

Abstract
Objective
Hereditary spastic paraplegia (HSP) has been reported rarely because of a monoallelic variant in ERLIN2. The present study aimed at describing a novel autosomal dominant ERLIN2 pedigree in a Chinese family and exploring the possible mechanism of HSP caused by ERLIN2 variants.
Methods
The proband and his family underwent a comprehensive medical history inquiry and neurological examinations. Whole‐exome sequencing was performed on the proband, and Sanger sequencing was performed on some family members. HeLa cell lines and mouse primary cortical neurons were used for immunofluorescence (IF) and reverse transcription‐PCR (RT‐PCR).
Results
Seven patients were clinically diagnosed with pure spastic paraplegia in four consecutive generations with the autosomal dominant inheritance model. All patients presented juvenile‐adolescent onset and gradually worsening pure HSP phenotype. Whole‐exome sequencing of the proband and Sanger sequencing of all available family members identified a novel heterozygous c.212 T>C (p.V71A) variant in exon 8 of the ERLIN2 gene. The c.212 T>C demonstrated a high pathogenic effect score through functional prediction. RT‐PCR and IF analysis of overexpressed V71A revealed an altered ER morphology and increased XBP‐1S mRNA levels, suggesting the activation of ER stress. Overexpression of V71A in primary cultured cortical neurons promoted axon growth.
Interpretation
The novel c.212 T>C heterozygous variant in human ERLIN2 caused pure HSP. Moreover, c.212 T>C heterozygous variant in ERLIN2 increased ER stress and affected axonal development.
Introduction
Hereditary spastic paraplegia (HSP) is a monogenic neurological disease with progressive lower extremity spasms and weakness. 1 The prevalence of HSP ranges from 2 to 5 per 100,000 individuals worldwide. 2 , 3 HSP phenotypes and genetics are highly heterogeneous. Different clinical symptoms indicate that HSP can be divided into pure or complicated forms. 4 Over 79 spastic paraplegia genes (SPG) have been identified. 5 Most patients have autosomal dominant, autosomal recessive, or X‐linked models, but no causative genes have been identified in 13–40% of cases. 6 Therefore, novel gene discovery is crucial to establish a definitive diagnosis.
Endoplasmic reticulum lipid raft‐associated 2 (ERLIN2) variants have been identified in HSP patients with spastic paraplegia 18 (SPG18). 5 SPG18 was considered an autosomal recessive disorder before 2018. So far, six SPG18 families presenting with autosomal dominant (AD) inheritance have been reported, 7 , 8 , 9 , 10 , 11 and the exact pathogenic mechanism has yet to be determined.
The ERLIN2 protein is a 40‐kDa transmembrane glycoprotein belonging to the prohibitin family with Stomatin‐prohibitin‐flotillin‐HflC/K (SPFH) domain. 12 , 13 ERLIN2, a key component of the endoplasmic reticulum (ER)‐associated protein degradation (ERAD) pathway, binds to activated inositol trisphosphate receptors (IP3Rs) and mediates polyubiquitination and subsequent degradation of IP3Rs or 3‐hydroxy‐3‐methylglutaryl‐CoA reductase. 14 , 15 , 16 The ERLIN2 variant causes continuous IP3R signal transduction activation and increases calcium release from ER, causing neuron hyperactivity and eventually leading to neuron death. 12 , 17 , 18 The relationship between ERLIN2 function and the ERAD pathway has been studied. Studies have also suggested that ERLIN2 may mediate the ER stress response to protein misfolding in breast cancer cells. 19
Protein accumulation frequently triggers ER stress under pathological circumstances. 20 Uncontrolled ER stress eventually causes cell death, including neuronal death in neurodegenerative diseases. 21 , 22 , 23 Motor neurons are particularly sensitive to ER stress because they are highly active. 24 , 25 , 26 Studies have suggested that ER stress increases in the early stage of amyotrophic lateral sclerosis (ALS). 27 , 28 , 29 The absence of membralin‐ERLIN2 interaction increases basal ER stress, which results in ER stress‐induced cell death. 30 Although ER stress pathogenicity has been studied in several motor neuron diseases, little is known regarding the relationship between ERLIN2 and ER stress in HSP.
Our study identified a Chinese AD‐HSP family with a novel heterozygous missense variant of ERLIN2 (p.V71A) that increased ER stress and altered axonal length.
Materials and Methods
Clinical assessment
A five‐generation family with HSP was recruited at the First Hospital of Shanxi Medical University, Taiyuan, China. The present study involved nine participants (four male and five female), including seven affected and two unaffected. Pedigree analysis suggested autosomal dominant inheritance. Informed consent was obtained from all individuals included in the study. Detailed medical history and physical examinations were obtained by a qualified investigator (J.G.). The severity of all HSP patients was rated using the Spastic Paraplegia Rating Scale (SPRS), with scores ranging from 0 to 52, and higher scores indicate a more severe disease. 31 Genomic DNA was extracted from peripheral blood (II‐3, II‐4, III‐2, III‐5, III‐6, IV‐3, IV‐6, and V‐1) and saliva (IV‐2) samples through standard procedures.
This study was reviewed and approved by the Ethics Institutional Committee of the First Hospital of Shanxi Medical University (K185).
Whole‐exome sequencing and data analysis
Whole‐exome capture (Running Gene, Beijing, China) of subject IV‐3 was performed as described previously. 32 Our data analysis procedure used the Illumina Sequence Control Software (SCS) and the BWA Aligner to align our exome sequencing data with the human reference genome (hg19). SNPs and INDELs were analyzed with the Genome Analysis Toolkit (GATK) and annotated using ANNOVAR. The clinically significant variants were filtered in the following ways: (1) retaining variants with a minor allele frequency (MAF) of less than 0.5% according to the dbSNP, genomAD, and EXAC databases; (2) remaining variants of exonic non‐synonymous SNVs, splice site SNVs, and INDELs; (3) classifying deleterious variants as related or unrelated to the disease.
