

Abstract
The intracellular bacterial pathogen Chlamydia trachomatis is a major cause of sexually transmitted disease worldwide. While protective immunity does appear to develop following natural chlamydial infection in humans, early vaccine trials using heat-killed C. trachomatis resulted in limited and transient protection with possible enhanced disease during follow-up. Thus, immunity following natural infection with live chlamydia may differ from immune responses induced by immunization with inactivated chlamydia. To study this differing immunology, we used murine bone marrow-derived dendritic cells (DC) to examine DC maturation and immune effector function induced by live and UV-irradiated C. trachomatis elementary bodies (live EBs and UV-EB, respectively). DC exposed to live EBs acquired a mature DC morphology; expressed high levels of major histocompatibility complex (MHC) class II, CD80, CD86, CD40, and ICAM-1; produced elevated amounts of interleukin-12 and tumor necrosis factor alpha; and were efficiently recognized by Chlamydia-specific CD4+ T cells. In contrast, UV-EB-pulsed DC expressed low levels of CD40 and CD86 but displayed high levels of MHC class II, ICAM-1, and CD80; secreted low levels of proinflammatory cytokines; and exhibited reduced recognition by Chlamydia-specific CD4+ T cells. Adoptive transfer of live EB-pulsed DC was more effective than that of UV-EB-pulsed DC at protecting mice against challenge with live C. trachomatis. The expression of DC maturation markers and immune protection induced by UV-EB could be significantly enhanced by costimulation of DC ex vivo with UV-EB and oligodeoxynucleotides containing cytosine phosphate guanosine; however, the level of protection was significantly less than that achieved by using DC pulsed ex vivo with viable EBs. Thus, exposure of DC to live EBs results in a mature DC phenotype which is able to promote protective immunity, while exposure to UV-EB generates a semimature DC phenotype with less protective potential. This result may explain in part the differences in protective immunity induced by natural infection and immunization with whole inactivated organisms and is relevant to rational chlamydia vaccine design strategies.
Chlamydia trachomatis is a major cause of sexually transmitted disease worldwide and is estimated to cause over 90 million cases annually (54). In women, more than 70% of cases are asymptomatic, and if left untreated, the infection can spread throughout the reproductive tract and cause complications, including tubal factor infertility, ectopic pregnancy, chronic pelvic inflammation, and fallopian tube scarring (5, 18). C. trachomatis is an obligate intracellular pathogen with a developmental cycle that includes two distinct forms: a metabolically inactive elementary body (EB) that infects epithelial cells and a larger metabolically active reticulate body that divides and differentiates into EBs.
Immunity to chlamydial infection involves both humoral and cell-mediated immune responses (31). Studies in animal models have established that both Chlamydia-specific CD4+ T cells producing gamma interferon (IFN-γ) (18, 38) and antibody production (32, 55) are critically involved in the clearance of a chlamydial infection and in resistance to reinfection, while the role of CD8+ T cells appears to be less important (32). Although immunoepidemiological studies suggest that similar patterns of immunity occur in humans (4), the immune effector mechanisms elicited in humans remain less well understood.
Observations of both humans and mice suggest that exposure to live and dead chlamydia induce distinct immunological effects in the mammalian host. After recovery from infection, for example, patients usually develop a strain-specific but short-lived resistance to reinfection (12, 50). However, individuals immunized with whole heat-inactivated chlamydia develop little protection and upon subsequent infections may develop symptoms of disease exacerbation (12, 13). Similarly, mice infected with C. trachomatis mouse pneumonitis (MoPn) develop almost sterile protective immunity (23, 49), while immunization with killed chlamydia or with the chlamydial major outer membrane protein (MOMP) induces responses ranging from little to no protection (23, 43). Furthermore, under some circumstances it appears that live chlamydia may actually impair immune recognition of infected cells or alter antigen-presenting cells by down-regulating both major histocompatibility complex (MHC) class I and MHC class II, thereby promoting a state of persistent infection (56, 57). It is apparent that seemingly opposite effects are induced by exposure of mammalian hosts to live and dead chlamydia. However, to date a direct comparison of the effects of live and dead chlamydia on cellular immunity has not been carried out.
Dendritic cells (DC) are key players in immunity that dictate the type and quality of the immune response that is generated to a particular antigen. DC are professional antigen-presenting cells that have an extraordinary capacity to stimulate naïve T cells and initiate primary immune responses (1, 30). Although the diverse functions of DC in immune regulation depend in part on the diversity of DC subsets and lineages, it has also become clear that the state of DC maturation is critical for the initiation of immune responses (22, 47). Mature DC promote efficient and specific protective immunity (11, 20), whereas immature DC are associated with immunological unresponsiveness (8, 9) and tolerance to self antigens (46, 51).
In the last few years, the role of DC in chlamydial immunobiology has been actively investigated (16, 17, 25, 27, 34, 35, 43, 44). Both live (27, 34, 35) and dead (49) chlamydia are effectively taken up by DC. However, after internalization, inclusions harboring replicating chlamydia are not formed within DC (27, 35); instead, chlamydia appear to be killed within the first few hours postinfection by a mechanism that involves fusion of the inclusion with host cell lysosomes (34), an event that is normally inhibited in chlamydia-infected epithelial cells (41). In the murine model, both live and dead chlamydia appear to activate and modulate DC function. For example, DC infected with live Chlamydia pneumoniae produce interleukin-12 (IL-12) through a mechanism that involves TLR2 and TLR4 (35), while DC pulsed with live C. trachomatis efficiently presented chlamydial antigen to T cells in vitro (27). Furthermore, DC isolated from the lungs of mice infected with live MoPn promoted a TH1 immune response to a model antigen (ovalbumin), whereas DC isolated from naïve mice induced a TH2 immune response (16). Exposure to dead chlamydia similarly affects DC function. DC exposed to either heat-killed or UV-irradiated chlamydia expressed inflammatory and immunomodulatory molecules, including CCR-7, IL-12, and IFN-γ-induced protein 10 (44), and upon intravenous adoptive transfer these DC conferred resistance to chlamydial challenge (44, 49). Furthermore, intraperitoneal administration of UV-inactivated chlamydia together with adenovirus expressing granulocyte-macrophage colony-stimulating factor (GM-CSF) resulted in the accumulation of DC in the peritoneum of infected mice, which correlated with the development of protective immunity (23).
While it appears that both live and dead chlamydia modulate DC immunobiology, it is not clear whether the DC maturation statuses or the patterns of DC immunogenicity induced in response to live and dead organisms are identical. The purpose of this study was to examine the effect of live and dead chlamydia on the activation and maturation of bone marrow-derived DC and to determine whether DC incubated with live and dead chlamydia are equally able to induce protective immunity upon adoptive transfer. This study serves as the first direct comparison of the effects of live and dead chlamydia on DC maturation and immunogenicity and may shed light on why candidate vaccines using inactivated chlamydia have thus far proved unsuccessful.
MATERIALS AND METHODS
Reagents and antibodies.
