

Keywords: GABA, glutamate, motoneuron, reflex, sensory axon
Abstract
When a muscle is stretched, sensory feedback not only causes reflexes but also leads to a depolarization of sensory afferents throughout the spinal cord (primary afferent depolarization, PAD), readying the whole limb for further disturbances. This sensory-evoked PAD is thought to be mediated by a trisynaptic circuit, where sensory input activates first-order excitatory neurons that activate GABAergic neurons that in turn activate GABAA receptors on afferents to cause PAD, though the identity of these first-order neurons is unclear. Here, we show that these first-order neurons include propriospinal V3 neurons, as they receive extensive sensory input and in turn innervate GABAergic neurons that cause PAD, because optogenetic activation or inhibition of V3 neurons in mice mimics or inhibits sensory-evoked PAD, respectively. Furthermore, persistent inward sodium currents intrinsic to V3 neurons prolong their activity, explaining the prolonged duration of PAD. Also, local optogenetic activation of V3 neurons at one segment causes PAD in other segments, due to the long propriospinal tracts of these neurons, helping to explain the radiating nature of PAD. This in turn facilitates monosynaptic reflex transmission to motoneurons across the spinal cord. In addition, V3 neurons directly innervate proprioceptive afferents (including Ia), causing a glutamate receptor-mediated PAD (glutamate PAD). Finally, increasing the spinal cord excitability with either GABAA receptor blockers or chronic spinal cord injury causes an increase in the glutamate PAD. Overall, we show the V3 neuron has a prominent role in modulating sensory transmission, in addition to its previously described role in locomotion.
NEW & NOTEWORTHY Locomotor-related propriospinal neurons depolarize sensory axons throughout the spinal cord by either direct glutamatergic axoaxonic contacts or indirect innervation of GABAergic neurons that themselves form axoaxonic contacts on sensory axons. This depolarization (PAD) increases sensory transmission to motoneurons throughout the spinal cord, readying the sensorimotor system for external disturbances. The glutamate-mediated PAD is particularly adaptable, increasing with either an acute block of GABA receptors or chronic spinal cord injury, suggesting a role in motor recovery.
INTRODUCTION
Sensory feedback is essential for maintaining posture and producing accurate movement (1–4), without which incoordination, joint injury, and falling occur, especially with peripheral neuropathies, aging, and spinal cord injuries (4–9). Accordingly, the spinal cord has evolved complex sensory control systems to integrate the many sensory inputs that come from the limbs into the motor systems (4, 10–14), allowing appropriate reflexes to support movements and respond rapidly to external disturbances, like when tripping or falling. The first line of sensory control is a direct modulation of the sensory afferents themselves within the spinal cord, by axoaxonic connections onto afferents from specialized GABAergic neurons (GAD2+, abbreviated here GABAaxo neurons) (15–20). This activates GABAA receptors on afferents, which ultimately leads to a large depolarization, termed primary afferent depolarization (PAD), mediated by outward chloride currents produced by the unusually high intracellular chloride concentrations in sensory neurons, compared to in other adult neurons (21–25).
Although the phenomena of PAD have been known for nearly a century (21), the underlying neuronal circuits that cause it are uncertain and its function has recently been disputed, especially with the advent of imaging and optogenetic methods that allow direct activation of the neurons causing PAD (19, 22). For example, GABAA receptors that produce PAD are not found at the Ia afferent terminals in the motor nucleus and are instead located electrotonically far away at nodes of Ranvier, where they have no influence on the terminals. Thus, PAD does not cause presynaptic inhibition of these afferent terminals. Indeed, while optogenetic activation of GABAaxo neurons causes GABAA-mediated PAD, this PAD does not directly produce presynaptic inhibition of proprioceptive sensory feedback to motoneurons, though it may be indirectly involved in postactivation depression of synaptic transmission (19, 26). Instead, activation of GABAB receptors on afferent terminals likely causes presynaptic inhibition independently of PAD, as it is blocked by GABAB antagonists, and mainly only GABAB, and not GABAA, receptors are expressed at proprioceptive afferent terminals near motoneurons (16, 19, 27). Unexpectedly, PAD in proprioceptive afferents is caused by GABAaxo neuron contacts onto GABAA receptors near nodes of Ranvier in the many myelinated branches of these afferents within the spinal cord, completely contrary to the long-standing assumption that PAD arises from afferent terminals and produces presynaptic inhibition (19, 22, 28, 29). This nodal GABA action paradoxically facilitates spike propagation at branch points (termed nodal facilitation), with branch-point failure otherwise common without GABA innervation (19). How this complex mixture of inhibition and facilitation is regulated by GABAaxo neurons is even more uncertain, and it may well differ at rest compared with during locomotion (30). Furthermore, we know little about even the neurons that directly innervate GABAaxo neurons that cause PAD in group Ia proprioceptive afferents, though considerable progress has recently been made in understanding the similar circuits that mediate PAD in low-threshold mechanoreceptors (LTMRs), including the CCK+ neurons that innervate GABAergic neurons that contact LTMRs (31–33).
PAD and associated GABAaxo neuron activity are well established to be tightly regulated during tasks like resting postural maintenance, walking, and reaching, but how and why this occurs remain unclear (4, 14, 23, 30, 34). We know that when a muscle at rest is stretched it not only responds with a reflex but also the proprioceptive and cutaneous feedback caused by the imposed movement elicits a PAD in many afferents, readying the whole body for further disturbances, though the balance of presynaptic inhibition and nodal facilitation remains unclear (19, 23). The PAD evoked by such sensory feedback is thought to be caused by a minimally trisynaptic circuit, where sensory inputs activate first-order excitatory neurons that activate GABAaxo neurons that in turn produce PAD in afferents (19, 23). Other than knowing the approximate location of the first-order neurons involved in PAD evoked in Ia afferents (35, 36), we know little about them, unlike the first-order neurons involved in PAD evoked in LTMRs (CCK+ neurons, mentioned earlier). We can surmise that they may be excitatory propriospinal commissural neurons (37, 38) to account for the widespread radiating nature of this PAD, where a single nerve stimulation can evoke PAD many segments away and across the midline (21, 22). Furthermore, these first-order neurons likely express specialized persistent intrinsic currents that allow them to produce very long responses, as PAD far outlasts the brief sensory activation needed to trigger it (21, 22).
The local spinal central pattern generator (CPG) circuits that produce walking or other complex movements like flexion withdrawal reflexes also strongly modulate PAD (4, 30, 39, 40). However, the details of the circuits involved also remain uncertain, other than knowing that GABAaxo neurons also produce this PAD, and likely some propriospinal neuron population allows this PAD to also be widespread across many segment levels and associated muscles (across the lumbosacral and sacrocaudal cord, which are involved in locomotion like walking and swimming). We do, however, know a lot about the neurons in the CPG itself (10, 12, 41, 42). Of these CPG-related neurons, the V3 neuron is a good candidate to be a first-order neuron generating PAD, as it has extensive propriospinal and commissural axons and is excitatory (43). Indeed, in the course of our recent studies of spasticity and locomotor activity in mice (44), we noticed that optogenetic activation V3 neurons not only activate motoneurons but also produce a pronounced PAD, which we now report here. Previous unpublished work has suggested that V3 neurons directly receive sensory input (published in abstract form) (45), consistent with our understanding of commissural propriospinal neurons (37, 38, 46); thus, we started here by verifying this, which would allow these neurons to not only be involved in PAD during locomotion but also more generally evoke PAD in response to sensory stimulation. We find that V3 neurons are not only involved in PAD but also are essential for a large portion of sensory-evoked PAD in proprioceptive afferents, even in the absence of locomotion, suggesting that the illusive first-order neuron on the proprioceptive trisynaptic PAD circuit may include the V3 neuron. We also unexpectedly find that V3 neurons produce a strong PAD that does not depend on GABA, but instead is in part mediated by N-methyl-d-aspartate (NMDA) receptors. Although such NMDA-dependent PAD has been observed before during stimulation of high-threshold C or Aδ afferents (31, 47), its origin is unclear, and thus we also examined how V3 neurons contributed NMDA-related PAD. These findings suggest that PAD circuits are not an isolated sensory control system, but a part of the overall sensorimotor system that controls movement, as V3 neurons have many motor functions, including directly driving motoneurons, switching on locomotor activity, and coordinating interlimb activity (43, 44, 48, 49).
METHODS
Adult Mice Strains Used
Recordings were made from V3 neurons, group Ia afferents, dorsal roots (DRs), and ventral roots in the sacrocaudal spinal cord of adult mice, maintained in vitro (3.4–6 mo old, both female and male equally). The adult sacrocaudal spinal cord was examined because it survives whole in vitro, unlike the lumbar cord, leaving neuronal circuits intact, including commissural V3 neurons (19). However, this isolated cord is devoid of supraspinal control. All experimental procedures were approved by the University of Alberta Animal Care and Use Committee, Health Sciences division. We evaluated V3 neurons in mice with Cre recombinase expressed under the Sim1 promotor region, since the definition of V3 neurons is expression of Sim1 during development (43, 44). These V3 neurons were visualized with Cre-driven fluorophores (tdTom or EYFP), silenced with Cre-driven VGLUT2 knockout (V3 neurons use VGLUT2 for vesicular glutamate transport), activated optogenetically using Cre-driven channelrhodopsin-2 (ChR2), or inhibited optogenetically with Cre-driven archaerhodopsin-3 (AchT).
The Sim1-Cre mice were obtained from two sources: 1) Sim1-Cre-ki mice, where Cre-recombinase is knocked in (ki) under the endogenous Sim1 promoter of the host genome (43), and so expresses Cre under the control of Sim1 expression (obtained courtesy of Dr. M. Goulding, Salk Inst.), and 2) Sim1-Cre-tg mice, where an artificially generated Sim1-Cre transgene (tg) is inserted into the mouse genome (The Jackson Laboratory, Strain No.: 006395) and transgene expression is observed in most areas that endogenously express Sim1, including the sacral spinal cord that we study here. Results obtained from the sacral spinal cord with Sim1-Cre-ki and Sim1-Cre-tg were similar and combined, and hereafter both these mice were abbreviated Sim1-Cre mice.
The following floxed reporter strains were used (50, 51): 1) B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze and B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze mice (abbreviated R26LSL-tdTom mice; The Jackson Laboratory, Stock Nos. 007914 and 007909; tdTomato fluorescent protein expressed under the R26::CAG promotor in cells that coexpress Cre), 2) B6J.129S6(129S4)-Slc17a6tm1Lowl/RujfJ mice where the VGLUT2 gene (Exon 2) is flanked by loxP sites and Cre recombinase excises the VGLUT2 exon to generate a knockout (abbreviated VGLUT2flox mice), 3) B6;129S-Gt(ROSA)26Sortm32(CAG-COP4*H134R/EYFP)Hze mice (abbreviated R26LSL-ChR2-EYFP mice; The Jackson Laboratory, Stock No. 012569; ChR2-EYFP fusion protein expressed under the R26::CAG promotor in cells that coexpress Cre because a loxP-flanked STOP cassette, LSL, that otherwise prevents transcription of the downstream ChR2-EYFP gene), and 4) B6;129S-Gt(ROSA)26Sortm35.1(CAG-aop3/GFP)Hze mice (abbreviated R26LSL-Arch3-GFP mice; The Jackson Laboratory Stock No. 012735; Arch3-GFP fusion protein expressed under the R26::CAG promotor in cells that coexpress Cre). Offspring without the Sim1-cre or mutation, but with the effectors tdTom, VGLUT2flox, ChR2, or Arch3 were used as controls.
Sim1-cre mice were crossed with homozygous reporter strains to generate Sim1-cre+;R26LSL-tdTom, Sim1-cre;VGLUT2flox, Sim1-cre;R26LSL-ChR2-EYFP, and Sim1-cre; R26LSL-Arch3-GFP mice that we abbreviate: Sim1//tdTom, Sim1//VGLUT2KO, Sim1//ChR2, and Sim1//Arch3 mice, respectively.
We also studied GABAergic neurons in mice with GFP expressed under the GAD1 promotor (termed GAD1-GFP, obtained courtesy of Dr. Peter Smith and Yuchio Yanagawa, specifically with GFP knocked in at the endogenous promoter for GAD1 in one allele) (52) and crossed these with Sim1//tdTom mice. In addition, we used mice with Cre inserted after GAD2, the latter as previously detailed (19), using Gad2tm1(cre/ERT2)Zjh mice (abbreviated Gad2CreER mice; The Jackson Laboratory, Stock No. 010702; CreERT2 fusion protein expressed under control of the endogenous Gad2 promotor). From these, we generated GAD2//ChR2 mice to optogenetically activate them as for Sim1//ChR2 mice detailed above.
Chronic Spinal Cord Injury
Some mice were studied after a chronic S2 spinal transection, described previously (44). Briefly, adult mice were transected at the S2 sacral spinal level at ∼2 mo of age (adult mice), and recordings were made 1.5–3 mo after injury when their affected muscles became spastic, as detailed previously (53). Under general anesthesia (100 mg/kg ip ketamine hydrochloride and 10 mg/kg ip xylazine) and using aseptic technique, the L2 lumbar vertebra was exposed and a laminectomy was performed to expose the S2 sacral spinal cord. The dura was cut transversely, and ∼0.1 mL of Xylocaine (1%) was applied to the exposed spinal cord. With the use of a surgical microscope, the exposed pia was held with fine forceps and the spinal cord was transected by aspirating a short section of the spinal cord using a fine suction tip. Caution was needed to avoid damaging the anterior artery and dorsal vein because the sacrocaudal spinal cord dies without these vessels. The dura was closed with 8-0 silk sutures and the muscle and skin were sutured in layers using 5-0 silk. Immediately following surgery, the mouse was placed in a recovery cage located on a heating blanket and allowed to recover.
In Vitro Recording in Whole Adult Spinal Cords
Mice were anesthetized with urethane (for mice 0.11 g/100 g, with a maximum dose of 0.065 g), a laminectomy was performed, and then the entire sacrocaudal spinal cord was rapidly removed and immersed in oxygenated modified artificial cerebrospinal fluid (mACSF), as detailed previously (44). The mice were then euthanized with Euthanyl (BimedaMTC; 700 mg/kg). Spinal roots were removed, except the sacral S2, S3, and S4 and caudal Ca1 ventral and dorsal roots on both sides of the cord. After 1.5 h in the dissection chamber (at 20°C), the cord was transferred to a recording chamber containing normal ACSF (nACSF) maintained at 23°C, with a flow rate >3 mL/min. A 1-h period in nACSF was given to wash out the residual anesthetic before recording, at which time the nACSF was recycled in a closed system. The cord was secured onto tissue paper at the bottom of a rubber (Silguard) chamber by insect pins in connective tissue and cut root fragments. The dorsal side of the cord was usually oriented upward when making intracellular recording from Ia afferents in the dorsal horn, whereas the cord was oriented with its left side upward when making recordings from motoneurons or V3 neurons. The laser beam used for optogenetics was focused vertically downward on the V3 neurons or GAD2 neurons, as detailed below.
Optogenetic Regulation of V3 Neurons
The Sim1//ChR2 or Sim1//Arch3 mice were used to optogenetically excite or inhibit V3 neurons (with 447 nm D442001FX and 532 nm LRS-0532-GFM-00200-01 lasers from Laserglow Technologies, Toronto), respectively, using methods we previously described (19, 44). GAD2//ChR2 mice were likewise used to excite GABAergic neurons. Light was derived from the laser passed through a fiber optic cable and then a half-cylindrical prism the length of about two spinal segments (8 mm; 3.9 mm focal length, Thor Labs, Newton), which collimated the light into a narrow long beam (200-µm wide and 8-mm long). This narrow beam was usually focused longitudinally on the left side of the spinal cord to target many V3 neurons at once. ChR2 rapidly depolarizes neurons (54), and thus we used 5–10-ms light pulses to activate V3 neurons. The light was kept at an intensity of 3xT, where T is the threshold light to evoke a dorsal root response, and 3xT is near the maximal response. This ensured that the light was not too intense to cause local heating, but thoroughly penetrated the spinal cord. In control experiments with light applied perpendicular to the dorsal surface, we found that monosynaptic activation of motoneurons occurred at 2.3xT, which we know is mediated by the most ventral V3 neurons that directly synapse on motoneurons. Thus, the intensity of 3xT that we used thoroughly penetrated through the entire width of the spinal cord, albeit with less intensity deeper in the spinal cord. Arch3 is a proton pump that is activated by green light, leading to hyperpolarization and slowly increased pH (over seconds), both of which inhibit the neurons (54, 55). Thus, we used longer light pulses to inhibit V3 neurons.
To directly confirm the presence of functional ChR2 expression in V3 neurons of Sim1//ChR2 mice, we recorded from them with similar methods and intracellular electrodes that we used to record from afferents (see below). Electrodes were advanced into these cells through the lateral edge of the cord, and their identity was established by a direct response to light activation of the ChR2 construct (5–10-ms light pulse, 447 nm), without a synaptic delay (<1 ms) and continued light response after blocking synaptic transmission.
Dorsal Root Stimulation
During intracellular recordings, all dorsal roots were mounted on silver-silver chloride wires above the nASCF of the in vitro chamber and covered with grease (a 3:1 mixture of petroleum jelly and mineral oil) for monopolar stimulation (22, 44). This grease was surrounded by a more viscous synthetic high-vacuum grease to prevent oil from leaking into the bath flow. Bipolar stimulation was also used at times to reduce the stimulus artifact. DRs were stimulated with a constant current stimulator (Isoflex, Israel) with short pulses (0.1 ms). During grease-gap recording (detailed below), only the Ca1 (or S4) DR was mounted for stimulation when we recorded PAD from other DRs (S2-S4 DRs).
