

Abstract
The growing incidence of infections caused by multi-drug resistant Gram-negative bacteria has led to an increased use of last-resort antibiotics such as the polymyxins. Polymyxin therapy is limited by toxicity concerns, most notably nephrotoxicity. Recently we reported the development of a novel class of semisynthetic polymyxins with reduced toxicity wherein the N-terminal lipid and diaminobutyric acid residue are replaced by a cysteine-linked lipid featuring a reductively labile disulfide bond. In the present study we further explored the potential of this approach by also varying the amino acid residue directly adjacent to the polymyxin macrocycle. This led to the identification of new semisynthetic polymyxins that maintain the potent antibacterial activity of the clinically used polymyxin B while exhibiting a further reduction in toxicity toward human proximal tubule epithelial cells. Furthermore, these new polymyxins were found to effectively synergize with novobiocin, rifampicin, and erythromycin against mcr-positive, polymyxin resistant E. coli.
A novel series of polymyxin analogues bearing a disulfide linked lipid tail are reported. The most promising variant identified exhibits antibacterial activity equipotent to that of polymyxin B and is >10-fold less toxic towards kidney cells.
Introduction
Among serious infections requiring antibacterial therapy, those due to drug-resistant Gram-negative bacteria form a particularly dangerous group. This is reflected in the World Health Organization's list of priority pathogens wherein drug-resistant A. baumannii, P. aeruginosa, and Enterobacteriaceae are the only members categorized as critical.1 In addition, a recent report indicates that more than half of the 1.27 M deaths registered in 2019 as attributable to drug-resistant bacterial infections were due to Gram-negative species.2 Despite the clear need, the development of new anti-Gram-negative antibiotics is limited due to both scientific and economic challenges.3 Among the compounds successfully developed to address Gram-negative pathogens in the past 20 years, the vast majority have been analogues of known classes discovered in the mid-20th century.4 A shining exception to this is GSK's gepotidacin, a new oral antibiotic for urinary tract infections, which recently performed well in phase III trials.5 In general, however, most new antibacterial drug discovery programs in recent years have failed to deliver novel therapies, often due to safety issues.6 A notable example in this case is the recently developed murepavadin which held great promise as a first-in-class narrow spectrum treatment for P. aeruginosa lung infections but which eventually failed in phase III trials due to excessive nephrotoxicity.7
With few new therapies available, older drugs for treating multi-drug resistant (MDR) Gram-negative infections remain a key component of the clinical antibiotic arsenal. This includes the polymyxins, a family of cyclic lipopeptide natural products that exclusively act against Gram-negative bacteria.8–10 Mechanistically, the polymyxins target lipid A, the central membrane-embedded anchor of bacterial lipopolysaccharide (LPS).11–13 While the activity of many antibiotics is limited by poor permeability across the Gram-negative outer membrane (OM), this is not an issue for the polymyxins, as their target is found on the outer surface of the OM. Upon binding to LPS in the OM, polymyxins effectively destabilize the OM and in the process enhance their own uptake to the periplasmic space by so-called ‘self-promoted uptake’.14 While polymyxin B (Fig. 1A) and the closely-related colistin both exhibit potent anti-Gram-negative activity and are used clinically, their systemic use is notoriously associated with toxicity, most notably nephrotoxicity.15–19 This nephrotoxicity is generally associated with the presence of the N-terminal lipid tail based on the finding that the polymyxin B nonapeptide (PMBN), resulting from removal of the lipid and N-terminal diaminobutric acid (Dab) residue, is much less toxic.20,21 With this knowledge, many medicinal chemistry campaigns have investigated the effect of modifying the exocyclic tripeptide and lipid moieties in pursuit of polymyxin analogues with reduced kidney cell toxicity.22–25 Notably, analogues that maintain the structure of the peptide macrocycle found in polymyxin B or colistin also allow for convenient semisynthetic strategies starting from readily available polymyxin B or colistin. To this end our group recently reported a new series of semisynthetic polymyxins wherein the lipidated Dab residue at the N-terminus was replaced by a Cys residue linked to a variety of lipids via a side chain disulfide (Fig. 1B).26 The rationale for doing so was based upon the hypothesis that such analogues should be unstable in the more highly reducing environment found inside renal proximal tubule cells,27 thereby degrading to less toxic metabolites analogous to PMBN. Using this approach, we successfully generated a number of novel polymyxins with antibacterial activity on par with polymyxin B and reduced toxicity towards human renal proximal tubular epithelial cells (PTECs). In the present study we expand upon this approach by exploring the impact of additional structural variations in the exocyclic tripeptide of polymyxin B, most notably at the 3-position. The 3-position is known to be a site of variation among naturally occurring polymyxin variants such as polymyxin A and polymyxin D which contain d-Dab or d-Ser at this position, respectively.10 This led researchers at Pfizer to further explore such substitutions leading to the identification of polymyxin derivatives with equal potency and reduced toxicity.24 Variation of the 3-position has subsequently been evaluated in other recently reported polymyxins now in preclinical/clinical development including: NAB-739 (containing d-Ser at position 3),28 and SPR206 and QPX9003 (both containing diaminopropionic acid (Dap) at position 3).23,29
Fig. 1. A) Clinically used polymyxin B with common residue number-ing indicated. B) Semisynthetic polymyxins containing reductively labile disulfide-linked lipid at the N-terminus.

Given the structural variability of the polymyxin 3-position in generating analogues with reduced nephrotoxicity, we set out to examine such substitutions in combination with our previously validated disulfide-linked lipidation strategy. In doing so we applied a robust semisynthetic strategy that provided ready access to a panel of novel polymyxins that were subsequently screened for antibacterial activity and toxicity towards PTECs. This led to the identification of a subset of compounds displaying potent activity equivalent to that of polymyxin B along with a further reduction in kidney cell toxicity relative to our first-generation compounds. In addition, we confirmed the capacity for these new polymyxins to synergize with Gram-positive specific antibiotics in overcoming polymyxin resistance among strains positive for plasmid-borne mobile colistin resistance (mcr).