Sanger sequencing
To confirm the accuracy of the variants, we performed Sanger sequencing on the clinically significant gene ERLIN2. The ERLIN2 clone primers were designed using the Primer3 software (http://frodo.wi.mit.edu/). The forward primer was 5′‐ACCCTGTGAGGAAGGAGGAT‐3′, and the reverse primer was 5′‐TCCATCACCTGGTCAAATCA‐3′. The co‐segregation analysis between genes and disease phenotypes was conducted using Sanger‐sequenced DNA samples from all available family members. A total of 200 unrelated subjects, recruited from Shanxi, China, where the disease family originated, were used as controls. All controls had no history of dyskinesia or other HSP‐related phenotypes. Sequence analysis was conducted using the BigDye Terminator Cycle Sequencing Kit. PCR products were evaluated using the ABI PRISM 3730 Analyzer (Applied Biosystems, USA).
Plasmids construction and mRNA synthesis
The full‐length human ERLILN2 cDNA (NM_007175.6) was obtained using RT‐PCR from human total RNA isolated from HeLa cells using the Total RNA Isolation Kit (RC101‐01, Vazyme, China). Human ERLILN2 cDNA was amplified and cloned into pCDNA3.1+ and pCAGGS‐IRES‐GFP vectors, with double restriction sites BamHI/XbaI and XbaI/NotI (New England Biolabs), respectively. The missense variant p.V71A was introduced into the hERLIN2 plasmid using site‐direct mutagenesis PCR (C214‐01, Vazyme, China). All generated plasmids were validated through Sanger sequencing. The primer sequences are listed in Table S1. Expression plasmids were extracted using the EndoFree Plasmid Mini Kit (CW2106, ComWin Biotech Co, China).
Cell culture and transfections
HeLa cells were grown in Dulbecco's modified Eagle medium, DMEM (11965092, Thermo Fisher Scientific, Waltham, MA, USA), supplemented with 10% fetal bovine serum (10091, Thermo Fisher Scientific, Waltham, MA, USA) at 37°C and 5% CO2 atmosphere. The cells were incubated for 24 h with the growth medium with or without 2 μg/mL tunicamycin (IT2670, Solarbio, China). The cells were transfected with a transfection reagent (101000046, jetPRIME, USA). After 48 h of incubation, protein and total RNA were extracted from the cells.
Primary cortical neurons were isolated from embryonic day 16.5 (E16.5) ICR mice, maintained in neurobasal medium (21103049, Invitrogen, Carlsbad, CA, USA) containing 1% Glutamax (35050061, Invitrogen, Carlsbad, CA, USA) and 2% B27 (17504044, Invitrogen, Carlsbad, CA, USA), and cultured on poly‐D‐lysine‐coated coverslips. Cortical neurons were transfected using the Calcium Phosphate Cell Transfection Kit (40803ES70, Beyotime Biotechnology, Shanghai, China) on Day 2 in vitro (DIV2) and collected on Day 5 in vitro (DIV5) for analysis of axon outgrowth or total RNA extraction from the neurons.
Western blot analysis
HeLa cells were cultured in six‐well plates and transfected with a transfection reagent (101000046, jetPRIME, USA). After 48 h, samples were collected from the six‐well plates and lysed in SDS buffer (P0013G, Beyotime, China). The protein extract was obtained after centrifugation. Protein quantitation was performed using the BCA method. Equal amounts of protein (30 μg) were boiled in sample buffer, ran on SDS‐PAGE gels, transferred onto a PVDF membrane, and immunoblotted with anti‐IP3R (ab264281, Abcom, 1:2000), anti‐HA (C29F4, Cell Signaling Technology, 1:1000), and α‐tubulin (bs‐0159R, Bioss, 1:2000) antibodies. Immunoblots were developed using the ECL reagent (AR1170, BOSTER, China) and imaged in the BIO‐RAD Imager.
Immunofluorescence
HeLa cells and neurons (DIV5) were fixed with 4% PFA and 0.5% Triton X‐100 in PBS. After blocking with 10% fetal calf serum in 0.5% Triton X‐10 PBS for 60 min, the cells were stained with HA‐Tag Rabbit primary antibody (C29F4, Cell Signaling Technology, 1:1000) and Calnexin (sc‐23954, Santa Cruz Biotechnology Inc., 1:50) probed with Alexa fluor‐conjugated secondary antibodies for subcellular localization of ERLIN2 and ER. DAPI was used to stain the nuclei. Images were captured under a confocal microscope (Leica, TSC‐SP8).
Reverse transcription‐PCR
Total RNA was extracted from the cells using the Total RNA Isolation Kit (RC101‐01, Vazyme, China). Total RNA from each sample was reverse transcribed into cDNA using the Revert Aid First Strand cDNA Synthesis Kit (R323‐01, Vazyme, China). PCR was then performed using the 2 × Phanta Max Master Mix (P515‐01, Vazyme, China). According to the manufacturer's instructions, each PCR reaction mixture comprised 1 μL of cDNA and 10 pm/μL of the appropriate forward and reverse primers in a final volume of 10 μL. Primer sequences are illustrated in Table S2. Primers were synthesized by Sangon Biotech (Shanghai, China). The resulting products were subjected to 2% agarose gel electrophoresis. The mRNA level in each sample was normalized to that of β‐Actin or XBP‐1U mRNA.