Intracellular Recording and Labeling Sensory Axons in the Dorsal Horn
Intracellular recordings were made from group Ia afferents with electrodes made from glass capillary tubes (1.5 mm and 0.86 mm outer and inner diameters, respectively; with filament; 603000 A-M Systems; Sequim) pulled with a Sutter P-87 puller (Flaming-Brown; Sutter Instrument, Novato), filled with either 1 M K-acetate and 1 M KCl or 500 mM KCl in 0.1 Trizma buffer with 5–10% neurobiotin; Vector Labs, Newark), and beveled to 30–40 MΩ using a rotary beveller (Sutter BV-10) (19). Intracellular recording was performed with an Axoclamp2B amplifier (Axon Inst. and Molecular Devices, San Jose). Recordings were low-pass filtered at 10 kHz and sampled at 30 kHz (Clampex and Clampfit; Molecular Devices, San Jose). Electrodes were advanced into afferents of the sacrocaudal spinal cord with a stepper motor (Model 2662, Kopf, 10 µm steps at maximal speed, 4 mm/s), usually at the boundary between the dorsal columns and dorsal horn gray matter, where axon bundle together densely, as they branch and descend to the ventral horn. Before penetrating afferents, we recorded the extracellular (EC) afferent volley following dorsal root (DR) stimulation (0.1 ms pulses, 3xT, T: afferent volley threshold, where T = ∼3 μA, repeated at 1 Hz), to determine the minimum latency and threshold of afferents entering the spinal cord. The group Ia afferent volley occurs first with a latency of 0.5–1.0 ms, depending on the root length (which was kept as long as possible, 10–20 mm). Upon penetration, afferents were identified with direct orthodromic spikes evoked from DR stimulation. We focused on the lowest threshold proprioceptive group Ia afferents, identified by their direct response to DR stimulation, very low threshold (< 1.5 x T, T: afferent volley threshold), and shortest latency following stimulation (group Ia latency, coincident with onset of afferent volley). Group Ib, II, and cutaneous afferents on average have slower conduction velocities and higher thresholds than Ia afferents (56, 57), allowing most of them to be excluded by only studying the fastest, lowest threshold afferents. Possibly some Ib afferents were included, though anatomically they are distinguishable when filled with neurobiotin (see below). Clean axon penetrations without injury occurred abruptly with the membrane potential settling rapidly to near −70 mV, and >70 mV spikes usually readily evoked by DR stimulation or brief current injection pulses (1–3 nA, 20 ms, 1 Hz). Sensory axons also had a characteristic >100-ms long depolarization following stimulation of a dorsal root (PAD) and short spike afterhyperpolarization (AHP ∼ 10 ms), which further distinguished them from other axons or neurons (not afferents). Injured axons had higher resting potentials (>−60 mV), poor spikes (<60 mV), and low resistance (to current pulse; Rm < 10 MΩ), and were discarded.
Some of the proprioceptive afferents that we recorded intracellularly were subsequently filled with neurobiotin by passing a very large positive 2–4 nA current with a 90% duty cycle (900 ms on, 100 ms off) for 10–20 min. The identity of group Ia proprioceptive afferents was then confirmed anatomically by their unique extensive innervation of motoneurons (22). Before penetrating and filling axons with neurobiotin-filled electrodes, a small negative holding current was maintained on the electrodes to avoid spilling neurobiotin outside axons.
Dorsal and Ventral Root Grease-Gap Recording
In addition to recording directly from single proprioceptive axons, we used a grease-gap method to record the composite intracellular response of many sensory axons or motoneurons by recording from dorsal and ventral roots, respectively, as previously detailed (19). We mounted the freshly cut roots onto silver-silver chloride wires just above the bath and covered them in grease over about a 2–5 mm length (longer length provides an improved seal), as detailed above. The grease was applied on the roots as close as possible to the spinal cord, maximizing the recorded signal. Return and ground wires were in the bath and likewise made of silver-silver chloride. Specifically for sensory axons, we recorded from the central ends of dorsal roots (S2–S4) cut within ∼2–4 mm of their entry into the spinal cord, to give the compound potential from all afferents in the root (dorsal roots potential, DRP), which has previously been shown to correspond to PAD, though it is attenuated compared with the intracellular recordings of PAD (22). In this arrangement, the majority of the afferents recorded are the largest afferents (including Ia afferents), as detailed previously (19). For optogenetic experiments, we additionally added silicon carbide powder (9% wt, Tech-Met, Markham) to the grease to make it opaque to light and minimize light-induced artifact current in the silver-silver chloride recording wire during optogenetic activation of ChR2 (detailed below). Likewise, we covered our bath ground and recording-return wires with a plastic shield to prevent stray light artifacts. The dorsal root recordings were amplified (2,000 times), high-pass filtered at 0.1 Hz to remove drift, low-pass filtered at 10 kHz, and sampled at 30 kHz (Axoscope 8; Axon Instruments/Molecular Devices, Burlingame, CA).
The composite EPSPs in many motoneurons were likewise recorded from the central cut end of ventral roots (S3–S4) mounted in grease (grease gap), which has also previously been shown to yield reliable estimates of the EPSPs, though again attenuated by the distance from the motoneurons (19). The monosynaptic EPSPs were again identified as monosynaptic by their rapid onset (first component, ∼1 ms after afferent volley arrives in the ventral horn; see below), lack of variability in latency (<1 ms jitter), persistence at high rates (10 Hz) and appearance in isolation at the threshold (T) for evoking EPSPs with DR stimulation (< 1.1xT, T ∼ afferent volley threshold), unlike polysynaptic reflexes which varying in latency, disappear at high rates, and mostly need stronger DR stimulation to activate. Usually, the Ca1 or S4 roots were stimulated to evoke the EPSPs.
Drugs and Solutions
Two kinds of artificial cerebrospinal fluid (ACSF) were used: a modified ACSF (mACSF) in the dissection chamber before recording and a normal ACSF (nACSF) in the recording chamber. The mACSF was composed of (in mM) 118 NaCl, 24 NaHCO3, 1.5 CaCl2, 3 KCl, 5 MgCl2, 1.4 NaH2PO4, 1.3 MgSO4, 25 d-glucose, and 1 kynurenic acid. Normal ACSF was composed of (in mM) 122 NaCl, 24 NaHCO3, 2.5 CaCl2, 3 KCl, 1 MgCl2, and 12 d-glucose. Both types of ACSF were saturated with 95% O2-5% CO2 and maintained at pH 7.4. The drugs sometimes added to the ACSF were APV (NMDA receptor antagonist), CNQX (AMPA antagonist), strychnine (glycine antagonist), tubocurarine (ACh antagonist), gabazine (GABAA antagonist, all from Tocris), and TTX (TTX-citrate; Toronto Research Chemicals, Toronto). Drugs were first dissolved as a 10–50 mM stock in water or DMSO before final dilution in ACSF.
Immunohistochemistry
Sim1//tdTom or Sim1//ChR2-EYFP mice were euthanized with Euthanyl (BimedaMTC; 700 mg/kg) and perfused intracardially with 10 mL of saline for 3–4 min, followed by 40 mL of 4% paraformaldehyde (PFA; in 0.1 M phosphate buffer at room temperature), over 15 min (Gabra5-KO mice also fixed similarly). Then, spinal cords of these mice were postfixed in PFA for 1 h at 4°C, and then cryoprotected in 30% sucrose in phosphate buffer (∼48 h). Alternatively, spinal cords where sensory axons were injected with neurobiotin in vitro were left in the recording chamber in oxygenated nACSF for an additional 4–6 h to allow time for diffusion of the neurobiotin throughout the axon and then the spinal cord was immersed in 4% paraformaldehyde (PFA; in phosphate buffer) for 20–22 h at 4°C, cryoprotected in 30% sucrose in phosphate buffer for 24–48 h. Following cryoprotection, all cords were embedded in OCT (Sakura Finetek, Torrance, CA), frozen at −60°C with 2-methylbutane, cut on a cryostat NX70 (Fisher Scientific) in sagittal or transverse 25-µm sections, and mounted on slides. Slides were frozen until further use.
The tissue sections on slides were first rinsed with phosphate-buffered saline (PBS, 100 mM, 10 min) and then again with PBS containing 0.3% Triton X-100 (PBS-TX, 10 min rinses used for all PBS-TX rinses). Next, for all tissue, nonspecific binding was blocked with a 1 h incubation in PBS-TX with 10% normal goat serum (NGS; S-1000, Vector Laboratories, Burlingame) or normal donkey serum (NDS; ab7475, Abcam, Cambridge, UK). Sections were then incubated for at least 20 h at room temperature with a combination of the following primary antibodies in PBS-TX with 2% NGS or NDS: guinea pig anti-VGLUT1 (1:1,000; AB5905, Sigma-Aldrich, St. Louis), guinea pig anti-VGLUT2 (1:20,000, AB225-I, Sigma-Aldrich, St. Louis), guinea pig anti-GAD2/GAD65 (1:500; 198 104; Synaptic Systems), chicken anti-VGAT (1:500; 131 006, Synaptic Systems, Gottingen, Germany), rabbit anti-VGAT (1:500; AB5062P, Sigma-Aldrich, St. Louis), rabbit anti-EYFP (1:500; orb256069, Biorbyt, Riverside, UK), mouse anti-bassoon (1:400, ENZO SAP7F407, MJS Biolynx Inc.), goat anti-RFP (1:500; orb334992, Biorbyt, Riverside, UK), rabbit anti-RFP (1:500; PM005, MBL International, Woburn), and rabbit anti-GFP (1:500, A11122, Thermo Fisher Scientific, Waltham). Genetically expressed EYFP (labeled by GFP-ab), tdTom (labeled by RFP-ab), and GFP were amplified with the above antibodies, rather than relying on endogenous fluorescence. When anti-mouse antibodies were applied in mice tissue, the M.O.M (Mouse on Mouse) immunodetection kit was used (M.O.M; BMK-2201, Vector Laboratories, Burlingame) before applying antibodies. This process included 1 h incubation with a mouse Ig-blocking reagent. Primary and secondary antibody solutions were diluted in a specific M.O.M diluent.
The following day, tissue was rinsed with PBS-TX (3 times for 10 min) and incubated with fluorescent secondary antibodies. The secondary antibodies used included: goat anti-rabbit Alexa Fluor 555 (1:200; A32732, Thermo Fisher Scientific, Waltham), goat anti-rabbit Alexa Fluor 647 (1:500, ab150079, Abcam, Cambridge, UK), goat ant-rabbit Pacific orange (1:500; P31584, Thermo Fisher Scientific, Waltham), goat anti-mouse Alexa Fluor 647 (1:500; A21235, Thermo Fisher Scientific, Waltham), goat anti-mouse Alexa Fluor 488 (1:500; A11001, Thermo Fisher Scientific, Waltham), goat anti-mouse Alexa Fluor 555 (1:500; A28180, Thermo Fisher Scientific, Waltham), goat anti-guinea pig Alexa Fluor 647 (1:500; A21450, Thermo Fisher Scientific, Waltham), goat anti-chicken Alexa Fluor 405 (1:200; ab175674, Abcam, Cambridge, UK), goat anti-chicken Alexa Fluor 647 (1:500; A21449, Thermo Fisher Scientific, Waltham), donkey anti-goat Alexa Fluor 555 (1:500; ab150130, Abcam, Cambridge, UK), donkey anti-rabbit Alexa Fluor 488 (1:500; A21206, Thermo Fisher Scientific, Waltham), Streptavidin-conjugated Alexa Fluor 488 (1:200; 016–540-084, Jackson immunoResearch, West Grove), or Streptavidin-conjugated Cyanine Cy5 (1:200; 016–170-084, Jackson ImmunoResearch, West Grove) in PBS-TX with 2% NGS or NDS, applied on slides for 2 h at room temperature. The latter streptavidin antibodies were used to label neurobiotin-filled afferents. After rinsing with PBS-TX (2 times × 10 min/each) and PBS (2 times × 10 min/each), the slides were covered with Fluoromount-G (00–4958-02, Thermo Fisher Scientific, Waltham) and coverslips (#1.5, 0.175 mm, 12–544-E; Fisher Scientific, Pittsburg).
Standard negative controls in which the primary antibody was either 1) omitted or 2) blocked with its antigen (quenching) were used to confirm the selectivity of the antibody staining, and no specific staining was observed in these controls. Previous tests detailed by the manufacturers further demonstrate the antibody specificity, including quenching, immunoblots (Western blots), coimmunoprecipitation, and/or receptor knockout.
Image acquisition was performed by confocal microscopy (Leica TCS SP8 Confocal System). All the confocal images were taken with a ×63 (1.4 NA) oil immersion objective lens or a ×20 water emersion lens and 0.1-µm optical sections that were collected into a z-stack over 10–20 µm. Excitation and recording wavelengths were set to optimize the selectivity of imaging the fluorescent secondary antibodies. Large areas were imaged with the Tilescan option in Leica Application Suite X software (Leica Microsystems CMS GmbH, Germany).
Statistical Analysis
Data were analyzed in Clampfit 8.0 (Axon Instruments) and Sigmaplot (Systat Software, USA). A Student’s t test or ANOVA (as appropriate) was used to test for statistical differences between variables, with a significance level of P < 0.05 (two-tailed). Power of tests was computed with α = 0.05 to design experiments. A Kolmogorov–Smirnov test for normality was applied to the dataset, with a P < 0.05 level set for significance. Most datasets were found to be normally distributed, as is required for a t test. For those that were not normal, a Wilcoxon signed-rank test was instead used with P < 0.05. Effects in male and female animals were similar and grouped together in analysis. Axons (roots) and motoneurons were recorded in vitro from widely separated locations (one segment apart or contralateral) within the whole spinal cord and are considered independent; so statistics were performed across all neurons or dorsal roots (n) from all animals, though comparing across animal averages also showed similar significant changes, with three or more animals used per condition. Data are indicated by mean ± standard deviation.
RESULTS
We began by genetically labeling V3 neurons in Sim1-cre//tdTom mice (Fig. 1A) or Sim1-cre//ChR2-EYFP mice to examine the role of V3 neurons in the putative trisynaptic circuit underlying sensory-evoked PAD (Fig. 1). These neurons were extensively innervated with sensory afferent terminals labeled with VGLUT1 (Fig. 1, A and B) (58), including terminals from group Ia afferents filled with neurobiotin (Fig. 1B). Overall, 91% of V3 neuron somas examined were densely innervated by afferents (VGLUT1+; n = 21/23 neurons in the intermediate zone). We mostly focused on the sacral spinal cord, as only this portion of the spinal cord survives whole in vitro for functional studies (see methods), though similar dense VGLUT1+ innervation of V3 neurons was seen in the lumbar cord. Furthermore, V3 neurons and their extensive processes were predominantly located at intermediate, ventral, and deep dorsal spinal levels near the central canal (∼laminae 6, 7, 9, and 10, as well as the sacral dorsal commissure), as previously detailed for the sacral cord (59), well positioned to interact with nearby GABAergic neurons that mediate PAD (detailed later) (19). Furthermore, this extensive afferent input onto V3 neurons is consistent with the dense VGLUT1 innervation seen more generally in intermediate zone excitatory neurons (46). The density of the VGLUT1 contacts on V3 neurons in the dorsal and intermediate regions suggests that many other afferents, in addition to the few Ia afferent terminals in this region, innervate V3 neurons.
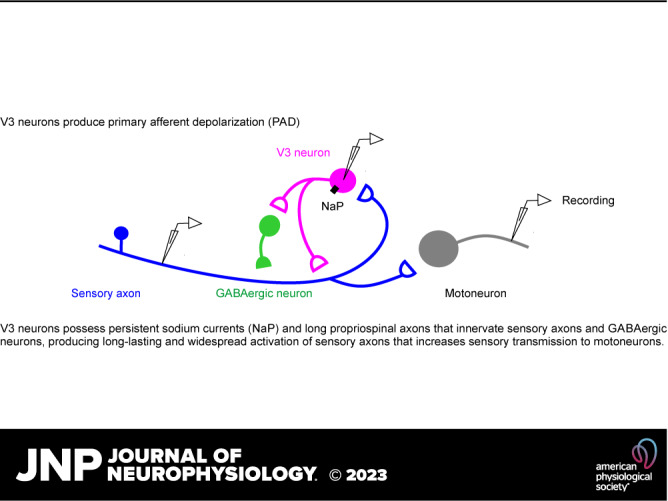
Figure 1.
Monosynaptic connections from sensory afferents to V3 neurons. A: V3 neurons in the spinal cord intermediate laminae decorated with VGLUT1+ contacts from afferents, some of which are Ia afferent contacts labeled by an intracellular injection of neurobiotin (blue). Spinal cord from Sim1//tdTom mouse. B: expanded image from A with 3-D reconstructed VGLUT1+ afferent contacts on V3 neurons labeled in yellow. Top: VGLUT1 contacts; middle: neurobiotin contacts; bottom: merge of above two panels. C: schematic of putative trisynaptic circuit where Ia afferents (blue) innervate V3 neurons, which in turn innervate GABAergic neurons, which return to innervate Ia afferents, ultimately producing PAD. Experimental setup indicated where Ia afferents are activated by dorsal root (DR) stimulation (0.1 ms pulse, 2xT, sensory threshold) and response recorded with a sharp intracellular electrode in or near V3 neurons or afferents. D: monosynaptic EPSP recorded intracellularly in V3 neuron in response to dorsal roots stimulation (S4 root of in vitro sacrocaudal cord; average of 10 trials at 0.3 Hz). E: same V3 EPSP as in D, but population response recorded nearby, extracellularly to V3 neurons (EC field). F: intra-axonal recording from Ia afferent during same DR stimulation, where the afferent is directly activated, as evident by an orthodromic spike, and following this a primary afferent depolarization (PAD) arises at a polysynaptic latency consistent with a circuit like in C. G: central latencies and durations of V3 EPSPs, EC fields, and PAD, where that latter was recorded either intracellularly in Ia afferents as in F (though in afferents of not directly activated by the DR stimulation, so the PAD onset can be judged) or by grease-gap recordings from DR (dorsal root potential, DRP). Central latency measured relative to arrival time of the orthodromic spike at the spinal cord, as in F, though measured from the extracellular afferent volley (not shown, though see Ref. 22). V3 EPSP latencies were minimally near 1 ms (0.8–3 ms), which is monosynaptic as the in vitro adult spinal cord has a synaptic delay of ∼1 ms at 23°C. PAD latencies were 2–4 ms, with an average of 3 ms, consistent with the trisynaptic circuit of C, though possibly with also some disynaptic innervation. *Significantly longer latency of PAD (Ia PAD or DRP) compared with V3 EPSP or its EC field latency, P < 0.05, n = 6 V3 neurons, 6 EC fields, 44 DRPs, and 5 Ia afferents, in 3–8 mice each. PAD, primary afferent depolarization.