Antibodies for fluorescence-activated cell sorting (FACS) analysis and enzyme-linked immunosorbent assay (ELISA) were purchased from Pharmingen (Mississauga, Ontario, Canada). The following monoclonal antibodies (MAbs) were used for FACS: rat anti-mouse FcR Ab (2.4G2); phycoerythrin-conjugated hamster anti-mouse CD11c (HL3); fluorescein isothiocyanate (FITC)-conjugated hamster anti-mouse CD40 (HM40-3); FITC-conjugated hamster anti-mouse CD80 (16-10A1); FITC-conjugated rat anti-mouse CD86 (GL1); FITC-conjugated hamster anti-mouse CD54 (3E2); and FITC-conjugated rat anti-mouse I-A/I-E (2G9). The following capture and detection antibody pairs were used for ELISA: IFN-γ (R4-6A2 and XMG2.2), IL-12 (C15.6 and C17.8), tumor necrosis factor alpha (TNF-α) (G281-2626 and MP5-X13), and IL-10 (JES5-2A5 and SXC-1). Iscove's modified minimal essential medium (IMDM), fetal calf serum (FCS), and GM-CSF were purchased from Stem Cell Technologies, Vancouver, Canada. Hybridoma X63 producing IL-4 was provided by F. Melchers, Basilea Institute, Basel, Switzerland. Oligodeoxynucleotide (ODN) sequence 1826 (37) containing two cytosine phosphate guanosine (CpG) motifs (TCCATGACGTTCCTGACGTT; the underlined bases represent the CpG motifs) and control ODN sequence 1982 (TCCAGGACTTCTCTCAGGTT) without CpG motifs were purchased from Coley Pharmaceutical group, Ottawa, Canada. LPS was from Sigma, St. Louis, Mo.
Generation and purification of DC.
Myeloid DC were generated from bone marrow progenitors in vitro by using GM-CSF and IL-4 as described previously (19) with minor modifications. Briefly, bone marrow cells flushed from the femurs of 8- to 12-week-old female C57BL/6 mice were cultured at a concentration of 7 × 105 cells/ml in 10-cm-diamater dishes (Falcon). The DC growth medium consisted of IMDM supplemented with 10% FCS, 15 ng of GM-CSF/ml, 2 mM l-glutamine, 2-mercaptoethanol, penicillin, streptomycin, and 5% IL-4 culture supernatant of Hybridoma X63 (see above). Fresh GM-CSF was added to the cultures at day 4. On day 7, nonadherent cells were harvested and purified by using anti-CD11c magnetic beads (Miltenyi Biotech Ltd.). Purities of >98% CD11c+ cells were routinely achieved, as determined by FACS (not shown).
Mice.
Female C57BL/6 or BALB/c mice were purchased from Charles River (St. Constant, Canada) and kept under pathogen-free conditions at the Animal Facility of the Jack Bell Research Centre. All animal procedures used in the study were approved by the animal care committee of the University of British Columbia.
Chlamydiae.
C. trachomatis MoPn strain Nigg (also known as Chlamydia muridarum) was used in this study. MoPn was grown in HeLa 229 cells in Eagle's minimal essential medium (Invitrogen) supplemented with 10% FCS. EBs were purified from HeLa cells on discontinuous density gradients of Renografin-76 (Nycomed Imaging, Brampton, Ontario, Canada) as described previously (23). Purified EBs were aliquoted and stored at −80°C in sucrose-phosphate-glutamic acid buffer. Infectivity and the number of inclusion-forming units (IFU) of purified EBs were assessed by immunostaining. Briefly, HeLa cell monolayers were infected with serial dilutions of EBs and incubated for 24 to 36 h at 37°C. Cells were fixed in methanol and stained with an antibody raised against chlamydial MOMP (ViroStat, Portland, Maine). Detection was carried out as previously described (23). Inactivation of EBs was carried out either by heating to 56°C for 30 min (49) or by exposure to UV light from a G15T8 UV lamp (D. William Fuller, Inc., Chicago, Ill.) at a distance of 5 cm for 45 min at room temperature as previously described (23). To ensure that treated EBs were completely inactivated, viability was tested on HeLa 229 cells as indicated above. No recoverable IFU was found after incubation of inactivated EBs on HeLa cells for a period of 24 or 36 h (data not shown). The IFU for both live EBs and UV-irradiated EBs (UV-EB) were calculated from the titers determined on original chlamydial purified stocks as described above. In preliminary experiments, we found that heat-inactivated and UV-irradiated EBs induced similar effects on DC activation in terms of expression of costimulatory molecules and MHC class II (data not shown). Therefore, throughout this work UV-irradiated EBs were used as the source of dead MoPn.
FACS analysis.
DC were resuspended at a cell density of 5 × 106 cells/ml and incubated on ice for 30 min with 10 μg of the murine anti-mouse immunoglobulin G FcR Ab/ml. Cells were added to FITC- or phycoerythrin-conjugated antibodies and incubated for 30 min. Cells were washed once with phosphate-buffered saline (PBS) containing 2% FCS and then with PBS containing 1 μg of propidium iodide/ml to stain dead cells (Sigma Chemicals). Analysis was carried out on a FACS Calibur (Becton-Dickinson, San Jose, Calif.). Only live cells were analyzed.
Allogeneic T-cell proliferation assays.
Purified DC were incubated for 4 h with medium alone, lipopolysaccharide (LPS), or either live EBs or UV-EB at a multiplicity of infection (MOI) of 1. Unbound bacteria were washed away with PBS containing 2% FCS, and cells were incubated for a total of 48 h. DC were harvested, washed once with IMDM, and irradiated with 1,500 rads for 10 min (RT250 X-ray machine; Philips). DC were seeded into U-bottom 96-well plates at concentrations ranging between 5 × 103 and 0.3 × 103 cells per well. Allogeneic T cells were purified from the spleens of BALB/c mice by negative selection with the MACS CD4+ T-cell isolation kit (Miltenyi Biotech Ltd.). T-cell density was adjusted to 106 cells/ml in IMDM, and 100 μl of the cell suspension was added to wells containing purified DC as described above. Plates were incubated for 48 h at 37°C. Tritiated thymidine (1 μCi/well; New England Nuclear) was added for 24 h, and cells were harvested for analysis of thymidine incorporation by liquid scintillation counting.
Cytokine production by DC.
Purified DC were cultured in 24-well plates at 106 cells/ml in the presence of either live EBs or UV-EB at an MOI of 3. DC were also incubated with either medium alone, LPS (1 μg/ml), or CpG (30 μg/ml). After 48 h of incubation, cell supernatants were collected and stored at −80°C for cytokine profiling. IFN-γ, IL-12 (p40/p70), TNF-α, and IL-10 determination in culture supernatants was carried out by ELISA (Pharmingen) as previously described (23) with minor modifications. Briefly, ELISA plates were coated with capture antibody at 4 μg/ml in bicarbonate buffer for 16 h at 4°C. After being washed three times with PBS containing 0.5% Tween 20, the plates were blocked with 1% bovine serum albumin in PBS for 2 h at room temperature. Culture supernatants were added, incubated for 3 h, and washed three times with PBS. Detection was carried out by standard procedures as previously described (23).
Presentation of chlamydial antigens by DC.
Presentation of antigens by DC was determined as described previously (33). Chlamydia-specific CD4+ T cells were generated by immunizing 8- to 12-week-old female C57BL/6 mice intraperitoneally (i.p.) with 106 live EBs and boosting 2 weeks later. Spleens were isolated from mice 1 week after the second immunization, and CD4+ T cells were isolated with a MACS CD4+ T-cell isolation kit (Miltenyi Biotech). As a control, CD4+ T cells from nonimmunized animals were isolated in parallel. Freshly purified chlamydia-specific CD4+ T cells or irrelevant control CD4+ T cells (4 × 105 T cells in 100 μl of medium) were added to DC prepared as follows. Immature purified DC were incubated with either live EBs or UV-EB at an MOI of 3 for 4 h. Cells were washed three times with PBS plus 2% FCS to remove unbound bacteria and plated in 96-well dishes at a concentration of 105 cells/well in 100 μl of IMDM containing 10% FCS. Cells were incubated for 48 h at 37°C. The amount of IFN-γ present in the supernatants as assayed by ELISA was used as a measure of antigen-specific T-cell recognition.
