponding entropie compensation.
Figure 4.

Isothermal titration calorimetry (ITC) analysis of human PNMT with the diastereomeric mixture of inhibitor 4. The reaction was carried out at 25 °C. The cell contained 40 μM PNMT in 50 mM KP04 buffer (pH 8.0) containing 1 mM ethylenediaminetetraacetic acid (EDTA), 0.5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP), and 15% glycerol The syringe contained 1000 μM inhibitor 4 prepared in the same buffer. A total of 19 injections were conducted at 150 s intervals. Left panel (a) indicates raw heat data and right panel (b) shows the integrated heat injections which have been normalized per mole of the injectant as a function of the molar ratio. The results were best fitted to a two-site binding model, (c) Thermodynamic constants for binding of TS analogue inhibitor 4 to PNMT.
Cellular Activity of the Diastereomeric Mixture of Inhibitor 4.
The cellular activity of PNMT and its expression often uses rat pheochromocytoma cells, PC12, or the mouse pheochromocytoma cell line (MPC).33,34 In our hands, hPheol failed to produce detectable levels of the PNMT protein even upon differentiation induced by dexamethasone. We overexpressed hPNMT in HEK293T cells using a lentiviral vector expressing GFP in addition to flag-tagged hPNMT. Western blots of transfected cell lysates and fluorescent imaging for GFP expression confirmed the overexpression of hPNMT (Figure 5a,b). HEK293T cells do not produce significant norepinephrine, hence exogenous norepinephrine and SAM were provided in the assay culture medium. The linear increase in epinephrine formation with increasing norepinephrine and SAM concentrations in intact cultured HEK293T cells overexpressing hPNMT confirmed the expression and hPNMT catalytic activity in this system (Figure 5c). Addition of TSA inhibitor 4 to cells in the presence of norepinephrine and SAM inhibited the activity of hPNMT in a concentration-dependent manner with an IC50 value of 81 ± 10 nM. Similarly, a positive control experiment with SK&F 64139, gave an IC50 value of 15 ± 2 nM (Figure 5d,e). Despite studies on the inhibition of PNMT in PC12 cells28 and animal models,27,48 we found no other reports of dose titration with SK&F 64139 in a cell-based assay that supports its known cellular availability. The cell permeability of inhibitor 4 suggests that it can act as a candidate for physiological PNMT inhibition. Although SK&F 64139 shows a lower cellular IC50, it is non-selective by blocking both adrenaline biosynthesis and the biological activity of α2-adrenoceptors, thereby complicating the interpretation of biological results when tested in vivo.49
Figure 5.

Mammalian cell overexpression of hPNMT and cellular activity of inhibitor 4 (diastereomeric mixture), (a) Plasmid concentration-dependent GFP fluorescence puncta in transformed HEK293T cells, (b) Western blot of hPNMT as a function of transforming plasmid concentration in HEK293T cells using anti-flag antibodies. AlphaTubulin was used as a loading control, (c) The linear production of epinephrine with increasing concentration of norepinephrine (with equimolar SAM) in intact cultured HEK293T cells overexpressing hPNMT. (d) Inhibition of hPNMT in intact cultured HEK293T cells by inhibitor 4. (e) Inhibition of hPNMT in intact cultured HEK293T cells by SK&F 64139.
Specificity of Bisubstrate SAM-Methyltransferase Inhibitors.
The enzyme specificity of inhibitor 4 can he compared to SAM bisubstrate inhibitor analogues for nicotinamide N-methyltransferase (NNMT) and protein N-terminal methyltransferase 1 (NTMT1) reported earlier (Figure 6).21,50 Compound LL320 and the chemically related NAH-C4-GPKRIA have been reported to be bisubstrate analogue inhibitors of NNMT and NTMT1, respectively. LL320 was reported to be a 1.6 nM inhibitor for NNMT. As LL320 contains elements of the SAM methyl donor and the aromatic acceptor group, it was also screened against PNMT, where the apparent IC50 was >100 μM. Therefore, compound LL320 shows a >62,000 preference for NNMT relative to PNMT, despite the common elements.21 Inhibitor NAH-C4-GPKRIA was reported to be a 130 pM inhibitor for NTMT1 and showed a >3000-fold selectivity when compared to euchromatic histone-lysine N-methyltransferase (G9a) domain-containing protein 7 (SETD7), NNMT, protein arginine methyltransferase 1 (PRMTl), PRMT3, Trypanosoma brucei PRMT7, or S-adenosylhomocysteine hydrolase (SAHH). These reports suggest the inhibitor scaffolds of SAM-based methyltransferases are highly specific for their targets.
Figure 6.

Bisubstrate inhibitors based on SAM-substituted scaffolds. LL320 and NAH-C4-GPKRIA are from refs 21 and 50.
Inhibitors 3 and 4 were tested against DNA methyltransferases, DNMT1 and DNMT3b. Inhibitor 3 gave IC50 values of 35 and 19 μM for DNMT1 and DNMT3b, respectively, demonstrating a specificity of 2900 and 1600, respectively, for PNMT relative to the DNA methyltransferases (Figure S2a,c, details in the Supporting Information). Inhibitor 4 gave IC50 values of 61 and 17 μM for DNMT1 and DNMT3b, demonstrating a specificity of 51,000 and 14,000 respectively for PNMT relative to the DNA methyltransferases (Figure S2b,d).