To examine the direct action of this sensory innervation, we made intracellular recordings from V3 neurons (Fig. 1, C–G). This revealed large, long-lasting EPSPs (V3 EPSPs) in response to low-threshold sensory stimulation of the dorsal roots (DR, 1.0 - 1.5xT, 2.8–4.2 µA, group I intensity), with a rapid onset latency (Fig. 1G), consistent with a minimally monosynaptic innervation from large proprioceptive or cutaneous afferents, as suggested by our confocal imaging (Fig. 1A; the in vitro adult spinal cord has a synaptic latency of ∼0.8–1 ms near room temperature) (19). Just outside of V3 neurons a pronounced extracellular field (EC V3 field, negative, Fig. 1E) was observed with a similar duration and monosynaptic latency as the V3 EPSP (Fig. 1G), suggesting large numbers of V3 neurons were activated coincidently by the sensory innervation.
These long-lasting V3 EPSPs were similar in duration and shape to the depolarization of primary sensory afferents evoked by the same DR stimulation (sensory-evoked PAD), either measured directly by intracellular recording from group Ia afferents or indirectly by grease-gap recording from the DRs, the latter which gives the population response of many large afferents (DRP, Fig. 1, F and G). When the same DR was stimulated that contained the recorded Ia afferent (Fig. 1C), there was a direct orthodromic spike evoked if the stimulus intensity was above the afferent threshold, and this was followed by PAD (homonymous PAD), as depicted in Fig. 1, C and F. Such direct spikes compose the afferent volley that drives the V3 neurons monosynaptically in Fig. 1D. Alternatively, when we stimulated an adjacent DR so there was no orthodromic spike, a similar PAD arose (heteronymous PAD, figures detailed below, and see Ref. 22). Either way, the sensory-evoked PAD activated by stimulating any DR (abbreviated drPAD) had a similar duration to the V3 response to the same DR stimulation (V3 EPSP; Fig. 1G). As previously reported, the central latency of drPAD was ∼2–4 ms (Fig. 1G) (22), longer than the V3 EPSP latency, and consistent with the classical assumption that PAD arises from a trisynaptic loop, such as in Fig. 1C. This provides sufficient time for the V3 neurons to contribute to PAD, as their V3 EPSP has a central latency of half the PAD latency, as mentioned above, even taking into account possible delays in spike initiation in V3 neurons (0.5–4 ms; not shown).
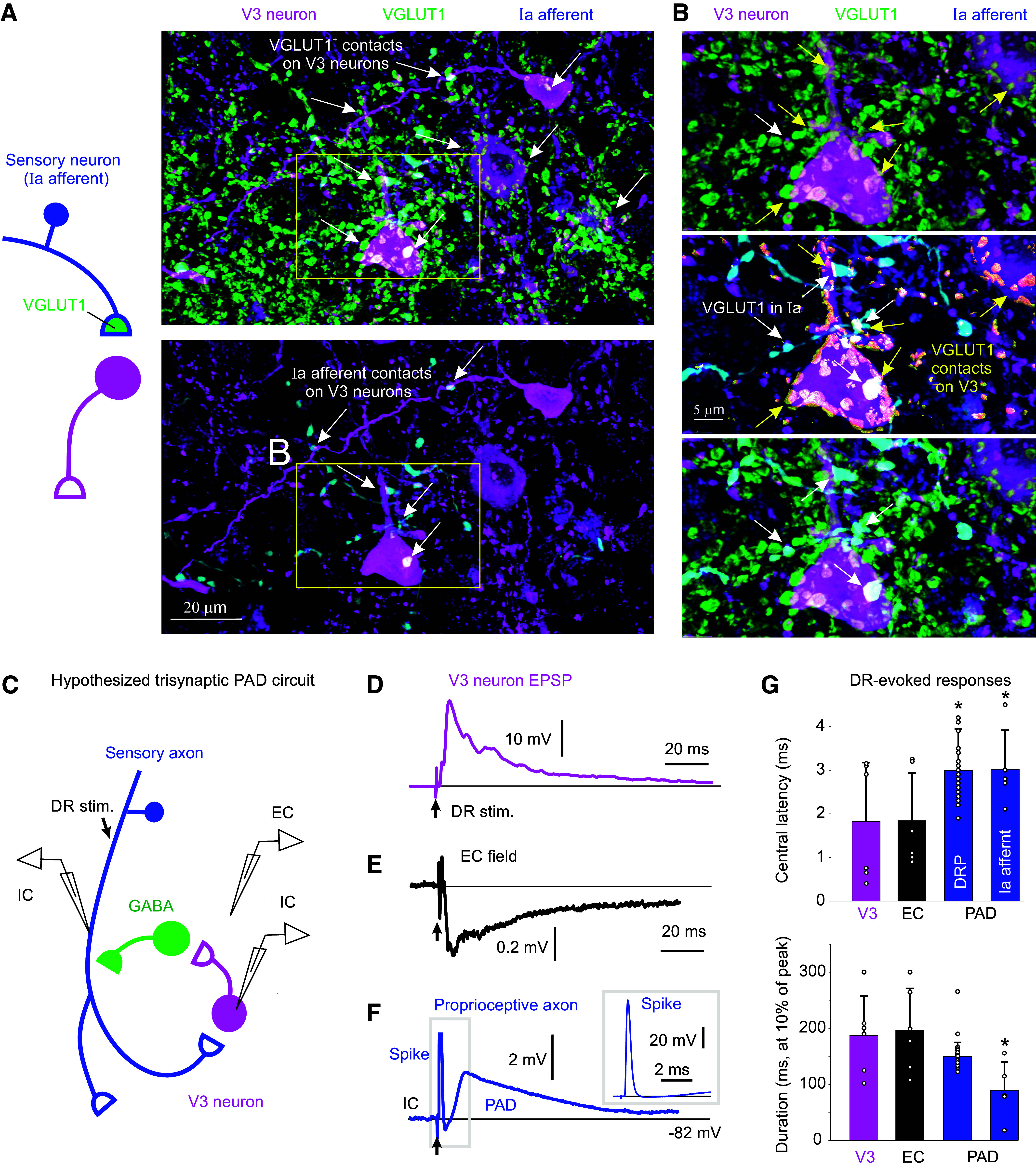
For V3 neurons to be part of the trisynaptic drPAD circuit, they need to directly contact GABAergic neurons (Fig. 1C). To establish this, we crossed Sim1//tdTom mice with GAD1-GFP mice to label both V3 and GABAergic neurons, the latter including GAD2+ neurons which are the subset of GAD1+ neurons that form axoaxonic connections on afferents (Fig. 2) (15). As expected, we found that V3 neurons extensively innervated GABAergic neurons, with presynaptic VGLUT2+ and Bassoon+ in these glutamatergic V3 neurons and postsynaptic GAD2 expression in GABAergic neurons labeled in GAD-GFP mice (Fig. 2, A–C). The majority of these V3 contacts were on GABAergic neurons in the intermediate and deep dorsal laminae (Fig. 2E). Presynaptic bassoon together with GAD2 was also expressed in the GABAergic neuron boutons near V3 contacts onto these neurons, showing that the GABAergic neurons release GABA very near to where they receive V3 neuron innervation (Fig. 2C), likely on a dendrite, a microcircuit arrangement that is common for GABAergic neurons (22, 47, 60). This suggests that GABAergic neurons may also innervate V3 neurons, an opposite arrangement to what we expected. Indeed, we found many GABAergic (GAD+) contacts onto V3 neurons, though these were more ventrally located than the V3 neuron contacts on GABAergic neurons (Fig. 2, D and E). Taken together, our anatomical results demonstrate that V3 neurons are part of the PAD circuit receiving sensory input and innervating GABAergic GAD2+ neurons that are known to in turn innervate sensory afferents (Figs. 1C and 2F). However, this PAD circuit is in part inhibited by GABAergic input (Fig. 2F), complicating the interpretation of the action of GABA receptor antagonists commonly used to study PAD, which would disinhibit the V3 neurons in the PAD circuit and spinal cord in general, as detailed below.
Figure 2.
Connections between V3 neurons and GABAergic neurons that produce PAD. A and B: V3 neurons (tdTom) contact onto GABAergic neurons, where presynaptic V3 neuron contacts are VGLUT2+(red), the vesicular transporter expressed in these glutamatergic V3 neurons. Spinal cord from mouse expressing Sim1//tdTom (magenta, V3) and GAD1-GFP (green, GABAergic neurons). Images shown both as 3-D reconstructions (3-D) and raw images (maximum projection of image stack). C: close up of V3 neuron contacts (Sim1//tdTom) onto GABAergic neuron (GAD1-GFP), with V3 presynaptic terminal labeled with bassoon and GABAergic neuron labeled with GAD2, the latter to show that it is a GAD2+ neuron. Bassoon is also expressed in the GABAergic neuron boutons near the GAD2 clusters and the V3 contacts. D: GABAergic (GAD1-GFP) neurons also innervate V3 neurons, with GAD2+ presynaptic contacts. E: neuron soma distributions for V3 (n = 52) and GABAergic (n = 146) neurons in the deep dorsal, intermediate (VII), central canal (cc, X) and ventral laminae, and incidence of contacts (VGLUT2+ or bassoon+) on soma from either V3 neurons or GABAergic neurons, from 3 mice. F: schematic summarizing trisynaptic circuit mediating PAD with the addition of a contact from GABAergic neurons onto V3 neurons that inhibits the circuit. PAD, primary afferent depolarization.
To quantify how important V3 neurons are to the drPAD evoked by proprioceptive sensory stimulation, we used two approaches to block their function. First, we selectively knocked out (KO) glutamate function in V3 neurons in Sim1-cre//VGLUT2-flox mice (Sim1//VGLUT2KO), and second, we inhibited V3 neurons optogenetically by applying light to Sim1//ArchT+ mice, where the ArchT construct selectively expressed in V3 neurons hyperpolarizes these neurons and inhibits a proton pump (Fig. 3). Both methods led to a substantial reduction of drPAD evoked by stimulation of proprioceptors in the DR, which has previously shown to be largely mediated by GABAA receptors in this preparation (GABA PAD, detailed later) (22). Mice lacking ArchT had no response to the light (ArchT−; Fig. 3C), ruling out the effects of light on other non-V3 neurons or damage from the light. The ArchT silencing of V3 neurons was likely incomplete, due to a lack of sufficient inhibition, but allowed within animal comparisons of changes in drPAD with light, yielding about a 50% reduction in drPAD (Fig. 3, A–C), suggesting that V3 neurons are responsible for at least half the PAD. The KO silencing of V3 neurons was likely complete (Fig. 3, D and E), but can only be compared across animals where drPAD variability was large, depending on the root size and quality of grease seal, but nevertheless on average led to an even larger reduction in drPAD than with ArchT. Importantly, with both these ArchT and KO experiments, we evoked the PAD with very low-threshold DR stimuli to ensure that only large proprioceptive Ia afferents were activated, which has previously been shown to mostly evoke PAD in proprioceptive afferents themselves, and not higher threshold cutaneous afferents (13, 19, 23, 31, 33, 35). Together, these results indicate that the majority (at least half) of sensory-evoked PAD in proprioceptive afferents is mediated through V3 neurons that act as the first-order neurons (Fig. 1C).
Figure 3.

V3 neurons are essential for a large fraction of the PAD evoked by proprioceptive stimulation (drPAD). A and B: typical PAD evoked by proprioceptive sensory stimulation of a DR (S4 DR, 0.1 ms, 1.1xT, T sensory threshold) in adjacent DR (DRP measured in S3 DR) in Sim1//ArchT+ mice, before and after silencing V3 neurons with light (532 nm, 5 mW/mm2). Averages of 9 trials at 0.1 Hz. Light had no effect on drPAD in mice without ArchT (ArchT−). Lower trace: V3-dependent portion of drPAD including dorsal root reflexes (DRRs), estimated from the change with light. C: group averages of drPAD evoked by selective proprioceptive DR stimulation (1.1–1.3xT), with and without ArchT expression; *Significant reduction with light, P < 0.05, n = 8 roots from 3 mice. D: typical drPAD evoked by proprioceptive stimulation (DR 2xT) as in A, but in mice without and with V3 neurons silenced by VGLUT2 KO; top: control mouse (Sim1-Cre lacking flx-VGLUT1); bottom: Sim1-Cre//flx-VGLUT2 mouse. E: group averages of drPAD, *Significantly smaller with KO, P < 0.05, n = 15 and 36 roots tested for 6 control and 7 KO mice, respectively. DR, dorsal root; PAD, primary afferent depolarization.
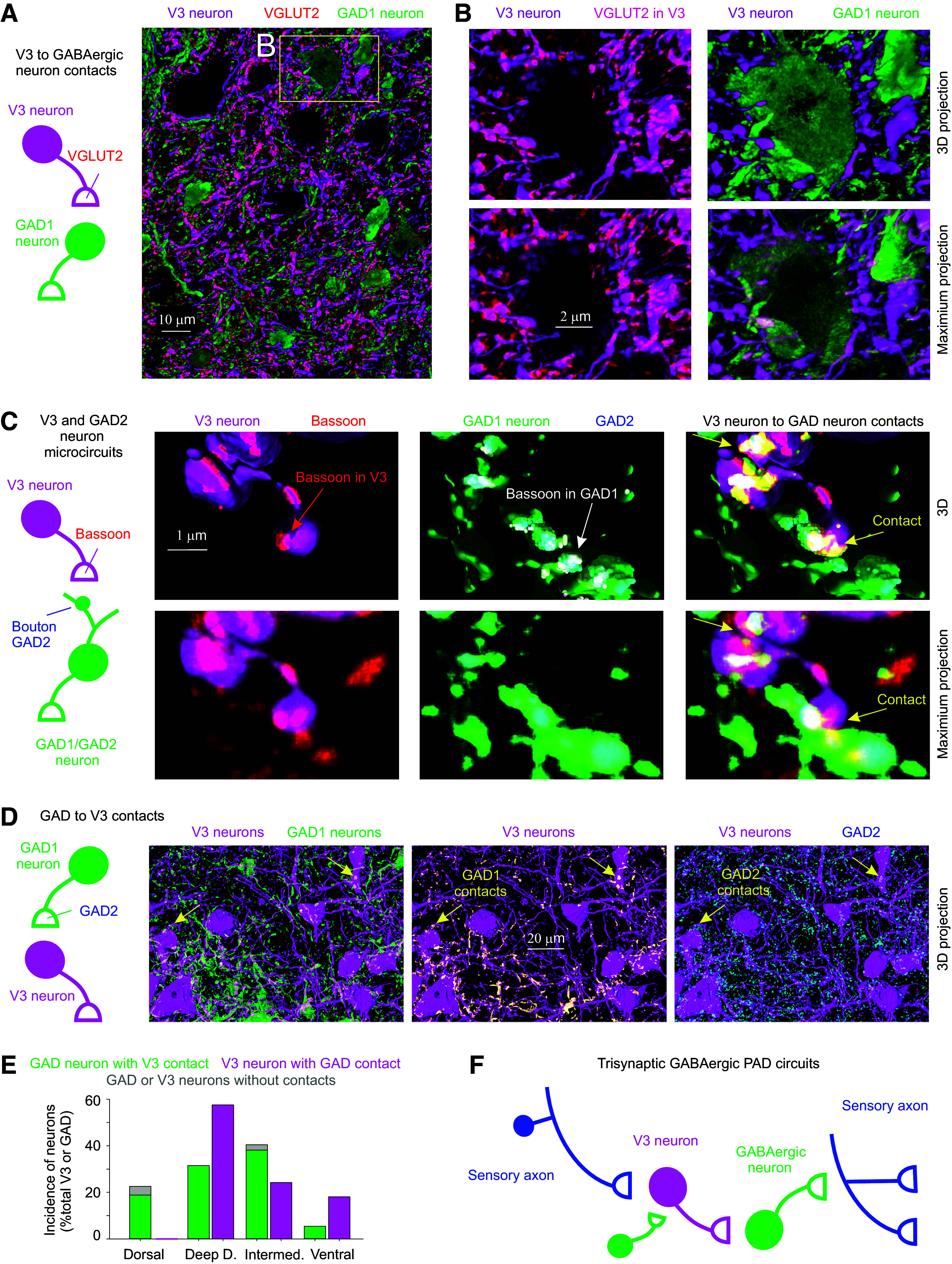
Considering that V3 neurons are part of the sensory PAD circuit, specifically innervating GABAergic neurons, we next examined Sim1//ChR2 mice where V3 neurons contained ChR2 so we could directly activate V3 neurons with a light pulse to examine their intrinsic properties and action on PAD. As mentioned earlier, V3 neurons in the sacral spinal cord were mostly located in the intermediate lamina with extensive arbors (Fig. 4A). Thus, we recorded from neurons this region (as in Fig. 4B), with V3 neurons penetrated through the lateral edge of the cord and identified by direct (non-delayed) responses to light activation of ChR2. Just like the long V3 EPSP observed with a brief DR stimulation (Figs. 1D and 4C), a brief light pulse evoked a long-lasting response in V3 neurons of Sim1//ChR2+ mice (but not control Sim1//ChR2- mice; Fig. 4D). These light responses were so large and characteristically long that they were also readily observed with extracellular recording (EC V3 field, seen as negative deflection). Individual trials in both the light and DR-evoked V3 responses showed considerable long-latency synaptic events (Fig. 4, C and D), and so we initially thought that perhaps the long-lasting nature of these V3 responses was due to reverberating synaptic circuits. Although these synaptic events were eliminated by a complete block of all fast synaptic transmission (with CNQX, APV, gabazine, and strychnine), this unexpectedly did not reduce the long-lasting V3 response to light, not decreasing the overall amplitude or duration (Fig. 4, D, E, and G–I). Instead, the sustained part of the light response (at 200 ms) increased in synaptic block, consistent with the V3 neurons receiving long-lasting recurrent synaptic inhibition (n = 11/11). Subsequent application of TTX markedly reduced the light response amplitude and duration (Fig. 4, F–I), indicating that intrinsic sodium-mediated persistent inward currents (Na PICs) contributed to the long-lasting V3 responses. This Na PIC may involve Nav1.8 and Nav 1.9 channels, as blockade required two times more TTX than needed to block axon conduction, and these channels are known to be somewhat TTX-resistant (47, 61). We struggled to activate this Na PIC by direct intracellular current injection, rather than light, possibly because the Na channels involved are located electrotonically distant to the somatic electrode on the many long propriospinal axons (or dendrites) of V3 neurons (with only 50-ms tail currents evoked by depolarizing current steps, not shown), whereas the ChR2 is widely distributed.
