

Abstract
Numerous potential amyotrophic lateral sclerosis (ALS)‐relevant pathways have been hypothesized and studied preclinically, with subsequent translation to clinical trial. However, few successes have been observed with only modest effects. Along with an improved but incomplete understanding of ALS as a neurodegenerative disease is the evolution of more sophisticated and diverse in vitro and in vivo preclinical modeling platforms, as well as clinical trial designs. We highlight proposed pathological pathways that have been major therapeutic targets for investigational compounds. It is likely that the failures of so many of these therapeutic compounds may not have occurred because of lack of efficacy but rather because of a lack of preclinical modeling that would help define an appropriate disease pathway, as well as a failure to establish target engagement. These challenges are compounded by shortcomings in clinical trial design, including lack of biomarkers that could predict clinical success and studies that are underpowered. Although research investments have provided abundant insights into new ALS‐relevant pathways, most have not yet been developed more fully to result in clinical study. In this review, we detail some of the important, well‐established pathways, the therapeutics targeting them, and the subsequent clinical design. With an understanding of some of the shortcomings in translational efforts over the last three decades of ALS investigation, we propose that scientists and clinicians may choose to revisit some of these therapeutic pathways reviewed here with an eye toward improving preclinical modeling, biomarker development, and the investment in more sophisticated clinical trial designs.
Introduction
Numerous therapeutic strategies for treating amyotrophic lateral sclerosis (ALS) have been studied in individuals with the disease. However, few successes have been recorded, and results in slowing disease progression have been modest. Our knowledge about the genetic and pathophysiological underpinnings of the disease has certainly grown in the last three decades, and it is clear that ALS is much more heterogeneous in its presentation and progression than was appreciated with the positive clinical trials testing riluzole for ALS during the 1990s, a drug with various other important mechanisms for neuroprotection in addition to its antiglutamatergic action. 1 , 2 With important discoveries regarding the genetic underpinnings of some patients with ALS, it is now well accepted that numerous biological pathways are involved in disease onset and progression. These discoveries require careful thought regarding therapeutic applications and decisions about how ALS therapeutics will be targeted to specific, but largely undefined, subsets of the disease.
Along with an improved, but incomplete, understanding of ALS as a neurodegenerative disease is the evolution of more sophisticated and diverse in vitro and in vivo preclinical modeling platforms, as well as clinical trial designs. This is highlighted as we revisit proposed pathological pathways that have been major therapeutic targets for investigational compounds and whose results have been reported in the published literature (Fig. 1). It is likely that the failures of so many of these therapeutic compounds may not have occurred because of lack of efficacy but rather because of a lack of preclinical studies that would help define an appropriate disease pathway and a failure to ensure target engagement. These failures also extend to the evolution of clinical trial design in which numerous studies lacked sufficient statistical power to obtain a meaningful signal, did not stratify ALS patients who might best respond to a particular therapeutic, or lacked a biomarker that might offer clues to a therapeutic's potential efficacy (Table S1). The assertion that certain pathways are not involved in ALS pathophysiology may be the result of an inadequate or incomplete intervention that accounts for these failures. In light of this, scientists and clinicians may choose to revisit some of these therapeutic pathways and modalities reviewed here with an eye toward improving preclinical modeling, biomarker development, and the investment in more sophisticated clinical trial designs.
Figure 1.
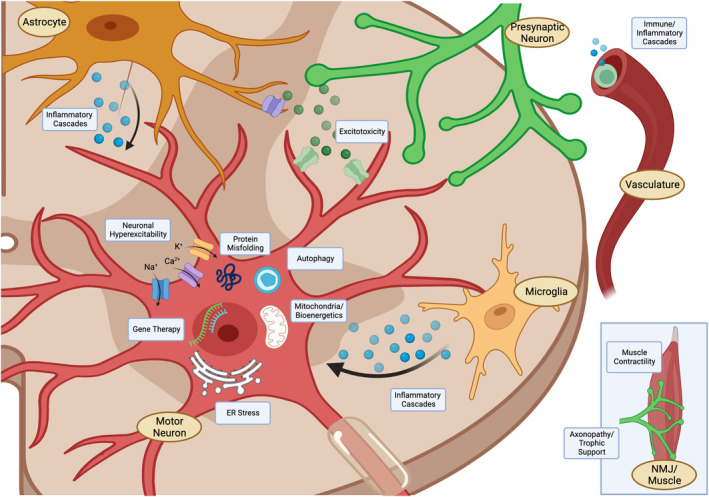
Major targets of ALS therapeutic trials. A number of ALS‐relevant pathways involving neuronal and glial cells, muscle, and systemic targets have been explored.
Targeted Pathways in ALS Pathogenesis
Antiglutamatergics
Because of the early observations that glutamate metabolism and glutamate neurotoxicity play a role in ALS pathogenesis, agents that targeted these pathways have been among the most well studied, both from the perspective of preclinical science as well as clinical trials in ALS. 3 , 4 The relative success of riluzole, whose mechanisms of action are proposed to include the inhibition of glutamate release, blockade of amino acid receptors, and inhibition of voltage‐dependent sodium channels on dendrites and cell bodies, in early ALS clinical trials resulted in the study of other agents believed to modulate glutamate excitotoxicity. 1 , 2 Importantly, riluzole provided a meaningful survival in ALS patients, and its efficacy, although modest, has been subsequently demonstrated over the last number of years. 5 In particular, it seems that riluzole slows the transit between milder to more advanced stages of ALS, as confirmed more recently. 6 , 7 Interestingly, only two preclinical studies (an in vivo model of seizure induction and reduction and an in vitro hippocampal slice model) were referenced in the original clinical publication justifying the use of the drug in ALS. 2 , 8 , 9 Confoundingly, since the original two clinical trial publications in ALS participants demonstrating the efficacy of riluzole, numerous in vitro and in vivo studies have been performed examining the mechanism and potential efficacy of riluzole in ALS models, with very mixed results regarding its neuroprotective capacity and the mechanisms of its neuroprotection. 10 , 11 This leaves open the question as to whether riluzole would have advanced to clinical trial for ALS today, armed with the data from these models that we now possess.
Given the modest but enduring success of riluzole in ALS, it is noteworthy that other agents modulating glutamatergic pathways have not met with success in any ALS measures. This raises the possibility that the mechanism of action and neuroprotection of riluzole may not be related to its antiglutamatergic effects alone but perhaps other modes of action, as well as its inhibition of persistent sodium current. 12 This is highlighted by the fact that other compounds studied in ALS involved in targeting glutamate receptor subtypes like α‐amino‐3‐hydroxyl‐5‐methyl‐4‐isoxazole‐propionate (AMPA) (talampanel and topiramate) or N‐methyl‐d‐aspartic acid (NMDA) (dextromethorphan, memantine) have not been successful. 13 , 14 , 15 , 16 The proposed attempt to increase glutamate transporter expression, with the goal of reducing extracellular glutamate in astrocytes by using ceftriaxone, was also not successful in a large Phase 3 study. 17 Unfortunately, in none of these trials was a biofluid or other biomarker available to help guide our understanding of target engagement or adequate inhibition of these proposed receptor subtypes.
How do we reconcile these early successes using riluzole with subsequent compounds affecting glutamatergic neurotransmission that have subsequently failed? As a group, these compounds were some of the earliest to have been studied as ALS therapies and, as such, have not benefitted from improvements in clinical trial design, including modified outcome measures and the emerging availability of biofluid biomarkers. Most of these compounds in this class underwent studies with very small sample sizes that, in the absence of dramatic clinical responses, make it difficult to assess potential signals of efficacy that would have led to larger studies. As outlined above, it may be that the efficacy of riluzole is multifactorial in its capacity to reduce excitotoxicity that subsequent compounds have been unable to reproduce. What this particular class of compounds has also lacked is a clear measure of target engagement that we are increasingly seeing in newer clinical trial designs for other compounds. Indeed, there have been no active studies of compounds directly affecting glutamatergic pathways in nearly a decade. In light of the failure of several antiglutamatergic compounds to deliver some measure of therapeutic success since riluzole, it may be challenging to expect further development of other compounds in this class without improvements in biomarker development, measures of target engagement, and/or more sensitive measures of clinical outcomes.
Modulators of hyperexcitability
Neuronal hyperexcitability, with some interplay related to glutamate toxicity, has a long history as a postulated mechanism common to all forms of ALS. Given that there are a number of ion channel agonists and antagonists—some of which also have antiglutamatergic activity—these compounds had drawn significant attention in the late 1990s as potential mediators of disease. The broad experience with their use for other medical disorders and known side effect profiles made them attractive for study.
Compounds including gabapentin, lamotrigine, topiramate, and valproic acid not only affect glutamatergic transmission but are also ion channel blockers, reducing inward sodium current and decreasing neuronal excitability. For this reason, they have been used for disorders with well‐described patterns of neuronal hyperexcitability, including epilepsy and pain. 14 , 18 , 19 , 20
Calcium dysregulation and its subsequent downstream cascades resulting in cell death have been studied in numerous in vitro and in vivo ALS models. 21 Therefore, reducing calcium influx via calcium channel blockade seems a reasonable approach for reducing hyperexcitability. Verapamil and nimodipine were both studied in 1996, in relatively small trials without significant changes in clinical measures, including respiratory function. 22 , 23 Compared with the existence of more substantial preclinical ALS‐relevant data generated today, neither of these compounds were directly studied using in vitro models of motor neuron death but relied heavily on the previously published relationships between hyperexcitability and glutamate release.
Two Phase 2 studies of mexiletine, a sodium channel blocker, failed to show any clinical or electrophysiological response in ALS, whereas another showed modest effects on measures of cortical and axonal hyperexcitability. 24 , 25 , 26 Valproic acid is widely used as an antiepileptic compound because of its activity as a sodium, potassium, and calcium channel blocker, as well as its effects on γ‐aminobutyric acid (GABA) levels. 27 Like many other compounds studied in ALS, however, it was chosen because, as a histone deacetylase (HDAC) inhibitor, it also reduces apoptosis, oxidative stress, and glutamate excitotoxicity. Despite its proposed effects on a number of potential ALS‐relevant pathways, ALS clinical measures were unaffected. 20
As preclinical modeling in ALS has evolved, we have seen the emergence of stem cell platforms for drug discovery. This was elegantly illustrated through the selection of ezogabine from a human‐induced pluripotent stem cell motor neuron (iPSC‐MN) platform because of its capacity for reducing hyperexcitability by activating certain voltage‐gated potassium channels and improving survival of human mutant superoxide dismutase 1 (mSOD1) and C9orf72 motor neurons. 28 Using hyperexcitability as a clinically relevant electrophysiological measure, Wainger et al. 29 demonstrated, in an efficient 10‐week clinical trial focused on electrophysiology, that ezogabine reduced hyperexcitability by several electrophysiological measures in ALS, in particular, improving the abnormal cortical inhibitory pathway's dysfunction in patient treated with higher doses. This trial was particularly relevant because it reinvigorates potential interest in hyperexcitability as a modulator of ALS physiology; reinforces the potential preclinical predictability of using human iPSC‐MN for further development; and, because of its short duration, results in an outcome that can be pursued for further development in a later‐stage trial. 29 Whether these preclinical and clinical electrophysiological measures predict meaningful clinical outcomes remains to be seen but provides a rational foundation for the further development of compounds that reduce neuronal hyperexcitability.
Inflammatory cascades
Although inflammatory cascades are often considered together, it is clear from the therapeutics discussed here that the specific targets within those cascades are quite varied. Indeed, inflammatory processes of resident cells including microglia, and to some degree astrocytes, can promote a feed‐forward mechanism for the induction of an inflammatory response. However, there has also been a resurgence of the hypotheses that peripheral inflammatory processes, including those of T lymphocytes and other immune cells, may contribute to disability. These cascades are among the most varied and actively investigated ALS therapeutic targets, with a body of preclinical in vitro and in vivo modeling data to support these hypotheses. 30
Bridging the relationship between glutamate excitotoxicity and the emerging role of inflammatory cascades in ALS, celecoxib, a cyclooxygenase‐2 (COX‐2) inhibitor of prostaglandin E2 (PGE2) synthesis, was studied for its role in reducing PGE2‐mediated release of glutamate from astroglia as well as COX‐2–induced release of cytokines. 31 Initially, the finding that ALS patients had elevated levels of PGE2 in the cerebrospinal fluid provided the rationale for use and served as one of the early investigations of a biomarker for the disease. 32 The study of this compound for 12 months in a double‐blinded fashion did not reach its clinical endpoint for success. 33 However, the more interesting observation from the study was that the elevated PGE2 levels previously reported in ALS patients were not reproducible in this study. Additionally, target engagement with lowering of PGE2 levels was not obtained by celecoxib, suggesting that the drug either did not act as predicted, did not achieve adequate concentrations in the central nervous system (CNS), or that the dose/metabolism of the compound was not enough to have an effect. In retrospect, patients with elevated PGE2 levels could have been selected to be enrolled in the study, allowing for stratification of the ALS population.
NP001, a pH‐adjusted intravenous formulation of sodium chlorite, hypothesized to regulate inflammation through reduction of nuclear factor‐κB and inhibition of interleukin (IL)‐1β within monocytes/macrophages, was not found to be efficacious in clinical outcome measures, but, in an emerging effort to examine biomarkers, a subset of ALS participants who had slowing of progression (responders) were also noted to have elevated IL‐18, IL‐6, interferon gamma, and C‐reactive protein (CRP) levels when compared to nonresponders. 34 , 35 This spawned another Phase 2B study in which only participants with high CRP values were enrolled. Again, this compound failed to reach its primary clinical endpoint, although, as with the first study, the participants who benefitted had higher levels of CRP at enrollment. 36 Notably, it was also discovered that CRP levels increase with age, and therefore, there may have been an overrepresentation of older ALS participants in this study.
Ibudilast is a nonselective inhibitor of phosphodiesterase that results in the reduction of leukotriene, cytokines, and other small molecules from microglia and monocytes. 37 , 38 A more recent preclinical study has also suggested that ibudilast may also act by inducing autophagy via mammalian target of rapamycin complex 1 (mTORC1)–transcription factor EB signaling in vitro, resulting in clearance of SOD1 and TAR DNA‐binding protein 43 (TDP‐43) aggregates. 39 In a continued evolution for the incorporation of biomarkers, a study by Babu et al, using PBR‐28 as a PET marker of neuroinflammation combined with neurofilament light chain (NfL) as a proposed biomarker of ALS disease progression, completed an open‐label study of ibudilast that failed to show any effect on these markers. 40 A single‐center, Phase 1b/2a study in 51 participants for 6 months, followed by a 6‐month open‐label extension, showed no change in disease progression, but a post hoc responder analysis showed that a subset of participants with a short history of ALS and progressive disease did respond. 41 This has spawned a current Phase 2b/3 trial with those key criteria required for enrollment. 42
Masitinib selectively inhibits the tyrosine‐kinase mast/stem cell growth factor receptor (c‐KIT) and reduces microglial activation through the blockade of colony‐stimulating factor 1 receptor. 43 , 44 In a study examining mSOD1 microglia in vitro accompanied by an in vivo cohort of mSOD1 rats, administration of masitinib reduced the expression of inflammatory mediators and prolonged duration of disease when administered after the onset of hindlimb paralysis. 44 Using these data to support further development, a clinical study of this compound showed a slowing of the ALS Functional Rating Scale–revised (ALSFRS‐R) decline by approximately 20%. 45 A confirmatory Phase 3 study is now underway (NCT 03127267).
