

Abstract
Background
Presence of cytotoxic T lymphocytes (CTL) in the tumor microenvironment (TME) predicts the effectiveness of cancer immunotherapies. The ability of toll-like receptor 3 (TLR3) ligands, interferons (IFNs) and COX2 inhibitors to synergistically induce CTL-attracting chemokines (but not regulatory T cell (Treg)-attractants) in the TME, but not in healthy tissues, observed in our preclinical studies, suggested that their systemic application can reprogram local TMEs.
Methods
Six evaluable patients (33–69 years) with metastatic triple-negative breast cancer received six doses of systemic chemokine-modulating (CKM) regimen composed of TLR3 ligand (rintatolimod; 200 mg; intravenous), IFN-α2b (20 MU/m2; intravenous) and COX2 inhibitor (celecoxib; 2×200 mg; oral) over 2 weeks. The predetermined primary endpoint was the intratumoral change in the expression of CTL marker, CD8α, in the post-CKM versus pre-CKM tumor biopsies. Patients received follow-up pembrolizumab (200 mg, intravenously, every 3 weeks), starting 3–8 days after completion of CKM.
Results
Post-CKM biopsies showed selectively increased CTL markers CD8α (average 10.2-fold, median 5.5-fold, p=0.034) and granzyme B (GZMB; 6.1-fold, median 5.8-fold, p=0.02), but not FOXP3 (Treg marker) relative to HPRT1 expression, resulting in the increases in average CD8α/FOXP3 ratio and GZMB/FOXP3 ratio. CKM increased intratumoral CTL-attractants CCL5 and CXCL10, but not Treg-attractants CCL22 or CXCL12. In contrast, CD8+ T cells and their CXCR3+ subset showed transient decreases in blood. One clinical response (breast tumor autoamputation) and three stable diseases were observed. The patient with clinical response remains disease free, with a follow-up of 46 months as of data cut-off.
Conclusions
Short-term systemic CKM selectively increases CTL numbers and CTL/Treg ratios in the TME, while transiently decreasing CTL numbers in the blood. Transient effects of CKM suggest that its simultaneous application with checkpoint blockade and other forms of immunotherapy may be needed for optimal outcomes.
Keywords: Immunotherapy, Breast Neoplasms, T-Lymphocytes, Tumor Microenvironment
WHAT IS ALREADY KNOWN ON THIS TOPIC
Our preclinical work demonstrated that the combination of toll-like receptor 3 (TLR3) ligands and interferon (IFN)-α has two unique levels of selectivity: (1) Enhancement of the desirable cytotoxic T lymphocytes (CTL) attractants and decrease of regulatory T cell /myeloid-derived suppressor cell attractants in the tumor microenvironment (TME); and (2) preferential impact on TME, rather than healthy tissues.
WHAT THIS STUDY ADDS
This proof-of-concept study is the first demonstration that systemic infusion of TLR3 ligands and IFN-α has such two levels of selectivity in clinical settings, offering an alternative to local treatments.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Since immunotherapies depend on the presence of intratumoral CTLs, our demonstration that systemic chemokine-modulating (CKM) can selectively enhance CTL homing to multiple tumor lesions provides rationale for its use as an immunotherapy-sensitizing regimen. The transient nature of its effects suggests that CKM should be used simultaneously with anti-programmed death-1 (anti-PD-1) or other immunotherapies for optimal outcomes.
Background
Intratumoral CD8+ T cell (cytotoxic T lymphocytes (CTL)) infiltration predicts clinical outcomes in patients with multiple cancer types,1–3 including breast,4–6 and their responsiveness to programmed death-1 (PD-1)/programmed cell death ligand 1 (PD-L1) blockade7–12 and chemotherapy.11–15 In contrast, intratumoral regulatory T cells (Tregs) predict poor outcomes,6 16–19 indicating the need for new means to selectively enhance intratumoral CTL densities, relative to Tregs.6 20 While local (intratumoral) injection of cytokines,21 22 TLR agonists23 24 or STING activators,25–29 were shown to promote local infiltration of CTLs and suppress tumor growth and/or enhance the effectiveness of PD-1 and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) blockade in mouse models and early phase clinical trials, there is a need for systemic treatments to promote local and selective accumulation of CTLs in the tumor microenvironment (TME) of patients with multiple lesions which cannot be targeted individually. Since CTLs versus Tregs are known to be attracted by different sets of chemokines, we developed strategies to selectively enhance intratumoral production of chemokines attracting CTLs (such as CXCL9, CXCL10, CXCL11, and CCL5, which bind to CTL-expressed receptors CXCR3 and CCR5),16 30–33 while suppressing the Treg-attractants and myeloid-derived suppressor cell (MDSC)-attractants (such as CCL22 and CXCL12; respective ligands for Treg/MDSC-expressed CCR4 and CXCR4).34–36
Our preclinical data from ex vivo human tumor models34–37 and mouse in vivo studies38 demonstrated that a chemokine modulatory regimen (CKM), composed of toll-like receptor 3 (TLR3) agonists (such as poly-IC or rintatolimod), type-1 interferons (such as IFN-α) and COX2 blockers (such as celecoxib or indomethacin), synergistically induces selective production of CTL attractants (but not Treg- attractants) in tumor tissues, promoting selective attraction of CTL, without Treg attraction.35 37 38 Moreover, the preferential activation of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) system in the TME (compared with surrounding tissues) allows for preferential induction of the CTL attractants in the TME, rather than surrounding tissues.35 This suggests that CKM may induce local intratumoral effects, even on its systemic application, eliminating the need for direct intratumoral injection and allowing simultaneous targeting of multiple tumor lesions in patients with metastatic disease. We have recently shown that local administration of the CKM regimen (rintatolimod, IFN-α and celecoxib) in patients with ovarian cancer receiving intraperitoneal chemotherapy (cisplatin) can elevate the local expression of interferon-stimulated genes, including CTL-attracting chemokines (CXCL9, CXCL10, CXCL11), major histocompatibility complex I/II, perforin and granzymes,39 but the local impact of systemic application of CKM has not been evaluated in the clinical settings. This pilot study provides proof of principle that systemic CKM can indeed be used to enhance local CTL infiltration in advanced cancer lesions, without the need for its local administration. Further, to evaluate the potential of CKM for prospective use jointly with PD-1 inhibitors, as the initial step, we tested if it can be safely applied before PD-1 blockade.
Methods
Patient eligibility
Eligible patients (≥18 years) had histologically confirmed, surgically unresectable, metastatic triple-negative breast cancer (mTNBC), Eastern Cooperative Oncology Group performance status ≤1, measurable disease per Response Evaluation Criteria in Solid Tumors (RECIST) V.1.1, no cancer directed therapy for at least 3 weeks prior to study treatment, biopsy-accessible lesion, adequate bone marrow reserve and hepatic and renal function. Patients were excluded if they had received prior anti-PD-1/PD-L1 therapy, were on systemic immunosuppressive agents (including steroids), had active autoimmune disease or history of transplantation, or had known serious mood disorders. Patients were also excluded if they had cardiac event within 3 months of signing consent (including acute coronary syndrome, myocardial infarction, or ischemia), New York Heart Association III or IV heart failure, history of upper gastrointestinal ulceration, bleeding or perforation within 3 years, prior allergic reaction, or hypersensitivity to nonsteroidal anti-inflammatory drugs (NSAIDs) or any drugs administered on the protocol. An amendment to exclude patients with positive antinuclear antibodies was introduced in September 2019.
