

Abstract
IL-17 and its receptor are founding members of an emerging family of cytokines and receptors with many unique characteristics. IL-17 is produced primarily by T cells, particularly those of the memory compartment. In contrast, IL-17 receptor is ubiquitously expressed, making nearly all cells potential targets of IL-17. Although it has only limited homology to other cytokines, IL-17 exhibits proinflammatory properties similar to those of tumor necrosis factor-α, particularly with respect to induction of other inflammatory effectors. In addition, IL-17 synergizes potently with other cytokines, placing it in the center of the inflammatory network. Strikingly, IL-17 has been associated with several bone pathologies, most notably rheumatoid arthritis.
Keywords: bone, cytokines, interleukin-17, inflammation, synergy
Introduction
The cytokine IL-17, originally termed CTLA-8, was isolated as a CD4-specific transcript from a rodent cDNA library [1]. Shortly thereafter, IL-17 was discovered in humans, and its receptor (IL-17R) was cloned and characterized [2-4]. The most striking feature of both IL-17 and IL-17R is that they are distinct in sequence from previously described cytokine/receptor families. However, they are highly homologous among mice, rats, and humans. In addition, an IL-17R homolog in zebrafish (termed SEF [similar expression of FGF genes]) has been described that functions in embryonic development [5], and mammalian homologs of SEF were also recently identified [6,7]. Consequently, IL-17 and IL-17R are now recognized to be the founding members of an emerging new family that, in mammals, contains at least six cytokines and five receptors (Table 1[8,9]). This review focuses primarily on the original IL-17 cytokine (also known as IL-17A), because its roles in bone physiology and arthritis are most clearly defined, but the biology of the remaining family members promises to be a fascinating emerging story within the field of 'high numbered' cytokines.
Table 1.
The IL-17 superfamily: cellular sources, receptors, and major functions
| Cytokine | Other names | Cellular source | Receptor | Major functions |
| IL-17 | IL-17A, CTLA-8 | T cells (memory) | IL-17R (also known as, IL-17AR) | Inflammation, neutrophil recruitment, cytokine secretion, bone metabolism |
| IL-17B | Multiple organs | IL-17BR (also known as, IL-17Rh1/Evi27) | Cytokine secretion, inflammation | |
| IL-17C | Unknown | Unknown | Regulation of Th1 cytokines | |
| IL-17D | IL-27 | Multiple organs | Unknown | Cytokine secretion |
| IL-17E | IL-25 | Th2 | IL-17BR (also known as, IL-17Rh1/Evi27) | Regulation of Th2 cytokines |
| IL-17F | ML-1 | CD4+ T cells and monocytes | IL-17R? | Angiogenesis, cytokine secretion, regulation of Th1 cytokines |
| HVS13 | vIL-17 | Herpesvirus saimiri infected cells | IL-17R? | Unknown (not required for cellular transformation) |
References and further information on this family can be found in the report by Moseley and coworkers [24]. CTLA, cytotoxic T-lymphocyte associated antigen; IL, interleukin; IL-17R, interleukin-17 receptor; Th, T-helper.
Interleukin-17 and interleukin-17 receptor structure
Even though IL-17 and IL-17R have been recognized for many years, there is still much to learn about their respective structures and functions. IL-17 is secreted primarily by CD4+ T cells in a mix of both nonglycosylated and N-glycosylated forms, which migrate in SDS-PAGE at 28 kDa and 33 kDa, respectively [2]. Secreted IL-17 apparently exists as a homodimer, but the specific contact points between IL-17 subunits or between IL-17 and IL-17R have never been defined [2,10]. IL-17B and IL-17F also exist as dimers [10,11]. While the amino acid sequence of IL-17 did not permit it to be classified as a member of any known cytokine families, X-ray crystallographic studies of IL-17F – its closest homolog – have been performed. Interestingly, the three-dimensional structure of IL-17F takes on a 'cystine knot fold', and hence resembles the neurotrophin family of growth factors, the canonical member of which is nerve growth factor [10].
The IL-17R is also particularly interesting because of its unique primary structure. It contains a single transmembrane domain and has an unusually large cytoplasmic tail [4,12]. This receptor is expressed in most cell types. One exception is in naïve T cells in mice, which do not bind IL-17 detectably (Dong C, personal communication). However, several mouse and human T cell lines do contain detectable mRNA encoding IL-17R, and so this receptor may be present in at least low levels in T cells (Gaffen SL, unpublished data) [12]. As a result of its ubiquitous expression, nearly all cells are potential targets of this cytokine, but it is still unclear which cells in vivo are the most physiologically relevant targets. Most studies to date have been performed in cells of fibroblast/osteoblast or epithelial origin, because these appear to be particularly responsive to IL-17. Although there was originally thought to be a unique cytokine–receptor relationship between IL-17 and IL-17R, more recent studies indicate that IL-17F binds, albeit weakly, to IL-17R [10]. Whereas IL-17 is composed of a homodimer of identical subunits, the configuration and stoichiometry of the receptor remain undefined. In this regard, discrepancies between IL-17 binding constants and concentrations needed to elicit biologic responses have hinted that an additional subunit might be involved in IL-17 signaling [10,12]. However, IL-17R is clearly an essential subunit, because cells from IL-17R-/- mice fail to bind to IL-17.
Sources, regulation, and biologic functions of interleukin-17
IL-17 is produced almost exclusively by T lymphocytes, primarily those of the CD4+ memory (CD45RO+) compartment [2,13,14]. Consequently, IL-17 does not obviously polarize to either the T-helper-1 or -2 lineages, although the literature is somewhat inconsistent in this regard [15-19]. Consistent with its production by memory cells, several recent studies have shown that IL-23, which is produced by dendritic cells (DCs) and acts mainly on memory T cells, is a potent stimulator of IL-17 secretion [20,21]. However, it should be noted that signaling through the T-cell receptor alone is sufficient to promote IL-17 production even in the absence of DCs or IL-23 (Liu X, Clements J, Gaffen S, unpublished data), and IL-23 deficient mice are still capable of producing IL-17, albeit at reduced levels [22]. In addition, IL-15 has been shown to induce IL-17 production [23].