Results and discussion
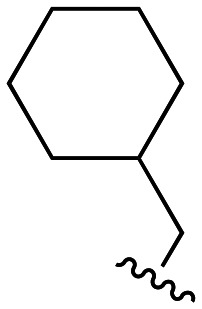
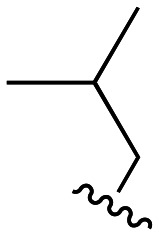
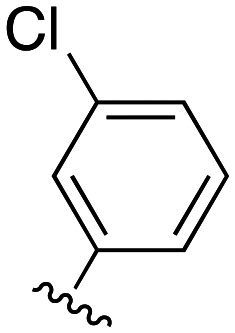
Our previous investigations involving polymyxin variants containing reductively labile lipids revealed that the incorporation of lipidated d-Cys based building blocks at the N-terminus consistently gave the most active compounds.26 Also, among the lipids screened in these studies we identified three moieties that were generally associated with potent antibacterial activity: ethylcyclohexyl, isopentyl, and m-chlorobenzyl. In the present study we therefore elected to focus on position 3 analogues wherein the N-terminal moiety was based on d-Cys linked to these three lipids by means of a disulfide bond. In considering options for the preparation of our new position 3 analogues we elected for a semisynthetic strategy employing the appropriately tri-Boc protected heptapeptide PMBH(Boc)3 building block accessible via enzymatic degradation of polymyxin B (Scheme 1A).30 In doing so, a two-step/one-pot procedure was followed wherein all Dab side chains were first Boc protected by treatment with Boc anhydride in a water/acetonitrile mixture. After complete protection (as indicated by LC/MS analysis) the industrial enzyme Savinase® was directly added as aqueous solution leading to selective cleavage between the Dab residue at position 3 and the heptapeptide macrocycle. While we initially followed the chromatographic purification of PMBH(Boc)3 reported in the literature, we subsequently developed an operationally simpler extractive work up of the crude digestion mixture resulting in isolation of the product in better yield and purity.
Scheme 1. Semisynthesis of polymyxin analogues 7a–e, 8a–e, and 9a–e: A) synthesis of PMBH(Boc)3 (3). Conditions: a) (Boc)2O, Et3N, H2O/ACN, RT, 3 h; b) Savinase®, H2O/ACN, pH 9, RT (till completed), followed by extractive work-up; B) synthesis of protected tripeptides 4a–e, 5a–e, 6a–e. Conditions: c) i. Fmoc-protected AA, DIPEA, CTC resin, RT, 2 h; ii. piperidine/DMF; iii. Fmoc-Thr(OtBu)-OH, BOP, DIPEA; d) i. piperidine/DMF; ii. d-Cys-disulfide building block I, II, or III, BOP, DIPEA; e) HFIP/DCM, 1.5 h, RT; C) synthesis of polymyxin analogues 7a–e, 8a–e, 9a–e. Conditions: f) HCTU, HOBt, DIPEA, DMF, RT, 3 h; g) TFA/TIPS/H2O, RT, 1.5 h followed by RP-HPLC. *Synthesis previously reported.26.

We next turned our attention to the synthesis of the required protected tripeptides containing the disulfide-linked acyl tail, which were prepared via solid phase peptide synthesis on 2-chlorotrityl chloride resin (Scheme 1B). In doing so a series of tripeptides was prepared bearing either d-Ser, Gly, d-Dap, l-Dap or d-Dab at position 3 along with ethylcyclohexyl, isopentyl, or m-chlorobenzyl moieties connected to the N-terminal d-Cys via a disulfide linkage. The requisite d-Cys lipid-disulfide building blocks were prepared as previously reported.26 Resin cleavage using mildly acidic conditions yielded protected tripeptides 4a–e, 5a–e, and 6a–e in purities suitable for direct use in the subsequent coupling to PMBH(Boc)33 (Scheme 1C). The solution phase coupling was performed using HCTU/HOBt after which the peptides were fully deprotected by treatment with TFA and purified by RP-HPLC to give 7a–e, 8a–e, and 9a–e. Of note, analogues 7f, 8f, and 9f, all bearing the native Dab at position 3, were prepared using our previously reported semisynthetic route making use of PMBN(Boc)4.26
Antibacterial assays
The antibacterial activities of polymyxins 7a–f, 8a–f, and 9a–f were assessed against a panel of Gram-negative bacteria and compared with that of polymyxin B. Table 1 presents these data grouped based on the nature of the N-terminal hydrophobic moiety with compounds 7a–f bearing the cyclohexyl, 8a–f the isopentyl, and 9a–f the m-chlorobenzyl groups respectively. Compounds 7a–e, containing alternate amino acids at position 3, exhibit some enhancement of activity relative to 7f which bears a Dab residue at this position as in polymyxin B. In general, the minimum inhibitory concentration (MIC) values measured for compounds 7a–e were comparable and slightly elevated relative to those measured for polymyxin B. By comparison, compounds 8a–f bearing the N-terminal isopentyl motif, exhibited enhanced antibacterial activities with some compounds exhibiting MICs equal to or lower than those measured for polymyxin B. Most notable in this regard is analogue 8d where position 3 is substituted with a Dap residue. Compounds 9a–f containing the m-chlorobenzyl motif, were also generally more active than 7a–f but slightly less active than 8a–f. Overall, these data indicate that among the d-Cys disulfide-linked lipid polymyxins here evaluated, those bearing the isopentyl moiety are most active with compound 8d exhibiting consistently potent antimicrobial activity (highlighted in bold in Table 1).
