

Abstract
Four new pentadentate N5-donor ligands, [N-(1-methyl-2-imidazolyl)methyl-N-(2-pyridyl)-methyl-N-(bis-2-pyridylmethyl)-amine] (L1), [N-bis(1-methyl-2-imidazolyl)methyl-N-(bis-2-pyridylmethyl)amine] (L2), (N-(isoquinolin-3-ylmethyl)-1,1-di(pyridin-2-yl)-N-(pyridin-2-ylmethyl)methanamine (L3), and N,N-bis(isoquinolin-3-ylmethyl)-1,1-di(pyridin-2-yl)methanamine (L4), have been synthesized based on the N4Py ligand framework, where one or two pyridyl arms of the N4Py parent are replaced by (N-methyl)imidazolyl or N-(isoquinolin-3-ylmethyl) moieties. Using these four pentadentate ligands, the mononuclear complexes [FeII(CH3CN)(L1)]2+ (1a), [FeII(CH3CN)(L2)]2+ (2a), [FeII(CH3CN)(L3)]2+ (3a), and [FeII(CH3CN)(L4)]2+ (4a) have been synthesized and characterized. The half-wave potentials (E1/2) of the complexes become more positive in the order: 2a < 1a < 4a ≤ 3a ≤ [Fe(N4Py)(CH3CN)]2+. The order of redox potentials correlates well with the Fe–Namine distances observed by crystallography, which are 2a > 1a ≥ 4a > 3a ≥ [Fe(N4Py)(CH3CN)]2+. The corresponding ferryl complexes [FeIV(O)(L1)]2+ (1b), [FeIV(O)(L2)]2+ (2b), [FeIV(O)(L3)]2+ (3b), and [FeIV(O)(L4)]2+ (4b) were prepared by the reaction of the ferrous complexes with isopropyl 2-iodoxybenzoate (IBX ester) in acetonitrile. The greenish complexes 3b and 4b were also isolated in the solid state by the reaction of the ferrous complexes in CH3CN with ceric ammonium nitrate in water. Mössbauer spectroscopy and magnetic measurements (using superconducting quantum interference device) show that the four complexes 1b, 2b, 3b, and 4b are low-spin (S = 1) FeIV=O complexes. UV/vis spectra of the four FeIV=O complexes in acetonitrile show typical long-wavelength absorptions of around 700 nm, which are expected for FeIV=O complexes with N4Py-type ligands. The wavelengths of these absorptions decrease in the following order: 721 nm (2b) > 706 nm (1b) > 696 nm (4b) > 695 nm (3b) = 695 nm ([FeIV(O) (N4Py)]2+), indicating that the replacement of the pyridyl arms with (N-methyl) imidazolyl moieties makes L1 and L2 exert weaker ligand fields than the parent N4Py ligand, while the ligand field strengths of L3 and L4 are similar to the N4Py parent despite the replacement of the pyridyl arms with N-(isoquinolin-3-ylmethyl) moieties. Consequently, complexes 1b and 2b tend to be less stable than the parent [FeIV(O)(N4Py)]2+ complex: the half-life sequence at room temperature is 1.67 h (2b) < 16 h (1b) < 45 h (4b) < 63 h (3b) ≈ 60 h ([FeIV(O)(N4Py)]2+). Compared to the parent complex, 1b and 2b exhibit enhanced reactivity in both the oxidation of thioanisole in the oxygen atom transfer (OAT) reaction and the oxygenation of C–H bonds of aromatic and aliphatic substrates, presumed to occur via an oxygen rebound process. Furthermore, the second-order rate constants for hydrogen atom transfer (HAT) reactions affected by the ferryl complexes can be directly related to the C–H bond dissociation energies of a range of substrates that have been studied. Using either IBX ester or H2O2 as an oxidant, all four new FeII complexes display good performance in catalytic reactions involving both HAT and OAT reactions.
Short abstract
Four FeIV=O complexes of pentadentate ligands based on the N4Py ligand framework have been synthesized and characterized. Their abilities to act as reagents for oxygen atom and hydrogen atom transfer reactions have been investigated.
Introduction
Oxidations of C–H bonds via hydrogen and/or oxygen atom transfer (HAT/OAT) are very important processes in biology. It has been conclusively demonstrated that the active intermediates in several nonheme iron enzymes involve high-valent FeIV=O moieties. For example, mononuclear nonheme FeIV=O cores have been detected in the catalytic cycles of α-ketoglutarate-dependent oxygenases (taurine dioxygenase1 and propyl-4-hydroxylase2), halogenases (cytochrome c3 halogenases3,4 with chlorine or bromine and SyrB2 halogenase5,6), and pterin-dependent hydroxylases (tyrosine7 and phenylalanine hydroxylases8). In order to gain mechanistic insights into their functions in biology, chemists have mounted a significant effort to mimic biological FeIV=O entities. Thus, several FeIV=O complexes have been synthesized to shed light on the electronic structures and spectroscopic properties of these high-valent iron–oxo intermediates.9−11
Unlike the FeIV=O intermediates found in nonheme enzymes, which have been proven to be in the high-spin (S = 2) state, most of the synthesized complexes are found to be in the low-spin (S = 1) state, with only a few exceptions being in the high-spin state.9,11,12 Density functional theory (DFT) calculations indicate that high-spin S = 2 FeIV=O complexes should be more reactive than the corresponding low-spin versions in hydrogen atom transfer,13,14 although it has been demonstrated that access to the S = 2 spin state is not required for HAT reactivity.15 For pseudo-octahedral complexes, a small energy gap between the dx2-y2 and dxy orbitals in the equatorial plane may facilitate the formation of a high-spin complex; this might be achieved by introducing weak-field equatorial donor entities in pentadentate ligands.9,16 A number of studies have shown that weakening of the ligand field will also enhance the reactivities of the pseudo-octahedral low-spin FeIV=O complexes (vide infra),17 which may be rationalized by so-called two-state reactivity, i.e., spin-crossover to a high-spin state in the transition state of an HAT reaction.18−20 Furthermore, recent investigations have described a number of highly reactive ferryl complexes.21,22
Among synthesized FeIV=O complexes, [FeIV(O)(N4Py)]2+ has been shown to exhibit powerful oxidative reactivity toward both alkanes and arenes, enabling it to cleave strong C–H bonds while possessing considerable thermal stability that makes it possible to study the complex in detail.23 Considering this, many derivatives of the N4Py ligand have been synthesized24 to investigate the influence of ligand modifications on the steric and electronic properties of their iron complexes and (thus) the reactivities of the FeIV=O complexes.25 It is apparent that both structural (steric) and electronic factors influence the reactivities of such ferryl–oxo complexes, but it remains difficult to predict such reactivities.
In a previous study, we explored the chemistry of the FeIV=O complexes of (N-methyl)benzimidazolyl derivatives of N4Py, viz., N3Py-(NMB) and N2Py2B (Figure 1).26 The complexes [FeIV(O)(L)]2+ (L = N3Py-(NMB), N2Py2B) were found to be considerably more reactive than the parent [FeIV(O)(N4Py)]2+ complex, with each replacement of a pyridyl unit with an (N-methyl)benzimidazolyl unit leading to an increase in HAT rate by almost 1 order of magnitude for a range of benchmark substrates. At the same time, the half-lives of the ferryl–oxo complexes were found to decrease for each (N-methyl)benzimidazolyl unit introduced so that the half-life at room temperature of [FeIV(O)(N2Py2B)]2+ was found to be only ca. 2.5 h, as compared to 60 h for the parent complex. The reactivities could also be correlated to a weakening of the equatorial field exerted by the pentadentate ligands, as manifested by the frequency of a long-wavelength absorption in the vicinity of 700 nm that is characteristic for this kind of complex (vide infra). Despite the relatively short half-life of [FeIV(O)(N2Py2B)]2+ and related [FeIV(O)(N2Py2Qn)]2+ (cf. Figure 1), Que and co-workers managed to crystallize and determine the solid-state structure of these two complexes.25 The crystal structures illustrated how the ligand modifications influence the FeIV=O environment relative to that of the parent N4Py complex. For example, the steric bulk of the quinolyl donors causes a tilt of the FeIV=O unit away from a linear N–FeIV=O arrangement by 10°, and the (N-methyl)benzimidazolyl moieties in [FeIV(O)(N2Py2B)]2+ also cause a relatively minor tilt of 3°. It was also concluded that longer Fe–N distances correlate linearly with log k2′ values for O- and H- atom transfer rates and that the electrophilicity of the FeIV=O center can be increased by the weakening of the ligand field. The steric effect on ligand coordination, and thus effective ligand field, has been explored in several other studies on the FeIV=O complexes of ligands based on the N4Py framework.24,27,28
Figure 1.

Structures of previously synthesized and new pentadentate ligands (L1–L4).
We decided to prepare new pentadentate ligands based on the same framework, where the steric influence of the ligand moieties is expected to be minimal, and the effective ligand field therefore is expected to primarily be related to the electronic properties of the various N-donor entities. Based on the ligands N2Py2Qn and N2Py2B, four new ligands (L1–L4, Figure 1) were designed and synthesized. Because of the sizes and orientations of the imidazolyl and isoquinolinyl moieties in L1–L4, it was expected that their coordination to a metal ion will lead to relatively “open” structures, where the steric influence of the ligands is minor and similar to that of the N4Py ligand, and this was indeed found to be the case (vide infra). The mononuclear FeII complexes of these four new pentadentate ligands have been synthesized, and the corresponding FeIV=O complexes have also been prepared and characterized. The reactivities of the new FeIV=O units toward external substrates have been investigated and are discussed below.
Results and Discussion
Synthesis and Characterization of Ligands
The syntheses of the four ligands are summarized in Scheme 1. Syntheses of L1 and L2 were achieved by a previously reported method,29 involving reductive amination to achieve condensation of 1-methyl-1H-imidazole-2-carbaldehyde (B, Scheme 1) with the secondary amine of N-[di(2-pyridinyl)methyl]-N-(2-pyridinylmethyl)methylamine (N3Py) to form L1, or the primary amine of bis(2-pyridyl)methylamine (A, Scheme 1) to form L2. Similarly, ligands L3 and L4 were synthesized by reaction of N3Py or bis(2-pyridyl)methylamine with one or two equivalents of 3-(chloromethyl)isoquinoline (C, Scheme 1) in the presence of K2CO3 (NMR Figures S1–S7, Supporting Information).
Scheme 1. Schematic of the Synthetic Routes for Ligands L1–L4.

Synthesis and Characterization of FeII Complexes
The ferrous complexes [FeII(CH3CN)(L1)](ClO4)2 (1a·(ClO4)2), [FeII(CH3CN)(L2)](ClO4)2 (2a·(ClO4)2), [FeII(CH3CN)(L3)](ClO4)2 (3a·(ClO4)2), and [FeII(CH3CN)(L4)](ClO4)2 (4a·(ClO4)2) (Figure 2) were prepared by reaction of the relevant ligand with Fe(ClO4)2·xH2O in acetonitrile solvent in a glovebox. The triflate and tetrafluoroborate salts [FeII(CH3CN)(L1)](OTf)2 (1a·(OTf)2), [FeII(CH3CN)(L2)](OTf)2 (2a·(OTf)2), [FeII(L3)(CH3CN)](OTf)2 (3a·(OTf)2), [FeII(CH3CN)(L4)](OTf)2 (4a·(OTf)2), and [FeII(CH3CN)(L4)](BF4)2 (4a·(BF4)2) were also prepared from the appropriate starting materials. The high-resolution mass spectra of 1a–4a were all consistent with those of M-CH3CN ions. Both [FeII(Ln)X]+ and [FeII(Ln)]2+ (n = 1–4, X = ClO4– or OTf–) peaks were observed (cf. the Experimental Section and Supporting Information). A ferric oxo-bridged dinuclear complex [FeIII2(L2)2(μ-O)](OTf)2 (5·(OTf)2) was obtained by the reaction of L2 with Fe(OTf)2·2MeCN under an ambient atmosphere and was identified via single-crystal X-ray diffraction (cf. the Experimental Section and Supporting Information).
Figure 2.

Schematic drawings of the cationic FeII complexes 1a–4a.
NMR Spectroscopy
The proton nuclear magnetic resonance (1H NMR) spectrum of 1a·(ClO4)2 in CD3CN indicates a low-spin state at room temperature, while the analogous spectrum in dimethyl sulfoxide-d6 (DMSO-d6) shows the presence of a mixture of high-spin and low-spin states for the same complex. The 1H NMR spectra of 2a·(ClO4)2 show the presence of a high-spin complex in both CD3CN and DMSO-d6 at room temperature. The 1H NMR spectra furthermore indicate that 3a·(ClO4)2 and 4a·(ClO4)2 are low-spin in CD3CN solution at ambient temperature; however, the recordings of the corresponding spectra of these two complexes in DMSO-d6 show the presence of high-spin complexes (see Figures S8–S15, Supporting Information). We anticipate that the spectra recorded in DMSO-d6 contain DMSO coordinated via its oxygen atom to the iron ion. The σ-donor DMSO ligand exerts a weaker field than acetonitrile, which is a σ-donor and π-acceptor.
Mössbauer Spectroscopy and Magnetic Measurements
The 57Fe-enriched samples [57FeII(CH3CN)(LX)] (OTf)2 (X = 1–4) were prepared from 57Fe(OTf)2. Zero-field Mössbauer spectra of solid samples of these 57Fe-enriched complexes were recorded at 80 K (Figure S16). The isomer shift (δ) and quadrupole splitting (ΔEQ) values are listed in Table 1. These values confirm the low-spin state of the FeII ions in 1a, 3a, and 4a; however, a mixture of high- and low-spin states for 2a is noted. As shown in Figure 3, the zero-field Mössbauer spectrum of 2a·(OTf)2 at 80 K can be fitted to the presence of a low-spin FeII main species (82%) and a second species (18%) containing FeII in the high-spin state. Mössbauer measurements at different temperatures revealed that the fraction of FeII in the high-spin state increases from 18% at 80 K to 35% at 295 K. It may be concluded that an incomplete spin-crossover transition occurs in the solid sample.
Table 1. Mössbauer Parameters for the Triflate Salts of Solid 1a–4a.
| complex | relative area (%) | δ (mm s–1) | ΔEQ (mm s–1) |
|---|---|---|---|
| 1a (80 K) | 100 | 0.43 | 0.17 |
| 2a (80 K) | 82 | 0.45 | 0.24 |
| 18 | 1.19 | 2.88 | |
| 2a (295 K) | 65 | 0.37 | 0.23 |
| 35 | 0.98 | 1.76 | |
| 3a (80 K) | 94 | 0.39 | 0.24 |
| 4a (80 K) | 100 | 0.38 | 0.36 |
Figure 3.