Figure 4.
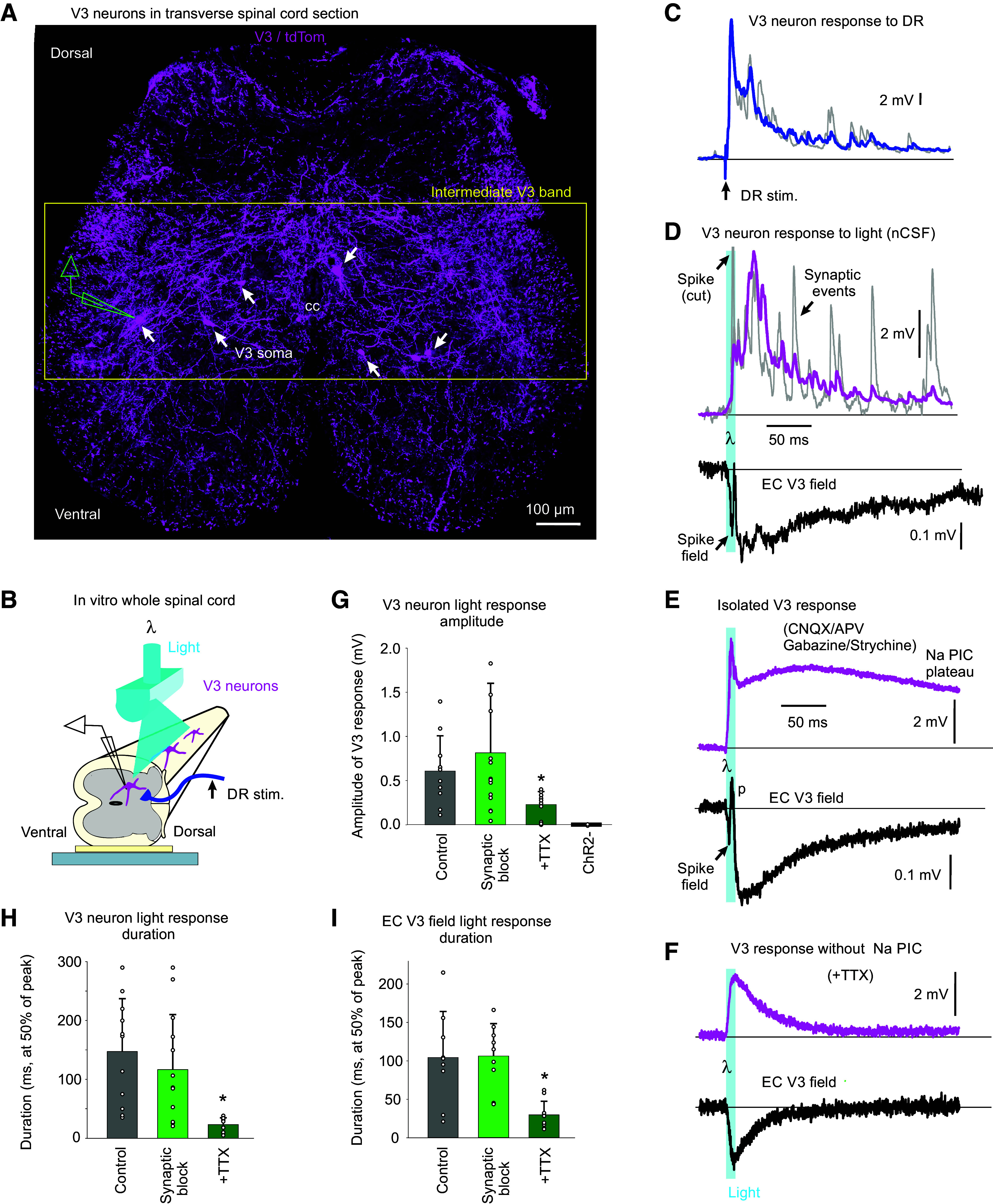
Long-lasting response evoked in V3 neurons by brief activation, mediated by persistent sodium currents. A: transverse section of sacral S3 spinal cord of Sim1//tdTom mice showing V3 neurons with their extensive arborizations throughout much of the intermediate lamina, where we recorded from V3 neurons intracellularly, as depicted. Image reproduced from Fig. 7A. where details are given. B: arrangement for intracellular or extracellular recording from V3 neurons in Sim1//ChR2 mice, with neurons identified by a direct response to a light pulse (10 ms pulse, λ = 447 nm laser, 0.7 mW/mm2, 3xT, T light threshold to evoke response, laser columnated and aligned axially along multiple segments), with no synaptic delay. C: long-lasting V3 neuron EPSP in response to DR stimulation (0.1 ms, 2xT; as in Fig. 1D), with blue line average of 10 trials (at 0.3 Hz), and gray line individual trial showing many synaptic events. D: long-lasting V3 neuron response to the light pulse (10 ms, 1.5xT), with magenta line average of 10 trials, and gray line individual trial showing many fast synaptic events superimposed, as well as a fast spike at the onset of the response. Lower black line is the extracellular recording (EC field, averages of 10 trials) of the population response of V3 neurons to the same light pulse, which also showed a fast spike. E: same as D, but after blocking synaptic transmission (with 50 µM CNQX, 50 µM APV, 50 µM gabazine, and 5 µM strychnine), showing a long-lasting V3 neuron depolarization (plateau potential) from voltage-gated currents intrinsic to the V3 neurons. F: same as E, but after also blocking sodium channels with TTX (2–3 µM), which eliminated the long-lasting response (and fast spike), showing that the plateau potential in E was due to persistent sodium currents (Na PICs). G–I: group averages of V3 neuron responses to the brief light pulse, with amplitudes (quantified at 60 ms latency) and durations shown in control conditions, after blocking synaptic transmission, and then after also applying TTX. Lack of light response in Sim1//ChR2- mice also shown. Duration measured at 50% peak amplitude. *Significant reduction with TTX but not synaptic blockade, P < 0.05, n = 11 intracellular V3 recordings from 5 mice, and n = 16 EC V3 fields from 5 mice. D–F on same time scale. DR, dorsal root; EC, extracellular.
The intracellular and extracellular V3 responses to light consistently exhibited fast synchronous spikes that arose at about a 2–5 ms latency, peaked at ∼5–6 ms, and were unaffected by blocking synaptic transmission (Fig. 4, G and H). This suggests that V3-evoked responses in postsynaptic neurons should have a minimally 2-ms latency, with on an average 5–6 ms latency, even with monosynaptic connections, which makes latencies of PAD-evoked by light slower than those evoked by DR stimulation, as detailed below. Likely, ChR2 is less effective at depolarizing V3 neurons than natural synaptic input, accounting for its slower action [as we have also seen in other neurons (19)], and so caution must be used in interpreting latencies of ChR2 responses.
Interestingly, the light-evoked fast synchronous spikes recorded intracellularly in V3 neurons of Sim1//ChR2 mice were sometimes not full height (Fig. 4E), suggesting that they arose somewhere in the long propriospinal axons of V3 neurons but did not always fully propagate back to the soma where we usually recorded, and so we observed only its passively attenuated spike (failure potential) (19). Indeed, the extracellular recording of this fast synchronous V3-evoked spike often had a large positive component, rather than the usual negative component expected for a spike arising near an electrode (Fig. 4E, positive field p) (19). Again, this indicates that the spike arose far away in a portion of the V3 neuron’s axon illuminated by the light, but did not always actively propagate to the recording site, causing only a passive outward current near the electrode (a current sink rather than source of spike), previously detailed (19). Considering that we typically applied the light mostly at and above (rostral) to the electrode during intracellular recording (for practical reasons with getting the electrode and laser in place), and that V3 neurons in the sacral cord tend to have a predominantly ascending propriospinal axons (as we detail below), we suggest that the light evoked spikes in many ascending propriospinal tracts and these spikes did not propagate antidromically to their parent cell bodies more caudally where the electrode was placed, which is not unusual because spike propagation failure is theoretically most likely when traveling from small distal branches to larger higher conductance branches (62).
To directly observe the action of V3 neurons on PAD, we returned to recording from DR afferents in Sim1//ChR2 mice (Fig. 5). As expected, a brief light activation of V3 neurons produced a long-lasting depolarization of afferents (termed V3 PAD; Fig. 5A). This V3 PAD was similar to classic sensory-evoked PAD produced by stimulating the dorsal roots (drPAD), in that it had a long duration, large amplitude, and short latency (Fig. 5, C–E; PAD estimated from the DRP with grease-gap methods, which is ∼10% of actual PAD size; see details in Ref. 22). Also, V3 PAD and drPAD were both sensitive to the GABAA receptor antagonist gabazine, consistent with both being in large part mediated by GABAA receptors (Fig. 5, A and B). Graded light activation of V3 neurons produced a graded PAD (like with graded DR stimulation, not shown), but we only quantified the near maximal PAD evoked by light (2–3 xT, where T is threshold light to evoked PAD).
Figure 5.
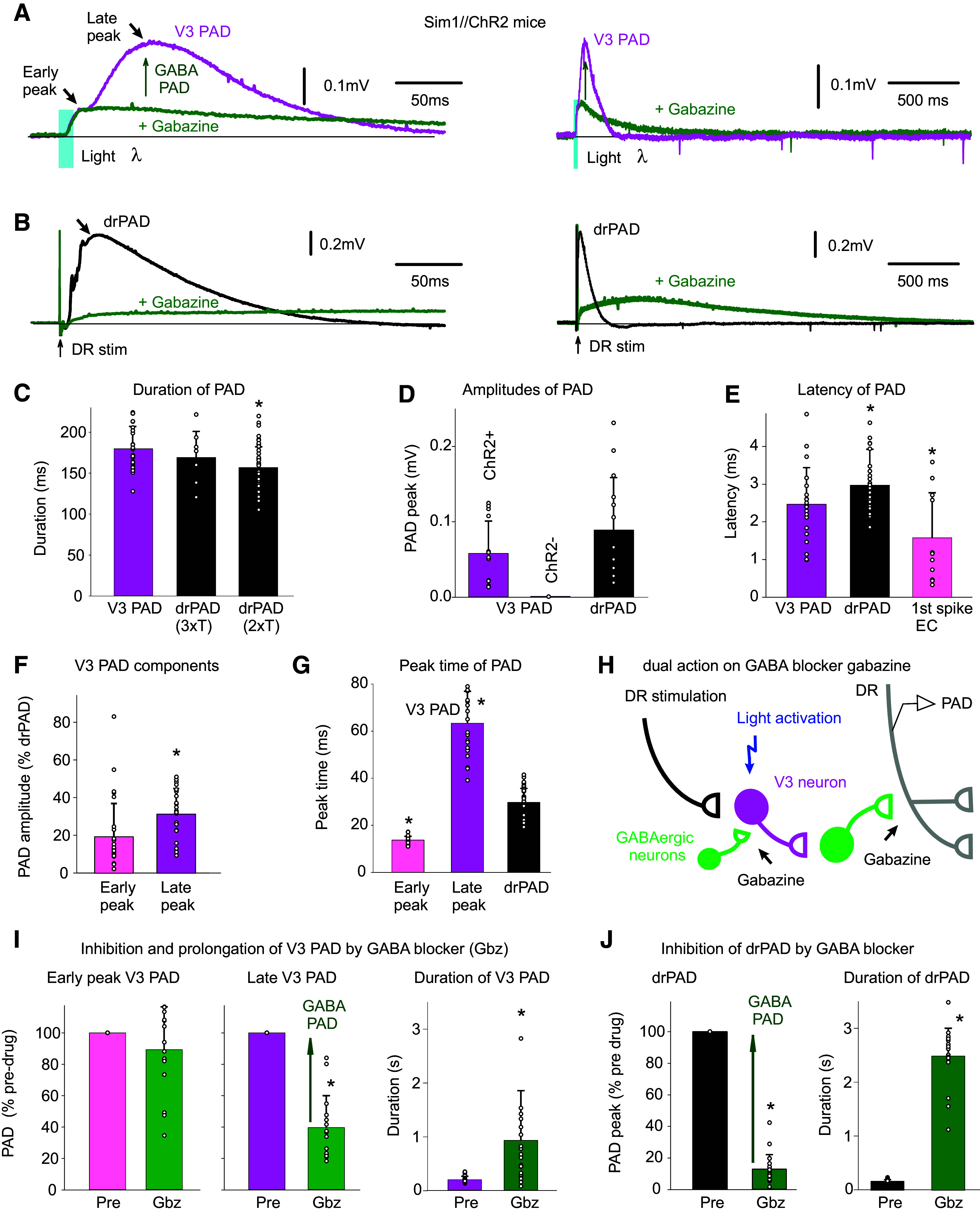
V3 neurons evoke a PAD (V3 PAD), partly mediated by GABAA receptors. A: brief optogenetic activation of V3 neurons in Sim1//ChR2 mice (10 ms pulse, λ = 447 nm laser, 0.7 mW/mm2, 3xT, columnated laser light aligned axially to maximally activate V3 neurons over multiple segments, as in Fig. 4B) evokes a PAD (termed V3 PAD) recorded in DR (S3 DR, DRP recorded with grease-gap method), with an early peak and a late peak, as indicated. Blocking GABAA receptors with gabazine (50 µM) blocks the late V3 PAD, but reveals a very long-lasting PAD that starts at the early peak, as shown on the expanded time scale on the right. B: similar to A, but brief DR stimulation evokes a drPAD that is mostly blocked by gabazine, but again reveals a very long-lasting PAD. C: group averages of durations of V3 PAD and drPAD evoked by 2xT and 3xT DR stimulation, before gabazine. *drPAD significantly less than V3 PAD, n = 28 V3 PADs and 50 drPADs from 6 and 9 mice, respectively, P < 0.05. D: group averages of maximum PAD amplitudes (late peak for V3 PAD), in Sim1//ChR2+ (same mice) and Sim1ChR2- mice (n = 12 from 4 mice), *drPAD significantly larger, P < 0.05. E: central latency of V3 PAD, drPAD, and first spike evoked in V3 neurons (the latter as in Fig. 4G. n = 11 in 6 mice), *significantly different than V3 PAD latency, P < 0.05. F and G: amplitude and peak time of the early and late peaks in V3 PAD, *late peak amplitude significantly larger than early peak, or V3 peak times significantly different than drPAD peak time, same mice, P < 0.05. H: schematic of trisynaptic circuit underlying PAD with GABAergic inhibition of V3 neurons, explaining disinhibition of late PAD with gabazine. I and J: changes in V3 PAD and drPAD with gabazine, *significant change in V3 PAD (n = 16, from 6 mice) and drPAD (n = 21 from 5 mice) amplitude and duration, P < 0.05. DR, dorsal root; PAD, primary afferent depolarization.
V3 PAD had a slightly shorter latency (2.5 ms) compared with drPAD (3 ms), consistent with its putative position two rather than three synapses away from the afferents in the trisynaptic circuit (Fig. 5H). However, considering that spikes arise in V3 neurons slower with light compared with DR activation (right of Fig. 5E and see above), these results suggest that the V3 neurons may also have an even more direct action on afferents (monosynaptic). Furthermore, unlike with drPAD, there were often two clearly distinguishable events in V3 PAD: one fast event with a peak at ∼10 ms (early peak) and a second with a much later peak at ∼60 ms (late peak; Fig. 5, A, F, and G). The sensory-evoked PAD (drPAD) for these same afferents had a single peak at ∼30 ms, between the early and late V3 PAD peaks (Fig. 5, A and G).
Interestingly, the late peak of V3 PAD was blocked by gabazine (the portion which we refer to as V3 GABA PAD), as was most of the drPAD, whereas the early peak of V3 PAD was unaffected, and so the former is mediated by GABA, and the latter is not. As mentioned, GABAA receptor antagonists like gabazine should disinhibit V3 neurons, and make their actions on PAD more pronounced, providing a dual excitatory and inhibitory action of gabazine (Fig. 5H). Consistent with this, we found that as the peak of drPAD or late peak of V3 PAD dropped in gabazine, there was a slow emergence of a very long-lasting PAD with a duration much beyond the normal end of PAD (seconds; Fig. 5B). Overall, in gabazine, this left an early depolarization followed by a very long-lasting PAD, both in V3- and DR-evoked PAD. Thus, even the classic drPAD has a non-GABA-mediated component that is revealed by disinhibiting the associated spinal circuits. The slow emergence, small size, and late peak (at 500 ms) of the PAD remaining after blocking GABAA receptors may have led to it being neglected in past studies of drPAD (19, 22). However, this non-GABA-mediated component of PAD appears to be larger and more accessible to direct V3 neuron activation (Fig. 5A, V3 PAD), suggesting that circuits that activate V3 neurons independently of sensory input (e.g., CPGs) may use this V3 PAD. Even before adding gabazine, we see that the V3 PAD has a similar duration to the drPAD evoked by a strong DR stimulus adequate to activate cutaneous afferents (3xT), whereas the drPAD evoked by only lower-threshold DR stimulation that mainly only activates proprioceptive afferents (2xT) is of shorter duration (Fig. 5C), consistent with the V3 PAD perhaps involving a non-GABAergic PAD mediated by NMDA as cutaneous afferent stimulation evokes a longer PAD (22) that has recently been shown to be partly NMDA mediated (31).
