

Abstract
We announce the assembly of the first de novo reference genome for the California Scrub-Jay (Aphelocoma californica). The genus Aphelocoma comprises four currently recognized species including many locally adapted populations across Mesoamerica and North America. Intensive study of Aphelocoma has revealed novel insights into the evolutionary mechanisms driving diversification in natural systems. Additional insights into the evolutionary history of this group will require continued development of high-quality, publicly available genomic resources. We extracted high molecular weight genomic DNA from a female California Scrub-Jay from northern California and generated PacBio HiFi long-read data and Omni-C chromatin conformation capture data. We used these data to generate a de novo partially phased diploid genome assembly, consisting of two pseudo-haplotypes, and scaffolded them using inferred physical proximity information from the Omni-C data. The more complete pseudo-haplotype assembly (arbitrarily designated “Haplotype 1”) is 1.35 Gb in total length, highly contiguous (contig N50 = 11.53 Mb), and highly complete (BUSCO completeness score = 97%), with comparable scaffold sizes to chromosome-level avian reference genomes (scaffold N50 = 66.14 Mb). Our California Scrub-Jay assembly is highly syntenic with the New Caledonian Crow reference genome despite ~10 million years of divergence, highlighting the temporal stability of the avian genome. This high-quality reference genome represents a leap forward in publicly available genomic resources for Aphelocoma, and the family Corvidae more broadly. Future work using Aphelocoma as a model for understanding the evolutionary forces generating and maintaining biodiversity across phylogenetic scales can now benefit from a highly contiguous, in-group reference genome.
Keywords: California Conservation Genomics Project, CCGP, long-read sequencing, N50, Omni-C, PacBio
Introduction
The scrub-jays (genus Aphelocoma) are part of a broader clade of jays that radiated in the Americas, which includes other iconic North American species like the Blue Jay (Cyanocitta cristata) and the Steller’s Jay (Cyanocitta stelleri). Beyond their charm to backyard birdwatchers, these “American jays” have also emerged as a model system for studying the mechanisms driving and maintaining phenotypic and genetic variation across complex landscapes (McCormack et al. 2011; DeRaad et al. 2019; Venkatraman et al. 2019; Cicero et al. 2022; DeRaad et al. 2022), the evolution of cooperative behavior (Brown 1974; Peterson and Burt 1992; Berg et al. 2012), vocal repertoires (Rosa et al. 2016), and ecological differentiation (Rice et al. 2003; McCormack et al. 2008). The scrub-jays offer a particularly compelling system for studying speciation genomics because the species complex is distributed across North America and is composed of seven discrete, parapatric lineages with varying levels of genetic divergence (DeRaad et al. 2022). These discrete lineages allow for multiple comparisons of different stages in the speciation process within a single species complex, making this group an important target for future studies seeking to reconstruct and understand the generation of biodiversity.
The scrub-jays also provide a textbook example of ecological specialization (Pitelka 1951), which is of particular interest for understanding speciation and adaptation. Specifically, the large, hooked bills of the California and Island scrub-jays are ideal for prying open acorns of oak (Quercus) species, while the thin, straight, tweezer-like bill of the closely related Woodhouse’s Scrub-Jay (Aphelocoma woodhouseii) is adapted for removing seeds from pinyon pine (genus Pinus) cones (Peterson 1993; Bardwell et al. 2001). These ecologically and genetically divergent species have a narrow zone of secondary contact in eastern California and western Nevada, where moderate rates of hybridization have been documented (Gowen et al. 2014; Vinciguerra et al. 2019). The reticulate evolution in this system offers great potential to increase our understanding of the role of ecological adaptation in both the generation and maintenance of biodiversity, but this effort will require extensive genomic resources.
Additionally, the conservation genetics of two isolated and range-restricted scrub-jays—the federally threatened Florida Scrub-Jay (Aphelocoma coerulescens) and the Island Scrub-Jay (Aphelocoma insularis)—have been studied intensively. Cutting-edge genomic research in these species of conservation concern includes intensive pedigree analyses of Florida Scrub-Jays (Chen et al. 2016; Aguillon et al. 2017) and an investigation of the genomic signatures of ecological adaptation at microgeographic scales in Island Scrub-Jays (Cheek et al. 2022). This ever-growing wealth of fruitful research into the evolutionary history of the scrub-jay species complex relies on shared genomic resources, which to this point have been limited to a single, scaffold-level reference genome assembly of the Florida Scrub-Jay (NCBI BioSample SAMN12253992; Feng et al. 2020). Here, we present the first de novo genome assembly for the California Scrub-Jay (NCBI BioSample SAMN31837216) as part of the California Conservation Genomics Project (CCGP; Shaffer et al. 2022; Toffelmier et al. 2022).
Methods
Generating a range map
We used the R package ebirdst (Strimas-Mackey et al. 2021) to download and process observational occurrence data for the California Scrub-Jay from the year 2020. Because the California Scrub-Jay is nonmigratory, the database does not discriminate between breeding and wintering observations, and year-round occurrence points were used to visualize the species’ distribution. We downloaded mean relative abundance at a medium resolution and converted it into quantiles of nonzero values. We plotted these data as a heatmap (Fig. 1) on top of a continental basemap generated using the R packages sf (Pebesma 2018) and rnaturalearth (South 2017).
Fig. 1.
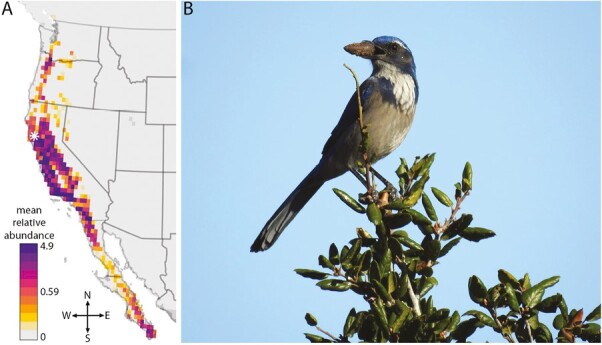
A) Mean relative abundance of California Scrub-Jays throughout North America, i.e. the average number of birds seen by an individual observer performing a traveling count at an optimal time of day. Sampling locality of the individual used to generate this reference genome is denoted with a white asterisk. B) California Scrub-Jay (Aphelocoma californica) photographed at Occidental College, Los Angeles, CA by John McCormack.
Biological materials
We collected an adult female California Scrub-Jay on 12 October 2020 at Cow Mountain Recreation Management Area, Lake County, California (39.10238, −123.07647). The specimen was collected under authorization of the California Department of Fish and Wildlife (permit # SC-458), U. S. Fish and Wildlife Service (permit # MB153526), and the University of California Animal Care and Use Committee (protocol # AUP-2015-10-8045-1). This vouchered specimen is deposited at the Museum of Vertebrate Zoology, University of California, Berkeley (MVZ:Bird:193978; https://arctos.database.museum/guid/MVZ:Bird:193978). DNA extractions for both HiFi and Omni-C sequencing were performed using flash frozen liver tissue.