Pioglitazone is an oral agent used for treatment of diabetes that was chosen for study in ALS because of its anti‐inflammatory properties, as well as preclinical data from three independent groups showing protection in mSOD1 mice. 46 , 47 , 48 , 49 A large Phase 2 study of the drug was stopped because of futility in extending survival in ALS participants. 50 Unfortunately, no inflammatory biomarkers were included in the study to be able to understand whether target engagement had been obtained. The study leaned heavily on the preclinical successes in mouse models of the disease, a preclinical bias that also supported the rationale behind the failed clinical trial of minocycline, a compound believed to have anti‐inflammatory and antiapoptotic effects. For minocycline, the results were both troubling and confusing for this pathway because the compound actually appeared to accelerate disease in ALS participants. 51
ALS pathogenesis has been associated with peripheral circulating regulatory T‐cell (Treg) levels, because their reduction promotes an increase in proinflammatory effector T cells and macrophage activation. 52 Supported by positive results in a mSOD1 mouse model, autologous infusion of expanded Tregs in ALS patients has shown positive clinical effects. 53 Dimethyl fumarate has been used in patients with multiple sclerosis by enhancing Treg levels in humans and reducing proinflammatory T cells. 54 However, a randomized, placebo‐controlled, double‐blind, Phase 2 trial in ALS did not prove effective. 55 Fingolimod is an immunomodulatory compound that antagonizes the sphingosine 1‐phosphate (S1P) receptors, blocking migration of lymphocytes from lymph organs and reducing circulating lymphocytes. 56 Tocilizumab, a humanized monoclonal antibody antagonist of the IL‐6R, was chosen for its peripheral effects on inflammation. 57 Both fingolimod and tocilizumab were evaluated in relatively small clinical studies that proved to be safe and well tolerated. However, of greater importance was the finding that both compounds showed evidence of target engagement allowing for future study of these compounds as mediators of peripheral as well as central inflammatory cascades.
Targeting both central and peripheral inflammatory pathways remains among the most active preclinical and clinical areas of research in ALS. Several clinical studies are underway or in planning stages, suggesting that despite some ALS failures, neuroinflammatory pathways are attractive as targeted therapeutics for neurodegeneration. 58 Compounds targeting these pathways were historically some of the first to utilize biomarkers for patient stratification and measurements of target engagement. The more routine incorporation of biomarkers into clinical trials for ALS today owe much to the evolving attempts to incorporate neuroinflammatory biomarkers into ALS study design.
It may be that the complexity of the inflammatory response in ALS requires either combinatorial therapy or the study of additional targets. Therefore, the successes or shortcomings of results from ALS clinical trials should take into consideration that inflammatory mediators show large individual variability and do not converge on a single pathway, cell type, or even distribution within the patient. Neuroinflammatory pathways may intersect with each other, interacting with both positive and negative mechanisms for neuronal pathology. Their influences on other cell death pathways can be unpredictable, suggesting the utility of a combinatorial strategy for therapeutics.
Trophic factors
The preclinical studies employing trophic factor administration to ALS models in vitro and in vivo have been extensive over the last three decades. The delivery of trophic factors to the CNS has remained a challenge but still provides attractive targets for investigations of viral vector delivery of these compounds to the CNS. 59 A host of these factors have been studied in ALS participants, most notably in the late 1990s, with a number of clinical trials attempting to directly evaluate their efficacy.
The preclinical evidence that supported the advancement of ciliary neurotrophic factor (CNTF) was based on its ability to support the survival of embryonic motor neurons from stochastic cell death, prevent death of facial motor neurons from axotomy in neonatal mice, prevent death of facial motor neurons in the PMN mouse model neuronopathy, and its overall effect in the wobbler mouse of motor neuronopathy. 60 , 61 , 62 , 63 , 64 CNTF underwent two relatively large clinical studies. 65 , 66 A major limitation of these studies was the development of significant side effects with peripheral administration. 67 , 68 Furthermore, neither of these studies showed any efficacy, which largely resulted in the abandonment of CNTF as a potential ALS therapeutic. Whether mitigating the side effect profile or a more directed delivery of the compound to the CNS without the systemic side effects could prove useful but has never been explored.
Perhaps the most thoroughly studied trophic factor was insulin‐like growth factor (IGF)‐1, which, in the first North American study, in 1997, showed an effect of slowing symptom progression. 69 These results, using a similar clinical trial methodological design, were not reproduced in a subsequent European study. 70 A large Phase 3 study of IGF‐1 that included a 2‐year evaluation failed to show any benefit, thus resulting in a significant shift in the field away from these targets. 71 All three of these studies utilized subcutaneous delivery of IGF‐1, and it is unknown to what degree CNS penetration was obtained. In an attempt to bypass concerns about blood–brain barrier (BBB) penetration, a small cohort of 12 participants was administered IGF‐1 via an intrathecal route, with modest slowing of disease in this very small sample size. 72
With preclinical data mirroring those using CNTF and with concerns about BBB penetrability, Ochs et al. 73 performed a limited study (Phase 1/2 trial) over 12 weeks in which brain‐derived neurotrophic factor (BDNF) was infused into ALS participants intrathecally. Importantly, investigators were able to measure cerebrospinal fluid (CSF) levels of BDNF, providing some pharmacokinetics (PK) data to support the rationale behind its intrathecal (IT) administration. However, given the short timeframe of the double‐blinded portion of the study, the investigators could not make conclusions regarding efficacy of the strategy.
Vascular endothelial growth factor (VEGF) has undergone extensive preclinical studies in a number of ALS models using varied forms of delivery. One Phase 1 trial investigated the tolerability, safety, and PK of intracerebroventricularly delivered VEGF in a small group of participants, showing sustained higher levels of VEGF in CSF after treatment. 73
Trophic factors need not be delivered systemically. Based on substantial literature about hepatocyte growth factor (HGF) and its neurotrophic effects, as well as some preclinical evidence in ALS mouse models of its role as a neuroprotectant, the hypothesis that a novel genomic complementary DNA (cDNA) hybrid human HGF injected via plasmid directly into muscle can result in the local production of HGF and subsequent maintenance of the neuromuscular junction was explored. 74 A study incorporating injections into targeted limb muscles showed safety and tolerability. 75 A Phase 2 study of HGF that includes sampling of muscle tissue as a biomarker has been completed (NCT04632225).
It was found that the myelin‐associated protein, Nogo‐C, is decreased in the muscle of animal models with different causes of denervation, but only in ALS patients is there a striking increase in Nogo‐A protein levels, which supports a disease‐specific mechanism causing neurite outgrowth inhibition. 76 In a study including participants with lower motor neuron diseases, it was described that the detection of Nogo‐A in muscle biopsy samples from patients correctly diagnosed those progressing to ALS with high sensitivity and specificity. 77 A preliminary, double‐blind trial included 40 ALS patients to receive intravenous ozanezumab (a humanized monoclonal antibody against Nogo‐A). PK results were consistent with monoclonal antibody treatments. The medication was well tolerated, but no treatment effects were observed for functional endpoints or muscle biomarkers. 78 A larger, randomized, double‐blind, and placebo‐controlled trial with ozanezumab was negative for the primary outcome. 79
Xaliproden, a serotonin‐1A agonist promoting release of neurotrophic factors from astrocytes, has been investigated as a trophic factor in ALS. This drug was investigated in a large, 18‐month trial including two studies: The first study, without riluzole, and a second study, with riluzole, with a total of 1210 participants. There was no influence on survival but a slightly significant positive impact on vital capacity. 80
The potential for any future evaluation of trophic factors, including those listed above, may have to wait for more sophisticated methods of delivery to the CNS via infusion or viral vector delivery of these compounds that allow for sustained production of these factors while mitigating side effects. Furthermore, there has been the suggestion from the preclinical literature that the timing of administration of these compounds during disease may be particularly relevant. Important to the field of ALS clinical research is that trophic factors like those discussed above have also been, or are being, studied as treatments in other neurodegenerative diseases. Therefore, if improvements in delivery, CNS expression, tolerability, or efficacy are found in other patient populations, these lessons could be translated to ALS.
Non‐cell autonomous effects in ALS and the use of stem cells as therapeutics
“Stem cell therapies” are often considered as a single therapeutic approach, but, in reality, there are numerous cell types that could be used as potential ALS therapeutics. For example, neural stem cells (NSC) with the capacity to differentiate into neurons, astrocytes, and oligodendrocytes that have been transplanted intraparenchymally are very different from mesenchymal stem cells (MSC) derived from bone marrow that have been infused intrathecally. Therefore, understanding these fundamental differences in cell types, their origin, and proposed mechanisms of action as ALS therapeutics is critical. This is especially true because the success or failure in efficacy, or a complication from stem cell therapy, could inappropriately affect the field moving forward. A complete list of the stem cell transplantation strategies for ALS is beyond the scope of this review but nicely reviewed by Je et al. 81 Rather, this section will highlight some of the NSC and MSC delivery strategies buoyed by preclinical approaches.
Some of the earliest studies with autologous bone marrow MSC involved intraspinal transplantation into the thoracic spinal cords of ALS participants. 82 These studies were then accompanied by a number of studies using MSC from a variety of autologous sources (bone marrow, adipose, umbilical cord). 81 These have all been early Phase 1–2 studies focusing on safety, with small sample sizes. Many are open label and not powered to see an efficacy signal. Encouragingly, most appear to have been safe, although the results of some of these clinicaltrials.gov‐registered studies remain unpublished. The results are also complicated by the lack of biomarker data and incomplete datasets regarding cell survival and engraftment efficiency.
More recent studies of autologous bone marrow‐derived MSC that express neurotrophic factors (MSC‐NTF) deserve mention given that they were studied in a randomized, double‐blinded fashion. The first study was a Phase 2 study demonstrating safety following a single intrathecal dosing. 83 The second study, a larger Phase 3 study utilizing repeated dosing of these cells and examining clinical efficacy, did not reach its primary endpoint. 84 Of particular interest in these studies was the longitudinal measurements of a number of neurotrophic factors and inflammatory biomarkers that showed some patterns suggesting activity of these MSC‐NTF cells. Nonetheless, intrathecal administration of unprogrammed, autologous bone marrow‐derived MSC (including NeuroNata‐R in South Korea) is a tested option, indicating that that programmed neurotrophic factor release is not an indispensable step. 85 , 86
MSC seek, in theory, to supply neurotrophic factors, anti‐inflammatory molecules, and, overall, to provide a supportive milieu. NSC, however, have the capacity to differentiate into a number of neuronal and non‐neuronal cell subtypes and, in addition to the factors possessed by MSC, could engage in the recapitulation of neural networks and possibly neuronal replacement/regeneration. There are few data to suggest that the intrathecal delivery of NSC or their more mature subtypes incorporate into the brain and spinal cord in a meaningful way. Therefore, most approaches have resulted in direct intraparenchymal transplantation of these cells into the spinal cord or brain. There are a number of studies using human NSC or their derivatives in animal models of ALS (as well as other neurodegenerative diseases). 87 However, because of the neurosurgical challenges, few have resulted in clinical trials for people with ALS. Human spinal cord‐derived NSC underwent Phase 1 and 2 studies in ALS, both of which were open label. Encouragingly, these studies showed safety of the surgical and cellular therapy, with some cells found at sites of transplantation at the time of autopsy. 88 Combining the potential of NSC to incorporate into networks as astrocytes with an engineered overexpression of glial cell‐derived neurotrophic factor (GDNF), Baloh et al. reported the results of a Phase 1–2a trial of CNS10‐NPC‐GDNF cells into the spinal cords of ALS participants. Up to 42 months following transplantation, cells were noted at the transplant site and had GDNF expression without any significant side effects. 89
Stem cell transplantation, particularly those incorporating NSC with the capacity to differentiate into neurons or glia, offers the potential for recapitulating neural networks and—perhaps someday—offers hope for regeneration of motor neurons. However, the current challenges for meaningful benefits from these strategies are still numerous. At the earliest preclinical stages, the transplantation and interactions of human cells with rodent cells (xenograft) for modeling therapeutic efficacy is a fundamental problem that may fall short of predicting true efficacy. Clinically, as a disease that affects the entire neuraxis, intraparenchymal delivery of these cells is limited to localized delivery. Still, proof‐of‐principle studies that examine the local efficacy of these cells can provide insights. Other hurdles both preclinically and clinically include a true understanding of cell survival following transplantation, the invasiveness of the procedures, immunosuppressive strategies, potential tumorigenicity, and others reviewed elsewhere. 90 Whether these challenges can be overcome likely awaits advances in technological approaches for delivery and cell integration. A much more robust effort, and potentially more impactful result for ALS, is more likely to come from the utilization of stem cells, specifically human iPSC that can be differentiated into a host of ALS‐relevant cell subtypes, as platforms for understanding mechanisms of disease, biomarker development, and drug screening. Indeed, these platforms are being realized preclinically for ALS therapy development. 91
Gene therapies
The use of gene therapies, particularly the use of antisense oligonucleotides (ASO) acting via RNase H‐dependent cleavage of targeted RNA, has generated significant enthusiasm in the field. Importantly for ALS, the preclinical rationale behind ASO knockdown of SOD1 was strong and resulted from a number of years of methodical analyses of RNA transcript and protein levels in ALS fibroblasts, normal rats and monkeys, mSOD1G93A rats and mice, and the correlation with histopathology and behavior. 92 , 93 , 94
The rigorous design of the clinical trials for ASO delivery for knockdown of SOD1 established the safety of this novel strategy for intrathecal delivery to the CNS. Equally as important, however, was the incorporation of SOD1 protein measurements in the CSF as biomarkers for future study design. 95 Several years later, a dose‐finding study of a SOD1 ASO was studied in a larger cohort of ALS participants harboring SOD1 mutations. In an evolution of design, a fast‐progressing ALS group was identified post hoc for additional analysis. The contributions of this study to the field of ALS were felt more broadly because it demonstrated a dose‐dependent reduction in SOD1 protein levels in the CSF as well as a reduction in the exploratory biomarker: NfL. Importantly, these findings seemed to correlate with a slowing of declines in functional measures, including ALSFRS‐R and slow vital capacity (SVC). These findings were particularly evident in those SOD1 ALS participants with more rapidly progressing disease. 96 In the Phase 3 study, the stratification of SOD1 participants into fast and slow progressors was prespecified, based on the analysis of the Phase 1–2 study. Unfortunately, despite good tolerance, the primary outcome measure for efficacy, through a slowing of the ALSFRS‐R, was not realized; interestingly, however, reductions of SOD1 protein in the CSF and a reduction in NfL levels were once again noted. Results from 95 participants who entered the open‐label extension were shared. At 52 weeks, rate of ALSFRS‐R decline was significantly smaller in early treated participants compared to a delayed treated group, and a better response was also detected for SVC and handheld dynamometry. 97 This compound, tofersen, was approved by the US Food and Drug Administration (FDA) in 2023 for treating ALS patients with SOD1 mutations. With the question of temporal administration of SOD1 ASO delivery in mind, another Phase 3 study of SOD1 ASO in asymptomatic SOD1 mutation carriers who develop early changes in biomarkers, but preceding motor deficits, is now underway (NCT04856982). Together, the preclinical data providing a strong rationale for SOD1 reduction as a therapeutic approach, combined with the rigorous and adaptive trial designs over several studies that have included patient stratification by gene mutation and progression, PK, pharmacodynamics, and biomarker data, are certain to provide a blueprint for both understanding the importance of preclinical study and also for clinical trial design.
Preclinical studies examining ASO targeting of C9orf72 hexanucleotide repeat expansion (HRE) initially leaned heavily on the use of human iPSC models in which ASOs were found to reduce C9orf72 HRE transcripts and clear intranuclear RNA foci. 98 , 99 , 100 Similarly, transgenic mice with bacterial artificial chromosomes (BAC) HRE containing the C9orf72 gene were utilized to demonstrate that ASOs targeting C9orf72 reduced HRE containing C9orf72 in the CNS and also decreased dipeptide repeat (DPR) levels. 101 , 102 Importantly, DPR reduction by ASOs were identified as a potential therapeutic readout with an eye toward clinical trial design. A Phase 1 trial (NCT03626012) was undertaken to deliver the ASO BIIB078 to participants with C9orf72 ALS. Although the investigational product was well tolerated, none of the primary endpoints were met. Interestingly, participants who received study drug trended toward a more rapid clinical decline. One limitation, yet to be fully evaluated, is the consideration from preclinical data that C9orf72‐targeting ASOs target only the sense strand of RNA, without targeting the antisense strand. This may lead to an incomplete reduction of RNA foci and, potentially, pathology. 103 Another possible explanation for the resulting clinical data suggest that the loss of function related to knockdown of C9orf72 may be exacerbating symptomatology. Other clinical trials, based on their efficacy in animal models, utilizing ASOs and targeting mutations in the FUS 104 (NCT04768972) and ATXN2 genes, 105 (NCT04494256) are currently underway.