Study design
This is a pilot, open-label, single-arm, single-center clinical trial (NCT03599453) approved by the Roswell Park Institutional Review Board (IRB; STUDY00000629) that primarily evaluated the intratumoral efficacy of systemic CKM regimen in selectively enhancing CD8+ T-cell infiltration into the TME in patients with mTNBC. The predetermined primary endpoint of the NCT03599453 trial was the inpatient (fold) change in CD8α relative to HPRT1, comparing post-treatment versus pretreatment tumor tissues from each patient. The secondary endpoints included assessment of overall response rate (ORR) to CKM plus pembrolizumab treatment per RECIST V.1.1, progression-free survival (PFS), overall survival (OS), and disease-control rate (DCR). In addition, the safety profile of CKM was assessed using the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE) V.5.0. The attribution to CKM and/or pembrolizumab (unrelated, unlikely, possible, probable, definite) was based on the side effect profiles in the Investigator Brochure. The exploratory endpoints include examination of the immune profile, and correlation of the immune analysis results with clinical outcomes and comparison of response assessments using RECIST V.1.1 and immune-related RECIST (irRECIST).
Drug administration
Figure 1A shows the study schema. All eligible patients underwent a pretreatment biopsy from an accessible lesion. This was followed by 200 mg celecoxib orally, 20 million units/m2 IFN-α2b (over 20 min), 200 mg intravenous rintatolimod (over 2.5 hours). A second daily dose of 200 mg celecoxib was given 12 hours after the initial dose. This CKM regimen was administered on days 0, 1, 2±1 day and then repeated on days 7, 8, 9±1 day. All doses of CKM were administered within 14 days of initiation. Prior to each CKM treatment, patients were administered 500 mL normal saline with pre-medications (acetaminophen, prochlorperazine). Post-treatment biopsy was performed on day 10, 11 or 12, prior to pembrolizumab administration (1–3 days after completion of the second course of the CKM). Follow-up pembrolizumab (200 mg intravenous) was administered, starting on days 3–8 after CKM (3–14 days allowed as per-protocol), in all cases after the post-CKM tumor biopsy, every 3 weeks for two cycles until toxicities or disease progression. After two cycles, patients were considered off active study but followed for adverse events for 90 days. Additional off-study treatment was per physician discretion. Patients who do not have both pretreatment and post-treatment biopsies or were unable to complete at least five doses of CKM were not evaluable for the primary endpoint. Figure 1B shows the Consolidated Standards of Reporting Trials (CONSORT) diagram of screening, enrollment, and follow-up of participants.
Figure 1.

Study design. (A) Systemic chemokine modulation (CKM) was given as six doses over 2 weeks (days 0, 1, 2, 7, 8, and 9). Each daily CKM consisted of intravenous IFN-α2b (20 MU/m2; over 30 min) followed by rintatolimod (200 mg; over 2.5 hours) and oral celecoxib (two doses of 200 mg; 12 hours apart). Tumor biopsies were performed before and after CKM, with the second biopsy performed 1–3 days post-CKM. As a follow up to CKM, patients received two cycles of pembrolizumab (200 mg intravenous), starting 3–8 days after completion of the second course of CKM. Following completion of pembrolizumab, tumors were assessed using RECIST V.1.1 guidelines. Patients continued treatment per physician discretion following completion of the trial. (B) Consolidated Standards of Reporting Trials flow diagram depicting screening, enrollment, and follow up of participants in the trial. The trial enrolled eight patients, six of whom were evaluable for the primary endpoint of CD8α changes in pre-CKM and post-CKM biopsies. IFN, interferon; RECIST, Response Evaluation Criteria for Solid Tumors.
Patients’ assessment
Adverse events were evaluated throughout the treatment period using CTCAE V.5.0. Tumor measurements were obtained using the CT at baseline (within 31 days prior to treatment initiation), after two cycles of pembrolizumab, and when the study was completed. End of treatment assessment was performed at the end of study treatment or at the time of treatment discontinuation. Imaging schedule off-study was per physician discretion and the date of the first scan documenting disease progression was recorded. Tumor response was evaluated per RECIST V.1.1, which was used to inform treatment decision, and PFS of each patient was assessed based on RECIST V.1.1 guidelines. ORR was defined as the proportion of patients with the best overall response of complete response (CR) or partial response (PR). In addition, response was also assessed using irRECIST which was used as an exploratory endpoint for correlation between RECIST and irRECIST for response assessment. PFS was defined as the time from the date of study initiation until the date of disease progression or the date of death due to any cause, whichever occurred first. OS was defined as the time from the date of study initiation until the date of death due to any cause. DCR was defined as the proportion of patients with the best overall response of CR, PR, or stable disease (SD). Tumor biopsy was conducted before treatment initiation and after completion of CKM pretreatment, on days 10, 11 or 12 prior to starting pembrolizumab. To capture possible delayed immune-related adverse events, follow-up safety assessments were performed at least monthly for 90 days after the last dose of pembrolizumab or until resolution of any drug-related toxicity. Survival status was monitored every 6 months until January 24, 2023.
Patient samples
Fresh biopsy was performed at baseline and after completion of CKM regimen, prior to starting pembrolizumab. Four tumor cores and one core of non-tumor surrounding tissue were obtained. Three cores of tumor and one core of non-tumor surrounding tissue were placed in Dulbecco’s phosphate-buffered saline. One core of the tumor was processed to formalin-fixed paraffin-embedded (FFPE) block per institutional standards. Peripheral blood was obtained at baseline; on day 0 prior to start of CKM, 2 hours post completion of rintatolimod on day 2 (after three doses of CKM), prior to start of CKM on day 7, and before pembrolizumab administration on day 1 of cycles 1 and 2.
Quantitative gene expression
Gene expression was evaluated using real-time quantitative PCR (RT-qPCR) using TaqMan platform. Biopsy strings of tumor or non-tumor tissue are cut into three pieces and placed into Lysing Matrix E Tubes (MP Biologicals) containing RLT buffer (RNeasy Kit; Qiagen) and agitated using an FP120 homogenizer (MP Biologicals). The total RNA was extracted using the RNeasy kit (Qiagen), 250 ng of RNA were used for complementary DNA (cDNA) synthesis (qScript; QuantaBio), and 25–50 ng of subsequent cDNA were used to quantify messenger RNA (mRNA) expression. All analysis was performed on the CFX 96 system (Bio-Rad). Commercially available TaqMan primers (Thermo Fisher Scientific Life Technologies) were used to evaluate local expression of immune cells markers (CD8α, granzyme B, FOXP3) and key chemokines involved in the attraction of the effector cells (CCL5 and CXCL10), Treg (CCL22) and MDSCs (CXCL12). The expression of each gene was normalized to the HPRT1 housekeeping gene.
Multicolor flow cytometry
Peripheral blood mononuclear cells from eight subjects who received interventions at day 0, day 2, day 7, and cycle 1 day 1 were cryopreserved. The frozen cells were thawed, processed, and acquired on the same day by flow cytometry. Frozen cells were thawed in RPMI 1640 with L-glutamine (Corning Cellgro) supplemented with DNase (12 units/mL; Worthington Biochemical) at 37°C for 20 min, washed, and resuspended in FCM buffer (Leinco Technologies) containing 0.5% bovine serum albumin, 0.1% sodium azide, and 0.04 g/L tetrasodium EDTA in phosphate-buffered saline at pH 7.2. Samples were stained for flow cytometry as previously described.40 Briefly, for each sample, three 12×75 mm polystyrene round-bottom tubes were prepared to include an autofluorescence control, backbone panel, and fully stained panel. To each tube, 200 mL of washed cells were transferred and incubated with mouse IgG (Invitrogen) for 10 min to block Fc receptors. The cells were surface labeled for 30 min at room temperature in the dark. All antibodies were tittered and used at saturating concentrations. The cells were washed and resuspended in 500 µL of FCM buffer and immediately acquired on a Cytek Aurora full spectrum flow cytometer (Cytek Biosciences) equipped with 355 nm (20 mW), 405 nm (100 mW), 488 nm (50 mW), 638 nm (80 mW) excitation lasers. The instrument was quality-controlled using SpectroFlow QC Beads (Cytek Biosciences) daily to ensure consistent performance on a day-to-day basis. The acquired composite fluorescence data were spectrally unmixed into the individual contributing fluorescent markers using the SpectroFlo software V.2.2.04 (Cytek BioSciences). All flow cytometry results presented in this study were analyzed using FCS Express V.7.