MIC data for polymyxin analogues 7a–f, 8a–f, and 9a–f. Values are expressed in μg mL−1 (backgrounds of all strains used are provided in Table S3†).
| E. coli | K. pneumoniae | A. baumannii | P. aeruginosa | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lipid | Position 3 residue | ATCC 25922 | 1313 | NCTC 13846a | ATCC 13883 | JS-123 | NCTC 13443 | ATCC 19606 | ATCC 17978 | MDR | 2018–006 | ATCC 27853 | NRZ 08418 | NRZ 03961 | |
| Polymyxin B | Dab | 1 | 0.125 | 4 | 0.25 | 0.25 | 0.5 | 0.25 | 0.25 | 0.5 | 0.125 | 1 | 1 | 1 | |
| 7a |
|
d-Ser | 2 | 1 | 8 | 1 | 1 | 1 | 0.25 | 1 | 1 | 0.5 | 4 | 4 | 4 |
| 7b | Gly | 2 | 0.5 | 8 | 1 | 0.5 | 1 | 0.5 | 0.5 | 1 | 0.5 | 2 | 2 | 4 | |
| 7c | d-Dap | 2 | 0.5 | 4 | 1 | 1 | 1 | 0.5 | 1 | 1 | 0.5 | 2 | 2 | 2 | |
| 7d | Dap | 1 | 0.5 | 2 | 1 | 0.5 | 1 | 0.5 | 1 | 1 | 0.5 | 2 | 2 | 2 | |
| 7e | d-Dab | 2 | 0.5 | 4 | 1 | 1 | 1 | 0.5 | 1 | 1 | 0.5 | 2 | 2 | 2 | |
| 7f | Dab | 1 | 2 | 8 | 1 | 2 | 2 | 0.5 | 1 | 1 | 0.5 | 2 | 4 | 4 | |
| 8a |
|
d-Ser | 2 | 0.5 | 16 | 0.5 | 0.5 | 1 | 0.25 | 0.25 | 0.5 | 0.5 | 4 | 8 | 8 |
| 8b | Gly | 2 | 0.25 | 16 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 4 | 4 | 4 | |
| 8c | d-Dap | 2 | 0.5 | 8 | 0.5 | 0.5 | 0.5 | 0.25 | 0.25 | 0.25 | 0.25 | 1 | 4 | 2 | |
| 8d | Dap | 1 | ≤0.125 | 4 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 1 | 1 | 1 | |
| 8e | d-Dab | 2 | 0.25 | 8 | 0.5 | 0.25 | 0.5 | 0.25 | 0.25 | 0.5 | 0.25 | 1 | 2 | 2 | |
| 8f | Dab | 1 | ≤0.125 | 8 | 0.5 | 0.25 | 0.5 | 0.5 | 1 | 1 | 0.5 | 1 | 1 | 2 | |
| 9a |
|
d-Ser | 2 | 0.5 | 8 | 1 | 0.25 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 4 | 4 | 8 |
| 9b | Gly | 2 | 0.25 | 8 | 1 | 0.5 | 0.5 | 1 | 1 | 1 | 1 | 4 | 4 | 4 | |
| 9c | d-Dap | 2 | 0.5 | 16 | 0.5 | 2 | 8 | 0.5 | 0.5 | 0.5–1 | 0.5 | 1 | 0.5 | 2 | |
| 9d | Dap | 2 | ≤0.125 | 2 | 0.5 | 0.5 | 2 | 0.5 | ≤0.25 | 8 | 2 | 4 | 2 | 2 | |
| 9e | d-Dab | 2 | 0.5 | 4 | 1 | 0.5 | 0.5 | 0.5 | 1 | 1 | 0.5 | 2 | 2 | 2 | |
| 9f | Dab | 1 | 0.125 | 4 | 0.5 | 0.25 | 0.25 | 1 | 1 | 4 | 2 | 4 | 2 | 1 | |
Multi-drug resistant strain, mcr-1 positive, making it intermediate resistant to polymyxin B.
Evaluation of kidney cell toxicity
The nephrotoxicity of the polymyxins is primarily based on their effects on proximal tubule cells (PTECs) which are known to accumulate polycationic agents.30 As such, PTECs are regularly used for evaluating the toxicity of polymyxins and related compounds.23,24,26 In the present study, differentiated PTECs were therefore exposed to 7a–f, 8a–f, 9a–f, as well as polymyxin B, at concentrations of 100, 250, and 500 μM for 24 hours, followed by viability read-out. In line with expectation, incubation with polymyxin B at 250 μM led to pronounced cytotoxic effect with an associated cellular viability of only 15% (Fig. 2, S1 and S2†). By comparison, the majority of the new analogues tested exhibited much lower levels of toxicity (Fig. 2). When grouped based on the N-terminal hydrophobic moiety, a clear trend is observed with the isopentyl substituted analogues 8a–f exhibiting the lowest level of toxicity towards PTECs. The importance of the amino acid featured at position 3 is also evident from these data: notably compounds 7f, 8f, and 9f which all contain Dab at position 3, as for polymyxin B, exhibited >50% cytotoxicity. The effect of variation at this position is striking with Gly-for-Dab substitution, as in 7b, 8b, and 9b, consistently leading to reduced toxicity. Especially noteworthy in this regard is compound 8d, which bears a Dap residue at position 3 and showed the lowest toxicity among all analogues prepared. Based on these findings, a more comprehensive analysis of the toxicity of 8d was conducted revealing it to have a TC50 of 1.48 mM on PTECs versus 0.13 mM for polymyxin B (Fig. S3†). Such a >10-fold reduction in cellular toxicity relative to polymyxin B is comparable to that seen for other polymyxin analogues that have previously been progressed for (pre)clinical development.23,24
Fig. 2. Viability of PTECs upon incubation with polymyxin analogues. Cells were exposed to the compound at a concentration of 250 μM for 24 hours, followed by washing, and incubation with resazurin (PrestoBlueTM) and fluorescence read-out. Data are normalized to positive control (medium treated cells). Represented date are from triplicates.