The gene encoding human IL-17 resides on human chromosome 6, adjacent to the gene encoding IL-17F [10], whereas other IL-17 family members are located elsewhere in the genome [24]. We recently showed that a minimal regulatory promoter element exists about 250 bases upstream of the transcriptional starting point [25]. In this regard, the signaling pathways leading to IL-17 gene regulation by any of these stimuli are poorly defined, although several studies indicated that the calcineurin/ NFAT (nuclear factor of activated T cells) pathway is essential [23,25] (Liu X, Clements J, Gaffen S, unpublished data). Other studies also indicate a role for the cAMP/protein kinase A pathway, although this signal may ultimately converge on NFAT signaling [13,14,26]. Like many cytokines, IL-17 gene expression is likely to be at least partially controlled at the level of mRNA stability, because AU-rich elements exist in the 3'-untranslated region that could target the transcript for rapid degradation [2,27,28]. Clearly, much still remains to be learned about how IL-17 expression is controlled biologically.
Functionally, IL-17 has been classified as a pro-inflammatory mediator, based on its ability to induce a wide array of inflammatory effectors in target cells (Fig. 1). Among these are cytokines (e.g. IL-6, tumor necrosis factor [TNF]-α, IL-1β, IFN-γ, and granulocyte colony-stimulating factor), chemokines (e.g. CXC chemokine ligand [CXCL]2/MIP-2/IL-8, CXCL1/Groα /KC, CC chemokine ligand [CCL]2/MCP-1, CCL5/RANTES, and CXCL5/LIX), and other effectors (e.g. cyclo-oxygenase-2, prostaglandin E2, nitric oxide, and intercellular adhesion molecule-1; for review [8]). Moreover, IL-17 cooperates either additively or synergistically with various inflammatory cytokines or agonists, thus placing this cytokine in the midst of a complex network that amplifies inflammation (see below). In this sense, IL-17 appears to function as an activator of the innate immune system, analogous to TNF-α and IL-1β, with which it shares many target genes. However, because IL-17 is produced by T cells rather than by monocytes or other innate cells, it presumably comes into play during adaptive or memory immune responses. Consequently, the function of IL-17 may be to trigger innate immune responses shortly after a second encounter with antigen, when the memory response is activated but when concentrations of antigen are still too low to trigger a full-scale innate immune response.
Figure 1.
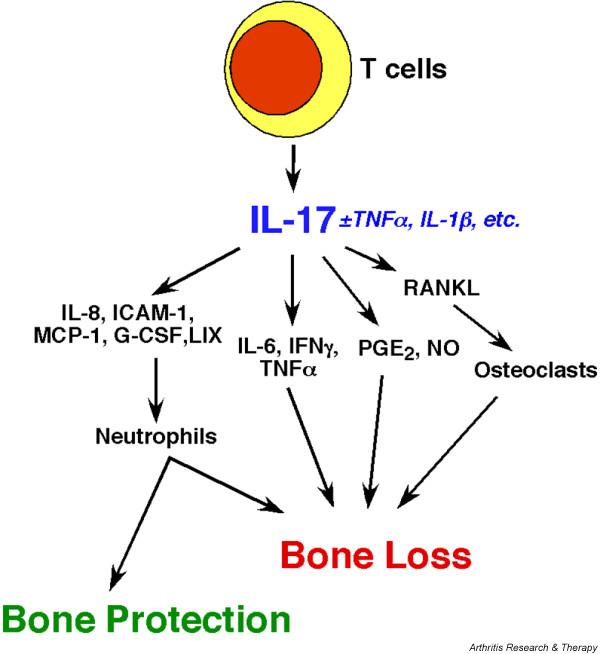
Opposing roles of IL-17 in bone turnover. IL-17 is produced by T cells (particularly memory T cells), and acts on a wide variety of target cells to trigger expression of inflammatory effectors. Most of these effectors have been shown to have an impact on bone metabolism. Those factors that promote osteoclastogenesis indirectly favor bone destruction. Conversely, chemotactic factors promote neutrophil recruitment and activation, which can exert both bone protective and bone destructive effects. G-CSF, granulocyte colony-stimulating factor; ICAM, intercellular adhesion molecule; IFN, interferon; IL, interleukin; LIX, LPS-inducible CXC chemokine; MCP, monocyte chemotactic protein; PGE2, prostaglandin E2; RANKL, receptor activator of nuclear factor-κB ligand; TNF, tumor necrosis factor.
Interleukin-17 as a synergistic cytokine
A prominent feature of IL-17 is its ability to synergize with other cytokines to enhance inflammation (for review [29]). In particular, IL-17 has been shown to synergize with IL-1β and TNF-α to drive expression of numerous inflammatory effectors [18,30-35]. IL-17 also synergizes with CD40 ligand, a TNF receptor family member, to upregulate target gene expression [36]. Similarly, IL-17 synergizes with IFN-γ to promote chemokine gene expression [37]. Microarray analysis of an osteoblast cell line examining synergy between IL-17 and TNF-α revealed that all genes induced by IL-17 alone were induced more potently in cooperation with TNF-α. This finding suggests that a primary function of IL-17 may be to amplify ongoing inflammatory responses [34,35].
Although the molecular mechanisms that mediate cytokine synergy are not fully understood, several have been proposed. For example, IL-17 cooperates with TNF-α or IL-1β to enhance mRNA stabilization of the CXCL1/Groα/KC chemokine transcript in peritoneal mesothelial cells [33]. In its synergy with CD40 ligand, IL-17 upregulates expression of CD40, thus enhancing all CD40 ligand dependent responses [36]. However, this is not true of IL-17 synergy with TNF-α, because IL-17 does not appear to enhance TNF receptor expression in osteoblasts [35]. Although IL-17 synergy with IFN-γ has been reported to occur via enhancement of the nuclear factor-κB (NF-κB) pathway [37], this is not the mechanism by which IL-17 synergizes with TNF-α [35]. Rather, we recently showed that IL-17 synergizes with TNF-α to promote IL-6 production by upregulating expression of CCAAT/enhancer binding protein (C/EBP)δ (also known as NF-IL-6β), a member of the bZIP family of transcription factors. The conserved C/EBP site in the IL-6 proximal promoter is essential for expression of IL-6, and thus cooperative upregulation of C/EBPδ mediated by IL-17 and TNF-α helps in turn to enhance transcription of the IL-6 gene [35,38]. Another report suggested that p38/mitogen-activated protein kinase (MAPK) may be a target of cooperative signaling between IL-17 and TNF-α [39]. In addition to transcription and RNA stability, synergistic signaling may affect regulation of chromatin remodeling, cytokine secretion, and possibly other levels of gene or protein regulation. Given the proclivity of IL-17 to function in concert with other cytokines, it will be very important to dissect the multiple mechanisms by which this cytokine promotes cooperative/synergistic signaling.