Phagocytosis and viability assays.
Purified DC were incubated with various MOIs of live EBs or UV-EB for 4 h at 37°C, and unbound bacteria were removed by washing. Infected DC were incubated for another 2 h at 37°C. DC were fixed with 2% paraformaldehyde and washed three times with permeabilization buffer containing 0.5% saponin, 2% PBS, and 0.2% sodium azide in PBS. Nonspecific sites were blocked with goat serum, and cells were stained with goat FITC-labeled antichlamydia MAb (ViroStat) in permeabilization buffer. Analysis was carried out by FACS. Results are expressed as the percentage of CD11c+ cells containing chlamydia. Less than 2% of DC were found to bind live EBs or UV-EB, as determined by staining with FITC-labeled antichlamydia antibody without permeabilization (data not shown). For DC viability, cells were incubated for 2 h with either live EBs or UV-EB at various MOIs. Unbound bacteria were removed by washing, and DC were incubated at 37°C for a total of 48 h. DC were stained with trypan blue (Sigma), and 100 cells were counted by visualization under a microscope. Results represent the percentage of living cells in the experimental group compared with the untreated controls.
Immunization and challenge.
Purified DC were incubated at 106 cells/ml with one of the following: (i) normal medium, (ii) live EBs (MOI of 1), (iii) UV-EB (MOI of 1), or (iv) UV-EB plus 30 μg of CpG/ml. Incubation was carried out for 4 h at 37°C prior to washing with PBS containing 2% FCS, and DC were incubated for an additional 42 h in IMDM containing 10% FCS. CpG was maintained in the culture medium for the duration of the 48-h incubation. Treated and untreated DC were collected and washed three times with endotoxin-free PBS (Stem Cell Technologies). DC (0.5 × 106 in PBS) were injected i.p. into C57BL/6 mice. Two groups of mice were immunized with 2 × 105 live EBs or UV-EB as controls. Two weeks later, mice were given a second immunization, and 7 days after the last immunization mice were challenged intranasally with 3,000 live EBs. Protection was assessed by measuring body weight loss on a daily basis. Eight days after the intranasal challenge, the lungs were removed and homogenized in 5 ml of sucrose-phosphate-glutamic acid buffer, and the suspension was used to determine the IFU numbers in HeLa cells as described for EB titration (see above).
Statistical analysis.
Statistical analysis was carried out by using Student's t test.
RESULTS
Effect of live EBs and UV-EB on DC.
To examine the effect of chlamydia on the maturation of DC, bone marrow-derived DC were generated and incubated with either live EBs or UV-EB. DC exposed to MoPn were evaluated for the following: (i) the ability to phagocytose MoPn, (ii) the viability of DC following phagocytosis, (iii) changes in morphology, and (iv) the ability to support chlamydial replication. Figure 1A shows that at low MOIs, both live EBs and UV-EB were internalized at relatively similar levels, while at MOIs greater than 6, DC internalized live EBs slightly more efficiently. Figure 1B demonstrates that low ratios of live EBs (MOIs of 1 to 3) did not significantly affect DC viability, whereas at high ratios (MOI > 3), only exposure to live EBs resulted in a significant reduction of DC viability (P < 0.001). At an MOI of 1 or 3, DC demonstrated equivalent levels of phagocytosis, and these MOIs had no discernible effect on DC viability when DC were exposed to either live EBs or UV-EB; MOIs of 1 to 3 were used throughout the rest of this work, as noted in the text. Microscopic examination 48 h after incubation showed that exposure to live EBs induced morphological features similar to the ones induced by LPS and known to be displayed by mature DC (53), namely, large floating cell aggregates, cells that were loosely attached to the plastic, and cells with rounded shapes that displayed large veils and long dendrites (data not shown). In contrast, minimal morphological changes were observed in DC incubated with UV-EB, and these DC more closely resembled the uninfected DC controls (data not shown).
FIG. 1.

Effect of live EBs and UV-EB on viability and phagocytic ability of DC. (A) DC were exposed to live EBs or UV-EB for 6 h at the indicated MOIs. Internalized bacteria were visualized by FACS after intracellular staining with antichlamydia FITC-labeled antibody. Results are given as the percentage of cells that were positive for both chlamydia and CD11c. (B) DC viability was determined by trypan blue exclusion and was calculated as the percentage of viable cells in the experimental group divided by the number of viable cells in the uninfected controls. Results represent the means ± standard deviations (SD) from four experiments. *, P < 0.05; **, P < 0.001.
We next examined whether MoPn survived and formed inclusions within DC. Purified DC were incubated with live EBs at an MOI of either 1 or 3 and incubated for between 1 and 72 h prior to harvesting and screening for inclusions. No inclusions were found within infected DC, and no infectious progeny were observed within HeLa cells incubated with infected DC lysates (data not shown). These results confirm those of previous studies indicating that chlamydia do not replicate within DC (34).
Live EBs and UV-EB induce different patterns of expression of DC activation markers.
It has been previously shown that mature DC express high levels of MHC class II and costimulatory molecules (15). Therefore, we examined whether exposure to MoPn alters DC surface marker expression. Purified DC were either left untreated or exposed to live EBs, UV-EB, or LPS. Table 1 shows that the expression of ICAM-1 and MHC-II was slightly higher in DC infected with live EBs than in cells incubated with UV-EB; however, this difference was not statistically significant. In contrast, the expression of CD40 and CD86 was highly up-regulated in response to live EBs and was nearly equivalent to levels found in LPS-stimulated DC (Table 1), while CD40 and CD86 expression was significantly lower following exposure to UV-EB (P < 0.001). In addition, increasing the MOI of UV-EB to 6 had no effect on the expression of CD40 and CD86 (data not shown). Importantly, all DC markers examined were expressed at relatively low levels in untreated, control DC and were highly expressed upon LPS stimulation (Table 1). This result demonstrates that exposure of DC to live EBs but not UV-EB induced full expression of costimulatory molecules and suggests that live EBs generated a fully mature DC phenotype, whereas exposure to UV-EB generated a semimature DC phenotype.
TABLE 1.
Phenotypes of BM-DC pulsed with Chlamydia trachomatis MoPn
| DC treatment | Proportion of CD11c-positive cells expressing the markera:
|
||||
|---|---|---|---|---|---|
| CD86 | CD80 | CD40 | ICAM-1 | IA (MHC class II) | |
| None | 21 ± 3 | 74 ± 8 | 23 ± 7 | 76 ± 9 | 65 ± 7 |
| Live EB | 88 ± 3 | 94 ± 7 | 89 ± 5 | 92 ± 8 | 86 ± 9 |
| UV-EB | 38 ± 4b | 91 ± 3 | 46 ± 5b | 87 ± 7 | 78 ± 10 |
| LPS | 82 ± 6 | 93 ± 5 | 92 ± 10 | 94 ± 5 | 82 ± 9 |
The proportions of CD11c-positive cells expressing the marker ± the standard errors are given.
P < 0.001.
Functional maturation of DC upon exposure to live EBs or UV-EB.