Pathology
Multispectral immunofluorescent (mIF) staining was performed on 4 µm sections of FFPE tumor tissue which was cut and placed on charged slides. Slides were dried at 65°C for 2 hours. After drying, the slides were placed on the BOND RXm Research Stainer (Leica Biosystems) and deparaffinized with BOND Dewax Solution (AR9222, Lecia Biosystems). The mIF staining process involved serial applications of the following for each biomarker: epitope retrieval/stripping with ER1 (citrate buffer pH 6, AR996, Leica Biosystems), blocking buffer (Akoya Biosciences,), primary antibody, Opal Polymer horseradish peroxidase (HRP) secondary antibody (Akoya Biosciences), Opal Fluorophore (Akoya Biosciences). All Akoya reagents used for mIF staining come as a kit (NEL821001KT). Spectral 4',6-diamidino-2-phenylindole (DAPI) stain (Akoya Biosciences) was applied once slides were removed from the BOND. They were cover slipped using an aqueous method and Diamond Antifade mounting medium (Invitrogen). The mIF panel consisted of the following antibodies (clone, company, and opal fluorophores): CD3 (P7, Abcam, Opal Polaris 480), CD8 (C8/144B, Agilent DAKO, Opal 690), granzyme B1 (11F1, Leica Biosciences, Opal 570), FOXP3 (236A/E7, Abcam, Opal 520), PD-1 (EH33, Cell Signaling, Opal 620), Pan Cytokeratin (AE1AE3, Agilent DAKO, Opal Polaris 780). The antibody-stained slides were imaged using the Vectra Polaris Automated Quantitative Pathology Imaging System (Akoya Biosciences). Image analysis was performed using inForm Software V.2.4.11 (Akoya Biosciences). Whole slide spectral unmixing was performed using the synthetic spectral library within inForm. From the unmixed images, representative regions of interest (ROIs) were selected by the study pathologist and used to train tissue and cell segmentation. A unique algorithm was created for each biomarker, using a machine learning technique, in which the operator selects positive and negative cell examples. These algorithms were then batch applied across all ROIs across the project slides.
Statistics
Sample size determination
The primary objective of the study was to determine the impact of CKM on CD8+T cells in the TME pre-CKM and post-CKM, measured by CD8α expression. An interim analysis was planned at N=3, with a final analysis at N=6. The overall α was set at 0.10, with the Fleming-Harrington-O’Brien spending function used to determine the levels at the interim (α1=0.003) and final (α2=0.084) analyses. Based on our recent study34 we assumed a coefficient of variation of 0.75 for CD8α expression and a pre to post-treatment correlation of 0.5. Under these conditions, if the post-treatment mean CD8+ T-cell count at least triples relative to pretreatment, then the study design (N1=3 and N2=3) has a power of 0.48 at the interim analysis (N=3) and >0.99 at the final analysis (N=6) to detect such an effect.
Statistical analyses
Patient characteristics, safety data, and antitumor activity were summarized using the appropriate descriptive statistics. OS was summarized using standard Kaplan-Meier methods. The biomarker expressions and cell percentages were averaged across replicates (N=2 or 3) to create a single score. The pretreatment and post-treatment mRNA expression levels were compared using a two-sided one-sample t-test about the log-fold change. The normality of the log-fold change was assessed using the Wilks-Shapiro test. Correlations between biomarker expression or cell percentages were evaluated using the Spearman correlation coefficient and displayed graphically using scatter plots. A longitudinal assessment of CTLs was done by modeling each log-CTL expression as a function of time and a random subject effect using a linear mixed model. Tests about the appropriate contrasts of model estimates were used to compare mean expression between time points. All model assumptions were verified graphically. Statistical analyses were performed using SAS V.9.4 software (SAS Institute, Cary, North Carolina, USA). Since the interim analysis of CD8α was non-significant at α1=0.03, all final analyses were conducted at a significance level of α2=0.084 (per the Fleming-Harrington-O’Brien spending function). Therefore, p values≤0.084 are considered statistically significant.
Results
Patient characteristics, study design and tolerability
Eight patients with mTNBC with lesions amenable to biopsy were enrolled in the study between March 2019 and July 2020 to receive two 3-day-long cycles of systemic CKM (see figure 1A for study schema and figure 1B for CONSORT diagram). Table 1 shows the patient characteristics at baseline.
Table 1.
Baseline characteristics of the study population
| Characteristic | Total (%) |
| Number of patients | 8 (100) |
| Median age in years (min – max) | 52.9 (32.8–74.5) |
| Race | |
| Caucasian | 7 (87.5) |
| African-American | 1 (12.5) |
| ECOG PS | |
| 0 | 7 (87.5) |
| 1 | 1 (12.5) |
| Prior systemic treatments for metastatic disease | |
| 0 | 6 (75) |
| 1 | 2 (25) |
| PD-L1≥1%* | 5 (62.5) |
*Measured by Ventana SP142 assay.
ECOG PS, Eastern Cooperative Oncology Group performance status; PD-L1, programmed cell death ligand 1.
Six patients (PT1, PT2, PT3, PT5, PT6, PT7) had received prior anticancer therapy (neoadjuvant/adjuvant therapy) and two patients had received prior anticancer therapy in the metastatic setting (PT2, PT3). Patients had a median follow-up time of 33.8 months (90% CI: 20.6 to 46.1 months) at the time of reporting. Six patients (PT3, PT4, PT5, PT6, PT7, PT8) were evaluable for primary endpoint as they underwent both pre-CKM and post-CKM treatment biopsies. Two patients were not evaluable because post-treatment tumor tissue was necrotic resulting in lack of intact RNA (PT1) or refusal of post-treatment biopsy (PT2). All eight patients were evaluable for toxicities and blood correlates. Follow-up pembrolizumab was started in all patients 3–8 days after the end of the second cycle of CKM and was associated with mostly grade 1 and 2 adverse events. Four grade 3 adverse events (neutropenia, fatigue and malaise, immune thrombocytopenic purpura, pneumonitis) were reported of which only two were clinically significant (pneumonitis and immune thrombocytopenic purpura), leading to pembrolizumab discontinuation in two patients (table 2).
Table 2.
Treatment-related adverse events
| Adverse event | Any grade (%) | Grade 3/4 (%) |
| Blood and lymphatic | ||
| ITP | 12.5 | 12.5 |
| Leukopenia | 12.5 | – |
| Lymphopenia | 12.5 | – |
| Neutropenia | 12.5 | 12.5 |
| Thrombocytopenia | 12.5 | 12.5 |
| GI/nutrition | ||
| Decreased appetite | 12.5 | – |
| Mouth hemorrhage | 12.5 | – |
| Nausea | 50 | – |
| Taste disorder | 12.5 | – |
| Vomiting | 12.5 | – |
| Investigations | ||
| ALT increase | 37.5 | – |
| AST increase | 37.5 | – |
| General | ||
| Chills | 75 | – |
| Eye disorder | 12.5 | – |
| Fatigue | 62.5 | – |
| Headache | 12.5 | – |
| Hot flashes | 37.5 | – |
| Malaise | 12.5 | 12.5 |
| Night sweats | 12.5 | – |
| Pain | 12.5 | – |
| Fever | 12.5 | – |
| Vascular | ||
| Dizziness | 12.5 | – |
| Hypotension | 12.5 | – |
| Respiratory | ||
| Dyspnea | 12.5 | 12.5 |
| Hypoxia | 12.5 | 12.5 |
| Pneumonitis | 12.5 | 12.5 |
| Cutaneous | ||
| Petechiae | 12.5 | – |
| Pruritus | 25 | – |
| Rash | 25 | – |
ALT, alanine aminotransferase; AST, aspartate transaminase; GI, gastrointestinal; ITP, immune thrombocytopenic purpura.