Zero-field Mössbauer spectra of the solid sample of complex 2a·(OTf)2 at 80 K (left) and 295 K (right).
Magnetic measurements [using a superconducting quantum interference device (SQUID)] and the Mössbauer spectrum of 2a·(OTf)2 in frozen acetonitrile were also recorded to investigate the spin-crossover properties in solution. As shown in Figure 4, 2a is predominantly diamagnetic (i.e., low-spin) at temperatures below 200 K in CH3CN. Above 200 K, the fraction of FeII in the high-spin state increases, which is attributed to the spin-crossover. At 350 K, about 50% of the sample is in the high-spin state. The Mössbauer spectrum at 80 K, with parameters for the main signal δ = 0.43 mm s–1 and ΔEQ = 0.22 mm s–1 (Figure S17), confirms that 2a is also predominantly in the low-spin state (92%) in frozen solution with only a small fraction in the high-spin FeII form (8%, δ = 1.20 mm s–1 and ΔEQ = 3.12 mm s–1). The Mössbauer spectra in both solid state and frozen acetonitrile solution and the magnetic measurements are all in agreement, indicating that the spin-crossover takes place for 2a·(OTf)2.
Figure 4.
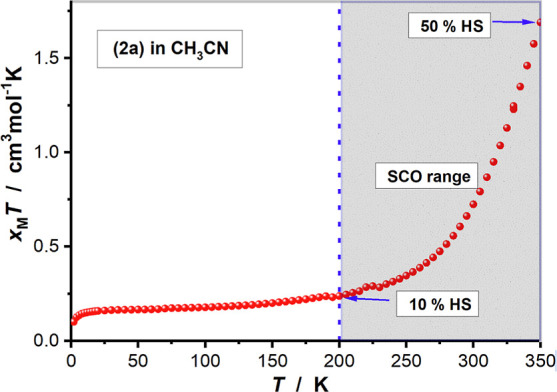
Plot of χMT versus T for 2a·(OTf)2 in acetonitrile (SCO = spin-crossover, HS = high-spin).
Absorption Spectroscopy
The UV/vis spectra of ligands L1–L4 in an acetonitrile solution show high-intensity bands in the UV region. Upon coordination to FeII, two new charge transfer (CT) bands appear in the visible region for all of the complexes/ligands (Figure 5). These CT bands can be assigned as metal-to-ligand charge transfer (MLCT) bands arising from electron transfer from the FeII t2g orbitals to the π* orbitals of the ligand. Complex 2a·(ClO4)2 exhibits significantly weaker absorbance (smaller observed extinction coefficients) relative to those of the other three complexes. The absorbance values for 2a·(ClO4)2 in acetonitrile increase when the temperature is lowered from 298 to 235 K (Figure S18). These observations can be rationalized by 2a·(ClO4)2 undergoing a spin-crossover, as discussed above, i.e,. the complex exists as a mixture of high-spin and low-spin forms in solution and the population of the low-spin state increases as the temperature decreases, in agreement with the SQUID and Mössbauer results for 2a·(OTf)2 in CH3CN (vide supra). Variable-temperature NMR spectroscopy of 2a·(ClO4)2 (Figure S19) also confirmed the spin-crossover of the complex.
Figure 5.
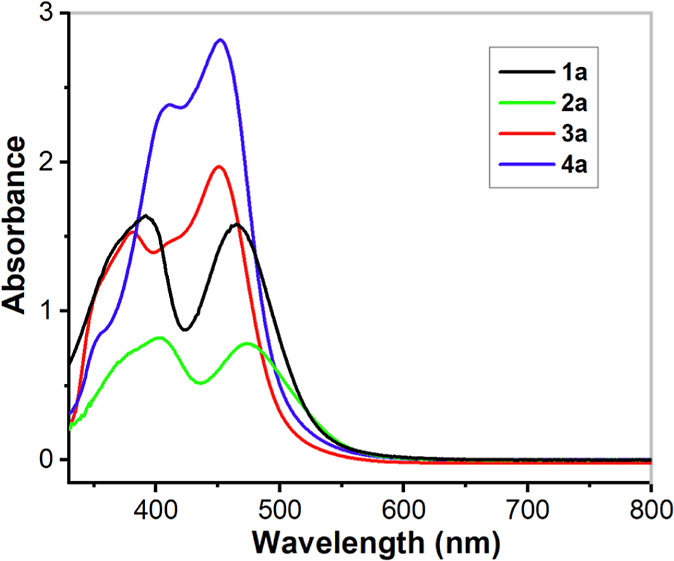
UV/vis spectra of complexes 1a·(ClO4)2, 2a·(ClO4)2, 3a·(ClO4)2, and 4a·(ClO4)2 (0.25 mM) in acetonitrile at 298 K. CT bands appear at λmax = 393 nm (ε ≈ 6280 M–1 cm–1) and 466 nm (ε ≈ 6000 M–1 cm–1) for 1a, at λmax = 380 nm (ε ≈ 6080 M–1 cm–1) and 451 nm (ε ≈ 7880 M–1 cm–1) for 3a, at λmax = 410 nm (ε ≈ 9560 M–1 cm–1) and 452 nm (ε ≈ 11,280 M–1 cm–1) for 4a, while complex 2a shows weaker absorbance and the CT bands appear at λmax = 404 nm (ε ≈ 3300 M–1 cm–1) and 475 nm (ε ≈ 3160 M–1 cm–1).
On the other hand, complexes 3a and 4a, which contain isoquinoline substituents in the framework of the pentadentate ligands, only show low-spin FeII ions. This observation is in contrast to that for the analogous quinoline complexes, [FeII(CH3CN)(N3Py-Qn)]2+ and [FeII(CH3CN)(N2Py2Qn)]2+, which are found to be in a mixture of high- and low-spin states.28 Furthermore, the absorption spectra of 3a and 4a differ from those of other FeII complexes of N4Py and its derivatives in that the longer-wavelength CT absorption (around 450 nm) exhibits a larger extinction coefficient than that of the shorter-wavelength CT band. This phenomenon is clearly attributable to the presence of the isoquinoline substituents in ligands L3 and L4 and suggests that the lowering of the π* orbitals on the isoquinoline substituents leads to enhanced mixing between the metal d(π) and ligand (π*) orbitals, therefore making it a better π-acceptor than pyridine or even quinoline. In order to elucidate how the isoquinoline substituents affect the absorptions, a time-dependent DFT (TD-DFT) analysis of 4a was undertaken, and a full account of this analysis is given in the Supporting Information (Table S1).
Figure 6 shows the calculated frontier orbitals for 4a. The highest occupied molecular orbital (HOMO), HOMO–1, and HOMO–2 are largely derived from the dxy, dxz, and dyz orbitals, respectively, of the FeII center. At the same time, HOMO–3 is a π orbital spread over the isoquinoline moiety, with a small contribution of the dxz orbital of the FeII metal. The lowest unoccupied molecular orbital (LUMO) and LUMO+2 orbitals are π* orbitals located over the isoquinoline moiety, and LUMO+1 is a π* orbital spread over the coordinated pyridines. The LUMO+9 and LUMO+10 are the empty dz2 and dx2–y2, respectively.
Figure 6.
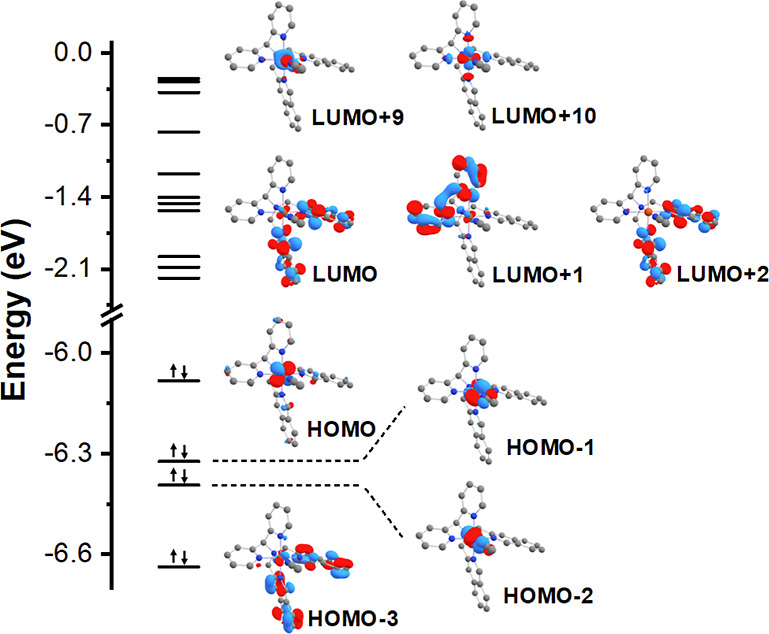
Calculated frontier orbitals for complex 4a within the PBE0/Def2-TZVP(-f) level of theory.
Transitions involving mostly the frontier orbitals described above are located in the 400–500 nm region. The transitions with higher oscillator strength are MLCT transitions from the dxz orbital of the FeII center to both the isoquinoline and pyridine moieties in the lower energy band (S6, f = 0.2326) and only to the isoquinoline in the higher energy band (S11, f = 0.1375). As the acceptor orbital is spread through an extensive π system, it increases the transition dipole moment (proportional to the oscillator strength), and the intensity of this transition is therefore shifted to higher values than usually found for FeII -pyridine systems. The remaining transitions in this region are also MLCT transitions but with a small contribution of d–d transitions involving the FeII center. Figure 7 shows a fit of the calculated transitions in the 400–500 nm region to the observed spectrum.
Figure 7.
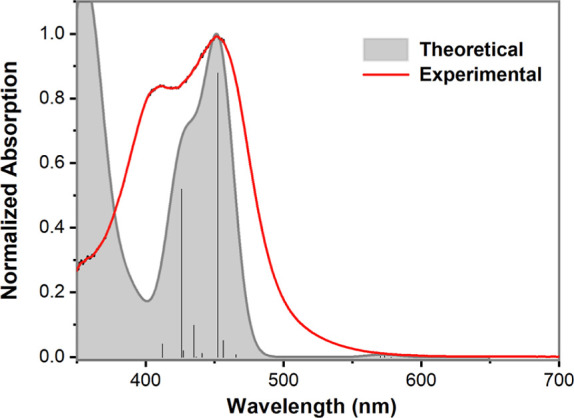
Experimental absorption spectrum of complex 4a in CH3CN and the theoretical absorption spectrum convoluted with Gaussians of 30 nm width. The theoretical spectrum was shifted by 64 nm (0.45 eV) from 381 nm (3.25 eV) to 445 nm (2.80 eV) so that the maximum of the first absorption matches with the one observed experimentally.
For comparison, the same TD-DFT analysis was also performed for the analogous quinoline-based complex [FeII(N2Py2Qn)(CH3CN)]2+. The frontier orbitals are roughly equivalent to those found for complex 4a (cf. Figure S20). For [FeII(N2Py2Qn)(CH3CN)]2+, two prominent bands that are similar to those of 1a/2a are observed. The first, at lower energy, is mainly related to MLCT from the FeII center to the quinoline moieties. In this case, both LUMO and LUMO+1 are centered on the quinoline moieties. Therefore, the transition is not spread through an extensive π system, and lower oscillator strength values are observed for these transitions than that for the isoquinoline complex. The second absorption band is mainly an MLCT transition from the FeII center to the pyridine moieties.
Crystal and Molecular Structures of Complexes 1a·(OTf)2, 2a·(ClO4)2, 3a·(OTf)2, 4a·(BF4)2, and 5·(OTf)2.
It was possible to grow single crystals suitable for X-ray diffraction of the perchlorate salt of 2a, trifluoromethanesulfonate salts of 1a, 3a, and 5, and tetrafluoroborate salt of 4a, and their crystal structures were determined (Tables S2–S6 and S8). The molecular structures of complexes 1a–4a are shown in Figure 8, and selected bond distances and bond angles are collated in Table 2. The crystal structures are similar to that of the “parent” complex [FeII(N4Py)(CH3CN)](ClO4)230 and show that the pentadentate ligands coordinate as envisaged, with the sixth coordination site at the iron ion being occupied by an acetonitrile molecule (solvent).
Figure 8.

Mercury plots of the molecular structures of the cations of 1a·(OTf)2, 2a·(ClO4)2, 3a·(OTf)2, and 4a·(BF4)2 showing the atom labeling scheme. Thermal ellipsoids are plotted at 30% probability; hydrogen atoms have been omitted for clarity.
Table 2. Selected Bond Distances (Å) and Bond Angles (deg) for FeII Complexes 1a–4a, [FeII(N2Py2B)(CH3CN)](ClO4)2,26 [FeII(N4Py)(CH3CN)](ClO4)2,30 and [FeII(N2Py2Qn)(CH3CN)](OTf)228,32a.
| 1a | 2a | 3a | 4a | [FeII(N4Py) (CH3CN)]2+ | [FeII(N2Py2B) (CH3CN)]2+ | [FeII(N2Py2Qn) (CH3CN)]2+ | |
|---|---|---|---|---|---|---|---|
| Fe–N (Å) | 2.049(18) N2A | 1.966(3) N1 | 1.963(2) N2 | 1.979(6) N1 | 1.976(3) N2 | 1.964(3) N1 | 2.103(5) N2 |
| 2.039(19) N2B | 1.967(3) N1B | ||||||
| Fe–N (Å) | 1.875(19) N4A | 1.956(2) N4 | 1.964(3) N3 | 1.971(5) N2 | 1.967(3) N3 | 1.983(3) N3 | 2.092(5) N3 |
| 1.87(2) N4B | 1.959(2) N4B | ||||||
| Fe–N (Å) | 1.967(3) N5 | 1.957(2) N5 | 1.970(2) N4 | 1.990(5) N4 | 1.968(3) N4 | 1.979(2) N4 | 2.019(5) N4 |
| 1.962(3) N5B | |||||||
| Fe–N (Å) | 1.964(3) N6 | 1.963(2) N6 | 1.953(3) N5 | 1.980(5) N5 | 1.975(3) N5 | 1.983(3) N2 | 2.010 (5) N5 |
| 1.968(3) N6B | |||||||
| Fe–NCH3CN(Å) | 1.924(3) N7 | 1.920(2) N8 | 1.919(3) N6 | 1.942(6) N6 | 1.915(3) N6 | 1.901(3) N6 | 1.951(6) N1 |
| 1.920(2) N8B | |||||||
| Fe–Namine (Å) | 1.987(3) N1 | 2.013(2) N3 | 1.965(2) N1 | 1.982(5) N3 | 1.961(3) N1 | 2.028(3) N5 | 2.017(5) N6 |
| 2.014(2) N3B | |||||||
| Fe–Nequatorial (avg.) (Å) | 1.964 A | 1.9605 | 1.9625 | 1.980(7) | 1.971 | 1.977 | 2.056 |
| 1.96 B | 1.964 B | ||||||
| Fe–Npyridine (avg.) (Å) | 1.935 A | 1.9565 | 1.962 | 1.980(8) | 1.971 | 1.974 | 2.0145 |
| 1.934 B | 1.9605 B | ||||||
| Fe–NNMI/IQ/NMB/Qn (avg.) | 2.049(18) | 1.9645 | 1.963(2) | 1.975(6) | 1.981 | 2.0975 | |
| 2.039(19) | 1.9675 B | ||||||
| Angleamine-Fe-CH3CN (deg) | 179.16(12) | 179.48(10) | 177.74(11) | 176.7(2) | 177.3 | 175.7 | 166.7 |
| 179.05(11) |
NMI: (N-methyl)imidazolyl moiety. IQ: N-(isoquinolin-3-ylmethyl) moiety.