Inhibitors 3 and 4 were also tested for their effects on protein methyltransferase function in cultured cells. HT-29 colorectal cells expressing protein arginine and protein lysine methyltransferases (PRMTs and PKMTs) were treated with inhibitor 3 or inhibitor 4 for 6 days before the soluble protein was extracted and blotted for protein symmetric dimethylarginine (SDMA), asymmetric dimethylarginme (ADMA), and trimethyl lysine (Me3-lysine) post-translational methylations. Positive controls included PRMT5-specific inhibitor GSK3326595 (SDMA) and Type I PRMT5 (PRMT1/4) specific inhibitor MS023 (ADMA).51 Depletion of protein methylation marks occurs on a biological scale of hours to days. Cells treated with GSK3326595 showed no band corresponding to SDMA while cells treated with MS023 showed no evidence of ADMA after 6 days of treatment (Figure S3). Western blot analysis showed no significant change in total SDMA, ADMA, and Me3-lysine in samples treated with 20 μM inhibitor 3 or inhibitor 4, indicating that neither of these molecules influenced protein methylation (Figure S3). TSA inhibitors 3 and 4 are highly specific for their target with a specificity of > 1600-fold for PNMT over protein methyltransferases.
Binding of Inhibitors 3 and 4 to Adrenergic Receptors.
Previously described inhibitors of PNMT showed significant affinity for α2-adrenoceptors complicating the interpretation of biological outcomes. The specificity of TSA inhibitors 3 and 4 for PNMT was compared to their binding to the human α2A adrenergic receptor (ADRA2a). A membrane preparation of ADRA2a was mixed with [3H]-RX821002, a selective α2-adrenoceptor antagonist with a binding affinity of 1.6 nM, approximately 1000-fold tighter than the binding affinity of ADRA2a for epinephrine (l μM).52 Inhibitors 3 and 4 were used as unlabeled competitors in the binding assay at 160 μM to the ADRA2a receptor (Figures 7a and S4). Variable concentrations of epinephrine and inhibitors 3 and 4 were used to estimate the binding affinity (Figures 7b and S5). With 1.6 mM inhibitor 4, less ligand was displaced from ADRA2a than with epinephrine.
Figure 7.

(a) Binding affinity and (b) dose dependence of the binding of inhibitors 3 and 4 to the ADRA2a receptor. ADRA2a = 4.13 nM, (+) 16 μM, (++) 160 μM, and (+++) 1600 μM (epinephrine, inhibitor 3, inhibitor 4).
The Kd value of epinephrine for the ADRA2a receptor was calculated to be 10.0 ± 0.2 μM, approximately 10-fold higher than previously reported (see the Supporting Information for calculation details). The Kd value of inhibitors 3 and 4 for the ADRA2a receptor was calculated to be 14.6 ± 1.1 and 14.3 ± 0.2 μM, respectively. These experiments demonstrate that TSA inhibitors 3 and 4 bind to PNMT orders of magnitude tighter than to the ADRA2a receptor. With a Kd value of 1.2 nM for PNMT, inhibitor 4 binds its target enzyme 12,000 times tighter than the ADRA2a receptor.
Structure of PNMT with the Diastereomeric Mixture of Inhibitor 4.
X-ray crystallography experiments with purified PNMT without added ligands produced a structure showing SAH bound to both catalytic sites. The PNMT complex with inhibitor 4 was obtained by soaking experiments. Crystals of the wild-type PNMT-SAH complex were soaked in crystallization buffer containing 1 mM inhibitor 4. PNMT crystals soaked in inhibitor 4 for 2 h yielded a structure with two subunits in the asymmetric unit, one bound with inhibitor 4 and the second bound with SAH. Crystals soaked for 12 h produced structures with inhibitor 4 in both subunits. The structure of PNMT in complex with inhibitor 4 and SAH was determined at 2.10 Å resolution in the P43212 space group (Figure 8a). The structure of PNMT in complex with inhibitor 4 in both subunits was determined at a resolution of 2.43 Å, in the P43212 space group (Figure 8a). The electron density for SAH and inhibitor 4 can be clearly seen in the corresponding binding pocket of both structures (Figure S6). Comparing structures with first-generation inhibitor 3 and inhibitor 4, reveals substantial protein conformational changes between the PNMT-SAH and PNMT-inhibitor 3 structures, but not between PNMT-SAH and PNMT-inhibitor 4 structures (Figures 8b and S7).24,50 The accessible surface area of the PNMT dimer interface is 11481 Å2 by PISA analysis.53 A Cα-superposition of the SAH-bound subunit with the inhibitor 4-bound subunit shows an RMSD of 0.144 Å, demonstrating no significant structural difference between the two chains. The PNMT monomer contains an α/β domain with 7-stranded mixed β-sheets (β1–β7) flanked on either side by 4 helices (Figure 8a). The binding sites for norepinephrine and SAM are buried and located at the edge of the β-sheet. Both binding sites are tightly covered by surrounding motifs.54
Figure 8.