Data presentation and Statistical analysis
All experiments were repeated at least three times. Quantitative data were expressed as mean ± SEM. Statistical analyses were conducted using the GraphPad Prism software (version 6). A Student t‐test or one‐way ANOVA with Dunnett's test was performed to compare the means between two groups or more than two groups, respectively. Differences with a p‐value <0.05 were considered significant.
Results
An autosomal dominant HSP family
Figure 1A presents an overview of the family pedigree. Since the age of 14, the proband (IV‐3), a 24‐year‐old male, complained of progressive worsening spasticity. At the age of 21, he was unable to run or walk fast because of leg stiffness. The neurological examination revealed hyperactive deep tendon reflexes in the lower limbs, ankle clonus, contracted Achilles tendon, and the Babinski sign. The patient had no cranial nerve palsy, cognitive functions, muscle weakness, and atrophy (Video S1).
Figure 1.
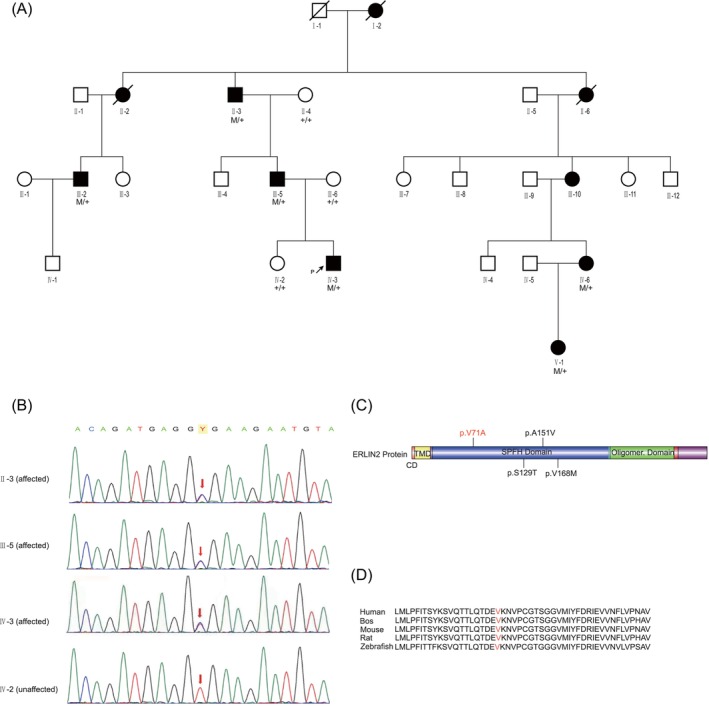
Pedigree of affected family, ERLILN2 variants, and conservation among species. (A) Family pedigree. Black filled symbol, affected; white symbol, unaffected; and black arrows, proband. Sanger sequencing was performed in some subjects (+/+: normal, M/+: heterozygous), demonstrating complete segregation of the ERLIN2 missense variant c.212 T>C, p.V71A with the disease. (B) The partial nucleotide sequences of exon 7 of ERLIN2 show the c.212 T>C variant in the affected or unaffected family members (II‐3, III‐5, IV‐3, and IV‐2). (C) Schematic representation of the basic structure and domain organization of the ERLIN2 protein. The figure was generated using the Illustrator for Biological Sciences (IBS). The observed variants were labeled. The site of the novel variant p.V71A identified in this study is indicated by red color. (D) Sequence alignment of ERLIN2 proteins from various species. The arrows indicate the amino acid changes in this study. Emboldened amino acids are conserved.
The proband's father (III‐5) was 47 years old. He began walking abnormally at 14, developed leg stiffness in his 20s, and eventually could not run by his 30s. Since the age of 40, he could not walk without crutches. Neurological examinations revealed lower limb spasticity, hyperreflexia, and pathologic reflexes. There was no muscle atrophy. Nerve conduction studies revealed no abnormal findings of the peripheral nerves. Needle electromyography showed no evidence of fibrillation, fasciculation potentials, or chronic neurogenic changes in the gastrocnemius muscle and anterior tibialis (Table S3).
The proband's grandfather (II‐3) had gait disturbance as a teenager, running difficulties in his 30s, and began using a cane in his 50s (Video S2). He had a severely rigid spinal deformity and lower limb contractures without muscle atrophy. Neurological examinations revealed hyperreflexia and pathologic reflexes in all extremities. The lower limbs were uncooperative because of contractures, whereas the upper limbs had normal muscle strength.
Other family members (III‐2, III‐10, and IV‐6) exhibited similar symptoms and neurological findings. The proband's uncle (III‐2), a 46‐year‐old man, began complaining of stiffness in the legs at the age of 20. Neurological examination revealed hypertonia, active or even hyperreflexia, and a positive Babinski sign but no muscle weakness. The proband's sister (IV‐6), a 43‐year‐old woman, had slowly progressive spasticity of the lower limbs at the age of 17. She can still walk without assistance but cannot walk on her heel. Neurological examination showed active hyperreflexia and a positive Babinski sign but normal muscle strength. The proband's family member (III‐10), a 63‐year‐old woman, had slowly progressive spasticity of the lower limbs at the age of 40. Until now, she can walk without assistance. She could not attend the neurology examination because of a scheduling conflict. V‐1, a 22‐year‐old woman, had no clinical symptoms; however, neurological examinations revealed hyperactive deep tendon reflexes in the lower limbs and the Babinski sign. Table 1 presents the details of the neurological examinations. The proband's 28‐year‐old sister (IV‐2) and mother (III‐6) are normal walkers; neurological examinations revealed no signs of muscle weakness, hypertonia, hyperreflexia, or the Babinski sign.