The Fe–N bond lengths of the equatorial plane observed for the complexes vary in the range of 1.9–2.0 Å, which is in agreement with those observed for [FeII(N4Py)(CH3CN)](ClO4)230 and some previously reported related FeII complexes.17,18 There are very slight shortenings of the Fe–N bond distances in the equatorial plane by approximately 0.01 Å for both 1·(ClO4)2 and 2·(ClO4)2 relative to the parent complex, while the axial Fe–N bond lengths are slightly longer than those for [FeII(N4Py)(CH3CN)](ClO4)2. The replacement of two pyridyl moieties with (N-methyl)imidazolyl moieties in 2a·(ClO4)2 results in a lengthening of the Fe–Namine bond distance, trans to the acetonitrile ligand, by approximately 0.05 Å. Similar axial elongations have been observed for the Mn(II) analogue of 2a(31) and for the complex [FeII(N2Py2B)(CH3CN)]2+26 (cf. Figure 1 and Table 2). The tilt angles for the axial positions (Namine–Fe–NCH3CN) are small for all four complexes — 4a·(BF4)2 (3.30°) > [FeII(N4Py)(CH3CN)](ClO4)2 (2.68°) > 3a·(OTf)2 (2.26°) > 1a·(OTf)2 (0.84°) > 2a·(ClO4)2 (0.52° or 0.95°) — indicating very little steric interaction between the equatorial donor groups and the axial acetonitrile ligands. A comparison between different Fe–N bond distances in 1a–4a, [FeII(N2Py2B)(CH3CN)](ClO4)2,26 [FeII(N4Py)(CH3CN)](ClO4)2, and [FeII(N2Py2Qn)](OTf)2 is shown in Table 2.
Cyclic Voltammetry
Cyclic voltammetry (CV) in acetonitrile was used to measure the half-wave potentials (E1/2) for the FeII complexes. The cyclic voltammograms for 1a–4a display a single reversible one-electron oxidation wave with peak separations comparable to that of ferrocene added to the same solution (Table 3 and Figure 9). Peak heights are linearly dependent on the square root of the scan rate examined from 5 to 3200 mV/s, as expected for Nernstian behavior (Figures S21–S24 in the Supporting Information.) The peaks are best assigned to the FeIII/FeII redox couple. Irreversible reduction waves were observed below −2.0 V for all complexes (Figure S26.) Scanning negative produced new small oxidation waves below 0 V even if no reduction peak was observed (See Figure S27 for an example.)
Table 3. CV Data for Complexes 1a–4a and [FeII(N4Py)(CH3CN)]2+.
| complex | E1/2 (V)a | ΔE (V) at scan rate of 100 mV/sb |
|---|---|---|
| 1a | 0.452 | 0.063 |
| 2a | 0.298 | 0.085 |
| 3a | 0.609 | 0.075 |
| 4a | 0.596 | 0.067 |
| [FeII(N4Py)(CH3CN)]2+ | 0.625 | 0.070 |
| Fc/Fc+ | 0 | 0.087 |
Voltages measured relative to the ferrocene ([Fe(C5H5)2])/ferrocenium ([Fe(C5H5)2]+) redox couple (Fc/Fc+). No difference was observed between Pt and glassy carbon working electrodes.
Observed ΔE in 0.10 M (Bu4N)PF6/CH3CN at room temperature, indicating that one electron is reversibly transferred.
Figure 9.

Cyclic voltammograms of 1a–4a and [Fe(N4Py)(CH3CN)]2+ in acetonitrile at 50 mV/s vs Fc/Fc+.
The E1/2 of the complexes become more positive in the following sequence: 2a < 1a < 4a ≤ 3a ≤ [Fe(N4Py)(CH3CN)]2+. These results demonstrate that successive replacement of the pyridyl moieties by (N-methyl)imidazolyl moieties leads to a significant lowering of the potentials for the oxidation of the corresponding FeII complexes, while substitution of pyridyl donor moieties with isoquinolines results in only very minor changes in the redox potentials. In addition, the order of potentials correlates well with the Fe–Namine distances observed by crystallography, which are in the order 2a > 1a ≥ 4a > 3a ≥ [Fe(N4Py)(CH3CN)]2+. In general, strong ligand fields result in higher redox potential values for the FeIII/FeII redox couple. The correlation suggests that the Fe–Namine distance may be used as an indicator of the ligand field strength in these complexes. As discussed above, 2a is expected to be in a mixture of high- and low-spin states in the solid state, and its longer Fe–Namine distance may reflect the population of the dz2 orbital in the high-spin form; the Fe–Namine distance of 2a is ≈ 2.01 Å, while that of the other three (low-spin) complexes is approximately 1.97–1.98 Å.
Generation and Characterization of FeIV=O Complexes
Upon addition of three equivalents of IBX ester as an oxidant, the reddish-brown solutions of the FeII complexes 1a–4a turned pale green (cf. graphical abstract) instantaneously, which is in agreement with the expected color of the corresponding FeIV=O complexes 1b–4b (Figure 10), by analogy with previous studies on FeIV=O complexes based on the N4Py ligand scaffold.24,25 High-resolution mass spectrometry (HRMS) analyses of the pale green solutions confirmed the formation of the FeIV=O complexes. Both [FeIV=O(Ln)X]+ and [FeIV=O(Ln)]2+ (n = 1–4, X = ClO4– or OTf–) peaks were observed (cf. the Experimental Section and Figures S36–S39, Supporting Information).
Figure 10.
Schematic drawings of FeIV=O complexes 1b–4b.
FeIV=O complexes 1b–4b could also be prepared by treating concentrated CH3CN solutions of the corresponding FeII precursors with 4 equiv of ceric ammonium nitrate as the oxidant, in water. Subsequent addition of excess sodium perchlorate to the reaction mixtures resulted in precipitation of the solid ferryl complexes in the case of 3b and 4b.
Mössbauer Spectroscopy and Magnetic Measurements on 1b–4b
Mössbauer spectroscopy was employed to confirm the oxidation states of the in situ generated FeIV=O complexes 1b–4b (Figure 11). The isomer shift and quadrupole splitting values are given in Table 4. The Mössbauer parameters for 1b–4b are very similar to those of previously studied low-spin FeIV=O species,11,25 strongly indicating the presence of low-spin (S = 1) FeIV=O units in all four complexes. The remaining (minor) absorptions are ascribed to FeIII contaminants; their isomer shifts lie in the range 0.43–0.47 mm s–1 with quadropole splittings of 1.27–1.39 mm s–1—parameters that are typical for high-spin ferric complexes. These contaminants may be (high-spin) FeIII–O–FeIII complexes,33,34 such as complex 5.
Figure 11.

Zero-field Mössbauer spectra of FeIV=O complexes 1b–4b in frozen acetonitrile at 80 K.
Table 4. Mössbauer Parameters for In Situ Generated Complexes 1b–4b and Related FeIV=O Complexes.
| complex | δ (mm s–1) | ΔEQ (mm s–1) |
|---|---|---|
| 1b (80 K), 84% | –0.03 | 1.13 |
| 2b (80 K), 77% | –0.03 | 1.38 |
| 3b (80 K), 70% | –0.04 | 0.87 |
| 4b (80 K), 83% | –0.04 | 0.85 |
| [FeIV(O)(N4Py)]2+35 | –0.04 | 0.93 |
| [FeIV(O)(N2Py2B)]2+26 | –0.02 | 1.34 |
| [FeIV(O)(N3Py-(NMB))]2+26 | –0.03 | 1.1 |
| [FeIV(O)(TMC) (CH3CN)]2+36 | 0.14 | 0.78 |
| [FeIV(O)(Bn-tpen)]2+35 | 0.01 | 0.87 |
| [FeIV(O)(N2Py2Qn)]2+32 | 0.03 | 0.56 |
Magnetic measurements at ambient temperature using the Evans method were performed on the four FeIV=O complexes 1b–4b in acetonitrile solution, and the results indicated that all four complexes are in the low-spin (S = 1) state (cf. Figures S40–S42, Supporting Information). In addition, SQUID magnetic susceptibility measurements in the temperature range 80–295 K on solid samples of 3b·(OTf)2 and 4b·(OTf)2 gave χMT values of ca. 0.76 and 0.92 cm3 mol–1K, respectively, slightly lower than that expected for the spin-only value of 1.0 cm3 mol–1K for low-spin (S = 1) FeIV=O complexes (Figure S28, Supporting Information). When the temperature was decreased below 80 K, the χMT value was found to decrease slowly due to the presence of zero-field splitting. Simulation of the magnetic susceptibility data and variable-temperature variable-field measurements according to the spin Hamiltonian in eq 1 leads to the best fit parameters g = 1.77, D = 16.6 cm–1 for 3b·(OTf)2 and g = 1.95, D = 20.9 cm–1 for 4b·(OTf)2 (see Supporting Information for more details); the low χMT and g values should be considered with caution, as they may reflect the presence of diamagnetic impurities.
| 1 |
To gain more insight into the electronic structure and to verify the D value by an independent method, solid 4b·(OTf)2 was investigated by applied magnetic field Mössbauer spectroscopy between 2 and 20 K and up to 7 T magnetic field (Figure 12).
Figure 12.
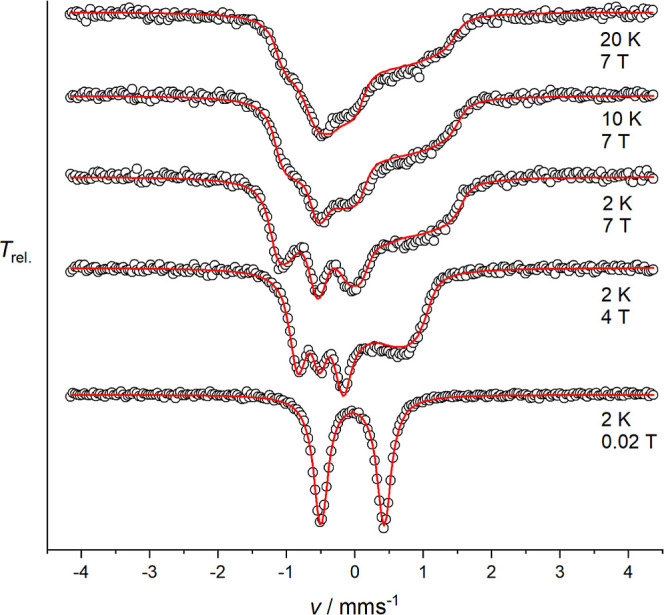
Magnetic Mössbauer spectra of solid 4b·(OTf)2 recorded at the indicated temperatures and fields applied perpendicular to the γ-rays. The red lines represent the best fit to a spin Hamiltonian for S = 1 in the fast spin relaxation regime.
The magnetically split Mössbauer spectra of 4b·(OTf)2 could be well simulated using the spin Hamiltonian for S = 1 with fit parameters D = 20.8 cm–1 (g = 1.95, fixed from magnetic susceptibility data), the magnetic hyperfine coupling constants A/gNμN = [−19.8, −16.8, −2.5] T, as well as δ = −0.04 mm s–1 and ΔEQ = 0.92 mm s–1. Therefore, the sign and the size of the zero-field splitting parameter D are in excellent agreement with those derived from the magnetic susceptibility data. Two large negative values and one small negative value of A/gNμN are in accordance with results previously reported for a tetragonal S = 1 FeIV=O species.37
Absorption Spectroscopy on FeIV=O Complexes
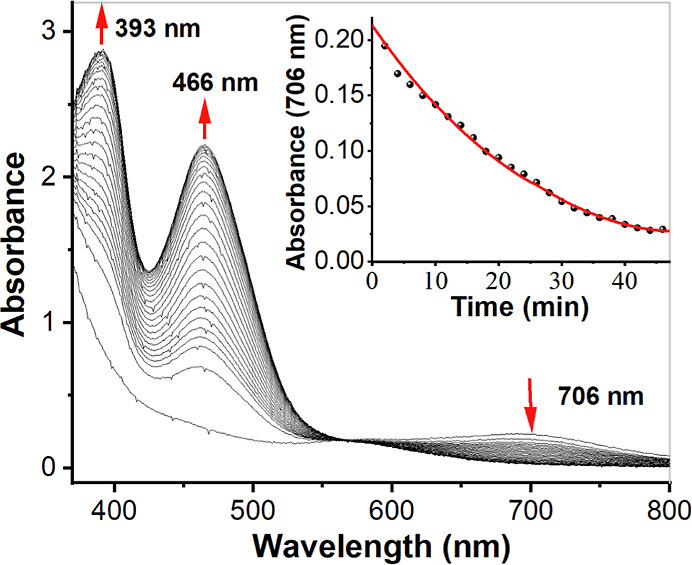
The absorption spectra of FeII complexes 1a–4a have been discussed above. The UV/vis spectra of pale green 1b–4b resemble those of related FeIV=O complexes of pentadentate nitrogen-donor ligands.24,38 In particular, a characteristic weak absorption with λmax in the vicinity of 700 nm was observed for all four complexes (Figure 13). This absorption in the near-IR region has been studied in detail by Solomon, Que and co-workers for the parent complex [FeIV(O)(N4Py)]2+,39 and it is related to transitions originating from the occupied dxy orbital and constitutes an indirect measurement of the strength of the equatorial ligand field. The time courses for decay of the FeIV=O complexes were also monitored at 298 K (Figure 14). As calculated, complex 1b has a t1/2 ≈ 16 h; t1/2 (2b) ≈1.67 h; t1/2 (3b) ≈ 63 h; t1/2 (4b) ≈ 45 h. In Table 5, the half-lives and the characteristic near-IR wavelengths of these four new FeIV=O complexes are compared to some previously reported low-spin FeIV=O complexes bearing pentadentate ligands.
