

Abstract
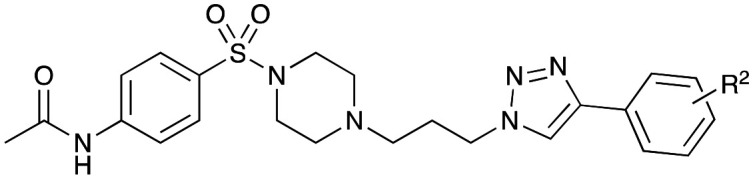
From lead 1, (N-(4-((4-(3-(4-(3-methoxyphenyl)-1H-1,2,3-triazol-1-yl)propyl)piperazin-1-yl)sulfonyl)-phenyl)acetamide), a S100A2–p53 protein–protein interaction inhibitor based on an in silico modelling driven hypothesis, four focused libraries were designed and synthesised. Growth inhibition screening was performed against 16 human cancer cell lines including the pancreatic cell lines MiaPaCa2, BxPC3, AsPC-1, Capan-2, HPAC, PANC-1 and the drug resistant CFPAC1. Modification of 1’s phenylacetamide moiety, gave Library 1 with only modest pancreatic cancer activity. Modification of the 3-OCH3Ph moiety (Library 2) gave 4-CH3 (26), 4-CH2CH3 (27), 4-CF3 (31) and 4-NO2 (32) with sterically bulky groups more active. A 4-CF3 acetamide replacement enhanced cytotoxicity (Library 3). The 4-C(CH3)336 resulted in a predicted steric clash in the S100A2–p53 binding groove, with a potency decrease. Alkyl moieties afforded more potent analogues, 34 (4-CH3) and 35 (CH2CH3), a trend evident against pancreatic cancer: GI50 3.7 (35; BxPC-3) to 18 (40; AsPC-1) μM. Library 4 analogues with a 2-CF3 and 3-CF3 benzenesulfonamide moiety were less active than the corresponding Library 3 analogues. Two additional analogues were designed: 51 (4-CF3; 4-OCH3) and 52 (4-CF3; 2-OCH3) revealed 52 to be 10–20 fold more active than 51, against the pancreatic cancer cell lines examined with sub-micromolar GI50 values 0.43 (HPAC) to 0.61 μM (PANC-1). MOE calculated binding scores for each pose are also consistent with the observed biological activity with 52. The obtained SAR data is consistent with the proposed interaction within the S100A2–p53 bonding groove.
Tight binding in the S100A2–p53 interaction groove = sub micromolar potencies against pancreatic cancer cell lines.
Introduction
Pancreatic cancer (PC) is predicted to become the second most common cause of cancer related mortality by 2030.1 With a projected 5-year survival rate of ∼10%, a PC diagnosis is essentially a death sentence.2,3 The highly metastatic nature of PC presents a major challenge for improving patient outcomes. These heterogeneous tumours are composed of cancer cells with differing morphologies and phenotypic profiles, making them extremely difficult to treat as they evade targeted therapies.
Currently, surgical resection is the only potentially curative treatment option for PC patients. However, despite this, surgery is only an option for <20% of patients as they do not present in a disease state conducive to successful surgery.3 In efforts to enhance patient outcomes, both chemotherapy and radiation therapy are used as adjuvant therapies following surgery. Disappointingly, this has had little effect on reducing PC related mortality, with a 5-year survival rates no greater than 20%.4–9 Indeed, there have been no outcome improvements in the past two decades. New targeted therapies and new screening approaches to PC diagnosis are urgently required.10–12
Our laboratory, and others, have invested considerable efforts in the identification of potential therapeutic targets for the treatment of PC.4,13–15 The heterogeneous nature of this cancer largely stymies efforts. However, multi-omic analyses suggests that the calcium binding protein, S100A2 is highly upregulated in PC and is associated with the aggressive, poor prognostic squamous subtype. Consequently, the development of small molecule S100A2 inhibitors offers a potential clinical pipeline in this space.16–21
The S100 protein family comprises the largest subgroup of calcium binding EF-hand protein types.10,22,23 At least 25 members of this family are known, spanning S100A1–S100A18, trichohylin, filaggrin, repetin, S100B, S100G, S100P and S100Z.10,11,23–26 S100 proteins function via a myriad of processes such as proliferation, migration and/or invasion, inflammation and differentiation.12 The proteins form both homodimers and heterodimers and are regulated by Ca2+, enabling the proteins to act as sensors to variations in Ca2+ concentration. Multiple cancer subtypes exhibit distinct S100 protein profile which has led to a decade long investigation of their role(s) in cancer.12
Of significance to PC is the S100 calcium binding protein A2 isoform (S100A2),12 which is comprised of two monomers. Each monomer contains an N-terminal S100-specific EF hand with helix I and helix II and a C-terminal classical EF hand consisting of helix III and helix IV, as well as two helices connected via the so-called hinge loop.27 Upregulation of S100A2 in PC is indicative of the aggressive squamous subtype, which constitutes ∼50% of PC patients in the advanced setting. It has been demonstrated that moderate and high levels of S100A2 expression in PC is a poor prognosis marker even after surgical resection. Patients displaying low/no levels of S100A2 in PC are known to have a survival benefit post-surgery, even in the presence of lymph node metastases.14,28 S100A2 levels also serve as a predictive biomarker of this disease state.14,29,30
Unique to the S100 protein family, S100A2 is found in high concentrations in the nucleus of cells; and is associated with the regulation of the cell cycle.31,32 It modulates the tumour suppressor p53 by binding with its transactivation domain. In PC, over expression of S100A2 inhibits p53, prevents p53 tumour suppression, and results in aberrant cancer cell proliferation.28,31 Thus, the S100A2 is regarded as a novel p53 target protein.12,16,17,21,33–36
Results and discussion
From 1 and 2, inhibitors of a range of PC cell lines were developed, e.g.3 and 4.16,17 Analogues from both leads showed good levels of activity across a panel of PC cell lines with growth inhibition (GI50) values of 1.6–4.9 and 1.1–3.1 μM for 3 and 4 respectively (Fig. 1). Building on these efforts, we envisaged hybridisation of 3 and 4 to analogues such as 5 aided by molecular modelling guided focused library synthesis. Our efforts thus far suggested that the inclusion of lipophillic/electron withdrawing moieties at this position was favoured.
Fig. 1. A. Lead compounds (1 and 2) with the chemical structure colour coded to highlight the three key fragments used in the development of pancreatic cancer active analogues 3 and 4.16,17 B. Proposed fragments to be used for focused library development herein.