Table 1.
Clinical characteristics of the affected individuals in the family.
| Individual | II‐3 | III‐2 | III‐5 | IV‐3 | IV‐6 | V‐1 |
|---|---|---|---|---|---|---|
| Gender | M | M | M | M | F | F |
| Age at examination | 71 | 46 | 47 | 24 | 43 | 22 |
| Age at onset | 21 | 20 | 14 | 14 | 17 | – |
| Disease duration | 50 | 26 | 33 | 10 | 26 | – |
| Age when walking needs assistant | 50 | – | 42 | – | ||
| Disability stagea | 5 | 3 | 4 | 3 | 3 | 1 |
| SPRS | 48 | 5 | 40 | 13 | 20 | 0 |
| Spastic gait | ++++ | + | ++++ | ++ | ++ | – |
| Increased muscle tone in LL | ++++ | + | ++++ | + | + | – |
| Hyperreflexia in LL | – | ++++ | ++++ | ++++ | +++ | +++ |
| Weakness in LL | NA | + | + | – | – | – |
| Babinski sign | + | + | + | + | + | + |
| Foot deformity | ++ | – | ++ | – | – | – |
| Bladder disturbances | – | – | – | – | – | – |
| Mental retardation | – | – | – | – | – | – |
| Seizure | – | – | – | – | – | – |
| Scoliosis | +++ | – | ++ | – | + | – |
–, absent; +, mild; ++, moderate; +++, severe; ++++, extremely severe; NA, not available; LL, lower limbs; SPRS: Spastic Paraplegia Rating Scale, range 0–52, higher scores indicating more severe disease; a: disability stage (SPATAX‐EUROSPA): 0, no functional handicap; 1, no functional handicap but signs at examination; 2, able to run, walking unlimited; 3, unable to run, limited walking without aid; 4, walking with one stick; 5, walking with two sticks; 6, unable to walk, requiring wheelchair; 7, confined to bed;
Causal variant identification for the HSP pedigree
We performed whole‐exome sequencing on the proband to precisely localize the disease gene. An average of 99.8% of targeted regions was covered at a mean read depth of 141.42×. As every generation exhibited similar symptoms, autosomal dominant inheritance was considered. After the filtering procedure, 356 small nucleotide variants (SNPs) and 95 indels were identified. In this study, we focused on variants discovered in genes that have been reported to cause HSP. ERLIN2 has been reported to cause HSP. Our study identified a novel variant in the ERLIN2 gene (c.212 T>C), but the other causative variants were not discovered in the ERLIN2 gene.
The c.212 T>C variant was confirmed in exon 7 of ERLIN2 through Sanger sequencing. The HSP‐affected family members were heterozygous for the c.212 T>C variant, whereas the unaffected family member had only the ERLIN2 reference sequence, suggesting the co‐segregation of genotype and phenotype within the family (Fig. 1B). In 200 unrelated controls from the same ethnic background, the variant was not detected. Furthermore, the identified variant was not documented in the Single Nucleotide Polymorphism Database (dbSNP), the 1000 Genomes Project, the Exome Aggregation Consortium (ExAC), or the Genome Aggregation Database (genomeAD).
The c.212 T>C single nucleotide variant converted amino acid 71 from valine to alanine (p.V71A), located in the SPFH domain of the ERLIN2 protein (Fig. 1C). The c.212 T>C demonstrated a high pathogenic effect score, predicted to be deleterious through SIFT (score: −3.255), Polyphen‐2 (possibly damaging, score: 0.711, sensitivity: 0.86, specificity: 0.92), and Mutation Taster (disease‐causing, score: 0.999). The p.V71A is also located in a region highly conserved in other species (Fig. 1D).
Studies have identified ERLIN2 variants that cause autosomal recessive HSP, namely SPG18 (OMIM 611225). 5 Heterozygous ERLIN2 variants also cause a rare autosomal dominant HSP. Studies have reported six AD‐SPG18 families, including 4 ERLIN2 variants 7 , 8 , 9 , 10 , 11 (Table 2). As with previous AD ERLIN2 variants, p.V71A was identified in the SPFH domain (Fig. 1C). AD‐SPG18 presents as juvenile‐ or adolescent‐onset pure HSP in these six families. A minority of patients with ERLIN2 variants initially present with spastic paraplegia before developing ALS 30–40 years later (Table 2).
Table 2.
A summary of all reported autosomal dominant spastic paraplegia 18 (AD‐SPG18) families.
| F1 | F2 | F3 | F4 | F5 | F6 | F7 | |
|---|---|---|---|---|---|---|---|
| Race | Germany | Norway | France | Korea | Chinese | NA | Chinese |
| Variants | c.386 G>C | c.386 G>C | c.502 G>A | c.452 C>T | c.502 G>A | c.206 A>T | c.212 T>C |
| Protein change | p.S129T | p.S129T | p.V168M | p.A151V | p.V168M | p.D69V | p.V71A |
| Age at examination, years | 25–76 | 35–59 | NA | 23–77 | 30–63 | 41–69 | 24–71 |
| Disease duration, years | 2–50 | 2–50 | Conversion to ALS (25–30 years later) | 1–39 | 10–48 | 16–50 | 10–50 |
| Age at onset, years | 13–46 | 9–28 | 25–45 | 15–38 | 8–15 | 20–25 | 14–21 |
| Symptoms at onset | Stiffness in the legs | Stiffness in the legs | NA | Awkward gait or Gait disturbance | Abnormal walking | Difficulty in descending stairs or moving his legs | Gait disturbance |
| Phenotype | Pure | Pure | Pure, convert to ALS | Pure | Pure | Pure, convert to ALS | Pure |
| Reference | Rydning et al. 7 | Rydning et al. 7 | Amador et al. 10 | Park et al. 8 | Chen et al. 9 | Kume et al. 11 | Our family |
ALS, amyotrophic lateral sclerosis; NA, not available.
