

Abstract
Background:
In EMBRACA, talazoparib prolonged progression-free survival versus chemotherapy (hazard ratio [HR] 0.542 [95% CI 0.413–0.711]; P < 0.0001) and improved patient-reported outcomes (PRO) in germline BRCA1/2 (gBRCA1/2)-mutated advanced breast cancer (ABC). We report final overall survival (OS) results.
Patients and methods:
This randomized phase III trial enrolled patients with gBRCA1/2-mutated HER2-negative ABC. Patients were randomized 2:1 to talazoparib or physician’s choice of chemotherapy. OS was analyzed using a stratified HR and log-rank test and prespecified rank-preserving structural failure time model to account for subsequent therapies.
Results:
431 patients were randomized (287 talazoparib/144 chemotherapy) with 412 patients treated (286 talazoparib/126 chemotherapy). By September 30, 2019, 216 deaths (75.3%) occurred for talazoparib and 108 (75.0%) for chemotherapy; median follow-up was 44.9 and 36.8 months, respectively. HR for OS was 0.848 (95% CI 0.670–1.073; P = 0.17). Kaplan-Meier survival percentages (95% CI) for talazoparib versus chemotherapy were: Month 12, 71% (66–76)/74% (66–81); Month 24, 42% (36–47)/38% (30–47); Month 36, 27% (22–33)/21% (14–29). Most patients received subsequent therapies: in the talazoparib and chemotherapy arms, respectively, 46.3%/41.7% received platinum therapy and 4.5%/32.6% received a poly(ADP-ribose) polymerase (PARP) inhibitor. Adjusting for subsequent PARP and/or platinum use, HR for OS was 0.756 (95% bootstrap CI 0.503–1.029). Grade 3–4 adverse events occurred in 69.6% (talazoparib) and 64.3% (chemotherapy) patients, consistent with previous reports. Extended follow-up showed significant overall improvement and delay in time to definitive clinically meaningful deterioration in both global health status/quality of life and breast symptoms favoring talazoparib versus chemotherapy (P < 0.01 for all), consistent with initial PRO analyses.
Conclusions:
In gBRCA1/2-mutated HER2-negative ABC, talazoparib did not significantly improve OS over chemotherapy, although subsequent treatments may have impacted analysis. Safety was consistent with previous observations. PRO continued to favor talazoparib.
ClinicalTrials.gov number:
Keywords: breast cancer, germline BRCA mutation, overall survival, PARP inhibitor, talazoparib
INTRODUCTION
Talazoparib is an oral inhibitor of poly(ADP-ribose) polymerase (PARP) catalytic activity,1 and it also has potent trapping activity of PARP at sites of damaged DNA.2 Deleterious mutations in breast cancer susceptibility genes 1 or 2 (BRCA1/2) leave cancer cells unable to repair damaged DNA through homologous recombination repair, and non-conservative repair mechanisms predominate causing DNA alterations and tumor cell death.3
Phase I/II studies (ABRAZO; NCT02034916) demonstrated that talazoparib has clinical benefit in patients with BRCA1/2-mutated advanced breast cancer (ABC).4, 5 The phase III EMBRACA trial (NCT01945775) compared the efficacy and safety of talazoparib with physician’s choice of chemotherapy for the treatment of HER2-negative ABC in patients with a germline BRCA1/2 mutation.6 EMBRACA is the largest trial of PARP monotherapy to date in ABC. Treatment with talazoparib significantly prolonged progression-free survival (PFS) versus chemotherapy (hazard ratio [HR] 0.542 [95% confidence interval (CI) 0.413–0.711; P < 0.0001]; median 8.6 months [95% CI 7.2–9.3] versus 5.6 months [95% CI 4.2–6.7]).6 At the time of the primary analysis, the HR for interim overall survival (OS; 163 events) was 0.761 (95% CI 0.547–1.060; P = 0.11). Patient-reported outcomes (PRO) favored talazoparib, with statistically significant overall improvement and significant delay in time to definitive clinically meaningful deterioration on the global health status/quality of life (GHS/QoL) and breast symptoms scales.6, 7 Most grade 3–4 adverse events (AEs) associated with the use of talazoparib were hematologic (primarily anemia), and were managed by supportive care and dose modifications.8, 9 Drug discontinuation due to AEs occurred in 5.9% of patients on talazoparib (in 1.4% due to hematologic AEs) and 8.7% of patients on chemotherapy. Talazoparib is approved in the US, EU, and other countries for the treatment of patients with HER2-negative, ABC with a germline BRCA1/2 mutation.10, 11
We report findings from the final analysis of OS with talazoparib compared with chemotherapy in EMBRACA, as well as updated safety and PRO.
METHODS
Trial design and patients
Details of the EMBRACA trial have been reported.6 This was an open-label, randomized, phase III trial in patients aged ≥18 years with HER2-negative locally advanced or metastatic breast cancer and a deleterious or suspected deleterious germline BRCA1/2 mutation detected by central testing with BRACAnalysis® (Myriad Genetics). Patients had received ≤3 previous cytotoxic regimens for advanced disease and previous treatment with a taxane, an anthracycline, or both, unless contraindicated. A protocol amendment (December 2015) expanded prior platinum use restrictions to permit enrolment if a patient had ≥6 months of stable disease following use of platinum in the neoadjuvant/adjuvant setting (versus ≥12 months in the prior version of protocol). Patients were randomized 2:1 to talazoparib (1 mg orally once daily) or a protocol-specified single-agent chemotherapy (capecitabine, eribulin, gemcitabine, or vinorelbine). Treatment was continued until disease progression or unacceptable toxicity. After treatment discontinuation, patients were followed every 12 weeks for survival status and subsequent use of anticancer therapy. The protocol and statistical analysis plan are available in the supplementary material, available at Annals of Oncology online.
End points and trial assessments
The primary endpoint, PFS, has been reported.6 OS, a prespecified secondary endpoint, was defined as the time from randomization to death due to any cause. For patients without a death date at the time of data cut-off or permanently lost to follow-up, OS was censored at the date the patient was last known to be alive. Prespecified exploratory subgroup analyses were performed at the final OS analysis to investigate treatment effects according to baseline characteristics and demographic factors. Updated safety (with long-term follow-up) and PRO (with the latest potential assessment occurring 27–40 days after the last dose of study drug) were performed (see supplementary material available online).