ASO are not the only approaches being considered for gene therapy. Preclinical study and development of RNA‐based therapeutics include small activating RNAs, adenine‐to‐inosine RNA base editing oligonucleotides, adeno‐associated virus (AAV)‐mediated gene silencing, AAV‐mediated gene correction, and AAV‐mediated gene expression activation. These approaches bring a host of additional opportunities and challenges to the therapeutic landscape. 106 Perhaps one of the most challenging hurdles will be to address host immunogenicity to AAV administration. The translational efforts that have brought gene therapies to clinical trial have become more sophisticated with regard to their preclinical and clinical design. Utilizing a combination of human iPSC and other in vitro models, mouse and other models of disease, and the identification of potential biomarker readouts of target engagement have all spurred the advancement of these compounds to the clinic. Furthermore, these gene therapy trials have all incorporated the stratification of participants (both genetic and phenotypic), biomarker development, and advanced ALS clinical trial outcome measures. The results of these studies, even if they fail to meet primary clinical endpoints, are sure to spur conversation regarding the utility and predictability of both the preclinical and clinical models and measures.
Modulators of oxidative stress and mitochondrial dysfunction
Inflammation and oxidative stress are related and contribute to neuronal degeneration. In particular, activation of glial cells induces oxidative stress and reactive oxygen species (ROS) resulting from multiple cellular processes and thus promoting inflammation. 107 ROS are generated by oxidative phosphorylation, from cellular defense mechanisms and the electron transport chain complex in the mitochondria. ROS excess can cause cell death resulting from oxidative damage to nucleic acids, lipids, and proteins. Markers of oxidative stress are increased in ALS, as identified in blood, CSF, muscle, and the CNS. 108 Antioxidant enzymes (catalase, glutathione peroxidase, and SOD), several molecules (uric acid, taurine, creatine, carotene, and flavonoids), and vitamins (C and E) can protect cells from ROS. 109 Mitochondrial dysfunction can interfere with apoptosis, in particular concerning respiratory chain and mitochondrial permeability pore function. For this reason, it could be an interesting new target for treating ALS.
Early studies of both N‐acetylcysteine, acting as a reduced glutathione (GSH), and selegiline, a monoamine oxidase inhibitor with antioxidant properties, had no ALS‐relevant preclinical data, were studied in participants with ALS, and neither had any beneficial effect. 110 , 111
One of the earliest preclinical studies using the mSOD1 mouse as a rationale for therapeutic development included the study of vitamin E. 112 Vitamin E (α‐tocopherol) was then tested in a study complicated by a high dropout rate, and although functional decline was smaller in the treated group, this did not reach significance. 113 Biological evaluation of oxidative stress was performed in a subset of participants after 3 months, measuring markers of cell membrane lipid peroxidation, erythrocyte SOD activity, glutathione peroxidase (GPX) activity, and vitamin E levels. Those treated with vitamin E were observed to have higher vitamin E plasma levels, a reduction in markers of membrane lipid peroxidation, an increase in plasma GPX, but no difference in erythrocyte GPX activity nor in erythrocyte SOD activity. Using high‐dose vitamin E also failed to produce an effect on ALS survival. 114
Creatine, as a readily available supplement, was also among the first in this category to be studied because it has antioxidant, anti‐inflammatory properties, and plays an important role in mitochondrial adenosine triphosphate production, a hallmark finding in mSOD1 mice in which the efficacy of creatine was demonstrated. 115 , 116 , 117 However, in ALS participants, there was no clinical effect with either high or low doses. 118 , 119
Armed with both in vitro data showing its antioxidant effect as a modulator of mitochondrial activity in cell culture models and in vivo data from ALS models demonstrating a behavioral and survival effect, dexpramipexole advanced to a Phase 2 study in ALS participants, with a significant difference between groups demonstrated in a joint rank test (functional decline and mortality), thus supporting a Phase 3 trial. The large Phase 3 study failed to meet any of the endpoints, highlighting the challenges of translating smaller Phase 2 studies into Phase 3 successes. Furthermore, the absence of a biofluid biomarker further handcuffed interpretation of the results, which were initially buoyed by preclinical evidence of an effect. 120 , 121
Olesoxime is a small molecule with a cholesterol‐like structure, with neuroprotective properties for motor neurons in cell culture and in rodents by controlling mitochondrial permeability pore function, as well as a behavioral effect in mSOD1 mice. 122 , 123 A large study was subsequently undertaken without clinical benefit. 124
Although vitamins and supplements received the most attention in the 1990s, methylcobalamin has been recently revisited. Methylcobalamin (vitamin B12) acts as a methyl donor for homocysteine remethylation with neuroprotective properties by decreasing the levels of neuronal homocysteine. 125 Moreover, methylcobalamin activates extracellular signal‐regulated kinases 1 and 2 and Akt, favoring neuronal survival. 126 Preclinical studies indicated that this compound has antiglutamatergic properties. 127 Animal studies (wobbler mouse) indicate an effect on survival. 128 Clinical studies in ALS have been promising regarding ventilation‐free survival in ALS participants. 129 A large Phase 2/3 clinical trial disclosed that ultrahigh‐dose methylcobalamin (25 mg or 50 mg) was safe and well tolerated but without significant positive effects. A later post hoc analysis indicated that early affected patients (treated within 1 year of onset) with a 1‐ to 2‐point decrease in ALSFRS‐R over a 12‐week lead‐in period had a slower functional decay and longer survival. 130 , 131 A recent Phase 3 trial with the above features showed that, at Week 16 of the randomized period, ALSFRS‐R was 1.97 points greater in the treated group. 131
In a demonstration of the continued evolution of preclinical ALS modeling, ropinirole was identified as a potential ALS therapeutic, through a screen of more than 1200 compounds, using cell death in hiPSC‐MN from sporadic ALS (SALS) patients as a functional readout. 132 This compound appeared to be most active as an antioxidant but also had effects on apoptosis and protein aggregation, consistent with the multiple actions of several other ALS‐relevant compounds studied. A Phase 1/2a trial has suggested that the compound is safe and well tolerated (UMIN000034954). 133 Whether using hiPSC‐MN from ALS patients as a preclinical tool to predict clinical success remains to be validated in a larger clinical study.
Edaravone is a free radical scavenger, capable of reducing lipid peroxides and hydroxyl radicals after neuronal insult, which was approved for treating stroke in Japan in 2001. 134 Preclinically, this compound was studied in three ALS rodent models. In particular, work in a mSOD1 rat model showed that this drug reduced motor disability, preserved lumbar motoneurons, and resulted in a decreased 3‐nitrotyrosine (3NT)/tyrosine ratio dose dependently. 135 In a preliminary, open clinical trial, 15 participants treated with 60 mg of edaravone intravenously showed a lesser functional decline over a 6‐month treatment period compared with a similar period before treatment. This finding was accompanied by a reduction in 3NT levels in the CSF. 136 In a larger, 24‐week Phase 2 trial, treatment with edaravone did not prove to be effective. 137 A Phase 3 trial with edaravone recruited a very specific Japanese population of ALS patients, with normal respiratory function, disease duration of 2 years or less, definite, or probable ALS according to the revised El Escorial criteria, scores of at least 2 points on all 12 items of ALSFRS‐R, with Grade 1 or 2 in the Japan ALS severity classification, and a decrease of 1–4 points in the ALSFRS‐R score during a 12‐week observation period before randomization. The trial revealed an effect on ALSFRS‐R decline in the treated group but without a significant impact on other functional measures. 138 Although this trial's results permitted approval in Japan, Korea, the United States, and some other countries, it was not approved by the European Medicines Agency. Consequent population studies in the United States and Europe did not confirm a positive impact of this drug in disease progression and survival, and it could be associated with more frequent hospitalization episodes. 139 , 140 , 141
The preparation of sodium phenylbutyrate (SP)/taurursodiol (TURSO), like many therapeutics discussed in this review, likely has many mechanisms of action, but an effect on endoplasmic reticulum (ER) stress and mitochondrial function is hypothesized to be the primary feature of this drug combination approved by the FDA in 2022 for the treatment of ALS. Several in vitro studies have examined these compounds separately, with an in vivo study in mSOD1 mice showing a dose‐dependent prolongation of survival and a reduction in cytochrome c release and caspase activation. 142 , 143 , 144 The combination of these compounds also showed a specific transcriptional effect in SALS‐derived fibroblasts. 145 With this proposed mechanism in mind, a Phase 2 study was conducted over 6 months showing a slowing of functional decline in the ALSFRS‐R in ALS participants taking SP/TURSO. 146 A subsequent post hoc analysis of the data from that study also suggested a significant survival effect in ALS participants taking this compound. 147 This compound is particularly interesting yet challenging to interpret from the standpoint of preclinical data supporting its progression to clinical trial. Although both of the compounds, studied separately, have mechanisms of action that could support their use in ALS patients, the combination of the compounds was not extensively studied preclinically in the published literature to inform about confidence in other combinatorial therapies moving forward. A large, Phase 3 study examining this compound is ongoing (NCTl05021536).
Autophagy
The pathogenic mechanisms by which defects in autophagy pathways lead to impaired protein homeostasis in ALS remain incompletely understood. Although there is evidence that specific genetic mutations important for autophagy lead to neurodegenerative conditions including ALS, the details by which these specific mutations cause neuronal injury and a clear understanding of the neuroprotective effects of autophagy have not been determined. 148 A few recent clinical trials have targeted aspects of autophagy, so far with largely disappointing results.
Lithium carbonate has been shown to have neuroprotective properties in vivo, in part by acting as an autophagy inducer. 149 , 150 Much of the original enthusiasm for lithium came from a publication in 2008 demonstrating an effect of this compound in mSOD1 mice, with a significant prolongation of survival in a relatively large cohort of ALS mice. 151 Although specific molecular autophagic pathways were not investigated in more detail, investigators did demonstrate an increase in the number of autophagic vacuoles and the clearance of α‐synuclein, ubiquitin, and SOD1 in vitro as well as in vivo. A small study reported by the group demonstrated a remarkable effect, with all participants taking lithium alive at 15 months. However, several randomized controlled studies in ALS have not shown benefit. 152 , 153 , 154 , 155 , 156 , 157 The largest of these trials, the LiCALS study, lasted over 18 months but failed to show a difference in the rate of survival. 156
Tamoxifen is another autophagy enhancer through both mTOR‐dependent and ‐independent pathways that has also been shown to reduce TDP43 protein aggregation in motor neurons and animal studies. 158 In a small randomized controlled trial, tamoxifen was found to slow progression of ALS in a modest fashion. 159 A comparison of 40‐ and 80‐mg daily doses of tamoxifen with 30 mg a day of creatine in a unique selective‐design Phase 2 study showed superiority of the higher dose of tamoxifen compared to the lower dose and creatine, suggesting that high‐dose tamoxifen should be compared to placebo in a future study. 160 Other compounds currently being developed and in clinical trial with abundant preclinical evidence in several ALS models of autophagy include rapamycin (NCT03359538) and colchicine (NCT03693781). 161 , 162 , 163 , 164 , 165 , 166 , 167 , 168 , 169
One barrier to studying this class of therapeutics in ALS is that the most effective methods for inducing autophagy are not certain. As has been previously posited, future autophagy inducers for neurodegenerative disease will require a better understanding of cell type‐specific regulatory mechanisms controlling autophagy in neuronal and nonneuronal cells in the CNS, methods to accurately measure the induction of autophagy, high‐throughput screening for autophagy induction, and alternative‐model systems such as iPSCs to test the effectiveness of these compounds. 148
Protein misfolding
ALS is associated with cytosolic aggregates containing specific misfolded proteins in both neuronal and glial cells that may lead to disease initiation and propagation, including SOD1 aggregates in SOD1 familial ALS and TDP43 aggregates in C9orf72 and most other forms of familial ALS, as well as SALS. 170 Recent evidence also suggests that clinical heterogeneity in ALS may result from polyconformational misfolding of ALS‐related aggregated proteins causing prion‐like activity. 171 A few novel inhibitors of SOD1 protein misfolding and aggregation have been studied in mSOD1 mice, most notably arimoclomol, an amplifier of heat shock proteins, which has shown the ability to rescue motor neurons from cell death in vitro and reduce protein aggregation. 172 Much of the enthusiasm for arimoclomol was based on an improvement in mSOD1 mouse survival by a reported 22% from untreated mSOD1 mice. However, it is notable that the mSOD1 mouse cohorts included only seven mice treated with arimoclomol—a study that is considered significantly underpowered. 173 A much larger study in 2008 showed that arimoclomol‐treated mSOD1 mice as late as 75 days of age showed a benefit when compared with untreated mSOD1 mice. 172 Later studies using this same model showed that arimoclomol showed beneficial effects at the neuromuscular junction. 174 A study of 38 SOD1 participants with rapidly progressing disease showed a nonsignificant trend toward a therapeutic benefit. 175 However, a Phase 3 study of arimoclomol in a large cohort of participants with SALS did not show any benefit. 176 Therefore, in order to determine whether arimoclomol's effect could be most appreciated in SOD1‐mediated disease—as may have been predicted by preclinical mouse modeling and early clinical studies—a larger trial specific to participants harboring SOD1 mutations would likely have to be undertaken.
Guanabenz also likely exerts its effect on protein misfolding and ER stress and was shown to be efficacious in mSOD1 mice by two separate groups. 177 , 178 In a Phase 2 study in which the treatment groups at three different doses were compared against historical ALS controls, the compound showed a potential benefit in the ALSFRS‐R, with fewer participants progressing to a more advanced stage of the disease. However, side effects from the drug confounded the study through a large dropout rate among individuals taking guanabenz. 179
Muscle
Although muscle weakness is the ultimate source of disability in ALS patients, it has largely been overlooked as a therapeutic target, likely because weakness and atrophy are thought to be bystanders of motor neuron loss rather than initiators of the process. 180 However, given that so much is known about muscle physiology, an attempt to improve muscle contraction force at submaximal stimulation frequencies, increase power, and diminish the degree of muscle fatigue by sensitizing the troponin complex in fast‐twitch skeletal muscle fibers to calcium could improve functional motor performance in ALS despite ongoing denervation. 181 Investigators utilized an in vitro model of human skeletal muscle to show the improvements in the response of muscle to nervous input, after which they used a rat model, which demonstrated improvements in grip strength. 182 To address whether modulation of this target would act similarly in denervated and weakened muscle, Hwee et al. treated moderately weak mSOD1 mice with tirasemtiv and were able to demonstrate improvements in submaximal isometric force and behavioral measures in female mSOD1 mice. In a unique and informative attempt to anticipate how this compound might translate to the maintenance of respiratory function, the compound also improved both diaphragmatic contractility in these mice using an ex vivo preparation as well as pulmonary tidal capacity. 183
With this framework in mind, several studies examined tirasemtiv, a skeletal muscle activator, for its potential in modifying functional changes related to ALS. Through a number of well‐designed studies examining dosing tolerability, interactions with riluzole, and functional measures, the compound showed some trend toward a treatment benefit in pulmonary SVC but was generally not well tolerated secondary to side effects. 184 Therefore, in an attempt to continue to target the mechanism of action while reducing side effects, reldesemtiv, a similar compound with much fewer CNS side effects, was designed but was discontinued due to futility. Another compound with a similar, but not completely overlapping, mechanism of action, levosimendan, was also studied in a large, Phase 3 study but failed to yield any meaningful benefits. 185
Other pathways involving muscle have attracted preclinical study that has not necessarily resulted in the study of therapeutics in ALS. Most notably, myostatin is secreted by myocytes and negatively regulates skeletal muscle growth through activin receptors. 186 A number of companies have developed myostatin inhibitors with the intention of treating a variety of neuromuscular disorders. 187 Preclinical studies have mostly been conducted in mSOD1 mice, with evidence of increases in muscle mass and, in some cases, strength, but not necessarily dramatic improvements in survival. 188 , 189 , 190 It may be that results from other neuromuscular disorders or other preclinical models for myostatin inhibition will invigorate an interest in myostatin inhibition for ALS.