Crystal structure of human PNMT (stereo-view) in complex with the diastereomeric mixture of inhibitor 4. (a) Structural fold of PNMT with inhibitor 4. (b) Superposition of PNMT bound to SAH (red), SK&F 64139 (blue), inhibitor 3 (cyan) and the inhibitor 4-bound structure (yellow). Binding of inhibitor 3 opens two loops from Glu218 to Val234 (3.2 Å, highlighted with a black asterisk) and His261 to Lys270 (2.8 Å, highlighted with a red asterisk). However, these loops are in close confirmation in the inhibitor 4-bound structure, (c) PNMT binding site in complex with inhibitor 4. Amino acid residues interacting with inhibitor 4 are displayed (gray). Selected hydrogen bond interactions are shown in black dotted lines. The dichloro-THIQ_ mimic of inhibitor 4 is bound in the hydrophobic pocket. The aromatic ring of this moiety is in a π–π stacking interaction with Phe182.
Catalytic Site Interaction and Structure Comparison.
Binding of inhibitor 4 at the catalytic site of PNMT fills both the SAM and norepinephrine binding pockets (details in Figure 8c). The N-13 and N-15 atoms of inhibitor 4 form hydrogen bonds with the peptide nitrogen of Val159 and the carboxylate of Asp158, respectively (Figure S8). The 0–39 of inhibitor 4 is in hydrogen bond interaction with the carboxylate of Asp101 and carboxamide of Asn106, whereas 0–40 is hydrogen bonded to the carboxylate of AsplOl. The carboxyl group in inhibitor 4 is hydrogen bonded to three tyrosine residues including the hydroxyl groups of Tyr35, Tyr40, and Tyr85. The N-23 of inhibitor 4 is hydrogen bonded to peptide carbonyls of Gly79, Ser80, and Ala181 (Figure 8c). The adenine base is sandwiched between the side chains of Phe102 and Vai187 providing strong hydrophobic interactions. Tyr27 and Phe30 residues create a hydrophobic pocket facilitating inhibitor binding. The dichloro-THIQ. moiety of inhibitor 4 is bound in the norepinephrine binding site where a pocket is formed by Tyr35, Val53, Lys57, Phe182, Glu219, Tyr222, Met258, Val269, and Val272. The aromatic ring in the norepinephrine site is in a π–π stacking interaction with Phe182. The binding of dichloro-THIQ, is stabilized by the hydrophobic interaction in the binding pocket. The N-33 atom of dichloro-THIQ, is in strong hydrogen bonding interaction with the carboxylate of GIu219 (Figure 9a,b).
Figure 9.

Binding site comparison of PNMT complex structures. Panel (a) and (b) show the binding interactions of SK&F 64139 (dark cyan, PDB ID: 1YZ3) and inhibitor 3 (green, PDB ID: 6WS1), respectively. Hydrogen binding interactions are highlighted with black dotted lines. Panel (c) superimposes SK&F 64139 (dark cyan), inhibitor 3 (green), and inhibitor 4 (gray) at the catalytic site. The SAH moiety has a similar binding conformation in all structures. The dichloro-THIQ_moiety of inhibitor 3 is pulled towards the SAM binding pocket. However, the dichloro-THIQ. moiety of inhibitor 4 is positioned similarly to SK& 641139 as the sole ligand. Movement of the loops caused by the binding of inhibitor 3 are highlighted with black and red asterisks. These loops are in close conformation in the inhibitor 4-bound structure.
The structure of the PNMT-inhibitor 4 complex was compared with the SK&F 64139 and inhibitor 3-bound PNMT structures (Figure 9c). The SAM mimic of inhibitor 3 and 4 binds in the same conformation in the cofactor (SAM) binding pocket (Figure 9c). Unlike the inhibitor 3-bound structure, binding of inhibitor 4 does not induce conformational changes in Glu218–Val234 and His261–Lys270 loops relative to the SK&F 64139-bound structure (Figure 9b). With inhibitor 3 bound, conformational changes are observed in residues Asn39 (1.2 Å), Glu219 (1.7 Å), Tyr222 (1.2 Å), and Asp267 (1.5 Å). However, in the inhibitor 4-hound structure, these residues have the same conformation as in the SK&F 64139- or SAH-bound PNMT complexes (Figure 8b). The side chain of Arg44 is disordered in the inhibitor 3-bound complex but not in the inhibitor 4-bound structure. When compared to the SK&F 64139-bound structure (PDB ID: 1YZ3),24 the dichloro-THIQ_moiety in inhibitor 4 is bound to the substrate (norepinephrine) or SK&F 64139 binding pocket (Figure 9c). The N-2 atom of SK&F 64139 is hydrogen bonded to the carboxylate of Glu219. A similar interaction is observed with the N-33 atom of inhibitor 4. Unlike the inhibitor 3-PNMT complex, the other interactions of SK&F 64139 with PNMT active site residues are similar in the inhibitor 4-bound structure (Figure 9c).