High molecular weight DNA extraction
Flash frozen liver (20 mg) was homogenized in 2 mL of homogenization buffer (10 mM Tris–HCl pH 8.0 and 25 mM Ethylenediaminetetraacetic acid, i.e., EDTA) using TissueRuptor II (Qiagen, Germany; Cat. # 9002755). In total, 2 mL of lysis buffer (10 mM Tris, 25 mM EDTA, 200 mM NaCl, and 1% Sodium Dodecyl Sulfate, i.e, SDS) and proteinase K (100 µg/mL) were added to the homogenate and incubated overnight at room temperature. Lysate was treated with RNase A (20 µg/mL) at 37 °C for 30 min and was cleaned with equal volumes of phenol/chloroform using phase-lock gels (Quantabio, Beverly, Massachusetts; Cat. # 2302830). The DNA was precipitated by adding 0.4× volume of 5 M ammonium acetate and 3× volume of ice-cold ethanol. The DNA pellet was washed twice with 70% ethanol and resuspended in an elution buffer (10 mM Tris, pH 8.0). We assessed the purity of genomic DNA (gDNA) using a NanoDrop ND-1000 spectrophotometer and recovered a 260/280 ratio of 1.82 and a 260/230 ratio of 2.02. The DNA was quantified by a Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Massachusetts), and a total yield of 50 µg was obtained. The integrity of the high molecular weight (HMW) gDNA was verified on a Femto pulse system (Agilent Technologies, Santa Clara, California), where a majority of DNA was observed in fragments above 125 kb or more.
HiFi library prep and sequencing
The HiFi SMRTbell library was constructed using the SMRTbell Express Template Prep Kit v2.0 (Pacific Biosciences—PacBio, Cat. #100-938-900) according to the manufacturer’s instructions. HMW gDNA was sheared to a target DNA size distribution between 15 and 18 kb. The sheared gDNA was concentrated using 0.45× of AMPure PB beads (PacBio, Menlo Park, California; Cat. #100-265-900) for the removal of single-strand overhangs at 37 °C for 15 min, followed by further enzymatic steps of DNA damage repair at 37 °C for 30 min end repair and A-tailing at 20 °C for 10 min and 65 °C for 30 min, and ligation of overhang adapter v3 at 20 °C for 60 min. The SMRTbell library was purified and concentrated with 1× AMPure PB beads (PacBio, Cat. #100-265-900) for nuclease treatment at 37 °C for 30 min and size selection using the BluePippin/PippinHT system (Sage Science, Beverly, Massachusetts; Cat. #BLF7510/HPE7510) to collect fragments greater than 7 to 9 kb. The 15 to 20 kb average HiFi SMRTbell library was sequenced at the UC Davis DNA Technologies Core (Davis, California) using two 8M SMRT cells, Sequel II sequencing chemistry 2.0, and 30-h movies each on a PacBio Sequel II sequencer.
Omni-C library prep and sequencing
The Omni-C library was prepared using the Dovetail Omni-C Kit (Dovetail Genomics, Scotts Valley, California) according to the manufacturer’s protocol with slight modifications. First, liver tissue was thoroughly ground with a mortar and pestle while cooled with liquid nitrogen. Subsequently, chromatin was fixed in place in the nucleus. The suspended chromatin solution was then passed through 100 and 40 µm cell strainers to remove large debris. Fixed chromatin was digested under various conditions of DNase I until a suitable fragment length distribution of DNA molecules was obtained. Chromatin ends were repaired and ligated to a biotinylated bridge adapter followed by proximity ligation of adapter containing ends. After proximity ligation, crosslinks were reversed, and the DNA was purified from proteins. Purified DNA was treated to remove biotin that was not internal to ligated fragments. An NGS library was generated using an NEB Ultra II DNA Library Prep kit (New England Biolabs—NEB, Ipswich, Massachusetts) with an Illumina compatible y-adaptor. Biotin-containing fragments were then captured using streptavidin beads. The post-capture product was split into two replicates prior to PCR enrichment to preserve library complexity, with each replicate receiving unique dual indices. The library was sequenced at Vincent J. Coates Genomics Sequencing Lab (Berkeley, California) across two separate flow cells on an Illumina NovaSeq platform (Illumina, San Diego, California) generating 150 basepair paired-end reads.
Genome assembly
We assembled the genome of the California Scrub-Jay following the CCGP assembly pipeline Version 6.0 (Table 1 lists the tools and nondefault parameters used in the assembly). The pipeline uses PacBio HiFi reads and Omni-C data to produce a high-quality, highly contiguous genome assembly, with heterozygous regions assigned arbitrarily to one of two pseudo-haplotypes. First, we removed the remnant adapter sequences from the PacBio HiFi dataset using HiFiAdapterFilt (Sim et al. 2022) and generated the initial dual or partially phased diploid assembly (http://lh3.github.io/2021/10/10/introducing-dual-assembly) using HiFiasm (Cheng et al. 2022) on Hi-C mode, with the filtered PacBio HiFi reads and the Omni-C dataset. We then aligned the Omni-C data to each pseudo-haplotype following the Arima Genomics Mapping Pipeline (https://github.com/ArimaGenomics/mapping_pipeline) and then scaffolded them with SALSA (Ghurye et al. 2017, 2019).
Table 1.
Software used for all the analyses presented in the paper with their corresponding version and nondefault parameters.
| Purpose | Software and optionsa | Version |
|---|---|---|
| Generating the range map | ||
| Processing of observational occurrence data | ebirdst | 1.2020.1 |
| Map plotting with occurrence data | sf | 1.0-7 |
| rnaturalearth | 0.1.0 | |
| Assembly | ||
| Filtering PacBio HiFi adapters | HiFiAdapterFilt | Commit 64d1c7b |
| K-mer counting | Meryl (k = 21) | 1 |
| Estimation of genome size and heterozygosity | GenomeScope | 2 |
| De novo assembly (contiging) | HiFiasm (Hi-C mode, –primary, output p_ctg.hap1, p_ctg.hap2) | 0.16.1-r375 |
| Scaffolding | ||
| Omni-C data alignment | Arima Genomics Mapping Pipeline | Commit 2e74ea4 |
| Omni-C scaffolding | SALSA (-DNASE, -i 20, -p yes) | 2 |
| Gap closing | YAGCloser (-mins 2 -f 20 -mcc 2 -prt 0.25 -eft 0.2 -pld 0.2) | Commit 0e34c3b |
| Omni-C contact map generation | ||
| Short-read alignment | BWA-MEM (-5SP) | 0.7.17-r1188 |
| SAM/BAM processing | samtools | 1.11 |
| SAM/BAM filtering | pairtools | 0.3.0 |
| Pairs indexing | pairix | 0.3.7 |
| Matrix generation | cooler | 0.8.10 |
| Matrix balancing | hicExplorer (hicCorrectmatrix correct --filterThreshold -2 4) | 3.6 |
| Contact map visualization | HiGlass | 2.1.11 |
| PretextMap | 0.1.4 | |
| PretextView | 0.1.5 | |
| PretextSnapshot | 0.0.3 | |
| Manual curation tools | Rapid curation pipeline (Wellcome Trust Sanger Institute, Genome Reference Informatics Team) | Commit 4ddca450 |
| Genome quality assessment | ||
| Basic assembly metrics | QUAST (--est-ref-size) | 5.0.2 |
| Assembly completeness | BUSCO (-m geno, -l aves) | 5.0.0 |
| Merqury | 2020-01-29 | |
| Circos Assembly Consistency (Jupiter) plot | JupiterPlot (ng = 95, m = 5000000) | 1.0 |
| Identification of repetitive elements | RepeatMasker | 4.0.5 |
| Organelle assembly | ||
| Mitogenome assembly | MitoHiFi (-r, -p 50, -o 1) | 2 Commit c06ed3e |
| Contamination screening | ||
| Local alignment tool | BLAST+ (-db nt, -outfmt “6 qseqid staxids bitscore std,” -max_target_seqs 1, -max_hsps 1, -evalue 1e-25) | 2.1 |
| General contamination screening | BlobToolKit | 2.3.3 |
Citations for the software used are detailed in the text.
aOptions detailed for nondefault parameters.