Conclusion
With our increased understanding of ALS as a heterogeneous neurodegenerative disorder based on genetic insights, it is perhaps not surprising that most therapies for ALS as a single disease have met with failure. It is also clear that “successes” in ALS therapies have been modest at best and that better drugs should be developed. However, it would also be naïve to dismiss specific pathways or therapeutic approaches as failures based on their study in SALS over the last four decades. The efficacies of riluzole, and more recently edaravone (as a free radical scavenger) and SP/tauroursodeoxycholic acid (AMX0035) (as an antiapoptotic therapy) highlight that, although the proposed cellular pathways are not new, the efficacy of the drugs that target these pathways is relevant to current therapeutic strategies. Indeed, the mechanism of action for edaravone, approved in 2017, is hypothesized to act as a free radical scavenger. 191 These mechanisms as mediators of ALS date back nearly 30 years. AMX0035 has been proposed as a mediator of ER stress and mitochondrial function, cascades believed to have origins in original publications from the early 1990s. 142 , 192 , 193 , 194 , 195 How we uncover these potential successes through a combination of preclinical modeling and trial design is the challenge moving forward. If these mechanisms of action, all three distinct, are still relevant, why have other pharmacotherapies targeting these pathways failed? There are, of course, a number of possible reasons, many of which may not have been explored. It is possible that these drugs have additional mechanisms of action. This has been postulated with riluzole, for example. 11
Drug development in ALS has been very dependent on the results of preclinical studies from the mSOD1 mouse, a model for ALS that unfortunately has inherent flaws, leading to a high failure rate in patients. 196 The recent change to a proper pathophysiological approach will hopefully result in positive outcomes, as indicated by some recent trials.
It is noteworthy that the vast majority of both in vitro and in vivo studies have focused on spinal motor neuron pathology. There are many fewer studies examining the potential for therapeutics targeting corticospinal motor neurons (CSMN). Part of this is by virtue of the fact that rodent corticospinal tracts are anatomically unique from those of humans. 197 Furthermore, in vitro models do not necessarily distinguish CSMN subtypes from nonspecific cortical neurons, in part because of a relative paucity of CSMN‐specific markers. However, there are preclinical studies of therapeutic compounds now beginning to address some of these shortcomings in targeting CSMN dysfunction. 198
By the same token that we have assessed interventions in well‐established ALS‐relevant pathways, new trials, including those that target nuclear export, TDP‐43 mislocalization, and other RNA processing defects, among others, should not be judged solely on the basis of early phase studies that are upcoming.
Encouragingly, the relative successes of gene therapy for SOD1 could be attributed to the targeting of a disease‐causing mutant protein (SOD1) that was complemented by rigorous in vitro and in vivo preclinical studies in model systems overexpressing this specific mutant protein. Finally, the stratification of ALS participants with those carrying known mutations with biomarker data supporting the efficacy of the compound supports the goal of the clinical program. Conversely, the abundant preclinical data supporting ASO use in C9orf72 with similar, if not greater, in vitro and in vivo modeling using hiPSC and murine models did not predict failure of the first trial of participants in this trial. It has been hypothesized that the lack of an ASO targeting the antisense strand resulted in an incomplete knockdown of pathologically produced DPRs. Whether approaching a more complete silencing of both RNA strands will be pursued or whether other downstream targets of C9orf72 HRE will be more fruitful remains to be seen. What is evident is that the available preclinical tools for investigating these pathways is now robust.
As this review has sought to highlight, the path toward designing therapeutics targeting certain ALS pathways is littered with shortcomings that should not preclude revisiting these potential mechanisms of disease. The diversification of preclinical in vitro and in vivo tools combined with an increased understanding of genetic influences should add additional fidelity toward selecting agents that move to clinical trial. Although not a focus of this review, improvements in genotypic and phenotypic ALS patient stratification, as well as in clinical trial design, allow for more confidence in measures that suggest efficacy. Biomarker development, however, remains a challenge in bridging preclinical studies to clinical efficacy. It may be a mistake to say that a pathway is not relevant because it failed in ALS studies 20 years ago. Doing so would risk losing further development of potential drugs that might target those same pathways more effectively.
Author Contribution
NJM JDC, and MdC wrote the manuscript.
Conflict of Interest
NJM is a consultant to Apellis and Cytokinetics and on the scientific advisory boards of Nura Bio and Akava. He receives clinical research support from Biogen/Idec, Apellis, Helixmith, Healey Center for ALS, Calico, and Sanofi. MdC has received consulting honoraria from Cytokinetics and Kedrion. He receives clinical research support from Cytokinetics, Pfizer, Ono, and Biogen/Idec. MDW has received honoraria for serving on scientific advisory boards for Alexion, UCB‐Ra, Argenx, Biogen, Mitsubishi Tanabe Pharma, and Amylyx; consulting honoraria from Cytokinetics and CSL Behring; and speaker honoraria from Soleo Health. He also serves as a special government employee for the US Food and Drug Administration.
Supporting information
Table S1. Clinical trials for targeted ALS pathways.
Acknowledgments
National Institutes of Health, USA 5R01NS117604‐03 to NJM. Editorial support (funded by Cytokinetics) was provided by iLuma Medical Communications (Tiffannie Nguyen, PharmD, and Ilona Kravtsova, PharmD, RPh).
Funding Statement
This work was funded by Cytokinetics ; National Institutes of Health, USA grant 5R01NS117604‐03.
References
- 1. Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. Dose‐ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet. 1996;347(9013):1425‐1431. doi: 10.1016/s0140-6736(96)91680-3 [DOI] [PubMed] [Google Scholar]
- 2. Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330(9):585‐591. doi: 10.1056/NEJM199403033300901 [DOI] [PubMed] [Google Scholar]
- 3. Plaitakis A, Caroscio JT. Abnormal glutamate metabolism in amyotrophic lateral sclerosis. Ann Neurol. 1987;22(5):575‐579. doi: 10.1002/ana.410220503 [DOI] [PubMed] [Google Scholar]
- 4. Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med. 1992;326(22):1464‐1468. doi: 10.1056/NEJM199205283262204 [DOI] [PubMed] [Google Scholar]
- 5. Andrews JA, Jackson CE, Heiman‐Patterson TD, Bettica P, Brooks BR, Pioro EP. Real‐world evidence of riluzole effectiveness in treating amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2020;21(7–8):509‐518. doi: 10.1080/21678421.2020.1771734 [DOI] [PubMed] [Google Scholar]
- 6. Fang T, Al Khleifat A, Meurgey JH, et al. Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: a retrospective analysis of data from a dose‐ranging study. Lancet Neurol. 2018;17(5):416‐422. doi: 10.1016/S1474-4422(18)30054-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Riviere M, Meininger V, Zeisser P, Munsat T. An analysis of extended survival in patients with amyotrophic lateral sclerosis treated with riluzole. Arch Neurol. 1998;55(4):526‐528. doi: 10.1001/archneur.55.4.526 [DOI] [PubMed] [Google Scholar]
- 8. Mizoule J, Meldrum B, Mazadier M, et al. 2‐Amino‐6‐trifluoromethoxy benzothiazole, a possible antagonist of excitatory amino acid neurotransmission—I: Anticonvulsant properties. Neuropharmacology. 1985;24(8):767‐773. doi: 10.1016/0028-3908(85)90011-5 [DOI] [PubMed] [Google Scholar]
- 9. Martin D, Thompson MA, Nadler JV. The neuroprotective agent riluzole inhibits release of glutamate and aspartate from slices of hippocampal area CA1. Eur J Pharmacol. 1993;250(3):473‐476. doi: 10.1016/0014-2999(93)90037-I [DOI] [PubMed] [Google Scholar]
- 10. Hogg MC, Halang L, Woods I, Coughlan KS, Prehn JHM. Riluzole does not improve lifespan or motor function in three ALS mouse models. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19(5–6):438‐445. doi: 10.1080/21678421.2017.1407796 [DOI] [PubMed] [Google Scholar]
- 11. Bellingham MC. A review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: what have we learned in the last decade? CNS Neurosci Ther. 2011;17(1):4‐31. doi: 10.1111/j.1755-5949.2009.00116.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pieri M, Carunchio I, Curcio L, Mercuri NB, Zona C. Increased persistent sodium current determines cortical hyperexcitability in a genetic model of amyotrophic lateral sclerosis. Exp Neurol. 2009;215(2):368‐379. doi: 10.1016/j.expneurol.2008.11.002 [DOI] [PubMed] [Google Scholar]
- 13. Pascuzzi RM, Shefner J, Chappell AS, et al. A phase II trial of talampanel in subjects with amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2010;11(3):266‐271. doi: 10.3109/17482960903307805 [DOI] [PubMed] [Google Scholar]
- 14. Cudkowicz ME, Shefner JM, Schoenfeld DA, et al. A randomized, placebo‐controlled trial of topiramate in amyotrophic lateral sclerosis. Neurology. 2003;61(4):456‐464. doi: 10.1212/wnl.61.4.456 [DOI] [PubMed] [Google Scholar]
- 15. Gredal O, Werdelin L, Bak S, et al. A clinical trial of dextromethorphan in amyotrophic lateral sclerosis. Acta Neurol Scand. 1997;96(1):8‐13. doi: 10.1111/j.1600-0404.1997.tb00231.x [DOI] [PubMed] [Google Scholar]
- 16. de Carvalho M, Pinto S, Costa J, Evangelista T, Ohana B, Pinto A. A randomized, placebo‐controlled trial of memantine for functional disability in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2010;11(5):456‐460. doi: 10.3109/17482968.2010.498521 [DOI] [PubMed] [Google Scholar]
- 17. Cudkowicz ME, Titus S, Kearney M, et al. Safety and efficacy of ceftriaxone for amyotrophic lateral sclerosis: a multi‐stage, randomised, double‐blind, placebo‐controlled trial. Lancet Neurol. 2014;13(11):1083‐1091. doi: 10.1016/S1474-4422(14)70222-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miller RG, Moore DH, Gelinas DF, et al. Phase III randomized trial of gabapentin in patients with amyotrophic lateral sclerosis. Neurology. 2001;56(7):843‐848. doi: 10.1212/wnl.56.7.843 [DOI] [PubMed] [Google Scholar]
- 19. Ryberg H, Askmark H, Persson LI. A double‐blind randomized clinical trial in amyotrophic lateral sclerosis using lamotrigine: effects on CSF glutamate, aspartate, branched‐chain amino acid levels and clinical parameters. Acta Neurol Scand. 2003;108(1):1‐8. doi: 10.1034/j.1600-0404.2003.00111.x [DOI] [PubMed] [Google Scholar]
- 20. Piepers S, Veldink JH, de Jong SW, et al. Randomized sequential trial of valproic acid in amyotrophic lateral sclerosis. Ann Neurol. 2009;66(2):227‐234. doi: 10.1002/ana.21620 [DOI] [PubMed] [Google Scholar]
- 21. Grosskreutz J, Van Den Bosch L, Keller BU. Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium. 2010;47(2):165‐174. doi: 10.1016/j.ceca.2009.12.002 [DOI] [PubMed] [Google Scholar]
- 22. Miller RG, Smith SA, Murphy JR, et al. A clinical trial of verapamil in amyotrophic lateral sclerosis. Muscle Nerve. 1996;19(4):511‐515. doi: 10.1002/mus.880190405 [DOI] [PubMed] [Google Scholar]
- 23. Miller RG, Shepherd R, Dao H, et al. Controlled trial of nimodipine in amyotrophic lateral sclerosis. Neuromuscul Disord. 1996;6(2):101‐104. doi: 10.1016/0960-8966(95)00024-0 [DOI] [PubMed] [Google Scholar]
- 24. Shibuya K, Misawa S, Kimura H, et al. A single blind randomized controlled clinical trial of mexiletine in amyotrophic lateral sclerosis: efficacy and safety of sodium channel blocker phase II trial. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16(5–6):353‐358. doi: 10.3109/21678421.2015.1038277 [DOI] [PubMed] [Google Scholar]
- 25. Weiss MD, Macklin EA, Simmons Z, et al. A randomized trial of mexiletine in ALS: safety and effects on muscle cramps and progression. Neurology. 2016;86(16):1474‐1481. doi: 10.1212/WNL.0000000000002507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weiss MD, Macklin EA, McIlduff CE, et al. Effects of mexiletine on hyperexcitability in sporadic amyotrophic lateral sclerosis: preliminary findings from a small phase II randomized controlled trial. Muscle Nerve. 2021;63(3):371‐383. doi: 10.1002/mus.27146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ghodke‐Puranik Y, Thorn CF, Lamba JK, et al. Valproic acid pathway: pharmacokinetics and pharmacodynamics. Pharmacogenet Genomics. 2013;23(4):236‐241. doi: 10.1097/FPC.0b013e32835ea0b2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wainger BJ, Kiskinis E, Mellin C, et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient‐derived motor neurons. Cell Rep. 2014;7(1):1‐11. doi: 10.1016/j.celrep.2014.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wainger BJ, Macklin EA, Vucic S, et al. Effect of Ezogabine on cortical and spinal motor neuron excitability in amyotrophic lateral sclerosis. JAMA Neurol. 2021;78(2):1‐12. doi: 10.1001/jamaneurol.2020.4300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Crisafulli SG, Brajkovic S, Cipolat Mis MS, Parente V, Corti S. Therapeutic strategies under development targeting inflammatory mechanisms in amyotrophic lateral sclerosis. Mol Neurobiol. 2018;55(4):2789‐2813. doi: 10.1007/s12035-017-0532-4 [DOI] [PubMed] [Google Scholar]
- 31. Drachman DB, Frank K, Dykes‐Hoberg M, et al. Cyclooxygenase 2 inhibition protects motor neurons and prolongs survival in a transgenic mouse model of ALS. Ann Neurol. 2002;52(6):771‐778. doi: 10.1002/ana.10374 [DOI] [PubMed] [Google Scholar]
- 32. Almer G, Teismann P, Stevic Z, et al. Increased levels of the pro‐inflammatory prostaglandin PGE2 in CSF from ALS patients. Neurology. 2002;58(8):1277‐1279. doi: 10.1212/wnl.58.8.1277 [DOI] [PubMed] [Google Scholar]
- 33. Cudkowicz ME, Shefner JM, Schoenfeld DA, et al. Trial of celecoxib in amyotrophic lateral sclerosis. Ann Neurol. 2006;60(1):22‐31. doi: 10.1002/ana.20903 [DOI] [PubMed] [Google Scholar]