The immune system and bone homeostasis
Bone undergoes a continuous cycle of remodeling that is required for its maintenance and healing, and recent advances have elucidated many of the molecular mechanisms that regulate or have an impact on this process (for review [40,41]). Two major types of cells are involved in bone remodeling. Osteoblasts, cells that are crucially involved in bone formation, are derived from mesenchymal stem cells and are closely related to fibroblasts, adipocytes, and muscle cells [42]. Osteoclasts, the cells responsible for bone degradation, are derived from hematopoietic precursors, and are thus related to macrophages and DCs [43]. In normal physiology, osteoblasts trigger the formation of osteoclasts, thus helping to maintain homeostasis in bone remodeling. Conversely, the bone resorbing activity of osteoclasts causes the release of various growth factors and bone cell mitogens that induce proliferation and differentiation of osteoblasts [40]. Importantly, a number of pathologic conditions adversely affect bone by altering the balance between osteoblast and osteoclast activity, causing localized or systemic osteoporosis (or, less frequently, osteopetrosis) [41,44]. Such conditions may have severe medical and economic consequences. For example, it is estimated that as many as 15% of adults suffer from periodontal disease severe enough to cause tooth loss, and the acute crippling in advanced rheumatoid arthritis (RA) can have devastating consequences for the quality of life of its victims. Therefore, it is paramount to understand the network of factors that control bone homeostasis, in order to develop optimal avenues of intervention and treatment in diseases that involve bone loss.
Recent discoveries have significantly advanced our understanding of the molecular basis for bone turnover (for review [41,45]). At a molecular level, osteoblasts express a receptor called RANKL (receptor activator of NF-κB ligand; also termed osteoprotegerin [OPG] ligand). RANKL is a member of the TNF receptor superfamily and is central in controlling osteoclastogenesis, and hence bone degradation [46,47]. RANKL acts by engaging its counter-receptor RANK (receptor activator of NF-κB) on osteoclast precursors, thereby triggering their maturation and activation in conjunction with signals from the growth factor macrophage colony-stimulating factor [48]. The interaction between RANK and RANKL can be further modulated by a soluble 'decoy' receptor called OPG, which also binds to RANK but does not induce osteoclastogenesis [49]. The relative balance between OPG and RANKL dictates the magnitude of osteoclastogenesis. For many years it has been recognized that the immune system exerts a profound effect on bone cell activity, explaining why infectious diseases such as periodontal disease or autoimmune diseases such as RA are associated with bone destruction (for review [50]). In particular, both T cells and inflammatory cytokines have been implicated in this process. Interestingly, activated T cells inducibly express RANKL, and can thus bypass osteoblasts in triggering osteoclastogenesis, ultimately tipping the balance in favor of bone destruction [51]. Inflammatory cytokines such as TNF-α or IL-1β (and IL-17; see below) act on osteoblasts to upregulate RANKL, either directly or indirectly through the production of other cytokines/chemokines [52]. Clinical strategies to block cytokines such as TNF-α and IL-1β have been quite effective in treating RA, and efforts are underway to influence the RANK–RANKL axis directly through the therapeutic use of OPG [45,53].
Evidence for a role of interleukin-17 in bone and arthritis
A number of studies have implicated IL-17 in bone metabolism. Most prominently, IL-17 is found at significantly elevated levels in the synovial fluid of patients with RA, and is present in osteoarthritic joints as well [54]. IL-17 has also been found in patients with relatively severe periodontitis, where it could potentially contribute to bone destruction [55]. In addition, IL-17 exerts many of its effects on bone cells in culture [54,56], including induction of both membrane-bound and soluble RANKL in primary mouse osteoblast/stromal cell cultures [52]. IL-17 is strongly implicated in several mouse models of RA. Enhancement of RANKL following IL-17 stimulation was not observed in several osteoblast or stromal cell lines, including MC3T3-E1 or ST-2 cells (Kirkwood KL, personal communication). However, in vivo bone erosion mediated by over-expression of IL-17 has been shown to occur through alterations in the RANKL/OPG ratio [57]. Furthermore, IL-17 knockout mice are highly resistant to collagen induced arthritis (CIA) [58], and blocking IL-17 reduces inflammatory symptoms and bone loss in mice with CIA [59,60]. Conversely, excess IL-17, as provided by adenovirus-mediated gene vectors, exacerbates disease [61-64]. Remarkably, mice deficient in the T cell costimulatory molecule ICOS (inducible co-stimulator) are also profoundly resistant to CIA, and the only cytokine deficiency detected in these mice was a reduction in IL-17 [65].
It is also striking that most IL-17-induced factors tend to be bone resorptive in nature (Fig. 1; for review [66]). For example, IL-6 has been shown to be a contributing factor to estrogen mediated bone loss [67] as well as bone loss due to periodontal disease [68]. Similarly, CXCL8/IL-8, prostaglandin E2, and nitric oxide have all been implicated in the pathogenesis of periodontitis [69]. However, the role played by neutrophils in bone turnover is more complex. During chronic inflammation, neutrophils are thought to contribute to bone destruction. However, neutrophils are generally considered to be bone protective in the context of periodontal disease-induced bone loss (for review [70,71]). IL-17 is a potent activator of neutrophil recruitment and activation, due in large part to its ability to promote chemokine secretion. Thus, IL-17 could potentially play a positive role in situations where neutrophil activity is bone protective.