The pseudo-haplotypes were manually curated by iteratively generating and analyzing their corresponding Omni-C contact maps. To generate the contact maps, we aligned the Omni-C data with BWA-MEM (Li 2013), identified ligation junctions, and generated Omni-C pairs using pairtools (Goloborodko et al. 2018). We generated a multiresolution Omni-C matrix with cooler (Abdennur an Mirny 2020) and balanced it with hicExplorer (Ramírez et al. 2018). We used HiGlass (Kerpedjiev et al. 2018) and the PretextSuite (https://github.com/wtsi-hpag/PretextView; https://github.com/wtsi-hpag/PretextMap; https://github.com/wtsi-hpag/PretextSnapshot) to visualize the contact maps where we identified mis-assemblies and mis-joins, and finally modified the assemblies using the Rapid Curation pipeline from the Wellcome Trust Sanger Institute, Genome Reference Informatics Team (https://gitlab.com/wtsi-grit/rapid-curation). Some of the remaining gaps (joins generated during scaffolding and curation) were closed using the PacBio HiFi reads and YAGCloser (https://github.com/merlyescalona/yagcloser). Finally, we checked for contamination using the BlobToolKit Framework (Challis et al. 2020).
Mitochondrial genome assembly
We assembled the mitochondrial (mtDNA) genome of A. californica from the PacBio HiFi reads using the reference-guided pipeline MitoHiFi (Allio et al. 2020; Uliano-Silva et al. 2021). The mtDNA genome of A. coerulescens (NCBI:NC_051467.1; Feng et al. 2020; B10K Project Consortium) was used as the starting sequence. After completion of the nuclear genome, we searched for matches of the resulting mitochondrial assembly sequence in the nuclear genome assembly using BLAST+ (Camacho et al. 2009) and filtered out contigs and scaffolds from the nuclear genome with a percentage of sequence identity >99% and size smaller than the mtDNA assembly sequence.
Evaluating the assembly
We generated k-mer counts from the PacBio HiFi reads using meryl (https://github.com/marbl/meryl). The k-mer counts were then used in GenomeScope2.0 (Ranallo-Benavidez et al. 2020) to estimate genome features including genome size, heterozygosity, and repeat content. We ran QUAST (Gurevich et al. 2013) to obtain general contiguity metrics. To evaluate genome quality and functional completeness, we used BUSCO (Manni et al. 2021) with the Aves ortholog database (aves_odb10) which contains 8,338 genes. Assessment of base level accuracy (QV) and k-mer completeness was performed using the previously generated meryl database and merqury (Rhie et al. 2020). We further estimated genome assembly accuracy via BUSCO gene set frameshift analysis using the pipeline described in Korlach et al. (2017). Measurements of the size of the phased blocks are based on the size of the contigs generated by HiFiasm on HiC mode. We follow the quality metric nomenclature established by Rhie et al. (2021), with the genome quality code x·y·P·Q·C, where x = log10[contig NG50]; y = log10[scaffold NG50]; P = log10[phased block NG50]; Q = Phred base accuracy QV (quality value); C = % genome represented by the first “n” scaffolds, following a karyotype of 2n = 80 estimated as the median number of chromosomes from other species in the same family (Genome on a Tree—GoaT; tax_tree(28725)). Quality metrics for the notation were calculated on the assembly for Haplotype 1.
We used RepeatMasker (Chen 2004) to identify repetitive elements throughout the assembly using a transposable element (TE) library generated from de novo repeat annotation of the closely related Steller’s Jay (C. stelleri) reference genome (Benham et al. 2023). We used the output from RepeatMasker as input for the included perl script “calcDivergencefromalign.pl” to classify repetitive regions into “families” of transposable elements. We generated visualizations of repetitive content across the genome and between assemblies using the R package (R Core Team 2019) ggplot2 (Wickham et al. 2020).
We then used the program Jupiter Plot (Chu 2018) to map the scaffolds of our pseudo-haplotype genome assemblies to each other to compare relative synteny and completeness. We repeated this procedure for additional pairwise comparisons between Haplotype 1 of our assembly and the chromosomally scaffolded New Caledonian Crow (Corvus moneduloides; NCBI BioSample SAMN12368441; https://www.ncbi.nlm.nih.gov/assembly/GCF_009650955.1/; Rhie et al. 2021) and the Florida Scrub-Jay reference genome (A. coerulescens; NCBI BioSample SAMN12253992; https://www.ncbi.nlm.nih.gov/assembly/GCA_013398375.1/; Feng et al. 2020). In all comparisons, we included only scaffolds >5 Mb from the genome used as the reference genome (i.e. the left side of each plot). For the comparison with the New Caledonian Crow and the comparison between our two pseudo-haplotype assemblies, we also specified that only the largest scaffolds making up 95% of the mapped genome assembly (i.e. the right side of each plot) should be used for visualization.
Results
Sequence data
The Omni-C and PacBio HiFi sequencing libraries generated 360.02 million read pairs and 5.01 million reads, respectively. The latter yielded 51.4-fold coverage (N50 read length 12,728 bp; minimum read length 89 bp; mean read length 12,578 bp; maximum read length of 51,606 bp) based on the GenomeScope2.0 genome size estimation of 1.23 Gb. Based on PacBio HiFi reads, we estimated 0.139% sequencing error rate and 0.663% nucleotide heterozygosity rate. The k-mer spectrum based on PacBio HiFi reads recovers a bimodal distribution with two major peaks at ~13 and ~26 (Fig. 2A), where peaks correspond to heterozygous and homozygous states of a diploid species.
Fig. 2.