Systemic CKM selectively increases CD8+ T-cell infiltration in the TME, and local ratios between CTLs and Tregs
Since CD8+ T-cell infiltration is an important prognostic marker that predicts improved disease outcomes in triple-negative breast cancer (TNBC),41 42 our predetermined primary endpoint of efficacy was the change in the treatment-associated CD8α as the reliable average marker of CTL infiltration in the total biopsy volume35 (figure 2A). Comparison of the mRNA in baseline biopsies with the biopsies performed after systemic CKM infusion showed an average 10.2-fold increase in CD8α (p=0.034; median 5.5-fold). Quantitative mRNA analysis of the key cytotoxic granule component granzyme B (GZMB), expressed by effector-type CD8+ T cells and natural killer cells, demonstrated an average 6.1-fold increase (p=0.02), confirming the increased CD8+ T-cell signature in the post-CKM biopsies. In contrast, the levels of a Treg marker, FOXP3, showed a downward trend but were not significantly reduced by CKM (p=0.203). These reciprocal changes resulted in an average 358.1-fold average increase (p=0.06; median 8.6-fold) in the ratio of CD8α/FOXP3 and 97.8-fold average increase (p=0.04; median 4.9-fold) in the GZMB/FOXP3 ratio in post-CKM versus pre-CKM biopsies (figure 2A and online supplemental table 1). No consistent changes in PD-L1 expression in post-treatment versus pretreatment tumor samples (measured by real-time quantitative polymerase chain reaction [RT-qPCR]) were observed (online supplemental figure 1 and table 1).
Figure 2.

Local increases in CTL markers in tumor lesions after systemic application of the CKM. (A) CD8⍺ and granzyme B (GZMB) transcripts (normalized for HPRT1 and shown as mean and median (MED) fold-increases over pre-CKM baseline for each patient; N=6); were measured using RT-qPCR (see online supplemental table 1 for raw data). Statistically significant increases in CD8α mRNA and GZMB mRNA following CKM treatment were observed. In contrast, FOXP3 message trended down following CKM. Statistically significant increases in ratios of CD8⍺/FOXP3 and GZMB/FOXP3 were also observed. Statistical significance by a two-sided one-sample t-test of the log-fold change was determined to be p≤0.084. (B) (top) Representative biopsy image stained with a multispectral panel consisting of GZMB, CD8, CD3, PD-1, and FOXP3 and tumor compartment marker, AE1AE3. The multiplex image depicts the spatial layout of the tumor environment, with monoplex images shown below. CTLs were defined as CD3+CD8+, Tregs were defined as CD3+FOXP3+. Examples of colocalization of biomarkers are depicted on the right. (bottom) Image analysis of biopsy pairs demonstrating a statistically significant difference is shown as mean (SD). The table describes the changes in the ratios of CTL/Treg and PD-1+CTL/Treg significantly increased in the post-treatment versus the pretreatment tumor tissues. P value≤0.084, determined by a two-sided one-sample t-test of the log-fold change was considered as statistically significant. CKM, chemokine modulatory; CTL, cytotoxic T lymphocytes; mRNA, messenger RNA; PD-1, programmed death-1; RT-qPCR, real-time quantitative polymerase chain reaction; Treg, regulatory T cells.
jitc-2023-007381supp001.pdf (449.2KB, pdf)
In three patients, we could additionally obtain paired pre-CKM and post-CKM biopsies of peritumoral tissues. Among these three paired samples, we did not observe significant post-CKM increases in CD8α levels, although the numbers are too small to be conclusive (online supplemental figure 2). In paired tumor biopsies, we performed multispectral imaging with a panel consisting of GZMB, CD8, CD3, PD-1 and FOXP3 and the tumor component marker, AE1AE3 (figure 2B). While this method did not allow us to demonstrate statistically significant absolute increases of CD3+CD8+ cell counts, the ratios of CD3+CD8+ cells to CD3+CD8–FOXP3+ cells increased by the average of 4.5-fold (p=0.009; median 3.29-fold) from 11.5±3.26 to 51.6±42.94. The average ratios of CD3+CD8+PD-1+ cells to CD3+CD8–FOXP3+ cells also significantly increased 4.05-fold (p=0.062; median 2.32-fold) from 7.2±3.82 to 29.24±30.77.
Systemic CKM selectively enhances CTL-attracting chemokines in the TME
To gain insight into the mechanism of the CKM-driven enhancement of the CTL signature, we analyzed changes in expression of CD8+ T cell-attracting and Treg-attracting chemokines. The average intratumoral expression levels of CCL5 mRNA (chemokine binding to the CTL-expressed CCR5) increased 4.1-fold (p=0.019; median 4.5-fold) (figure 3A and online supplemental table 1), accompanied by a minor 1.9-fold increase in CXCL9 (p=0.767; median 1.2-fold); and a 4.1-fold average increase in CXCL10 (ligand for CTL-expressed CXCR3) expression (p=0.104; median 2.7-fold). In contrast, the levels of both CCL22 (ligand for Treg expressed CCR4) and CXCL12 (ligand for CXCR4) remained unchanged (figure 3A and online supplemental table 1). These changes were associated with an increase in the ratios of CCL5/CCL22 and CXCL10/CCL22 (p=0.044, p=0.172, respectively). Interestingly, the CXCL10 increases post-CKM strongly correlated with the increases in CD8α and GMZB, while CCL5 increases post-CKM showed a reduced correlation with these markers of enhanced CTL infiltration (figure 3B). Because intratumoral CTLs do not produce CXCR3 ligands, such as CXCL10,35 the enhanced CXCL10—CD8α correlations in post-CKM tumor biopsies may suggest the dominant role of enhanced intratumoral CXCL10 production in CKM-driven increase in CTL attraction to the TME. Since effector CD8+ T cells (which also express CCR530) can be attracted by CCL5, but also produce this factor themselves,35 43 44 intratumoral increases of CCL5 may be both causative but also secondary to CTL enhancement.
Figure 3.

Selective increases in CTL-attracting chemokines, but not Treg attractants, in post CKM tumor biopsies. Chemokine expression in pre-CKM and post-CKM tumor biopsies was measured using RT-qPCR. (A) Transcripts (normalized to HPRT1 and shown as mean and median (MED) fold-increases over pre-CKM baseline for each patient) of CCR5 ligand (CCL5) and CXCR3 ligands (CXCL9 and CXCL10) increased following CKM treatment (N=6 patients). See online supplemental table 1 for raw data. P value≤0.084, determined by a two-sided one-sample t-test of the log-fold change was considered as statistically significant. (B) Pretreatment and post-treatment CD8⍺ and GZMB expression levels in the tumor biopsies were correlated with CXCL10 and CCL5 expression at each of these time points. CKM, chemokine modulatory; CTL, cytotoxic T lymphocytes; GZMB, granzyme B; RT-qPCR, real-time quantitative polymerase chain reaction; Treg, regulatory T cells.
CKM induces transient selective decreases in blood CXCR3+ CTLs, but not CXCR4+ Tregs
In contrast to the increases in CTL markers in CKM-treated patients, the longitudinal flow cytometry and RT-qPCR analysis of circulating blood cells (figure 4 and online supplemental figure 3A) demonstrated transient decreases in the numbers of circulating effector-type CD3+CD4–CD8+ CTLs and the CXCR3+ CTL subset (figure 4A), consistent with CTLs transition from blood to tumor tissues. In contrast to CTLs, the numbers of circulating CD3+CD4+CD8–CD25brightCD127Dim/neg Tregs and the CXCR4+ subset of Tregs were not affected by CKM treatment (figure 4B), confirming the selectivity of CKM action. The decreases in CTL post three consecutive doses of CKM (between day 0 and day 2) were transient and returned to pretreatment levels by day 7 (directly before the second cycle of CKM), as shown by flow cytometry (online supplemental figure 3A) and RT-qPCR analysis (online supplemental figure 3B). Interestingly, PT3, who showed elevated numbers of peripheral CD8+ T cells at baseline and after CKM, developed grade 3 immune thrombocytopenic purpura with platelet count <5000/mm3 after subsequent initiation of pembrolizumab, resulting in discontinuation of treatment.