Considering that axonal NMDA receptors contribute to PAD in cutaneous afferents (31) and that we find the action of V3 neurons may be faster than disynaptic, we next examined whether the glutamatergic V3 neurons themselves contact Ia proprioceptive afferents and contribute to a glutamate-mediated PAD in these afferents. Indeed, we found that neurobiotin-filled Ia afferents receive VGLUT2+ V3 innervation (Fig. 6A). These V3 contacts were at branch points where nodes are located in large myelinated branch points of these afferents (at 28.8% of nodes examined, n = 52) and at the terminals (at 12.7% of afferent terminal boutons, n = 55), both examined in intermediated laminae and similar to the GABAergic innervation of these myelinated afferents that we have detailed previously (19).
Figure 6.
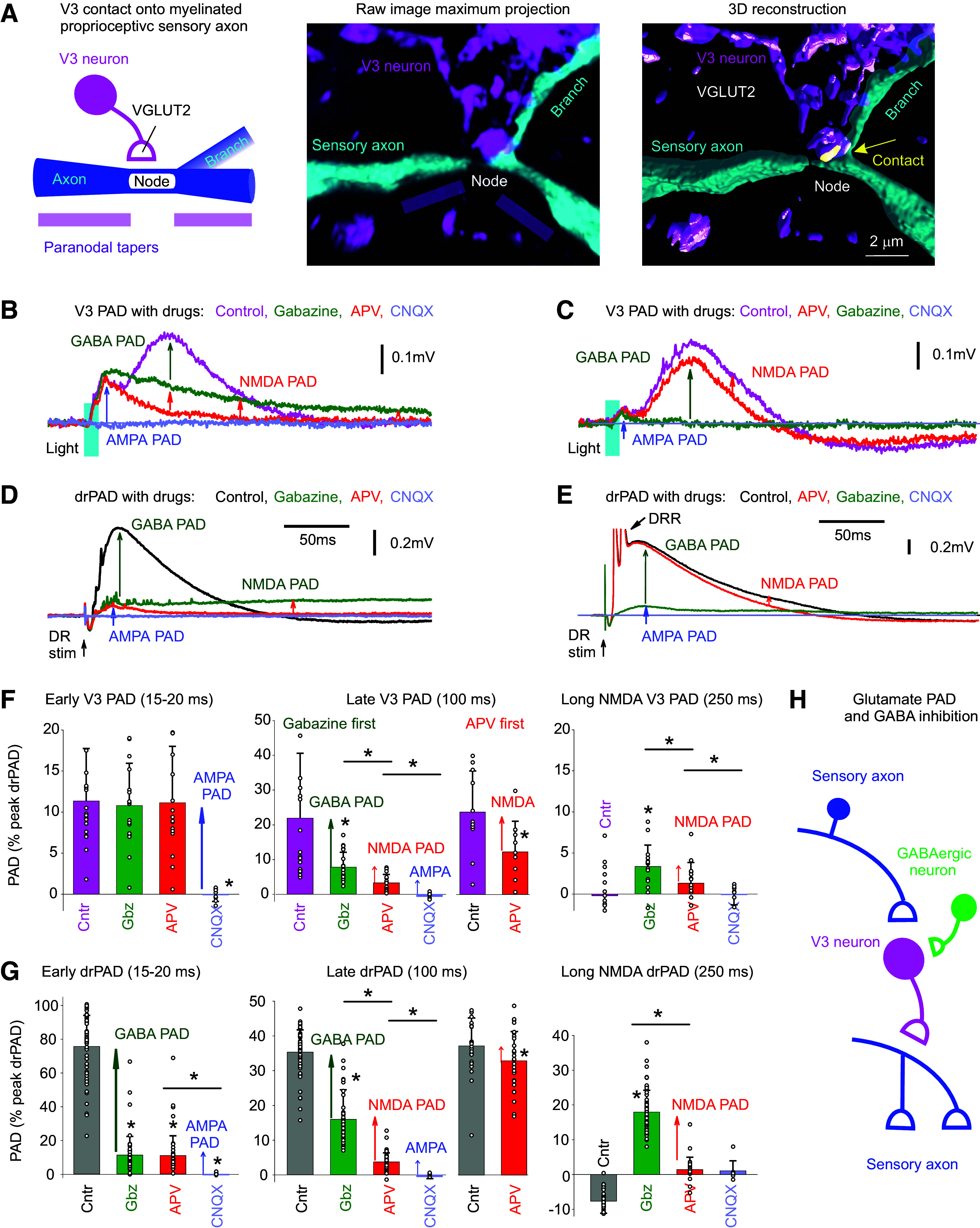
V3 neurons evoke a glutamate-mediated PAD, independent of GABA, by direct innervation of afferents. A: V3 neuron contact (VGLUT2+) on large myelinated Ia afferent branch in the intermediate spinal cord, at a branch point where the node of Ranvier is located (node also identified by the paranodal taper; afferent filled with neurobiotin). V3 neurons labeled with EYFP in Sim1//ChR2-EYFP mouse. Contact shown in yellow computed from 3-D reconstruction on right. B: PAD evoked by a brief light pulse (10 ms pulse, λ = 447 nm laser, 0.7 mW/mm2, 3xT, laser aligned as in Fig. 4B) in Sim1//ChR2-EYFP mouse, and GABA PAD, NMDA PAD, and AMPA PAD components estimated by the change with sequential application of gabazine, APV, and CNQX, respectively (50 µM each). Recorded in S3 DR by grease gap (DRPs). C: same as B, but APV applied first, with sequential application of APV, gabazine, and CNQX. D and E: same as B and C, but for drPAD evoked by DR stimulation (Ca1 DR 0.1 ms, 2xT). F: group averages of V3 PAD early peak (at 15–20 ms), late peak (at 100 ms), and long-lasting NMDA PAD (at 250 ms), with sequential application of gabazine, APV CNQX (gabazine first condition), or APV first (only detailed for late V3 PAD since early PAD unaffected, not shown). *Significant PAD reduction with drug application, P < 0.05, n = 20 gabazine first recordings (of which 6 also had CNQX), and n = 11 APV first recordings, in S4 and S3 DR combined from 5 mice each. G: group averages of drPAD amplitudes at same times as measured for V3 PAD in F, for comparison. *Significant PAD reduction with drug application, P < 0.05, n = 20 gabazine first recordings, and n = 28 APV first recordings from 10 mice each. H: schematic of the putative disynaptic V3 neuron circuit mediating PAD independently of GABA, but with GABA tonically inhibiting V3 neurons, leading to a disinhibition of this glutamate-mediated PAD with gabazine. DR, dorsal root; NMDA, N-methyl-d-aspartate; PAD, primary afferent depolarization.
Consistent with anatomical connections of V3 neurons to afferents, we found that the NMDA antagonist APV blocked the long-lasting component of both V3 PAD (Fig. 6, B, C, and F) and drPAD (Fig. 6, D, E, and G), indicating the presence of an APV-sensitive PAD mediated by NMDA (termed NMDA PAD). This NMDA PAD was best seen after applying GABA antagonists (gabazine first, Fig. 6, B, D, F, and G), which again disinhibited the spinal cord circuits and so amplified the remaining PAD. However, APV also directly reduced PAD without gabazine present (Fig. 6, C and E–G), indicating some resting-state NMDA PAD. Together these results indicate the existence of an NMDA-mediated PAD that is independent of GABA and unmasked during the disinhibition of the spinal cord by gabazine. This NMDA PAD had a long-onset latency (12.4 ± 7.1 ms) and lasted for up to 3 s (Fig. 5, I and J).
After blocking GABA PAD with gabazine and NMDA PAD with APV, there still remained an early rising short PAD (Fig. 6, B–G), corresponding to the early peak in the V3 PAD detailed above. This is likely also mediated by glutamatergic V3 neuron contacts on afferents, based on its very short latency (relative to the V3 spike latency), and its sensitivity to the AMPA/kainate receptor blocker CNQX [Fig. 6, B and C, consistent with it being mediated by AMPA/kainate receptors (abbreviated AMPA PAD)]. Because CNQX also blocks afferent transmission, this PAD could alternatively be mediated by another transmitter like glycine or ACh, though antagonists to these receptors (strychnine or tubocurarine) (63) did not reduce this early PAD (not shown; n = 4/4). Thus, tentatively we call this AMPA PAD. AMPA PAD is seen in V3 PAD (Fig. 6, B and C), as well as drPAD (Fig. 6, D and E), but the latter AMPA drPAD is small (Fig. 6, D and E) and was likely previously missed in studies of drPAD. Taken together, AMPA PAD and NMDA PAD are likely mediated by a common glutamate innervation of afferents, in a minimally disynaptic circuit depicted in Fig. 6H.
To confirm that functional NMDA receptors were on the afferents in the spinal cord, we applied NMDA (100 µM for 2 min) to the synaptically isolated afferents (recorded as usual from central end of DRs close to the cord, as in Fig. 3B, but in the presence of 2 μM TTX, 50 μM CNQX, 50 μM gabazine, and 5 μM strychnine, to block spikes, glutamate, GABA, and glycine respectively), and indeed this caused a significant depolarization (0.78 ± 0.174 mV DRP, P < 0.05, n = 6 roots from 4 mice, not shown). Furthermore, when we applied TTX (1 μM), APV (50 μM), and gabazine (50 μM) to block all spike-mediated circuit activity, but leaving AMPA receptors available, there still remained very small V3 PAD (< 1% of pre-drug PAD) that was blocked by CNQX, confirming the presence of an AMPA PAD induced by direct connections from V3 neurons to afferents, in this case, caused by ChR2-induced release of glutamate from V3 terminals on the afferents, unaided by sodium spikes (n = 2/2 similar). This also indicates that the vast majority of the action of ChR2 in V3 neurons is via sodium spikes that propagate from the cell body to the terminals (>99%), and not terminal ChR2 action.
Radiating Intersegmental PAD Caused by Long Propriospinal V3 Axons
V3 neurons are propriospinal and commissural neurons with long axons that ascend and descend the spinal cord in the ventral and ventrolateral white matter, as we confirmed in Fig. 7A (43, 64). Thus, these long projecting axons may well help explain the characteristic radiating nature of PAD, where one nerve or dorsal root stimulation can activate PAD in many muscle afferents, including in afferents many segments away and across the midline (22), though afferents themselves also project in the dorsal columns across segments, but do not cross the midline (65). In contrast, GABAergic GAD2+ neurons involved in PAD are small interneurons that cannot explain this radiating PAD (15, 23). To examine the details of how V3 neurons activate PAD across spinal segments, we recorded PAD from a given dorsal root while selectively activating V3 neurons at varying spinal segments on one side of the cord, by focal light application in Sim1//ChR2 mice. Overall, the GABA-mediated late peak of V3 PAD was best evoked by light applied at or below, but not above, the DR where PAD was measured, and the peak was equally large ipsilateral and contralateral to the light (Fig. 7, B, D, and E). This is consistent with GABA PAD being mediated by ascending V3 propriospinal tracts in the sacral cord, as depicted by a single ascending axon in the schematic of Fig. 7A. As ChR2 is expressed all along the propriospinal V3 axons as well as at the cell body (seen with EYFP expression in Sim1//ChR2-EYFP mice; not shown), the finding that light applied above a given root does not evoke a large V3 PAD in the root indicates that spikes evoked in ascending propriospinal axons by light do not travel antidromically back down to parent V3 neurons at or below the root, otherwise there should be a similar PAD with light above or below the root. This lack of antidromic spike conduction in ascending propriospinal axons is consistent with our observation that a rostral activation of these V3 axons produces a spike propagation failure in the antidromic direction, as detailed above (Fig. 4E).
Figure 7.
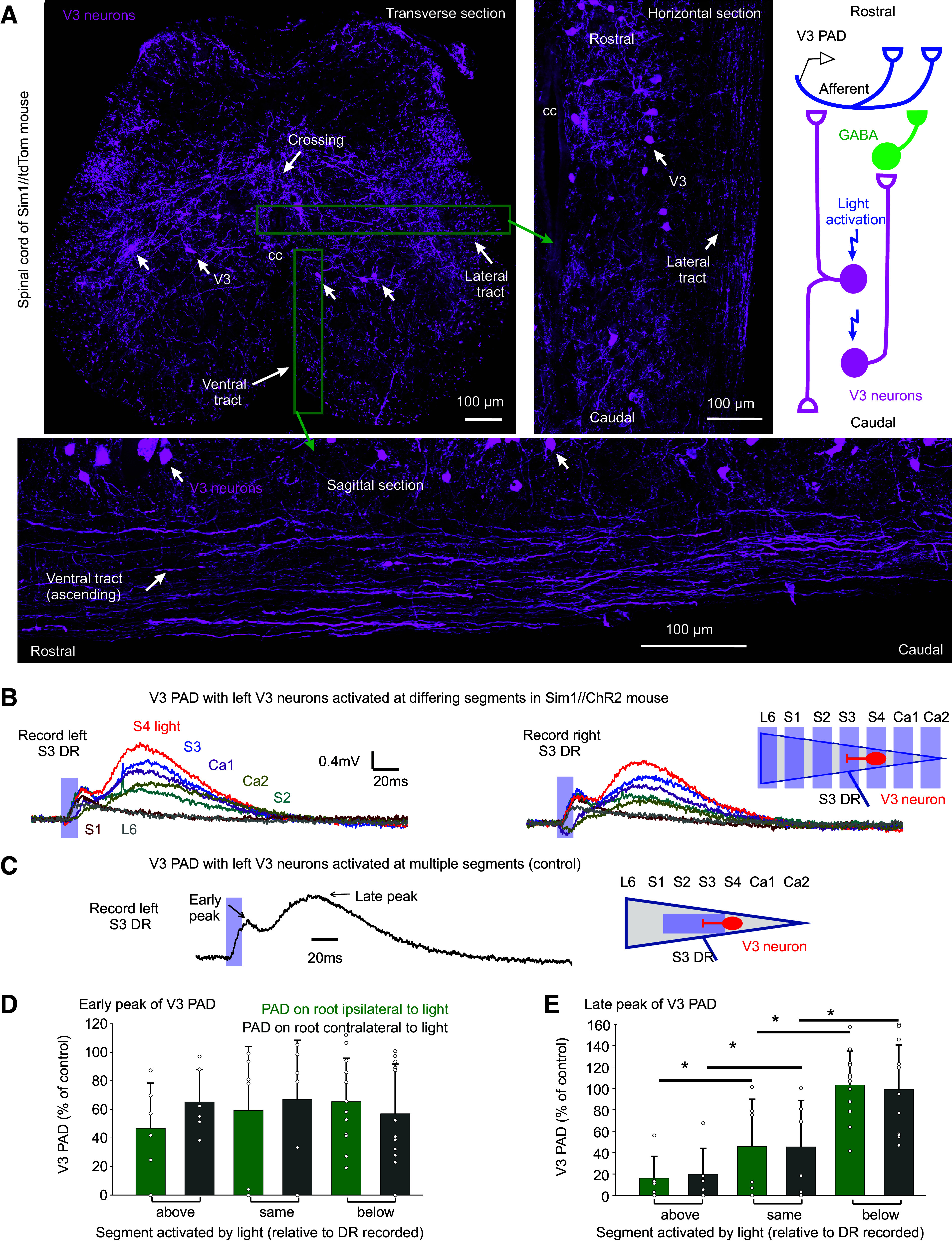
Intersegmental propriospinal projections mediating PAD. A: V3 neurons in sacral S3 spinal cord of Sim1//tdTom mice shown in transverse (top left, repeated from Fig. 4A), sagittal (bottom), and horizontal (right) planes (the latter two at approximate locations indicated by green boxes), showing axonal tracts in the white matter formed by V3 neurons. Also, some V3 axons crossed the midline, as indicated. Schematic of proposed axonal projections of V3 neurons, with V3 neurons involved in GABA PAD (late peak of V3 PAD) mainly ascending from the lower sacral V3 neurons, and V3 neurons involved in glutamatergic PAD (AMPA PAD, early peak) both ascending and descending. B and C: V3 PAD in sacral S3 DRs evoked by light focused on the left lateral face of the cord in only one segment at a time (B, by turning the narrow columnated beam of Fig. 4B across the cord, as shown; 10 ms pulse, λ = 447 nm laser, 0.7 mW/mm2, 3xT). V3 PAD was similar in left and right S3 DRs (commissural). Note that S3 PAD evoked in the S3 DR by applying light at S4 (red) was largest, sometimes even larger than PAD evoked by light applied across multiple segments at and above the S3 level (C; with columnated light turned to align with cord over S2–S3, as was usual arrangement elsewhere; Fig. 4B), the latter used as our control PAD with which to normalized responses. D and E: normalized group averages of early and late peaks of V3 PAD, grouped by whether the light was applied above (rostral to), at, or below (caudal to) the segment of the root where PAD was recorded (S3 or S4 DRs). *Significant difference with applied light above or below root segmental level, P < 0.05, n = 6–12 roots per condition. DR, dorsal root; PAD, primary afferent depolarization.
Interestingly, the glutamate-mediated early peak of V3 PAD in a given root was equally large when we activated V3 neurons with light above, at, or below the segment of the root (Fig. 7, B and D), suggesting that both ascending and descending V3 neurons cause this glutamate-mediated PAD, and consistent with a differing underlying circuit, as depicted by a bipolar V3 neuron on Fig. 7A schematic. In contrast, direct activation of GABAergic GAD2 neurons in GAD2//ChR2 mice led to a PAD that was mainly only large in roots directly under the light application site (not shown; n = 4/4), consistent with the small spatial extent of these neurons (<1 mm) (18).