Synergist potential of polymyxin analogue 8d
The low toxicity of 8d towards PTECs, combined with its potent antibacterial activity, prompted us to further assess its antimicrobial activity against an expanded panel of Gram-negative bacteria, consistently showing it to be on par with polymyxin B (ESI,† Table S1). Recent studies have also revealed the capacity for polymyxin B to behave as a synergist in potentiating the activity of other antibiotics against polymyxin resistant strains.31 Specifically, antibiotics that are typically only effective on Gram-positive bacteria can be made active against Gram-negative bacteria in the presence of a compound that enhances their passage across the OM.32–34 Among the best studied such synergists is PMBN which, though inactive itself, strongly potentiates the activity of antibiotics like novobiocin, rifampicin, and erythromycin on Gram-negative bacteria.33,35,36 Interestingly, while the potentiating effect of PMBN is severely diminished against polymyxin-resistant bacteria, full-length polymyxins do exhibit synergistic activity against such strains.13,31 We were therefore interested in also assessing the capacity of our novel polymyxin 8d to potentiate the activity of Gram-positive specific antibiotics against polymyxin-resistant bacteria. Using checkerboard assays with an mcr-2 positive E. coli isolate, we found novobiocin, rifampicin, and erythromycin to be strongly potentiated by 8d, with calculated fractional inhibitory concentration index (FICi) values well below 0.5 in all cases (Fig. 3). Notably, low concentrations of 8d (0.5–1.0 μg mL−1) were sufficient to effectively potentiate the activity of these antibiotics against this polymyxin resistant strain. Furthermore, the synergy exhibited by 8d is comparable to that observed for polymyxin B (see ESI† Fig. S4, Table S2). The capacity for sub-MIC concentrations of polymyxins to synergize with other clinically used antibiotics may provide a means of safer administration of these potent but nephrotoxic agents. In this regard, the finding that our novel polymyxins show similar synergistic activity while also exhibiting lower kidney cell toxicity is noteworthy.
Fig. 3. Checkerboard assays with 8d, combined with antibiotics typically used for Gram-positive infections only: novobiocin (left), rifampicin (middle), erythromycin (right) against an mcr-2 positive polymyxin-resistant E. coli isolate. Color intensity corresponds to growth (as read by OD600 measurements), with white areas indicating no growth. Each square represents data from a technical triplicate.

Conclusion
Polymyxin B and colistin are increasingly used to treat infections caused by MDR Gram-negative bacteria. Clinical application of these agents for systemic use is hindered by their dose-limiting nephrotoxicity. To address this issue, our group recently reported a new series of semisynthetic polymyxins that contain a reductively labile disulfide linkage between the peptide and lipid moieties leading to reduced kidney cell toxicity relative to polymyxin B.26 We here describe efforts directed at further developing this concept by introducing various amino acid substitutions at position 3 which has previously been shown to be amenable to modifications that can attenuate polymyxin activity and toxicity. In general, substituting the parent Dab residue at position 3 in our disulfide containing polymyxins with l-Dap, d-Dap, d-Ser, Gly or d-Dab was found to further reduce in vitro kidney toxicity. Most notably, compound 8d including l-Dap at position 3, in combination with a disulfide-linked isopentyl moiety attached to the N-terminal d-Cys, was found to outperform all the other analogues, both in terms of antibacterial activity and kidney cell toxicity. Compound 8d exhibits the same potency as polymyxin B towards Gram-negative pathogens while being >10-fold less toxic towards PTECs. Also of note is the finding that 8d effectively synergizes with novobiocin, rifampicin, and erythromycin – antibiotics typically used for the treatment of Gram-positive infections – against polymyxin-resistant mcr-positive E. coli. Taken together, the improved balance between antibacterial activity and kidney cell toxicity observed for these next-generation polymyxins is encouraging and suggests that such design strategies may serve to address the shortcomings associated with the polymyxins as they remain an important class of last-resort antibiotics.
Experimental section
Reagents
All reagents employed were of American Chemical Society (ACS) grade or finer and were used without further purification unless otherwise stated. Commercially sourced polymyxin B was obtained as a mixture of isomers (Combi-Blocks, San Diego, USA), with polymyxin B1, B2, and B3 accounting for >90% of the isomers. The Boc-d-Cys based building blocks I, II and III were prepared as previously described.26
General procedures
For compound characterization, HRMS analysis was performed on a Shimadzu Nexera X2 UHPLC system with a Waters Acquity HSS C18 column (2.1 × 100 mm, 1.8 μm) at 30 °C and equipped with a diode array detector. The following solvent system, at a flow rate of 0.5 mL min−1, was used: solvent A, 0.1% formic acid in water; solvent B, 0.1% formic acid in acetonitrile. Gradient elution was as follows: 95 : 5 (A/B) for 1 min, 95 : 5 to 15 : 85 (A/B) over 6 min, 15 : 85 to 0 : 100 (A/B) over 1 min, 0 : 100 (A/B) for 3 min, then reversion back to 95 : 5 (A/B) for 3 min. This system was connected to a Shimadzu 9030 QTOF mass spectrometer (ESI ionization) calibrated internally with Agilent's API-TOF reference mass solution kit (5.0 mM purine, 100.0 mM ammonium trifluoroacetate and 2.5 mM hexakis(1H,1H,3H-tetrafluoropropoxy)phosphazine) diluted to achieve a mass count of 10 000.
Purity of the peptides was confirmed to be ≥95% by analytical RP-HPLC using a Shimadzu Prominence-i LC-2030 system with a Dr. Maisch ReproSil Gold 120 C18 column (4.6 × 250 mm, 5 μm) at 30 °C and equipped with a UV detector monitoring at 214 nm. The following solvent system, at a flow rate of 1 mL min−1, was used: solvent A, 0.1% TFA in water/acetonitrile, 95/5; solvent B, 0.1% TFA in water/acetonitrile, 5/95. Gradient elution was as follows: 100 : 0 (A/B) for 3 min, 100 : 0 to 0 : 100 (A/B) over 47 min, 0 : 100 (A/B) for 4 min, then reversion back to 100 : 0 (A/B) over 1 min, 100 : 0 (A/B) for 5 min.