In summary, IL-17 clearly has an impact on bone metabolism, and in the context of arthritis it appears to be a bone destructive cytokine.
Interleukin-17 in other diseases
IL-17 has been implicated in numerous other disease settings. Intriguingly, IL-17 is highly homologous to an open reading frame found in the T cell tropic Herpesvirus saimiri, although its physiologic significance within the context of this virus remains unknown [12,72]. However, addition of the gene encoding murine IL-17 into the vaccinia virus enhanced its virulence significantly, suggesting a possible pathogenic role for this cytokine in viral infections [73]. The role played by IL-17 in tumorigenesis is complex. IL-17 was shown to promote growth and tumorigenicity of human cervical tumors in athymic (nude) mice [74]. In contrast, IL-17 also inhibited the growth of hematopoietic tumors in immunocompetent but not nude mice [75]. IL-17 has also been found at elevated levels in the context of bacterial infections, such as periodontitis [55] and Helicobacter pylori infections [76]. Finally, IL-17 plays an important role in immune responses in the lung. Specifically, IL-17R-/- mice are highly susceptible to lung airway infections due to a failure to recruit neutrophils [77]. Human bronchial epithelial cells induce chemokines following IL-17 stimulation, and local administration of IL-17 in mouse lung tissue causes neutrophil recruitment and increases in elastase and myeloperoxidase activities (for review [78,79]). Finally, data from IL-17-/- and IL-17R-/- mice indicate that this cytokine is also involved in a variety of other T cell dependent events. For example, delayed type hypersensitivity and contact hypersensitivity responses are severely impaired in IL-17-/- mice [80]. Interestingly, attempts to over-express IL-17 transgenically have not been successful, perhaps because of a generalized inflammation that is lethal to developing embryos [81]. Thus, IL-17 is important for numerous immune functions related to regulation of inflammation, and can play both pathogenic and protective roles in vivo.
Interleukin-17 and interleukin-17 receptor signaling
The signaling mechanisms used by IL-17 to regulate its downstream targets are surprisingly poorly defined. As indicated above, the IL-17R is the founding member of a new subclass of cytokine receptors that do not bear homology to type I or II cytokine receptors, TNF receptors, or other receptor families [12,82]. Because so little is known about signaling pathways induced by this class of receptor, few predictions can be made based its primary amino acid structure.
Recently, however, it was suggested that IL-17 receptors may contain a putative TIR (Toll/IL-1 receptor) domain in the intracellular region [7], and the IL-17R tail also contains at least two putative TNF receptor-associated factor (TRAF)-binding domains (Gaffen SL, unpublished observations) [83]. Although early reports suggested that IL-17 activates the transcription factor NF-κB [12], careful comparisons show that NF-κB induction is generally quite modest as compared with that triggered by TNF-α or Toll-like receptor agonists [35]. Other signaling pathways implicated in IL-17 signaling include the MAPK, protein kinase A and JAK/STAT (Janus kinase/signal transducer and activator of transcription) pathways (for review [8]). However, only in a few cases have these pathways been linked to specific signaling outcomes. One study showed convincingly that IL-17 recruits the adaptor molecule TRAF6 in murine embryonic fibroblast cells, which are among the few cell types that induce NF-κB strongly. In these cells, TRAF6 lies upstream of signaling leading to IL-6 and intercellular adhesion molecule-1 expression [84]. Based on paradigms in the TNF and Toll-like receptors, TRAF6 probably also lies upstream of MAPK signaling, although this remains to be proven for the IL-17R [85]. In another study, the IL-17-induced MAPK pathway was linked to IL-6 gene expression via stabilization of IL-6 mRNA [39]. Similarly, IL-17 alone mediates cyclo-oxygenase-2 mRNA stability in a p38-MAPK dependent manner [86]. To date, no detailed mutagenesis studies of IL-17R have been performed, and so regions of the receptor required for activation of various signaling pathways have not yet been determined.
Conclusion
IL-17 is the prototypical member of a fascinating new family of cytokines. Although it is clear that IL-17 is proinflammatory in nature, its physiologic significance is only just beginning to be elucidated. The unique structure of IL-17 and its receptor hint at exciting new discoveries in the area of signal transduction as well as potential therapeutic intervention strategies. With respect to arthritis, IL-17 appears to be largely pathogenic. However, findings in IL-17 and IL-17R knockout mice indicate a nonredundant role for this cytokine in regulating host immunity to infection. Future work on the IL-17 family will no doubt yield many surprises, and will probably establish new paradigms for cytokine biology.
Abbreviations
CCL = CC chemokine ligand; C/EBP = CCAAT/enhancer binding protein; CIA = collagen induced arthritis; CXCL = CXC chemokine ligand; DC = dendritic cell; IFN = interferon; IL = interleukin; IL-17R = IL-17 receptor; MAPK = mitogen-activated protein kinase; NF-κB = nuclear factor-κB; OPG = osteoprotegerin; RA = rheumatoid arthritis; RANKL = receptor activator of nuclear factor-κB ligand; TNF = tumor necrosis factor; TRAF = tumor necrosis factor receptor-associated factor.
Competing interests
The author has received travel reimbursement and an honorarium from Amgen Corporation, Seattle, WA, USA.
Note
This article is the second in a review series on Biology of recently discovered cytokines edited by John O'Shea
Other articles in the series can be found at http://arthritis-research.com/articles/review-series.asp
Acknowledgments
Acknowledgments
We thank Drs Austin Gurney (Genentech, South San Francisco, CA, USA) and Joel Tocker (Amgen Corporation, Seattle, WA, USA) for helpful suggestions. We also thank Drs Chen Dong (University of Washington, Seattle, WA, USA) and KL Kirkwood (University of Michigan, Ann Arbor, MI, USA) for sharing unpublished information.
This work was supported by the US National Institutes of Health (AI49329) and the Arthritis Foundation.