CONCLUSIONS
A second-generation TSA inhibitor of PNMT (inhibitor 4) was designed by comparing the crystal structure of PNMT in complex with inhibitor 3 and a PNMT structure with the well-studied SK&F 64139. It uses the dichloro-THIQ. analogue of norepinephrine, and the cationic transition-state analogue of SAM covalently linked by a spacer to mimic the catalytic site dimensions. A 10-fold improvement was observed with inhibitor 4 when compared with the first-generation inhibitor 3. Comparative steady-state, ITC, and structural studies establish a negative cooperative binding mechanism for inhibitor 4 with large favorable entropic contributions. Inhibitor 4 occupies the catalytic site without the protein distortion caused by the first-generation inhibitor 3 complex. The improved catalytic site fit enjoyed by inhibitor 4 more than accounts for the increased entropic factor added by a new methylene bridge in the inhibitor 4 structure. A new cell-based assay was implemented to investigate the biological activity of TSA inhibitor 4. An IC50 value of 81 nM was measured when transiently transfected HEK293T cells were incubated with inhibitor 4. Inhibitor 4 demonstrates up to 51,000-fold specificity for PNMT relative to DNA and protein methyltransferases. Moreover, inhibitor 4 exhibits a 12,000-fold specificity for PNMT over α2-adrenoceptor.
This study highlights the significance of rational inhibitor design and the potential for future studies of TSA inhibitor 4 in mouse models of PTSD.
METHODS
Synthesis of Inhibitor 4.
Experimental details for the synthesis and characterization of TSA inhibitor 4 are provided in the Supporting Information.
Isothermal Titration Calorimetry (ITC) Experiments.
A MicroCAL PEAQ-ITC instrument (Malvern Panalytical) was used to measure the thermodynamic parameters of PNMT binding to inhibitor 4. The PNMT was stored in a buffer containing 50 mM potassium phosphate (pH 8.0), 1 mM EDTA, 0.5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP), and 15% glycerol. A stock solution of the desired inhibitor was prepared in D2O and the concentration was measured using 1H NMR spectroscopy. The volume of the NMR sample was measured precisely before it was freeze-dried. The dry sample was resuspended in an identical volume of enzyme storage buffer prior to the ITC experiment. This extra step helps eliminate the heat exchange resulting from buffer mismatch. The concentration of protein was determined using nanodrop (λ280, ε = 44.92 cm−1 mM−1). The titration solutions were prepared by dilution of stock solutions into the enzyme storage buffer. In a typical ITC experiment, 300 μL of protein was placed in the cell, and varying concentrations of the inhibitor solution was taken with a syringe. An initial 0.4 μL injection was followed by 18 injections of 1.5 μL at 25 °C at 150 s intervals. The reference power was 10 kcal/s and the stirring rate was 750 rpm. Control ITC experiments involved the titration of the ligand into buffer solution, buffer to buffer, and the working solution of PNMT to buffer to account for heat of dilution and mixing. These titrations were subtracted from the experimental results before the data were analyzed.
Cell Culture.
HEK293T cells were maintained in a complete medium comprising of Gibco high glucose DMEM with L-glutamine (Thermofisher, Waltham, MA) supplemented with 10% Gibco heat-inactivated fetal bovine serum (FBS) (Thermofisher, Waltham, MA). Cells were cultured at 37 °C in a 5% CO2 atmosphere. Cells were subcultured at a 1:3 ratio every other day to maintain a monolayer of cells. Cells were detached with 2 mL of 0.025% (w/v) EDTA-free trypsin (Gibco, Thermofisher, Waltham, MA) per 10 cm dish for 3 min at room temperature. The reaction was stopped by adding 3 volumes of complete medium and cells were recovered by centrifugation at 300g for 2 min at room temperature. A Nexelcom Cellometer was used to count cells using trypan blue to assess cell density and viability.
Transient Transfection and PNMT Overexpression.
The lentiviral vector pLV[Exp]-EGFP:T2A:Puro-EFlA > 3xFLAG/PNMT was synthesized using VectorBuilder (Chicago, IL). This was designed to express EGFP and puromycin-resistant genes separated by the T2A self-cleaving peptide sequence. 3xFlag-tagged PNMT was cloned into this vector. The plasmid was produced by transformation into E. coli DH5α and purified using a Qiagen midi prep kit (Qjagen). Cells were transfected using the calcium phosphate transfection protocol in a 96-well tissue culture-treated plate. Briefly, DNA calculated to 1 μg final in each well was mixed with 62 μL of 2 M CaCl2 and sterile water was added to a total volume of 500 μL. 500 μL of 2× HEPES buffered saline (HBS) was added to this mixture in 100 μL increments with mixing. The DNA mixture was incubated for 30 min at room temperature after which the mixture was added directly to the wells containing approximately 50,000 cells per well at 70–90% confluency. Plates were incubated at 37 °C under 5% CO2 for 7 h after which fresh media was added and cultured under the same conditions overnight. Cells were monitored for PNMT overexpression by fluorescent imaging and Western blotting using the anti-flag primary antibody (Abcam) and a goat anti-mouse horse radish peroxidase (HRP) conjugated secondary antibody (BD Biosciences). α-Tubulin was used as a loading control using the anti-α -tubulin antibody (1:10000) (R&D Systems).
Cellular Inhibition of PNMT.
The inhibition assay was performed approximately 24 h after transfection in 96-well tissue culture-treated plates. The assay was performed in a 50 μL volume in phosphate-buffered saline (PBS) for 4 h at 37 °C under 5% CO2. The linear production of epinephrine in this system was monitored by varying norepinephrine concentrations from 0 to 150 μM with equimolar amounts of SAM. For the inhibition assay, 50 μM each of norepinephrine and SAM were added to each well. Inhibitor 4 concentration was varied from 0 to 10 μM. Controls used the known PNMT inhibitor, SK&F 64139, and a non-transfected cell in the presence of inhibitor, norepinephrine, and SAM. Non-transfected cells incubated with norepinephrine and SAM were used to determine background epinephrine levels, which were subtracted from all values.