- 34. Miller RG, Zhang R, Block G, et al. NP001 regulation of macrophage activation markers in ALS: a phase I clinical and biomarker study. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(7–8):601‐609. doi: 10.3109/21678421.2014.951940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miller RG, Block G, Katz JS, et al. Randomized phase 2 trial of NP001‐a novel immune regulator: safety and early efficacy in ALS. Neurol Neuroimmunol Neuroinflamm. 2015;2(3):e100. doi: 10.1212/NXI.0000000000000100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Miller RG, Zhang R, Bracci PM, et al. Phase 2B randomized controlled trial of NP001 in amyotrophic lateral sclerosis: pre‐specified and post hoc analyses. Muscle Nerve. 2022;66(1):39‐49. doi: 10.1002/mus.27511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Suzumura A, Ito A, Yoshikawa M, Sawada M. Ibudilast suppresses TNFalpha production by glial cells functioning mainly as type III phosphodiesterase inhibitor in the CNS. Brain Res. 1999;837(1–2):203‐212. doi: 10.1016/s0006-8993(99)01666-2 [DOI] [PubMed] [Google Scholar]
- 38. Mizuno T, Kurotani T, Komatsu Y, et al. Neuroprotective role of phosphodiesterase inhibitor ibudilast on neuronal cell death induced by activated microglia. Neuropharmacology. 2004;46(3):404‐411. doi: 10.1016/j.neuropharm.2003.09.009 [DOI] [PubMed] [Google Scholar]
- 39. Chen Y, Wang H, Ying Z, Gao Q. Ibudilast enhances the clearance of SOD1 and TDP‐43 aggregates through TFEB‐mediated autophagy and lysosomal biogenesis: the new molecular mechanism of ibudilast and its implication for neuroprotective therapy. Biochem Biophys Res Commun. 2020;526(1):231‐238. doi: 10.1016/j.bbrc.2020.03.051 [DOI] [PubMed] [Google Scholar]
- 40. Babu S, Hightower BG, Chan J, et al. Ibudilast (MN‐166) in amyotrophic lateral sclerosis‐ an open label, safety and pharmacodynamic trial. Neuroimage Clin. 2021;30:102672. doi: 10.1016/j.nicl.2021.102672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brooks B, Bravver E, Sanjak M, et al. A single‐center, randomized, double‐blind, placebo‐controlled, six‐month clinical trial followed by an open‐label extension to evaluate the safety, tolerability and clinical endpoint responsiveness of the phosphodiesterase type 4 (PDE4) inhibitor Ibudilast (MN‐166) in subjects with amyotrophic lateral sclerosis (ALS) – STEP‐IBUDILAST‐ALS‐DB‐OLE‐1 (I8‐1B). Neurology. 2015;84(14 Supplement):I8-1B. https://n.neurology.org/content/84/14_Supplement/I8‐1B [Google Scholar]
- 42. Oskarsson B, Maragakis N, Bedlack RS, et al. MN‐166 (ibudilast) in amyotrophic lateral sclerosis in a phase IIb/III study: COMBAT‐ALS study design. Neurodegener Dis Manag. 2021;11(6):431‐443. doi: 10.2217/nmt-2021-0042 [DOI] [PubMed] [Google Scholar]
- 43. Dubreuil P, Letard S, Ciufolini M, et al. Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS One. 2009;4(9):e7258. doi: 10.1371/journal.pone.0007258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Trias E, Ibarburu S, Barreto‐Núñez R, et al. Post‐paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J Neuroinflammation. 2016;13(1):177. doi: 10.1186/s12974-016-0620-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mora JS, Genge A, Chio A, et al. Masitinib as an add‐on therapy to riluzole in patients with amyotrophic lateral sclerosis: a randomized clinical trial. Amyotroph Lateral Scler Frontotemporal Degener. 2020;21(1–2):5‐14. doi: 10.1080/21678421.2019.1632346 [DOI] [PubMed] [Google Scholar]
- 46. Ceriello A. Thiazolidinediones as anti‐inflammatory and anti‐atherogenic agents. Diabetes Metab Res Rev. 2008;24(1):14‐26. doi: 10.1002/dmrr.790 [DOI] [PubMed] [Google Scholar]
- 47. Shibata N, Kawaguchi‐Niida M, Yamamoto T, Toi S, Hirano A, Kobayashi M. Effects of the PPARgamma activator pioglitazone on p38 MAP kinase and IkappaBalpha in the spinal cord of a transgenic mouse model of amyotrophic lateral sclerosis. Neuropathology. 2008;28(4):387‐398. doi: 10.1111/j.1440-1789.2008.00890.x [DOI] [PubMed] [Google Scholar]
- 48. Schütz B, Reimann J, Dumitrescu‐Ozimek L, et al. The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis‐like symptoms in superoxide dismutase‐G93A transgenic mice. J Neurosci. 2005;25(34):7805‐7812. doi: 10.1523/JNEUROSCI.2038-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kiaei M, Kipiani K, Chen J, Calingasan NY, Beal MF. Peroxisome proliferator‐activated receptor‐gamma agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2005;191(2):331‐336. doi: 10.1016/j.expneurol.2004.10.007 [DOI] [PubMed] [Google Scholar]
- 50. Dupuis L, Dengler R, Heneka MT, et al. A randomized, double blind, placebo‐controlled trial of pioglitazone in combination with riluzole in amyotrophic lateral sclerosis. PLoS One. 2012;7(6):e37885. doi: 10.1371/journal.pone.0037885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gordon PH, Moore DH, Miller RG, et al. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol. 2007;6(12):1045‐1053. doi: 10.1016/S1474-4422(07)70270-3 [DOI] [PubMed] [Google Scholar]
- 52. Chen X, Feng W, Huang R, et al. Evidence for peripheral immune activation in amyotrophic lateral sclerosis. J Neurol Sci. 2014;347(1–2):90‐95. doi: 10.1016/j.jns.2014.09.025 [DOI] [PubMed] [Google Scholar]
- 53. Thonhoff JR, Beers DR, Zhao W, et al. Expanded autologous regulatory T‐lymphocyte infusions in ALS: a phase I, first‐in‐human study. Neurol Neuroimmunol Neuroinflamm. 2018;5(4):e465. doi: 10.1212/NXI.0000000000000465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ghadiri M, Rezk A, Li R, et al. Dimethyl fumarate‐induced lymphopenia in MS due to differential T‐cell subset apoptosis. Neurol Neuroimmunol Neuroinflamm. 2017;4(3):e340. doi: 10.1212/NXI.0000000000000340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vucic S, Henderson RD, Mathers S, Needham M, Schultz D, Kiernan MC. Safety and efficacy of dimethyl fumarate in ALS: randomised controlled study. Ann Clin Transl Neurol. 2021;8(10):1991‐1999. doi: 10.1002/acn3.51446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Berry JD, Paganoni S, Atassi N, et al. Phase IIa trial of fingolimod for amyotrophic lateral sclerosis demonstrates acceptable acute safety and tolerability. Muscle Nerve. 2017;56(6):1077‐1084. doi: 10.1002/mus.25733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Milligan C, Atassi N, Babu S, et al. Tocilizumab is safe and tolerable and reduces C‐reactive protein concentrations in the plasma and cerebrospinal fluid of ALS patients. Muscle Nerve. 2021;64(3):309‐320. doi: 10.1002/mus.27339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mifflin L, Hu Z, Dufort C, et al. A RIPK1‐regulated inflammatory microglial state in amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2021;118(13):e2025102118. doi: 10.1073/pnas.2025102118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Boulis NM, Federici T, Glass JD, Lunn JS, Sakowski SA, Feldman EL. Translational stem cell therapy for amyotrophic lateral sclerosis. Nat Rev Neurol. 2011;8(3):172‐176. doi: 10.1038/nrneurol.2011.191 [DOI] [PubMed] [Google Scholar]
- 60. Oppenheim RW, Prevette D, Yin QW, Collins F, MacDonald J. Control of embryonic motoneuron survival in vivo by ciliary neurotrophic factor. Science. 1991;251(5001):1616‐1618. doi: 10.1126/science.2011743 [DOI] [PubMed] [Google Scholar]
- 61. Sendtner M, Kreutzberg GW, Thoenen H. Ciliary neurotrophic factor prevents the degeneration of motor neurons after axotomy. Nature. 1990;345(6274):440‐441. doi: 10.1038/345440a0 [DOI] [PubMed] [Google Scholar]
- 62. Sendtner M, Schmalbruch H, Stöckli KA, Carroll P, Kreutzberg GW, Thoenen H. Ciliary neurotrophic factor prevents degeneration of motor neurons in mouse mutant progressive motor neuronopathy. Nature. 1992;358(6386):502‐504. doi: 10.1038/358502a0 [DOI] [PubMed] [Google Scholar]
- 63. Mitsumoto H, Ikeda K, Holmlund T, et al. The effects of ciliary neurotrophic factor on motor dysfunction in wobbler mouse motor neuron disease. Ann Neurol. 1994;36(2):142‐148. doi: 10.1002/ana.410360205 [DOI] [PubMed] [Google Scholar]
- 64. Longo FM. Will ciliary neurotrophic factor slow progression of motor neuron disease? Ann Neurol. 1994;36(2):125‐127. doi: 10.1002/ana.410360202 [DOI] [PubMed] [Google Scholar]
- 65. Miller RG, Petajan JH, Bryan WW, et al. A placebo‐controlled trial of recombinant human ciliary neurotrophic (rhCNTF) factor in amyotrophic lateral sclerosis. rhCNTF ALS study group. Ann Neurol. 1996;39(2):256‐260. doi: 10.1002/ana.410390215 [DOI] [PubMed] [Google Scholar]
- 66. Group ACTS . A double‐blind placebo‐controlled clinical trial of subcutaneous recombinant human ciliary neurotrophic factor (rHCNTF) in amyotrophic lateral sclerosis. Neurology. 1996;46(5):1244‐1249. doi: 10.1212/WNL.46.5.1244 [DOI] [PubMed] [Google Scholar]
- 67. Penn RD, Kroin JS, York MM, Cedarbaum JM. Intrathecal ciliary neurotrophic factor delivery for treatment of amyotrophic lateral sclerosis (phase I trial). Neurosurgery. 1997;40(1):94‐99; discussion 99–100. doi: 10.1097/00006123-199701000-00021 [DOI] [PubMed] [Google Scholar]
- 68. Aebischer P, Pochon NA, Heyd B, et al. Gene therapy for amyotrophic lateral sclerosis (ALS) using a polymer encapsulated xenogenic cell line engineered to secrete hCNTF. Hum Gene Ther. 1996;7(7):851‐860. doi: 10.1089/hum.1996.7.7-851 [DOI] [PubMed] [Google Scholar]
- 69. Lai EC, Felice KJ, Festoff BW, et al. Effect of recombinant human insulin‐like growth factor‐I on progression of ALS. A placebo‐controlled study. The North America ALS/IGF‐I Study Group. Neurology. 1997;49(6):1621‐1630. doi: 10.1212/wnl.49.6.1621 [DOI] [PubMed] [Google Scholar]
- 70. Borasio GD, Robberecht W, Leigh PN, et al. A placebo‐controlled trial of insulin‐like growth factor‐I in amyotrophic lateral sclerosis. European ALS/IGF‐I Study Group. Neurology. 1998;51(2):583‐586. doi: 10.1212/wnl.51.2.583 [DOI] [PubMed] [Google Scholar]
- 71. Sorenson EJ, Windbank AJ, Mandrekar JN, et al. Subcutaneous IGF‐1 is not beneficial in 2‐year ALS trial. Neurology. 2008;71(22):1770‐1775. doi: 10.1212/01.wnl.0000335970.78664.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nagano I, Shiote M, Murakami T, et al. Beneficial effects of intrathecal IGF‐1 administration in patients with amyotrophic lateral sclerosis. Neurol Res. 2005;27(7):768‐772. doi: 10.1179/016164105X39860 [DOI] [PubMed] [Google Scholar]
- 73. Van Damme P, Tilkin P, Mercer KJ, et al. Intracerebroventricular delivery of vascular endothelial growth factor in patients with amyotrophic lateral sclerosis, a phase I study. Brain Commun. 2020;2(2):fcaa160. doi: 10.1093/braincomms/fcaa160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kadoyama K, Funakoshi H, Ohya W, Nakamura T. Hepatocyte growth factor (HGF) attenuates gliosis and motoneuronal degeneration in the brainstem motor nuclei of a transgenic mouse model of ALS. Neurosci Res. 2007;59(4):446‐456. doi: 10.1016/j.neures.2007.08.017 [DOI] [PubMed] [Google Scholar]
- 75. Sufit RL, Ajroud‐Driss S, Casey P, Kessler JA. Open label study to assess the safety of VM202 in subjects with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(3–4):269‐278. doi: 10.1080/21678421.2016.1259334 [DOI] [PubMed] [Google Scholar]
- 76. Dupuis L, Gonzalez de Aguilar JL, di Scala F, et al. Nogo provides a molecular marker for diagnosis of amyotrophic lateral sclerosis. Neurobiol Dis. 2002;10(3):358‐365. doi: 10.1006/nbdi.2002.0522 [DOI] [PubMed] [Google Scholar]
- 77. Pradat PF, Bruneteau G, Gonzalez de Aguilar JL, et al. Muscle Nogo‐a expression is a prognostic marker in lower motor neuron syndromes. Ann Neurol. 2007;62(1):15‐20. doi: 10.1002/ana.21122 [DOI] [PubMed] [Google Scholar]
- 78. Meininger V, Pradat PF, Corse A, et al. Safety, pharmacokinetic, and functional effects of the nogo‐a monoclonal antibody in amyotrophic lateral sclerosis: a randomized, first‐in‐human clinical trial. PLoS One. 2014;9(5):e97803. doi: 10.1371/journal.pone.0097803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Meininger V, Genge A, van den Berg LH, et al. Safety and efficacy of ozanezumab in patients with amyotrophic lateral sclerosis: a randomised, double‐blind, placebo‐controlled, phase 2 trial. Lancet Neurol. 2017;16(3):208‐216. doi: 10.1016/S1474-4422(16)30399-4 [DOI] [PubMed] [Google Scholar]
- 80. Meininger V, Bensimon G, Bradley WR, et al. Efficacy and safety of xaliproden in amyotrophic lateral sclerosis: results of two phase III trials. Amyotroph Lateral Scler Other Motor Neuron Disord. 2004;5(2):107‐117. doi: 10.1080/14660820410019602 [DOI] [PubMed] [Google Scholar]
- 81. Je G, Keyhanian K, Ghasemi M. Overview of stem cells therapy in amyotrophic lateral sclerosis. Neurol Res. 2021;43(8):616‐632. doi: 10.1080/01616412.2021.1893564 [DOI] [PubMed] [Google Scholar]
- 82. Mazzini L, Fagioli F, Boccaletti R, et al. Stem cell therapy in amyotrophic lateral sclerosis: a methodological approach in humans. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4(3):158‐161. doi: 10.1080/14660820310014653 [DOI] [PubMed] [Google Scholar]
- 83. Berry JD, Cudkowicz ME, Windebank AJ, et al. NurOwn, phase 2, randomized, clinical trial in patients with ALS: safety, clinical, and biomarker results. Neurology. 2019;93(24):e2294‐e2305. doi: 10.1212/WNL.0000000000008620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cudkowicz ME, Lindborg SR, Goyal NA, et al. A randomized placebo‐controlled phase 3 study of mesenchymal stem cells induced to secrete high levels of neurotrophic factors in amyotrophic lateral sclerosis. Muscle Nerve. 2022;65(3):291‐302. doi: 10.1002/mus.27472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Nam JY, Lee TY, Kim K, et al. Efficacy and safety of Lenzumestrocel (Neuronata‐R® inj.) in patients with amyotrophic lateral sclerosis (ALSUMMIT study): study protocol for a multicentre, randomized, double‐blind, parallel‐group, sham procedure‐controlled, phase III trial. Trials. 2022;23(1):415. doi: 10.1186/s13063-022-06327-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Oh KW, Noh MY, Kwon MS, et al. Repeated intrathecal mesenchymal stem cells for amyotrophic lateral sclerosis. Ann Neurol. 2018;84(3):361‐373. doi: 10.1002/ana.25302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Vercelli A, Mereuta OM, Garbossa D, et al. Human mesenchymal stem cell transplantation extends survival, improves motor performance and decreases neuroinflammation in mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2008;31(3):395‐405. doi: 10.1016/j.nbd.2008.05.016 [DOI] [PubMed] [Google Scholar]