V3 Neurons Facilitate Proprioceptive Sensory Transmission to Motoneurons
We next examined the functional action of V3 neurons on sensory transmission. Recently, we have shown that PAD acts to directly facilitate spike transmission in afferents, including both antidromically and orthodromically propagated spikes (19, 22). Considering that our ArchT inhibition of V3 neurons demonstrated that at least half of drPAD is mediated through V3 action, we returned to this ArchT data to examine whether V3 neurons also contribute to spike propagation (Fig. 3A). For this, we examined spikes produced in afferents by drPAD, which propagate both to the motoneurons and antidromically out the DR, the latter which we measured (termed the dorsal root reflex, DRR, Figs. 8, A and B, and 3A) (22). These spikes were markedly reduced in incidence (Fig. 8B) and on average reduced by half with the ArchT inhibition of V3 neurons (Fig. 8C), consistent with a similar reduction in drPAD itself. Thus, V3 neurons facilitate spike transmission in sensory axons, likely via their depolarizing action (PAD) helping sodium channel spike initiation, similar to the general function of PAD (19).
Figure 8.
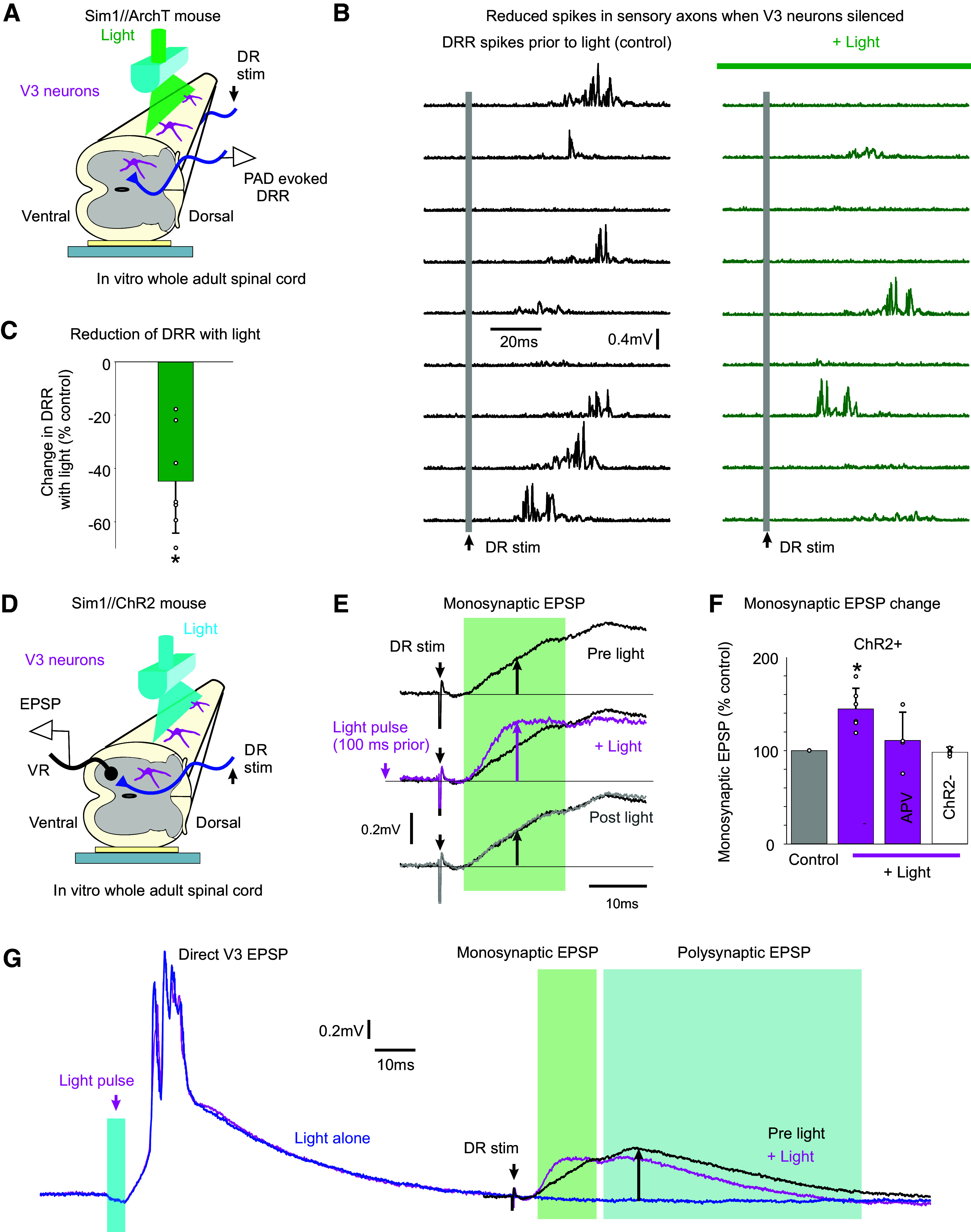
V3 neurons increase spike transmission in sensory afferents. A and B: dorsal root reflex (DRR) recorded in S4 DR from stimulating the Ca1 DR (0.1 ms, 2xT; raster plot of 9 trials at 0.1 Hz) in Sim1//ArchT mice, before and after silencing V3 neurons with light (532 nm, 5 mW/mm2). The drPAD evoked by this stimulation is shown in Fig. 3A (same recording), where the rising phase of PAD produced the DRR, but here the DRR is shown in isolation by removing the slow PAD response with a 100 Hz high-pass filter and then rectifying. C: change in integrated area under rectified DRR with silencing of V3 neurons, *significant change, P < 0.05, n = 8 roots from 3 mice. D and E: motoneuron monosynaptic EPSP evoked by dorsal root stimulation (S4 DR, 0.1 ms pulse, 1.1xT) measured with grease-gap recordings from ventral root (S4 VR, giving composite EPSP of motoneuron pool; top trace in E). Light activation of the V3 neurons (10 ms pulse, λ = 447 nm laser, 0.7 mW/mm2, 3xT, laser aligned as in Fig. 4B) applied 100 ms before the same DR stimulation increased the resulting monosynaptic EPSP (magenta trace in E). F: group averages of change in monosynaptic EPSP with light, both without (as in E) and with APV (50 µM) in the bath. *Significant increase, P < 0.05, n = 6 roots without APV, n = 4 roots with APV present, from 4 mice each. G: same data as in E, but on longer time base to show direct response to the V3 activation (left) and decrease in polysynaptic reflex evoked by the DR stimulation (right, at upward arrow). PAD, primary afferent depolarization.
We also examined changes in orthodromic afferent spike transmission induced by V3 neurons, by examining the monosynaptic transmission of Ia afferents to motoneurons, with recordings from motoneurons while evoking monosynaptic EPSPs with DR stimulation pulses (Fig. 8, D and E; composite EPSP recorded on ventral root). When we activated V3 neurons optogenetically in Sim1//ChR2 mice to produce a V3 PAD, the monosynaptic EPSPs in motoneurons were as expected increased during the V3 PAD (Fig. 8, E and F). Thus, the V3 PAD increases proprioceptive afferent transmission to motoneurons. This facilitation was somewhat reduced when APV was applied to block the NMDA receptors and associated NMDA PAD, though further experiments are needed to confirm this (not significant).
These monosynaptic EPSP experiments were somewhat confounded by the strong postsynaptic EPSPs that V3 neurons themselves directly produced in motoneurons, which lasted ∼50–100 ms (Fig. 8G) (44). Thus, to avoid postsynaptic actions of these long motoneuron EPSPs, we tested the DR-evoked monosynaptic EPSP at 100 ms after the light application, a time when V3 PAD was still present (Fig. 5C, PAD lasts ∼150 ms), but the V3-evoked motoneuron EPSP had subsided. Thus, the facilitation of the monosynaptic EPSP by V3 neurons at this time (Fig. 8, E and F) is likely from the V3 neuron actions on the sensory axons, and not its postsynaptic action on motoneurons. Nevertheless, the polysynaptic pathways directly activated by V3 neurons likely became refractory to subsequent reactivation, as we observed that the DR-evoked polysynaptic EPSP decreased with light (Fig. 8G), though we did not quantify this more complex process. To complicate matters further, possibly V3-evoked PAD and DRRs contribute to the polysynaptic EPSPs evoked by light, with the latter leading to postactivation depression of the monosynaptic afferent transmission (19), though this remains to be explored.
Increased Glutamate PAD and Decreased GABA PAD after SCI
Considering that glutamate-dependent PAD is unmasked by bringing up the excitability of spinal circuits with gabazine, we wondered whether the marked increase in spinal cord excitability with chronic spinal cord injury (SCI) (44, 66) also unmasked glutamate-mediated PAD. For this, we examined Sim1//ChR2 mice with an S2 sacral transection ∼1.5–3 mo previously and measured NMDA, AMPA, and GABA PAD components with drug applications (APV, gabazine, and CNQX-sensitive components, respectively; Fig. 9, A–D). Interestingly, the absolute size of the drPAD did not change with SCI, whereas the glutamate-dependent early peak of the V3 PAD markedly increased with SCI (Fig. 9E). The late peak of the V3 PAD tended also to increase, though not significantly due to high variability in absolute PAD sizes between animals and dorsal roots (Fig. 9E). Furthermore, the balance of GABA and glutamate PAD components shifted to increased glutamate contribution with SCI, as seen by normalizing these components as a fraction of the peak PAD recorded for each root (pre-drug drPAD; Fig. 9G). For the V3 PAD evoked by light, its APV-sensitive NMDA PAD component made up a larger proportion of the total PAD after SCI (Fig. 9, A–C and G). This occurred regardless of whether (Fig. 9B) or not (Fig. 9D) we added gabazine before APV, indicating that there is a large NMDA PAD in the resting cord with SCI (without gabazine), unlike without injury, suggesting increased direct V3 action on afferents. In contrast, NMDA PAD made up only a minor faction of V3 PAD without injury, even when measured after disinhibition with gabazine (Fig. 9, A and G). AMPA PAD likewise made up a larger fraction of the total PAD after SCI (Fig. 9, A–D, and G; measured after NMDA and GABA receptor blockade) and had a longer duration. In contrast, the gabazine-sensitive GABA V3 PAD was unchanged after SCI, though it tended to increase, consistent with the larger overall V3 PAD after SCI.
Figure 9.
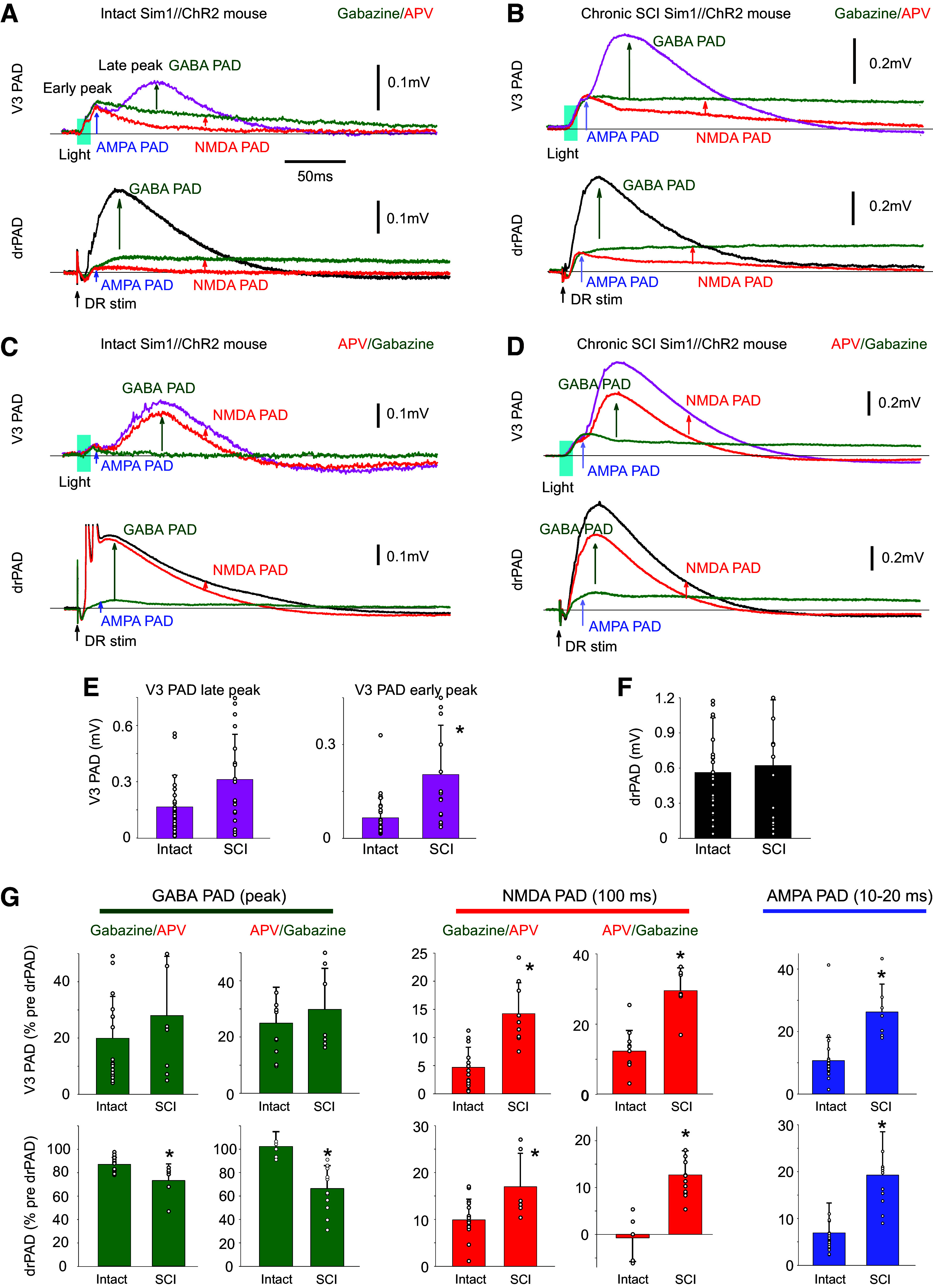
Chronic spinal cord injury (SCI) increases NMDA and AMPA PAD while reducing GABA PAD. A: changes in V3 PAD and drPAD recorded in S3 DR (as detailed in Fig. 6, B and D) with sequential application of gabazine and APV in Sim1//ChR2 mouse (50 µM each). GABA and NMDA PAD measured by drug-induced reductions, and AMPA PAD measured as remaining PAD in both drugs. V3 PAD and drPAD evoked by a brief light or DR stimulation, respectively (as detailed in Fig. 6, B and D). B: same as A, but in Sim1//ChR2 mouse with an S2 spinal transection 1 mo previously (chronic SCI). C and D: same as A and B, but with APV added before gabazine to estimated resting state NMDA PAD. E and F: group averages of changes in PAD with SCI, with significant change (*) in early peak of V3 PAD, but not the late peak of V3 PAD or drPAD, P < 0.05, n = 31 and 15 from 6 control and 6 SCI mice. G: group averages of changes in GABA PAD (at peak), NMDA PAD (recorded at 100 ms latency), and AMPA PAD (recorded at peak between 10 and 20 ms) with SCI, *significant change, P < 0.05, n = 20 and 6–9 roots tested with gabazine given first, without and with SCI, respectively, and n = 12 and 6–9 roots with APV given first, without and with SCI, respectively; from 4–6 mice each condition. All changes with drugs normalized by predrug drPAD peak. NMDA, N-methyl-d-aspartate; PAD, primary afferent depolarization.
The glutamate and GABA components of the drPAD showed similar changes to the V3 PAD (Fig. 9, A–D). In this case, the NMDA and AMPA drPAD components made up a larger and longer-lasting proportion of the total drPAD after SCI (Fig. 9G). Even the small or absent NMDA PAD measured in the uninjured animal (without disinhibiting cord with gabazine) increased with SCI (Fig. 9G), again consistent with SCI leading to increased excitability of the V3 circuits that directly produce PAD. In contrast, the GABA drPAD decreased with SCI from >85% of the total peak drPAD without injury, to 60–70% of the total drPAD (Fig. 9G).
DISCUSSION
Our results demonstrate that propriospinal V3 neurons play a major role in depolarizing group Ia proprioceptive sensory afferents across many spinal segments, facilitating afferent spike propagation, and ultimately modulating proprioceptive sensory transmission to motoneurons. They do this by both GABA-dependent and GABA-independent pathways. Specifically, V3 neurons appear to serve as the first-order neurons in the classic trisynaptic circuit that causes GABA-mediated PAD in Ia afferents in response to proprioceptive sensory stimulation (23, 35), as silencing V3 neurons eliminates a large part of this PAD (drPAD), and these neurons have the appropriate anatomical and functional connections. That is, V3 neurons receive extensive sensory innervation, including proprioceptive Ia afferent input (and likely many other afferents), project over many segments, and in turn, activate GAD2+ GABAergic neurons that are known to form axoaxonic connections back onto Ia sensory afferents (19). This circuit modulates sensory transmission to motoneurons, as evidenced by the action of PAD evoked by optogenetic activation of V3 neurons (V3 PAD), and importantly acts over many segments, explaining the radiating nature of PAD and its reflex modulation (21–23, 28, 29). Although we show that V3 neurons induce PAD in single Ia afferents (Fig. 1), we did not specifically examine other afferents. Thus, part of the composite V3 PAD we measured from dorsal roots may arise from other low-threshold afferents (LTMRs, i.e., cutaneous afferents), in addition to Ia afferents. Glutamatergic CCK+ interneurons serve as first-order neurons in the GABA-dependent PAD evoked in LTMRs (31), but unlike V3 neurons, are not propriospinal neurons, having only a limited extent of synaptic contacts confined to the LTMR-recipient zone (67). Thus, it remains to be determined whether the radiating intersegmental nature of sensory-evoked PAD observed in cutaneous afferents (68) is also mediated by a propriospinal V3-like neuron, similar to sensory-evoked PAD in proprioceptive afferents.