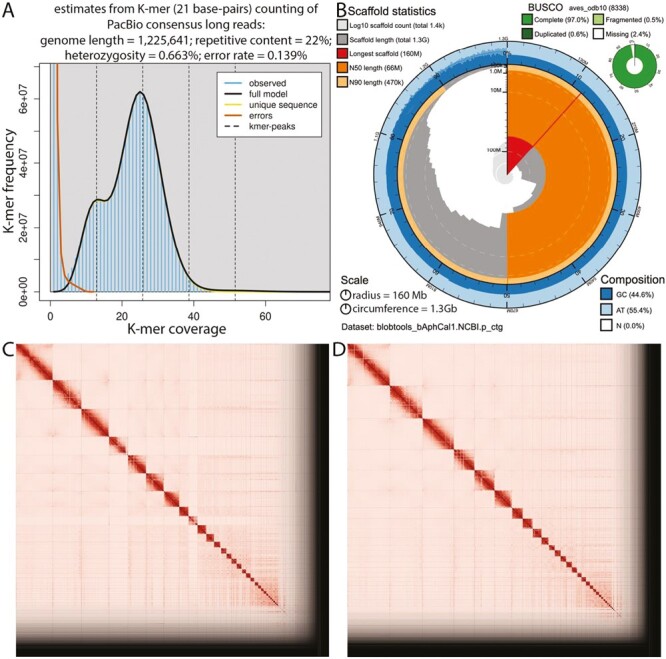
Visual overview of genome assembly metrics. A) K-mer spectra output generated from PacBio HiFi data without adapters using GenomeScope2.0, which indicates low (<1%) heterozygosity. The lower shoulder of the distribution corresponds to haplotype-specific K-mers (i.e. K-mers including a heterozygous site), which should have half the read depth of the taller peak, which corresponds to K-mers shared identically by both haplotypes in the diploid individual we sequenced. B) BlobToolKit Snail plot showing a graphical representation of the quality metrics presented in Table 2 for the Aphelocoma californica "Haplotype 1" assembly (bAphCali1.0.hap1). The plot circle represents the full size of the assembly. The red line represents the size of the longest scaffold; all other scaffolds are arranged in size-order moving clockwise around the plot and drawn in gray starting from the outside of the central plot. Dark and light orange arcs show the scaffold N50 and scaffold N90 values. The central light gray spiral shows the cumulative scaffold count with a white line at each order of magnitude. White regions in this area reflect the proportion of Ns in the assembly; the dark versus light blue area around it shows mean, maximum, and minimum GC versus AT content at 0.1% intervals (Challis et al. 2020). Omni-C contact maps for C) Haplotype 1 and D) Haplotype 2 genome assemblies generated with PretextSnapshot. Omni-C contact maps translate proximity of genomic regions in 3D space to contiguous linear organization. Each cell in the contact map corresponds to sequencing data supporting the linkage (or join) between two genomic regions.
Genome assembly
The final assembly (bAphCal1) consists of two pseudo-haplotypes, both with assembly sizes similar to the estimated value from GenomeScope2.0 (Fig. 2A, Pflug et al. 2020). Haplotype 1 assembly consists of 1,393 scaffolds spanning 1.35 Gb with a contig N50 of 11.53 Mb, scaffold N50 of 66.14 Mb, longest contig spanning 77.93 Mb, and largest scaffold spanning 158.41 Mb. The Haplotype 2 assembly consists of 1,631 scaffolds spanning 1.19 Gb with a contig N50 of 11.67 Mb, scaffold N50 of 65.72 Mb, largest contig spanning 52.3 Mb, and largest scaffold spanning 160.92 Mb. Detailed assembly statistics are reported in Table 2, and a graphical representation for the Haplotype 1 assembly is depicted in Fig. 2B. The Haplotype 1 assembly has a BUSCO completeness score of 97% using the Aves gene set, a per-base quality (QV) of 45.89, a k-mer completeness of 92.49, and a frameshift indel QV of 41.16; the Haplotype 2 assembly has a BUSCO completeness score of 93.3% using the same BUSCO gene set, a per-base quality (QV) of 48.23, a k-mer completeness of 85.91, and a frameshift indel QV of 41.16.
Table 2.
Sequencing, assembly statistics, and accession numbers.
| BioProjects and Vouchers | CCGP NCBI BioProject | PRJNA720569 | |||||
| Genera NCBI BioProject | PRJNA766263 | ||||||
| Species NCBI BioProject | PRJNA777134 | ||||||
| NCBI BioSample | SAMN31837216 | ||||||
| Specimen identification | MVZ:Bird:193978 | ||||||
| NCBI Genome accessions | Haplotype 1 | Haplotype 2 | |||||
| Assembly accession | GCA_028536675.1 | GCA_028536645.1 | |||||
| Genome sequences | JAQMYR000000000 | JAQMYS000000000 | |||||
| Genome Sequence | PacBio HiFi reads | Run | 1 PACBIO_SMRT (Sequel II) run: 5 M spots, 63.1 G bases, 35.3 Gb | ||||
| Accession | SRR23446543 | ||||||
| Omni-C Illumina reads | Run | 2 ILLUMINA (Illumina NovaSeq 6000) run: 360.1 M spots, 108.7 G bases, 36.9 Gb | |||||
| Accession | SRR23446541, SRR23446542 | ||||||
| Genome Assembly Quality Metrics | Assembly identifier (quality codea) | bAphCal1(7.7.P7.Q46.C85) | |||||
| HiFi read coverageb | 51.48× | ||||||
| Haplotype 1 | Haplotype 2 | ||||||
| Number of contigs | 1,665 | 1,799 | |||||
| Contig N50 (bp) | 11,534,525 | 11,670,509 | |||||
| Contig NG50b | 13,901,234 | 11,191,859 | |||||
| Longest contigs | 77,933,725 | 52,308,079 | |||||
| Number of scaffolds | 1,393 | 1,631 | |||||
| Scaffold N50 | 66,144,195 | 65,728,546 | |||||
| Scaffold NG50b | 76,801,313 | 65,728,546 | |||||
| Largest scaffold | 158,410,743 | 160,920,769 | |||||
| Size of final assembly | 1,348,500,425 | 1,191,279,315 | |||||
| Phased block NG50b | 11,313,027 | 12,152,900 | |||||
| Gaps per Gbp (# gaps) | 201 (272) | 141 (168) | |||||
| Indel QV (frameshift) | 41.16 | 41.16 | |||||
| Basepair QV | 45.8906 | 48.2358 | |||||
| Full assembly = 46.8365 | |||||||
| K-mer completeness | 92.4996 | 85.916 | |||||
| Full assembly = 99.5694 | |||||||
| BUSCO completeness (aves_odb10) n = 8,338 |
C | S | D | F | M | ||
| H1c | 97.00% | 96.40% | 0.60% | 0.50% | 2.50% | ||
| H2c | 93.30% | 92.80% | 0.50% | 0.60% | 6.10% | ||
| Organelles | # Partial mitochondrial sequence | JAQMYR010001393.1 | |||||
aAssembly quality code x·y·P·Q·C derived notation, from Rhie et al. (2021). x = log10[contig NG50]; y = log10[scaffold NG50]; P = log10 [phased block NG50]; Q = Phred base accuracy QV (quality value); C = % genome represented by the first “n” scaffolds, following a karyotype of 2n = 80 for A. californica, estimated as the median number of chromosomes from other species in the same family (Genome on a Tree—GoaT; tax_tree(28725)). Quality code for all the assembly denoted by haplotype 1 assembly (bAphCal1.0.hap1).
bRead coverage and NGx statistics have been calculated based on the estimated genome size of 1.225 Gb.
cH1: Haplotype 1; H2: Haplotype 2.
During manual curation, we generated 8 breaks and 71 joins in Haplotype 1 as well as 10 breaks and 54 joins in Haplotype 2. We were able to close a total of 13 gaps, 7 in the Haplotype 1 assembly and 6 in the Haplotype 2 assembly. Finally, we filtered out five contigs from Haplotype 2, four corresponding to Arthropod and Baccilota contaminants and one corresponding to mitochondrial contamination. No further contigs were removed. The Omni-C contact maps show that both pseudo-haplotype assemblies are highly contiguous (Fig. 2C and D; both assemblies are available on NCBI, see Data availability for details). We separately assembled a mtDNA genome with MitoHiFi (NCBI sequence JAQMYR010001393.1; https://www.ncbi.nlm.nih.gov/nuccore/JAQMYR010001393.1/). The final mtDNA genome size was 18,021 bp. The base composition of the final assembly version was A = 29.35%, C = 21.33%, G = 20.23%, T = 29.08%, and the mtDNA genome assembly includes 10 unique transfer RNAs.