Statistical analysis
The trial was designed with adequate power to detect certain effect sizes for the primary endpoint, PFS, and for the secondary endpoint, OS. A total of 321 deaths would give the trial 80% power (at a two-sided alpha level of 5%) to detect a 39% increase in OS, with a targeted HR for death of 0.72. Assuming an exponential distribution of OS, this corresponds to an increase in median OS from 20 months to 27.8 months. An interim analysis of OS at a significance level of 0.0001 was conducted at the time of PFS primary analysis (data cut-off: September 15, 2017). The final analysis, using a significance level of 0.0499, was performed using the intent-to-treat population (ITT) when 324 deaths had been observed (data cut-off: September 30, 2019), and the OS analysis was conducted using the stratified two-sided log-rank test. Median OS was estimated using the Kaplan-Meier method, with 95% CIs calculated, and the HR was estimated using a stratified Cox regression model with treatment group as the only main effect. To maintain the overall two-sided type-I error rate of 5%, the analyses of PFS and OS were protected under a multiplicity adjustment schema using gate-keeping methodology (see supplementary material available online). No additional adjustments for multiplicity or repeated tests were implemented. A multi-covariate analysis was performed for OS. The HR was estimated using a stratified Cox regression model, with treatment group and selected prognostic factors as the main effects and a backward elimination process used to determine the final model. The prognostic factors included Eastern Cooperative Oncology Group (ECOG) score (0 versus >0), BRCA status (BRCA1 versus BRCA2), prior platinum treatment (yes versus no), and time from initial diagnosis of breast cancer to initial diagnosis of ABC (<12 months versus ≥12 months). Subgroup analyses of OS were conducted similarly to the analysis of the whole ITT population and displayed in a forest plot. Two analyses using the rank-preserving structural failure time model (RPSFTM) method were performed to estimate the treatment effect on OS accounting for subsequent treatment with a PARP inhibitor and/or platinum therapy, and PARP inhibitor therapy alone.12 Statistical methodology for the PRO, carried out without adjustment for multiplicity, was previously described.6, 7
RESULTS
Patients
Overall, 431 patients were randomized between October 2013 and April 2017, and included in the ITT population (supplementary Figure S1). Of these, 287 patients were randomized to talazoparib (286 treated) and 144 to chemotherapy (126 treated with capecitabine [55], eribulin [50], gemcitabine [12], and vinorelbine [9]). One patient randomized to talazoparib and 18 patients randomized to chemotherapy withdrew consent before receiving the drug. Eighteen patients (17 on talazoparib [5.9%], one on capecitabine [0.7%]) were ongoing at the data cut-off (September 30, 2019).
While the baseline characteristics were generally similar, the talazoparib group included a higher proportion of patients with a baseline ECOG performance status (PS) of 1 or 2 (46.4% versus 41.0%) and a higher proportion of patients whose breast cancer progressed to advanced disease within 12 months of initial diagnosis (37.6% versus 29.2%) than the chemotherapy group (supplementary Table S1). Further information regarding the baseline characteristics of the ITT population have been reported previously.6
Overall survival and subsequent therapies
At the time of the final OS analysis, 324 patients had died (216 [75.3%] in the talazoparib group and 108 [75.0%] in the chemotherapy group) after a median follow-up of 44.9 months (95% CI 37.9–47.0) and 36.8 months (95% CI 34.3–43.0), respectively.
The HR for OS with talazoparib versus chemotherapy was 0.848 (95% CI 0.670–1.073; P = 0.17) (Figure 1A). Kaplan-Meier survival percentages (95% CI) for talazoparib versus chemotherapy were 71% (66–76) versus 74% (66–81) at Month 12, 42% (36–47) versus 38% (30–47) at Month 24, and 27% (22–33) versus 21% (14–29) at Month 36. Covariate OS analysis showed a HR (95% CI) for treatment of 0.799 (95% CI 0.629–1.014; P = 0.06), and that a poorer ECOG PS and shorter time from initial diagnosis of breast cancer to initial diagnosis of ABC were associated with an increased risk of death (HR 0.772; 95% CI 0.616–0.968; P = 0.02 for ECOG score of 0 versus >0, and HR 1.488; 95% CI 1.177–1.882; P = 0.0009 for <12 months versus ≥12 months) (supplementary Table S2).
Figure 1. Final OS (A) in the overall population, (B) accounting for subsequent PARP inhibitor and/or platinum therapy, or (C) accounting for subsequent PARP inhibitor only (ITT population).

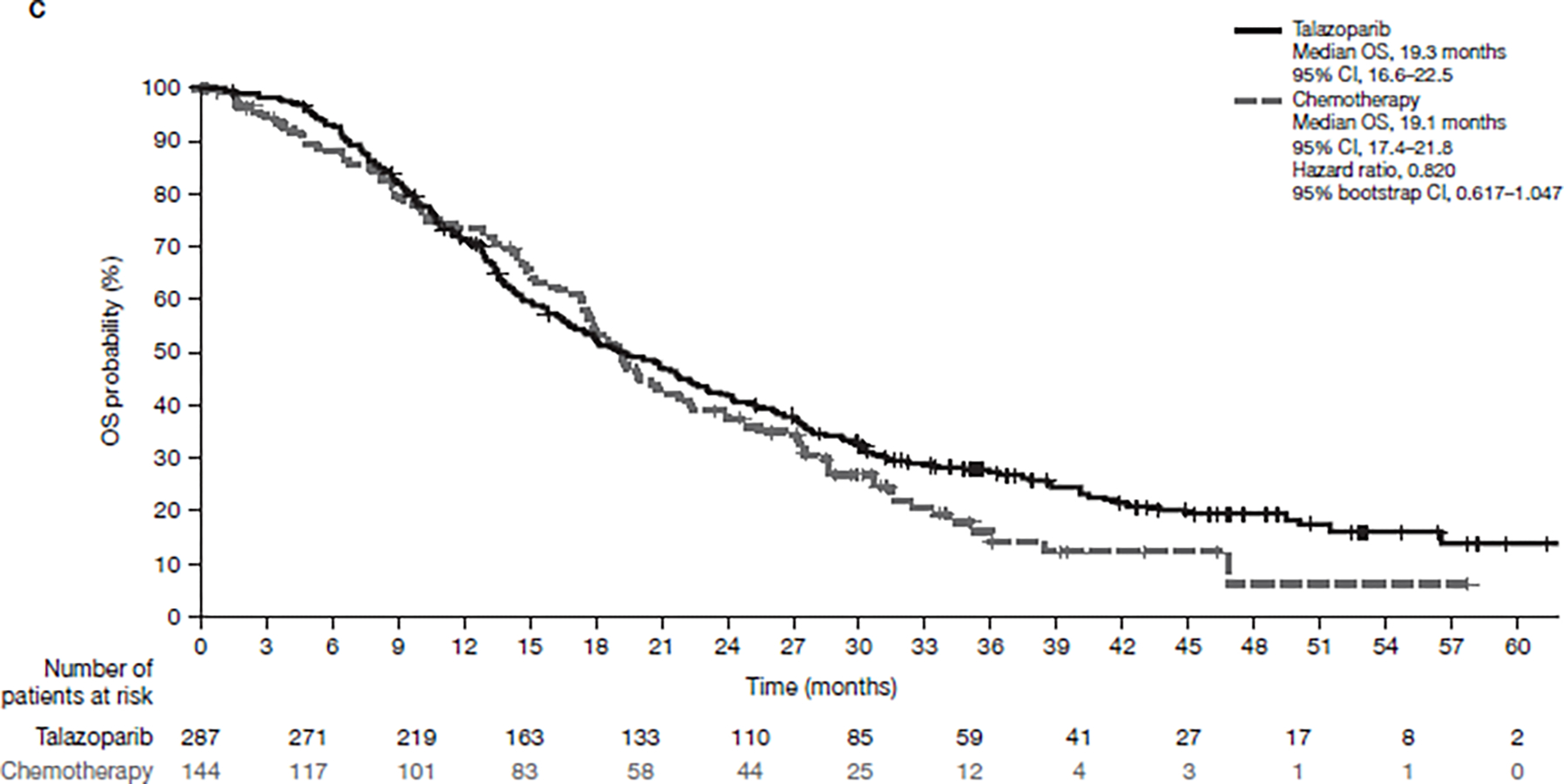
CI, confidence interval; ITT, intent-to-treat; OS, overall survival; PARP, poly(ADP-ribose) polymerase.