Inhibition was assayed by using the norepinephrine/epinephrine ELISA kit from Abnova following the manufacturer’s protocol. Briefly, cells were lysed using Pierce IP lysis buffer with the Halt protease inhibitor cocktail (Thermofisher, MA.) following the manufacturer’s protocol. 10 μL of cell lysate was analyzed for epinephrine levels using ELISA. IC50 values were determined by plotting the inhibitor concentration against epinephrine levels using the 4-parameter equation in GraphPad Prism. All inhibition assays were performed three times in duplicate.
General Binding Assay of the ADRA2a Receptor.
To 100 μL of binding assay buffer containing 50 mM HEPES pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1 mM CaCl2, and 0.2% BSA was added 16 nM [3H]-RX821002. 1 unit of membrane lysate containing ADRA2a (5 μL, 4.13 nM ADRA2a protein) was added, followed by cold epinephrine (chase) or inhibitor 3 or inhibitor 4. Samples were adjusted to 140 μL, mixed, and incubated at room temperature for 5 min before spinning through a 10 kDa MWCO filter at 15,000g for 5 min and 30–40 μL of filtrate was analyzed by liquid scintillation counting.
PNMT Crystallization and Soaking with the Diastereomeric Mixture of Inhibitor 4.
Purified PNMT contained bound SAH. Initial X-ray crystallography experiments with PNMT produced SAH-bound structures. To obtain structures containing only inhibitor 4, we first crystallized PNMT in complex with SAH. Subsequently, the SAH was replaced through time course soaking of the crystals with inhibitor 4. Crystallization of PNMT was achieved by using hanging drop vapor diffusion at 22 °C. Then, 10 mg/mL protein was screened with previously known crystallization conditions.55 Crystallization trials were done in 24-well VDXm hanging drop plates (Hampton Research). Crystallization drops contained 1 μL of PNMT and 1 μL of well solution. The volume of the well solution was 1 mL. Diffraction-quality crystals were obtained in two weeks under crystallization conditions containing 100 mM sodium cacodylate pH 5.5, 170 mM LiCl, and 18% PEG 6000. These crystals were washed in mother liquor and incubated in soaking buffer containing 100 mM sodium cacodylate pH 5.5, 170 mM LiCl, 20% PEG 6000, and 1.0 mM inhibitor 4 for different time periods. After soaking, crystals were cryoprotected with 20% ethylene glycol and flash-frozen for diffraction data collection.
Structure Determination and Refinement.
Crystal structures of PNMT in complex with TSA inhibitor 4 were solved by molecular replacement using the PHASER program.56 Chain-A of the wild-type PNMT (PDB ID: 4MIK) structure was used as the initial search model. The model obtained from PHASER was manually built and completed using the COOT program.57 Structure refinement was performed using PHENIX-REFINE, using standard protocols for NCS refinement.58 The inhibitor molecule was deleted from the model to initiate the refinement. After the addition of water molecules, the inhibitor molecule was fitted to its electron density (Table S1).
Structure Analysis.
Crystal structures of PNMT in complex with SK&F 64139; (PDB ID: 1YZ3, chain: B), inhibitor 3 (PDB ID: 6WS1, chain: B), and inhibitor 4 (PDB ID: 7TX2, chain: B) were used in the structure comparison.1,24 Structural superimpositions were carried out using the SSM protocol of COOT and the geometry analyses of the final model was achieved using MolProbity.59 Additional structural analyses, including the calculation of the B-factor profiles, were accomplished using BAVERAGE of the CCP4 suite.60 Structural figures were generated using the molecular graphics program PyMOL. For PNMT-inhibitor 4 complex structure (PDB ID: 7TX2), subunit-B was used for all the structural analyses and comparisons.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge the assistance of Dr. Heather Snell, Department of Neuroscience, Albert Einstein College of Medicine for the growth of the hPhe01 cells, Dr. Emmanuel Burgos and Dr. Maxim Maron, Department of Biochemistry, Albert Einstein College of Medicine for assistance in culturing and conversion of the HEK293T cell line to PNMT-expressing transfected cells, and Dr. Philip Provencher, Department of Chemistry, Princeton University, for insightful conversations regarding the chemical synthesis of the inhibitor. Data collection also involved resources of the Advanced Photon Source; a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Company, which operates the facility.
Funding
This work was supported by NIH research grant R01 GM041916 (VLS). The Bruker 600 MHz NMR instrument in the Structural NMR Resource at the Albert Einstein College of Medicine was purchased using funds from NIH award 1S100D016305 and is supported by a Cancer Center Support Grant (P30 CA013330). The Albert Einstein College of Medicine Crystallographic Core X-Ray Diffraction facility is supported by NIH Shared Instrumentation Grant S10 OD020068.
Footnotes
The authors declare no competing financial interest.
Contributor Information
Niusha Mahmoodi, Department of Biochemistry, Albert Einstein College of Medicine, Bronx, New York 10461, United States.