Comparing our partially phased pseudo-haplotype assemblies
Synteny mapping revealed overall synteny between our two phased pseudo-haplotype assemblies (here and on NCBI, these two assemblies are consistently, but arbitrarily, designated Haplotypes 1 and 2; Fig. 3A). Notably, synteny mapping indicates that both sex-linked scaffolds (i.e. scaffolds corresponding to the Z and W chromosomes in the New Caledonian Crow genome assembly) are present in the Haplotype 1 assembly and absent from the Haplotype 2 assembly. The Haplotype 2 assembly exhibited both a smaller assembly size and a reduced proportion of repetitive elements (Fig. 3B), likely because of this lack of sex chromosome scaffolds, which are enriched for repetitive elements in birds (Davis et al. 2010). Across both pseudo-haplotype assemblies, three classes of repetitive elements dominate the repetitive landscape: long interspersed nuclear elements (LINEs), long terminal repeat retrotransposons (LTRs), and satellite DNA (Fig. 3C). The most frequent repetitive element class (LTRs) spans >8% of the Haplotype 1 assembly and >6% of the Haplotype 2 assembly.
Fig. 3.
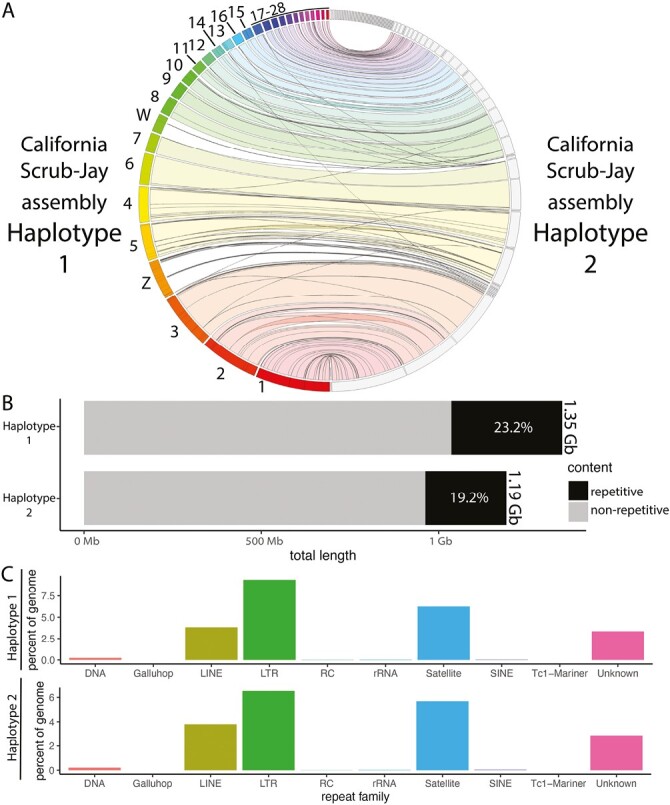
Comparison of the two pseudo-haplotype assemblies. A) Jupiter plot displaying the synteny between the two assemblies. Scaffolds on the left are ordered by size and labeled based on synteny with the chromosomally assembled New Caledonian Crow reference genome. Note that scaffolds without matches on the top of the plot are due to a limit in the number of comparisons that can be drawn with the Jupiter Plot software, not necessarily a lack of syntenic matches. Meanwhile scaffolds in the middle of the assembly without a match (e.g., chromosomes Z and W), reflect a real lack of a syntenic match between assemblies. B) Bar plot comparing the genome size and proportion of repetitive content between the two pseudo-haplotype assemblies. C) Bar plots show the proportion of the genome spanned by each repeat family for each assembly.
Comparing our “Haplotype 1” assembly to other Corvid assemblies
Synteny mapping reveals strong overall synteny between the chromosome-level reference genome for the New Caledonian Crow and Haplotype 1 of our de novo assembled reference genome for the California Scrub-Jay (Fig. 4A). This synteny comparison also highlights the overall contiguity of our Haplotype 1 assembly, with all visualized chromosome-assigned scaffolds from the New Caledonian Crow genome assembly represented by a single scaffold in our California Scrub-Jay assembly.
Fig. 4.
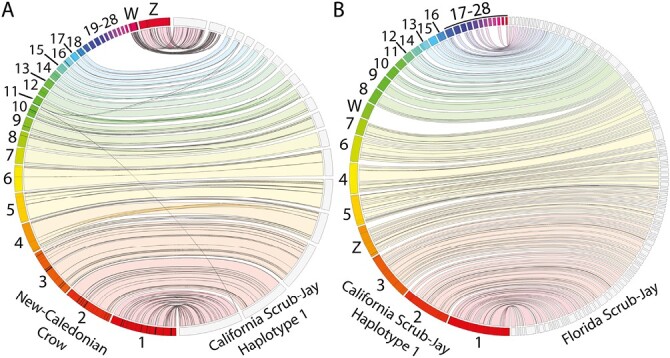
Assessing synteny between publicly available reference genomes from the avian family Corvidae. A) Jupiter plot visually displaying synteny between our “Haplotype 1” California Scrub-Jay genome assembly and the chromosomally assembled New Caledonian Crow reference genome. B) Jupiter plot mapping the scaffolds of a reference assembly for the closely related Florida Scrub-Jay to our “Haplotype 1” assembly. Scaffolds in our Haplotype 1 assembly are labeled as in Fig. 3A. The lack of homology with the W chromosome in our assembly was expected because a male (lacking the W chromosome) individual was sequenced to generate the Florida Scrub-Jay assembly.
Visualizing synteny with the Florida Scrub-Jay reference genome revealed limited evidence for structural rearrangements within the scrub-jay species complex, as expected among closely related avian species (Fig. 4B). This synteny comparison also highlights the increased contiguity of our assembly compared with the only previously available reference genome for the genus Aphelocoma. Specifically, our novel assembly offers a contig N50 nearly two orders of magnitude greater (11.53 Mb versus 134 kb; Table 2) than the current publicly available Florida Scrub-Jay reference genome (Feng et al. 2020).
Discussion
We present the first de novo reference genome assemblies for the California Scrub-Jay (Aphelocoma californica). We used state-of-the-art approaches for long-read sequencing (PacBio HiFi) and chromatin conformation capture (Dovetail Omni-C) to generate a highly contiguous and complete reference genome that reaches the threshold for “gold quality” avian genome assemblies (Kapusta and Suh 2017). The ~11.5 Mb contig N50 of both assemblies (primary and alternate) places them within the top 10% (top 100 overall) of all publicly available avian reference genomes in terms of contiguity (as of 13 February 2023; referenced from https://www.ncbi.nlm.nih.gov/data-hub/genome/?taxon=8782). Meanwhile, the BUSCO scores for our Haplotype 1 and 2 assemblies (97% and 93.3% of avian genes fully present, respectively) indicate that a large proportion of conserved avian genes can be found in their complete states in both assemblies. The exceptional completeness and contiguity statistics of these assemblies indicate that they will serve as an important public resource for investigators seeking to understand the genomics and evolutionary history of the scrub-jays and their allies, and more generally all birds, for years to come.