Prespecified analyses of OS by subgroups showed that the effect of talazoparib was generally consistent across subgroups (Figure 2). As the population was either triple-negative or hormone receptor-positive within EMBRACA, the effect of talazoparib on OS in these two subgroups was generally consistent, with HR 0.899 (95% CI 0.634–1.276) for triple-negative patients and HR 0.827 (95% CI 0.597–1.143) for hormone receptor-positive patients. Accordingly, the effect of talazoparib on OS by BRCA status was similar, with BRCA2 patients having a HR for OS of 0.794 (95% CI 0.571–1.106) and BRCA1 patients having a HR for OS of 0.772 (95% CI 0.539–1.104). All other subgroups are shown in Fig 2.
Figure 2. Subgroup analysis for OS (ITT population).
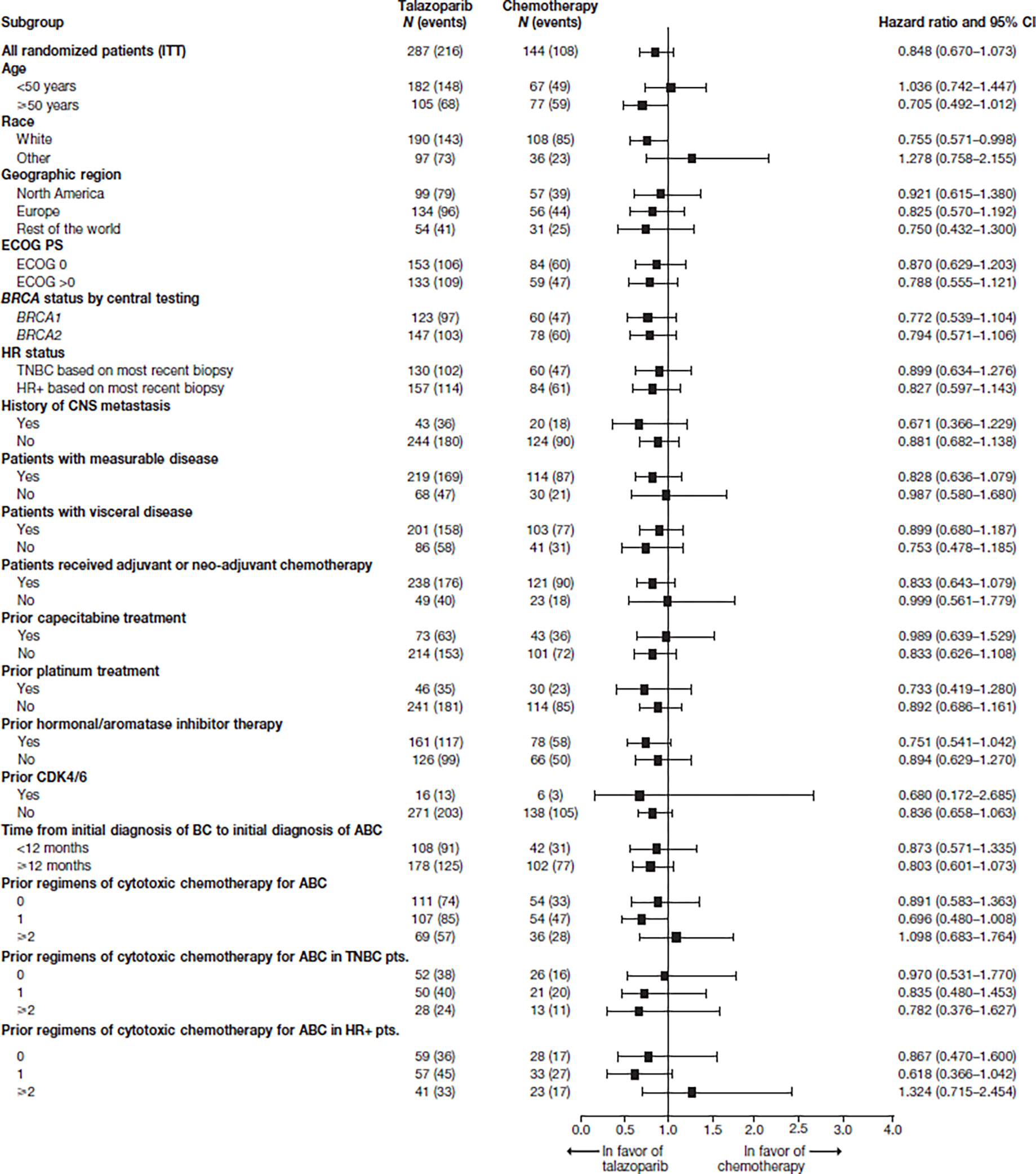
ABC, advanced breast cancer; BRCA, breast cancer susceptibility gene; CI, confidence interval; CNS, central nervous system; ECOG, Eastern Cooperative Oncology Group; HR+, hormone-receptor positive; ITT, intent-to-treat; OS, overall survival; PS, performance status; pts., patients; TNBC, triple-negative breast cancer.
Exploratory analysis revealed that patients with a longer prior platinum treatment-free interval (prior to study entry), were more likely to have a longer duration of survival particularly in the talazoparib-treated arm (Figure 3).
Figure 3. Duration of OS by prior platinum treatment-free interval in patients treated with (A) talazoparib (N = 46) and (B) chemotherapy (N = 29) (ITT population with prior platinum treatment in the talazoparib/chemotherapy arm).