Synthesis of protected tripeptides 4a–e, 5a–e, 6a–e
The protected tripeptides 4a–e, 5a–e, and 6a–e were synthesized on 2-chlorotrityl chloride (CTC) resin, employing general solid phase peptide synthesis methods. To begin, CTC resin was functionalized with either Fmoc-l-Dap(Boc), Fmoc-d-Dap(Boc), Fmoc-d-Dab(Boc), Fmoc-d-Ser(tBu) or Fmoc-Gly, all coupled to the resin via the carboxyl moiety. Typical loadings were 0.4–0.7 mmol per gram of resin. Peptides were synthesized on a 0.12 mmol scale. Fmoc deprotections were carried out using 20% piperidine in DMF (5 minutes + 25 minutes), followed by washings (DCM, DMF). Coupling of Fmoc-Thr(OtBu) was done in DMF for 1 hour with an amino acid : BOP : DIPEA ratio of 4 : 4 : 8, relative to the resin. The Boc-d-Cys based building blocks I, II and III (prepared as previously reported26) were coupled in DMF for 3 hours, with an amino acid : BOP : HOBt : collidine (2,4,6-trimethylpyridine) ratio of 2 : 2 : 2 : 4, relative to the resin. After synthesis completion, peptides were washed (DMF, DCM), followed by resin detachment with 20% hexafluoroisopropanol (HFIP) in DCM (1.5 hours). HFIP was removed by rotary evaporation, yielding the protected peptides which were used directly in the next step.
Synthesis of PMBH(Boc)3
The protected cyclic peptide PMBH(Boc)3 was obtained after enzymatic digestion of Boc protected polymyxin B.30 Polymyxin B sulfate (1.5 g, 1.0 mmol) was dissolved in a mixture of acetonitrile (15 mL) and water (7.5 mL). Triethylamine (0.89 mL, 6.4 mmol) was added, and the mixture was stirred for 5 minutes. Di-tert-butyl dicarbonate (1.4 g, 6.4 mmol) was added in one portion and mixture was stirred at RT for 1.5 hours. Savinase® 16 L (4.5 mL) was added and pH was adjusted to 9 by addition of NaOH (1 M). After overnight digestion, additional Savinase® (2 mL) and NaOH (1 M, till mixture achieved pH 9) was added and the digestion was again left to run overnight. After completion, water (30 mL) was added and the mixture was extracted with MTBE (2 × 60 mL). Combined organics were washed with NaOH (1 M, 30 mL) and water (30 mL), three times. The organic layer was washed with brine, dried over Na2SO4 and concentrated in vacuo to give PMBH(Boc)3 as a yellowish powder. Yield: 0.9 g, 0.85 mmol, 85%.
General procedure for synthesis of polymyxin analogues 7a–e, 8a–e, and 9a–e
The relevant protected tripeptide (4a–e, 5a–e, 6a–e, 2 eq.) was dissolved in DCM and pre-activated by addition of collidine (2,4,6-trimethylpyridine, 3.8 eq.), O-(6-chlorobenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HCTU, 1.9 eq.), and HOBt (1.9 eq.) (both dissolved in DMF). After 5 minutes, the mixture was added to PMBH(Boc)3 (1 eq.) previously dissolved in DCM and the reaction left for 3 hours at RT. After completion, the mixture was concentrated by rotary evaporation, followed directly by deprotection with TFA/TIPS/H2O (95/2.5/2.5, 90 min). The mixture was then added drop-wise to ice-cold MTBE (60 mL) to precipitate the peptide. After lyophilization from tBuOH/H2O, the crude peptide was purified by RP-HPLC.
Optimized synthesis of analogue 8d
Protected peptide 5d (80 mg, 0.12 mmol, 1 eq.) and PMBH(Boc)3 (0.17 g, 0.16 mmol, 1.3 eq.) were dissolved in DCM/DMF (8/3 mL). To the stirred solution was added DIPEA (63 μL, 0.36 mmol, 3 eq.), and after stirring for 5 min, O-(7-azabenzotriazol-1-yl)-N,N,N′N′-tetramethyluronium hexafluorophosphate (HATU, 46 mg, 0.12 mmol, 1 eq.) was added. The reaction was left to proceed at RT for 18 h. The reaction mixture was then diluted with water (15 mL) and extracted with MTBE (40 mL). The organic layer was washed with KHSO4 (1 M, 15 mL), NaHCO3 (sat. aq., 15 mL) and brine (10 mL), followed by drying over Na2SO4 and concentration via rotary evaporation. The crude peptide was then deprotected by treatment with TFA/TIPS/H2O (95/2.5/2.5, 15 mL, 2 h). The sample was partially concentrated and the crude peptide precipitated addition to ice-cooled MTBE/PE (2/1). After lyophilization from tBuOH/H2O, the crude peptide was purified by RP-HPLC. Yield: 0.10 g, 0.059 mmol, 49% (based on penta-TFA salt).
RP-HPLC purification
Final compounds were purified via preparative HPLC using a BESTA-Technik system with a Dr. Maisch Reprosil Gold 120 C18 column (25 × 250 mm, 10 μm) and equipped with an ECOM Flash UV detector monitoring at 214 nm. The following solvent system, at a flow rate of 12 mL min−1, was used: solvent A, 0.1% TFA in water/acetonitrile 95/5; solvent B, 0.1% TFA in water/acetonitrile 5/95. Gradient elution was as follows: 100 : 0 (A/B) for 3 min, 100 : 0 to 85 : 15 (A/B) over 2 min, 85 : 15 to 40 : 60 (A/B) over 45 min, 40 : 60 to 0 : 100 (A/B) over 2 min, 0 : 100 (A/B) for 3 min, then reversion back to 100 : 0 (A/B) over 1 min, 100 : 0 (A/B) for 3 min.