Mature DC are able to provide effective signals to T, B, and NK cells and to produce proinflammatory cytokines (15). Three sets of functional assays were performed to examine the functional maturation status of DC upon incubation with live EBs or UV-EB. First, chlamydia-treated DC were tested for their ability to induce allogeneic T-cell proliferation (42). Purified DC were incubated with live EBs or UV-EB and cocultured with allogeneic T cells purified from the spleens of BALB/c mice, and T-cell proliferation was assessed by incorporation of tritiated thymidine. As shown in Fig. 2A, DC pulsed with live EBs induced proliferation of allogeneic T cells, comparable to the proliferation induced by LPS-treated DC. In contrast, proliferation induced by DC treated with UV-EB was significantly lower and not different from the negative control.
FIG. 2.



Phenotypic and functional maturation of DC upon exposure to live EBs and UV-EB. (A) Allogeneic T-cell proliferation assays. DC from C57BL/6 mice were left untreated, incubated with LPS (1 μg/ml), or exposed to either live EBs or UV-EB for 48 h prior to irradiation. Irradiated DC were cocultured with purified BALB/c T cells for 48 h, and tritiated thymidine was added for an additional 24 h prior to harvesting and analysis for thymidine incorporation. Results represent the number of counts per minute (cpm) calculated per 100 DC. (B) Cytokine profiling. DC were untreated, incubated with LPS (1 μg/ml), or exposed to live EBs or UV-EB for 48 h before the supernatants were analyzed for cytokine production by ELISA. (C) IFN-γ production by Chlamydia-specific T cells. CD4+ T cells isolated from mice immunized against MoPn were cultured in isolation or cocultured with DC pulsed with either live EBs or UV-EB. Naïve DC and DC exposed to either live EBs or UV-EB were cultured in the absence of T cells to serve as DC negative controls. T cells isolated from naïve mice were either cultured alone or cocultured with DC pulsed with either live EBs or UV-EB and represent T-cell negative controls. Cells were incubated for 48 h, and the amount of IFN-γ secreted by T cells was determined by ELISA. All experiments were performed in triplicate and repeated on three separate occasions, with similar results. Results represent the mean ± SD from one representative experiment. *, P < 0.01; **, P < 0.001; ***, P < 0.01.
Second, live EBs and UV-EB were tested for their ability to induce secretion of proinflammatory (IL-12, TNF-α, and IFN-γ) or regulatory (IL-10) cytokines by DC (14, 39, 48, 52). Figure 2B shows that the secretion levels of IL-12 and TNF-α by DC in response to live EBs were 16 ± 1.5 and 1.5 ± 0.1 ng/ml, respectively. These amounts were 3 (P < 0.01) and 15 (P < 0.001) times higher than the corresponding cytokine levels produced by DC in response to UV-EB. Interestingly, both live EBs and UV-EB induced similarly low levels of IL-10 (Fig. 2B). No IFN-γ production was detected in response to either live EBs or UV-EB (data not shown). Production of IL-12 and TNF-α by DC in response to LPS was similar to the production induced by UV-EB and far less than the amounts induced by live EBs. In contrast, LPS was a much more potent inducer of IL-10 than was chlamydia (P < 0.001) (Fig. 2B).
Third, we compared the abilities of live EB- and UV-EB-pulsed DC to process and present antigens to chlamydia-specific CD4+ T cells (33). DC exposed to live EBs or UV-EB were incubated with chlamydia-specific CD4+ T cells isolated from mice immunized against MoPn. Production of IFN-γ was used to measure an antigen-specific TH1 response. In terms of IFN-γ production, chlamydia-specific CD4+ T cells recognized DC pulsed with live EBs significantly more efficiently (14.3 ± 1.1 ng/ml) than did DC pulsed with UV-EB (7.1 ± 1.3 ng/ml) (P < 0.01). IFN-γ production required immune T cells, since neither naïve DC nor DC pulsed with either live EBs or UV-EB produced IFN-γ. Similarly, naïve CD4+ T cells did not secrete IFN-γ upon coculture with live EB- or UV-EB-pulsed DC (Fig. 2C).
Collectively, these data show that live EBs induced functional maturation of DC compared to UV-EB. These findings suggest that live EB-pulsed DC might be more effective at inducing protective immunity in vivo than DC pulsed with UV-irradiated or otherwise inactivated EBs.
Resistance to chlamydial infection induced by live EB- or UV-EB-pulsed DC.
To determine whether DC pulsed with live EBs induced better protective immunity in vivo than UV-EB-pulsed DC, unstimulated DC or DC pulsed with either live EBs or UV-EB were adoptively transferred by intraperitoneal injection into naïve mice. Protection was assessed by monitoring body weight loss after intranasal challenge with a lethal dose of MoPn. Mice were sacrificed at 8 days postchallenge, and pulmonary bacterial load was determined. As shown in Fig. 3A, mice adoptively immunized with live EB-pulsed DC developed a mild course of disease as determined by body weight loss. The disease was rapidly controlled, and by day 7 the mice had completely recovered their original body weights. As shown in Fig. 3B, 0.3 × 103 IFU were detected in the lungs of these mice 8 days after challenge. In contrast, mice immunized i.p. with UV-EB-pulsed DC exhibited progressive weight loss (P < 0.001) during the 8-day period (Fig. 3A), and 4.8 × 106 IFU were detected in their lungs (P < 0.001 compared to live EB-pulsed DC) (Fig. 3B). Of note, mice i.p. immunized with live EBs and allowed to recover for 2 weeks prior to pulmonary challenge lost more weight and also had a higher pulmonary IFU count than mice immunized i.p. with live EB-pulsed DC (P < 0.01 and P < 0.001) (Fig. 3A and B, respectively). Taken together, these results suggest that live EB-pulsed DC are more effective than UV-EB-pulsed DC in conferring protective immunity when administered by i.p. adoptive transfer.
FIG. 3.

Body weight loss and pulmonary bacterial load of mice immunized with live EB- or UV-EB-pulsed DC. DC were left untreated or incubated with either live EBs or UV-EB for a total of 48 h at 37°C. DC were washed with endotoxin-free PBS, and a total of 0.5 × 106 DC were injected into the intraperitoneal cavities of mice, which were separated into experimental groups of five mice. Two groups of mice were immunized intraperitoneally with either 2 × 105 live EBs or UV-EB as controls. Animals were boosted with an identical dose of DC or EBs on day 14 and finally challenged intranasally on day 21 with 3,000 IFU of live EBs. Immune protection was determined by daily measurement of body weight after the final challenge (A) and the determination of pulmonary IFU counts on day 8 postchallenge (B). All experiments were performed on three separate occasions, with similar results. Results represent the mean ± SD from one representative experiment. *, P < 0.001 (comparing live EB-pulsed DC and UV-EB-pulsed DC); **, P < 0.01; and ***, P < 0.001 (comparing live EBs and live EB-pulsed DC).
CpG enhances the DC maturation levels induced by UV-EB alone.
Oligodeoxynucleotides containing CpG motifs have been used as adjuvants to induce TH1 type immune responses (36, 37). We examined whether UV-EB-pulsed DC could be induced to mature by costimulation with CpG ODN sequence 1826 (37). Purified DC were either untreated, incubated with UV-EB (MOI of 3) and CpG (30 μg/ml), or incubated with CpG alone. Cells were then tested for the expression of CD40 and CD86, cytokine secretion, and the ability to induce allogeneic T-cell proliferation. As shown in Fig. 4, CpG alone was effective at promoting DC maturation according to all three criteria and also induced a mature DC morphology (data not shown). Thus, in contrast to UV-EB-pulsed DC, the combination of UV-EB and CpG enhanced the expression of both CD86 and CD40 (P < 0.01) (Fig. 4A) and significantly stimulated allogeneic T-cell proliferation (P < 0.01) (Fig. 4B). Furthermore, in contrast to UV-EB-pulsed DC, costimulation with CpG and UV-EB promoted the production of IL-12 (P < 0.01) and TNF-α (P < 0.001) but not IL-10 (Fig. 4C). Surprisingly, UV-EB appeared to interfere with the ability of CpG to induce full DC maturation, since CpG-induced expression of CD86 or production of IL-12 and TNF-α were significantly lower when DC were coincubated with both CpG and UV-EB than when DC were incubated with CpG alone (Fig. 4C). Nevertheless, these results suggest that DC activation by UV-EB can be enhanced by adjuvants such as CpG.