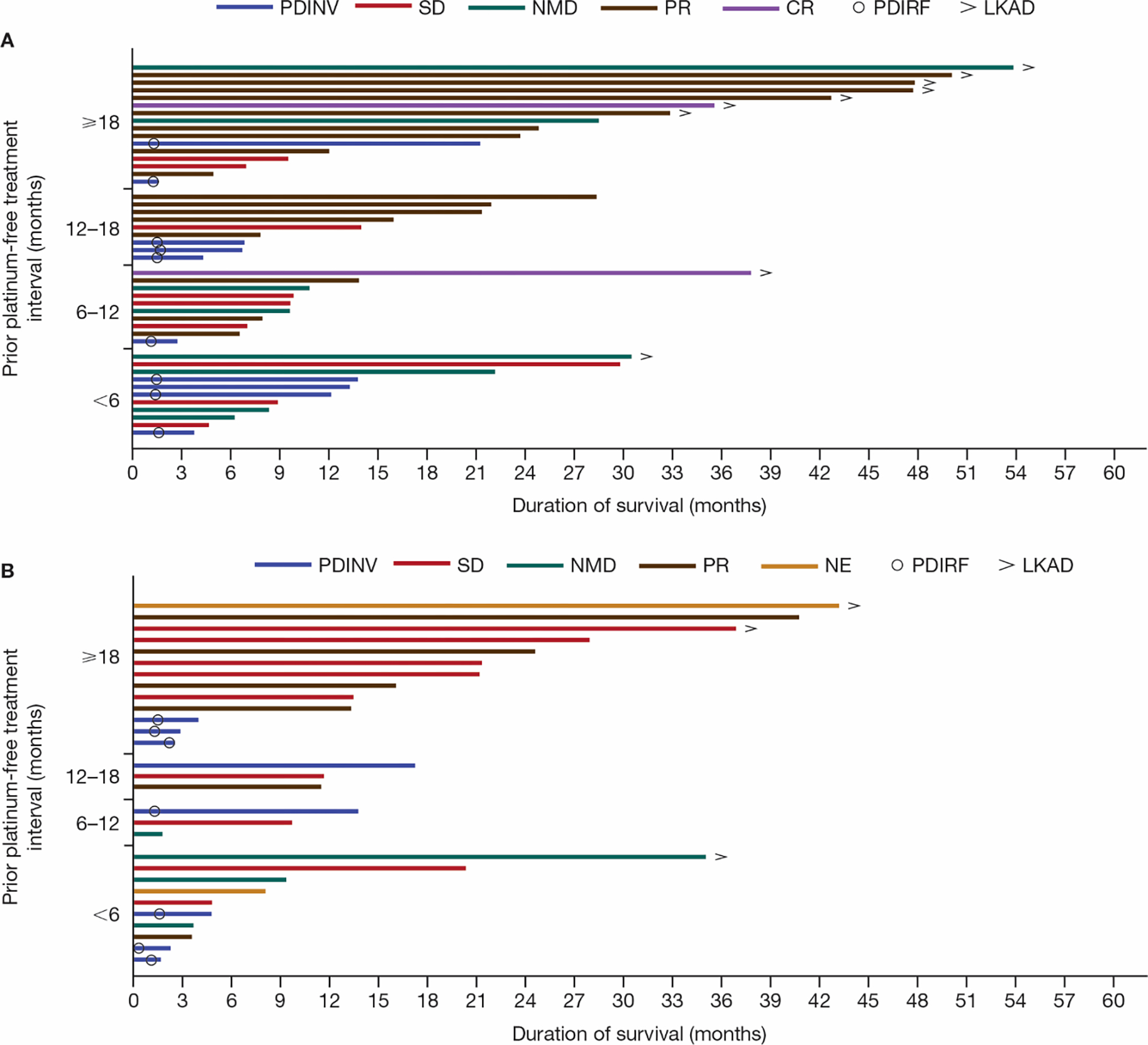
Of the 21 patients who had a prior platinum treatment-free interval of <6 months, 20 patients had received prior platinum therapy for locally advanced or metastatic disease (without objective disease progression during the platinum therapy) and 1 patient had received platinum therapy in the neoadjuvant/adjuvant setting. CR, complete response; ITT, intent-to-treat; LKAD, last known alive date; NE, not evaluable; NMD, no measurable disease; OS, overall survival; PDINV, progressive disease by investigator assessment; PDIRF, progressive disease by Independent Radiology Facility assessment; PR, partial response; SD, stable disease.
Altogether, a similar percentage of patients received subsequent systemic antineoplastic therapy in the talazoparib and chemotherapy groups (80.8% and 76.4%, respectively) (Table 1), with a median of two subsequent lines (range, 1–8). The most common subsequent therapies (≥15% overall) were carboplatin (talazoparib: 38.7%; chemotherapy: 34.0%), capecitabine (33.8% and 15.3%), gemcitabine (27.2% and 25.7%), eribulin (26.1% and 18.1%), and paclitaxel (22.3% and 19.4%) across all lines. The most common subsequent therapies (≥10%) by line after study therapy were: first line, carboplatin (20.9% and 17.4%), capecitabine (16.0% and 7.6%), and olaparib (0.7% and 11.8%), and; second line, capecitabine (10.1% and 2.8%) (Table 1). Subsequent therapies for triple-negative or hormone receptor-positive patients are shown in supplementary Tables S3 and S4.
Table 1.
Subsequent systemic antineoplastic therapies (ITT population)a
| Talazoparib (N = 287) | Chemotherapy (N = 144) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
||||||||||
| Antineoplastic therapy | Line of subsequent therapy | Line of subsequent therapy | ||||||||
|
|
||||||||||
| Any | First | Second | Third | ≥Fourth | Any | First | Second | Third | ≥Fourth | |
|
| ||||||||||
| Any | 232 (80.8) | 232 (80.8) | 154 (53.7) | 105 (36.6) | 64 (22.3) | 110 (76.4) | 110 (76.4) | 74 (51.4) | 50 (34.7) | 30 (20.8) |
| Cytotoxic | ||||||||||
| Carboplatin | 111 (38.7) | 60 (20.9) | 26 (9.1) | 19 (6.6) | 9 (3.1) | 49 (34.0) | 25 (17.4) | 12 (8.3) | 7 (4.9) | 6 (4.2) |
| Capecitabine | 97 (33.8) | 46 (16.0) | 29 (10.1) | 12 (4.2) | 11 (3.8) | 22 (15.3) | 11 (7.6) | 4 (2.8) | 4 (2.8) | 3 (2.1) |
| Gemcitabine | 78 (27.2) | 26 (9.1) | 24 (8.4) | 16 (5.6) | 12 (4.2) | 37 (25.7) | 13 (9.0) | 8 (5.6) | 9 (6.3) | 7 (4.9) |
| Eribulin | 75 (26.1) | 22 (7.7) | 15 (5.2) | 19 (6.6) | 19 (6.6) | 26 (18.1) | 10 (6.9) | 8 (5.6) | 4 (2.8) | 4 (2.8) |
| Paclitaxel | 64 (22.3) | 18 (6.3) | 21 (7.3) | 11 (3.8) | 17 (5.9) | 28 (19.4) | 10 (6.9) | 10 (6.9) | 2 (1.4) | 7 (4.9) |
| Vinorelbine | 40 (13.9) | 9 (3.1) | 8 (2.8) | 7 (2.4) | 16 (5.6) | 13 (9.0) | 5 (3.5) | 1 (0.7) | 4 (2.8) | 3 (2.1) |
| Cisplatin | 29 (10.1) | 15 (5.2) | 11 (3.8) | 2 (0.7) | 2 (0.7) | 10 (6.9) | 7 (4.9) | 2 (1.4) | 1 (0.7) | 0 |
| CDK4/6 inhibitor | ||||||||||
| Palbociclib | 39 (13.6) | 17 (5.9) | 14 (4.9) | 7 (2.4) | 2 (0.7) | 15 (10.4) | 7 (4.9) | 5 (3.5) | 1 (0.7) | 2 (1.4) |
| Hormonal treatment | ||||||||||
| Fulvestrant | 35 (12.2) | 14 (4.9) | 11 (3.8) | 3 (1.0) | 7 (2.4) | 17 (11.8) | 8 (5.6) | 7 (4.9) | 0 | 2 (1.4) |
| Letrozole | 29 (10.1) | 17 (5.9) | 7 (2.4) | 4 (1.4) | 2 (0.7) | 9 (6.3) | 5 (3.5) | 3 (2.1) | 1 (0.7) | 0 |
| PARP inhibitor | ||||||||||
| Olaparib | 8 (2.8) | 2 (0.7) | 3 (1.0) | 2 (0.7) | 1 (0.3) | 36 (25.0) | 17 (11.8) | 8 (5.6) | 6 (4.2) | 5 (3.5) |
Counts include all subsequent treatments regardless of whether the therapy is used as monotherapy or in a combination therapy. Gemcitabine includes generic names gemcitabine and gemcitabine hydrochloride, eribulin includes generic names eribulin and eribulin mesylate, paclitaxel includes generic names paclitaxel and paclitaxel albumin, and vinorelbine includes generic names vinorelbine and vinorelbine tartrate.