Yacoba V. T. Minnow, Department of Biochemistry, Albert Einstein College of Medicine, Bronx, New York 10461, United States
Rajesh K. Harijan, Department of Biochemistry, Albert Einstein College of Medicine, Bronx, New York 10461, United States
Gabriel T. Bedard, Department of Biochemistry, Albert Einstein College of Medicine, Bronx, New York 10461, United States
Vern L. Schramm, Department of Biochemistry, Albert Einstein College of Medicine, Bronx, New York 10461, United States
REFERENCES
- (1).Mahmoodi N; Harijan RK; Schramm VL. Transition-State Analogues of Phenylethanolamine N-Methyltransferase. J. Am. Chem. Soc. 2020, 142, 14222–14233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Westfall TC; Westfall DP Adrenergic Agonists and Antagonists; McGraw-Hill: New York, 2006; pp 215–264. [Google Scholar]
- (3).Costa MV; Carvalho F; Bastos ML; Carvalho RA; Carvalho M; Remiao F Adrenaline and Noradrenaline: Partners and Actors in the Same Play; InTechOpen, 2012; p 442. [Google Scholar]
- (4).Kitahama K; Denoroy L; Goldstein M; Jouvet M; Pearson J K Immunohistochemistry of Tyrosine Hydroxylase and Phenylethanolamine N-Methyltransferase in the Human Brain Stem: Description of Adrenergic Perikarya and Characterization of Longitudinal Catecholaminergic Pathways. Neuroscience 1988, 25, 97–111. [DOI] [PubMed] [Google Scholar]
- (5).Ziegler MG; Bao X; Kennedy BP; Joyner A; Enns R Location, Development, Control, and Function of Extraadrenal Phenylethanolamine N -Methyltransferase. Ann. N. Y. Acad. Sci. 2002, 971, 76–82. [DOI] [PubMed] [Google Scholar]
- (6).Stolk JM; U’Prichard DC; Fuxe K Epinephrine in the Central Nervous System; Oxford University Press, 1988. [Google Scholar]
- (7).Peltsch H; Khurana S; Byrne CJ; Nguyen P; Khaper N; Kumar A; Tai TC Cardiac phenylethanolamine N-methyltransferase: localization and regulation of gene expression in the spontaneously hypertensive rat. Can. J. Physiol. Pharmacol 2016, 94, 363–372. [DOI] [PubMed] [Google Scholar]
- (8).Martinho R; Oliveira A; Correia G; Marques M; Seixas R; Serrão P; Moreira-Rodrigues M Epinephrine May Contribute to the Persistence of Traumatic Memories in a Post-traumatic Stress Disorder Animal Model. Front. Mol. Neurosci. 2020, 13, No. 588802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lymperopoulos A; Rengo G; Gao E; Ebert SN; Dorn GW; Koch WJ Reduction of Sympathetic Activity via Adrenal-targeted GRK2 Gene Deletion Attenuates Heart Failure Progression and Improves Cardiac Function after Myocardial Infarction. J. Biol Chem. 2010, 285, 16378–16386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kennedy BP; Bottiglieri T; Anting E; Ziegler MG; Hansen LA; Masliah E Elevated S-adenosylhomocysteine in Alzheimer brain: Influence on methyltransferases and cognitive function. J. Neural Transm. 2004, 111, 547–567. [DOI] [PubMed] [Google Scholar]
- (11).Gearhart DA; Neafsey EJ; Collins M A Phenylethanolamine N-methyltransferase has beta-carboline 2N-methyltransferase activity: hypothetical relevance to Parkinson’s disease. Neurochem. Int. 2002, 40, 611–620. [DOI] [PubMed] [Google Scholar]
- (12).Mazzio EA; Close F; Soliman KF A The Biochemical and Cellular Basis for Nutraceutical Strategies to Attenuate Neurodegeneration in Parkinson’s Disease. Int. ]. Mol. Sci. 2011, 12, 506–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Georgieva P; Wu Q; McLeish MJ; Himo F The reaction mechanism of phenylethanolamine N-methyltransferase: A density functional theory study. Biochim. Biophys. Acta, Proteins Proteomics 2009, 1794, 1831–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hou QQ,; Wang JH.; Gao J.; Liu YJ; Liu CB. QM/MM studies on the catalytic mechanism of Phenylethanolamine N-methyltransferase. Biochim. Biophys. Acta, Proteins Proteomics 2012, 1824, 533–541. [DOI] [PubMed] [Google Scholar]
- (15).Stratton CF; Poulin MB; Du, Qy Schramm VL. Kinetic Isotope Effects and Transition State Structure for Human Phenyl-ethanolamine N-Methyltransferase. ACS Chem. Biol 2017, 12, 342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Krakoff LR; Axelrod J Inhibition of phenylethanolamine-N-methyl transferase. Biochem. Pharmacol. 1967, 16, 1384–1386. [DOI] [PubMed] [Google Scholar]
- (17).Fuller RW; Hunt JM Substrate specificity of phenethanol-amine N-methyl transferase. Biochem. Pharmacol 1965, 14, 1896–1897. [DOI] [PubMed] [Google Scholar]
- (18).Fuller RW; Mills J; Marsh MM Inhibition of Phenethanolamine N-Methyl Transferase by Ring-Substituted α-Methylphenethylamines (Amphetamines). J. Med. Chem. 1971, 14, 322–325. [DOI] [PubMed] [Google Scholar]