FIG. 4.
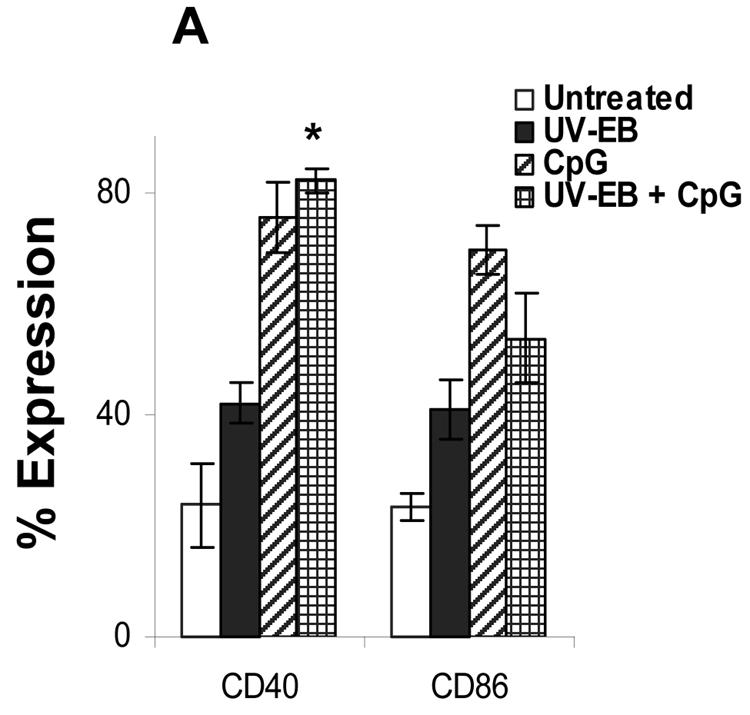


CpG induces maturation of UV-EB-pulsed DC. (A) Effect of CpG on the surface expression of CD40 and CD86. DC were left untreated or incubated with either UV-EB, UV-EB and CpG, or CpG alone. After 48 h, DC were stained for CD11c and either CD40 or CD86 and analyzed by FACS. Results shown are the percentage of CD11c+ cells expressing either CD40 or CD86. Results represent the mean ± SD from five independent experiments. (B) Allogeneic T-cell proliferation induced by DC upon exposure to UV-EB and CpG. Purified DC were left untreated or incubated with CpG (30 μg/ml), UV-EB alone, or UV-EB and CpG (30 μg/ml) and incubated for 48 h. Tritiated thymidine was added for an additional 24 h before cells were harvested and analyzed for thymidine incorporation. Experiments were performed on three separate occasions, with similar results. Results represent the mean ± SD from one experiment and show the number of cpm calculated per 100 DC. (C) Cytokine production by DC upon exposure to UV-EB and CpG. DC were left untreated or incubated with CpG (30 μg/ml), UV-EB, or UV-EB and CpG (30 μg/ml) for 48 h before the supernatants were analyzed for cytokine production by ELISA. Experiments were performed on three separate occasions, with similar results. Results represent the mean ± SD from one representative experiment. *, P < 0.01; **, P < 0.001 (both comparing UV-EB-pulsed DC and DC exposed to UV-EB and CpG).
Adoptive transfer of DC exposed to UV-EB and CpG enhances immune protection.
In order to test the immunogenicity conferred by CpG to UV-EB-pulsed DC, cells were costimulated ex vivo with UV-EB plus CpG for a total of 48 h and injected i.p. into naïve mice. Control groups received unstimulated DC, DC pulsed with UV-EB, or DC treated with CpG alone. Protection was measured by body weight loss and bacterial load in the lungs 8 days postchallenge. Mice immunized with DC costimulated with UV-EB and CpG showed significantly less body weight loss than mice given DC pulsed with UV-EB (P < 0.01) (Fig. 5A). As shown in Fig. 5B, the pulmonary bacterial loads of these mice by day 8 were somewhat reduced (1 × 106), whereas those of mice immunized with UV-EB-pulsed DC were significantly higher (5.7 × 106) (P < 0.001). Thus, although the immunogenic potential of DC induced by UV-EB can be enhanced by CpG, this only marginally enhanced protective immunity.
FIG. 5.

Body weight loss and pulmonary bacterial load of mice immunized with UV-EB- and CpG-pulsed DC. DC were left untreated or incubated with live EBs, UV-EB, CpG, or UV-EB and CpG for a total of 48 h at 37°C. DC were washed with endotoxin-free PBS, and a total of 0.5 × 106 DC were injected into the intraperitoneal cavities of mice, which were separated into experimental groups of five mice. Animals were boosted with an identical dose of DC on day 14 and finally challenged intranasally on day 21 with 3,000 IFU of live EBs. Immune protection was determined by daily measurement of body weight after the final challenge (A) and the determination of pulmonary IFU counts on day 8 postchallenge (B). All experiments were performed on three separate occasions, with similar results. Results represent the mean ± SD from one representative experiment. *, P < 0.01; and **, P < 0.001 (both comparing UV-EB-pulsed DC and UV-EB- and CpG-pulsed DC).
DISCUSSION
Observations from both clinical data from human infections and studies using murine infection models suggest that exposure to live and dead chlamydial EBs generate distinct immune responses. While both humans and mice demonstrate protective immunity following infection (4, 31), murine immunization studies using whole inactivated chlamydia organisms resulted in little or no protection against infection (23), and related human vaccine trials were similarly unsuccessful (7, 12). Since DC are decisive effectors of innate immunity and dictate the type of acquired immune response to a particular antigen, and this ability depends in part on the maturation status of DC (11, 20), we reasoned that live and dead chlamydia could induce different patterns of DC immunogenicity by differentially inducing DC maturation. This could explain, in part, the apparent disparate immune effects induced by live and dead chlamydia in vivo.
In this study, we compared the DC maturation statuses and immune effector functions induced by exposure to live or dead C. trachomatis MoPn EBs. We found that although both live and dead chlamydia are efficiently phagocytosed by DC, live EBs were more effective than UV-EB at inducing optimal phenotypic and functional maturation of DC as determined by the following results: (i) the induction of mature DC morphology; (ii) an increased ability to induce allogeneic T-cell proliferation; (iii) increased expression of CD40, CD80, CD86, MHC class II, and ICAM-1; (iv) enhanced production of proinflammatory cytokines IL-12 and TNF-α but not IL-10; and (v) an enhanced ability to promote protective immunity to challenge infection upon adoptive transfer. According to the criteria defining DC maturation types outlined by Lutz and Schuler (26), these observations indicate that exposure to UV-EB induces a semimature DC phenotype, while exposure to live EBs generates a mature DC phenotype.