In ≥5% of patients for a particular line of therapy in any group.
CDK, cyclin-dependent kinase; ITT, intent-to-treat; PARP, poly(ADP-ribose) polymerase.
Overall, 47 patients (32.6%) in the chemotherapy group received subsequent PARP inhibitor compared with 13 patients (4.5%) in the talazoparib group (Table 2). 133 patients (46.3%) in the talazoparib group received subsequent platinum treatment compared with 60 patients (41.7%) in the chemotherapy group (38.7%/34.0% for carboplatin and 10.1%/6.9% for cisplatin, respectively, in the two arms).
Table 2.
Subsequent PARP inhibitor or platinum therapy (ITT population)
| Talazoparib (N = 287) | Chemotherapy (N = 144) | |
|---|---|---|
|
| ||
| Received subsequent PARP inhibitor or platinum, n (%) | 139 (48.4) | 86 (59.7) |
| PARP inhibitor | 13 (4.5) | 47 (32.6) |
| Olaparib | 8 (2.8) | 36 (25.0) |
| PARP inhibitor | 3 (1.0)a | 8 (5.6) |
| Veliparib | 2 (0.7) | 5 (3.5) |
| Platinum | 133 (46.3) | 60 (41.7) |
| Carboplatin | 111 (38.7) | 49 (34.0) |
| Cisplatin | 29 (10.1) | 10 (6.9) |
| Oxaliplatin | 0 | 2 (1.4) |
Three patients who took commercial talazoparib after discontinuation of talazoparib in the study are shown in the PARP inhibitor class.
ITT, intent-to-treat; PARP, poly(ADP-ribose) polymerase.
For the RPSFTM analysis accounting for subsequent use of PARP inhibitor and/or platinum therapy, the HR for OS was 0.756 (95% bootstrap CI 0.503–1.029) (Figure 1B). For the subsequent use of PARP inhibitor-only analysis, the adjusted HR was 0.820 (95% bootstrap CI 0.617–1.047) (Figure 1C). For the chemotherapy group, patients receiving neither subsequent PARP inhibitor nor platinum therapy had a shorter OS and total treatment duration than those who did (Figure 4).
Figure 4. Swimmers plot of OS and treatment duration according to whether patients received subsequent PARP inhibitor or platinum therapy in patients treated with (A) talazoparib and (B) chemotherapy (ITT population).
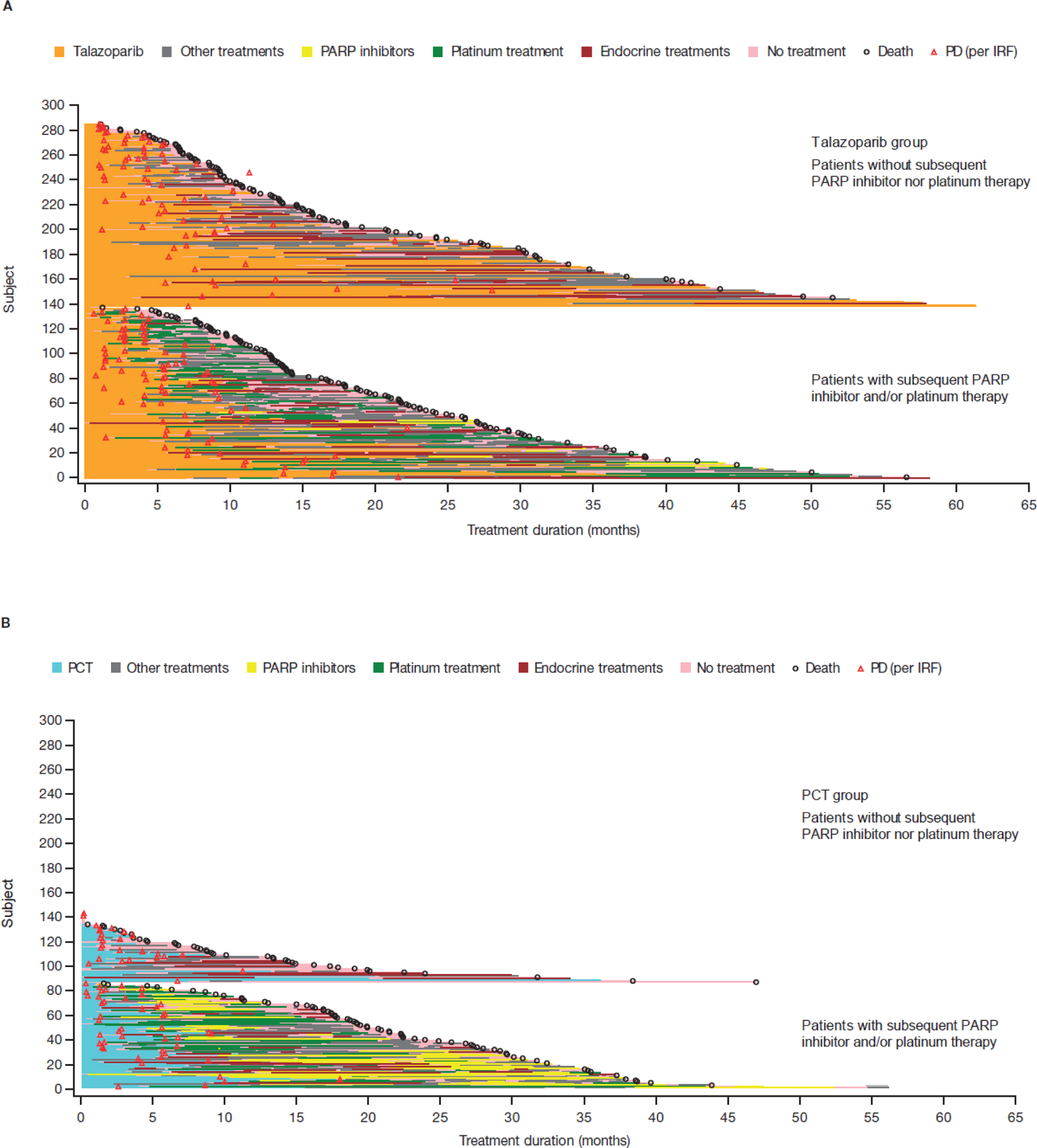
The analysis data cut-off date for OS is September 30, 2019, and for PD is September 15, 2017. For patients whose subsequent treatment end date was missing, the earliest date (of death date, end of study date, and OS data cut-off date) was used.