Figure 4.
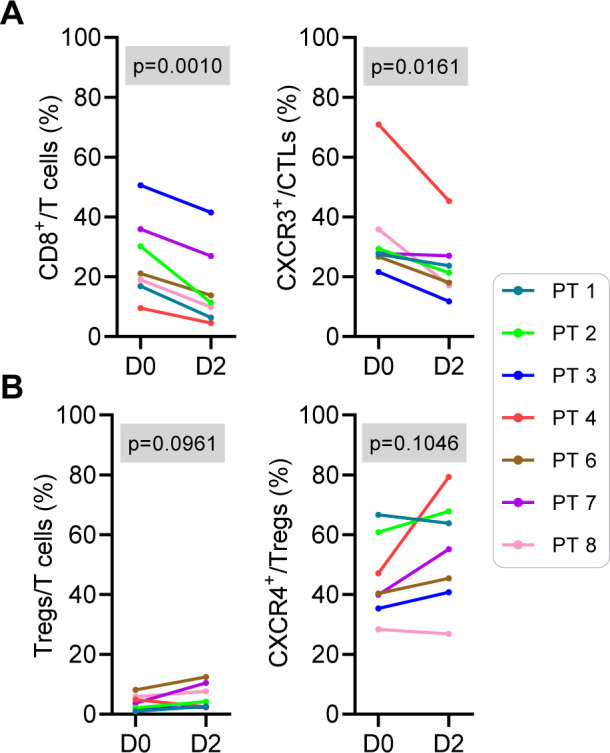
CKM induces transient decreases of circulating CTLs in the blood. Changes in the immune cell subsets circulating in the peripheral blood were measured by multiparameter flow cytometry (N=7). A statistically significant decrease in the percentages of (A) CD8+ T cells and CXCR3+CD8+ T cells, but not (B) Tregs or a subset of CXCR4+ Tregs, 2 days post-CKM compared with day 0 was observed. P value≤0.084, determined by t-test was considered as significant. CKM, chemokine modulatory; CTL, cytotoxic T lymphocytes; Treg, regulatory T cells.
CKM pretreatment does not change the toxicity profile of subsequent pembrolizumab treatment
Data cut-off for the safety and efficacy analyses was January 24, 2023. Seven patients discontinued follow-up pembrolizumab during the study period: two due to treatment-related adverse events (pneumonitis and immune thrombocytopenic purpura) and five due to disease progression. No unexpected toxicities of the follow-up pembrolizumab therapy were observed in any of the eight patients. The most common treatment-related adverse events of any grade were chills (75%), fatigue (62.5%), nausea (50%), hot flashes (37.5%), alanine aminotransferase increase (37.5%) and aspartate transaminase increase (37.5%), all grade 3 or below. One event of non-clinically significant grade 3 neutropenia was also observed, possibly related to combination of rintatolimod and IFN-α. One event of grade 3 pneumonitis resulted in hospitalization and was described as definitely related to pembrolizumab. The only grade 3 event attributed to the combination was immune thrombocytopenic purpura in which the patient’s platelets decreased from 222×103/mm3 to 64×103/ mm3 after CKM and partially rebounded to 68×103/mm3 but decreased to <5000/mm3 following pembrolizumab implementation. It was treated with steroids, intravenous immunoglobulin and rituximab and fully resolved. This patient was diagnosed with a positive antinuclear antibody, suggestive of autoimmunity or potential unmasking of an underlying autoimmune condition. Online supplemental tables 2 and 3 show the treatment-related adverse events (all toxicities) in all eight patients in the study that were definitely, probably, or possibly related to CKM or pembrolizumab, respectively.
Clinical status of CKM-pretreated patients receiving follow-up pembrolizumab
Each of the eight CKM-pretreated patients received follow-up pembrolizumab, starting 3–8 days after the end of the second CKM cycle. Online supplemental table 4 shows the details of the demographic, tumor characteristics and tumor response to treatment in the intention-to-treat population. One patient (PT1) with initial progressive disease (PD) per RECIST V.1.1, showed PR with continued pembrolizumab (figure 5). Three patients (PT3, PT6, PT7) developed transient SD and continued follow-up pembrolizumab until progression, although the duration of stable disease was limited to 3.5 months or less (figure 5B,C). PD-L1 expression at baseline was measured by Ventana SP142 assay. Interestingly, all the three patients with SD (PT3, PT6, PT7) had PD-L1≥5% and the only other patient with PR (PT1) had PD-L1 10%. There were no responses observed among patients with PD-L1 0 (PT2, PT4, PT5) or 1% (PT8). DCR was 37.5%. Figure 5A shows the maximum per cent change in tumor size from baseline measured according to RECIST V.1.1. The longitudinal changes in tumor size are shown in figure 5B. The timelines of each individual patient’s clinical status are shown in figure 5C and the OS in figure 5D. Except for PT1, other patients eventually progressed and started the next line, standard of care treatment, per physician discretion. One patient with SD died, while the other two continue to receive standard treatments. At the data cut-off (January 24, 2023), four patients were alive and median OS was not reached (NR; 90% CI: 12.9 to NR).
Figure 5.

Clinical status of CKM-treated patients receiving follow-up pembrolizumab. (A) Depiction of the maximum per cent change in tumor size from baseline. Patients with PD according to RECIST V.1.1 are shown with hashed bars while those with stable disease are shown with solid bars. (B) Change in tumor burden over time shown as longitudinal change in RECIST percentage from baseline. Patient 1 had a chest wall resection at week 17 as indicated by an asterisk. (C) The length of treatment duration and outcome for each patient is displayed. As of data cut-off of January 24, 2023, four patients remain alive with PT1 remaining disease-free. (D) Overall survival is shown for all eight patients on the trial. (E) Longitudinal changes in clinical status of PT1. (top) Timeline indicating the treatment regimen administered to PT1 who was chemo-refractory (to doxorubicin, cyclophosphamide, and paclitaxel) before starting the trial. The patient received 2 weeks of CKM, followed by pembrolizumab every 3 weeks. The left breast tumor mass persisted on day 45 but underwent subsequent autoamputation (see day 108 scan). By day 164, a smaller mass re-emerged on the left chest wall and was surgically resected on day 197. Pembrolizumab was discontinued due to pneumonitis on day 744, but the patient remains disease-free as of the last evaluation on day 1400, 46 months since the start of treatment. (middle, center) Computer-assisted tomography scans of PT1 before (middle, left), after CKM plus one dose of pembrolizumab, and CKM plus three doses of pembrolizumab (middle, right). H&E-staining of tumor tissue at 40× pre-CKM (bottom, left) and post-CKM before starting pembrolizumab (bottom, right) showing massive necrosis and no viable tumor in post-CKM biopsy at day 21. CKM, chemokine modulatory; PD, progressive disease; PR, partial response; RECIST, Response Evaluation Criteria in Solid Tumors; SD, stable disease.
PT1 showed evidence of tumor necrosis in post-CKM biopsy, prior to initiation of pembrolizumab. She initially developed PD by RECIST V.1.1, but subsequent PR evidenced by breast tumor mass autoamputation and continued shrinkage in the pulmonary nodules (figure 5E and online supplemental table 4). However, this patient was not evaluable for primary endpoint due to lack of viable tissue in post-treatment tumor biopsy. The patient underwent resection of a recurrent chest wall mass at 6.5 months. The pulmonary nodules continued to respond to pembrolizumab, and the patient remains disease-free at data cut-off (currently 46 months since the start of treatment).