References
- Rouvier E, Luciani M-F, Mattei M-G, Denizot F, Golstein P. CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a Herpesvirus Saimiri gene. J Immunol. 1993;150:5445–5456. [PubMed] [Google Scholar]
- Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, Pin J-J, Garrone P, Garcia E, Saeland S, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. 1996;183:2593–2603. doi: 10.1084/jem.183.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Painter SL, Fanslow WC, Ulrich D, Macduff BM, Spriggs MK, Armitage RJ. Cutting Edge: Human IL-17: A novel cytokine derived from T cells. J Immunol. 1995;155:5483–5486. [PubMed] [Google Scholar]
- Yao Z, Spriggs MK, Derry JMJ, Strockbine L, Park LS, VandenBos T, Zappone J, Painter SL, Armitage RJ. Molecular characterization of the human interleukin-17 receptor. Cytokine. 1997;9:794–800. doi: 10.1006/cyto.1997.0240. [DOI] [PubMed] [Google Scholar]
- Tsang M, Friesel R, Kudoh T, Dawid IB. Identification of Sef, a novel modulator of FGF signalling. Nat Cell Biol. 2002;4:165–169. doi: 10.1038/ncb749. [DOI] [PubMed] [Google Scholar]
- Yang RB, Ng CK, Wasserman SM, Komuves LG, Gerritsen ME, Topper JN. A novel interleukin-17 receptor-like protein identified in human umbilical vein endothelial cells antagonizes basic fibroblast growth factor-induced signaling. J Biol Chem. 2003;278:33232–33238. doi: 10.1074/jbc.M305022200. [DOI] [PubMed] [Google Scholar]
- Xiong S, Zhao Q, Rong Z, Huang G, Huang Y, Chen P, Zhang S, Liu L, Chang Z. hSef inhibits PC-12 cell differentiation by interfering with Ras-mitogen-activated protein kinase MAPK signaling. J Biol Chem. 2003;278:50273–50282. doi: 10.1074/jbc.M306936200. [DOI] [PubMed] [Google Scholar]
- Aggarwal S, Gurney AL. IL-17: A prototype member of an emerging cytokine family. J Leukoc Biol. 2002;71:1–8. [PubMed] [Google Scholar]
- Eberl M. Don't count your interleukins before they've hatched. Trends Immunol. 2002;23:341–342. doi: 10.1016/S1471-4906(02)02239-1. [DOI] [PubMed] [Google Scholar]
- Hymowitz SG, Filvaroff EH, Yin JP, Lee J, Cai L, Risser P, Maruoka M, Mao W, Foster J, Kelley RF, et al. IL-17s adopt a cystine knot fold: structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. EMBO J. 2001;20:5332–5341. doi: 10.1093/emboj/20.19.5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Ullrich SJ, Zhang J, Connolly K, Grzegorzewski KJ, Barber MC, Wang W, Wathen K, Hodge V, Fisher CL, et al. A novel cytokine receptor-ligand pair. Identification, molecular characterization, and in vivo immunomodulatory activity. J Biol Chem. 2000;275:19167–19176. doi: 10.1074/jbc.M910228199. [DOI] [PubMed] [Google Scholar]
- Yao Z, Fanslow WC, Seldin MF, Rousseau A-M, Painter SL, Comeau MR, Cohen JI, Spriggs MK. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. 1995;3:811–821. doi: 10.1016/1074-7613(95)90070-5. [DOI] [PubMed] [Google Scholar]
- Shin HC, Benbernou N, Fekkar H, Esnault S, Guenounou M. Regulation of IL-17, IFN-gamma and IL-10 in human CD8 + T cells by cyclic AMP-dependent signal transduction pathway. Cytokine. 1998;10:841–850. doi: 10.1006/cyto.1998.0375. [DOI] [PubMed] [Google Scholar]
- Shin HC, Benbernou N, Esnault S, Guenounou M. Expression of IL-17 in human memory CD45RO+ T lymphocytes and its regulation by protein kinase A pathway. Cytokine. 1999;11:257–266. doi: 10.1006/cyto.1998.0433. [DOI] [PubMed] [Google Scholar]
- Teunissen MB, Koomen CW, de Waal Malefyt R, Wierenga EA, Bos JD. Interleukin-17 and interferon-γ synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol. 1998;111:645–649. doi: 10.1046/j.1523-1747.1998.00347.x. [DOI] [PubMed] [Google Scholar]
- Aarvak T, Chabaud M, Miossec P, Natvig JB. IL-17 is produced by some proinflammatory Th1/Th0 cells but not by Th2 cells. J Immunol. 1999;162:1246–1251. [PubMed] [Google Scholar]
- Chabaud M, Page G, Miossec P. Enhancing effect of IL-1, IL-17, and TNF-alpha on macrophage inflammatory protein-3alpha production in rheumatoid arthritis: regulation by soluble receptors and Th2 cytokines. J Immunol. 2001;167:6015–6020. doi: 10.4049/jimmunol.167.10.6015. [DOI] [PubMed] [Google Scholar]
- Albanesi C, Cavani A, Girolomoni G. IL-17 is produced by nickel-specific T lymphocytes and regulates ICAM-1 expression and chemokine production in human keratinocytes: synergistic or antagonistic effects with IFN-γ and TNF-α. J Immunol. 1999;162:494–502. [PubMed] [Google Scholar]
- Infante-Duarte C, Horton HF, Byrne MC, Kamradt T. Microbial lipopeptides induce the production of IL-17 in Th cells. J Immunol. 2000;165:6107–6115. doi: 10.4049/jimmunol.165.11.6107. [DOI] [PubMed] [Google Scholar]
- Happel KI, Zheng M, Young E, Quinton LJ, Lockhart E, Ramsay AJ, Shellito JE, Schurr JR, Bagby GJ, Nelson S, et al. Cutting edge: roles of Toll-like receptor 4 and IL-23 in IL-17 expression in response to Klebsiella pneumoniae infection. J Immunol. 2003;170:4432–4436. doi: 10.4049/jimmunol.170.9.4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal S, Ghilardi N, Xie MH, De Sauvage FJ, Gurney AL. Interleukin 23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin 17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- Ghilardi N, Kljavin N, Chen Q, Lucas S, Gurney A, de Sauvage FJ. Compromised humoral and delayed-type hypersensitivity responses in IL-23-deficient mice. J Immunol. 2004;172:2827–2833. doi: 10.4049/jimmunol.172.5.2827. [DOI] [PubMed] [Google Scholar]