IRF, Independent Radiology Facility; ITT, intent-to-treat; OS, overall survival; PARP, poly(ADP-ribose) polymerase; PCT, physician’s choice of chemotherapy; PD, progressive disease.
Exposure to trial intervention
Median study-drug exposure was 6.9 months (range 0.03–61.4 months) for talazoparib and 3.9 months (range 0.2–36.3 months) for chemotherapy (Table 3). Seventy-four patients (25.9%) received talazoparib for ≥12 months, while nine patients (7.1%) received chemotherapy for ≥12 months. Evaluation of the intervals beyond one year showed that some patients had prolonged exposure to talazoparib versus chemotherapy: at 12 to <24 months, 36 (12.6%) versus 8 (6.3%), respectively; at 24 to <36 months, 25 (8.7%) versus 0; and at ≥36 months, 13 (4.5%) versus 1 (0.8%). In patients randomized to chemotherapy, only those receiving capecitabine (n = 8, seven with durations between 12 to <24 months, and one ≥36 months) or eribulin (n = 1 duration between 12 to <24 months) had study-drug exposure beyond one year.
Table 3.
Exposure by study drug (safety population)
| Talazoparib (N = 286) | Chemotherapy (N = 126) | Chemotherapy (N = 126) |
||||
|---|---|---|---|---|---|---|
| Capecitabine (N = 55) | Eribulin (N = 50) | Gemcitabine (N = 12) | Vinorelbine (N = 9) | |||
|
| ||||||
| Study-drug exposure, mo | ||||||
| Median | 6.9 | 3.9 | 4.1 | 2.9 | 5.5 | 4.2 |
| Min, max | 0.0, 61.4 | 0.2, 36.3 | 0.3, 36.3 | 0.5, 18.1 | 0.5, 6.9 | 0.2, 9.2 |
| Study-drug exposure (mo), n (%) | ||||||
| <1 | 9 (3.1) | 9 (7.1) | 3 (5.5) | 4 (8.0) | 1 (8.3) | 1 (11.1) |
| 1 to <3 | 43 (15.0) | 47 (37.3) | 21 (38.2) | 21 (42.0) | 2 (16.7) | 3 (33.3) |
| 3 to <6 | 76 (26.6) | 34 (27.0) | 11 (20.0) | 15 (30.0) | 5 (41.7) | 3 (33.3) |
| 6 to <12 | 84 (29.4) | 27 (21.4) | 12 (21.8) | 9 (18.0) | 4 (33.3) | 2 (22.2) |
| 12 to <24 | 36 (12.6) | 8 (6.3) | 7 (12.7) | 1 (2.0) | 0 | 0 |
| 24 to <36 | 25 (8.7) | 0 | 0 | 0 | 0 | 0 |
| ≥36 | 13 (4.5) | 1 (0.8) | 1 (1.8) | 0 | 0 | 0 |
| Relative dose intensity, %a | ||||||
| Median | 85.4 | 86.2 | 95.6 | 87.2 | 64.3 | |
| Min, max | 26.2, 3000.0b | 33.3, 106.9 | 49.0, 101.6 | 65.6, 100.0 | 37.0, 100.0 | |
Relative dose intensity defined as actual dose intensity divided by planned dose intensity.
Maximum value due to a talazoparib dose of 30 mg ingested as an accidental overdose in one patient.
max, maximum; min, minimum; mo, month.
Safety
AEs in the two groups were consistent with the primary analysis6, 9 (Table 4; supplementary Tables S5 and S6). The most common AEs (>30% of patients) were anemia, fatigue, nausea, neutropenia, and headache in the talazoparib group and nausea, fatigue, and neutropenia in the chemotherapy group (supplementary Tables S5 and S6). Grade 3 or 4 AEs occurred in 69.6% of patients receiving talazoparib and 64.3% of patients receiving chemotherapy, with hematologic grade 3–4 AEs in 56.6% and 38.9% of patients, respectively. Anemia was reported in 54.9% of patients who received talazoparib compared with 19.0% of patients who received chemotherapy, and was grade 3 or 4 in 40.2% and 4.8% of patients, respectively. At least one blood transfusion was received by 39.2% of patients receiving talazoparib compared with 5.6% receiving chemotherapy (supplementary Table S7); this may have been partly due to the protocol requirements for talazoparib interruption and/or restarting talazoparib according to hemoglobin level, whereas investigators followed local prescribing information in the chemotherapy arm.9
Table 4.
Summary of AEsa (safety population)
| Talazoparib (N = 286) | Chemotherapy (N = 126) | |
|---|---|---|
| Number of patients (%) | ||
|
| ||
| Any AE | 282 (98.6) | 123 (97.6) |
| Serious AE | 101 (35.3) | 39 (31.0) |
| Serious and drug-related AE | 30 (10.5) | 11 (8.7) |
| Grade 3 or 4 serious AE | 81 (28.3) | 34 (27.0) |
| AE resulting in permanent drug discontinuationb | 22 (7.7) | 12 (9.5) |
AE grades were evaluated based on the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03.
Data obtained from the AE case report form, action taken; data includes those with progressive disease.
AE, adverse event.
AEs leading to permanent treatment discontinuation (excluding progressive disease) occurred in 5.9% of patients on talazoparib and 8.7% on chemotherapy. Hematologic AEs rarely led to permanent discontinuation of talazoparib: three patients (1%) discontinued due to anemia, one patient (0.3%) due to neutropenia or thrombocytopenia. Dose modifications of talazoparib were used if a patient experienced toxicity (supplementary Table S8).
There were no confirmed cases of myelodysplastic syndrome. As reported with the primary data analysis, one case of acute myeloid leukemia (AML) occurred in a patient who received capecitabine6 and a previously unreported case of AML occurred in a patient who received talazoparib (supplementary Table S5).
Patient-reported outcome
With extended follow-up, favorable PRO remained consistent with the initial analysis. A significant improvement in the estimated overall change from baseline in GHS/QoL scores (European Organization for Research and Treatment of Cancer [EORTC] Quality of Life Questionnaire [QLQ-C30]) was observed in the talazoparib group while a significant deterioration was observed in the chemotherapy group (2.1 [95% CI 0.1–4.1] versus −5.7 [95% CI −10.0– −1.4]; P = 0.001). There was also a significant improvement in the estimated overall change from baseline in the breast symptoms (EORTC QLQ-BR23) in the talazoparib group, with non-significant change in the chemotherapy group (−4.9 [95% CI −6.5– −3.2] versus 0.1 [95% CI −3.2–3.5], P = 0.009). Compared with chemotherapy, treatment with talazoparib resulted in significant delay in time to definitive clinically meaningful deterioration in both the GHS/QoL (supplementary Figure S2) and in breast symptoms (supplementary Figure S3).