Figure 13.

(a) UV/vis spectra of complexes 1b, 2b, 3b, and 4b (0.5 mM) in acetonitrile recorded at room temperature (298 K). (b) Expansion of the 600–800 nm region of the spectra.
Figure 14.

(a) UV/vis spectra of 0.5 mM complex 1b·(ClO4)2 in acetonitrile. The inset shows the self-decay of complex 1b·(ClO4)2 at room temperature (298 K), as monitored at 706 nm; (b) UV/vis spectra of 0.5 mM complex 2b·(ClO4)2 in acetonitrile. The inset shows the self-decay of complex 2b·(ClO4)2 at room temperature (298 K), as monitored at 721 nm.
Table 5. Optical Spectral Properties and Stabilities (t1/2) of FeIV=O Complexes Supported by Pentadentate Ligands.
| complex | λmax, nm (ε, M–1 cm–1) | t1/2 at 298 K | ref. |
|---|---|---|---|
| 1b | 706 (379) | 16 h | this work |
| 2b | 721 (274) | 1.67 h | this work |
| 3b | 695 (540) | 63 h | this work |
| 4b | 696 (544) | 45 h | this work |
| [FeIV(O)(N4Py)]2+ | 696 (400) | 60 h | (35) |
| [FeIV(O)(Me3cyclam-CH2C(O)NMe2)]2+ | 810 (270) | 5 d | (40) |
| [FeIV(O)(N2Py2B]2+ | 725 (380) | 2.5 h | (26) |
| [FeIV(O)(Bn-tpen)]2+ | 739 (400) | 6 h | (35) |
| [FeIV(O)(TMC-Py)]2+ | 834 (260) | 7 h | (41) |
| [FeIV(O)(N2Py2Qn]2+ | 770 (380) | 2.5 h | (25) |
| [FeIV(O)(N4PyMe2)]2+ | 740 (220) | 14 min | (29) |
As shown in Table 5, both the sequence of λmax in the near IR region −721 nm (2b) > 706 nm (1b) > 696 nm (4b) > 695 nm (3b) = 695 nm ([FeIV(O)(N4Py)]2+) and the sequence of half-lives at room temperature −1.67 h (2b) < 16 h (1b) < 45 h (4b) < 63 h (3b) ≈ 60 h ([FeIV(O)(N4Py)]2+) indicate that the ligand fields exerted by the isoquinoline-substituted ligands L3 and L4 are roughly equivalent to that of N4Py, while the fields of L1 and L2 tend to become progressively weaker as (N-methyl)imidazolyl substituents are introduced in the N4Py ligand scaffold, and are significantly weaker than those of the other two ligands.23
Crystal and Molecular Structure of [FeIV(O)(L4)]2+
Crystals of 4b suitable for X-ray analysis could be obtained by slow evaporation of acetonitrile at 2–5 °C over the course of 8 h,25 and diffraction data were obtained at the MAX IV synchrotron (cf. the Experimental Section). Relevant crystallographic details are summarized in Table S2 (Supporting Information). As shown in Figure 15, the axial positions are occupied by the amine nitrogen (N5) and the oxygen atom (O1) with an almost linear N5–Fe1–O1 angle of 177.61(7)°. The tilt angle (2.4°) is much less than that observed in the FeIV=O complexes of the analogous N2Py2Qn ligand but is very similar to that for the parent N4Py complex. The Fe1–O1 bond distance of 1.6584(14) Å is very close to that reported for other crystallographically characterized S = 1 FeIV=O complexes. As expected, the steric influence of the ligand moieties in 4b is minimal, and the effective ligand field is therefore expected to primarily be related to the electronic properties of the various N-donor entities. A comparison of bond lengths and tilt angles, and the relation to the reactivities for 4b and other related complexes, are listed in Table 8 (vide infra).
Figure 15.

Mercury plots of the complex cation of 4b with 40% probability ellipsoids in two perspectives. Hydrogen atoms have been omitted for clarity with the exception of ligand hydrogens in direct vicinity of the oxido ligand (right-hand plot). Selected bond distances (Å) and angles (deg): Fe(1)–O(1) 1.6584(14), Fe(1)–N(1) 1.9546(17), Fe(1)–N(2) 1.9685(17), Fe(1)–N(3) 1.9709(17), Fe(1)–N(4) 1.9608(17), Fe(1)–N(5) 2.0486(16), N(4)–Fe(1)–N(2) 164.78(7), O(1)–Fe(1)–N(5) 177.61(7).
Table 8. Comparison of Bond Lengths and FeIV=O Tilt Angles with Observed Rate Constants for 4b and Related Complexes.
| ligand | L4 | N4Py | N2Py2B | N2Py2Qn | N3Py-Qn |
|---|---|---|---|---|---|
| Fe=O (Å) | 1.6584(14) | 1.639(5) | 1.656(4) | 1.677(5) | 1.642(5) |
| Fe–Namine (Å) | 2.0486(16) | 2.033(8) | 2.115(6) | 2.084(4) | 2.053(6) |
| Fe–NPy (avg.) (Å) | 1.9697(17) | 1.964(5) | 1.989 | 2.023 | 1.977 |
| Fe–NIQ/Py/NMB/Qn (avg.) (Å)a | 1.9577(17) | 1.949(5) | 1.952 | 2.070 | 2.025(5) |
| Fe–Nequatorial (avg.) | 1.9637(17) | 1.9565(5) | 1.9705 | 2.0465 | 1.989 |
| tilt angle (deg) | 2.4 | 0.6 | 3 | 9.57 | 5.8 |
| k2 for thioanisole oxidation (M–1 s–1) (temp., K) | 0.0345 (273 K) | 0.049 (273 K) | 0.31 (243 K) | 7.4 (233 K) | 0.023 (233 K) |
| k2 for cyclohexane oxidation (M–1 s–1) (298 K) | 0.0067 × 10–3 | 0.055 × 10–3 | 2.9 × 10–3 | 30 × 10–3 | 0.86 × 10–3 |
| ref. | this work | (25) | (26) | (29) | (28) |
IQ: N-(isoquinolin-3-ylmethyl) moiety.
HAT Reactions
The observed short lifetimes of complexes 1b and 2b indicate that the complexes may exhibit enhanced reactivity toward external substrates in comparison to the parent [FeIV(O)(N4Py)]2+ complex. A series of alkane and arene substrates having different C–H bond dissociation energies (BDEs), ranging from 81 to 99.3 kcal/mol, were studied, and the relative reactivities of complexes 1b–4b were evaluated. The reactions showed pseudo-first-order behavior under conditions of excess substrate (10–1000 equiv), and the observed rate constants (kobs) were linearly dependent on substrate concentration. From these linear plots, second-order rate constants (k2) were obtained. The k2 values thus obtained are listed in Table 6. A primary kinetic isotope effect (KIE) value of ∼14 for HAT by 2b was obtained by determining the second-order rate constants for separate reactions of toluene and its d8 isotopomer with the FeIV=O complex (Figure S29), using the same methodology described above; this KIE value is quite similar to that obtained for the (N-methyl)benzimidazolyl equivalent of 2b.26
Table 6. Comparison of Second-Order Rate Constants for HAT Reactions Affected by Complexes 1b–4b and [FeIV(O)(N4Py)]2+a.
| alkaneb | DC–Hc(kal/mol) | k2d (M–1 s–1) for 1b | k2d (M–1 s–1) for 2b | k2d (M–1 s–1) for 3b | k2d (M–1 s–1) for 4b | k2d (M–1 s–1) for [FeIV(O)N4Py]2+ |
|---|---|---|---|---|---|---|
| triphenylmethane (1) | 81 | 8.2 × 10–2 | 8.43 × 10–1 | 6.05 × 10–2 | 1.23 × 10–1 | 0.037 |
| cumene (1) | 84.5 | 3.56 × 10–3 | 2.13 × 10–2 | 3.0 × 10–3 | 3.6 × 10–3 | 2 × 10–3 |
| ethylbenzene (2) | 87 | 2.66 × 10–3 | 1.84 × 10–2 | 1.9 × 10–3 | 3.1 × 10–3 | 4 × 10–3 |
| toluene (3) | 90 | 5.78 × 10–4 | 4.7 × 10–3 | 3.3 × 10–4 | 4.5 × 10–4 | 6.3 × 10–4 |
| cyclooctane (16) | 95.3 | 2 × 10–3 | 1.2 × 10–2 | 3 × 10–4 | 6.7 × 10–6 | |
| 2,3-dimethylbutane (2) | 96.5 | 2.3 × 10–4 | 1.2 × 10–4 | |||
| cyclohexane (12) | 99.3 | 3.53 × 10–4 | 4.33 × 10–3 | 9.14 × 10–5 | 6.7 × 10–6 | 5.5 × 10–5 |
| self-decay | 1.17 × 10–5 | 1.17 × 10–4 | 2.8 × 10–6 | 4.3 × 10–6 | 2.3 × 10–6 |
All experiments were performed in triplicate, at a minimum.
Numbers in parentheses represent the number of H atoms on the substrate that would react with the FeIV=O species.
DC–H values were obtained from ref (29).
A plot of logarithmic values of second-order rate constants (log k2’) (k2’ is the second-order rate constant divided by the number of equivalent C–H bonds in the substrate) for absorbance decay versus C–H BDEs for the different substrates shows a roughly linear correlation (Figure 16). This correlation and the observed large KIE value for 2b constitute strong evidence of the reactions taking place via HAT.
Figure 16.
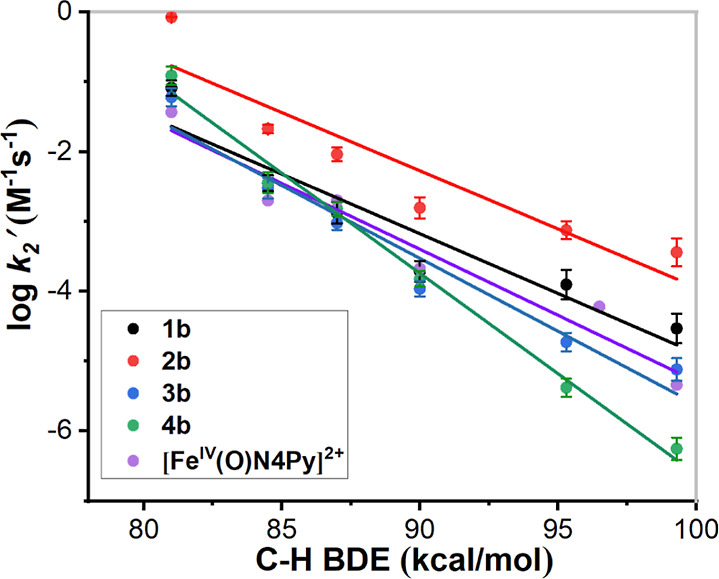
Plots of log k2’ versus C–H BDEs of different alkane substrates for 1b, 2b, 3b, 4b, and [FeIV(O)(N4Py)]2+; see Table 6 for the identities of the substrates.
Complexes 1b and 2b reacted readily with the C–H bonds of the different substrates at ambient temperature. On the other hand, for substrates with high BDEs, 3b and 4b reacted at slightly lower rates than [FeIV(O)(N4Py)]2+. The plots in Figure 16 clearly show that 2b consistently reacted with faster rates of reaction than the other ferryl complexes. The approximate collinearity of the plots/slopes for complexes 1b and 2b indicates that the effect on reaction rates upon introducing the second (N-methyl)imidazolyl arm is roughly the same irrespective of the substrate. At higher substrate BDEs—95.3 kcal/mol (cyclooctane), 99.3 kcal/mol (cyclohexane)—the HAT rates for the four different FeIV=O complexes are clearly differentiated, and the order is 2b > 1b > 3b > 4b in the order of decreasing HAT rates. Given that the steric influence of the ligands appears to be low, as reflected by the Namine–Fe–NCH3CN angles in 1a–4a (Table 2, vide supra), we interpret the differences in the reactivities of the FeIV=O complexes to be mainly attributable to the electronic (donor) properties of the various N-donor entities of the four ligands. It may be noted that while 4b displays the lowest reactivity of all FeIV=O complexes, the observed rate constant for this complex is on par with those of both 1b and 3b for HAT from triphenylmethane, which is the substrate with the lowest BDE but the greatest steric bulk around the C–H bond; 2b remains the most reactive species also for this substrate. Again, this suggests that it is the electronic contributions of the ligand donor moieties that mainly dictate the reactivities of the complexes rather than the steric influence of the ligands.
OAT Reactions
The OAT abilities of 1b–4b were also investigated, using thioanisole (PhSMe) as a substrate (Scheme 2). Because of the rapid reaction rates, the reactivities of 1b and 2b were studied at 243 K. Upon the addition of thioanisole, the characteristic near-IR band (λmax = 706 nm) decayed for complex 1b, indicating OAT from the FeIV=O complex to thioanisole to form methyl phenyl sulfoxide (vide infra). During the course of the reaction, complex 1b was converted into its FeII precursor (1a) with a clear isosbestic point at 565 nm as identified by UV/vis spectrophotometry (Figure 17). The reaction showed pseudo-first order behavior under conditions of excess substrate (100–500 equiv) and the observed rate constant (kobs) was linearly dependent on substrate concentration (Figure S30). From this linear plot, a second-order rate constant (k2) with a value of (2.6 ± 0.2) × 10–3 M–1 s–1 was obtained for the oxidation of thioanisole by complex 1b. Under the same conditions, 2b reacted with thioanisole at a rate considerably faster than 1b. The second-order rate constant (k2) for complex 2b was found to be (4.5 ± 0.2) × 10–1 M–1 s–1, meaning that 2b is more reactive by approximately two orders of magnitude than 1b in this OAT reaction (Figure S30). Similarly, the OAT reactivities of 3b and 4b were analyzed at 273 K (Figure S31). Table 7 shows a comparison of the reaction rates of the OAT (thianisole oxidation) processes of complexes 1b–4b with previously reported FeIV=O complexes. It can be seen that complex 2b is among the most reactive FeIV=O complexes for OAT (thianisole oxidation) and approximately on par with its (N-methyl)benzimidazolyl analogue [FeIV(O)(N2Py2B)]2+.26 Based on a previous study of a series of S = 1 FeIV=O complexes, the rates of OAT to thioanisole correlate linearly with the increase in the redox potentials, reflecting the relative electrophilicities of the FeIV=O complexes.38 It may thus be inferred that complex 2b has much higher electrophilicity than complexes 1b, 3b, 4b, [FeIV(O)(N4Py)]2+, and most other reported iron-oxo species supported by mixed amine/pyridine donor pentadentate ligands.