However, the question remains as to why live EB-pulsed DC produce a more effective protective immune response upon adoptive transfer than UV-EB-pulsed DC. We found that live EB-pulsed DC displayed significantly higher levels of the costimulatory molecules CD86 and CD40 and produced a stronger allogeneic T-cell response than UV-EB-pulsed DC. As the expression of DC costimulatory molecules is critical for effective DC-mediated T-cell activation (15), it stands to reason that live EB-pulsed DC would be more effective at stimulating a T-cell response due to increased surface expression of CD40 and CD86. In addition, live EB-pulsed DC may be better immune effectors than UV-EB-pulsed DC due to their ability to produce elevated levels of the proinflammatory cytokines IL-12 and TNF-α (Fig. 2B). Both populations of DC produced similar, low levels of IL-10, which has been found to suppress protective immunity against chlamydial infections (17). However, IL-12 is required to effectively control chlamydial infections (24), and TNF-α is known to promote DC migration (48). Therefore, these cytokine profiles and maturation phenotypes are consistent with live EB-pulsed DC being more immunogenic and therefore more effective at promoting an antichlamydial immune response than UV-EB-pulsed DC.
Our results are consistent with those of studies of other pathogens demonstrating that exposure to live microorganisms generates mature DC phenotypes and that these DC are highly immunogenic and generate a protective immune response upon adoptive transfer (28, 29). In particular, one study published while the manuscript was in preparation demonstrated that DC isolated from mice infected with Listeria monocytogenes expressed elevated levels of costimulatory molecules and produced IFN-γ and IL-12. Furthermore, upon adoptive transfer, DC exposed to live L. monocytogenes but not DC exposed to killed L. monocytogenes promoted resistance to a subsequent lethal infection through a mechanism that involves both CD4+ and CD8+ T cells (40). However, it should be noted that in contrast to our results and those observed with L. monocytogenes are observations with Bordetella bronchiseptica (45) and Legionella pneumophila showing that DC pulsed with killed microorganisms induced a more effective protective immune response than DC pulsed with live microorganisms (21). Overall, these findings suggest that the effects of live and dead bacteria on DC maturation are pathogen specific and are not easily predictable.
Our results demonstrating that UV-EB-pulsed DC induce poor protection upon adoptive transfer differ from those of a previous study by Su et al. (49), who demonstrated that DC exposed to heat-killed C. trachomatis and adoptively transferred intravenously induced effective protection against intravaginal chlamydial challenge. The present study used an intraperitoneal route for adoptive transfer, examined pulmonary chlamydial titers, and compared DC loaded with inactivated EBs to DC loaded with live EBs. These differences in experimental design likely explain the differences in the reported observations. In addition, we have observed that while intravenous adoptive transfer of UV-EB-pulsed DC protects against body weight loss after subsequent MoPn infection, pulmonary IFU titers were significantly higher than in animals immunized in a similar manner with live EB-pulsed DC (data not shown). Thus, while the route of adoptive transfer of loaded DC may play a role in the induction of effective protective immunity, our results demonstrate that factors associated with DC maturation status are also important for optimal protective effects.
In light of the critical importance of DC maturation for effective T-cell priming (1), the findings that live EBs and UV-EB induce differential DC maturation and distinct immunogenicity profiles have practical implications. For example, it has been recently shown that intravenous adoptive transfer of DC pulsed ex vivo with chlamydial MOMP generated a TH2 type of response and that immunized mice were not protected following vaginal challenge (43). Similarly, adoptive transfer of DC exposed to leishmania antigens did not protect mice upon subsequent challenge. However, adoptive transfer of DC pulsed with leishmania antigen in combination with CpG did result in protection against leishmanial challenge (36). In our studies, although UV-EB-pulsed DC showed partial DC maturation and partial protection against MoPn upon adoptive transfer, costimulation of these DC with CpG increased DC maturation to levels higher than those achieved with UV-EB alone, increased their ability to induce allogeneic T-cell proliferation, increased production of IL-12 and TNF-α, and conferred enhanced protection to challenge infection after adoptive transfer. However, CpG treatment did not induce full activation of UV-EB-treated DC and was not as efficient in protecting against chlamydial challenge as live EB-pulsed DC (Fig. 4 and 5). Overall, this result indicates that the choice of an appropriate adjuvant will be an important consideration for future efforts in chlamydial vaccine design and reinforces the fact that the examination of DC maturation markers is not sufficient for the identification of an efficacious candidate protein-adjuvant combination: the ultimate test remains protective studies.
This study raises several interesting questions as to the effect of chlamydia on DC and as to which bacterial proteins or components are responsible for the induction of either a tolerogenic or a protective immune response. The purpose of using UV-irradiated EBs as opposed to heat-inactivated EBs in this study was to maintain chlamydial surface proteins in a native conformation so that the interaction of the EB with the DC would be as similar as possible between the two treatment conditions. Therefore, it would be expected that a live EB would affect a DC in a manner similar to a UV-EB. The fact that the immune responses initiated by DC exposed to UV-EB and CpG are attenuated in comparison to those of DC pulsed with CpG alone suggests that UV-EB may induce repressive effects that cannot be overcome by CpG. Live EBs, on the other hand, promoted full DC maturation and efficiently promoted protective immunity. Even though our study and others have not found chlamydia to survive or replicate within DC, it is possible that upon infection, chlamydial proteins and/or factors are released intracellularly, which potentially activates DC. These include chlamydial type III secreted proteins (10), chlamydial chaperonins, and early chlamydial secreted factors used by the bacteria to survive inside the host cell (2). In fact, early events occurring in HeLa cells as a consequence of chlamydial infection include reorganization of the host cell cytoskeleton (6) and chlamydia-dependent phosphorylation of host cell proteins (3). These events are not entirely dependent on de novo chlamydial protein synthesis, suggesting that they are induced by preformed chlamydial factors. Whether similar events happen in chlamydia-infected DC but not in UV-EB-treated DC needs further investigation.
In conclusion, our results demonstrate that DC exposed to live EBs are phenotypically and functionally distinct from those generated upon exposure to UV-EB. Furthermore, live EB-pulsed DC are immunologically more effective than UV-EB-pulsed DC, as they strongly promoted protective immunity to chlamydial infection. These results suggest that live and dead chlamydia may differently stimulate endogenous DC in vivo and may explain the distinct outcomes observed between infection with live chlamydia and immunization with inactivated microorganisms. The study indicates that future directions for rational vaccine design require the use of strategies that cause full DC activation and that the ideal antigen-adjuvant combination would mimic the DC maturation effects induced by live EBs.
Acknowledgments
We thank Neil Reiner for critically reviewing the manuscript.
This work was supported by a grant to R.C.B. from the Canadian Institutes of Health Research.