Assessing the genome size and repetitive content of the California Scrub-Jay
The two pseudo-haplotype assemblies we present here differ in overall genome size (1.35 and 1.19 Gb for the Haplotype 1 and 2 assemblies, respectively), flanking the 1.23 Gb kmer-based genome size estimate. Synteny mapping between these assemblies clearly reveals that scaffolds corresponding to both avian sex chromosomes (Z and W) are present in Haplotype 1 but absent in Haplotype 2. The missing sex chromosome scaffolds in the Haplotype 2 assembly are an artifact of the pseudo-haplotype approach, reflecting the fact that both of these chromosomes (W and Z) are present only in a single copy in individuals of the heterogametic sex, and by chance, each of these hemizygous copies was assigned to the Haplotype 1 assembly. This difference explains the gaps in both overall assembly size and repetitive content between our two pseudo-haplotype assemblies. We encourage investigators interested in a California Scrub-Jay reference genome assembly to use the Haplotype 1 assembly (GenBank assembly accession: GCA_028536675.1) if they wish to perform analyses involving sex chromosomes.
Notably, our pseudo-haplotype assemblies represent the largest assembled reference genomes to date from the avian family Corvidae (as of 17 March 2023; accessed from: https://www.ncbi.nlm.nih.gov/assembly/?term=corvidae). This unusually large genome size in A. californica appears to result from an expansion in repetitive elements, specifically LINEs, LTRs, and satellite DNA compared with other Corvid genomes (Benham et al. 2023). While assessing genome size across different assembly approaches is difficult (Barrière et al. 2009), our large genome assembly size is bolstered by previous genome size estimates based on c-values, which indicated that the A. coerulescens likely has the largest genome among Corvids (Andrews et al. 2009). Together, these lines of evidence tentatively support an increased genome size in the genus Aphelocoma, that may have been driven by an expansion in repetitive elements in this lineage. As more high-quality avian reference genomes with accurate size estimations become publicly available, our understanding of the evolution of avian genome size will continue to increase.
Assessing synteny across the avian family Corvidae
Our results add support to the well-documented evidence for extensive synteny across the avian genome (Ellegren 2010), but also provide evidence for some small structural rearrangements between the California Scrub-Jay and the New Caledonian Crow (estimated split ~10 MYA; McCullough et al. 2022). Meanwhile, there is very little visual evidence for structural rearrangements between California and Florida scrub-jays (estimated split ~5 MYA; McCormack et al. 2011), although the differing contiguity between these assemblies makes it difficult to conclude with confidence that we are not missing rearrangements split between scaffolds. Overall, it is difficult to determine whether the mismatches in these synteny plots result from real structural rearrangements or whether they are technical artifacts from the difficult and variable process of de novo genome assembly. Future work using high-quality de novo assemblies for all scrub-jay species will be necessary to better understand the prevalence and evolutionary implications of structural variation among this species complex (e.g. Höök et al. 2023).
Conclusions
We generated a high-quality de novo reference genome assembly for the California Scrub-Jay, with two phased pseudo-haplotype assemblies made publicly available via NCBI (PRJNA9044314 and PRJNA9044313). This expands the number of taxa within Aphelocoma for which a de novo reference genome assembly has been generated and sets a new bar for contiguity among Aphelocoma reference genomes. Future studies of the genus Aphelocoma will now be able to use this chromosome-level reference genome for mapping genomic sequence data. This will increase the genomic resolution of any future investigations aimed at identifying the genomic regions associated with environmental adaptation, genetic incompatibilities, or adaptive trait divergence (e.g. Aguillon et al. 2021) among the scrub-jays. Future comparative studies will also be able to leverage this assembly to assess evolutionary constraints on genome size and repetitive content across corvids and beyond. Overall, this de novo assembly represents a crucial addition to the pool of shared genomic resources for the highly studied scrub-jay species complex, which is rapidly developing as a natural model system for understanding how evolutionary forces shape patterns of extant biodiversity.
Acknowledgments
This project was completed under the guidance of renowned UCLA professor Bob Wayne (1956–2022), who was passionate about using the genome to understand evolution and guide conservation efforts, especially in iconic large mammals such as dogs and pumas. Following Bob’s passing, the American Genetic Association established the Robert K. Wayne Conservation Scholarship and Research Fund to support graduate students whose research directly benefits threatened species. The endowment fund for this scholarship can be directly supported by visiting the following webpage (www.theaga.org/donate). This group of authors plans to carry out ongoing work on the evolutionary genomics of scrub-jays in memory of Bob Wayne. PacBio Sequel II library prep and sequencing were carried out at the DNA Technologies and Expression Analysis Cores at the UC Davis Genome Center, supported by NIH Shared Instrumentation Grant 1S10OD010786-01. Deep sequencing of Omni-C libraries used the Novaseq S4 sequencing platforms at the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 OD018174 Instrumentation Grant. We thank the staff at the UC Davis DNA Technologies and Expression Analysis Cores and the UC Santa Cruz Paleogenomics Laboratory for their diligence and dedication to generating high-quality sequence data.
Contributor Information
Devon A DeRaad, Department of Ecology and Evolutionary Biology and Biodiversity Institute, University of Kansas, Lawrence, KS, United States.
Merly Escalona, Department of Biomolecular Engineering, University of California Santa Cruz, Santa Cruz, CA, United States.
Phred M Benham, Museum of Vertebrate Zoology, University of California, Berkeley, Berkeley, CA, United States; Department of Integrative Biology, University of California, Berkeley, Berkeley, CA, United States.
Mohan P A Marimuthu, DNA Technologies and Expression Analysis Core Laboratory, Genome Center, University of California-Davis, Davis, CA, United States.
Ruta M Sahasrabudhe, DNA Technologies and Expression Analysis Core Laboratory, Genome Center, University of California-Davis, Davis, CA, United States.
Oanh Nguyen, DNA Technologies and Expression Analysis Core Laboratory, Genome Center, University of California-Davis, Davis, CA, United States.
Noravit Chumchim, DNA Technologies and Expression Analysis Core Laboratory, Genome Center, University of California-Davis, Davis, CA, United States.
Eric Beraut, Department of Ecology and Evolutionary Biology, University of California, Santa Cruz, Santa Cruz, CA, United States.
Colin W Fairbairn, Department of Ecology and Evolutionary Biology, University of California, Santa Cruz, Santa Cruz, CA, United States.
William Seligmann, Department of Ecology and Evolutionary Biology, University of California, Santa Cruz, Santa Cruz, CA, United States.
Rauri C K Bowie, Museum of Vertebrate Zoology, University of California, Berkeley, Berkeley, CA, United States; Department of Integrative Biology, University of California, Berkeley, Berkeley, CA, United States.
Carla Cicero, Museum of Vertebrate Zoology, University of California, Berkeley, Berkeley, CA, United States.
John E McCormack, Moore Laboratory of Zoology, Occidental College, Los Angeles, CA, United States.