ERLIN2‐V71A increases ER stress
ERLIN2 encodes a member of the SPFH domain‐containing family of lipid raft‐associated proteins. ERLIN2 is situated in lipid rafts of the endoplasmic reticulum and plays a critical role in ER stress and the ERAD pathway. HeLa cells were transfected with either ERLIN2‐WT or V71A to investigate the expression and subcellular localization of the cells. As shown in Figure 2A, the expression levels of ERLIN2‐WT and V71A were consistent. Previous studies have reported that ERLIN2 overexpression promoted IP3R degradation in the ERAD pathway. However, we found that overexpression of ERLIN2‐WT or V71A diminished IP3R protein levels, suggesting that V71A does not affect the ERAD pathway. To explore the effect of V71A on ER stress, we induced ER stress using tunicamycin (TM) as a positive control. One of the main pathways of ER stress is the splicing of X‐box binding protein 1 (XBP‐1S) mediated by inositol demand enzyme 1α (IRE1α). TM can induce ER stress by increasing XBP‐1S. 33 Immunofluorescence labeling ER with Calnexin (CANX) revealed partial colocalization of ERLIN2‐WT and V71A with ER. However, the V71A and TM treatment groups had altered ER morphology, with fragmented or bubble‐like tubular structures (Fig. 2B). In the V71A‐ and TM‐treated groups, RT‐PCR revealed increased transcription levels of XBP‐1S and the XBP‐1S/XBP‐1U ratio compared with the control group. Furthermore, the levels of XBP‐1S and the XBP‐1S/XBP‐1U ratio in the V71A group were significantly higher than those in the WT group (Fig. 2C–E). These findings suggest that V71A overexpression promotes ER stress through the IRE1 pathway. This finding was confirmed by performing an analogous experiment on mouse primary cortical neurons. Similarly, V71A increased XBP1 splicing (Fig. S1). These findings collectively suggest that V71A increases ER stress.
Figure 2.
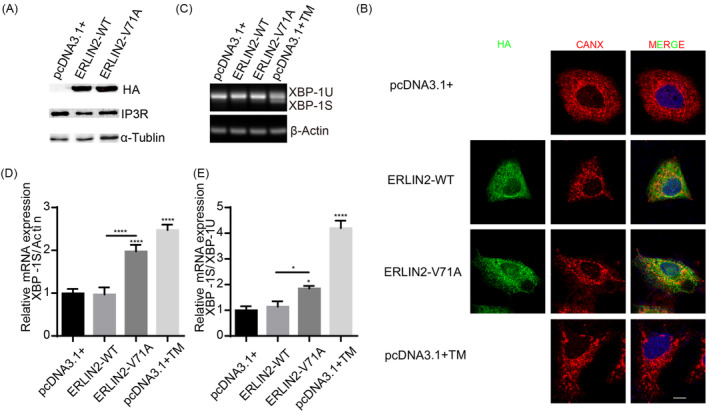
ERLIN2‐V71A overexpression alters ER morphology and increases XBP‐1 splicing. (A) The expression levels of ERLIN2‐WT‐HA, ERLIN2‐V71A‐HA, and IP3R were detected through WB. (B) HeLa cells were cultured with or without TM or transfected with ERLIN2‐WT‐HA or ERLIN2‐V71A‐HA and then immunostained for HA (green) and CANX (red) to detect ER. Merged images are to the right. Bars, 10 μm. (C–E) HeLa cells were cultured with or without TM or transfected with ERLIN2‐WT‐HA or ERLIN2‐V71A‐HA, and then, the relative mRNA level of XBP‐1S/Actin and XBP‐1S/XBP‐1U was detected using RT‐PCR. One‐way ANOVA test with Dunnett's test or unpaired t‐test; values are presented as mean n ± SEM. *p < 0.05, ****p < 0.0001.
ERLIN2‐V71A facilitates axonal outgrowth
Previous research has demonstrated that the XBP‐1S protein regulates neuritis growth in developing neurons. 34 We produced ERLIN2‐WT and V71A variant plasmids in the pCAGGS‐IRES‐GFP vector to transfect mouse primary cortical neurons and to investigate the role of ERLIN2 in neural development. At DIV5, immunofluorescence revealed that ERLIN2 V71A mutants were associated with longer axons and total neuritis than ERLIN2‐WT (Fig. 3).
Figure 3.