Editor: J. L. Flynn
REFERENCES
- 1.Banchereau, J., and R. M. Steinman. 1998. Dendritic cells and the control of immunity. Nature 392:245-252. [DOI] [PubMed] [Google Scholar]
- 2.Belland, R. J., G. Zhong, D. D. Crane, D. Hogan, D. Sturdevant, J. Sharma, W. L. Beatty, and H. D. Caldwell. 2003. Genomic transcriptional profiling of the developmental cycle of Chlamydia trachomatis. Proc. Natl. Acad. Sci. USA 100:8478-8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Birkelund, S., L. Bini, V. Pallini, M. Sanchez-Campillo, S. Liberatori, J. D. Clausen, S. Ostergaard, A. Holm, and G. Christiansen. 1997. Characterization of Chlamydia trachomatis l2-induced tyrosine-phosphorylated HeLa cell proteins by two-dimensional gel electrophoresis. Electrophoresis 18:563-567. [DOI] [PubMed] [Google Scholar]
- 4.Brunham, R. C., J. Kimani, J. Bwayo, G. Maitha, I. Maclean, C. Yang, C. Shen, S. Roman, N. J. Nagelkerke, M. Cheang, and F. A. Plummer. 1996. The epidemiology of Chlamydia trachomatis within a sexually transmitted diseases core group. J. Infect. Dis. 173:950-956. [DOI] [PubMed] [Google Scholar]
- 5.Brunham, R. C., D. J. Zhang, X. Yang, and G. M. McClarty. 2000. The potential for vaccine development against chlamydial infection and disease. J. Infect. Dis. 181(Suppl. 3):S538-S543. [DOI] [PubMed] [Google Scholar]
- 6.Carabeo, R. A., S. S. Grieshaber, E. Fischer, and T. Hackstadt. 2002. Chlamydia trachomatis induces remodeling of the actin cytoskeleton during attachment and entry into HeLa cells. Infect. Immun. 70:3793-3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clements, C., S. P. Dhir, J. T. Grayston, and S. P. Wang. 1979. Long term follow-up study of a trachoma vaccine trial in villages of northern India. Am. J. Ophthalmol. 87:350-353. [DOI] [PubMed] [Google Scholar]
- 8.Dhodapkar, M. V., R. M. Steinman, J. Krasovsky, C. Munz, and N. Bhardwaj. 2001. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J. Exp. Med. 193:233-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferguson, T. A., J. Herndon, B. Elzey, T. S. Griffith, S. Schoenberger, and D. R. Green. 2002. Uptake of apoptotic antigen-coupled cells by lymphoid dendritic cells and cross-priming of CD8(+) T cells produce active immune unresponsiveness. J. Immunol. 168:5589-5595. [DOI] [PubMed] [Google Scholar]
- 10.Fields, K. A., D. J. Mead, C. A. Dooley, and T. Hackstadt. 2003. Chlamydia trachomatis type III secretion: evidence for a functional apparatus during early-cycle development. Mol. Microbiol. 48:671-683. [DOI] [PubMed] [Google Scholar]
- 11.Fujii, S., K. Liu, C. Smith, A. J. Bonito, and R. M. Steinman. 2004. The linkage of innate to adaptive immunity via maturing dendritic cells in vivo requires CD40 ligation in addition to antigen presentation and CD80/86 costimulation. J. Exp. Med. 199:1607-1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grayston, J. T., and S. P. Wang. 1978. The potential for vaccine against infection of the genital tract with Chlamydia trachomatis. Sex. Transm. Dis. 5:73-77. [DOI] [PubMed] [Google Scholar]
- 13.Grayston, J. T., S. P. Wang, L. J. Yeh, and C. C. Kuo. 1985. Importance of reinfection in the pathogenesis of trachoma. Rev. Infect. Dis. 7:717-725. [DOI] [PubMed] [Google Scholar]
- 14.Gurunathan, S., C. Prussin, D. L. Sacks, and R. A. Seder. 1998. Vaccine requirements for sustained cellular immunity to an intracellular parasitic infection. Nat. Med. 4:1409-1415. [DOI] [PubMed] [Google Scholar]
- 15.Hackstein, H., and A. W. Thomson. 2004. Dendritic cells: emerging pharmacological targets of immunosuppressive drugs. Nat. Rev. Immunol. 4:24-34. [DOI] [PubMed] [Google Scholar]
- 16.Han, X., Y. Fan, S. Wang, J. Yang, L. Bilenki, H. Qiu, L. Jiao, and X. Yang. 2004. Dendritic cells from Chlamydia-infected mice show altered Toll-like receptor expression and play a crucial role in inhibition of allergic responses to ovalbumin. Eur. J. Immunol. 34:981-989. [DOI] [PubMed] [Google Scholar]
- 17.Igietseme, J. U., G. A. Ananaba, J. Bolier, S. Bowers, T. Moore, T. Belay, F. O. Eko, D. Lyn, and C. M. Black. 2000. Suppression of endogenous IL-10 gene expression in dendritic cells enhances antigen presentation for specific Th1 induction: potential for cellular vaccine development. J. Immunol. 164:4212-4219. [DOI] [PubMed] [Google Scholar]
- 18.Igietseme, J. U., C. M. Black, and H. D. Caldwell. 2002. Chlamydia vaccines: strategies and status. BioDrugs 16:19-35. [DOI] [PubMed] [Google Scholar]
- 19.Inaba, K., M. Inaba, N. Romani, H. Aya, M. Deguchi, S. Ikehara, S. Muramatsu, and R. M. Steinman. 1992. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 176:1693-1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inaba, K., J. P. Metlay, M. T. Crowley, and R. M. Steinman. 1990. Dendritic cells pulsed with protein antigens in vitro can prime antigen-specific, MHC-restricted T cells in situ. J. Exp. Med. 172:631-640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kikuchi, T., T. Kobayashi, K. Gomi, T. Suzuki, Y. Tokue, A. Watanabe, and T. Nukiwa. 2004. Dendritic cells pulsed with live and dead Legionella pneumophila elicit distinct immune responses. J. Immunol. 172:1727-1734. [DOI] [PubMed] [Google Scholar]
- 22.Liu, Y. J. 2001. Dendritic cell subsets and lineages, and their functions in innate and adaptive immunity. Cell 106:259-262. [DOI] [PubMed] [Google Scholar]
- 23.Lu, H., Z. Xing, and R. C. Brunham. 2002. GM-CSF transgene-based adjuvant allows the establishment of protective mucosal immunity following vaccination with inactivated Chlamydia trachomatis. J. Immunol. 169:6324-6331. [DOI] [PubMed] [Google Scholar]
- 24.Lu, H., X. Yang, K. Takeda, D. Zhang, Y. Fan, M. Luo, C. Shen, S. Wang, S. Akira, and R. C. Brunham. 2000. Chlamydia trachomatismouse pneumonitis lung infection in IL-18 and IL-12 knockout mice: IL-12 is dominant over IL-18 for protective immunity. Mol. Med. 6:604-612. [PMC free article] [PubMed] [Google Scholar]
- 25.Lu, H., and G. Zhong. 1999. Interleukin-12 production is required for chlamydial antigen-pulsed dendritic cells to induce protection against live Chlamydia trachomatis infection. Infect. Immun. 67:1763-1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lutz, M. B., and G. Schuler. 2002. Immature, semi-mature and fully mature dendritic cells: which signals induce tolerance or immunity? Trends Immunol. 23:445-449. [DOI] [PubMed] [Google Scholar]
- 27.Matyszak, M. K., J. L. Young, and J. S. Gaston. 2002. Uptake and processing of Chlamydia trachomatis by human dendritic cells. Eur. J. Immunol. 32:742-751. [DOI] [PubMed] [Google Scholar]
- 28.Mbow, M. L., N. Zeidner, N. Panella, R. G. Titus, and J. Piesman. 1997. Borrelia burgdorferi-pulsed dendritic cells induce a protective immune response against tick-transmitted spirochetes. Infect. Immun. 65:3386-3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller, G., S. Lahrs, V. G. Pillarisetty, A. B. Shah, and R. P. DeMatteo. 2002. Adenovirus infection enhances dendritic cell immunostimulatory properties and induces natural killer and T-cell-mediated tumor protection. Cancer Res. 62:5260-5266. [PubMed] [Google Scholar]