Scheme 2. OAT to Thioanisole by Complexes 1b–4b.

Figure 17.
Decay of 1b to its FeII precursor in the presence of thioanisole (250 mM) at 243 K. Inset: time course of the decay monitored at 706 nm.
Table 7. OAT (Thioanisole Oxidation) of Different FeIV=O Complexes Supported by Different N5 Ligands.
| complex | k2 (M–1 s–1) | temperature of measurement (K) | ref. |
|---|---|---|---|
| 1b | 0.00026 | 243 | this work |
| 2b | 0.45 | 243 | this work |
| 3b | 0.068 | 273 | this work |
| 4b | 0.034 | 273 | this work |
| [FeIV(O)(N4Py)]2+ | 0.014 | 263 | (25) |
| [FeIV(O)(N4Py)]2+ | 0.00024 | 233 | (25) |
| [FeIV(O)(Bn-tpen)]2+ | 0.33 | 263 | (38) |
| [FeIV(O)(Bn-tpen)]2+ | 0.014 | 233 | (38) |
| [FeIV(O)(N2Py2B)]2+ | 0.31 | 243 | (26) |
| [FeIV(O)(N3Py-(NMB))]2+ | 0.033 | 243 | (26) |
| [FeIV(O)(N2Py2Qn)]2+ | 7.4 | 233 | (25,29) |
| [FeIV(O)(N3Py-Qn)]2+ | 0.023 | 233 | (28) |
| [FeIV(O)(N4PyMe2)]2+ | 1.03 | 263 | (29) |
| [FeIV(O)(N4Py)]2+ | 0.049 | 273 | (25) |
Comparative parameters for a few structurally related ferryl complexes of N4Py and its derivatives are collated in Table 8. In contradiction to previous reports where a linear correlation has been proposed between the tilt angle and the reactivity,25 the reactivity patterns cannot be directly correlated with the tilt angle of the Fe–O moiety. For example, the tilt angle of 4b is 2.4°, which lies between the FeIV=O complexes of the N2Py2B and N4Py ligands. However, complex 4b is much less reactive than the FeIV=O complex of both N2Py2B and N4Py ligands in cyclohexane/thioanisole oxidation. Also, there is no correlation between the Fe=O bond lengths in the FeIV=O complexes and their reactivities. Based on the discussion above, it remains difficult to adequately predict reactivity only depending on steric/structural criteria.
Catalytic Oxidation Reactions Effected by the FeII Complexes 1a–4a
A series of aromatic and aliphatic substrates were used to analyze catalytic oxidations effected by 1a–4a, using IBX ester or H2O2 as the terminal oxidants. The catalytic data are collected in Tables 9 and 10. For OAT catalysis with thioanisole, all FeIV=O complexes in this work show excellent turnovers (yield ≈ 100%) with IBX ester. Similarly, for the HAT reaction with IBX ester as an oxidant, complexes 3a and 4a gave higher yields than 1a or 2a for substrates with high BDEs. Furthermore, complexes 3a and 4a could be recovered from the reaction mixtures after the completion of the catalytic reactions, but this was not the case for 1a and 2a. These observations may be rationalized by the established greater instability of the active species formed from 1a and 2a, which are presumed to be the corresponding ferryl complexes 1b and 2b. These unstable species may undergo degradation, leading to unproductive pathway(s) and low catalytic turnover. For the substrates toluene and cyclohexene, the total turnover numbers (all products) were less than 1 when IBX ester was used as an oxidant, and the reactions were thus not catalytic.
Table 9. Catalytic Activities of Complexes 1–4b with IBX Ester (Catalyst:IBX Ester = 1:10).
| substrate (mmol) | product | complex 1a |
complex 2a |
complex 3a |
complex 4a |
||||
|---|---|---|---|---|---|---|---|---|---|
| TONa | efficiencyb | TON | efficiency | TON | efficiency | TON | efficiency | ||
| thioanisole (0.25) | thioanisole oxide | 9.2 | 92 | 10 | 100 | 10 | 100 | 10 | 100 |
| 9,10-DHA (0.05) | anthracene | 9.6 | 96 | 10 | 100 | 10 | 100 | 9.6 | 96 |
| 1,4-CHD (0.25) | benzene | 9.3 | 93 | 9.7 | 97 | 10 | 100 | 10 | 100 |
| triphenylmethane (0.25) | triphenylmethanol | 9.8 | 98 | 10 | 100 | 9.3 | 93 | 8.65 | 86.5 |
| cumene (0.25) | 2-phenyl-2-propanol | 1.66 | 30.7 | 3.56 | 57.7 | ||||
| acetophenone | 1.41 | 2.21 | |||||||
| ethylbenzene (0.25) | 1-phenylethanol | 0.47 | 19.3 | 0.57 | 24 | 0.75 | 29.1 | 0.71 | 32.1 |
| 1-acetophenone | 1.46 | 1.826 | 2.16 | 2.5 | |||||
Turnover number (TON) = mol of product/mol of catalyst.
Efficiency (%) = moles of total products/mol of oxidant ×100.
Table 10. Catalytic Activities of Complexes with H2O2 (Catalyst:H2O2 = 1:100).
| substrate (mmol) | product | complex 1a |
complex 2a |
complex 3a |
complex 4a |
||||
|---|---|---|---|---|---|---|---|---|---|
| TONa | efficiencyb | TON | efficiency | TON | efficiency | TON | efficiency | ||
| thioanisole (0.25) | thioanisole oxide | 1.8 | 1.8 | 2.5 | 2.5 | 27.8 | 27.8 | 27 | 27 |
| 9,10-DHA (0.05) | anthracene | 15.9 | 15.9 | 23.2 | 23.2 | 28.2 | 28.2 | 26.7 | 26.7 |
| 1,4-CHD | benzene | 16.5 | 16.5 | 22.4 | 22.4 | 54.4 | 54.4 | 49.5 | 49.5 |
| triphenylmethane (0.25) | triphenylmethanol | 22.8 | 30.8 | 30.8 | 8 | 8 | 10 | 10 | |
| cumene (0.25) | 2-phenyl-2-propanol | 6.9 | 7.49 | 12.6 | 14.2 | ||||
| acetophenone | 0.59 | 1.6 | |||||||
| ethylbenzene (0.25) | 1-phenylethanol | 5.3 | 12.33 | 7 | 16.02 | 5.42 | 13.24 | 4.4 | 12.8 |
| 1-acetophenone | 7.03 | 9.02 | 7.82 | 8.4 | |||||
| toluene (0.25) | benzaldehyde | 4 | 4 | 6.1 | 6.1 | 4.5 | 4.5 | 3.5 | 3.5 |
| cyclohexane (0.5) | cyclohexanol | 4.38 | 8.98 | 6.2 | 13.4 | 5.86 | 12.33 | 5.6 | 11.87 |
| cyclohexanone | 4.6 | 7.2 | 6.47 | 6.27 | |||||
Turnover number (TON) = mol of product/mol of catalyst.
Efficiency (%) = moles of total products/mol of oxidant ×100.
When H2O2 was used as an oxidant, 3a and 4a gave much better yields than did 1a and 2a. The identities of the active oxidants were not determined in these reactions. Studies on the reactions of the parent complex [FeII(L)(N4Py)]n+ (L = monodentate ligand) with H2O2 show that an FeIII–OOH complex is formed initially,42 and this hydroperoxo complex may undergo O–O bond homolysis to generate the corresponding FeIV=O complex and a hydroxyl radical. However, at high concentrations of hydrogen peroxide, studies indicate that the formation of an FeIII(OH) species is favored over ferryl formation.43 A relatively low steady-state concentration of hydrogen peroxide should therefore favor the formation of oxidative species (FeIV=O, OH·). Considering the high concentrations of hydrogen peroxide used in our study and the low catalytic efficiencies (Table 9), substantial formation of an unproductive FeIII(OH) species cannot be excluded nor can the oxidation of substrates by hydroxyl radicals formed in Fenton reactions be excluded.
For triphenylmethane and 9,10-dihydroanthracene (9,10-DHA) — the substrates in this study that contain the greatest steric bulk around the C–H bond — the catalytic oxidations in this work give excellent yields. The small difference in yields between 9,10-DHA and 1,4-cyclohexadiene (1,4- CHD), which involve similar BDEs but very different steric environments, also indicates that the electronic contribution of the ligand donor moieties mainly dictates the reactivities of the complexes rather than the steric influence of the ligands.
Summary and Conclusions
We report the synthesis and characterization of four new FeII complexes and four corresponding FeIV=O complexes supported by four new pentadentate nonheme ligands based on the N4Py framework. As discussed above, the spin states and the redox potentials for the FeII complexes, the absorption wavelengths in the near-IR region, the half-lives, and the HAT/OAT reactivities of the FeIV=O complexes are all consistent with ligand L2, containing two (N-methyl)imidazolyl donors, exerting the weakest ligand field in these complexes. This is followed by L1, where only one pyridyl group is replaced with an (N-methyl)imidazolyl group. The sequences of λmax for the characteristic near-IR absorptions of the FeIV=O complexes and of the half-lives at room temperature are consistent with the replacement of pyridyl arms with (N-methyl)imidazolyl moieties rendering the ligand fields of L1 and L2 to be weaker than for the parent N4Py ligand. Compared to the parent complex [FeIV(O)(N4Py)]2+, complexes [FeIV(O)(L1)]2+ (1b) and [FeIV(O)(L2)]2+ (2b) exhibit enhanced reactivity in both the OAT and HAT reactions. The ferryl complexes 1b–4b are robust for the conversion of the OAT to thioanisole with excellent turnover numbers. Using H2O2 as the oxidant, complexes 1b–4b also displayed good performance in catalytic oxidation reactions (HAT and OAT). The crystal structures of FeII complexes 1a–4a and FeIV=O complex 4b and the reactivities of the ferryl complexes toward bulky substrates show that the steric influence of the four new ligands on the sixth (axial) position in the iron complexes is relatively minor.
Table 3 (vide supra) provides a clear indication of the effect the different N-donor substituents in the ligand framework have on the redox potential of their corresponding iron complexes and thus indicates the effective overall donor capacity of the pentadentate ligands. The half-wave potentials (E1/2) of the FeIII/FeII redox couple become more positive in the following sequence of the ligands: L2 (two (N-methyl)imidazolyl) < L1 (one (N-methyl)imidazolyl) < L4 (two isoquinolines) < L3 (one isoquinoline) < N4Py (cf. Figure 9 and Table 3). The trend in half-wave potentials can be interpreted as a decrease in the electron density over the iron center due to the electron-withdrawing effect of the groups. It is in agreement with the effect seen by introducing electron-withdrawing and electron-donating substituents in para-position of the central (axial) pyridyl moiety in FeII complexes of the pentadentate ligand Py5Me2,44 where the more electron-withdrawing groups give a more positive E1/2 for the FeIII/FeII couple. In this study, the (N-methyl)imidazolyl substituent is essentially a pure sigma-donor ligand and leads to the largest electron density on the ferrous ion and the weakest ligand field/highest-lying HOMOs. The isoquinoline and pyridyl substituents are σ-donors and π-acceptors; the metal π to ligand π* back-donation for these nitrogen donors leads to lower electron densities on the ferrous ions and stronger ligand fields/lower-lying HOMOs.
In summary, the observed HAT/OAT reactivities of the ferryl complexes in the present study may be directly correlated to the electronic properties of the ligand substituents rather than to steric effects leading to consequential electronic effects.
Experimental Section
Materials
All reagents and solvents were obtained from commercial sources and used without further purification unless otherwise noted. All solvents were of at least 99.5% purity. 1-Methyl-1-H-2-carbaldehyde,45 bis(2-pyridyl)methylamine,46 and N-[di(2-pyridinyl)methy]-N-(2-pyridinylmethyl)methylamine47 were prepared according to literature procedures.
Physical Methods
Preparation and handling of air-sensitive reactions were carried out under a nitrogen atmosphere in a glovebox. All room-temperature NMR spectra were collected on a Bruker Avance 400 MHz spectrometer in CD3CN or DMSO-d6 solvents. Variable-temperature NMR spectra were collected on Bruker Avance or Varian Inova 500 MHz spectrometers in CD3CN solvents. Fourier transform infrared spectroscopy on KBr pellets was performed on a Cary 630 FTIR Spectrometer (IR spectra of FeII and FeIV=O complexes are found in the Supporting Information). The electrochemical analyses were recorded using a Pine Research WaveNow potentiostat, a 2 mm glassy carbon working electrode wiped between scans, a platinum wire counter electrode, and Ag/Ag+ (silver wire in 0.005 M AgNO3, 0.10 M Bu4NPF6, and CH3CN) isolated by a frit used as a reference electrode and calibrated versus ferrocene. Anhydrous acetontrile (Carlo Erba) was used as the solvent with 0.10 M tetrabutylammonium hexafluorophosphate as the supporting electrolyte. All potentials are referenced to the ferrocene/ferrocenium couple (0.400 V vs NHE). The solutions for the electrochemical experiments were purged with solvent-saturated nitrogen and kept under an inert atmosphere throughout the measurements. HRMS was performed using a UHPLC/SFC-QTOF instrument. UV/vis spectra and all kinetic experiments were performed on a CARY100 UV/vis spectrophotometer (Agilent Technologies) equipped with a R928 PMT detector and a USP-203-B Unisoku cryostat, which permits monitoring of the temperature of the experiments from 193 to 373 K. All UV/vis spectra were measured using a four-side transparent quartz cuvette with a screw cap (path length: 10 mm). Product analyses were carried out by GC measurements on an Agilent Technologies 7820 A gas chromatograph with a 16-sample automatic liquid sampler and a flame ionization detector. The products passed through an SPB-35 capillary GC column and were identified by their different retention times. Zero-field and magnetic field Mössbauer measurements were carried out with 57Co sources in Rh matrices on alternating constant Wissel Mössbauer spectrometers equipped with Janis or OptiCool helium cryostats. Magnetic susceptibility measurements were made with a Quantum-Design MPMS3 SQUID magnetometer equipped with a 7 T magnet. A detailed description of the experimental setup and data treatment for Mössbauer and magnetic measurements is found in the Supporting Information.