- Ziolkowska M, Koc A, Luszczukiewicz G, Ksiezopolksa-Pietrzak K, Klimczak E, Chwalinska-Sadowska H, Maslinski W. High levels of IL-17 in rheumatoid arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporin A-sensitive mechanism. J Immunol. 2000;164:2832–2838. doi: 10.4049/jimmunol.164.5.2832. [DOI] [PubMed] [Google Scholar]
- Moseley TA, Haudenschild DR, Rose L, Reddi AH. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003;14:155–174. doi: 10.1016/S1359-6101(03)00002-9. [DOI] [PubMed] [Google Scholar]
- Liu X, Lin X, Gaffen SL. Crucial role for nuclear factor of activated T cells (NFAT) in T cell receptor-mediated regulation of the human interleukin-17 gene. J Biol Chem. 2004, Sep 30. [DOI] [PubMed]
- Sheridan CM, Heist EK, Beals CR, Crabtree GR, Gardner P. Protein kinase A negatively modulates the nuclear accumulation of NF-ATc1 by priming for subsequent phosphorylation by glycogen synthase kinase-3. J Biol Chem. 2002;277:48664–48676. doi: 10.1074/jbc.M207029200. [DOI] [PubMed] [Google Scholar]
- Yao Z, Timour M, Painter S, Fanslow W, Spriggs M. Complete nucleotide sequence of the mouse CTLA8 gene. Gene. 1996;168:223–225. doi: 10.1016/0378-1119(95)00778-4. [DOI] [PubMed] [Google Scholar]
- Lindsten T, June CH, Ledbetter JA, Stella G, Thompson CB. Regulation of lymphokine messenger RNA stability by a surface-mediated T-cell activation pathway. Science. 1989;244:339–343. doi: 10.1126/science.2540528. [DOI] [PubMed] [Google Scholar]
- Miossec P. Interleukin-17 in rheumatoid arthritis: if T cells were to contribute to inflammation and destruction through synergy. Arthritis Rheum. 2003;48:594–601. doi: 10.1002/art.10816. [DOI] [PubMed] [Google Scholar]
- Laan M, Cui A-H, Hoshino H, Lötvall J, Sjöstrand M, Gruenert DC, Skoogh B-E, Lindén A. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J Immunol. 1999;162:2347–2352. [PubMed] [Google Scholar]
- Maertzdorf J, Osterhaus AD, Verjans GM. IL-17 expression in human herpetic stromal keratitis: modulatory effects on chemokine production by corneal fibroblasts. J Immunol. 2002;169:5897–5903. doi: 10.4049/jimmunol.169.10.5897. [DOI] [PubMed] [Google Scholar]
- Van Bezooijen RL, Papapoulos SE, Lowik CW. Effect of interleukin-17 on nitric oxide production and osteoclastic bone resorption: is there dependency on nuclear factor-kappaB and receptor activator of nuclear factor kappaB (RANK)/RANK ligand signaling? Bone. 2001;28:378–386. doi: 10.1016/S8756-3282(00)00457-9. [DOI] [PubMed] [Google Scholar]
- Witowski J, Pawlaczyk K, Breborowicz A, Scheuren A, Kuzlan-Pawlaczyk M, Wisniewska J, Polubinska A, Friess H, Gahl GM, Frei U, et al. IL-17 stimulates intraperitoneal neutrophil infiltration through the release of GRO alpha chemokine from mesothelial cells. J Immunol. 2000;165:5814–5821. doi: 10.4049/jimmunol.165.10.5814. [DOI] [PubMed] [Google Scholar]
- Ruddy MJ, Shen F, Smith J, Sharma A, Gaffen SL. Interleukin-17 regulates expression of the CXC chemokine LIX/CXCL5 in osteoblasts: implications for inflammation and neutrophil recruitment. J Leukoc Biol. 2004;76:135–144. doi: 10.1189/jlb.0204065. [DOI] [PubMed] [Google Scholar]
- Ruddy MJ, Wong GC, Liu XK, Yamamoto H, Kasayama S, Kirkwood KL, Gaffen SL. Functional cooperation between interleukin-17 and tumor necrosis factor-alpha is mediated by CCAAT/enhancer binding protein family members. J Biol Chem. 2004;279:2559–2567. doi: 10.1074/jbc.M308809200. [DOI] [PubMed] [Google Scholar]
- Woltman AM, de Haij S, Boonstra JG, Gobin SJ, Daha MR, van Kooten C. Interleukin-17 and CD40-ligand synergistically enhance cytokine and chemokine production by renal epithelial cells. J Am Soc Nephrol. 2000;11:2044–2055. doi: 10.1681/ASN.V11112044. [DOI] [PubMed] [Google Scholar]
- Takaya H, Andoh A, Makino J, Shimada M, Tasaki K, Araki Y, Bamba S, Hata K, Fujiyama Y, Bamba T. Interleukin-17 stimulates chemokine (interleukin-8 and monocyte chemoattractant protein-1) secretion in human pancreatic periacinar myofibroblasts. Scand J Gastroenterol. 2002;37:239–245. doi: 10.1080/003655202753416948. [DOI] [PubMed] [Google Scholar]
- Eickelberg O, Pansky A, Mussmann R, Bihl M, Tamm M, Hilde-brand P, Perruchoud AP, Roth M. Transforming growth factor-beta1 induces interleukin-6 expression via activating protein-1 consisting of JunD homodimers in primary human lung fibroblasts. J Biol Chem. 1999;274:12933–12938. doi: 10.1074/jbc.274.18.12933. [DOI] [PubMed] [Google Scholar]
- Tokuda H, Kanno Y, Ishisaki A, Takenaka M, Harada A, Kozawa O. Interleukin (IL)-17 enhances tumor necrosis factor-alpha-stimulated IL-6 synthesis via p38 mitogen-activated protein kinase in osteoblasts. J Cell Biochem. 2004;91:1053–1061. doi: 10.1002/jcb.20004. [DOI] [PubMed] [Google Scholar]
- Baker P. The role of immune responses in bone loss during periodontal disease. Microbes Infect. 2000;2:1181–1192. doi: 10.1016/S1286-4579(00)01272-7. [DOI] [PubMed] [Google Scholar]
- Theill L, Boyle W, Penninger J. RANK-L and RANK: T cells, bone loss and mammalian evolution. Annu Rev Immunol. 2002;20:795–823. doi: 10.1146/annurev.immunol.20.100301.064753. [DOI] [PubMed] [Google Scholar]