MIC assays
Minimum inhibitory concentrations (MICs) on relevant Gram-negative bacteria were determined by broth microdilution. Indicated bacteria were taken from glycerol stocks and incubated overnight on blood agar at 37 °C. Well-isolated colonies were taken, suspended in TSB (5 mL) and grown to an OD600 of 0.5. To be tested compounds were dissolved in DMSO (6.4 mg mL−1), diluted in cation adjusted Mueller-Hinton Broth (CAMHB) (12.5 and 20 μg mL−1 Mg2+ and Ca2+, respectively) and diluted serially on polypropylene microtiter plates. To the diluted compounds (50 μL per well) was added 50 μL of relevant bacterial suspension, to yield a final concentration of 106 CFU mL−1. Plates were covered by adhesive gas permeable membranes and incubated at 37 °C. MIC was read out as the lowest concentration that inhibited visual bacterial growth. Shown values are consistent results from at least triplicate experiments.
Checkerboard assays
Combinations of antibiotics and synergists were assessed in an 8 × 8 format, evaluating 64 combinations. E. coli EQAS mcr-2 was taken from glycerol stocks and incubated overnight on blood agar at 37 °C. Well-isolated colonies were taken, suspended in TSB (5 mL) and grown to an OD600 of 0.5. To be tested antibiotics and synergists were diluted in cation adjusted Mueller-Hinton Broth (CAMHB) (12.5 and 20 μg mL−1 Mg2+ and Ca2+, respectively). Next, for each combination of antibiotic and synergist, one was diluted serially on polypropylene microtiter plates, whereas the other was diluted serially in multi-well reservoirs. Antibiotics (25 μL) and synergists (25 μL) were combined in checkerboard format on 96 well plates, yielding 64 individual combinations, each in triplicate. To the mixed compounds was added 50 μL of bacterial suspension, to yield a final concentration of 106 CFU mL−1. Plates were covered by adhesive gas permeable membranes and incubated at 37 °C for 18 hours. The resulting bacterial suspension was homogenized, and bacterial growth was read by OD600 measurements on a Tecan plate reader. The resulting values are represented in checkerboard format, normalized by the positive control (maximum growth) and negative control (no growth). The fractional inhibitory concentration index (FICi) is calculated as follows: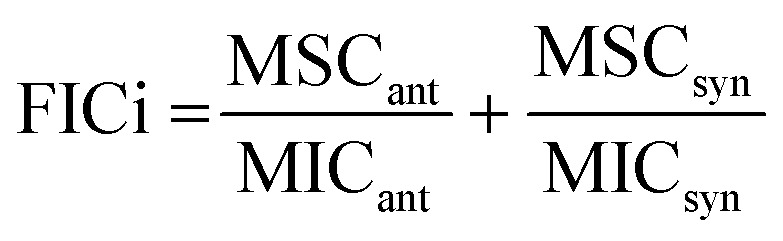 with MSCant and MSCsyn the optimal synergetic concentration of antibiotic and synergist respectively. MICant and MICsyn indicate the MICs of the individual compounds. An FICi ≤ 0.5 indicates synergy.37
with MSCant and MSCsyn the optimal synergetic concentration of antibiotic and synergist respectively. MICant and MICsyn indicate the MICs of the individual compounds. An FICi ≤ 0.5 indicates synergy.37
Assessment of cell-based toxicity on PTECs
Conditionally immortalized PTECs (ciPTECs) were used to assess the toxicity of the polymyxin analogues. The specific ciPTEC-14.4 cell line used (RRID: CVCL_W184) was purchased from Cell4Pharma (Oss, the Netherlands), obtained at passage 38, and cultured as reported previously.38 Mycoplasma contamination was checked monthly and was found to be negative in all cells used. Compounds were dissolved at 100, 250, 500 or 1000 μM in serum free medium [DMEM/F-12, supplemented with insulin (5 μg mL−1), transferrin (5 μg mL−1), selenium (5 μg mL−1), hydrocortisone (35 ng mL−1), epidermal growth factor (10 ng mL−1) and triiodothyronine (40 pg mL−1)]. Differentiated cells were washed once with HBSS and compounds were transferred to the ciPTECs containing plate (80 μL per well). Compounds were incubated for 24 hours at 37 °C. Cells were washed by HBSS, followed by incubation with 10% PrestoBlue™ cell viability reagent (Thermo Scientific, Vienna, Austria) in HBSS at 37 °C in the dark. Fluorescence was recorded (excitation: 530 nm, emission: 590 nm). The raw data are corrected for PrestoBlue™ background fluorescence and reported relative to the non-treatment control (cells treated with medium only).
Abbreviations
- ACN
Acetonitrile
- ATCC
American type culture collection
- BOP
(Benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate
- CAMHB
Cation adjusted Mueller-Hinton broth
- CFU
Colony forming units
- CTC
Chloro trityl chloride
- Dab
2,4-Diaminobutyric acid
- Dap
2,3-Diaminopropionic acid
- DIPEA
N,N-Di-isopropylethylamine
- DMEM
Dulbecco's modified Eagle medium
- FICi
Fractional inhibitory concentration index
- HATU
O-(7-Azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- HCTU
O-(6-Chlorobenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- HBSS
Hancks' balanced salt solution
- MIC
Minimal inhibitory concentration
- MSC
Minimal synergistic concentration
- NCTC
National collection of type cultures
- o/n
Overnight
- OM
Outer membrane
- PE
Petroleum ether
- PMBN
Polymyxin B nonapeptide
- PMBH
Polymyxin B heptapeptide
- PTEC
Proximal tubule epithelial cell
- RP-HPLC
Reversed phase high performance liquid chromatography
- TC50
Half-maximum toxicity concentration
Conflicts of interest
The authors declare no competing financial interest.
Supplementary Material
Acknowledgments
Funding was provided by the European Research Council (ERC consolidator grant to NIM, grant agreement no. 725523). Paolo Innocenti is kindly acknowledged for HRMS analysis and critical reading of the manuscript. TOC figure was created using BioRender, https://biorender.com.