Furthermore, we unexpectedly found that V3 neurons cause a pronounced GABA-independent PAD mediated by NMDA and AMPA/kainate receptors (NMDA and AMPA PAD), at least partly mediated by direct connections from V3 neurons to Ia afferents, with NMDA receptors on the afferents. This enables sensory stimulation to disynaptically activate a PAD, faster than the classic trisynaptic circuit that mediates GABA PAD. NMDA receptors and associated RNA are known to be expressed in low- and high-threshold mechanoreceptors and proprioceptors, including Ia afferents (69, 70). Presynaptic NMDA receptors (preNMDA) produce PAD in LTMR afferents (31), and these preNMDA receptors have been shown in other systems to facilitate synaptic transmission (70). This NMDA PAD in LTMRs is mediated by small dorsally oriented glutamatergic VGLUT3+ interneurons, and thus are again different from the large propriospinal V3 neurons that we observe mediate NMDA PAD (31, 71). A similar pre-NMDA arrangement likely exists in proprioceptive afferents as we know these afferents are innervated by V3 neurons that cause a direct fast glutamate-dependent PAD that can be mimicked by NMDA application in synaptic isolation. This could contribute to the increased sensory transmission we observed with V3 activation (which was somewhat APV-sensitive). Alternatively, NMDA receptors on neurons in polysynaptic pathways could also contribute to the NMDA PAD we observed. Indeed, likely, all forms of PAD (GABA, NMDA, and AMPA PAD) are additionally mediated by longer latency polysynaptic pathways than the minimally trisynaptic and disynaptic pathways that we describe here. For example, the complex central pattern generator (CPG) circuits that underlie walking and the corticospinal tracts involved in grasping are known to evoke PAD (4, 13, 14, 34, 39), and thus long-loop circuits may evoke the differing forms of PAD via V3 neurons, and could partly explain the long-lasting NMDA PAD (via the CPG). Furthermore, we cannot rule out fast AMPA PAD triggering spikes in afferents (DRRs) that in turn trigger further GABA PAD reverberatively in other afferents, perhaps explaining the delay in the peak of the GABA component in the V3 PAD.
Under resting conditions in vitro, NMDA and AMPA drPAD evoked by sensory stimulation is not very large, at least not the many-second-long component of NMDA PAD, explaining why it has not been well studied in the past. However, direct activation of V3 neurons evokes a more pronounced NMDA and AMPA PAD, suggesting that any system, including the CPG that engages the V3 neurons (43) may also evoke pronounced GABA-independent NMDA and AMPA PAD. Indeed, with increased spinal cord excitability, caused by either chronic spinal cord injury or blocking GABA receptors with gabazine, both NMDA and AMPA PAD become much more important. Gabazine likely causes disinhibition of neuronal circuits in the cord that drive the NMDA PAD (including V3 neurons), by blocking GABA receptors on these neurons, though we cannot rule out the dose of gabazine that we used also blocking glycine receptors (72), which would further disinhibit these neurons. Whether or not such glutamate-mediated PAD is important in awake animals remains an open question, though it may be best activated via cutaneous stimulation, as it seems to be associated with cutaneous evoked PAD (with stronger stimulation), and similar NMDA-associated PAD has been reported in other in vitro systems in their resting state (22, 31, 47). Furthermore, it may well be that during complex movements like locomotion, where PAD is increased by the CPG, NMDA PAD may play a larger role.
The dual action of GABA on PAD that we uncovered is particularly relevant to understanding studies of PAD that use GABA blockers. That is, GABAA receptors on the V3 neuron inhibit these neurons and their PAD action, whereas the GABAA receptors on afferents are indirectly activated by V3 neuron connection to GABAergic neurons. Thus, in our and other previous studies (16, 19, 22) where GABAA receptor blockers are used to quantify GABA’s role in PAD and sensory transmission, there is both a direct block of GABA PAD and a disinhibition of V3 neurons, the later which enhances NMDA and AMPA PAD. Thus, the size of the PAD mediated by afferent GABAA receptors or its underlying functional action may be underestimated. Perhaps a better estimate of GABA PAD is obtained by first blocking NMDA receptors (and NMDA PAD), before blocking GABAA receptors, as we previously also noted when evaluating the actions of GABAA antagonists on monosynaptic transmission (19).
Although GABA-mediated PAD (GABA PAD) is a major focus of this paper, GABA PAD can be viewed as a proxy measure for more general GABAergic action on afferents, regardless of whether that action produces PAD. That is, axoaxonic innervation of proprioceptive afferents by GABAergic neurons has two opposing actions: causing presynaptic inhibition of sensory transmission by activating GABAB receptors at afferent terminals that do not produce PAD and causing facilitation of sensory action conduction by activating GABAA receptors at or near nodes of Ranvier that cause PAD, which in turn assists local sodium spike conduction at the nodes (called nodal facilitations) (19). Thus, our finding that V3 neurons mediate GABA PAD on afferents, suggests that they may also activate GABAB receptors that cause presynaptic inhibition, though this remains to be examined. So far, we have only shown that V3 neuron action facilitates monosynaptic sensory transmission to motoneurons, consistent with them at least causing nodal facilitation. Whether they also produce GABAB-mediated actions, including the well-known rate-dependent depression (RDD) of the monosynaptic reflex (73, 74), remains an open question. Furthermore, the extent to which our results on GABAergic and glutamatergic control of PAD by sacral V3 neurons generalizes to lumbar V3 neurons is unknown. It is likely that because the sacral cord controls the tail and axial muscles that make left-right movements, the sacral V3 circuitry most closely resembles lumbar V3 circuitry controlling trunk axial muscles or lumbar V3 circuitry controlling flexor-extensor movement, though this remains to be investigated. We do know that the properties of sacral V3 neurons that we observe closely resemble properties of commissural propriospinal neurons in the lumbar cord (37, 38), including receiving monosynaptic sensory input, having commissural and segmental projections, and an intermediate laminae location.
Our demonstration that the PAD evoked by V3 neurons changes to being more glutamate dominated after chronic spinal transection suggests that descending V3 innervation from the higher spinal or supraspinal tracts (including V3 tracts) regulates V3 neuron action, consistent with the well-established finding that descending tracts modulate PAD in general (23, 75). Interestingly, we found that at least in the sacral cord, it was ascending intersegmental propriospinal V3 neurons that best evoked GABA PAD, suggesting less dependence on descending tracts. In contrast, the early glutamate-dependent PAD was evoked equally well by ascending or descending intersegmental V3 neurons, suggesting a differing underlying population mediating GABA PAD compared with AMPA PAD, with the latter more dependent on descending tracts (64). With chronic spinal cord injury, the AMPA and NMDA PAD increased markedly, whereas the GABA PAD decreased or remained unchanged, again suggesting different circuits underlying these types of PAD. Interestingly, the overall drPAD did not change much with injury, just the balance of GABA and glutamate-dependent PAD, consistent with previous observations of not much change in PAD with SCI, except for a higher sensory threshold to be evoked (76). The latter may be explained by the increased NMDA PAD, which seems to be evoked by higher threshold cutaneous afferents, compared with low-threshold Ia afferents, as detailed above. The functional outcomes of the increased NMDA PAD with SCI remain to the determined, though it may be a compensation for the loss of spinal GABAergic neurons with SCI (77).
Importantly, the long propriospinal projections of V3 neurons (64) allow local sensory or V3 neurons stimulation to evoke PAD in afferents in distant segments and across the midline, thus helping to explain the remarkable radiating nature of PAD (21, 68). We, however, cannot rule out PAD also being produced by coupled V3 neurons or other neurons connected in microcircuits contributing to the radiating long-distant action of PAD across multiple segments. This is in contrast to the small GABAergic neurons involved in PAD that only locally affect PAD (15, 23).
Furthermore, the intrinsic properties of V3 neurons, including Na PICs, allow them to respond with very long plateau-like depolarizations following a brief activation, thus explaining the long-lasting nature of PAD and its potent action on modulating sensory transmission over long time periods. Interestingly, the somewhat TTX-resistant nature of the sodium channels in V3 neurons suggests that the previous observation that an NMDA PAD triggered by high-threshold afferents (C fiber) is somewhat TTX resistant is not just explained by TTX-resistant channels in C fibers themselves (22, 47). Instead, it is likely also explained by TTX-resistant sodium channels in V3 neurons and the direct connections of V3 neurons to afferents that mediate NMDA PAD. This implies that the previously supposed microcircuit underlying this C fiber-mediated PAD may not actually be a microcircuit, but instead, a large-scale circuit where PAD is evoked by hypothetical C fiber inputs onto V3 neurons that in turn monosynaptically depolarize large myelinated sensory afferents, like Ia afferents. This is consistent with the intersegmental nature of this TTX-resistant PAD, occurring in one dorsal root in response to stimulation of another, and likely mediated by the propriospinal axons of V3 neurons (22, 47). However, the actual details of C fibers innervation of V3 neurons and the TTX-resistant sodium channels in V3 neurons remain to be explored to confirm these ideas. Taken together, these properties of V3 neurons allow them to form wide-ranging spatial temporal integration of their sensory and other inputs (CPG) to control PAD and sensory transmission.
V3 neurons have previously mainly been studied in the context of locomotion and general motoneuron function (43), and so our finding that they play a major role in sensory transmission needs to be reconciled with their motor functions. The underlying commonality may be that V3 neurons act like neuromodulators with their slow integrator properties, turning on and off sensory and motor systems during differing tasks. For example, we previously found that V3 neurons cause direct sustained depolarizations of motoneurons (see also Ref. 48) and seem to be crucial in turning on sustained locomotor-like activity and spasms, especially after spinal cord injury in mice (44). Furthermore, in zebrafish, V3-like neurons steadily depolarize during bouts of locomotion and are not rhythmically active (49). Instead, they serve to amplify and prolonging motor output, consistent with them causing a broad depolarization of the many CPG neurons and motoneurons they innervate, and thus acting as high-level neuromodulators that amplify and integrate activity, rather than controlling fine rhythm details (49). Although the latter study is in fish swimming and our studies are in the sacral cord that controls the tail during both walking and swimming, these suggested neuromodulatory actions of V3 neurons likely generalize to the lumbar control of hindlimb walking, though this remains to be directly tested. We do know that the sacral cord has a similar circuitry to the lumbar cord, with similar V3 neurons and classical circuits including monosynaptic Ia transmission, Ia reciprocal inhibitory interneurons (IaINs), and Renshaw cells, though likely most closely resembling axial trunk control circuits with extensive crossed actions (19, 43, 78). More generally, the long ascending and descending propriospinal tracts of V3 neurons have been suggested to be involved in coordinating reciprocal flexor-extensor and left-right multijoint muscle activity during locomotion, though they somehow do this without controlling the details of the movements (43, 49). That is, V3 neurons and more generally other propriospinal neurons are not normally needed for basic locomotor rhythm generation (i.e., silencing V3s does not stop the CPG or walking) and are often not rhythmically driven by the CPG (11). Instead, they may be involved in the high-level task of choosing which general motor action occurs, whether it is reciprocal stepping, hopping, or swimming (11, 43). With this view that V3 neurons act like neuromodulators that ready the motor system for movement, it makes sense that V3 neurons also modulate sensory neurons, readying sensory feedback during movement. Whether subpopulations of V3 neurons target these differing sensorimotor actions remains an open question (45, 48, 59), though this would allow flexibility in controlling sensory function during movement. In retrospect, the view that V3 neurons broadly supervise sensorimotor systems, taken together with V3 neurons causing PAD, suggests that PAD is just one of the many actions of V3 neurons involved in coordinating complex movements, and PAD is not part of an isolated system regulating sensory transmission.
DATA AVAILABILITY
Data will be made available upon reasonable request.
GRANTS
This research was supported by the Canadian Institutes of Health Research (MOP 14697 and PJT 165823 to D.J.B.) and the US National Institutes of Health (NIH, R01NS47567, to D.J.B. and K.F.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.L., K.H., S.B., K.F., M.A.G., Y.L., A.M.L-O., K.K.F., and D.J.B. conceived and designed research; S.L., K.H., S.B., A.K., K.F., Y.L., A.M.L-O., K.K.F., and D.J.B. performed experiments; S.L., K.H., S.B., A.K., K.F., Y.L., A.M.L-O., K.K.F., and D.J.B. analyzed data; S.L., K.H., S.B., A.K., Y.L., A.M.L-O., K.K.F., and D.J.B. interpreted results of experiments; S.L., K.H., S.B., A.K., Y.L., A.M.L-O., and D.J.B. prepared figures; S.L., K.H., and D.J.B. drafted manuscript; S.L., K.F., M.A.G., Y.L., A.M.L-O., K.K.F., and D.J.B. edited and revised manuscript; S.L., S.B., A.K., K.F., M.A.G., Y.L., A.M.L-O., K.K.F., and D.J.B. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Leo Sanelli for technical assistance. We thank Dr. Elzbieta Jankowska for many suggestions on the manuscripts.
Present address of Y. Li: Dept. of Physiology, Emory University, Atlanta, GA 30322, USA.