- 88. Glass JD, Boulis NM, Johe K, et al. Lumbar intraspinal injection of neural stem cells in patients with amyotrophic lateral sclerosis: results of a phase I trial in 12 patients. Stem Cells. 2012;30(6):1144‐1151. doi: 10.1002/stem.1079 [DOI] [PubMed] [Google Scholar]
- 89. Baloh RH, Johnson JP, Avalos P, et al. Transplantation of human neural progenitor cells secreting GDNF into the spinal cord of patients with ALS: a phase 1/2a trial. Nat Med. 2022;28(9):1813‐1822. doi: 10.1038/s41591-022-01956-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Haidet‐Phillips AM, Maragakis NJ. Neural and glial progenitor transplantation as a neuroprotective strategy for amyotrophic lateral sclerosis (ALS). Brain Res. 2015;1628(Pt B):343‐350. doi: 10.1016/j.brainres.2015.06.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Johns AE, Maragakis NJ. Exploring motor neuron diseases using iPSC platforms. Stem Cells. 2022;40(1):2‐13. doi: 10.1093/stmcls/sxab006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Smith RA, Miller TM, Yamanaka K, et al. Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Invest. 2006;116(8):2290‐2296. doi: 10.1172/JCI25424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. McCampbell A, Cole T, Wegener AJ, et al. Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J Clin Invest. 2018;128(8):3558‐3567. doi: 10.1172/JCI99081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Crisp MJ, Mawuenyega KG, Patterson BW, et al. In vivo kinetic approach reveals slow SOD1 turnover in the CNS. J Clin Invest. 2015;125(7):2772‐2780. doi: 10.1172/JCI80705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Miller TM, Pestronk A, David W, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first‐in‐man study. Lancet Neurol. 2013;12(5):435‐442. doi: 10.1016/S1474-4422(13)70061-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Miller T, Cudkowicz M, Shaw PJ, et al. Phase 1‐2 trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med. 2020;383(2):109‐119. doi: 10.1056/NEJMoa2003715 [DOI] [PubMed] [Google Scholar]
- 97. Miller TM, Cudkowicz ME, Genge A, et al. Trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med. 2022;387(12):1099‐1110. doi: 10.1056/NEJMoa2204705 [DOI] [PubMed] [Google Scholar]
- 98. Lagier‐Tourenne C, Baughn M, Rigo F, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A. 2013;110(47):E4530‐E4539. doi: 10.1073/pnas.1318835110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Sareen D, O'Rourke JG, Meera P, et al. Targeting RNA foci in iPSC‐derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5(208):208ra149. doi: 10.1126/scitranslmed.3007529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Donnelly CJ, Zhang PW, Pham JT, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80(2):415‐428. doi: 10.1016/j.neuron.2013.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Liu Y, Dodart JC, Tran H, et al. Variant‐selective stereopure oligonucleotides protect against pathologies associated with C9orf72‐repeat expansion in preclinical models. Nat Commun. 2021;12(1):847. doi: 10.1038/s41467-021-21112-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Tran H, Moazami MP, Yang H, et al. Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat Med. 2022;28(1):117‐124. doi: 10.1038/s41591-021-01557-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Boros BD, Schoch KM, Kreple CJ, Miller TM. Antisense oligonucleotides for the study and treatment of ALS. Neurotherapeutics. 2022;19(4):1145‐1158. doi: 10.1007/s13311-022-01247-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Korobeynikov VA, Lyashchenko AK, Blanco‐Redondo B, Jafar‐Nejad P, Shneider NA. Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat Med. 2022;28(1):104‐116. doi: 10.1038/s41591-021-01615-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Becker LA, Huang B, Bieri G, et al. Therapeutic reduction of ataxin‐2 extends lifespan and reduces pathology in TDP‐43 mice. Nature. 2017;544(7650):367‐371. doi: 10.1038/nature22038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Meijboom KE, Brown RH. Approaches to gene modulation therapy for ALS. Neurotherapeutics. 2022;19(4):1159‐1179. doi: 10.1007/s13311-022-01285-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Park HR, Yang EJ. Oxidative stress as a therapeutic target in amyotrophic lateral sclerosis: opportunities and limitations. Diagnostics (Basel). 2021;11(9):1546. doi: 10.3390/diagnostics11091546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Agar J, Durham H. Relevance of oxidative injury in the pathogenesis of motor neuron diseases. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4(4):232‐242. doi: 10.1080/14660820310011278 [DOI] [PubMed] [Google Scholar]
- 109. Orrell RW, Lane RJM, Ross M. A systematic review of antioxidant treatment for amyotrophic lateral sclerosis/motor neuron disease. Amyotroph Lateral Scler. 2008;9(4):195‐211. doi: 10.1080/17482960801900032 [DOI] [PubMed] [Google Scholar]
- 110. Louwerse ES, Weverling GJ, Bossuyt PM, Meyjes FE, de Jong JM. Randomized, double‐blind, controlled trial of acetylcysteine in amyotrophic lateral sclerosis. Arch Neurol. 1995;52(6):559‐564. doi: 10.1001/archneur.1995.00540300031009 [DOI] [PubMed] [Google Scholar]
- 111. Lange DJ, Murphy PL, Diamond B, et al. Selegiline is ineffective in a collaborative double‐blind, placebo‐controlled trial for treatment of amyotrophic lateral sclerosis. Arch Neurol. 1998;55(1):93‐96. doi: 10.1001/archneur.55.1.93 [DOI] [PubMed] [Google Scholar]
- 112. Gurney ME, Cutting FB, Zhai P, et al. Benefit of vitamin E, riluzole, and gabapentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann Neurol. 1996;39(2):147‐157. doi: 10.1002/ana.410390203 [DOI] [PubMed] [Google Scholar]
- 113. Desnuelle C, Dib M, Garrel C, Favier A. A double‐blind, placebo‐controlled randomized clinical trial of alpha‐tocopherol (vitamin E) in the treatment of amyotrophic lateral sclerosis. ALS riluzole‐tocopherol study group. Amyotroph Lateral Scler Other Motor Neuron Disord. 2001;2(1):9‐18. doi: 10.1080/146608201300079364 [DOI] [PubMed] [Google Scholar]
- 114. Graf M, Ecker D, Horowski R, et al. High dose vitamin E therapy in amyotrophic lateral sclerosis as add‐on therapy to riluzole: results of a placebo‐controlled double‐blind study. J Neural Transm (Vienna). 2005;112(5):649‐660. doi: 10.1007/s00702-004-0220-1 [DOI] [PubMed] [Google Scholar]
- 115. Klivenyi P, Ferrante RJ, Matthews RT, et al. Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nat Med. 1999;5(3):347‐350. doi: 10.1038/6568 [DOI] [PubMed] [Google Scholar]
- 116. Zhang W, Narayanan M, Friedlander RM. Additive neuroprotective effects of minocycline with creatine in a mouse model of ALS. Ann Neurol. 2003;53(2):267‐270. doi: 10.1002/ana.10476 [DOI] [PubMed] [Google Scholar]
- 117. Snow RJ, Murphy RM. Factors influencing creatine loading into human skeletal muscle. Exerc Sport Sci Rev. 2003;31(3):154‐158. doi: 10.1097/00003677-200307000-00010 [DOI] [PubMed] [Google Scholar]
- 118. Groeneveld GJ, Veldink JH, van der Tweel I, et al. A randomized sequential trial of creatine in amyotrophic lateral sclerosis. Ann Neurol. 2003;53(4):437‐445. doi: 10.1002/ana.10554 [DOI] [PubMed] [Google Scholar]
- 119. Shefner JM, Cudkowicz ME, Schoenfeld D, et al. A clinical trial of creatine in ALS. Neurology. 2004;63(9):1656‐1661. doi: 10.1212/01.wnl.0000142992.81995.f0 [DOI] [PubMed] [Google Scholar]
- 120. Wang H, Larriviere KS, Keller KE, et al. R+ pramipexole as a mitochondrially focused neuroprotectant: initial early phase studies in ALS. Amyotroph Lateral Scler. 2008;9(1):50‐58. doi: 10.1080/17482960701791234 [DOI] [PubMed] [Google Scholar]
- 121. Cudkowicz M, Bozik ME, Ingersoll EW, et al. The effects of dexpramipexole (KNS‐760704) in individuals with amyotrophic lateral sclerosis. Nat Med. 2011;17(12):1652‐1656. doi: 10.1038/nm.2579 [DOI] [PubMed] [Google Scholar]
- 122. Martin LJ. Olesoxime, a cholesterol‐like neuroprotectant for the potential treatment of amyotrophic lateral sclerosis. IDrugs. 2010;13(8):568‐580. [PMC free article] [PubMed] [Google Scholar]
- 123. Bordet T, Buisson B, Michaud M, et al. Identification and characterization of cholest‐4‐en‐3‐one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J Pharmacol Exp Ther. 2007;322(2):709‐720. doi: 10.1124/jpet.107.123000 [DOI] [PubMed] [Google Scholar]
- 124. Lenglet T, Lacomblez L, Abitbol JL, et al. A phase II‐III trial of olesoxime in subjects with amyotrophic lateral sclerosis. Eur J Neurol. 2014;21(3):529‐536. doi: 10.1111/ene.12344 [DOI] [PubMed] [Google Scholar]
- 125. Zoccolella S, Simone IL, Lamberti P, et al. Elevated plasma homocysteine levels in patients with amyotrophic lateral sclerosis. Neurology. 2008;70(3):222‐225. doi: 10.1212/01.wnl.0000297193.53986.6f [DOI] [PubMed] [Google Scholar]
- 126. Okada K, Tanaka H, Temporin K, et al. Methylcobalamin increases Erk1/2 and Akt activities through the methylation cycle and promotes nerve regeneration in a rat sciatic nerve injury model. Exp Neurol. 2010;222(2):191‐203. doi: 10.1016/j.expneurol.2009.12.017 [DOI] [PubMed] [Google Scholar]
- 127. Kikuchi M, Kashii S, Honda Y, Tamura Y, Kaneda K, Akaike A. Protective effects of methylcobalamin, a vitamin B12 analog, against glutamate‐induced neurotoxicity in retinal cell culture. Invest Ophthalmol Vis Sci. 1997;38(5):848‐854. [PubMed] [Google Scholar]
- 128. Ikeda K, Iwasaki Y, Kaji R. Neuroprotective effect of ultra‐high dose methylcobalamin in wobbler mouse model of amyotrophic lateral sclerosis. J Neurol Sci. 2015;354(1–2):70‐74. doi: 10.1016/j.jns.2015.04.052 [DOI] [PubMed] [Google Scholar]
- 129. Izumi Y, Kaji R. Clinical trials of ultra‐high‐dose methylcobalamin in ALS. Brain Nerve. 2007;59(10):1141‐1147. [PubMed] [Google Scholar]
- 130. Kaji R, Imai T, Iwasaki Y, et al. Ultra‐high‐dose methylcobalamin in amyotrophic lateral sclerosis: a long‐term phase II/III randomised controlled study. J Neurol Neurosurg Psychiatry. 2019;90(4):451‐457. doi: 10.1136/jnnp-2018-319294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Oki R, Izumi Y, Fujita K, et al. Efficacy and safety of ultrahigh‐dose methylcobalamin in early‐stage amyotrophic lateral sclerosis: a randomized clinical trial. JAMA Neurol. 2022;79(6):575‐583. doi: 10.1001/jamaneurol.2022.0901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Fujimori K, Ishikawa M, Otomo A, et al. Modeling sporadic ALS in iPSC‐derived motor neurons identifies a potential therapeutic agent. Nat Med. 2018;24(10):1579‐1589. doi: 10.1038/s41591-018-0140-5 [DOI] [PubMed] [Google Scholar]
- 133. Morimoto S, Takahashi S, Fukushima K, et al. Ropinirole hydrochloride remedy for amyotrophic lateral sclerosis ‐ protocol for a randomized, double‐blind, placebo‐controlled, single‐center, and open‐label continuation phase I/IIa clinical trial (ROPALS trial). Regen Ther. 2019;11:143‐166. doi: 10.1016/j.reth.2019.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Edaravone Acute Infarction Study Group . Effect of a novel free radical scavenger, edaravone (MCI‐186), on acute brain infarction. Randomized, placebo‐controlled, double‐blind study at multicenters. Cerebrovasc Dis. 2003;15(3):222‐229. doi: 10.1159/000069318 [DOI] [PubMed] [Google Scholar]
- 135. Aoki M, Warita H, Mizuno H, Suzuki N, Yuki S, Itoyama Y. Feasibility study for functional test battery of SOD transgenic rat (H46R) and evaluation of edaravone, a free radical scavenger. Brain Res. 2011;1382:321‐325. doi: 10.1016/j.brainres.2011.01.058 [DOI] [PubMed] [Google Scholar]
- 136. Yoshino H, Kimura A. Investigation of the therapeutic effects of edaravone, a free radical scavenger, on amyotrophic lateral sclerosis (phase II study). Amyotroph Lateral Scler. 2006;7(4):241‐245. doi: 10.1080/17482960600881870 [DOI] [PubMed] [Google Scholar]
- 137. Abe K, Itoyama Y, Sobue G, et al. Confirmatory double‐blind, parallel‐group, placebo‐controlled study of efficacy and safety of edaravone (MCI‐186) in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(7–8):610‐617. doi: 10.3109/21678421.2014.959024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Writing Group , Edaravone (MCI‐186) ALS 19 Study Group . Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double‐blind, placebo‐controlled trial. Lancet Neurol. 2017;16(7):505‐512. doi: 10.1016/S1474-4422(17)30115-1 [DOI] [PubMed] [Google Scholar]
- 139. Lunetta C, Moglia C, Lizio A, et al. The Italian multicenter experience with edaravone in amyotrophic lateral sclerosis. J Neurol. 2020;267(11):3258‐3267. doi: 10.1007/s00415-020-09993-z [DOI] [PubMed] [Google Scholar]
- 140. Vu M, Tortorice K, Zacher J, et al. Assessment of use and safety of edaravone for amyotrophic lateral sclerosis in the veterans affairs health care system. JAMA Netw Open. 2020;3(10):e2014645. doi: 10.1001/jamanetworkopen.2020.14645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Witzel S, Maier A, Steinbach R, et al. Safety and effectiveness of long‐term intravenous administration of edaravone for treatment of patients with amyotrophic lateral sclerosis. JAMA Neurol. 2022;79(2):121‐130. doi: 10.1001/jamaneurol.2021.4893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Kaur B, Bhat A, Chakraborty R, et al. Proteomic profile of 4‐PBA treated human neuronal cells during ER stress. Mol Omics. 2018;14(1):53‐63. doi: 10.1039/C7MO00114B [DOI] [PubMed] [Google Scholar]
- 143. Rodrigues CMP, Solá S, Sharpe JC, Moura JJG, Steer CJ. Tauroursodeoxycholic acid prevents Bax‐induced membrane perturbation and cytochrome C release in isolated mitochondria. Biochemistry. 2003;42(10):3070‐3080. doi: 10.1021/bi026979d [DOI] [PubMed] [Google Scholar]
- 144. Ryu H, Smith K, Camelo SI, et al. Sodium phenylbutyrate prolongs survival and regulates expression of anti‐apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J Neurochem. 2005;93(5):1087‐1098. doi: 10.1111/j.1471-4159.2005.03077.x [DOI] [PubMed] [Google Scholar]