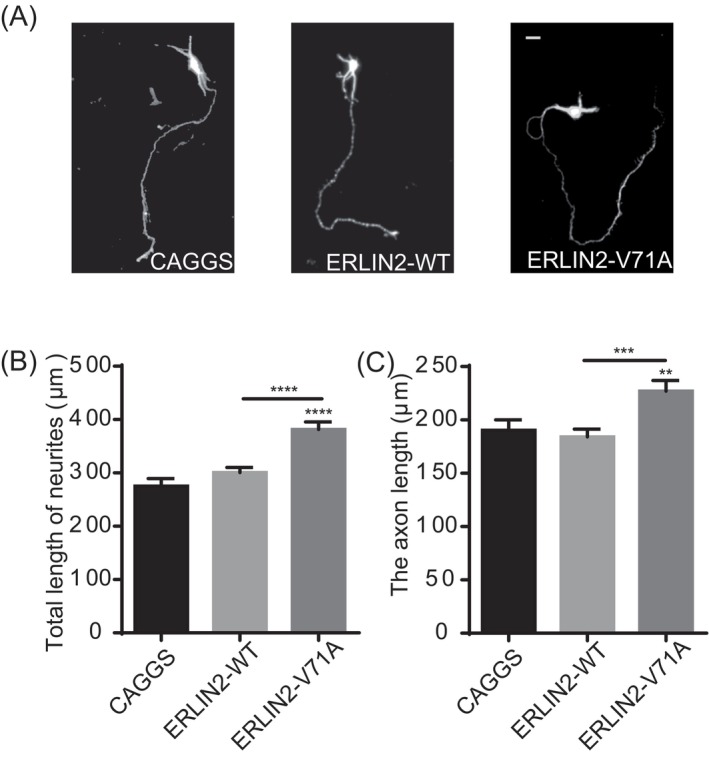
Overexpression of ERLIN2‐V71A facilitates axon outgrowth. (A) Representative images of primary cortical neurons at DIV 5. Neurons were transfected with pCAGGS‐IRES‐GFP (CAGGS), ERLIN2‐WT, or ERLIN2‐V71A at DIV2. Bars, 10 μm. (B and C) Quantification of total neurites length or the axon length. One‐way ANOVA test with Dunnett's test; values for more than 60 neurons from three independent experiments are presented as mean ± SEM. **p < 0.01, ***p < 0.001, ****p < 0.0001, unpaired t‐test.
Discussion
The present study reports a novel heterozygous ERLIN2 variant p.V71A in a Chinese family through whole‐exome sequencing. We discovered that ERLIN2‐V71A altered ER morphology and increased XBP1‐S expression, supporting the association between ER stress and HSP. Functional studies have indicated that ERLIN2‐V71A promotes axonal growth, suggesting ER stress is one of the mechanisms underlying HSP.
Almost all patients in the family presented with pure HSP symptoms, including progressive lower limb weakness and spasticity. DNA sequencing identified heterozygous variant V71A to ERLIN2 in the proband, and the variant was co‐segregated with the clinical phenotype in the family. As previously reported, ERLIN2 variants can cause AD and AR inheritance patterns. A heterozygous variant in ERLIN2 causes pure HSP, 7 , 8 , 9 , 10 , 11 whereas biallelic variants in ERLIN2 cause autosomal recessive complicated HSP. 17 , 35 , 36 The clinical symptoms of SPG18 may differ by gender, 8 , 37 with milder and later‐onset symptoms in female patients. The average age of male onset in the present study ranged from 14 to 25 years, consistent with previously reported cases. 7 , 8 , 9 , 10 , 11 Our family shown that the disorder appeared later and presented milder symptoms in women. A 22‐year‐old woman with the variant gene exhibited no clinical signs of the disease except for Babinski signs. Additional research is required to better understand inheritance modes and gender differences.
ERLIN2 is an ER membrane protein containing an evolutionarily conserved SPFH domain, which includes the determinants for binding to activated IP3Rs. 38 , 39 Notably, all autosomal dominant HSP cases, including ours, involve variants in ERLIN's SPFH domain. The ERLIN2‐T65I variant in the SPFH domain inhibits the IP3Rs ERAD pathway, causing abnormal protein accumulation. 18 Although overexpression of both ERLIN2‐WT and V71A decreased IP3R protein levels, we found that overexpression of V71A increased ER stress.
XBP‐1 serves as a significant transcription factor and is activated under ER stress. 40 , 41 As a key regulator of ER stress, XBP‐1 can be divided into the active isoform (XBP‐1S) and the inactive isoform (XBP‐1U). 42 The generation of XBP‐1S induces ER stress, whereas XBP‐1U is independent of ER stress activation. 43 , 44 ER stress was induced by targeting XBP‐1 splicing to increase the XBP‐1S/XBP‐1U ratio. 45 After overexpression of V71A, the XBP‐1S/XBP‐1U ratio was elevated compared with the control group, which was consistent with the TM group. Moreover, the levels of XBP‐1S and the XBP‐1S/XBP‐1U ratio in the V71A group were significantly higher than those in the WT group. Our findings confirm that overexpression of V71A increases ER stress. Moreover, we discovered that ERLIN2‐V71A facilitated axon growth. Our results demonstrated that multiple pathogenic mechanisms appeared to cause selective neurodegeneration of the corticospinal tracts in HSP. 46
Conclusion
We demonstrated that a novel ERLIN2 missense variant caused pure HSP in an autosomal dominant manner. Our findings reveal that the ERLIN2 variant increases ER stress, causing abnormal neuron development. These findings expand the mutational and inheritance spectrum of SPG18. Moreover, these findings shed light on the pathogenesis of the ERLIN2 variant.
Funding Information
This work was supported by the National Natural Science Foundation of China Youth Fund Project under grant no. 82001222, the Innovation Research Foundation of China International Medical Exchange Foundation under grant no. Z‐2016‐20‐1801, the Shanxi Science and Technology Department under grant no. 202103021223437 and 202203021212037, and the Provincial Doctor Foundation project of Shanxi Medical University under grant no. SD2248.
Author Contributions
GJH, CXL, WJ, and ZRJ designed the study; WJ and ZRJ performed the experiments and analyzed the data; YZX collected the clinical data; CHS and MJ helped analyzed the data; WJ and ZRJ wrote the manuscript.
Conflict of Interest
The authors state that there is no conflict of interest.
Consent for Publication
All authors agree with the content of the manuscript, and all patients or legal guardians consented to the publication.
Supporting information
Appendix S1.
Video S1.
Video S2.
Acknowledgements
We thank all participants for their interest and participation to the study.