Synthesis and Characterization of Ligands
Synthesis of N-(1-Methyl-2-imidazolyl)methyl-N-(2-pyridyl)-methyl-N-(bis-2-pyridylmethyl)-amine (L1)
A round-bottom flask was charged with N-[di(2-pyridinyl)methyl]-N-(2-pyridinylmethyl)methylamine (N3Py, cf.Scheme 1, 1.00 g, 3.6 mmol) and NaBH(CH3COO)3 (5.34 g, 25.2 mmol) in CH2Cl2. The solution was kept in an ice bath for 0.5 h under a nitrogen atmosphere. After that, 1-methyl-1H-imidazole-2-carboxaldehyde (B, 0.4 g, 3.6 mmol) was added. The ice bath was removed after 1 h, after which the solution was left stirring for 48 h at room temperature and under a nitrogen atmosphere. Subsequently, the solution was neutralized with saturated NaHCO3 solution until no bubbles were generated. The resultant solution was then extracted with CH2Cl2, washed with brine, and dried over Na2SO4. Evaporation of the organic solvent afforded the crude ligand L1 as a brown oil (cf. Scheme 1). The crude product was purified by passing it through a silica column using a mixture of ethyl acetate, dichloromethane, and triethylamine (2:1:1) as eluent. Yield 1.05 g (75%). 1H NMR (400 MHz, DMSO) δ 8.49 (dddd, J = 10.6, 4.8, 1.9, 0.9 Hz, 3H), 7.87–7.66 (m, 5H), 7.45 (d, J = 7.8 Hz, 1H), 7.32–7.18 (m, 3H), 6.96 (d, J = 1.2 Hz, 1H), 6.74 (d, J = 1.2 Hz, 1H), 5.25 (s, 1H), 3.79 (d, J = 17.9 Hz, 4H), 3.40 (s, 3H) (Figure S1).
Synthesis of N-bis(1-Methyl-2-imidazolyl)methyl-N-(bis-2-pyridylmethyl)amine (L2)
Ligand L2 was obtained by the reaction of bis(2-pyridyl)methylamine (A) with two equivalents of 1-methyl-1H-imidazole-2-carboxaldehyde (B) in the presence of NaBH(CH3COO)3 in CH2Cl2 (cf. Scheme 1). The subsequent workup was identical with that used for Ligand L1. Yield: 55%. 1H NMR (400 MHz, DMSO) δ 8.60–8.44 (m, 2H), 7.83–7.68 (m, 2H), 7.64–7.42 (m, 2H), 7.25 (dddd, J = 14.9, 7.5, 4.8, 1.2 Hz, 2H), 7.02 (dd, J = 6.9, 1.2 Hz, 2H), 6.77 (dd, J = 17.8, 1.2 Hz, 2H), 5.28–4.95 (m, 1H), 3.80 (s, 2H), 3.77–3.56 (m, 2H), 3.30 (s, 6H). 13C NMR (101 MHz, DMSO) δ 172.53, 161.86, 159.87, 149.23, 145.15, 137.07, 136.75, 126.89, 124.71, 122.66, 122.59, 122.28, 122.02, 70.02, 68.25, 55.37, 46.51, 40.46, 32.36, 21.60, 11.68 (Figures S2–S4).
Synthesis of N-(Isoquinolin-3-ylmethyl)-1,1-di(pyridin-2-yl)-N-(pyridin-2-ylmethyl)methanamine (L3)
A round-bottom flask was charged with a mixture of N-[di(2-pyridinyl)methyl]-N-(2-pyridinylmethyl)methylamine (N3Py, 1.00 g, 3.6 mmol) and potassium carbonate (2.3 g, 16 mmol) in CH3CN. The solution was stirred for several minutes at room temperature. After that, 3-(chloromethyl)isoquinoline (C, cf.Scheme 1, 0.7 g, 4 mmol) was added, and the mixture was then refluxed for 48 h. After cooling to room temperature, the solvent was evaporated, and the residue was purified by flash column chromatography (Al2O3 neutral activated, ethyl acetate/hexane/triethylamine (10:5:1, Rf = 0.3) to give N-(isoquinolin-3-ylmethyl)-1,1-di(pyridin-2-yl)-N-(pyridin-2-ylmethyl)methanamine (L3, 0.76 g, 51%) as a dark red oil. 1H NMR (400 MHz, CDCl3) δ 9.25 (s, 1H), 8.61 (ddd, J = 4.8, 1.9, 0.9 Hz, 2H), 8.55–8.46 (m, 1H), 7.96 (d, J = 8.2 Hz, 1H), 7.92 (s, 1H), 7.84–7.77 (m, 3H), 7.74–7.52 (m, 6H), 7.17 (ddd, J = 7.5, 4.8, 1.2 Hz, 2H), 7.09 (ddd, J = 7.5, 4.9, 1.2 Hz, 1H), 5.49 (s, 1H), 4.18 (s, 2H), 4.09 (s, 2H) (Figure S5).
Synthesis of N,N-bis(Isoquinolin-3-ylmethyl)-1,1-di(pyridin-2-yl)methanamine (L4)
A mixture of di(2-pyridyl)methylamine, (A, 0.67 g, 3.6 mmol), potassium carbonate (2.3 g, 16 mmol), and acetonitrile were stirred for several minutes at room temperature. To the mixture was added 3-(chloromethyl)isoquinoline (C, 1.4 g, 8 mmol). The subsequent workup was identical to that used for Ligand L3 to give L4 (1.3 g, 77%) as a dark red oil. 1H NMR (400 MHz, CDCl3) δ 9.17 (s, 2H), 8.58 (d, J = 4.8 Hz, 2H), 7.97 (s, 2H), 7.88 (d, J = 8.2 Hz, 2H), 7.84 (d, J = 7.9 Hz, 2H), 7.77 (d, J = 8.2 Hz, 2H), 7.68 (d, J = 7.6 Hz, 2H), 7.62 (d, J = 7.2 Hz, 2H), 7.51 (t, J = 7.5 Hz, 2H), 7.14 (td, 2H), 5.55 (s, 1H), 4.21 (s, 4H). 13C NMR (101 MHz, CDCl3) δ 160.4, 151.9, 149.3, 136.3, 130.1, 127.6, 127.4, 126.6, 126.5, 123.9, 121.9, 118.8, 72.2, 57.10 (Figures S6 and S7).
Synthesis and Characterization of Complexes
Complexes [FeII(CH3CN)(L1)](ClO4)2 (1a·(ClO4)2), [FeII(CH3CN)(L2)](ClO4)2 (2a·(ClO4)2), [FeII(L3)(CH3CN)](ClO4)2 (3a·(ClO4)2), and [FeII(CH3CN)(L4)](ClO4)2 (4a·(ClO4)2) were prepared by the reaction of one equivalent of L1/L2/L3/L4 with one equivalent of FeII salts in a minimum amount of dry acetonitrile at room temperature in a N2 atmosphere (Scheme 2, Supporting Information). CAUTION!Perchlorate salts are potentially explosive and should be handled with care.
Complexes [FeII(CH3CN)(L1)](OTf)2 (1a·(OTf)2), [FeII(CH3CN)(L2)](OTf)2 (2a·(OTf)2), [FeII(L3)(CH3CN)](OTf)2 (3a·(OTf)2), and [FeII(CH3CN)(L4)](OTf)2 (4a·(OTf)2) were prepared by the reaction of ligand (L1/L2/L3/L4) with one equivalent of Fe(OTf)2·2MeCN. Similarly, the reaction of L4 with Fe(BF4)2·6H2O in a glovebox gave [FeII(CH3CN)(L4)](BF4)2 (4a·(BF4)2). The methods of crystallizations of 1a–4a are described in the Supporting Information.
1a·(ClO4)2
1H NMR (400 MHz, CD3CN) δ 9.19–8.92 (m, 3H), 7.93 (d, J = 6.0 Hz, 4H), 7.70 (td, J = 7.6, 1.4 Hz, 1H), 7.41 (d, J = 13.9 Hz, 4H), 7.18 (d, J = 7.3 Hz, 1H), 7.09 (s, 1H), 6.49 (s, 1H), 4.55–4.12 (m, 4H), 3.41 (s, 3H) (Figure S8). HRMS: m/z = 213.0631 [FeII(L1)]2+(z = 2) calc. 213.0628, m/z = 525.0754 [FeII(L1)(ClO4)]+(z = 1) calc. 525.0741 (Figure S32).
2a·(ClO4)2
1H NMR (400 MHz, CD3CN) δ 79.29, 52.91, 37.38, 25.22, 16.73, 14.00 (s, 1H), 13.00, 11.89, 11.79, 10.19, 6.48, 5.43, 4.58, 3.3, 1.12, −1.60 (Figure S10). HRMS: m/z = 214.5684 [FeII(L2)]2+(z = 2) calc. 214.5628, m/z = 528.0861 [FeII(L2)(ClO4)]+(z = 1) calc. 528.0850 (Figure S33).
3a·(ClO4)2
1H NMR (400 MHz, CD3CN) δ 9.81 (s, 1H), 9.10–9.04 (m, 1H), 8.98 (ddt, J = 10.9, 5.5, 1.2 Hz, 2H), 8.23–8.16 (m, 1H), 8.04–7.90 (m, 4H), 7.86–7.72 (m, 3H), 7.64 (td, J = 7.7, 1.5 Hz, 1H), 7.49 (s, 1H), 7.47–7.30 (m, 3H), 7.05 (d, J = 7.9 Hz, 1H), 6.38 (s, 1H), 4.50–4.26 (m, 4H) (Figure S12). HRMS ([57FeII(L3)(CH3CN)](OTf)2): m/z = 237.0951 [57FeII(L3)]2+ (z = 2) calc. 237.0651, and m/z = 623.0323 [57FeII(L3)(OTf)]+ (z = 1) calc. 623.0828 (Figure S34, mass spectrum obtained on an 57Fe sample).
4a·(ClO4)2
1H NMR (400 MHz, CD3CN) δ 9.83 (s, 2H), 9.03 (d, J = 7.8 Hz, 2H), 8.15 (d, J = 8.5 Hz, 2H), 8.06–7.89 (m, 4H), 7.81–7.64 (m, 6H), 7.44 (s, 2H), 7.38 (ddd, J = 7.3, 5.5, 2.0 Hz, 2H), 6.40 (s, 1H), 4.49 (d, J = 17.1 Hz, 2H), 4.39–4.25 (m, 2H) (Figure S14). HRMS: m/z = 261.5743 [FeII(L4)]2+ (z = 2) calc. 261.5730, and m/z = 622.0962 [FeII(L4)(ClO4)]+ (z = 1) calc. 622.0944 (Figure S35).
4a·(BF4)2
Elemental analysis (C33H28B2F8FeN6): calc. C, 53.70; H, 3.82; N, 11.39, found C, 53.83; H, 3.85; N, 11.41. m/z = 568.1446 [FeII(L4) (HCOO)]+ (z = 1) (calc. 568.14).
Crystal Structure Determinations
The crystals of 2a·(ClO4)2 and 5·(OTf)2 were immersed in cryo-oil, mounted in a Nylon loop, and measured at a temperature of 120 K. The X-ray diffraction data were collected on Bruker Kappa Apex II and Bruker Kappa Apex II Duo diffractometers using Mo Kα radiation (λ = 0.710 73 Å). The APEX248 program package was used for cell refinements and data reductions. The structure was solved by the charge flipping technique (SUPERFLIP)49 or direct methods using the SIR2011 program50 with the Olex2 graphical user interface.51 A semiempirical numerical absorption correction based on equivalent reflections (SADABS)52 was applied to all data. Structural refinements were carried out using SHELXL-97.53 The crystal of 2·(ClO4)2 was diffracting only weakly, and therefore atoms N2, C16, C17, C18, C19, and C20 were restrained to have the same Uij components within the standard uncertainty of 0.02. In 2·(ClO4)2, one molecule of the acetonitrile of crystallization was disordered over two sites with equal occupancy. Hydrogen atoms were positioned geometrically and were also constrained to ride on their parent atoms, with C–H = 0.95–0.1.00 Å and Uiso = 1.2–1.5 Ueq (parent atom).
X-ray data for 1a·(OTf)2 and 3a·(OTf)2 were collected on a BRUKER D8-QUEST diffractometer (monochromated Mo–Kα radiation, λ = 0.71073 Å) by use of ω or ω and φ scans at low temperature. The structures were solved with SHELXT54 and refined on F2 using all reflections with SHELXL.55 Non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed in calculated positions and assigned to an isotropic displacement parameter of 1.5/1.2 Ueq(C). One CF3SO3– ion was found to be disordered about two positions in 1a·(OTf)2 (occupancy factors: 0.809(3)/0.191(3)) and about three positions in 3a·(OTf)2 (occupancy factors: 0.391(2)/0.305(2)/0.303(3)). SAME and RIGU restraints and EADP constraints were applied to model the disordered parts. In 1a,·(OTf)2 parts of the ligand were found to be disordered (occupancy factors: 0.506(7)/0.494(7)). SAME, SADI (d(Fe–N)), and SIMU restraints were used to model the disorder. The noncoordinating MeCN in 3a·(OTf)2 was refined at half occupancy. Absorption corrections were performed by the multiscan method with SADABS.52
X-ray diffraction data for 4a·(BF4)2 were collected at room temperature with an Oxford Diffraction Excalibur 3 system using ω-scans and Mo–Kα radiation (λ = 0.71073 Å). The data were extracted and integrated using Crysalis RED. The structure was solved by direct methods and refined by full-matrix least-squares calculations on F2 using SHELXT,55 SHELXL,54 integrated in OLEX2.49 Disordered electron density corresponding to 0.33 MeCN/Fe was handled using the solvent mask in OLEX2.
Because the crystals of 4b·(ClO4)2 were too small to be measured at a regular X-ray source, data were collected at the MAX IV synchrotron. The crystals were measured at the BioMAX beamline,56 equipped with a Dectrix Eiger X 16 M detector and an Arinax MD3 microdiffractometer. The beamline wavelength was set to 0.6199 for the data collection, and the beam size was set to 0.05 mm. A 900° data set was collected about a single axis at 22 ms per degree and a beam attenuation of 85% to avoid exceeding the count rate of the detector. The software program XDS56 was used for data processing. Data were integrated to a maximum resolution (sin θ/λ) of 0.655 (θmax = 28.832 deg), with a data completeness of 84.9%. The space group was determined with the CCP4 program Pointless57 and the structure determined ab initio with SHELXD.58
The crystallographic details are summarized in Tables S2–S8, Supporting Information.