- Ducy P, Schinke T, Karsenty G. The osteoblast: a sophisticated fibroblast under central surveillance. Science. 2000;289:1501–1504. doi: 10.1126/science.289.5484.1501. [DOI] [PubMed] [Google Scholar]
- Teitelbaum S. Bone resorption by osteoclasts. Science. 2000;289:1504–1508. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- Rodan GA, Martin TJ. Therapeutic approaches to bone diseases. Science. 2000;289:1508–1514. doi: 10.1126/science.289.5484.1508. [DOI] [PubMed] [Google Scholar]
- Nakashima T, Wada T, Penninger JM. RANKL and RANK as novel therapeutic targets for arthritis. Curr Opin Rheumatol. 2003;15:280–287. doi: 10.1097/00002281-200305000-00016. [DOI] [PubMed] [Google Scholar]
- Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/S0092-8674(00)81569-X. [DOI] [PubMed] [Google Scholar]
- Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli S, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- Li J, Sarosi I, Yan XQ, Morony S, Capparelli C, Tan HL, McCabe S, Elliott R, Scully S, Van G, et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci USA. 2000;97:1566–1571. doi: 10.1073/pnas.97.4.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/S0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- Grcevic D, Katavic V, Lukic IK, Kovacic N, Lorenzo JA, Marusic A. Cellular and molecular interactions between immune system and bone. Croat Med J. 2001;42:384–392. [PubMed] [Google Scholar]
- Kong Y-Y, Gelge U, Sarosi H, Bolon B, Tafuri A, Morony S, Cappareli C, Li J, Elliott R, McCabe S, et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402:304–309. doi: 10.1038/46303. [DOI] [PubMed] [Google Scholar]
- Nakashima T, Kobayashi Y, Yamasaki S, Kawakami A, Eguchi K, Sasaki H, Sakai H. Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-kappaB ligand: modulation of the expression by osteotropic factors and cytokines. Biochem Biophys Res Commun. 2000;275:768–775. doi: 10.1006/bbrc.2000.3379. [DOI] [PubMed] [Google Scholar]
- Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med. 2001;344:907–916. doi: 10.1056/NEJM200103223441207. [DOI] [PubMed] [Google Scholar]
- van Bezooijen RL, Farih-Sips HCM, Papapoulos SE, Löwik CWGM. Interleukin-17: a new bone acting cytokine in vitro. J Bone Miner Res. 1999;14:1513–1521. doi: 10.1359/jbmr.1999.14.9.1513. [DOI] [PubMed] [Google Scholar]
- Johnson RB, Wood N, Serio FG. Interleukin-11 and IL-17 and the pathogenesis of periodontal disease. J Periodontol. 2004;75:37–43. doi: 10.1902/jop.2004.75.1.37. [DOI] [PubMed] [Google Scholar]
- Chabaud M, Durand JM, Buchs N, Fossiez F, Page G, Frappart L, Miossec P. Human interleukin 17: a T cell-derived proinflam-matory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999;42:963–970. doi: 10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Lubberts E, van den Bersselaar L, Oppers-Walgreen B, Schwarzenberger P, Coenen-de Roo CJ, Kolls JK, Joosten LA, van den Berg WB. IL-17 promotes bone erosion in murine collagen-induced arthritis through loss of the receptor activator of NF-kappa B ligand/osteoprotegerin balance. J Immunol. 2003;170:2655–2662. doi: 10.4049/jimmunol.170.5.2655. [DOI] [PubMed] [Google Scholar]
- Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173–6177. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- Lubberts E, Joosten LAB, Chabaud M, van den Bersselaar L, Oppers B, Coenen-de Roo CJJ, Richards CD, Miossec P, van den Berg WB. IL-4 gene therapy for collagen arthritis suppresses synovial IL-17 and osteoprotegerin ligand and prevents bone erosion. J Clin Invest. 2000;105:1697–1710. doi: 10.1172/JCI7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubberts E, Koenders MI, Oppers-Walgreen B, van den Bersselaar L, Coenen-de Roo CJ, Joosten LA, van den Berg WB. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. 2004;50:650–659. doi: 10.1002/art.20001. [DOI] [PubMed] [Google Scholar]
- Chabaud M, Lubberts E, Joosten L, van Den Berg W, Miossec P. IL-17 derived from juxta-articular bone and synovium contributes to joint degradation in rheumatoid arthritis. Arthritis Res. 2001;3:168–177. doi: 10.1186/ar294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubberts E, Joosten LA, Oppers B, van den Bersselaar L, Coenen-de Roo CJ, Kolls JK, Schwarzenberger P, van de Loo FA, van den Berg WB. IL-1-independent role of IL-17 in synovial inflammation and joint destruction during collagen-induced arthritis. J Immunol. 2001;167:1004–1013. doi: 10.4049/jimmunol.167.2.1004. [DOI] [PubMed] [Google Scholar]
- Lubberts E, Joosten LA, van de Loo FA, Schwarzenberger P, Kolls J, van den Berg WB. Overexpression of IL-17 in the knee joint of collagen type II immunized mice promotes collagen arthritis and aggravates joint destruction. Inflamm Res. 2002;51:102–104. doi: 10.1007/BF02684010. [DOI] [PubMed] [Google Scholar]
- Lubberts E, Joosten LA, van de Loo FA, van den Gersselaar LA, van den Berg WB. Reduction of interleukin-17-induced inhibition of chondrocyte proteoglycan synthesis in intact murine articular cartilage by interleukin-4. Arthritis Rheum. 2000;43:1300–1306. doi: 10.1002/1529-0131(200006)43:6<1300::AID-ANR12>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Nurieva RI, Treuting P, Duong J, Flavell RA, Dong C. Inducible costimulator is essential for collagen-induced arthritis. J Clin Invest. 2003;111:701–706. doi: 10.1172/JCI17321. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- McCauley LK. Transgenic mouse models of metabolic bone disease. Curr Opin Rheumatol. 2001;13:316–325. doi: 10.1097/00002281-200107000-00014. [DOI] [PubMed] [Google Scholar]