- (19).Fuller RW; Roush BW; Snoddy HD; Molloy BB Inhibition of Phenylethanolamine N-Methyltransferase by Benzylamines. 2. in Vitro and in Vivo Studies with 2,3-Dichloro-α-methylbenzylamine. J. Med. Chem. 1973, 16, 106–109. [DOI] [PubMed] [Google Scholar]
- (20).Lu J; Bart AG.; Wu Q.; Criscione KR.; McLeish MJ.; Scott EE.; Grünewald. Structure-Based Drug Design of Bisubstrate Inhibitors of Phenylethanolamine N-Methyltransferase Possessing Low Nanomolar Affinity at Both Substrate Binding Domains. J. Med. Chem. 2020, 63, 13878–13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Chen D; Li L; Diaz K; Iyamu ID; Yadav R; Noinaj N; Huang R Novel Propargyl-Linked Bisubstrate Analogues as Tight-Binding Inhibitors for Nicotinamide N-Methyltransferase. J. Med. Chem. 2019, 62, 10783–10797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chen D; Dong G; Noinaj N; Huang R Discovery of Bisubstrate Inhibitors for Protein N-Terminal Methyltransferase 1. J. Med. Chem. 2019, 62, 3773–3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Pendleton RG; Kaiser C; Gessner G Studies on adrenal phenylethanolamine N-methyltransferase (PNMT) with S K & F 64139, a selective inhibitor. J. Pharmacol. Exp. Ther. 1976, 197, 623–632. [PubMed] [Google Scholar]
- (24).Wu Q; Gee CL; Lin F; Tyndall JD; Martin JL; Grunewald GL; McLeish MJ Structural, mutagenic, and kinetic analysis of the binding of substrates and inhibitors of human phenylethanolamine N-methyltransferase. J. Med. Chem. 2005, 48, 7243–7252. [DOI] [PubMed] [Google Scholar]
- (25).Oliveira A; Martinho R; Serrão P; Moreira-Rodrigues M Epinephrine Released During Traumatic Events May Strengthen Contextual Fear Memory Through Increased Hippocampus mRNA Expression of Nr4a Transcription Factors. Front. Mol. Neurosci. 2018, 11, 334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kennedy B; Elayan H; Ziegler MG Glucocorticoid induction of epinephrine synthesizing enzyme in rat skeletal muscle and insulin resistance. J. Clin. Invest. 1993, 92, 303–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kennedy B; Elayan H; Ziegler MG Glucocorticoid hypertension and nonadrenal phenylethanolamine N-methyltransferase. Hypertension 1993, 21, 415–419. [DOI] [PubMed] [Google Scholar]
- (28).Byrd JC; Hadjiconstantinou M; Cavalla D Epinephrine synthesis in the PC12 pheochromocytoma cell line. Eur. J. Pharmacol. 1986, 127, 139–142. [DOI] [PubMed] [Google Scholar]
- (29).Pendleton RG; Gessner G; Weiner G; Jenkins B; Sawyer J; Bondinell W; Intoccia A Studies on SK&F 29661, an organ-specific inhibitor of phenylethanolamine N-methyltransferase. J. Pharmacol. Exp. Ther. 1979, 208, 24–30. [PubMed] [Google Scholar]
- (30).Grunewald GL; Caldwell TM; Li Q; Slavica M; Criscione KR; Borchardt R T.; Wang, W. Synthesis and biochemical evaluation of 3-fluoromethyl-1,2,3, 4-tetrahydroisoquinolines as selective inhibitors of phenylethanolamine N-methyltransferase versus the aIpha(2)-adrenoceptor. J. Med. Chem. 1999, 42, 3588–3601. [DOI] [PubMed] [Google Scholar]
- (31).Ghayee HK; Bhagwandin VJ; Stastny V; Click A; Ding LH; Mizrachi D; Zou YS; Chari R; Lam WL; Bachoo RM.; Smith AL.; Story MD.; Sidhu S.; Robinson BG.; Nwariaku FE.; Gazdar AF.; Auchus RJ.; Shay JW. Progenitor cell line (hPheol) derived from a human pheochromocytoma tumor. PLoS One 2013, 8, No. e65624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Bayley JP; Devilee P Advances in paraganglioma-pheochromocytoma cell lines and xenografts. Endocr.-Relat. Cancer 2020, 27, R433–r450. [DOI] [PubMed] [Google Scholar]
- (33).Greene LA; Tischler A S. Estabhshment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. U.SA. 1976, 73, 2424–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Powers JF; Evinger MJ; Tsokas P; Bedri S; Alroy J; Shahsavari M; Tischler A S. Pheochromocytoma cell lines from heterozygous neurofibromatosis knockout mice. Cell Tissue Res. 2000, 302, 309–320. [DOI] [PubMed] [Google Scholar]
- (35).Ebert SN; Lindley SE; Bengoechea TG; Bain D; Wong DL Adrenergic differentiation potential in PC12 cells: influence of sodium butyrate and dexamethasone. Mol. Brain Res. 1997, 47, 24–30. [DOI] [PubMed] [Google Scholar]
- (36).Kim KT; Park DH; Joh TH Parallel up-regulation of catecholamine biosynthetic enzymes by dexamethasone in PC12 cells. J. Neurochem. 1993, 60, 946–951. [DOI] [PubMed] [Google Scholar]
- (37).Korpershoek E; Pacak K; Martiniova L Murine models and cell lines for the investigation of pheochromocytoma: applications for future therapies? Endocr. Pathol. 2012, 23, 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Powers JF; Schelling KH; Brachold JM; Tischler AS Plasticity of pheochromocytoma cell lines from neurofibromatosis knockout mice. Ann. N. Y. Acad. Sci. 2002, 971, 371–378. [DOI] [PubMed] [Google Scholar]