Generation of FeIV=O Complexes
To an acetonitrile solution (0.5 mM) of the perchlorate salts of 1a, 2a, and 4a, and the triflate salt of 3a, three equiv of IBX ester was added at 25 °C. Immediately, the reddish-brown solutions turned pale green. Based on HRMS of the pale green solution originating from 1a·(ClO4)2, the peaks at m/z = 221.1334 and m/z = 541.2484 correspond to the formulations of [FeIV=O(L1)]2+ (calc. 221.0602) and [FeIV=O(L1)(ClO4)]+ (calc. 541.0690), respectively (Figure S36). Based on HRMS of the pale green solution originating from 2a·(ClO4)2, the peaks at m/z = 222.5657 and m/z = 544.0801 correspond to the formulations of [FeIV=O(L2)]2+ (calc. 222.5657) and [FeIV=O(L2)(ClO4)]+ (calc. 544.0799.), respectively (Figure S37). Based on HRMS of the pale green solution originating from an enriched sample of [57FeII(L3)(CH3CN)](OTf)2, the peaks at m/z = 245.0928 ([57FeIV=O(L3)]2+ (calc. 245.0628) and m/z = 639.0225 ([57FeIV=O(L3)(OTf)]+ (calc. 639.0776)) correspond to complex 3b (Figure S38). Based on HRMS of the pale green solution originating from 4a·(ClO4)2, the peaks at m/z = 269.5718 and m/z = 638.0911 correspond to the formulations of [FeIV=O(L4)]2+ (calc. 269.5700) and [FeIV=O(L4)(ClO4)]+ (calc. 638.0900), respectively (Figure S39). After deaeration of the solutions of the FeIV=O complexes, UV/vis spectroscopy was performed at 25 °C with maximum absorption at 706 nm (ε = 379 M–1 cm–1) for [FeIV=O(L1)]2+(complex 1b), 721 nm (ε = 274 M–1 cm–1) for [FeIV=O(L2)]2+ (complex 2b)), 695 nm (ε = 540 M–1 cm–1) for [FeIV=O(L3)]2+ (complex 3b)), and 696 nm (ε = 544 M–1 cm–1) for [FeIV=O(L4)]2+ (complex 4b) separately. The time course decays of the FeIV=O complexes were then monitored, giving the half-lives for complex 1b (1.67 h), complex 2b (16 h), complex 3b (64 h), and complex 4b (45 h).
HAT Reactions
Kinetic experiments were performed by adding an appropriate amount of the relevant substrate (2.5–500 mM) to a 0.5 mM solution of complex 1b/2b/3b/4b in CH3CN (generated by addition of three equivalents of IBX ester to 1a·(ClO4)2/2a·(ClO4)2/3a·(ClO4)2/4a·(ClO4)2). After deaeration with argon, the time course decay of the FeIV=O complex was monitored at 25 °C by UV/vis spectroscopy. Rate constants, kobs, were evaluated by pseudo-first-order fitting of the absorbance decay at 706 nm/721 nm/695 nm/696 nm for complex 1b/2b/3b/4b. Second-order rate constants were evaluated from the slope of the linear fits of kobs versus concentration-dependence data. To isolate the organic products, the solutions after the end of the reaction were passed through a silica column, using ethyl acetate as the eluent, to remove the metal complex. The ethyl acetate solutions were then analyzed by GC using a known concentration of naphthalene solution as the quantification standard. Results obtained from these studies are listed in Table 6.
OAT (Thioanisole Oxidation) Reactions
The FeIV=O complexes 1b/2b/3b/4b solutions were prepared as described above. The solutions were placed in a cuvette, and the temperature of the UV/vis instrument was adjusted to −30 °C. Subsequently, appropriate amounts of the substrate, thioanisole, were added to the FeIV=O solution. After that, the subsequent decay was monitored. Time courses were subjected to pseudo-first-order fitting, and second-order rate constants were evaluated from the concentration-dependence data. The products were quantified following the procedure already described. Results obtained from these studies are listed in Table 7.
Analysis of the Products after Oxidation of Substrates
The FeII precursor complexes (1a–4a) (0.005 mmol) were dissolved in 3 mL of dry, deoxygenated acetonitrile. To the resulting solution, 10 equiv of IBX ester or 100 equiv of H2O2 was added along with the substrates at 298 K under an inert atmosphere. The reaction occurred under stirring for 4 h. Then, the resulting solution was passed through a silica column (60–120 mesh size) using ethyl acetate as an eluent. The combined organic phase was then analyzed by GC. For GC analyses, naphthalene was used as an internal standard, and the products were identified by comparison of their GC retention times with those of authentic compounds.
Theoretical Methods
Geometry optimization of complex 4a was performed in vacuum at the DFT level using the PBE059,60 and def2-TZVP(-f) basis set for all atoms.61−63 Dispersion effects (if existent) were included using Grimme’s D3 correction with Becke-Johnson (BJ) damping.64,65 The evaluation of the four-center integrals was accelerated with the RIJCOSX algorithm, using the resolution of identity approximation for the Coulomb part (RIJ) and the chain of spheres approach for the Fock exchange (COSX).66,67 RIJ requires the specification of an auxiliary basis set for the Coulomb part (Def2/J) and a numerical integration grid for the exchange part (DefGrid-2).68 Analytical harmonic vibrational frequency calculations were conducted to verify whether the ground state is the minima on the potential energy surface. TD-DFT69 was employed to obtain the first 25 singlet excited states, including the solvent correction, using the linear response conductor-like polarizable continuum model (LR-CPCM) method.70 All these calculations were performed using Orca 5.0.3,71 and the geometric representations of the complexes were obtained using the Chemcraft program.72
Acknowledgments
This paper is dedicated to the memory of Professor Sofi Elmroth, an outstanding inorganic chemist, an excellent teacher, and a respected colleague. The work has been supported by the Deutsche Forschungsgemeinschaft (DFG) in the framework of the Research Unit 5215 (FOR 5251 “Bioinspired Oxidation Catalysis with Iron Complexes”; project Me1313/18-1/445916766 to F.M.). Purchase of the Bruker D8-QUEST diffractometer as well as of the Mössbauer spectrometer with magnet cryostat was enabled by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) and the Niedersächsische Ministerium für Wissenschaft und Kultur (MWK), project numbers 423268549 (INST 186/1087 1327-1 FUGG) and 424956795 (INST 186/1332-1 FUGG), respectively. Y.L. and E.N. thank the Royal Physiographic Society for funding. S.Y. and O.F.W. thank the SIDA bilateral program for financial support. Y.L. thanks the Chinese Scholarship Council for a predoctoral fellowship. G.C.L. thanks the Wenner-Gren Foundation for a visiting fellowship to Lund University. The authors thank Sofia Essén for high-resolution mass spectrometry measurements.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.3c02526.
Synthesis methods; crystallographic data; UV/vis spectra; FTIR spectra; NMR spectra; Mössbauer spectra; SQUID measurements; cyclic voltammograms; tables of selected bond distances (Å) and bond angles (deg) of complexes; optimized ground-state geometries; and CCDC entries (1a.(OTf)2), (2a.(ClO4)2), (3a.(OTf)2), (4a.(BF4)2), (4b.(ClO4)2), and (5.(OTf)2) with the supplementary crystallographic data for the structures described in this paper (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Riggs-Gelasco P. J.; Price J. C.; Guyer R. B.; Brehm J. H.; Barr E. W.; Bollinger J. M.; Krebs C. EXAFS Spectroscopic Evidence for an Fe=O Unit in the Fe(IV) Intermediate Observed during Oxygen Activation by Taurine:α-Ketoglutarate Dioxygenase. J. Am. Chem. Soc. 2004, 126, 8108–8109. 10.1021/ja048255q. [DOI] [PubMed] [Google Scholar]
- Hoffart L. M.; Barr E. W.; Guyer R. B.; Bollinger J. M.; Krebs C. Direct Spectroscopic Detection of a C-H-Cleaving High-Spin Fe(IV) Complex in a Prolyl-4-Hydroxylase;. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 14738–14743. 10.1073/pnas.0604005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galonić D. P.; Barr E. W.; Walsh C. T.; Bollinger J. M.; Krebs C. Two Interconverting Fe(IV) Intermediates in Aliphatic Chlorination by the Halogenase CytC3. Nat. Chem. Biol. 2007, 3, 113–116. 10.1038/nchembio856. [DOI] [PubMed] [Google Scholar]
- Galonić Fujimori D.; Barr E. W.; Matthews M. L.; Koch G. M.; Yonce J. R.; Walsh C. T.; Bollinger J. M.; Krebs C.; Riggs-Gelasco P. J. Spectroscopic Evidence for a High-Spin Br-Fe(IV)-Oxo Intermediate in the α-Ketoglutarate-Dependent Halogenase CytC3 from Streptomyces. J. Am. Chem. Soc. 2007, 129, 13408–13409. 10.1021/ja076454e. [DOI] [PubMed] [Google Scholar]
- Matthews M. L.; Krest C. M.; Barr E. W.; Vaillancourt F. H.; Walsh C. T.; Green M. T.; Krebs C.; Bollinger J. M. Substrate-Triggered Formation and Remarkable Stability of the C-H Bond-Cleaving Chloroferryl Intermediate in the Aliphatic Halogenase, SyrB2. Biochemistry 2009, 48, 4331–4343. 10.1021/bi900109z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S. D.; Srnec M.; Matthews M. L.; Liu L. V.; Kwak Y.; Park K.; Bell III C. B.; Alp E. E.; Zhao J.; Yoda Y.; Kitao S.; Seto M.; Krebs C.; Bollinger J. M.; Solomon E. I. Elucidation of the Fe(iv)=O intermediate in the catalytic cycle of the halogenase SyrB2. Nature 2013, 499, 320–323. 10.1038/nature12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eser B. E.; Barr E. W.; Frantom P. A.; Saleh L.; Bollinger J. M. Jr.; Krebs C.; Fitzpatrick P. F. Direct Spectroscopic Evidence for a High-Spin Fe(IV) Intermediate in Tyrosine Hydroxylase. J. Am. Chem. Soc. 2007, 129, 11334–11335. 10.1021/ja074446s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panay A. J.; Lee M.; Krebs C.; Bollinger J. M. Jr.; Fitzpatrick P. F. Evidence for a High-Spin Fe(IV) Species in the Catalytic Cycle of a Bacterial Phenylalanine Hydroxylase. Biochemistry 2011, 50, 1928–1933. 10.1021/bi1019868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri M.; Que L. Toward the Synthesis of More Reactive S = 2 Non-Heme Oxoiron(IV) Complexes. Acc. Chem. Res. 2015, 48, 2443–2452. 10.1021/acs.accounts.5b00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. L.; Ross D. L.; Barman S. K.; Ziller J. W.; Borovik A. S. C-H Bond Cleavage by Bioinspired Nonheme Metal Complexes. Inorg. Chem. 2021, 60, 13759–13783. 10.1021/acs.inorgchem.1c01754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald A. R.; Que L. High-Valent Nonheme Iron-Oxo Complexes: Synthesis, Structure, and Spectroscopy. Coord. Chem. Rev. 2013, 257, 414–428. 10.1016/j.ccr.2012.08.002. [DOI] [Google Scholar]
- Warm K.; Paskin A.; Kuhlmann U.; Bill E.; Swart M.; Haumann M.; Dau H.; Hildebrandt P.; Ray K. A Pseudotetrahedral Terminal Oxoiron(IV) Complex: Mechanistic Promiscuity in C-H Bond Oxidation Reactions. Angew. Chem., Int. Ed. 2021, 60, 6752–6756. 10.1002/anie.202015896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaik S.; Chen H.; Janardanan D. Exchange-Enhanced Reactivity in Bond Activation by Metal-Oxo Enzymes and Synthetic Reagents. Nat. Chem. 2011, 3, 19–27. 10.1038/nchem.943. [DOI] [PubMed] [Google Scholar]
- Geng C.; Ye S.; Neese F. Analysis of Reaction Channels for Alkane Hydroxylation by Nonheme Iron(IV)-Oxo Complexes. Angew. Chem., Int. Ed. 2010, 49, 5717–5720. 10.1002/anie.201001850. [DOI] [PubMed] [Google Scholar]
- Kupper C.; Mondal B.; Serrano-Plana J.; Klawitter I.; Neese F.; Costas M.; Ye S.; Meyer F. Nonclassical Single-State Reactivity of an Oxo-Iron(IV) Complex Confined to Triplet Pathways. J. Am. Chem. Soc. 2017, 139, 8939–8949. 10.1021/jacs.7b03255. [DOI] [PubMed] [Google Scholar]
- Winkler J. R.; Gray H. B.. Electronic Structures of Oxo-Metal Ions. In Molecular Electronic Structures of Transition Metal Complexes I; Mingos D. M. P., Day P., Dahl J. P., Eds.; Springer: Berlin, 2012; pp 17–28. [Google Scholar]
- Deutscher J.; Gerschel P.; Warm K.; Kuhlmann U.; Mebs S.; Haumann M.; Dau H.; Hildebrandt P.; Apfel U. P.; Ray K. A Bioinspired Oxoiron(IV) Motif Supported on a N2S2 macrocyclic Ligand. Chem. Commun. 2021, 57, 2947–2950. 10.1039/D1CC00250C. [DOI] [PubMed] [Google Scholar]
- Shaik S. Two-State Reactivity: Personal Recounting of Its Conception and Future Prospects. Isr. J. Chem. 2020, 60, 938–956. 10.1002/ijch.202000002. [DOI] [Google Scholar]
- Shaik S.; Hirao H.; Kumar D. Reactivity of High-Valent Iron-Oxo Species in Enzymes and Synthetic Reagents: A Tale of Many States. Acc. Chem. Res. 2007, 40, 532–542. 10.1021/ar600042c. [DOI] [PubMed] [Google Scholar]
- Mandal D.; Mallick D.; Shaik S. Kinetic Isotope Effect Determination Probes the Spin of the Transition State, Its Stereochemistry, and Its Ligand Sphere in Hydrogen Abstraction Reactions of Oxoiron(IV) Complexes. Acc. Chem. Res. 2018, 51, 107–117. 10.1021/acs.accounts.7b00442. [DOI] [PubMed] [Google Scholar]