Robert K Wayne, Department of Ecology and Evolutionary Biology, University of California, Los Angeles, CA, United States.
Funding
This work was supported by the California Conservation Genomics Project, with funding provided to the University of California by the State of California, State Budget Act of 2019 [UC Award ID RSI-19-690224].
Data availability
The assembled reference genomes presented here have been made immediately publicly available at: NCBI BioProject PRJNA904314 (Pseudo-haplotype 1; https://www.ncbi.nlm.nih.gov/bioproject/PRJNA904314/) and NCBI BioProject PRJNA904313 (Pseudo-haplotype 2; https://www.ncbi.nlm.nih.gov/bioproject/PRJNA904313/). All raw sequence data generated here are also immediately publicly available via NCBI, including PacBio sequence data (accession: SRX19355857; https://trace.ncbi.nlm.nih.gov/Traces/?view=run_browser&acc=SRR23446543&display=metadata), OmniC data from NovaSeq flow cell 1 (accession: SRX19355858; https://www.ncbi.nlm.nih.gov/sra/SRX19355858), and flow cell 2 (accession: SRX19355859; https://www.ncbi.nlm.nih.gov/sra/SRX19355859). Assembly scripts and other data for the analyses presented can be found at the following GitHub repository: www.github.com/ccgproject/ccgp_assembly.
References
- Abdennur N, Mirny LA.. Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics. 2020:36:311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguillon SM, Fitzpatrick JW, Bowman R, Schoech SJ, Clark AG, Coop G, Chen N.. Deconstructing isolation-by-distance: the genomic consequences of limited dispersal. PLoS Genet. 2017:13:e1006911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguillon SM, Walsh J, Lovette IJ.. Extensive hybridization reveals multiple coloration genes underlying a complex plumage phenotype. Proc R Soc B. 2021:288:20201805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allio R, Schomaker-Bastos A, Romiguier J, Prosdocimi F, Nabholz B, Delsuc F.. MitoFinder: efficient automated large-scale extraction of mitogenomic data in target enrichment phylogenomics. Mol Ecol Resour. 2020:20:892–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews CB, Mackenzie SA, Gregory TR.. Genome size and wing parameters in passerine birds. Proc R Soc B Biol Sci. 2009:276:55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell E, Benkman CW, Gould WR.. Adaptive geographic variation in western scrub‐jays. Ecology. 2001:82:2617–2627. [Google Scholar]
- Barrière A, Yang SP, Pekarek E, Thomas CG, Haag ES, Ruvinsky I.. Detecting heterozygosity in shotgun genome assemblies: lessons from obligately outcrossing nematodes. Genome Res. 2009:19:470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benham PM, Cicero C, DeRaad DA, McCormack JE, Wayne R, Escalona M, Beraut E, Marimuthu MPA, Nguyen O, Nachman MW, et al. A highly contiguous reference genome for the Steller’s jay (Cyanocitta stelleri). J Hered. 2023:114:549–560. doi: 10.1093/jhered/esad042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg EC, Aldredge RA, Peterson AT, McCormack JE.. New phylogenetic information suggests both an increase and at least one loss of cooperative breeding during the evolutionary history of Aphelocoma jays. Evol Ecol. 2012:26:43–54. [Google Scholar]
- Brown JL. Alternate routes to sociality in jays—with a theory for the evolution of altruism and communal breeding. Am Zool. 1974:14:63–80. [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL.. BLAST+: architecture and applications. BMC Bioinformatics. 2009:10:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challis R, Richards E, Rajan J, Cochrane G, Blaxter M.. BlobToolKit—interactive quality assessment of genome assemblies. G3. 2020:10:1361–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheek RG, Forester BR, Salerno PE, Trumbo DR, Langin KM, Chen N, Scott Sillett T, Morrison SA, Ghalambor CK, Chris Funk W.. Habitat-linked genetic variation supports microgeographic adaptive divergence in an island-endemic bird species. Mol Ecol. 2022:31:2830–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics. 2004:5:4.10.1–4.10.14. [DOI] [PubMed] [Google Scholar]
- Chen N, Cosgrove EJ, Bowman R, Fitzpatrick JW, Clark AG.. Genomic consequences of population decline in the endangered Florida scrub-jay. Curr Biol. 2016:26:2974–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Jarvis ED, Fedrigo O, Koepfli K-P, Urban L, Gemmell NJ, Li H. Haplotype-resolved assembly of diploid genomes without parental data. Nat Biotech, 2022:40:1332–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J. Jupiter Plot: a Circos-based tool to visualize genome assembly consistency (1.0). Zenodo; 2018. doi: 10.5281/zenodo.1241235 [DOI] [Google Scholar]
- Cicero C, Mason NA, Oong Z, Title PO, Morales ME, Feldheim KA, Koo MS, Bowie RC.. Deep ecomorphological and genetic divergence in Steller’s Jays (Cyanocitta stelleri, Aves: Corvidae). Ecol Evol. 2022:12:1–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JK, Thomas PJ, Thomas JW; NISC Comparative Sequencing Program. A W-linked palindrome and gene conversion in New World sparrows and blackbirds. Chromosome Res. 2010:18:543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRaad DA, Maley JM, Tsai WL, McCormack JE.. Phenotypic clines across an unstudied hybrid zone in Woodhouse’s Scrub-Jay (Aphelocoma woodhouseii). The Auk. 2019:136:uky018. [Google Scholar]
- DeRaad DA, McCormack JE, Chen N, Peterson AT, Moyle RG.. Combining species delimitation, species trees, and tests for gene flow clarifies complex speciation in scrub-jays. Syst Biol. 2022:71:1453–1470. [DOI] [PubMed] [Google Scholar]
- Ellegren H. Evolutionary stasis: the stable chromosomes of birds. Trends Ecol Evol. 2010:25:283–291. [DOI] [PubMed] [Google Scholar]
- Feng S, Stiller J, Deng Y, Armstrong J, Fang Q, Reeve AH, Xie D, Chen G, Guo C, Faircloth BC, et al. Dense sampling of bird diversity increases power of comparative genomics. Nature. 2020:587:252–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghurye J, Pop M, Koren S, Bickhart D, Chin C-S.. Scaffolding of long read assemblies using long range contact information. BMC Genomics. 2017:18:527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghurye J, Rhie A, Walenz BP, Schmitt A, Selvaraj S, Pop M, Koren S, Koren S.. Integrating Hi-C links with assembly graphs for chromosome-scale assembly. PLoS Comput Biol. 2019:15:e1007273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goloborodko A, Abdennur N, Venev S, Brandao HB, Fudenberg G.. mirnylab/pairtools: v0.2.0. 2018. doi: 10.5281/zenodo.1490831 [DOI]
- Gowen FC, Maley JM, Cicero C, Peterson AT, Faircloth BC, Warr TC, McCormack JE.. Speciation in Western Scrub-Jays, Haldane’s rule, and genetic clines in secondary contact. BMC Evol Biol. 2014:14:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, Tesler G.. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013:29:1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höök L, Näsvall K, Vila R, Wiklund C, Backström N.. High-density linkage maps and chromosome level genome assemblies unveil direction and frequency of extensive structural rearrangements in wood white butterflies (Leptidea spp.). Chromosome Res. 2023:31:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapusta A, Suh A.. Evolution of bird genomes—a transposon’s-eye view. Ann N Y Acad Sci. 2017:1389:164–185. [DOI] [PubMed] [Google Scholar]