- (39).Xie L; Chen L; Gu P; Wei L; Kang X A Convenient Method for Extraction and Analysis with High-Pressure Liquid Chromatography of Catecholamine Neurotransmitters and Their Metabolites. J. Visualized Exp. 2018, 133, No. e5644S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Tsunoda M; Aoyama C; Ota S; Tamura T; Funatsu T Extraction of catecholamines from urine using a monolithic disk-packed spin column and high-performance liquid chromatography-electrochemical detection. Anal Methods 2011, 3, 582–585. [DOI] [PubMed] [Google Scholar]
- (41).He H; Carballo-Jane E; Tong X; Cohen LH Measurement of catecholamines in rat and mini-pig plasma and urine by liquid chromatography-tandem mass spectrometry coupled with solid phase extraction.J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2015, 997, 154–161. [DOI] [PubMed] [Google Scholar]
- (42).Dunand M; Gubian D; Stauffer M; Abid K; Grouzmann E High-throughput and sensitive quantitation of plasma catecholamines by ultraperformance liquid chromatography-tandem mass spectrometry using a solid phase microwell extraction plate. Anal. Chem. 2013, 85, 3539–3544. [DOI] [PubMed] [Google Scholar]
- (43).Ren W; Chang W; Dai J; Shi Y; Li J; Shi Y An Effective Pd-Catalyzed Regioselective Hydroformylation of Olefins with Formic Acid. J. Am. Chem. Soc. 2016, 138, 14864–14867. [DOI] [PubMed] [Google Scholar]
- (44).Mattson RJ; Pham KM; Leuck DJ; Cowen KA. An improved method for reductive alkylation of amines using titanium-(IV) isopropoxide and sodium cyanoborohydride. J. Org. Chem. 1990, 55, 2552–2554. [Google Scholar]
- (45).Burgos ES; Walters RO; Huffinan DM; Shechter D A simplified characterization of: S-adenosyl-l-methionine-consuming enzymes with 1-Step EZ-MTase: A universal and straightforward coupled-assay for in vitro and in vivo setting. Chem. Sci. 2017, 8, 6601–6612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Hermann J C; Marti-Arbona, R.; Fedorov, A A.; Fedorov, E.; Almo, S. C.; Shoichet, B. K; Raushel, F. M. Structure-based activity prediction for an enzyme of unknown function. Nature 2007, 448, 775–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Morrison JF Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim. Biophys. Acta, Enzymol. 1969, 185, 269–286. [DOI] [PubMed] [Google Scholar]
- (48).Coen CW; Gallo RV Effects of various inhibitors of phenylethanolamine N-methyltransferase on pulsatile release of LH in ovariectomized rats. J. Endocrinol 1986, 111, 51–59. [DOI] [PubMed] [Google Scholar]
- (49).Toomey RE; Homg JS; Hemrick-Luecke SK; Fuller RW alpha 2 Adrenoreceptor affinity of some inhibitors of norepinephrine N-methyltransferase. Life Sci. 1981, 29, 2467–2672. [DOI] [PubMed] [Google Scholar]
- (50).Chen D; Dong C; Dong G; Srinivasan K; Min J; Noinaj N; Huang R Probing the Plasticity in the Active Site of Protein N-terminal Methyltransferase 1 Using Bisubstrate Analogues. J. Med. Chem. 2020, 63, 8419–8431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Hwang JW; Cho Y; Bae GU; Kim SN; Kim YK Protein arginine methyltransferases: promising targets for cancer therapy. Exp. Mol. Med. 2021, 53, 788–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Wu Y; Zeng L; Zhao S Ligands of Adrenergic Receptors: A Structural Point of View. Biomolecules 2021, 11, No. 936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Krissinel E; Henrick K Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [DOI] [PubMed] [Google Scholar]
- (54).Martin JL; Begun J; McLeish MJ; Caine JM; Grunewald GL Getting the adrenaline going: crystal structure of the adrenaline-synthesizing enzyme PNMT. Structure 2001, 9, 977–985. [DOI] [PubMed] [Google Scholar]
- (55).Gee CL; Drinkwater N; Tyndall JD; Grunewald GL; Wu Q; McLeish MJ; Martin JL Enzyme adaptation to inhibitor binding: a cryptic binding site in phenylethanolamine N-methyltransferase. J. Med. Chem. 2007, 50, 4845–4853. [DOI] [PubMed] [Google Scholar]
- (56).McCoy AJ; Grosse-Xunstleve RW.; Adams PD.; Winn MD.; Storoni LC.; Read RJ. Phaser crystallographic software. /. Appl. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW.; Oeflher.; Read RJ.; Richardson DC.; Richardson JS.; Terwilliger TC.; Zwart PH. PHENIX: a comprehensive Python-based system for macro-molecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Chen VB; Arendall WB 3rd, Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Project Collaborative Computational. The CCP4 suite: programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994, 50, 760–763. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.