- Sheng Y.; Abelson C. S.; Prakash J.; Draksharapu A.; Young V. G.; Que L. Unmasking Steps in Intramolecular Aromatic Hydroxylation by a Synthetic Nonheme Oxoiron(IV) Complex. Angew. Chem., Int. Ed. 2021, 60, 20991–20998. 10.1002/anie.202108309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Eguía B. N.; Serrano-Plana J.; Company A.; Costas M. Catalytic O2 activation with Synthetic Models of α-Ketoglutarate Dependent Oxygenases. Chem. Commun. 2020, 56, 14369–14372. 10.1039/D0CC05942K. [DOI] [PubMed] [Google Scholar]
- Roelfes G.; Lubben M.; Leppard S. W.; Schudde E. P.; Hermant R. M.; Hage R.; Wilkinson E. C.; Que L. Jr.; Feringa B. L. Functional Models for Iron-Bleomycin. J. Mol. Catal. A: Chem. 1997, 117, 223–227. 10.1016/S1381-1169(96)00356-1. [DOI] [Google Scholar]
- Mukherjee G.; Alili A.; Barman P.; Kumar D.; Sastri C. V.; de Visser S. P. Interplay Between Steric and Electronic Effects: A Joint Spectroscopy and Computational Study of Nonheme Iron(IV)-Oxo Complexes. Chem.—Eur. J. 2019, 25, 5086–5098. 10.1002/chem.201806430. [DOI] [PubMed] [Google Scholar]
- Rasheed W.; Draksharapu A.; Banerjee S.; Young V. G. Jr.; Fan R.; Guo Y.; Ozerov M.; Nehrkorn J.; Krzystek J.; Telser J.; Que L. Jr. Crystallographic Evidence for a Sterically Induced Ferryl Tilt in a Non-Heme Oxoiron(IV) Complex that Makes it a Better Oxidant. Angew. Chem., Int. Ed. 2018, 57, 9387–9391. 10.1002/anie.201804836. [DOI] [PubMed] [Google Scholar]
- Mitra M.; Nimir H.; Demeshko S.; Bhat S. S.; Malinkin S. O.; Haukka M.; Lloret-Fillol J.; Lisensky G. C.; Meyer F.; Shteinman A. A.; Browne W. R.; Hrovat D. A.; Richmond M. G.; Costas M.; Nordlander E. Nonheme Fe(IV) Oxo Complexes of Two New Pentadentate Ligands and Their Hydrogen-Atom and Oxygen-Atom Transfer Reactions. Inorg. Chem. 2015, 54, 7152–7164. 10.1021/ic5029564. [DOI] [PubMed] [Google Scholar]
- Mukherjee G.; Lee C. W. Z.; Nag S. S.; Alili A.; Cantú Reinhard F. G.; Kumar D.; Sastri C. V.; de Visser S. P. Dramatic rate-Enhancement of oxygen atom transfer by an Iron(IV)-oxo species by equatorial ligand field perturbations. Dalton Trans. 2018, 47, 14945–14957. 10.1039/C8DT02142B. [DOI] [PubMed] [Google Scholar]
- Munshi S.; Sinha A.; Yiga S.; Banerjee S.; Singh R.; Hossain M. K.; Haukka M.; Valiati A. F.; Huelsmann R. D.; Martendal E.; Peralta R.; Xavier F.; Wendt O. F.; Paine T. K.; Nordlander E. Hydrogen-atom and oxygen-atom transfer reactivities of iron(IV)-oxo complexes of quinoline-substituted pentadentate ligands. Dalton Trans. 2022, 51, 870–884. 10.1039/D1DT03381F. [DOI] [PubMed] [Google Scholar]
- Singh R.; Ganguly G.; Malinkin S. O.; Demeshko S.; Meyer F.; Nordlander E.; Paine T. K. A Mononuclear Nonheme Iron(IV)-Oxo Complex of a Substituted N4Py Ligand: Effect of Ligand Field on Oxygen Atom Transfer and C-H Bond Cleavage Reactivity. Inorg. Chem. 2019, 58, 1862–1876. 10.1021/acs.inorgchem.8b02577. [DOI] [PubMed] [Google Scholar]
- Roelfes G.; Lubben M.; Chen K.; Ho R. Y. N.; Meetsma A.; Genseberger S.; Hermant R. M.; Hage R.; Mandal S. K.; Young V. G.; Zang Y.; Kooijman H.; Spek A. L.; Que L.; Feringa B. L. Iron Chemistry of a Pentadentate Ligand That Generates a Metastable Fe(III)-OOH Intermediate. Inorg. Chem. 1999, 38, 1929–1936. 10.1021/ic980983p. [DOI] [PubMed] [Google Scholar]
- Singh P.; Lee Y.; Mayfield J. R.; Singh R.; Denler M. C.; Jones S. D.; Day V. W.; Nordlander E.; Jackson T. A. Enhanced Understanding of Structure-Function Relationships for Oxomanganese(IV) Complexes. Inorg. Chem. 2023, 10.1021/acs.inorgchem.3c00600. [DOI] [PubMed] [Google Scholar]
- Rana S.; Biswas J. P.; Sen A.; Clémancey M.; Blondin G.; Latour J. M.; Rajaraman G.; Maiti D. Selective C-H Halogenation over Hydroxylation by Non-Heme Iron(IV)-Oxo. Chem. Sci. 2018, 9, 7843–7858. 10.1039/C8SC02053A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer S.; Klawitter I.; Demeshko S.; Bill E.; Meyer F. A Tetracarbene-Oxoiron(IV) Complex. Angew. Chem., Int. Ed. 2013, 52, 901–905. 10.1002/anie.201208044. [DOI] [PubMed] [Google Scholar]
- Kurtz D. M. Jr. Oxo -and Hydroxo-Bridged Diiron Complexes: A Chemical Perspective on a Biological Unit. Chem. Rev. 1990, 90, 585–606. 10.1021/cr00102a002. [DOI] [Google Scholar]
- Kaizer J.; Klinker E. J.; Oh N. Y.; Rohde J.-U.; Song W. J.; Stubna A.; Kim J.; Münck E.; Nam W.; Que L. Nonheme FeIVO Complexes That Can Oxidize the C-H Bonds of Cyclohexane at Room Temperature. J. Am. Chem. Soc. 2004, 126, 472–473. 10.1021/ja037288n. [DOI] [PubMed] [Google Scholar]
- Ray K.; England J.; Fiedler A. T.; Martinho M.; Münck E.; Que L. Jr. An Inverted and More Oxidizing Isomer of [FeIV(O)- (tmc)(NCCH3)]2+. Angew. Chem., Int. Ed. 2008, 47, 8068–8071. 10.1002/anie.200802219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer S.; Klawitter I.; Demeshko S.; Bill E.; Meyer F. A Tetracarbene-Oxoiron(IV) Complex. Angew. Chem., Int. Ed. 2013, 52, 901–905. 10.1002/anie.201208044. [DOI] [PubMed] [Google Scholar]
- Wang D.; Ray K.; Collins M. J.; Farquhar E. R.; Frisch J. R.; Gómez L.; Jackson T. A.; Kerscher M.; Waleska A.; Comba P.; Costas M.; Que L. Nonheme Oxoiron(IV) Complexes of Pentadentate N5 Ligands: Spectroscopy, Electrochemistry, and Oxidative Reactivity. Chem. Sci. 2013, 4, 282–291. 10.1039/C2SC21318D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker A.; Rohde J.-U.; Klinker E. J.; Wong S. D.; Que L.; Solomon E. I. Spectroscopic and Quantum Chemical Studies on Low-Spin FeIV=O Complexes: Fe-O Bonding and Its Contributions to Reactivity. J. Am. Chem. Soc. 2007, 129, 15983–15996. 10.1021/ja074900s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England J.; Bigelow J. O.; Van Heuvelen K. M.; Farquhar E. R.; Martinho M.; Meier K. K.; Frisch J. R.; Munck E.; Que L. Jr. An Ultra-stable Oxoiron(IV) Complex and its Blue Conjugate Base. Chem. Sci. 2014, 5, 1204–1215. 10.1039/C3SC52755G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibon A.; England J.; Martinho M.; Young V. G. Jr.; Frisch J. R.; Guillot R.; Girerd J.-J.; Münck E.; Que L. Jr.; Banse F. Proton- and Reductant-Assisted Dioxygen Activation by a Nonheme Iron(II) Complex to Form an Oxoiron(IV) Intermediate. Angew. Chem., Int. Ed. 2008, 47, 7064–7067. 10.1002/anie.200801832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelfes G.; Lubben M.; Chen K.; Ho R. Y. N.; Meetsma A.; Genseberger S.; Hermant R. M.; Hage R.; Mandal S. K.; Young V. G.; Zang Y.; Kooijman H.; Spek A. L.; Que L.; Feringa B. L. Iron Chemistry of a Pentadentate Ligand That Generates a Metastable FeIII-OOH Intermediate. Inorg. Chem. 1999, 38, 1929–1936. 10.1021/ic980983p. [DOI] [PubMed] [Google Scholar]
- Chen J.; Draksharapu A.; Angelone D.; Unjaroen D.; Padamati S. K.; Hage R.; Swart M.; Duboc C.; Browne W. R. H2O2 Oxidation by FeIII-OOH Intermediates and Its Effect on Catalytic Efficiency. ACS Catal. 2018, 8, 9665–9674. 10.1021/acscatal.8b02326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantarojsiri T.; Sun Y.; Long J. R.; Chang C. J. Water-Soluble Iron(IV)-Oxo Complexes Supported by Pentapyridine Ligands: Axial Ligand Effects on Hydrogen Atom and Oxygen Atom Transfer Reactivity. Inorg. Chem. 2015, 54, 5879–5887. 10.1021/acs.inorgchem.5b00658. [DOI] [PubMed] [Google Scholar]
- Ortega-Villar N.; Ugalde-Saldívar V. M.; Flores-Pérez B.; Flores-Alamo M.; Real J. A.; Moreno-Esparza R. A new N6 hexadentate ligand and a novel heptacoordinated N6O-type Fe(III) compounds: Synthesis, characterization and structure of [Fe(dimpyen)(OH)](A)2 (A=PF6– or ClO4−). Inorg. Chim. Acta 2011, 375, 213–219. 10.1016/j.ica.2011.05.008. [DOI] [Google Scholar]
- Renz M.; Hemmert C.; Meunier B. Synthesis of Bis[di(2-pyridyl)methyl]amine (BDPMA) by a Novel One-Pot Multi-Step Reductive Amination with Molecular Sieves and Zn/iPrOH. Eur. J. Org Chem. 1998, 1998, 1271–1273. . [DOI] [Google Scholar]
- Roelfes G.; Branum M. E.; Wang L.; Que L. Jr.; Feringa B. L. Efficient DNA Cleavage with an Iron Complex without Added Reductant. J. Am. Chem. Soc. 2000, 122, 11517–11518. 10.1021/ja001965l. [DOI] [Google Scholar]
- APEX2, in SAINT-Plus and SADABS; Bruker AXS Inc.: Madison, Wisconsin, USA, 2008.
- Palatinus L.; Chapuis G. SUPERFLIP—a Computer Program for the Solution of Crystal Structures by Charge Flipping in Arbitrary Dimensions. J. Appl. Crystallogr. 2007, 40, 786–790. 10.1107/S0021889807029238. [DOI] [Google Scholar]
- Camalli M.; Carrozzini B.; Cascarano G. L.; Giacovazzo C. Automated Determination of the Extinction Symbol. via Electron Diffraction Data. J. Appl. Crystallogr. 2012, 45, 351–356. 10.1107/S0021889812003317. [DOI] [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. 10.1107/S0021889808042726. [DOI] [Google Scholar]
- Bruker . SADABS; Bruker AXS Inc.: Madison, Wisconsin, USA, 2001.
- Sheldrick G. M.SHELXL-97—Program for Crystal Structure Refinement; University of Gottingen: Germany, 1997.
- Sheldrick G. M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. 2015, 71, 3–8. 10.1107/s2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. 2015, 71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursby T.; Åhnberg K. A.; Appio R.; Aurelius O.; Barczyk A.; Bartalesi A.; Bjelčić M.; Bolmsten F.; Cerenius Y.; Doak R. B.; Eguiraun M.; Eriksson T.; Friel R. J.; Gorgisyan I.; Gross A.; Haghighat V.; Hennies F.; Jagudin E.; Norsk Jensen B.; Jeppsson T.; Kloos M.; Lidon-Simon J.; de Lima G. M. A.; Lizatovic R.; Lundin M.; Milan-Otero A.; Milas M.; Nan J.; Nardella A.; Rosborg A.; Shilova A.; Shoeman R. L.; Siewert F.; Sondhauss P.; Talibov V. O.; Tarawneh H.; Thånell J.; Thunnissen M.; Unge J.; Ward C.; Gonzalez A.; et al. BioMAX—the First Macromolecular Crystallography Beamline at MAX IV Laboratory. J. Synchrotron Rad. 2020, 27, 1415–1429. 10.1107/s1600577520008723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R. An Introduction to Data Reduction: Space-Group Determination, Scaling and Intensity Statistics. Acta Crystallogr. 2011, 67, 282–292. 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. M. A Short History of SHELX. Acta Crystallogr. 2008, 64, 112–122. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple [Phys. Rev. Lett. 77, 3865 (1996)]. Phys. Rev. Lett. 1997, 78, 1396. 10.1103/PhysRevLett.78.1396. [DOI] [PubMed] [Google Scholar]
- Schäfer A.; Huber C.; Ahlrichs R. Fully Optimized Contracted Gaussian Basis Sets of Triple Zeta Valence Quality for Atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. 10.1063/1.467146. [DOI] [Google Scholar]
- Schäfer A.; Horn H.; Ahlrichs R. Fully Optimized Contracted Gaussian Basis Sets for Atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. 10.1063/1.463096. [DOI] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 15104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Ehrlich S.; Goerigk L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- Izsák R.; Neese F. An Overlap Fitted Chain of Spheres Exchange Method. J. Chem. Phys. 2011, 135, 144105. 10.1063/1.3646921. [DOI] [PubMed] [Google Scholar]
- Izsák R.; Neese F.; Klopper W. Robust Fitting Techniques in the Chain of Spheres Approximation to the Fock Exchange: The Role of the Complementary Space. J. Chem. Phys. 2013, 139, 094111. 10.1063/1.4819264. [DOI] [PubMed] [Google Scholar]
- Helmich-paris B.; de Souza B.; Neese F.; Izsák R. An Improved Chain of Spheres for Exchange Algorithm. J. Chem. Phys. 2021, 155, 104109. 10.1063/5.0058766. [DOI] [PubMed] [Google Scholar]
- Petrenko T.; Kossmann S.; Neese F. Efficient Time-Dependent Density Functional Theory Approximations for Hybrid Density Functionals : Analytical Gradients and Parallelization. J. Chem. Phys. 2011, 134, 054116. 10.1063/1.3533441. [DOI] [PubMed] [Google Scholar]
- Cammi R.; Mennucci B.; Tomasi J. Fast Evaluation of Geometries and Properties of Excited Molecules in Solution: A Tamm-Dancoff Model with Application to 4-Dimethylaminobenzonitrile. J. Chem. Phys. 2000, 104, 5631–5637. 10.1021/jp000156l. [DOI] [Google Scholar]
- Neese F. Software Update: The ORCA Program System, Version 4.0. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2018, 8, e1327 10.1002/wcms.1327. [DOI] [Google Scholar]
- Chemcraft—Graphical Software for Visualization of Quantum Chemistry Computations. https://www.chemcraftprog.com.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.