- Kerpedjiev P, Abdennur N, Lekschas F, McCallum C, Dinkla K, Strobelt H, Luber JM, Ouellette SB, Azhir A, Kumar N, et al. HiGlass: web-based visual exploration and analysis of genome interaction maps. Genome Biol. 2018:19:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korlach J, Gedman G, Kingan SB, Chin C-S, Howard JT, Audet J-N, Jarvis ED, Jarvis ED.. De novo PacBio long-read and phased avian genome assemblies correct and add to reference genes generated with intermediate and short reads. GigaScience. 2017:6:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM, arXiv, arXiv:1303.3997, 2013, doi: 10.48550/arXiv.1303.3997. [DOI]
- Manni M, Berkeley MR, Seppey M, Simao FA, Zdobnov EM.. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol Bio Evol, 2021:38:4647–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack JE, Heled J, Delaney KS, Peterson AT, Knowles LL.. Calibrating divergence times on species trees versus gene trees: implications for speciation history of Aphelocoma jays. Evolution. 2011:65:184–202. [DOI] [PubMed] [Google Scholar]
- McCormack JE, Peterson AT, Bonaccorso E, Smith TB.. Speciation in the highlands of Mexico: genetic and phenotypic divergence in the Mexican jay (Aphelocoma ultramarina). Mol Ecol. 2008:17:2505–2521. [DOI] [PubMed] [Google Scholar]
- McCullough JM, Oliveros CH, Benz BW, Zenil-Ferguson R, Cracraft J, Moyle RG, Andersen MJ.. Wallacean and Melanesian islands promote higher rates of diversification within the global passerine radiation Corvides. Syst Biol. 2022:71:1423–1439. [DOI] [PubMed] [Google Scholar]
- Pebesma E. Simple features for R: standardized support for spatial vector data. R J. 2018:10:439–446. [Google Scholar]
- Peterson AT. Adaptive geographical variation in bill shape of scrub jays (Aphelocoma coerulescens). Am Nat. 1993:142:508–527. [Google Scholar]
- Peterson AT, Burt DB.. Phylogenetic history of social evolution and habitat use in the Aphelocoma jays. Anim Behav. 1992:44:859–866. [Google Scholar]
- Pflug JM, Holmes VR, Burrus C, Johnston JS, Maddison DR.. Measuring genome sizes using read-depth, k-mers, and flow cytometry: methodological comparisons in beetles (Coleoptera). G3. 2020:10:3047–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitelka F. Speciation and ecologic distribution in American jays of the genus Aphelocoma. Univ Calif Publ Zool. 1951:50:195–464. [Google Scholar]
- R Core Team. R: a language and environment for statistical computing. Vienna (Austria): R Foundation for Statistical Computing; 2019. [Google Scholar]
- Ramírez F, Bhardwaj V, Arrigoni L, Lam KC, Grüning BA, Villaveces J, Habermann B, Akhtar A, Manke T, Manke T.. High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat Commun. 2018:9:1–15. doi: 10.1038/s41467-017-02525-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranallo-Benavidez TR, Jaron KS, Schatz MC.. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun. 2020:11:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhie A, McCarthy SA, Fedrigo O, Damas J, Formenti G, Koren S, Uliano-Silva M, Chow W, Fungtammasan A, Kim J, et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature. 2021:592:737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhie A, Walenz BP, Koren S, Phillippy AM.. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 2020:21:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice NH, Martínez-Meyer E, Peterson AT.. Ecological niche differentiation in the Aphelocoma jays: a phylogenetic perspective. Biol J Linn Soc. 2003:80:369–383. [Google Scholar]
- Rosa GL, Ellis JM, Bonaccorso E, dos Anjos L.. Friend or foe? Social system influences the allocation of signals across functional categories in the repertoires of the New World jays. Behaviour. 2016:153:467–524. [Google Scholar]
- Shaffer HB, Toffelmier E, Corbett-Detig RB, Escalona M, Erickson B, Fiedler P, Gold M, Harrigan RJ, Hodges S, Luckau TK, et al. Landscape genomics to enable conservation actions: the California Conservation Genomics Project. J Hered. 2022:113:577–588. [DOI] [PubMed] [Google Scholar]
- Sim SB, Corpuz RL, Simmonds TJ, Geib SM.. HiFiAdapterFilt, a memory efficient read processing pipeline, prevents occurrence of adapter sequence in PacBio HiFi reads and their negative impacts on genome assembly. BMC Genomics. 2022:23:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- South A. Rnaturalearth: world map data from natural earth. R package version 0.1.0. 2017.
- Strimas-Mackey M, Ligocki S, Auer T, Fink D.. ebirdst: tools for loading, plotting, mapping and analysis of eBird Status and Trends data products. R package version 1.0.0. 2021. https://cornelllabofornithology.github.io/ebirdst/ [Google Scholar]
- Toffelmier E, Beninde J, Shaffer HB.. The phylogeny of California, and how it informs setting multi-species conservation priorities. J Hered. 2022:113:597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uliano-Silva M, Nunes JGF, Krasheninnikova K, McCarthy SA.. marcelauliano/MitoHiFi: mitohifi_v2.0 (v2.0). Zenodo; 2021. doi: 10.5281/zenodo.5205678 [DOI] [Google Scholar]
- Venkatraman MX, DeRaad DA, Tsai WL, Zarza E, Zellmer AJ, Maley JM, Mccormack JE.. Cloudy with a chance of speciation: integrative taxonomy reveals extraordinary divergence within a Mesoamerican cloud forest bird. Biol J Linn Soc. 2019:126:1–15. [Google Scholar]
- Vinciguerra NT, Tsai WL, Faircloth BC, McCormack JE.. Comparison of ultraconserved elements (UCEs) to microsatellite markers for the study of avian hybrid zones: a test in Aphelocoma jays. BMC Res Notes. 2019:12:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H, Chang W, Henry L, Pedersen TL, Takahashi K, Wilke C, Woo K, Yutani H, Dunnington D. ggplot2: create elegant data visualisations using the grammar of graphics. R package, version 3.3.2. 2020. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The assembled reference genomes presented here have been made immediately publicly available at: NCBI BioProject PRJNA904314 (Pseudo-haplotype 1; https://www.ncbi.nlm.nih.gov/bioproject/PRJNA904314/) and NCBI BioProject PRJNA904313 (Pseudo-haplotype 2; https://www.ncbi.nlm.nih.gov/bioproject/PRJNA904313/). All raw sequence data generated here are also immediately publicly available via NCBI, including PacBio sequence data (accession: SRX19355857; https://trace.ncbi.nlm.nih.gov/Traces/?view=run_browser&acc=SRR23446543&display=metadata), OmniC data from NovaSeq flow cell 1 (accession: SRX19355858; https://www.ncbi.nlm.nih.gov/sra/SRX19355858), and flow cell 2 (accession: SRX19355859; https://www.ncbi.nlm.nih.gov/sra/SRX19355859). Assembly scripts and other data for the analyses presented can be found at the following GitHub repository: www.github.com/ccgproject/ccgp_assembly.