Funding Statement
This work was funded by Innovation Research Foundation of China International Medical Exchange Foundation grant Z‐2016‐20‐1801; National Natural Science Foundation of China Youth Fund Project grant 82001222; Provincial Doctor Foundation project of Shanxi Medical University grant SD2248; Shanxi Provincial Science and Technology Department grants 202103021223437 and 202203021212037.
Contributor Information
Xueli Chang, Email: changxueli841228@163.com.
Junhong Guo, Email: neuroguo@163.com.
References
- 1. Salinas S, Proukakis C, Crosby A, Warner TT. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 2008;7(12):1127‐1138. [DOI] [PubMed] [Google Scholar]
- 2. Ruano L, Melo C, Silva MC, Coutinho P. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology. 2014;42(3):174‐183. [DOI] [PubMed] [Google Scholar]
- 3. Coutinho P, Ruano L, Loureiro JL, et al. Hereditary ataxia and spastic paraplegia in Portugal: a population‐based prevalence study. JAMA Neurol. 2013;70(6):746‐755. [DOI] [PubMed] [Google Scholar]
- 4. Harding A. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1(8334):1151‐1155. [DOI] [PubMed] [Google Scholar]
- 5. Shribman S, Reid E, Crosby AH, Houlden H, Warner TT. Hereditary spastic paraplegia: from diagnosis to emerging therapeutic approaches. Lancet Neurol. 2019;18(12):1136‐1146. [DOI] [PubMed] [Google Scholar]
- 6. Schüle R, Wiethoff S, Martus P, et al. Hereditary spastic paraplegia: clinicogenetic lessons from 608 patients. Ann Neurol. 2016;79(4):646‐658. [DOI] [PubMed] [Google Scholar]
- 7. Rydning SL, Dudesek A, Rimmele F, et al. A novel heterozygous variant in ERLIN2 causes autosomal dominant pure hereditary spastic paraplegia. Eur J Neurol. 2018;25(7):943‐e71. [DOI] [PubMed] [Google Scholar]
- 8. Park JM, Lee B, Kim JH, et al. An autosomal dominant ERLIN2 mutation leads to a pure HSP phenotype distinct from the autosomal recessive ERLIN2 mutations (SPG18). Sci Rep. 2020;10(1):3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen S, Zou JL, He S, Li W, Zhang JW, Li SJ. More autosomal dominant SPG18 cases than recessive? The first AD‐SPG18 pedigree in Chinese and literature review. Brain Behav. 2021;11(12):e32395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Amador MD, Muratet F, Teyssou E, et al. Spastic paraplegia due to recessive or dominant mutations in ERLIN2 can convert to ALS. Neurol Genet. 2019;5(6):e374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kume K, Kamada M, Shimatani Y, Takata T, Izumi Y, Kawakami H. Novel monoallelic variant in ERLIN2 causes spastic paraplegia converted to amyotrophic lateral sclerosis. J Neurol Sci. 2021;430:119984. [DOI] [PubMed] [Google Scholar]
- 12. Manganelli V, Longo A, Mattei V, et al. Role of ERLINs in the control of cell fate through lipid rafts. Cell. 2021;10(9):2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hoegg MB, Browman DT, Resek ME, Robbins SM. Distinct regions within the erlins are required for oligomerization and association with high molecular weight complexes. J Biol Chem. 2009;284(12):7766‐7776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pearce MM, Wormer DB, Wilkens S, Wojcikiewicz RJ. An endoplasmic reticulum (ER) membrane complex composed of SPFH1 and SPFH2 mediates the ER‐associated degradation of inositol 1,4,5‐trisphosphate receptors. J Biol Chem. 2009;284(16):10433‐10445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pearce MM, Wang Y, Kelley GG, Wojcikiewicz RJ. SPFH2 mediates the endoplasmic reticulum‐associated degradation of inositol 1,4,5‐trisphosphate receptors and other substrates in mammalian cells. J Biol Chem. 2007;282(28):20104‐20115. [DOI] [PubMed] [Google Scholar]
- 16. Jo Y, Sguigna PV, DeBose‐Boyd RA. Membrane‐associated ubiquitin ligase complex containing gp78 mediates sterol‐accelerated degradation of 3‐hydroxy‐3‐methylglutaryl‐coenzyme A reductase. J Biol Chem. 2011;286(17):15022‐15031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alazami A, Adly N, al Dhalaan H, Alkuraya FS. A nullimorphic ERLIN2 mutation defines a complicated hereditary spastic paraplegia locus (SPG18). Neurogenetics. 2011;12(4):333‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Egorova PA, Bezprozvanny IB. Inositol 1,4,5‐trisphosphate receptors and neurodegenerative disorders. FEBS J. 2018;285(19):3547‐3565. [DOI] [PubMed] [Google Scholar]
- 19. Wang G, Liu G, Wang X, et al. ERLIN2 promotes breast cancer cell survival by modulating endoplasmic reticulum stress pathways. BMC Cancer. 2012;12:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7(12):1013‐1030. [DOI] [PubMed] [Google Scholar]
- 21. Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006;13(3):385‐392. [DOI] [PubMed] [Google Scholar]
- 22. Scheper W, Hoozemans JJ. Endoplasmic reticulum protein quality control in neurodegenerative disease: the good, the bad and the therapy. Curr Med Chem. 2009;16(5):615‐626. [DOI] [PubMed] [Google Scholar]