- 145. Fels JA, Dash J, Leslie K, Manfredi G, Kawamata H. Effects of PB‐TURSO on the transcriptional and metabolic landscape of sporadic ALS fibroblasts. Ann Clin Transl Neurol. 2022;9(10):1551‐1564. doi: 10.1002/acn3.51648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Paganoni S, Macklin EA, Hendrix S, et al. Trial of sodium phenylbutyrate–taurursodiol for amyotrophic lateral sclerosis. N Engl J Med. 2020;383(10):919‐930. doi: 10.1056/NEJMoa1916945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Paganoni S, Watkins C, Cawson M, et al. Survival analyses from the CENTAUR trial in amyotrophic lateral sclerosis: evaluating the impact of treatment crossover on outcomes. Muscle Nerve. 2022;66(2):136‐141. doi: 10.1002/mus.27569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Chua JP, De Calbiac H, Kabashi E, Barmada SJ. Autophagy and ALS: mechanistic insights and therapeutic implications. Autophagy. 2022;18(2):254‐282. doi: 10.1080/15548627.2021.1926656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Pasquali L, Longone P, Isidoro C, Ruggieri S, Paparelli A, Fornai F. Autophagy, lithium, and amyotrophic lateral sclerosis. Muscle Nerve. 2009;40(2):173‐194. doi: 10.1002/mus.21423 [DOI] [PubMed] [Google Scholar]
- 150. Motoi Y, Shimada K, Ishiguro K, Hattori N. Lithium and autophagy. ACS Chem Nerosci. 2014;5(6):434‐442. doi: 10.1021/cn500056q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Fornai F, Longone P, Cafaro L, et al. Lithium delays progression of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2008;105(6):2052‐2057. doi: 10.1073/pnas.0708022105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Aggarwal SP, Zinman L, Simpson E, et al. Safety and efficacy of lithium in combination with riluzole for treatment of amyotrophic lateral sclerosis: a randomised, double‐blind, placebo‐controlled trial. Lancet Neurol. 2010;9(5):481‐488. doi: 10.1016/S1474-4422(10)70068-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Chiò A, Borghero G, Calvo A, et al. Lithium carbonate in amyotrophic lateral sclerosis: lack of efficacy in a dose‐finding trial. Neurology. 2010;75(7):619‐625. doi: 10.1212/WNL.0b013e3181ed9e7c [DOI] [PubMed] [Google Scholar]
- 154. Miller RG, Moore DH, Forshew DA, et al. Phase II screening trial of lithium carbonate in amyotrophic lateral sclerosis: examining a more efficient trial design. Neurology. 2011;77(10):973‐979. doi: 10.1212/WNL.0b013e31822dc7a5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Verstraete E, Veldink JH, Huisman MHB, et al. Lithium lacks effect on survival in amyotrophic lateral sclerosis: a phase IIb randomised sequential trial. J Neurol Neurosurg Psychiatry. 2012;83(5):557‐564. doi: 10.1136/jnnp-2011-302021 [DOI] [PubMed] [Google Scholar]
- 156. UKMND‐LiCALS Study Group , Morrison KE, Dhariwal S, et al. Lithium in patients with amyotrophic lateral sclerosis (LiCALS): a phase 3 multicentre, randomised, double‐blind, placebo‐controlled trial. Lancet Neurol. 2013;12(4):339‐345. doi: 10.1016/S1474-4422(13)70037-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Boll MC, Alcaraz‐Zubeldia M, Rios C, González‐Esquivel D, Montes S. A phase 2, double‐blind, placebo‐controlled trial of a valproate/lithium combination in ALS patients. Neurologia (Engl Ed). 2022;S2173‐5808(22)00089‐X. doi: 10.1016/j.nrleng.2022.07.003 [DOI] [PubMed] [Google Scholar]
- 158. Wang IF, Guo BS, Liu YC, et al. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA‐binding protein 43. Proc Natl Acad Sci U S A. 2012;109(37):15024‐15029. doi: 10.1073/pnas.1206362109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Chen PC, Hsieh YC, Huang CC, Hu CJ. Tamoxifen for amyotrophic lateral sclerosis: a randomized double‐blind clinical trial. Medicine (Baltimore). 2020;99(22):e20423. doi: 10.1097/MD.0000000000020423 [DOI] [PubMed] [Google Scholar]
- 160. Babu S, Macklin EA, Jackson KE, et al. Selection design phase II trial of high dosages of tamoxifen and creatine in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2020;21(1–2):15‐23. doi: 10.1080/21678421.2019.1672750 [DOI] [PubMed] [Google Scholar]
- 161. Mandrioli J, D'Amico R, Zucchi E, et al. Rapamycin treatment for amyotrophic lateral sclerosis: protocol for a phase II randomized, double‐blind, placebo‐controlled, multicenter, clinical trial (RAP‐ALS trial). Medicine (Baltimore). 2018;97(24):e11119. doi: 10.1097/MD.0000000000011119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Barmada SJ, Serio A, Arjun A, et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol. 2014;10(8):677‐685. doi: 10.1038/nchembio.1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Madill M, McDonagh K, Ma J, et al. Amyotrophic lateral sclerosis patient iPSC‐derived astrocytes impair autophagy via non‐cell autonomous mechanisms. Mol Brain. 2017;10(1):22. doi: 10.1186/s13041-017-0300-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Cheng CW, Lin MJ, Shen CKJ. Rapamycin alleviates pathogenesis of a new drosophila model of ALS‐TDP. J Neurogenet. 2015;29(2–3):59‐68. doi: 10.3109/01677063.2015.1077832 [DOI] [PubMed] [Google Scholar]
- 165. Deivasigamani S, Verma HK, Ueda R, Ratnaparkhi A, Ratnaparkhi GS. A genetic screen identifies tor as an interactor of VAPB in a drosophila model of amyotrophic lateral sclerosis. Biol Open. 2014;3(11):1127‐1138. doi: 10.1242/bio.201410066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Lattante S, de Calbiac H, Le Ber I, Brice A, Ciura S, Kabashi E. Sqstm1 knock‐down causes a locomotor phenotype ameliorated by rapamycin in a zebrafish model of ALS/FTLD. Hum Mol Genet. 2015;24(6):1682‐1690. doi: 10.1093/hmg/ddu580 [DOI] [PubMed] [Google Scholar]
- 167. Nalbandian A, Ghimbovschi S, Wang Z, et al. Global gene expression profiling in R155H Knock‐In murine model of VCP disease. Clin Transl Sci. 2015;8(1):8‐16. doi: 10.1111/cts.12241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Mandrioli J, Crippa V, Cereda C, et al. Proteostasis and ALS: protocol for a phase II, randomised, double‐blind, placebo‐controlled, multicentre clinical trial for colchicine in ALS (Co‐ALS). BMJ Open. 2019;9(5):e028486. doi: 10.1136/bmjopen-2018-028486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Crippa V, Cicardi ME, Ramesh N, et al. The chaperone HSPB8 reduces the accumulation of truncated TDP‐43 species in cells and protects against TDP‐43‐mediated toxicity. Hum Mol Genet. 2016;25(18):3908‐3924. doi: 10.1093/hmg/ddw232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Ince PG, Lowe J, Shaw PJ. Amyotrophic lateral sclerosis: current issues in classification, pathogenesis and molecular pathology. Neuropathol Appl Neurobiol. 1998;24(2):104‐117. doi: 10.1046/j.1365-2990.1998.00108.x [DOI] [PubMed] [Google Scholar]
- 171. Ayers JI, Borchelt DR. Phenotypic diversity in ALS and the role of poly‐conformational protein misfolding. Acta Neuropathol. 2021;142(1):41‐55. doi: 10.1007/s00401-020-02222-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Kalmar B, Novoselov S, Gray A, Cheetham ME, Margulis B, Greensmith L. Late stage treatment with arimoclomol delays disease progression and prevents protein aggregation in the SOD1 mouse model of ALS. J Neurochem. 2008;107(2):339‐350. doi: 10.1111/j.1471-4159.2008.05595.x [DOI] [PubMed] [Google Scholar]
- 173. Kieran D, Kalmar B, Dick JRT, Riddoch‐Contreras J, Burnstock G, Greensmith L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med. 2004;10(4):402‐405. doi: 10.1038/nm1021 [DOI] [PubMed] [Google Scholar]
- 174. Kalmar B, Edet‐Amana E, Greensmith L. Treatment with a coinducer of the heat shock response delays muscle denervation in the SOD1‐G93A mouse model of amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2012;13(4):378‐392. doi: 10.3109/17482968.2012.660953 [DOI] [PubMed] [Google Scholar]
- 175. Benatar M, Wuu J, Andersen PM, et al. Randomized, double‐blind, placebo‐controlled trial of arimoclomol in rapidly progressive SOD1 ALS. Neurology. 2018;90(7):e565‐e574. doi: 10.1212/WNL.0000000000004960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Arimoclomol Fails to Show Efficacy in ALS . Neurology live. Published May 18 2021. Accessed February 24, 2023. https://www.neurologylive.com/view/arimoclomol‐fails‐to‐show‐efficacy‐in‐als
- 177. Wang L, Popko B, Tixier E, Roos RP. Guanabenz, which enhances the unfolded protein response, ameliorates mutant SOD1‐induced amyotrophic lateral sclerosis. Neurobiol Dis. 2014;71:317‐324. doi: 10.1016/j.nbd.2014.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Jiang HQ, Ren M, Jiang HZ, et al. Guanabenz delays the onset of disease symptoms, extends lifespan, improves motor performance and attenuates motor neuron loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neuroscience. 2014;277:132‐138. doi: 10.1016/j.neuroscience.2014.03.047 [DOI] [PubMed] [Google Scholar]
- 179. Dalla Bella E, Bersano E, Antonini G, et al. The unfolded protein response in amyotrophic later sclerosis: results of a phase 2 trial. Brain. 2021;144(9):2635‐2647. doi: 10.1093/brain/awab167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Shefner JM, Musaro A, Ngo ST, et al. Skeletal muscle in amyotrophic lateral sclerosis. Brain. 2023;awad202. Online ahead of print. doi: 10.1093/brain/awad202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Hinken A, Driscoll L, Lee K, et al. The fast skeletal troponin activator, CK‐1909178 reduces muscle fatigue in a model of peripheral artery disease in situ. Biophys J. 2010;98(3):543a. doi: 10.1016/j.bpj.2009.12.2945 20159150 [DOI] [Google Scholar]
- 182. Russell AJ, Hartman JJ, Hinken AC, et al. Activation of fast skeletal muscle troponin as a potential therapeutic approach for treating neuromuscular diseases. Nat Med. 2012;18(3):452‐455. doi: 10.1038/nm.2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Hwee DT, Kennedy A, Ryans J, et al. Fast skeletal muscle troponin activator tirasemtiv increases muscle function and performance in the B6SJL‐SOD1G93A ALS mouse model. PLoS One. 2014;9(5):e96921. doi: 10.1371/journal.pone.0096921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Shefner JM, Cudkowicz ME, Hardiman O, et al. A phase III trial of tirasemtiv as a potential treatment for amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20:584‐594. doi: 10.1080/21678421.2019.1612922 [DOI] [PubMed] [Google Scholar]
- 185. Cudkowicz M, Genge A, Maragakis N, et al. Safety and efficacy of oral levosimendan in people with amyotrophic lateral sclerosis (the REFALS study): a randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet Neurol. 2021;20(10):821‐831. doi: 10.1016/S1474-4422(21)00242-8 [DOI] [PubMed] [Google Scholar]
- 186. McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF‐beta superfamily member. Nature. 1997;387(6628):83‐90. doi: 10.1038/387083a0 [DOI] [PubMed] [Google Scholar]
- 187. Abati E, Manini A, Comi GP, Corti S. Inhibition of myostatin and related signaling pathways for the treatment of muscle atrophy in motor neuron diseases. Cell Mol Life Sci. 2022;79(7):374. doi: 10.1007/s00018-022-04408-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Chevalier‐Larsen E, Holzbaur ELF. Axonal transport and neurodegenerative disease. Biochim Biophys Acta. 2006;1762(11–12):1094‐1108. doi: 10.1016/j.bbadis.2006.04.002 [DOI] [PubMed] [Google Scholar]
- 189. Miller TM, Kim SH, Yamanaka K, et al. Gene transfer demonstrates that muscle is not a primary target for non‐cell‐autonomous toxicity in familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2006;103(51):19546‐19551. doi: 10.1073/pnas.0609411103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Morrison BM, Lachey JL, Warsing LC, et al. A soluble activin type IIB receptor improves function in a mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2009;217(2):258‐268. doi: 10.1016/j.expneurol.2009.02.017 [DOI] [PubMed] [Google Scholar]
- 191. Cho H, Shukla S. Role of edaravone as a treatment option for patients with amyotrophic lateral sclerosis. Pharmaceuticals (Basel). 2020;14(1):29. doi: 10.3390/ph14010029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Kolb PS, Ayaub EA, Zhou W, Yum V, Dickhout JG, Ask K. The therapeutic effects of 4‐phenylbutyric acid in maintaining proteostasis. Int J Biochem Cell Biol. 2015;61:45‐52. doi: 10.1016/j.biocel.2015.01.015 [DOI] [PubMed] [Google Scholar]
- 193. Wiley JC, Meabon JS, Frankowski H, et al. Phenylbutyric acid rescues endoplasmic reticulum stress‐induced suppression of APP proteolysis and prevents apoptosis in neuronal cells. PLoS One. 2010;5(2):e9135. doi: 10.1371/journal.pone.0009135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Khalaf K, Tornese P, Cocco A, Albanese A. Tauroursodeoxycholic acid: a potential therapeutic tool in neurodegenerative diseases. Transl Neurodegener. 2022;11(1):33. doi: 10.1186/s40035-022-00307-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Bowling AC, Schulz JB, Brown RH, Beal MF. Superoxide dismutase activity, oxidative damage, and mitochondrial energy metabolism in familial and sporadic amyotrophic lateral sclerosis. J Neurochem. 1993;61(6):2322‐2325. doi: 10.1111/j.1471-4159.1993.tb07478.x [DOI] [PubMed] [Google Scholar]
- 196. Bonifacino T, Zerbo RA, Balbi M, et al. Nearly 30 years of animal models to study amyotrophic lateral sclerosis: a historical overview and future perspectives. Int J Mol Sci. 2021;22(22):12236. doi: 10.3390/ijms222212236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Lemon RN. Descending pathways in motor control. Annu Rev Neurosci. 2008;31:195‐218. doi: 10.1146/annurev.neuro.31.060407.125547 [DOI] [PubMed] [Google Scholar]
- 198. Gautam M, Genç B, Helmold B, et al. SBT‐272 improves TDP‐43 pathology in ALS upper motor neurons by modulating mitochondrial integrity, motility, and function. Neurobiol Dis. 2023;178:106022. doi: 10.1016/j.nbd.2023.106022 [DOI] [PubMed] [Google Scholar]
- 199. Martin D, Bustos GA, Bowe MA, Bray SD, Nadler JV. Autoreceptor regulation of glutamate and aspartate release from slices of the hippocampal CA1 area. J Neurochem. 1991;56(5):1647‐1655. doi: 10.1111/j.1471-4159.1991.tb02063.x [DOI] [PubMed] [Google Scholar]
- 200. Skradski S, White HS. Topiramate blocks kainate‐evoked cobalt influx into cultured neurons. Epilepsia. 2000;41(s1):45‐47. doi: 10.1111/j.1528-1157.2000.tb02171.x [DOI] [PubMed] [Google Scholar]
- 201. Maragakis NJ, Dietrich J, Wong V, et al. Glutamate transporter expression and function in human glial progenitors. Glia. 2004;45(2):133‐143. doi: 10.1002/glia.10310 [DOI] [PubMed] [Google Scholar]