- Jilka RL, Hangoc G, Girasole G, Passeri G, Williams DC, Abrams JS, Boyce B, Broxmeyer H, Manolagas S. Increased osteoclast development after estrogen loss: mediated by interleukin-6. Science. 1992;257:88–91. doi: 10.1126/science.1621100. [DOI] [PubMed] [Google Scholar]
- Baker PJ, Dixon M, Evans RT, Dufour L, Johnson E, Roopenian DC. CD4+ T cells and the proinflammatory cytokines gamma interferon and interleukin-6 contribute to alveolar bone loss in mice. Infect Immun. 1999;67:2804–2809. doi: 10.1128/iai.67.6.2804-2809.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page R, Kornman K, editors . Periodontology 2000. Copenhagen: Munksgaard; 1997. [Google Scholar]
- Kantarci A, Oyaizu K, Van Dyke TE. Neutrophil-mediated tissue injury in periodontal disease pathogenesis: findings from localized aggressive periodontitis. J Periodontol. 2003;74:66–75. doi: 10.1902/jop.2003.74.1.66. [DOI] [PubMed] [Google Scholar]
- Baker PJ, DuFour L, Dixon M, Roopenian DC. Adhesion molecule deficiencies increase Porphyromonas gingivalis-induced alveolar bone loss in mice. Infect Immun. 2000;68:3103–3107. doi: 10.1128/IAI.68.6.3103-3107.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knappe A, Hiller C, Niphuis H, Fossiez F, Thurau M, Wittmann S, Kuhn EM, Lebecque S, Banchereau J, Rosenwirth B, et al. The interleukin-17 gene of herpesvirus saimiri. J Virol. 1998;72:5797–5801. doi: 10.1128/jvi.72.7.5797-5801.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patera AC, Pesnicak L, Bertin J, Cohen JI. Interleukin 17 modulates the immune response to vaccinia virus infection. Virology. 2002;299:56–63. doi: 10.1006/viro.2002.1400. [DOI] [PubMed] [Google Scholar]
- Tartour E, Fossiez F, Joyeux I, Galinha A, Gey A, Claret E, Sastre-Garau X, Couturier J, Mosseri V, Vives V, et al. Interleukin 17, a T-cell-derived cytokine, promotes tumorigenicity of human cervical tumors in nude mice. Cancer Res. 1999;59:3698–3704. [PubMed] [Google Scholar]
- Benchetrit F, Ciree A, Vives V, Warnier G, Gey A, Sautes-Fridman C, Fossiez F, Haicheur N, Fridman WH, Tartour E. Interleukin-17 inhibits tumor cell growth by means of a T-cell-dependent mechanism. Blood. 2002;99:2114–2121. doi: 10.1182/blood.V99.6.2114. [DOI] [PubMed] [Google Scholar]
- Luzza F, Parrello T, Monteleone G, Sebkova L, Romano M, Zarrilli R, Imeneo M, Pallone F. Up-regulation of IL-17 is associated with bioactive IL-8 expression in Helicobacter pylori-infected human gastric mucosa. J Immunol. 2000;165:5332–5337. doi: 10.4049/jimmunol.165.9.5332. [DOI] [PubMed] [Google Scholar]
- Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzen-berger P, Oliver P, Huang W, Zhang P, Zhang J, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden A, Adachi M. Neutrophilic airway inflammation and IL-17. Allergy. 2002;57:769–775. doi: 10.1034/j.1398-9995.2002.02164.x. [DOI] [PubMed] [Google Scholar]
- Schwarzenberger P, La Russa V, Miller A, Ye P, Huang W, Zieske A, Nelson S, Bagby GJ, Stoltz S, Mynatt RL, et al. IL-17 stimulates granulopoiesis in mice: Use of an alternate, novel gene therapy-derived method for in vivo evaluation of cytokines. J Immunol. 1998;161:6383–6389. [PubMed] [Google Scholar]
- Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–387. doi: 10.1016/S1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- Pan G, French D, Mao W, Maruoka M, Risser P, Lee J, Foster J, Aggarwal S, Nicholes K, Guillet S, et al. Forced expression of murine IL-17E induces growth retardation, jaundice, a Th2-biased response, and multiorgan inflammation in mice. J Immunol. 2001;167:6559–6567. doi: 10.4049/jimmunol.167.11.6559. [DOI] [PubMed] [Google Scholar]
- Ozaki K, Leonard WJ. Cytokine and cytokine receptor pleiotropy and redundancy. J Biol Chem. 2002;277:29355–29358. doi: 10.1074/jbc.R200003200. [DOI] [PubMed] [Google Scholar]
- Pullen SS, Dang TT, Crute JJ, Kehry MR. CD40 signaling through tumor necrosis factor receptor-associated factors (TRAFs). Binding site specificity and activation of downstream pathways by distinct TRAFs. J Biol Chem. 1999;274:14246–14254. doi: 10.1074/jbc.274.20.14246. [DOI] [PubMed] [Google Scholar]
- Schwandner R, Yamaguchi K, Cao Z. Requirement of tumor necrosis factor-associated factor (TRAF)6 in interleukin 17 signal transduction. J Exp Med. 2000;191:1233–1239. doi: 10.1084/jem.191.7.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S. Toll-like receptors and innate immunity. Adv Immunol. 2001;78:1–56. doi: 10.1016/S0065-2776(01)78001-7. [DOI] [PubMed] [Google Scholar]
- Faour WH, Mancini A, He QW, Di Battista JA. T-cell-derived interleukin-17 regulates the level and stability of cyclooxygenase-2 (COX-2) mRNA through restricted activation of the p38 mitogen-activated protein kinase cascade: role of distal sequences in the 3'-untranslated region of COX-2 mRNA. J Biol Chem. 2003;278:26897–26907. doi: 10.1074/jbc.M212790200. [DOI] [PubMed] [Google Scholar]