Discussion/conclusion
Our data demonstrate the ability of the systemically applied short-term combinatorial CKM regimen composed of rintatolimod, IFN-α2b and celecoxib, to reprogram local TME of mTNBC for selectively enhanced CTL accumulation, without concomitant enhancement of Treg infiltration, with the opposite effects on the CTLs in patient’s peripheral blood. These in vivo observations from our study are in line with the predictions from our preclinical studies (references35 38 and manuscript in preparation) which showed two levels of selectivity of action of the combinatorial CKM: (1) its selective induction of CTL attractants (but not Treg attractants) in the tumor tissues and (2) its preferential impact on tumor (rather than healthy tissues), due to selective NF-kB activation within tumor tissues.35 While the preliminary data from the current trial is in line with our preclinical observations35 that peritumoral non-cancer tissues do not respond to the CKM treatment (online supplemental figure 2) to the same extent as the tumor tissues, the limited number of non-tumor samples with detectable CD8α (only three of six biopsies), makes these data not conclusive and in need of further validation in a larger study. However, the reciprocal effect of CKM on blood versus the tumor tissues indicate its unique potential for its systemic use to reprogram local TME of multiple tumor lesions, even if not accessible to direct injections.
Additionally, these in vivo observations are in line with strong synergy between rintatolimod and IFN-α in the induction of CTL attractants: all three CXCR3 ligands, CXCL9, CXCL10, and CXCL11, as well as CCR5 ligand, CCL5, and suppression of CCL22 (Treg attractant), we observed in vitro our preclinical models.35 38 In vitro data from us and others indicate that these synergistic effects are mediated by (1) enhancement of TLR3 expression by IFN-α, and (2) the ability of rintatolimod to block the effects of tumor-associated suppressive factor, PGE2 (which enhances CCL22 and suppresses CTL attracting chemokines) by downregulation of PGE2 receptor, EP4 (refs.37 45 46 and data not shown). The addition of celecoxib further enhances the desirable immunomodulatory properties by suppressing the COX-2-dependent production of PGE2.35 38
Despite its small size, our study met its primary efficacy endpoint (increases in CD8α used as a compound marker of CTL infiltration in whole biopsy volume35) highlighting the consistency of the CKM-driven reprogramming of the breast cancer TME. In contrast to the striking immunological changes induced by systemic CKM in the TME (including increases in CD8α, GZMB, CD8α/FOXP3 and GZMB/FOXP3), we did not observe consistently improved clinical outcomes. One potential explanation of this discrepancy is an unclear antigen specificity of CKM-driven CD8+ T-cell infiltration. The second limiting factor may be a short duration of CKM-driven effects, as suggested by only transient decreases in blood CD8+ T cells. Based on these observations, our upcoming clinical trials will test the clinical activity of concomitant, rather than sequential, application of CKM combined with PD-1 blockade over a prolonged period of time, to evaluate its clinical benefit. While the low dose of rintatolimod used in this clinical trial represents the recommended dose in patients with chronic fatigue syndrome47 48 and, as shown in this trial, it was sufficient to consistently reprogram mTNBC TME, it also remains to be tested if higher doses may be beneficial.
We have previously observed that the addition of local CKM (intraperitoneal rintatolimod and IFN-α plus oral celecoxib) to intraperitoneal chemotherapy (cisplatin) in patients with ovarian cancer can elevate local CTL-attracting chemokines (CXCL9, CXCL10, CXCL11) and CTL markers perforin and granzymes.39 We have also shown that systemic CKM (intravenous) can be safely administered in combination with dendritic cell vaccine in patients with peritoneal carcinomatosis (appendiceal cancer, colorectal cancer, or peritoneal mesothelioma) after cytoreductive surgery and hyperthermic intraperitoneal chemotherapy,49 but that study did not involve TME analysis, precluding the evaluation of its local immunologic effects.
The current study demonstrates that even six doses of systemic CKM (rintatolimod, IFN-α2b, and celecoxib) administered over 2 weeks show local immunologic effectiveness in, at least transient, reprograming of the local immunologic milieu of mTNBC. Because mTNBC represents a particular clinical challenge, largely due to its single-agent anti-PD-1 resistance,20 50 this approach could significantly benefit patient care. The current demonstration that CKM can be safely applied prior to PD-1 blockade, paves the way to concomitant administration of CKM and anti-PD-1, the timing determined to be therapeutically optimal in our mouse studies (Kokolus et al manuscript in preparation).
Although one instance of a massive tumor necrosis triggered by CKM which resulted in breast tumor autoamputation following subsequent pembrolizumab infusion is provocative, the current study was not designed for, and does not allow conclusions to be drawn on the clinical activity of the current regimen.
Responses to single-agent checkpoint inhibitors in mTNBC are rare and seen mostly in the first-line rather than later-line settings, with KEYNOTE-086 demonstrating 21.4% ORR to first-line pembrolizumab (PD-L1 positive) but only 5.2% ORR after ≥1 systemic therapy51 52 (both PD-L1 positive and negative). Similarly, in KEYNOTE-119, the ORR to single agent pembrolizumab given in second-line and beyond was only 9.6%, which increased with higher PD-L1 expression.53 Interestingly, in our study, the four patients with SD or PR had PD-L1≥5%, while no responses were observed among the four patients with PD-L1 of 0 or 1%. It is therefore possible that pembrolizumab as a single agent would be similarly effective in this population. Although IFN-α has been reported to elevate the levels of PD-L1 expression, our preliminary TaqMan analyses did not identify any consistent changes in PD-L1 expression in the tumor biopsies obtained before and after CKM (online supplemental figure 1). The design of our current study where pembrolizumab was given sequentially after CKM reflected our primary goal to evaluate the local immunologic impact of CKM on the CTL signature in the TME prior to PD-1 administration, rather than its clinical activity. Our follow-up study with clinical endpoint will involve concomitant prolonged application of CKM and PD-1 blockade, due to the transient nature of the CKM-induced changes in CD8+ T cells, and a much larger number of patients. Poor median OS for mTNBC of only 17.2 months54 makes mTNBC a particularly compelling target of CKM-based therapies. On KEYNOTE-119, 14% patients developed grade 3–4 pembrolizumab-related adverse events, such as increased liver enzymes, fatigue, anemia, leukopenia, while 20% experienced serious adverse events, including, pleural effusion, pneumonia, and febrile neutropenia. On KEYNOTE-086, 12.9% patients experienced grade 3–4 pembrolizumab-related fatigue, nausea, diarrhea, pneumonitis and type 1 diabetes mellitus (DM). In our study, CKM-attributed grade 3–4 adverse events were observed in 37.5% (3/8) patients while pembrolizumab-related grade 3–4 adverse events were observed in 25% (2/8) patients. While these percentages are numerically higher, the interpretation is limited by small number of patients in this study.
Recent approval of PD-1 blockade as an addition to chemotherapy in mTNBC,50 54 55 raises the question whether CKM could be combined with chemotherapy to promote optimal responsiveness of mTNBC (including PD-L1 negative mTNBC) to PD-1 blockade; potentially also in earlier stages of TNBC. Recent clinical data showing that the effectiveness of chemotherapy and immune checkpoint inhibitor in mTNBC is enhanced in patients with high PD-L1 expression,11 12 known to correlate with CTL infiltration, and mouse data showing that tumor-infiltrating type-1 immune cells sensitize cancer to chemotherapy56 strongly supports this possibility.
Our current data demonstrate that even short-term systemic CKM can reprogram the local TME of advanced cancer lesions for selectively enhanced CTL attraction, circumventing the requirement for intratumoral delivery of TME-reprograming agents, which is possible only in a small proportion of patients with advanced cancers with biopsy-accessible lesions. Moreover, the enhanced CTL infiltration triggered by systemic CKM avoids the intratumoral increases of Tregs, observed after intratumoral injection of STING activators.26–29 These two desirable levels of CKM selectivity: preferential activation of tumor, rather than healthy tissues, and selective promotion of CTL (but not Treg) influx to the TMEs, makes CKM a particularly interesting option for patients with advanced multifocal cancers.