- 202. Canton T, Böhme GA, Boireau A, et al. RPR 119990, a novel alpha‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid antagonist: synthesis, pharmacological properties, and activity in an animal model of amyotrophic lateral sclerosis. J Pharmacol Exp Ther. 2001;299(1):314‐322. [PubMed] [Google Scholar]
- 203. Van Damme P, Van den Bosch L, Van Houtte E, Eggermont J, Callewaert G, Robberecht W. Na+ entry through AMPA receptors results in voltage‐gated K+ channel blockade in cultured rat spinal cord motoneurons. J Neurophysiol. 2002;88(2):965‐972. doi: 10.1152/jn.2002.88.2.965 [DOI] [PubMed] [Google Scholar]
- 204. Van Den Bosch L, Vandenberghe W, Klaassen H, Van Houtte E, Robberecht W. Ca(2+)‐permeable AMPA receptors and selective vulnerability of motor neurons. J Neurol Sci. 2000;180(1–2):29‐34. doi: 10.1016/s0022-510x(00)00414-7 [DOI] [PubMed] [Google Scholar]
- 205. Leach MJ, Marden CM, Miller AA. Pharmacological studies on lamotrigine, a novel potential antiepileptic drug: II. Neurochemical studies on the mechanism of action. Epilepsia. 1986;27(5):490‐497. doi: 10.1111/j.1528-1157.1986.tb03573.x [DOI] [PubMed] [Google Scholar]
- 206. Stefani A, Spadoni F, Bernardi G. Voltage‐activated calcium channels: targets of antiepileptic drug therapy? Epilepsia. 1997;38(9):959‐965. doi: 10.1111/j.1528-1157.1997.tb01477.x [DOI] [PubMed] [Google Scholar]
- 207. Casanovas A, Ribera J, Hukkanen M, Riveros‐Moreno V, Esquerda JE. Prevention by lamotrigine, MK‐801 and N omega‐nitro‐L‐arginine methyl ester of motoneuron cell death after neonatal axotomy. Neuroscience. 1996;71(2):313‐325. doi: 10.1016/0306-4522(95)00461-0 [DOI] [PubMed] [Google Scholar]
- 208. Church J, Lodge D, Berry SC. Differential effects of dextrorphan and levorphanol on the excitation of rat spinal neurons by amino acids. Eur J Pharmacol. 1985;111(2):185‐190. doi: 10.1016/0014-2999(85)90755-1 [DOI] [PubMed] [Google Scholar]
- 209. Choi D. Ionic dependence of glutamate neurotoxicity. J Neurosci. 1987;7(2):369‐379. doi: 10.1523/JNEUROSCI.07-02-00369.1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Peters S, Choi DW. Quinolinate is a weak excitant of cortical neurons in cell culture. Brain Res. 1987;420(1):1‐10. doi: 10.1016/0006-8993(87)90233-2 [DOI] [PubMed] [Google Scholar]
- 211. Rothstein JD, Patel S, Regan MR, et al. Beta‐lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433(7021):73‐77. doi: 10.1038/nature03180 [DOI] [PubMed] [Google Scholar]
- 212. Greenberg G. Calcium channels and neuromuscular disease. Ann Neurol. 1994;35(2):131‐132. doi: 10.1002/ana.410350203 [DOI] [PubMed] [Google Scholar]
- 213. Freedman DD, Waters DD. “Second generation” dihydropyridine calcium antagonists. Greater vascular selectivity and some unique applications. Drugs. 1987;34(5):578‐598. doi: 10.2165/00003495-198734050-00005 [DOI] [PubMed] [Google Scholar]
- 214. Fritz E, Izaurieta P, Weiss A, et al. Mutant SOD1‐expressing astrocytes release toxic factors that trigger motoneuron death by inducing hyperexcitability. J Neurophysiol. 2013;109(11):2803‐2814. doi: 10.1152/jn.00500.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Sugai F, Yamamoto Y, Miyaguchi K, et al. Benefit of valproic acid in suppressing disease progression of ALS model mice. Eur J Neurosci. 2004;20(11):3179‐3183. doi: 10.1111/j.1460-9568.2004.03765.x [DOI] [PubMed] [Google Scholar]
- 216. Leng Y, Liang MH, Ren M, Marinova Z, Leeds P, Chuang DM. Synergistic neuroprotective effects of lithium and valproic acid or other histone deacetylase inhibitors in neurons: roles of glycogen synthase kinase‐3 inhibition. J Neurosci. 2008;28(10):2576‐2588. doi: 10.1523/JNEUROSCI.5467-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Fujimoto T, Sakoda S, Fujimura H, Yanagihara T. Ibudilast, a phosphodiesterase inhibitor, ameliorates experimental autoimmune encephalomyelitis in Dark August rats. J Neuroimmunol. 1999;95(1):35‐42. doi: 10.1016/S0165-5728(98)00251-3 [DOI] [PubMed] [Google Scholar]
- 218. Wakita H, Tomimoto H, Akiguchi I, et al. Ibudilast, a phosphodiesterase inhibitor, protects against white matter damage under chronic cerebral hypoperfusion in the rat. Brain Res. 2003;992(1):53‐59. doi: 10.1016/j.brainres.2003.08.028 [DOI] [PubMed] [Google Scholar]
- 219. Kagitani‐Shimono K, Mohri I, Fujitani Y, et al. Anti‐inflammatory therapy by ibudilast, a phosphodiesterase inhibitor, in demyelination of twitcher, a genetic demyelination model. J Neuroinflammation. 2005;2(1):10‐12. doi: 10.1186/1742-2094-2-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Zhu S, Stavrovskaya IG, Drozda M, et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417(6884):74‐78. doi: 10.1038/417074a [DOI] [PubMed] [Google Scholar]
- 221. Van Den Bosch L, Tilkin P, Lemmens G, Robberecht W. Minocycline delays disease onset and mortality in a transgenic model of ALS. Neuroreport. 2002;13(8):1067‐1070. doi: 10.1097/00001756-200206120-00018 [DOI] [PubMed] [Google Scholar]
- 222. Kriz J, Nguyen MD, Julien JP. Minocycline slows disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2002;10(3):268‐278. doi: 10.1006/nbdi.2002.0487 [DOI] [PubMed] [Google Scholar]
- 223. Potenza RL, De Simone R, Armida M, et al. Fingolimod: a disease‐modifier drug in a mouse model of amyotrophic lateral sclerosis. Neurotherapeutics. 2016;13(4):918‐927. doi: 10.1007/s13311-016-0462-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Noda H, Takeuchi H, Mizuno T, Suzumura A. Fingolimod phosphate promotes the neuroprotective effects of microglia. J Neuroimmunol. 2013;256(1–2):13‐18. doi: 10.1016/j.jneuroim.2012.12.005 [DOI] [PubMed] [Google Scholar]
- 225. Mizwicki MT, Menegaz D, Zhang J, et al. Genomic and nongenomic signaling induced by 1α,25(OH)2‐vitamin D3 promotes the recovery of amyloid‐β phagocytosis by Alzheimer's disease macrophages. J Alzheimers Dis. 2012;29(1):51‐62. doi: 10.3233/JAD-2012-110560 [DOI] [PubMed] [Google Scholar]
- 226. Sendtner M, Carrol P, Holtmann B, Hughes RA, Thoenen H. Ciliary neurotrophic factor. J Neurobiol. 1994;25(11):1436‐1453. doi: 10.1002/neu.480251110 [DOI] [PubMed] [Google Scholar]
- 227. Corse AM, Bilak MM, Bilak SR, Lehar M, Rothstein JD, Kuncl RW. Preclinical testing of neuroprotective neurotrophic factors in a model of chronic motor neuron degeneration. Neurobiol Dis. 1999;6(5):335‐346. doi: 10.1006/nbdi.1999.0253 [DOI] [PubMed] [Google Scholar]
- 228. Kaspar BK, Lladó J, Sherkat N, Rothstein JD, Gage FH. Retrograde viral delivery of IGF‐1 prolongs survival in a mouse ALS model. Science. 2003;301(5634):839‐842. doi: 10.1126/science.1086137 [DOI] [PubMed] [Google Scholar]
- 229. Dodge JC, Haidet AM, Yang W, et al. Delivery of AAV‐IGF‐1 to the CNS extends survival in ALS mice through modification of aberrant glial cell activity. Mol Ther. 2008;16(6):1056‐1064. doi: 10.1038/mt.2008.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Ochs G, Penn RD, York M, et al. A phase I/II trial of recombinant methionyl human brain derived neurotrophic factor administered by intrathecal infusion to patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(3):201‐206. doi: 10.1080/14660820050515197 [DOI] [PubMed] [Google Scholar]
- 231. Koliatsos VE, Clatterbuck RE, Winslow JW, Cayouette MH, Price DL. Evidence that brain‐derived neurotrophic factor is a trophic factor for motor neurons in vivo. Neuron. 1993;10(3):359‐367. doi: 10.1016/0896-6273(93)90326-m [DOI] [PubMed] [Google Scholar]
- 232. Azzouz M, Le T, Ralph GS, et al. Lentivector‐mediated SMN replacement in a mouse model of spinal muscular atrophy. J Clin Invest. 2004;114(12):1726‐1731. doi: 10.1172/JCI22922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Storkebaum E, Lambrechts D, Dewerchin M, et al. Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci. 2005;8(1):85‐92. doi: 10.1038/nn1360 [DOI] [PubMed] [Google Scholar]
- 234. Bros‐Facer V, Krull D, Taylor A, et al. Treatment with an antibody directed against Nogo‐A delays disease progression in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Hum Mol Genet. 2014;23(16):4187‐4200. doi: 10.1093/hmg/ddu136 [DOI] [PubMed] [Google Scholar]
- 235. Duong FHT, Warter JM, Poindron P, Passilly P. Effect of the nonpeptide neurotrophic compound SR 57746A on the phenotypic survival of purified mouse motoneurons. Br J Pharmacol. 1999;128(7):1385‐1392. doi: 10.1038/sj.bjp.0702910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Sadan O, Melamed E, Offen D. Bone‐marrow‐derived mesenchymal stem cell therapy for neurodegenerative diseases. Expert Opin Biol Ther. 2009;9(12):1487‐1497. doi: 10.1517/14712590903321439 [DOI] [PubMed] [Google Scholar]
- 237. Glass JD, Hertzberg VS, Boulis NM, et al. Transplantation of spinal cord‐derived neural stem cells for ALS: analysis of phase 1 and 2 trials. Neurology. 2016;87(4):392‐400. doi: 10.1212/WNL.0000000000002889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Xu L, Yan J, Chen D, et al. Human neural stem cell grafts ameliorate motor neuron disease in SOD‐1 transgenic rats. Transplantation. 2006;82(7):865‐875. doi: 10.1097/01.tp.0000235532.00920.7a [DOI] [PubMed] [Google Scholar]
- 239. Mazzini L, Gelati M, Profico DC, et al. Results from phase I clinical trial with intraspinal injection of neural stem cells in amyotrophic lateral sclerosis: a long‐term outcome. Stem Cells Transl Med. 2019;8(9):887‐897. doi: 10.1002/sctm.18-0154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Zalfa C, Rota Nodari L, Vacchi E, et al. Transplantation of clinical‐grade human neural stem cells reduces neuroinflammation, prolongs survival and delays disease progression in the SOD1 rats. Cell Death Dis. 2019;10(5):1‐15. doi: 10.1038/s41419-019-1582-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Klein SM, Behrstock S, McHugh J, et al. GDNF delivery using human neural progenitor cells in a rat model of ALS. Hum Gene Ther. 2005;16(4):509‐521. doi: 10.1089/hum.2005.16.509 [DOI] [PubMed] [Google Scholar]
- 242. Suzuki M, Tork C, Shelley B, et al. Sexual dimorphism in disease onset and progression of a rat model of ALS. Amyotroph Lateral Scler. 2007;8(1):20‐25. doi: 10.1080/17482960600982447 [DOI] [PubMed] [Google Scholar]
- 243. Gowing G, Shelley B, Staggenborg K, et al. Glial cell line‐derived neurotrophic factor‐secreting human neural progenitors show long‐term survival, maturation into astrocytes, and no tumor formation following transplantation into the spinal cord of immunocompromised rats. Neuroreport. 2014;25(6):367‐372. doi: 10.1097/WNR.0000000000000092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Thomsen GM, Avalos P, Ma AA, et al. Transplantation of neural progenitor cells expressing glial cell line‐derived neurotrophic factor into the motor cortex as a strategy to treat amyotrophic lateral sclerosis. Stem Cells. 2018;36(7):1122‐1131. doi: 10.1002/stem.2825 [DOI] [PubMed] [Google Scholar]
- 245. Self W, Schoch K, Alex J, et al. Protein production is an early biomarker for RNA‐targeted therapies. Ann Clin Transl Neurol. 2018;5(12):1492‐1504. doi: 10.1002/acn3.657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Cudkowicz ME, van den Berg LH, Shefner JM, et al. Dexpramipexole versus placebo for patients with amyotrophic lateral sclerosis (EMPOWER): a randomised, double‐blind, phase 3 trial. Lancet Neurol. 2013;12(11):1059‐1067. doi: 10.1016/S1474-4422(13)70221-7 [DOI] [PubMed] [Google Scholar]
- 247. Danzeisen R, Schwalenstoecker B, Gillardon F, et al. Targeted antioxidative and neuroprotective properties of the dopamine agonist pramipexole and its nondopaminergic enantiomer SND919CL2x [(+)2‐amino‐4,5,6,7‐tetrahydro‐6‐Lpropylamino‐benzathiazole dihydrochloride]. J Pharmacol Exp Ther. 2006;316(1):189‐199. doi: 10.1124/jpet.105.092312 [DOI] [PubMed] [Google Scholar]
- 248. Vieira FG, LaDow E, Moreno A, et al. Dexpramipexole is ineffective in two models of ALS related neurodegeneration. PLoS One. 2014;9(12):e91608. doi: 10.1371/journal.pone.0091608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Ito S, Izumi Y, Niidome T, Ono Y. Methylcobalamin prevents mutant superoxide dismutase‐1‐induced motor neuron death in vitro. Neuroreport. 2017;28(2):101‐107. doi: 10.1097/WNR.0000000000000716 [DOI] [PubMed] [Google Scholar]
- 250. Ito H, Wate R, Zhang J, et al. Treatment with edaravone, initiated at symptom onset, slows motor decline and decreases SOD1 deposition in ALS mice. Exp Neurol. 2008;213(2):448‐455. doi: 10.1016/j.expneurol.2008.07.017 [DOI] [PubMed] [Google Scholar]
- 251. Ikeda K, Iwasaki Y. Edaravone, a free radical scavenger, delayed symptomatic and pathological progression of motor neuron disease in the wobbler mouse. PLoS One. 2015;10(10):e0140316. doi: 10.1371/journal.pone.0140316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Cudkowicz ME, Shefner JM, Simpson E, et al. Arimoclomol at dosages up to 300 mg/day is well tolerated and safe in amyotrophic lateral sclerosis. Muscle Nerve. 2008;38(1):837‐844. doi: 10.1002/mus.21059 [DOI] [PubMed] [Google Scholar]
- 253. Kieran D, Sebastia J, Greenway MJ, et al. Control of motoneuron survival by angiogenin. J Neurosci. 2008;28(52):14056‐14061. doi: 10.1523/JNEUROSCI.3399-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Shefner JM, Watson ML, Meng L, Wolff AA, Neals/Cytokinetics STUDY Team . A study to evaluate safety and tolerability of repeated doses of tirasemtiv in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(7–8):574‐581. doi: 10.3109/21678421.2013.822517 [DOI] [PubMed] [Google Scholar]
- 255. Shefner JM, Wolff AA, Meng L, et al. A randomized, placebo‐controlled, double‐blind phase IIb trial evaluating the safety and efficacy of tirasemtiv in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2016;17(5–6):426‐435. doi: 10.3109/21678421.2016.1148169 [DOI] [PubMed] [Google Scholar]
- 256. Al‐Chalabi A, Shaw P, Leigh PN, et al. Oral levosimendan in amyotrophic lateral sclerosis: a phase II multicentre, randomised, double‐blind, placebo‐controlled trial. J Neurol Neurosurg Psychiatry. 2019;90(10):1165‐1170. doi: 10.1136/jnnp-2018-320288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Shefner JM, Andrews JA, Genge A, et al. A phase 2, double‐blind, randomized, dose‐ranging trial of reldesemtiv in patients with ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2021;22(3–4):287‐299. doi: 10.1080/21678421.2020.1822410 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Clinical trials for targeted ALS pathways.