- 23. Hetz C, Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci. 2014;15(4):233‐249. [DOI] [PubMed] [Google Scholar]
- 24. Vinay L, Brocard F, Pflieger JF, Simeoni‐Alias J, Clarac F. Perinatal development of lumbar motoneurons and their inputs in the rat. Brain Res Bull. 2000;53(5):635‐647. [DOI] [PubMed] [Google Scholar]
- 25. Carrascal L, Nieto‐Gonzalez JL, Cameron WE, Torres B, Nunez‐Abades PA. Changes during the postnatal development in physiological and anatomical characteristics of rat motoneurons studied in vitro. Brain Res Brain Res Rev. 2005;49(2):377‐387. [DOI] [PubMed] [Google Scholar]
- 26. Li Y, Brewer D, Burke RE, Ascoli GA. Developmental changes in spinal motoneuron dendrites in neonatal mice. J Comp Neurol. 2005;483(3):304‐317. [DOI] [PubMed] [Google Scholar]
- 27. Atkin JD, Farg MA, Turner BJ, et al. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein‐disulfide isomerase with superoxide dismutase 1. J Biol Chem. 2006;281(40):30152‐30165. [DOI] [PubMed] [Google Scholar]
- 28. Nagata T, Ilieva H, Murakami T, et al. Increased ER stress during motor neuron degeneration in a transgenic mouse model of amyotrophic lateral sclerosis. Neurol Res. 2007;29(8):767‐771. [DOI] [PubMed] [Google Scholar]
- 29. Kanekura K, Suzuki H, Aiso S, Matsuoka M. ER stress and unfolded protein response in amyotrophic lateral sclerosis. Mol Neurobiol. 2009;39(2):81‐89. [DOI] [PubMed] [Google Scholar]
- 30. Yang B, Qu M, Wang R, et al. The critical role of membralin in postnatal motor neuron survival and disease. Elife. 2015;4:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schule R, Holland‐Letz T, Klimpe S, et al. The Spastic Paraplegia Rating Scale (SPRS): a reliable and valid measure of disease severity. Neurology. 2006;67(3):430‐434. [DOI] [PubMed] [Google Scholar]
- 32. Shi J, Zhao F, Pang X, et al. Whole‐exome sequencing identifies a heterozygous mutation in SLC12A6 associated with hereditary sensory and motor neuropathy. Neuromuscul Disord. 2021;31(2):149‐157. [DOI] [PubMed] [Google Scholar]
- 33. Bouchecareilh M, Higa A, Fribourg S, Moenner M, Chevet E. Peptides derived from the bifunctional kinase/RNase enzyme IRE1α modulate IRE1α activity and protect cells from endoplasmic reticulum stress. FASEB J. 2011;25(9):3115‐3129. [DOI] [PubMed] [Google Scholar]
- 34. Hayashi A, Kasahara T, Iwamoto K, et al. The role of brain‐derived neurotrophic factor (BDNF)‐induced XBP1 splicing during brain development. J Biol Chem. 2007;282(47):34525‐34534. [DOI] [PubMed] [Google Scholar]
- 35. Al‐Yahyaee S, Al‐Gazali LI, De Jonghe P, et al. A novel locus for hereditary spastic paraplegia with thin corpus callosum and epilepsy. Neurology. 2006;66(8):1230‐1234. [DOI] [PubMed] [Google Scholar]
- 36. Yıldırım Y, Kocasoy Orhan E, Ugur Iseri SA, et al. A frameshift mutation of ERLIN2 in recessive intellectual disability, motor dysfunction and multiple joint contractures. Hum Mol Genet. 2011;20(10):1886‐1892. [DOI] [PubMed] [Google Scholar]
- 37. Erfanian Omidvar M, Torkamandi S, Rezaei S, et al. Genotype‐phenotype associations in hereditary spastic paraplegia: a systematic review and meta‐analysis on 13,570 patients. J Neurol. 2021;268(6):2065‐2082. [DOI] [PubMed] [Google Scholar]
- 38. Browman DT, Hoegg MB, Robbins SM. The SPFH domain‐containing proteins: more than lipid raft markers. Trends Cell Biol. 2007;17(8):394‐402. [DOI] [PubMed] [Google Scholar]
- 39. Ande SR, Mishra S. Prohibitin interacts with phosphatidylinositol 3,4,5‐triphosphate (PIP3) and modulates insulin signaling. Biochem Biophys Res Commun. 2009;390(3):1023‐1028. [DOI] [PubMed] [Google Scholar]
- 40. Zhang K, Kaufman RJ. The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology. 2006;66(2 Suppl 1):S102‐S109. [DOI] [PubMed] [Google Scholar]
- 41. Lee AH, Iwakoshi NN, Glimcher LH. XBP‐1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23(21):7448‐7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881‐891. [DOI] [PubMed] [Google Scholar]
- 43. Glimcher LH, Lee AH, Iwakoshi NN. XBP‐1 and the unfolded protein response (UPR). Nat Immunol. 2020;21(9):963‐965. [DOI] [PubMed] [Google Scholar]
- 44. Ong HK, Soo BPC, Chu KL, Chao SH. XBP‐1, a cellular target for the development of novel anti‐viral strategies. Curr Protein Pept Sci. 2018;19(2):145‐154. [DOI] [PubMed] [Google Scholar]
- 45. Ge P, Gao M, du J, Yu J, Zhang L. Downregulation of microRNA‐512‐3p enhances the viability and suppresses the apoptosis of vascular endothelial cells, alleviates autophagy and endoplasmic reticulum stress as well as represses atherosclerotic lesions in atherosclerosis by adjusting spliced/unspliced ratio of X‐box binding protein 1 (XBP‐1S/XBP‐1U). Bioengineered. 2021;12(2):12469‐12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fink JK. Hereditary spastic paraplegia: clinico‐pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013;126(3):307‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1.
Video S1.
Video S2.