Electronic supplementary information (ESI) available: Molecular formula strings, analytical data including HRMS values, analytical HPLC traces, and supplemental tables and figures. See DOI: https://doi.org/10.1039/d3md00456b
References
- World Health Organization, Prioritization of pathogens to guide discovery, research and development of new antibiotics for drug-resistant bacterial infections, including tuberculosis, WHO, Geneva, 2017 [Google Scholar]
- Murray C. J. L. Ikuta K. S. Sharara F. Swetschinski L. Aguilar G. R. Gray A. Han C. Bisignano C. Rao P. Wool E. Johnson S. C. Browne A. J. Chipeta M. G. Fell F. Hackett S. Haines-Woodhouse G. Hamadani B. H. K. Kumaran E. A. P. McManigal B. Agarwal R. Akech S. Albertson S. Amuasi J. Andrews J. Aravkin A. Ashley E. Bailey F. Baker S. Basnyat B. Bekker A. Bender R. Bethou A. Bielicki J. Boonkasidecha S. Bukosia J. Carvalheiro C. Castañeda-Orjuela C. Chansamouth V. Chaurasia S. Chiurchiù S. Chowdhury F. Cook A. J. Cooper B. Cressey T. R. Criollo-Mora E. Cunningham M. Darboe S. Day N. P. J. De Luca M. Dokova K. Dramowski A. Dunachie S. J. Eckmanns T. Eibach D. Emami A. Feasey N. Fisher-Pearson N. Forrest K. Garrett D. Gastmeier P. Giref A. Z. Greer R. C. Gupta V. Haller S. Haselbeck A. Hay S. I. Holm M. Hopkins S. Iregbu K. C. Jacobs J. Jarovsky D. Javanmardi F. Khorana M. Kissoon N. Kobeissi E. Kostyanev T. Krapp F. Krumkamp R. Kumar A. Kyu H. H. Lim C. Limmathurotsakul D. Loftus M. J. Lunn M. Ma J. Mturi N. Munera-Huertas T. Musicha P. Mussi-Pinhata M. M. Nakamura T. Nanavati R. Nangia S. Newton P. Ngoun C. Novotney A. Nwakanma D. Obiero C. W. Olivas-Martinez A. Olliaro P. Ooko E. Ortiz-Brizuela E. Peleg A. Y. Perrone C. Plakkal N. Ponce-de-Leon A. Raad M. Ramdin T. Riddell A. Roberts T. Robotham J. V. Roca A. Rudd K. E. Russell N. Schnall J. Scott J. A. G. Shivamallappa M. Sifuentes-Osornio J. Steenkeste N. Stewardson A. J. Stoeva T. Tasak N. Thaiprakong A. Thwaites G. Turner C. Turner P. van Doorn H. R. Velaphi S. Vongpradith A. Vu H. Walsh T. Waner S. Wangrangsimakul T. Wozniak T. Zheng P. Sartorius B. Lopez A. D. Stergachis A. Moore C. Dolecek C. Naghavi M. Lancet. 2022;399:629–655. doi: 10.1016/S0140-6736(21)02724-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Årdal C. Balasegaram M. Laxminarayan R. McAdams D. Outterson K. Rex J. H. Sumpradit N. Nat. Rev. Microbiol. 2020;18:267–274. doi: 10.1038/s41579-019-0293-3. [DOI] [PubMed] [Google Scholar]
- Lewis K. Nat. Rev. Drug Discovery. 2013;12:371–387. doi: 10.1038/nrd3975. [DOI] [PubMed] [Google Scholar]
- GSK, Gepotidacin's positive phase III data shows potential to be the first in a new class of oral antibiotics for uncomplicated urinary tract infections in over 20 years, https://www.gsk.com/en-gb/media/press-releases/gepotidacin-s-positive-phase-iii-data-shows-potential-to-be-the-first-in-a-new-class-of-oral-antibiotics-for-uncomplicated-urinary-tract-infections/, (accessed 12 July 2023)
- Prasad N. K. Seiple I. B. Cirz R. T. Rosenberg O. S. Antimicrob. Agents Chemother. 2022;66:e00054-22. doi: 10.1128/aac.00054-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker S. S. Black T. A. Drug Discovery Today. 2021;26:2152–2158. doi: 10.1016/j.drudis.2021.03.027. [DOI] [PubMed] [Google Scholar]
- Ainsworth G. C. Brown A. M. Brownlee G. Nature. 1947;160:263. doi: 10.1038/160263a0. [DOI] [PubMed] [Google Scholar]
- Stansly P. G. Shepherd R. G. White H. J. Bull. Johns Hopkins Hosp. 1947;81:43–54. [PubMed] [Google Scholar]
- Brown P. Dawson M. J. J. Antibiot. 2017;70:386–394. doi: 10.1038/ja.2016.146. [DOI] [PubMed] [Google Scholar]
- Yin N. Marshall R. L. Matheson S. Savage P. B. J. Am. Chem. Soc. 2003;125:2426–2435. doi: 10.1021/ja0284456. [DOI] [PubMed] [Google Scholar]
- Brandenburg K. David A. Howe J. Koch M. H. J. Andrä J. Garidel P. Biophys. J. 2005;88:1845–1858. doi: 10.1529/biophysj.104.047944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slingerland C. J. Kotsogianni I. Wesseling C. M. J. Martin N. I. ACS Infect. Dis. 2022;8:2396–2404. doi: 10.1021/acsinfecdis.2c00307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock R. E. W. Annu. Rev. Microbiol. 1984;38:237–264. doi: 10.1146/annurev.mi.38.100184.001321. [DOI] [PubMed] [Google Scholar]
- Yow E. M. Moyer J. H. Arch. Intern. Med. 1953;92:248–257. doi: 10.1001/archinte.1953.00240200098012. [DOI] [PubMed] [Google Scholar]