DISCUSSION
Findings from this prespecified final analysis of OS in the EMBRACA trial comparing talazoparib with chemotherapy in patients with gBRCA1/2-mutated ABC found no statistically significant difference in OS between the treatment groups; the HR was 0.848 (95% CI 0.670–1.073; P = 0.17). Prespecified covariate analyses showed that a poorer ECOG PS and shorter time from initial diagnosis of breast cancer to initial diagnosis of ABC were associated with a statistically significant increased risk of death. Adjustment for these covariates reduced the HR for OS with talazoparib versus chemotherapy to 0.799 (95% CI 0.629–1.014; P = 0.06). The effect of talazoparib on OS was generally consistent across predefined subgroups. However, it is interesting to note that patients receiving talazoparib had a higher survival probability at later timepoints, and a higher percentage of patients on talazoparib than on chemotherapy continued therapy beyond 1, 2, and 3 years (Table 3).
Interpretation of the OS results may have been confounded by subsequent treatments. Analysis accounting for subsequent PARP inhibitor and/or platinum therapies showed that the primary OS analysis was impacted by subsequent treatment with platinum and/or a PARP inhibitor, even if subsequent treatment with a PARP inhibitor alone was less impactful than platinum and/or a PARP inhibitor.
Addressing the influence of subsequent therapies on the long survival post-progression (SPP; the time from progression to death) is essential in understanding the effects of the therapy evaluated within the trial. For cancers with a long SPP, the variability in SPP, influenced by subsequent therapies, can dilute the OS benefit so that the ability to detect statistical significance is minimized.13 However, there is continued justification for PFS as a surrogate for OS, with significant associations found between PFS and OS in patients with HER2-negative metastatic breast cancer.14–17 Several statistical methods have been applied to adjust for the potentially confounding effects of subsequent treatment in clinical trials. Here, we used the RPSFTM that estimates the counterfactual OS of the chemotherapy arm that would have been observed without platinum and/or a PARP inhibitor, assuming that treatment effects are constant and subsequent systemic therapy is the same between the two arms. Despite the caveats around RPSFTM assumptions,12 the HR (95% CI) obtained for OS after adjustment for subsequent platinum and/or a PARP inhibitor suggests that the primary OS analysis underestimated the treatment benefit of talazoparib. Additional sensitivity analyses were not done for subgroups with limited patient numbers in EMBRACA as they were not considered statistically robust.
In the phase III trial of olaparib (OlympiAD; NCT02000622) versus chemotherapy, a statistically significant benefit in PFS did not translate into a statistically significant improvement in OS, although, in contrast to EMBRACA, the trial was not powered to identify a difference in OS.18 In OlympiAD, a lower proportion of patients in the control group than in EMBRACA went on to receive a PARP inhibitor as subsequent therapy (8% and 33%, respectively): as OlympiAD was the first phase III PARP trial to read out, availability of subsequent PARP inhibitor therapy was limited. In both trials, over 40% of patients across treatment arms received subsequent platinum therapy.18 Reversion mutations in BRCA1/2 that restore DNA repair proficiency have been shown to lead to resistance to both PARP inhibitors and platinum therapy; thus, the possible impact of subsequent platinum therapy is of interest.3 Other differences existed between the two phase III trials’ results;6, 18 however, cross-trial comparisons should be made with caution due to differences in study design, patient characteristics, and the effects of subsequent therapies in the chemotherapy arm.
The safety profile of talazoparib was consistent with the primary analysis.6, 9 Most grade 3–4 AEs reported in the talazoparib group were hematologic and most were successfully managed by supportive care (including transfusion) and dose modifications. The rate of permanent treatment discontinuation due to AEs was 5.9%. The favorable PRO observed with extended follow-up were consistent with initial analyses;6, 7 patients treated with talazoparib had significant overall improvement and significant delay in the time to definitive clinically meaningful deterioration in both patient-reported GHS/QoL and breast symptoms.
The main limitation of this trial is the open-label design due to the use of both oral and intravenous agents, as reported previously.6 Other limitations are the lack of platinum-based agents as an option in the chemotherapy group and the possible confounding factor on the OS results by subsequent therapies. It is recognized that results of the phase III TNT trial involving patients with triple-negative breast cancer showed that the subset of 43 patients with gBRCA1/2 mutation had a greater response and PFS in favor of carboplatin over docetaxel (although no OS advantage) not seen in the overall ITT population.19 In addition, the results of the phase III BROCADE-3 trial in patients with HER2-negative gBRCA1/2-mutated ABC showed that the combination of the PARP inhibitor veliparib with carboplatin/paclitaxel significantly improved PFS compared with carboplatin/paclitaxel alone (no interim OS advantage was observed).20 However, at the time the EMBRACA study was designed, the chosen reference agents were considered to be the standard single-agent therapies for patients with ABC.
In conclusion, OS, evaluated as a key secondary endpoint in this trial, was not significantly improved with talazoparib compared with chemotherapy (HR 0.848; 95% CI 0.670–1.073; P = 0.17), although subsequent therapies may have impacted results. Talazoparib was generally well tolerated with manageable toxicity and no new safety signals. Improvements in PRO with extended follow-up supported results previously reported. The findings of this trial confirm that the incorporation of talazoparib in clinical practice is a favorable treatment option for patients with locally advanced or metastatic breast cancer with a gBRCA1/2 mutation.
Supplementary Material
Highlights.
In BRCA1/2-mutated advanced breast cancer, talazoparib did not significantly improve overall survival (OS) vs chemotherapy
OS results were generally consistent across subgroups including by prior platinum, hormone-receptor status, or line of treatment
Most patients received subsequent systemic therapies, which may have confounded the survival outcome
Toxicities were managed by supportive care medication/dose modifications; safety was consistent with previous observations
Extended follow-up of patient-reported outcomes continued to favor talazoparib over chemotherapy
ACKNOWLEDGMENTS
We thank the patients and their families and caregivers for their participation, as well as the trial centers and patient advocacy groups who supported this trial. We also wish to acknowledge the contributions of other colleagues who participated in the data analyses on OS, including Paul Bycott, Yiyun Tang, Kris Schuler, and Gerald Luscan. Medical writing support was provided by Annette Smith, PhD, of CMC AFFINITY, McCann Health Medical Communications and was funded by Pfizer.
FUNDING
This work was sponsored by Medivation, which was acquired by Pfizer Inc. in September 2016.
DISCLOSURES
JKL reports grant or research support from Novartis, Pfizer, Genentech, GSK, EMD-Serono, AstraZeneca, and Zenith Epigenetics; fees for speakers’ bureaus from Med Learning Group, Physician’s Education Resource, Prime Oncology, Medscape, Medpage, Clinical Care Options, and UpToDate; honoraria from UpToDate; membership on advisory committees or review panels, or board membership, for AstraZeneca, Pfizer, and Ayala Pharmaceuticals (all uncompensated); membership on review panels for NCCN, ASCO, and NIH PDQ; patent, royalties, or other intellectual properties from UpToDate; travel, accommodation, and expenses from Med Learning Group, Physician’s Education Resource, Medscape, and Clinical Care Options; and employment by the University of Texas MD Anderson Cancer Center.
SAH reports contracted research support and editorial assistance from Ambrx, Amgen, Arvinas, Bayer, Biomarin, Cascadian Therapeutics, Daiichi-Sankyo, Dignitana, Genentech/Roche, GSK, Immunomedics, Lilly, MacroGenics, Merrimack, Novartis, Pfizer, OBI Pharma, Pieris Pharmaceuticals, Puma Biotechnology, Radius Health, Sanofi, and Seattle Genetics.