Our study has met its primary efficacy objective to demonstrate the ability of systemic CKM administration to reprogram the local TME of patients with advanced TNBC for selectively enhanced CTL influx. The toxicities with the combination were comparable to those expected with pembrolizumab monotherapy. While the study was not designed to analyze clinical efficacy (small size and delayed rather than simultaneous administration of PD-1 blockade), we are planning a larger phase 2 study to analyze clinical efficacy of this combination and confirm the tolerability.
Acknowledgments
The authors thank AIM ImmunoTech for cost-free drug supply of rintatolimod and for access to rintatolimod’s FDA drug master file during IND filing.
Footnotes
Contributors: Study concept by PK. Study design by PK, MO, SG. Patient recruitment, clinical information collection, specimen collection and study coordination were done by SG, MO, LW, MLT, TLO, APE, EGL. Experimental design and interpretation were done by SG, MO, PK. Specimen collection and RNA analysis was performed by MJG, RTS and KMK. Flow cytometry was performed by PKW, KTS, HM and OM. Histology (IHC) and Vectra analysis was done by AW. Statistical analysis was performed by KA and AG. The manuscript was written by SG and PK, with input from all authors. Guarantor: PK.
Funding: This work has been supported by the NIH/NCI grants 1P01CA234212, 2P30A016056-42, KL2 TR001413, UL1 TR001412, 2R50CA211108, DOD grant W81XWH-19-1-0674, Rustum Family Foundation, Jacobs Family Foundation, Roswell Alliance Foundation and Roswell Park Institutional Funds.
Competing interests: No, there are no competing interests.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Not applicable.
Ethics approval
This study involves human participants and was approved by Roswell Park Comprehensive Cancer Center IRB; #STUDY00000629. Participants gave informed consent to participate in the study before taking part.
References
- 1. Zhang L, Conejo-Garcia JR, Katsaros D, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med 2003;348:203–13. 10.1056/NEJMoa020177 [DOI] [PubMed] [Google Scholar]
- 2. Pagès F, Berger A, Camus M, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med 2005;353:2654–66. 10.1056/NEJMoa051424 [DOI] [PubMed] [Google Scholar]
- 3. Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006;313:1960–4. 10.1126/science.1129139 [DOI] [PubMed] [Google Scholar]
- 4. Mahmoud SMA, Paish EC, Powe DG, et al. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J Clin Oncol 2011;29:1949–55. 10.1200/JCO.2010.30.5037 [DOI] [PubMed] [Google Scholar]
- 5. Denkert C, von Minckwitz G, Brase JC, et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J Clin Oncol 2015;33:983–91. 10.1200/JCO.2014.58.1967 [DOI] [PubMed] [Google Scholar]
- 6. Stanton SE, Disis ML. Clinical significance of tumor-infiltrating lymphocytes in breast cancer. J Immunotherapy Cancer 2016;4:59. 10.1186/s40425-016-0165-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54. 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Herbst RS, Soria J-C, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014;515:563–7. 10.1038/nature14011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Taube JM, Klein A, Brahmer JR, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res 2014;20:5064–74. 10.1158/1078-0432.CCR-13-3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71. 10.1038/nature13954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Emens LA, Molinero L, Loi S, et al. Atezolizumab and NAB-paclitaxel in advanced triple-negative breast cancer: biomarker evaluation of the impassion130 study. J Natl Cancer Inst 2021;113:1005–16. 10.1093/jnci/djab004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Emens LA, Adams S, Barrios CH, et al. First-line atezolizumab plus NAB-paclitaxel for unresectable, locally advanced, or metastatic triple-negative breast cancer: impassion130 final overall survival analysis. Ann Oncol 2021;32:1650. 10.1016/j.annonc.2021.10.002 [DOI] [PubMed] [Google Scholar]
- 13. Seo AN, Lee HJ, Kim EJ, et al. Tumour-infiltrating CD8+ lymphocytes as an independent predictive factor for pathological complete response to primary systemic therapy in breast cancer. Br J Cancer 2013;109:2705–13. 10.1038/bjc.2013.634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brown JR, Wimberly H, Lannin DR, et al. Multiplexed quantitative analysis of CD3, CD8, and CD20 predicts response to neoadjuvant chemotherapy in breast cancer. Clin Cancer Res 2014;20:5995–6005. 10.1158/1078-0432.CCR-14-1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mao Y, Qu Q, Zhang Y, et al. The value of tumor infiltrating lymphocytes (TILs) for predicting response to neoadjuvant chemotherapy in breast cancer: a systematic review and meta-analysis. PLoS ONE 2014;9:e115103. 10.1371/journal.pone.0115103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 2004;10:942–9. 10.1038/nm1093 [DOI] [PubMed] [Google Scholar]
- 17. Bates GJ, Fox SB, Han C, et al. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol 2006;24:5373–80. 10.1200/JCO.2006.05.9584 [DOI] [PubMed] [Google Scholar]
- 18. Ohara M, Yamaguchi Y, Matsuura K, et al. Possible involvement of regulatory T cells in tumor onset and progression in primary breast cancer. Cancer Immunol Immunother 2009;58:441–7. 10.1007/s00262-008-0570-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jiang D, Gao Z, Cai Z, et al. Clinicopathological and prognostic significance of FOXP3+ tumor infiltrating lymphocytes in patients with breast cancer: a meta-analysis. BMC Cancer 2015;15:727. 10.1186/s12885-015-1742-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Disis ML, Bernhard H, Jaffee EM. Use of tumour-responsive T cells as cancer treatment. Lancet 2009;373:673–83. 10.1016/S0140-6736(09)60404-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Freeman BE, Hammarlund E, Raué H-P, et al. Regulation of innate CD8+ T-cell activation mediated by cytokines. Proc Natl Acad Sci U S A 2012;109:9971–6. 10.1073/pnas.1203543109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jackaman C, Bundell CS, Kinnear BF, et al. IL-2 Intratumoral Immunotherapy enhances CD8+ T cells that mediate destruction of tumor cells and tumor-associated vasculature: a novel mechanism for IL-2. J Immunol 2003;171:5051–63. 10.4049/jimmunol.171.10.5051 [DOI] [PubMed] [Google Scholar]
- 23. Mullins SR, Vasilakos JP, Deschler K, et al. Intratumoral immunotherapy with TLR7/8 agonist MEDI9197 modulates the tumor microenvironment leading to enhanced activity when combined with other immunotherapies. J Immunother Cancer 2019;7:244. 10.1186/s40425-019-0724-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Veenstra JJ, Gibson HM, Littrup PJ, et al. Cryotherapy with concurrent CpG oligonucleotide treatment controls local tumor recurrence and modulates HER2/neu immunity. Cancer Res 2014;74:5409–20. 10.1158/0008-5472.CAN-14-0501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Demaria O, De Gassart A, Coso S, et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci U S A 2015;112:15408–13. 10.1073/pnas.1512832112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Flood BA, Higgs EF, Li S, et al. STING pathway agonism as a cancer therapeutic. Immunol Rev 2019;290:24–38. 10.1111/imr.12765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alvarez M, Molina C, De Andrea CE, et al. Intratumoral co-injection of the poly I:C-derivative BO-112 and a STING agonist synergize to achieve local and distant anti-tumor efficacy. J Immunother Cancer 2021;9:e002953. 10.1136/jitc-2021-002953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jiang X, Wang J, Zheng X, et al. Intratumoral administration of STING-activating nanovaccine enhances T cell immunotherapy. J Immunother Cancer 2022;10:e003960. 10.1136/jitc-2021-003960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Meric-Bernstam F, Sweis RF, Hodi FS, et al. Phase I dose-escalation trial of MIW815 (ADU-S100), an intratumoral STING agonist, in patients with advanced/metastatic solid tumors or lymphomas. Clin Cancer Res 2022;28:677–88. 10.1158/1078-0432.CCR-21-1963 [DOI] [PubMed] [Google Scholar]