REFERENCES
- 1. Bennett DJ, De Serres SJ, Stein RB. Gain of the triceps surae stretch reflex in decerebrate and spinal cats during postural and locomotor activities. J Physiol 496: 837–850, 1996. doi: 10.1113/jphysiol.1996.sp021731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Capaday C, Stein RB. Amplitude modulation of the soleus H-reflex in the human during walking and standing. J Neurosci 6: 1308–1313, 1986. doi: 10.1523/JNEUROSCI.06-05-01308.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bennett DJ, Gorassini M, Prochazka A. Catching a ball: contributions of intrinsic muscle stiffness, reflexes, and higher order responses. Can J Physiol Pharmacol 72: 525–534, 1994. doi: 10.1139/y94-076. [DOI] [PubMed] [Google Scholar]
- 4. Rossignol S, Dubuc R, Gossard J-P. Dynamic sensorimotor interactions in locomotion. Physiol Rev 86: 89–154, 2006. doi: 10.1152/physrev.00028.2005. [DOI] [PubMed] [Google Scholar]
- 5. Andrechek ER, Hardy WR, Girgis-Gabardo AA, Perry RLS, Butler R, Graham FL, Kahn RC, Rudnicki MA, Muller WJ. ErbB2 is required for muscle spindle and myoblast cell survival. Mol Cell Biol 22: 4714–4722, 2002. doi: 10.1128/MCB.22.13.4714-4722.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bouyer LJG, Rossignol S. Contribution of cutaneous inputs from the hindpaw to the control of locomotion. II. Spinal cats. J Neurophysiol 90: 3640–3653, 2003. doi: 10.1152/jn.00497.2003. [DOI] [PubMed] [Google Scholar]
- 7. Takeoka A, Arber S. Functional local proprioceptive feedback circuits initiate and maintain locomotor recovery after spinal cord injury. Cell Rep 27: 71–85.e3, 2019. doi: 10.1016/j.celrep.2019.03.010. [DOI] [PubMed] [Google Scholar]
- 8. Estilow T, Glanzman AM, Burns J, Harrington A, Cornett K, Menezes MP, Shy R, Moroni I, Pagliano E, Pareyson D, Bhandari T, Muntoni F, Laura M, Reilly MM, Finkel RS, Eichinger KJ, Herrmann DN, Troutman G, Bray P, Halaki M, Shy ME, Yum SW; CMTPedS STUDY GROUP. Balance impairment in pediatric charcot-marie-tooth disease. Muscle Nerve 60: 242–249, 2019. doi: 10.1002/mus.26500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ahmad I, Noohu MM, Verma S, Singla D, Hussain ME. Effect of sensorimotor training on balance measures and proprioception among middle and older age adults with diabetic peripheral neuropathy. Gait Posture 74: 114–120, 2019. doi: 10.1016/j.gaitpost.2019.08.018. [DOI] [PubMed] [Google Scholar]
- 10. Gosgnach S, Bikoff JB, Dougherty KJ, El Manira A, Lanuza GM, Zhang Y. Delineating the diversity of spinal interneurons in locomotor circuits. J Neurosci 37: 10835–10841, 2017. doi: 10.1523/JNEUROSCI.1829-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zholudeva LV, Abraira VE, Satkunendrarajah K, McDevitt TC, Goulding MD, Magnuson DSK, Lane MA. Spinal interneurons as gatekeepers to neuroplasticity after injury or disease. J Neurosci 41: 845–854, 2021. doi: 10.1523/JNEUROSCI.1654-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ziskind-Conhaim L, Hochman S. Diversity of molecularly defined spinal interneurons engaged in mammalian locomotor pattern generation. J Neurophysiol 118: 2956–2974, 2017. doi: 10.1152/jn.00322.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lalonde RL, Bui T. Do spinal circuits still require gating of sensory information by presynaptic inhibition after spinal cord injury? Curr Opin Physiol 19: 113–118, 2021. doi: 10.1016/j.cophys.2020.10.001. [DOI] [Google Scholar]
- 14. Ueno M, Nakamura Y, Li J, Gu Z, Niehaus J, Maezawa M, Crone SA, Goulding M, Baccei ML, Yoshida Y. Corticospinal circuits from the sensory and motor cortices differentially regulate skilled movements through distinct spinal interneurons. Cell Rep 23: 1286–1300.e7, 2018. doi: 10.1016/j.celrep.2018.03.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Betley JN, Wright CV, Kawaguchi Y, Erdelyi F, Szabo G, Jessell TM, Kaltschmidt JA. Stringent specificity in the construction of a GABAergic presynaptic inhibitory circuit. Cell 139: 161–174, 2009. doi: 10.1016/j.cell.2009.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fink AJ. Exploring a behavioral role for presynaptic inhibition at spinal sensory-motor synapses (PhD thesis). New York: Columbia University, 2013. [Google Scholar]
- 17. Fink AJP, Croce KR, Huang ZJ, Abbott LF, Jessell TM, Azim E. Presynaptic inhibition of spinal sensory feedback ensures smooth movement. Nature 509: 43–48, 2014. doi: 10.1038/nature13276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hughes DI, Mackie M, Nagy GG, Riddell JS, Maxwell DJ, Szabo G, Erdelyi F, Veress G, Szucs P, Antal M, Todd AJ. P boutons in lamina IX of the rodent spinal cord express high levels of glutamic acid decarboxylase-65 and originate from cells in deep medial dorsal horn. Proc Natl Acad Sci USA 102: 9038–9043, 2005. doi: 10.1073/pnas.0503646102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hari K, Lucas-Osma AM, Metz K, Lin S, Pardell N, Roszko DA, Black S, Minarik A, Singla R, Stephens MJ, Pearce RA, Fouad K, Jones KE, Gorassini MA, Fenrich KK, Li Y, Bennett DJ. GABA facilitates spike propagation through branch points of sensory axons in the spinal cord. Nat Neurosci 25: 1288–1299, 2022. doi: 10.1038/s41593-022-01162-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hari K, Lucas-Osma AM, Metz K, Lin S, Pardell N, Roszko DA, Black S, Minarik A, Singla R, Stephens MJ, Pearce RA, Fouad K, Jones KE, Gorassini MA, Fenrich KK, Li Y, Bennett DJ. GABA facilitates spike propagation at branch points of sensory axons in the spinal cord (Preprint). BioRxiv, 2021. doi: 10.1101/2021.01.20.427494. [DOI] [PMC free article] [PubMed]
- 21. Barron DH, Matthews BH. The interpretation of potential changes in the spinal cord. J Physiol 92: 276–321, 1938. doi: 10.1113/jphysiol.1938.sp003603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lucas-Osma AM, Li Y, Lin S, Black S, Singla R, Fouad K, Fenrich KK, Bennett DJ. Extrasynaptic α5GABAA receptors on proprioceptive afferents produce a tonic depolarization that modulates sodium channel function in the rat spinal cord. J Neurophysiol 120: 2953–2974, 2018. doi: 10.1152/jn.00499.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rudomin P, Schmidt RF. Presynaptic inhibition in the vertebrate spinal cord revisited. Exp Brain Res 129: 1–37, 1999. doi: 10.1007/s002210050933. [DOI] [PubMed] [Google Scholar]
- 24. Bardoni R, Takazawa T, Tong CK, Choudhury P, Scherrer G, Macdermott AB. Pre- and postsynaptic inhibitory control in the spinal cord dorsal horn. Ann N Y Acad Sci 1279: 90–96, 2013. doi: 10.1111/nyas.12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Szabadics J, Varga C, Molnar G, Olah S, Barzo P, Tamas G. Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science 311: 233–235, 2006. doi: 10.1126/science.1121325. [DOI] [PubMed] [Google Scholar]
- 26. Metz K, Matos IC, Hari K, Bseis O, Afsharipour B, Lin S, Singla R, Fenrich KK, Li Y, Bennett DJ, Gorassini MA. Post-activation depression from primary afferent depolarization (PAD) produces extensor H-reflex suppression following flexor afferent conditioning. J Physiol 601: 1925–1956, 2023. doi: 10.1113/JP283706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Curtis DR, Lacey G. GABA-B receptor-mediated spinal inhibition. Neuroreport 5: 540–542, 1994. doi: 10.1097/00001756-199401000-00002. [DOI] [PubMed] [Google Scholar]
- 28. Eccles JC, Eccles RM, Magni F. Central inhibitory action attributable to presynaptic depolarization produced by muscle afferent volleys. J Physiol 159: 147–166, 1961. doi: 10.1113/jphysiol.1961.sp006798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eccles JC, Magni F, Willis WD. Depolarization of central terminals of Group I afferent fibres from muscle. J Physiol 160: 62–93, 1962. doi: 10.1113/jphysiol.1962.sp006835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gossard JP. Control of transmission in muscle group IA afferents during fictive locomotion in the cat. J Neurophysiol 76: 4104–4112, 1996. doi: 10.1152/jn.1996.76.6.4104. [DOI] [PubMed] [Google Scholar]
- 31. Zimmerman AL, Kovatsis EM, Pozsgai RY, Tasnim A, Zhang Q, Ginty DD. Distinct modes of presynaptic inhibition of cutaneous afferents and their functions in behavior. Neuron 102: 420–434.e8, 2019. doi: 10.1016/j.neuron.2019.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Koch SC, Del Barrio MG, Dalet A, Gatto G, Gunther T, Zhang J, Seidler B, Saur D, Schule R, Goulding M. RORβ spinal interneurons gate sensory transmission during locomotion to secure a fluid walking gait. Neuron 96: 1419–1431.e5, 2017. doi: 10.1016/j.neuron.2017.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lidierth M, Wall PD. Dorsal horn cells connected to the lissauer tract and their relation to the dorsal root potential in the rat. J Neurophysiol 80: 667–679, 1998. doi: 10.1152/jn.1998.80.2.667. [DOI] [PubMed] [Google Scholar]
- 34. Moreno-Lopez Y, Bichara C, Delbecq G, Isope P, Cordero-Erausquin M. The corticospinal tract primarily modulates sensory inputs in the mouse lumbar cord. eLife 10: e65304, 2021. doi: 10.7554/eLife.65304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jankowska E, McCrea D, Rudomin P, Sykova E. Observations on neuronal pathways subserving primary afferent depolarization. J Neurophysiol 46: 506–516, 1981. doi: 10.1152/jn.1981.46.3.506. [DOI] [PubMed] [Google Scholar]
- 36. Rudomin P, Solodkin M, Jimenez I. Synaptic potentials of primary afferent fibers and motoneurons evoked by single intermediate nucleus interneurons in the cat spinal cord. J Neurophysiol 57: 1288–1313, 1987. doi: 10.1152/jn.1987.57.5.1288. [DOI] [PubMed] [Google Scholar]
- 37. Bannatyne BA, Liu TT, Hammar I, Stecina K, Jankowska E, Maxwell DJ. Excitatory and inhibitory intermediate zone interneurons in pathways from feline group I and II afferents: differences in axonal projections and input. J Physiol 587: 379–399, 2009. doi: 10.1113/jphysiol.2008.159129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jankowska E, Bannatyne BA, Stecina K, Hammar I, Cabaj A, Maxwell DJ. Commissural interneurons with input from group I and II muscle afferents in feline lumbar segments: neurotransmitters, projections and target cells. J Physiol 587: 401–418, 2009. doi: 10.1113/jphysiol.2008.159236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Anden NE, Jukes MG, Lundberg A, Vyklicky L. The effect of DOPA on the spinal cord. 1. Influence on transmission from primary afferents. Acta Physiol Scand 67: 373–386, 1966. doi: 10.1111/j.1748-1716.1966.tb03324.x. [DOI] [PubMed] [Google Scholar]
- 40. Jankowska E, Jukes MG, Lund S, Lundberg A. Reciprocal innervation through interneuronal inhibition. Nature 206: 198–199, 1965. doi: 10.1038/206198a0. [DOI] [PubMed] [Google Scholar]
- 41. Kiehn O. Decoding the organization of spinal circuits that control locomotion. Nat Rev Neurosci 17: 224–238, 2016. doi: 10.1038/nrn.2016.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Leblond H, L'Esperance M, Orsal D, Rossignol S. Treadmill locomotion in the intact and spinal mouse. J Neurosci 23: 11411–11419, 2003. doi: 10.1523/JNEUROSCI.23-36-11411.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang Y, Narayan S, Geiman E, Lanuza GM, Velasquez T, Shanks B, Akay T, Dyck J, Pearson K, Gosgnach S, Fan C-M, Goulding M. V3 spinal neurons establish a robust and balanced locomotor rhythm during walking. Neuron 60: 84–96, 2008. doi: 10.1016/j.neuron.2008.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lin S, Li Y, Lucas-Osma AM, Hari K, Stephens MJ, Singla R, Heckman CJ, Zhang Y, Fouad K, Fenrich KK, Bennett DJ. Locomotor-related V3 interneurons initiate and coordinate muscles spasms after spinal cord injury. J Neurophysiol 121: 1352–1367, 2019. doi: 10.1152/jn.00776.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Deska-Gauthier DA, Zhang H, Bennett L, Jones C, Bikoff JB, Zhang Y. Discrete spinal V3 interneuron subpopulations are specified through temporally and molecularly distinct developmental pathways. Program 151.16. In: 2018 Neuroscience Meeting Planner. San Diego, CA: Society for Neuroscience, 2018. [Google Scholar]
- 46. Liu TT, Bannatyne BA, Jankowska E, Maxwell DJ. Properties of axon terminals contacting intermediate zone excitatory and inhibitory premotor interneurons with monosynaptic input from group I and II muscle afferents. J Physiol 588: 4217–4233, 2010. doi: 10.1113/jphysiol.2010.192211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Russo RE, Delgado-Lezama R, Hounsgaard J. Dorsal root potential produced by a TTX-insensitive micro-circuitry in the turtle spinal cord. J Physiol 528: 115–122, 2000. doi: 10.1111/j.1469-7793.2000.00115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chopek JW, Nascimento F, Beato M, Brownstone RM, Zhang Y. Sub-populations of spinal V3 interneurons form focal modules of layered pre-motor microcircuits. Cell Rep 25: 146–156.e3, 2018. doi: 10.1016/j.celrep.2018.08.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bohm UL, Kimura Y, Kawashima T, Ahrens MB, Higashijima S-I, Engert F, Cohen AE. Voltage imaging identifies spinal circuits that modulate locomotor adaptation in zebrafish. Neuron 110: 1211–1222.e4, 2022. doi: 10.1016/j.neuron.2022.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Madisen L, Mao T, Koch H, Zhuo J-M, Berenyi A, Fujisawa S, Hsu Y-WA, Garcia AJ 3rd, Gu X, Zanella S, Kidney J, Gu H, Mao Y, Hooks BM, Boyden ES, Buzsaki G, Ramirez JM, Jones AR, Svoboda K, Han X, Turner EE, Zeng H. A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat Neurosci 15: 793–802, 2012. doi: 10.1038/nn.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng HA. robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13: 133–140, 2010. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tamamaki N, Yanagawa Y, Tomioka R, Miyazaki J, Obata K, Kaneko T. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J Comp Neurol 467: 60–79, 2003. doi: 10.1002/cne.10905. [DOI] [PubMed] [Google Scholar]
- 53. Bennett DJ, Gorassini M, Fouad K, Sanelli L, Han Y, Cheng J. Spasticity in rats with sacral spinal cord injury. J Neurotrauma 16: 69–84, 1999. doi: 10.1089/neu.1999.16.69. [DOI] [PubMed] [Google Scholar]
- 54. Zhang F, Vierock J, Yizhar O, Fenno LE, Tsunoda S, Kianianmomeni A, Prigge M, Berndt A, Cushman J, Polle J, Magnuson J, Hegemann P, Deisseroth K. The microbial opsin family of optogenetic tools. Cell 147: 1446–1457, 2011. doi: 10.1016/j.cell.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. El-Gaby M, Zhang Y, Wolf K, Schwiening CJ, Paulsen O, Shipton OA. Archaerhodopsin selectively and reversibly silences synaptic transmission through altered pH. Cell Rep 16: 2259–2268, 2016. doi: 10.1016/j.celrep.2016.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Boyd IA, Kalu KU. Scaling factor relating conduction velocity and diameter for myelinated afferent nerve fibres in the cat hind limb. J Physiol 289: 277–297, 1979. doi: 10.1113/jphysiol.1979.sp012737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Eccles JC, Eccles RM, Lundberg A. Synaptic actions on motoneurones in relation to the two components of the group I muscle afferent volley. J Physiol 136: 527–546, 1957. doi: 10.1113/jphysiol.1957.sp005778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Todd AJ, Hughes DI, Polgar E, Nagy GG, Mackie M, Ottersen OP, Maxwell DJ. The expression of vesicular glutamate transporters VGLUT1 and VGLUT2 in neurochemically defined axonal populations in the rat spinal cord with emphasis on the dorsal horn. Eur J Neurosci 17: 13–27, 2003. doi: 10.1046/j.1460-9568.2003.02406.x. [DOI] [PubMed] [Google Scholar]
- 59. Borowska J, Jones CT, Zhang H, Blacklaws J, Goulding M, Zhang Y. Functional subpopulations of V3 interneurons in the mature mouse spinal cord. J Neurosci 33: 18553–18565, 2013. doi: 10.1523/JNEUROSCI.2005-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shreckengost J, Calvo J, Quevedo J, Hochman S. Bicuculline-sensitive primary afferent depolarization remains after greatly restricting synaptic transmission in the mammalian spinal cord. J Neurosci 30: 5283–5288, 2010. doi: 10.1523/JNEUROSCI.3873-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. de Lera Ruiz M, Kraus RL. Voltage-gated sodium channels: structure, function, pharmacology, and clinical indications. J Med Chem 58: 7093–7118, 2015. doi: 10.1021/jm501981g. [DOI] [PubMed] [Google Scholar]
- 62. Goldstein SS, Rall W. Changes of action potential shape and velocity for changing core conductor geometry. Biophys J 14: 731–757, 1974. doi: 10.1016/S0006-3495(74)85947-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shreckengost J, Halder M, Mena-Avila E, Garcia-Ramirez DL, Quevedo J, Hochman S. Nicotinic receptor modulation of primary afferent excitability with selective regulation of Aδ-mediated spinal actions. J Neurophysiol 125: 568–585, 2021. doi: 10.1152/jn.00228.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Blacklaws J, Deska-Gauthier D, Jones CT, Petracca YL, Liu M, Zhang H, Fawcett JP, Glover JC, Lanuza GM, Zhang Y. Sim1 is required for the migration and axonal projections of V3 interneurons in the developing mouse spinal cord. Dev Neurobiol 75: 1003–1017, 2015. doi: 10.1002/dneu.22266. [DOI] [PubMed] [Google Scholar]
- 65. Li Y, Hari K, Lucas-Osma AM, Fenrich KK, Bennett DJ, Hammar I, Jankowska E. Branching points of primary afferent fibers are vital for the modulation of fiber excitability by epidural DC polarization and by GABA in the rat spinal cord. J Neurophysiol 124: 49–62, 2020. doi: 10.1152/jn.00161.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Murray KC, Nakae A, Stephens MJ, Rank M, D'Amico J, Harvey PJ, Li X, Harris RL, Ballou EW, Anelli R, Heckman CJ, Mashimo T, Vavrek R, Sanelli L, Gorassini MA, Bennett DJ, Fouad K. Recovery of motoneuron and locomotor function after spinal cord injury depends on constitutive activity in 5-HT2C receptors. Nat Med 16: 694–700, 2010. doi: 10.1038/nm.2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Abraira VE, Kuehn ED, Chirila AM, Springel MW, Toliver AA, Zimmerman AL, Orefice LL, Boyle KA, Bai L, Song BJ, Bashista KA, O'Neill TG, Zhuo J, Tsan C, Hoynoski J, Rutlin M, Kus L, Niederkofler V, Watanabe M, Dymecki SM, Nelson SB, Heintz N, Hughes DI, Ginty DD. The cellular and synaptic architecture of the mechanosensory dorsal horn. Cell 168: 295–310.e19, 2017. doi: 10.1016/j.cell.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Eccles JC, Krnjevic K. Potential changes recorded inside primary afferent fibres within the spinal cord. J Physiol 149: 250–273, 1959. doi: 10.1113/jphysiol.1959.sp006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wu H, Petitpre C, Fontanet P, Sharma A, Bellardita C, Quadros RM, Jannig PR, Wang Y, Heimel JA, Cheung KKY, Wanderoy S, Xuan Y, Meletis K, Ruas J, Gurumurthy CB, Kiehn O, Hadjab S, Lallemend F. Distinct subtypes of proprioceptive dorsal root ganglion neurons regulate adaptive proprioception in mice. Nat Commun 12: 1026, 2021. doi: 10.1038/s41467-021-21173-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Dedek A, Hildebrand ME. Advances and barriers in understanding presynaptic N-methyl-D-aspartate receptors in spinal pain processing. Front Mol Neurosci 15: 864502, 2022. doi: 10.3389/fnmol.2022.864502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Peirs C, Williams S-PG, Zhao X, Walsh CE, Gedeon JY, Cagle NE, Goldring AC, Hioki H, Liu Z, Marell PS, Seal RP. Dorsal horn circuits for persistent mechanical pain. Neuron 87: 797–812, 2015. doi: 10.1016/j.neuron.2015.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wang P, Slaughter MM. Effects of GABA receptor antagonists on retinal glycine receptors and on homomeric glycine receptor alpha subunits. J Neurophysiol 93: 3120–3126, 2005. doi: 10.1152/jn.01228.2004. [DOI] [PubMed] [Google Scholar]
- 73. Hultborn H, Illert M, Nielsen J, Paul A, Ballegaard M, Wiese H. On the mechanism of the post-activation depression of the H-reflex in human subjects. Exp Brain Res 108: 450–462, 1996. doi: 10.1007/BF00227268. [DOI] [PubMed] [Google Scholar]
- 74. Lev-Tov A, Meyers DE, Burke RE. Activation of type B gamma-aminobutyric acid receptors in the intact mammalian spinal cord mimics the effects of reduced presynaptic Ca2+ influx. Proc Natl Acad Sci USA 85: 5330–5334, 1988. doi: 10.1073/pnas.85.14.5330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Andersen P, Eccles JC, Sears TA. Cortically evoked depolarization of primary afferent fibers in the spinal cord. J Neurophysiol 27: 63–77, 1964. doi: 10.1152/jn.1964.27.1.63. [DOI] [PubMed] [Google Scholar]
- 76. Caron G, Bilchak JN, Cote M-P. Direct evidence for decreased presynaptic inhibition evoked by PBSt group I muscle afferents after chronic SCI and recovery with step-training in rats. J Physiol 598: 4621–4642, 2020. doi: 10.1113/JP280070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Meisner JG, Marsh AD, Marsh DR. Loss of GABAergic interneurons in laminae I-III of the spinal cord dorsal horn contributes to reduced GABAergic tone and neuropathic pain after spinal cord injury. J Neurotrauma 27: 729–737, 2010. doi: 10.1089/neu.2009.1166. [DOI] [PubMed] [Google Scholar]
- 78. Jankowska E, Padel Y, Zarzecki P. Crossed disynaptic inhibition of sacral motoneurones. J Physiol 285: 425–444, 1978. doi: 10.1113/jphysiol.1978.sp012580. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available upon reasonable request.