LAM, HR, Y-HI, and WE have nothing to disclose.
K-HL reports honoraria from Roche and AstraZeneca, and has participated in advisory boards for Bayer, Ono Pharmaceutical, Samsung Bioepis, Roche, Eisai, and AstraZeneca.
HSR reports research support to the University of California San Francisco from Eisai, Genentech, Lilly, MacroGenics, Merck, Novartis, OBI Pharma, Odonate Therapeutics, Immunomedics, Daiichi-Sankyo, and Pfizer; a consulting role with Samsung and Celtrion; and travel support from Pfizer, Novartis, MacroGenics, Mylan, Daiichi-Sankyo, AstraZeneca and Novartis.
AG reports travel/accommodation/meeting registration fees from Pfizer, AstraZeneca, Roche, and Novartis.
SD is a speaker and advisor for Pfizer, Novartis, Puma, Eli Lilly, Clovis, Genentech, AstraZeneca, Genomic Heath, and Agendia.
NW reports stock and ownership in CSL Behring, research funding (institution) from Medivation, honoraria for advisory boards from Novartis and Pfizer, and consultancy fees, honoraria for advisory boards and travel and accommodation fees from Roche.
AG reports honoraria from AstraZeneca and Pfizer for participation in advisory boards.
RY reports consulting fees from Roche, Pfizer, Novartis, and Eli Lilly, has been a speaker for Roche, Teva, Medison, MSD, AstraZeneca, Novartis, and Pfizer, and reports a grant from Roche.
RGWQ is an employee of Pfizer and reports ownership interest in Pfizer and Amgen.
TU, SL, and AC are employees of Pfizer and report ownership interest in Pfizer.
JLB reports consulting fees from Pfizer, Medivation, Amgen, Novartis, Genomic Health, Daiichi Sankyo, and Myriad Genetics.
MM reports research funding from Roche, and Novartis, and consulting or advisory role for Roche/Genentech, Novartis, AstraZeneca, Lilly, Taiho Pharmaceutical, PharmaMar, and Pfizer.
JE has received consulting fees from Pfizer, Novartis, Lilly, Roche, and Tesaro; contracted research from Pfizer, Lilly, Novartis, Seattle Genetics, AstraZeneca, Roche, and Odonate; and travel support from AstraZeneca, Celgene, Pfizer, Novartis, Lilly, and Tesaro.
DATA SHARING
Upon request, and subject to certain criteria, conditions, and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de-identified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices (1) for indications that have been approved in the US and/or EU or (2) in programs that have been terminated (i.e. development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de-identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
REFERENCES
- 1.Wang B, Chu D, Feng Y et al. Discovery and Characterization of (8S,9R)-5-Fluoro-8-(4-fluorophenyl)-9-(1-methyl-1H-1,2,4-triazol-5-yl)-2,7,8,9-te trahydro-3H-pyrido[4,3,2-de]phthalazin-3-one (BMN 673, Talazoparib), a Novel, Highly Potent, and Orally Efficacious Poly(ADP-ribose) Polymerase-1/2 Inhibitor, as an Anticancer Agent. J Med Chem 2016; 59: 335–357. [DOI] [PubMed] [Google Scholar]
- 2.Murai J, Huang SY, Renaud A et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther 2014; 13: 433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017; 355: 1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Bono J, Ramanathan RK, Mina L et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov 2017; 7: 620–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turner NC, Telli ML, Rugo HS et al. A Phase II Study of Talazoparib After Platinum or Cytotoxic Nonplatinum Regimens in Patients With Advanced Breast Cancer and Germline BRCA1/2 Mutations (ABRAZO). Clin Cancer Res 2019; 25: 2717–2724. [DOI] [PubMed] [Google Scholar]
- 6.Litton JK, Rugo HS, Ettl J et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N Engl J Med 2018; 379: 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ettl J, Quek RGW, Lee KH et al. Quality of life with talazoparib versus physician’s choice of chemotherapy in patients with advanced breast cancer and germline BRCA1/2 mutation: patient-reported outcomes from the EMBRACA phase III trial. Ann Oncol 2018; 29: 1939–1947. [DOI] [PubMed] [Google Scholar]
- 8.Ettl J, Quek RGW, Hurvitz SA et al. Hospitalization and supportive care medication (SCM) utilisation in patients (pts) with advanced breast cancer (ABC) and a germline BRCA1/2 mutation in EMBRACA. Ann Oncol 2019; 30 (suppl 3): iii47–iii64. [Google Scholar]
- 9.Hurvitz SA, Goncalves A, Rugo HS et al. Talazoparib in patients with a germline BRCA-mutated advanced breast cancer: detailed safety analyses from the phase III EMBRACA trial. Oncologist 2020; 25: e439–e450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Food and Drug Administration. TALZENNA® (talazoparib) prescribing information. 2019. Available at: https://www.talzenna.com. Last update September 2019. Accessed March 23, 2020.
- 11.European Medicines Agency. TALZENNA® (talazoparib) summary of product characteristics. 2019. Available at: https://www.ema.europa.eu/en/medicines/human/EPAR/talzenna. Last update July 2019. Accessed March 23, 2020.
- 12.Robins JM, Tsiatis AA. Correcting for non-compliance in randomized trials using rank preserving structural failure time models. Communications in Statistics - Theory and Methods 1991; 20: 2609–2631. [Google Scholar]
- 13.Broglio KR, Berry DA. Detecting an overall survival benefit that is derived from progression-free survival. J Natl Cancer Inst 2009; 101: 1642–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forsythe A, Chandiwana D, Barth J et al. Progression-free survival/time to progression as a potential surrogate for overall survival in HR+, HER2- metastatic breast cancer. Breast Cancer (Dove Med Press) 2018; 10: 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adunlin G, Cyrus JW, Dranitsaris G. Correlation between progression-free survival and overall survival in metastatic breast cancer patients receiving anthracyclines, taxanes, or targeted therapies: a trial-level meta-analysis. Breast Cancer Res Treat 2015; 154: 591–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beauchemin C, Cooper D, Lapierre ME et al. Progression-free survival as a potential surrogate for overall survival in metastatic breast cancer. Onco Targets Ther 2014; 7: 1101–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seidman AD, Bordeleau L, Fehrenbacher L et al. National Cancer Institute Breast Cancer Steering Committee Working Group Report on Meaningful and Appropriate End Points for Clinical Trials in Metastatic Breast Cancer. J Clin Oncol 2018; JCO1800242 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robson ME, Tung N, Conte P et al. OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann Oncol 2019; 30: 558–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tutt A, Tovey H, Cheang MCU et al. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: the TNT Trial. Nat Med 2018; 24: 628–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diéras VC, San HS, Kaufman B et al. Phase III study of veliparaib with carboplatin and paclitaxel in HER2-negative advanced/metastatic gBRCA-associated breast cancer. Ann Oncol 2019; 30: v857–v858. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Upon request, and subject to certain criteria, conditions, and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de-identified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices (1) for indications that have been approved in the US and/or EU or (2) in programs that have been terminated (i.e. development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de-identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.