- 30. Watchmaker PB, Berk E, Muthuswamy R, et al. Independent regulation of chemokine responsiveness and cytolytic function versus CD8+ T cell expansion by dendritic cells. J Immunol 2010;184:591–7. 10.4049/jimmunol.0902062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kunz M, Toksoy A, Goebeler M, et al. Strong expression of the lymphoattractant C-X-C chemokine Mig is associated with heavy infiltration of T cells in human malignant melanoma. J Pathol 1999;189:552–8. [DOI] [PubMed] [Google Scholar]
- 32. Musha H, Ohtani H, Mizoi T, et al. Selective infiltration of CCR5(+)CXCR3(+) T lymphocytes in human colorectal carcinoma. Int J Cancer 2005;116:949–56. 10.1002/ijc.21135 [DOI] [PubMed] [Google Scholar]
- 33. Ohtani H, Jin Z, Takegawa S, et al. Abundant expression of CXCL9 (MIG) by stromal cells that include dendritic cells and accumulation of CXCR3+ T cells in lymphocyte-rich gastric carcinoma. J Pathol 2009;217:21–31. 10.1002/path.2448 [DOI] [PubMed] [Google Scholar]
- 34. Muthuswamy R, Corman JM, Dahl K, et al. Functional reprogramming of human prostate cancer to promote local attraction of effector CD8(+) T cells. Prostate 2016;76:1095–105. 10.1002/pros.23194 [DOI] [PubMed] [Google Scholar]
- 35. Muthuswamy R, Berk E, Junecko BF, et al. NF-ΚB hyperactivation in tumor tissues allows tumor-selective reprogramming of the chemokine microenvironment to enhance the recruitment of cytolytic T effector cells. Cancer Res 2012;72:3735–43. 10.1158/0008-5472.CAN-11-4136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Muthuswamy R, Wang L, Pitteroff J, et al. Combination of IFNα and poly-I:C reprograms bladder cancer microenvironment for enhanced CTL attraction. J Immunother Cancer 2015;3:6. 10.1186/s40425-015-0050-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Theodoraki M-N, Yerneni S, Sarkar SN, et al. Helicase-driven activation of Nfkappab-Cox2 pathway mediates the immunosuppressive component of dsRNA-driven inflammation in the human tumor microenvironment. Cancer Res 2018;78:4292–302. 10.1158/0008-5472.CAN-17-3985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Obermajer N, Urban J, Wieckowski E, et al. Promoting the accumulation of tumor-specific T cells in tumor tissues by dendritic cell vaccines and chemokine-modulating agents. Nat Protoc 2018;13:335–57. 10.1038/nprot.2017.130 [DOI] [PubMed] [Google Scholar]
- 39. Orr B, Mahdi H, Fang Y, et al. Phase I trial combining chemokine-targeting with Loco-regional chemoimmunotherapy for recurrent, platinum-sensitive ovarian cancer shows induction of CXCR3 ligands and markers of type 1 immunity. Clin Cancer Res 2022;28:2038–49. 10.1158/1078-0432.CCR-21-3659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tario JD, Wallace PK. Reagents and cell staining for immunophenotyping by flow cytometry. In: McManus LM, Mitchell RN, eds. Pathobiology of Human Disease. San Diego: Elsevier, 2014: 3678–701. [Google Scholar]
- 41. Vihervuori H, Autere TA, Repo H, et al. Tumor-infiltrating lymphocytes and CD8(+) T cells predict survival of triple-negative breast cancer. J Cancer Res Clin Oncol 2019;145:3105–14. 10.1007/s00432-019-03036-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen Z, Chen X, Zhou E, et al. Intratumoral CD8(+) cytotoxic lymphocyte is a favorable prognostic marker in node-negative breast cancer. PLoS ONE 2014;9:e95475. 10.1371/journal.pone.0095475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Catalfamo M, Karpova T, McNally J, et al. Human CD8+ T cells store RANTES in a unique secretory compartment and release it rapidly after TCR stimulation. Immunity 2004;20:219–30. 10.1016/s1074-7613(04)00027-5 [DOI] [PubMed] [Google Scholar]
- 44. Wagner L, Yang OO, Garcia-Zepeda EA, et al. Beta-chemokines are released from HIV-1-specific cytolytic T-cell granules complexed to proteoglycans. Nature 1998;391:908–11. 10.1038/36129 [DOI] [PubMed] [Google Scholar]
- 45. Tissari J, Sirén J, Meri S, et al. IFN-alpha enhances TLR3-mediated antiviral cytokine expression in human endothelial and epithelial cells by up-regulating TLR3 expression. J Immunol 2005;174:4289–94. 10.4049/jimmunol.174.7.4289 [DOI] [PubMed] [Google Scholar]
- 46. Miettinen M, Sareneva T, Julkunen I, et al. Ifns activate toll-like receptor gene expression in viral infections. Genes Immun 2001;2:349–55. 10.1038/sj.gene.6363791 [DOI] [PubMed] [Google Scholar]
- 47. Mitchell WM. Efficacy of rintatolimod in the treatment of chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME). Expert Rev Clin Pharmacol 2016;9:755–70. 10.1586/17512433.2016.1172960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Strayer DR, Carter WA, Stouch BC, et al. A double-blind, placebo-controlled, randomized, clinical trial of the TLR-3 agonist rintatolimod in severe cases of chronic fatigue syndrome. PLoS ONE 2012;7:e31334. 10.1371/journal.pone.0031334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ramanathan R, Choudry H, Jones H, et al. Phase II trial of adjuvant dendritic cell vaccine in combination with celecoxib, interferon-alpha, and rintatolimod in patients undergoing cytoreductive surgery and hyperthermic intraperitoneal chemotherapy for peritoneal metastases. Ann Surg Oncol 2021;28:4637–46. 10.1245/s10434-020-09464-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Emens LA. Immunotherapy in triple-negative breast cancer. Cancer J 2021;27:59–66. 10.1097/PPO.0000000000000497 [DOI] [PubMed] [Google Scholar]
- 51. Adams S, Schmid P, Rugo HS, et al. Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: cohort A of the phase II KEYNOTE-086 study. Ann Oncol 2019;30:397–404. 10.1093/annonc/mdy517 [DOI] [PubMed] [Google Scholar]
- 52. Adams S, Loi S, Toppmeyer D, et al. Pembrolizumab monotherapy for previously untreated, PD-L1-positive, metastatic triple-negative breast cancer: cohort B of the phase II KEYNOTE-086 study. Ann Oncol 2019;30:405–11. 10.1093/annonc/mdy518 [DOI] [PubMed] [Google Scholar]
- 53. Winer EP, Lipatov O, Im S-A, et al. Pembrolizumab versus investigator-choice chemotherapy for metastatic triple-negative breast cancer (KEYNOTE-119): a randomised, open-label, phase 3 trial. Lancet Oncol 2021;22:499–511. 10.1016/S1470-2045(20)30754-3 [DOI] [PubMed] [Google Scholar]
- 54. Cortes J, Rugo HS, Cescon DW, et al. Pembrolizumab plus chemotherapy in advanced triple-negative breast cancer. N Engl J Med 2022;387:217–26. 10.1056/NEJMoa2202809 [DOI] [PubMed] [Google Scholar]
- 55. Cortes J, Cescon DW, Rugo HS, et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020;396:1817–28. 10.1016/S0140-6736(20)32531-9 [DOI] [PubMed] [Google Scholar]
- 56. Cecil D, Park KH, Curtis B, et al. Type I T cells sensitize treatment refractory tumors to chemotherapy through inhibition of oncogenic signaling pathways. J Immunother Cancer 2021;9:e002355. 10.1136/jitc-2021-002355 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2023-007381supp001.pdf (449.2KB, pdf)
Data Availability Statement
All data relevant to the study are included in the article or uploaded as supplementary information.