- 30.Moll, H. 2003. Dendritic cells and host resistance to infection. Cell. Microbiol. 5:493-500. [DOI] [PubMed] [Google Scholar]
- 31.Morrison, R. P., and H. D. Caldwell. 2002. Immunity to murine chlamydial genital infection. Infect. Immun. 70:2741-2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morrison, S. G., H. Su, H. D. Caldwell, and R. P. Morrison. 2000. Immunity to murine Chlamydia trachomatis genital tract reinfection involves B cells and CD4+ T cells but not CD8+ T cells. Infect. Immun. 68:6979-6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neild, A. L., and C. R. Roy. 2003. Legionella reveal dendritic cell functions that facilitate selection of antigens for MHC class II presentation. Immunity 18:813-823. [DOI] [PubMed] [Google Scholar]
- 34.Ojcius, D. M., Y. Bravo de Alba, J. M. Kanellopoulos, R. A. Hawkins, K. A. Kelly, R. G. Rank, and A. Dautry-Varsat. 1998. Internalization of Chlamydia by dendritic cells and stimulation of Chlamydia-specific T cells. J. Immunol. 160:1297-1303. [PubMed] [Google Scholar]
- 35.Prebeck, S., C. Kirschning, S. Durr, C. da Costa, B. Donath, K. Brand, V. Redecke, H. Wagner, and T. Miethke. 2001. Predominant role of toll-like receptor 2 versus 4 in Chlamydia pneumoniae-induced activation of dendritic cells. J. Immunol. 167:3316-3323. [DOI] [PubMed] [Google Scholar]
- 36.Ramirez-Pineda, J. R., A. Frohlich, C. Berberich, and H. Moll. 2004. Dendritic cells (DC) activated by CpG DNA ex vivo are potent inducers of host resistance to an intracellular pathogen that is independent of IL-12 derived from the immunizing DC. J. Immunol. 172:6281-6289. [DOI] [PubMed] [Google Scholar]
- 37.Rhee, E. G., S. Mendez, J. A. Shah, C. Y. Wu, J. R. Kirman, T. N. Turon, D. F. Davey, H. Davis, D. M. Klinman, R. N. Coler, D. L. Sacks, and R. A. Seder. 2002. Vaccination with heat-killed leishmania antigen or recombinant leishmanial protein and CpG oligodeoxynucleotides induces long-term memory CD4+ and CD8+ T cell responses and protection against leishmania major infection. J. Exp. Med. 195:1565-1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rothfuchs, A. G., M. R. Kreuger, H. Wigzell, and M. E. Rottenberg. 2004. Macrophages, CD4(+) or CD8(+) cells are each sufficient for protection against Chlamydia pneumoniae infection through their ability to secrete IFN-gamma. J. Immunol. 172:2407-2415. [DOI] [PubMed] [Google Scholar]
- 39.Sallusto, F., A. Langenkamp, J. Geginat, and A. Lanzavecchia. 2000. Functional subsets of memory T cells identified by CCR7 expression. Curr. Top. Microbiol. Immunol. 251:167-171. [DOI] [PubMed] [Google Scholar]
- 40.Sashinami, H., A. Nakane, Y. Iwakura, and M. Sasaki. 2003. Effective induction of acquired resistance to Listeria monocytogenes by immunizing mice with in vivo-infected dendritic cells. Infect. Immun. 71:117-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schramm, N., C. R. Bagnell, and P. B. Wyrick. 1996. Vesicles containing Chlamydia trachomatis serovar L2 remain above pH 6 within HEC-1B cells. Infect. Immun. 64:1208-1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schuler, G., and R. M. Steinman. 1985. Murine epidermal Langerhans cells mature into potent immunostimulatory dendritic cells in vitro. J. Exp. Med. 161:526-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shaw, J., V. Grund, L. Durling, D. Crane, and H. D. Caldwell. 2002. Dendritic cells pulsed with a recombinant chlamydial major outer membrane protein antigen elicit a CD4(+) type 2 rather than type 1 immune response that is not protective. Infect. Immun. 70:1097-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shaw, J. H., V. R. Grund, L. Durling, and H. D. Caldwell. 2001. Expression of genes encoding Th1 cell-activating cytokines and lymphoid homing chemokines by chlamydia-pulsed dendritic cells correlates with protective immunizing efficacy. Infect. Immun. 69:4667-4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skinner, J. A., A. Reissinger, H. Shen, and M. H. Yuk. 2004. Bordetella type III secretion and adenylate cyclase toxin synergize to drive dendritic cells into a semimature state. J. Immunol. 173:1934-1940. [DOI] [PubMed] [Google Scholar]
- 46.Steinman, R. M., D. Hawiger, and M. C. Nussenzweig. 2003. Tolerogenic dendritic cells. Annu. Rev. Immunol. 21:685-711. [DOI] [PubMed] [Google Scholar]
- 47.Steinman, R. M., and M. Pope. 2002. Exploiting dendritic cells to improve vaccine efficacy. J. Clin. Investig. 109:1519-1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stoitzner, P., M. Zanella, U. Ortner, M. Lukas, A. Tagwerker, K. Janke, M. B. Lutz, G. Schuler, B. Echtenacher, B. Ryffel, F. Koch, and N. Romani. 1999. Migration of langerhans cells and dermal dendritic cells in skin organ cultures: augmentation by TNF-alpha and IL-1beta. J. Leukoc. Biol. 66:462-470. [PubMed] [Google Scholar]
- 49.Su, H., R. Messer, W. Whitmire, E. Fischer, J. C. Portis, and H. D. Caldwell. 1998. Vaccination against chlamydial genital tract infection after immunization with dendritic cells pulsed ex vivo with nonviable chlamydiae. J. Exp. Med. 188:809-818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Taylor, H. R. 1990. Development of immunity to ocular chlamydial infection. Am. J. Trop. Med. Hyg. 42:358-364. [DOI] [PubMed] [Google Scholar]
- 51.'t Hart, B. A., and Y. van Kooyk. 2004. Yin-Yang regulation of autoimmunity by DCs. Trends Immunol. 25:353-359. [DOI] [PubMed] [Google Scholar]
- 52.Trinchieri, G. 2003. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 3:133-146. [DOI] [PubMed] [Google Scholar]
- 53.Winzler, C., P. Rovere, M. Rescigno, F. Granucci, G. Penna, L. Adorini, V. S. Zimmermann, J. Davoust, and P. Ricciardi-Castagnoli. 1997. Maturation stages of mouse dendritic cells in growth factor-dependent long-term cultures. J. Exp. Med. 185:317-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.World Health Organization. 2001. Global prevalence and incidence of selected curable sexually transmitted diseases: overview and estimates. World Health Organization, Geneva, Switzerland.
- 55.Yang, X., and R. C. Brunham. 1998. Gene knockout B cell-deficient mice demonstrate that B cells play an important role in the initiation of T cell responses to Chlamydia trachomatis (mouse pneumonitis) lung infection. J. Immunol. 161:1439-1446. [PubMed] [Google Scholar]
- 56.Zhong, G., T. Fan, and L. Liu. 1999. Chlamydia inhibits interferon gamma-inducible major histocompatibility complex class II expression by degradation of upstream stimulatory factor 1. J. Exp. Med. 189:1931-1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhong, G., L. Liu, T. Fan, P. Fan, and H. Ji. 2000. Degradation of transcription factor RFX5 during the inhibition of both constitutive and interferon gamma-inducible major histocompatibility complex class I expression in chlamydia-infected cells. J. Exp. Med. 191:1525-1534. [DOI] [PMC free article] [PubMed] [Google Scholar]