- Kelesidis T. Falagas M. E. Expert Opin. Drug Saf. 2015;14:1687–1701. doi: 10.1517/14740338.2015.1088520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nation R. L. Rigatto M. H. P. Falci D. R. Zavascki A. P. Antibiotics. 2019:8. doi: 10.3390/antibiotics8010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira M. S. Prado G. V. B. Costa S. F. Grinbaum R. S. Levin A. S. Diagn. Microbiol. Infect. Dis. 2009;65:431–434. doi: 10.1016/j.diagmicrobio.2009.07.018. [DOI] [PubMed] [Google Scholar]
- Liu X. Chen Y. Yang H. Li J. Yu J. Yu Z. Cao G. Wu X. Wang Y. Wu H. Fan Y. Wang J. Wu J. Jin Y. Guo B. Hu J. Bian X. Li X. Zhang J. J. Infect. 2021;82:207–215. doi: 10.1016/j.jinf.2021.01.006. [DOI] [PubMed] [Google Scholar]
- Keirstead N. D. Wagoner M. P. Bentley P. Blais M. Brown C. Cheatham L. Ciaccio P. Dragan Y. Ferguson D. Fikes J. Galvin M. Gupta A. Hale M. Johnson N. Luo W. McGrath F. Pietras M. Price S. Sathe A. G. Sasaki J. C. Snow D. Walsky R. L. Kern G. Toxicol. Sci. 2014;137:278–291. doi: 10.1093/toxsci/kft247. [DOI] [PubMed] [Google Scholar]
- Key reference: ; Danner R. L. Joiner K. A. Rubin M. Patterson W. H. Johnson N. Ayers K. M. Parrillo J. E. Antimicrob. Agents Chemother. 1989;33:1428–1434. doi: 10.1128/AAC.33.9.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quale J. Shah N. Kelly P. Babu E. Backer M. Rosas-Garcia G. Salamera J. George A. Bratu S. Landman D. Microb. Drug Resist. 2012;18:132–136. doi: 10.1089/mdr.2011.0163. [DOI] [PubMed] [Google Scholar]
- Brown P. Abbott E. Abdulle O. Boakes S. Coleman S. Divall N. Duperchy E. Moss S. Rivers D. Simonovic M. Singh J. Stanway S. Wilson A. Dawson M. J. ACS Infect. Dis. 2019;5:1645–1656. doi: 10.1021/acsinfecdis.9b00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Key reference: ; Magee T. V. Brown M. F. Starr J. T. Ackley D. C. Abramite J. A. Aubrecht J. Butler A. Crandon J. L. Dib-Hajj F. Flanagan M. E. Granskog K. Hardink J. R. Huband M. D. Irvine R. Kuhn M. Leach K. L. Li B. Lin J. Luke D. R. MacVane S. H. Miller A. A. McCurdy S. McKim J. M. Nicolau D. P. Nguyen T.-T. Noe M. C. O'Donnell J. P. Seibel S. B. Shen Y. Stepan A. F. Tomaras A. P. Wilga P. C. Zhang L. Xu J. Chen J. M. J. Med. Chem. 2013;56:5079–5093. doi: 10.1021/jm400416u. [DOI] [PubMed] [Google Scholar]
- Mingeot-Leclercq M. P. Tulkens P. M. Denamur S. Vaara T. Vaara M. Peptides. 2012;35:248–252. doi: 10.1016/j.peptides.2012.03.033. [DOI] [PubMed] [Google Scholar]
- Key reference: ; Slingerland C. J. Wesseling C. M. J. Innocenti P. Westphal K. G. C. Masereeuw R. Martin N. I. J. Med. Chem. 2022;65:15878–15892. doi: 10.1021/acs.jmedchem.2c01528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lash L. H. Toxicol. Appl. Pharmacol. 2005;204:329–342. doi: 10.1016/j.taap.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Vaara M. Vaara T. Lundberg C. V. J. Antimicrob. Chemother. 2018;73:452–455. doi: 10.1093/jac/dkx394. [DOI] [PubMed] [Google Scholar]
- Roberts K. D. Zhu Y. Azad M. A. K. Han M.-L. Wang J. Wang L. Yu H. H. Horne A. S. Pinson J.-A. Rudd D. Voelcker N. H. Patil N. A. Zhao J. Jiang X. Lu J. Chen K. Lomovskaya O. Hecker S. J. Thompson P. E. Nation R. L. Dudley M. N. Griffith D. C. Velkov T. Li J. Nat. Commun. 2022;13:1625. doi: 10.1038/s41467-022-29234-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B. Akin A. Magee T. V. Martinez C. Szeliga J. Vuong D. V. Synthesis. 2015;47:2088–2092. doi: 10.1055/s-0034-1380549. [DOI] [Google Scholar]
- MacNair C. R. Stokes J. M. Carfrae L. A. Fiebig-Comyn A. A. Coombes B. K. Mulvey M. R. Brown E. D. Nat. Commun. 2018;9:458. doi: 10.1038/s41467-018-02875-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz K. A. Hergenrother P. J. Acc. Chem. Res. 2021;54:1322–1333. doi: 10.1021/acs.accounts.0c00895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesseling C. M. J. Martin N. I. ACS Infect. Dis. 2022;8:1731–1757. doi: 10.1021/acsinfecdis.2c00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H. Drug Resistance Updates. 1998;1:93–98. doi: 10.1016/S1368-7646(98)80023-X. [DOI] [PubMed] [Google Scholar]
- Viljanen P. Vaara M. Antimicrob. Agents Chemother. 1984;25:701–705. doi: 10.1128/AAC.25.6.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French S. Farha M. Ellis M. J. Sameer Z. Côté J.-P. Cotroneo N. Lister T. Rubio A. Brown E. D. ACS Infect. Dis. 2020;6:1405–1412. doi: 10.1021/acsinfecdis.9b00159. [DOI] [PubMed] [Google Scholar]
- Odds F. C. J. Antimicrob. Chemother. 2003;52:1. doi: 10.1093/jac/dkg301. [DOI] [PubMed] [Google Scholar]
- Wilmer M. J. Saleem M. A. Masereeuw R. Ni L. van der Velden T. J. Russel F. G. Mathieson P. W. Monnens L. A. van den Heuvel L. P. Levtchenko E. N. Cell Tissue Res. 2010;339:449–457. doi: 10.1007/s00441-009-0882-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.