Our initial studies, Library 1, focused on modification of the terminal sulfonamide moiety of 2. To enable library synthesis, the key triazole 13 was accessed in four steps from piperazine 9.37,38 Mono-Boc piperazine 10 was prepared under flow chemistry conditions.39 Alkylation with 3-bromopropan-1-ol gave (10), and OH to OMs functional group interchange facilitated the required azide installation (12); (yield: 18%; 4 steps). In our hands access to 12 was scalable to gram quantities. Huisgen copper catalysed coupling of 12 with 3-ethynylanisole (8a) and removal of the Boc-protecting group gave the desired triazolopiperazine (13a) (yield 16%, 5 steps).40 Retention of the 3-OCH3 moiety allowed direct comparison of the obtained activity with the lead compounds. Huisgen coupling of a small library of 4-substituted benzene sulfonyl chlorides (8a–l) gave Library 1 (2, 14–24) in moderate (27%) to good yields (89%) (Scheme 1).
Scheme 1. Synthesis of compound Library 1, 2, 14–24. Reagents and conditions: (i) Boc2O, CH2Cl2, 0 °C to RT, 2 h; (ii) 3-bromo-1-propanol, K2CO3, CH3CN, 80 °C, 24 h; (iii) CH3SO2Cl, K2CO3, CH2Cl2, RT, 3 h; (iv) NaN3 CH3CN, 80 °C, 3 h; (v) CH3COCH3, 1-ethynyl-3-methoxybenzene, CuSO4·5H2O, sodium ascorbate, H2O, reflux, 24 h; (vi) EtOAc, HCl, 0.5 h; (vii) CH2Cl2, NaOH, RT, 1–3 h. 2 = CH3CONH, 14 = H, 15 = CH3, 16 = (CH3)2CH, 17 = (CH3)3C, 18 = OCH3, 19 = 4-F, 20 = Cl, 21 = Br, 22 = CF3, 23 = 4-NO2, 24 = CH3CO.

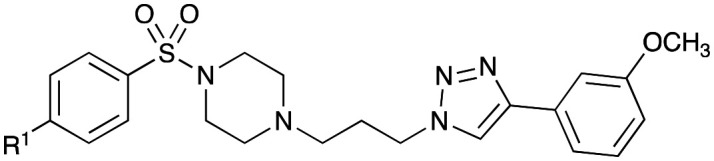
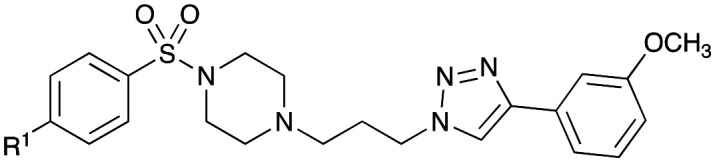
In our proposed Library 2 analogues (Table 3), the pendent bulky phenyl ring substituents (blue moiety in Fig. 1) were explored. Synthesis was conducted as per Scheme 2 with analogues accessed by coupling of 4-acetamidobenzene sulfonyl chloride (8a) and selected piperazines (13b–i) to give analogues 25–32 (see Scheme 2 and Table 2 for details) in moderate (27%) to good yields (77%).
Percentage growth inhibition evaluation (at 25 μM, n = 3) of Library 2 compounds 25–32, relative to an untreated control, in a panel of cancer cell lines and MCF10A cells. Higher values indicate higher compound potency.
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| R2 | HT29a | U87b | MCF-7c | A2780d | H460e | A431f | Du145g | BE2-Cb | SJ-G2h | MiaPaCa-2i | MCF10Aj |
| 25 H | 31 ± 4 | 10 ± 4 | 24 ± 9 | 47 ± 4 | 23 ± 2 | 31 ± 6 | 34 ± 3 | 32 ± 11 | 59 ± 41 | 30 ± 2 | 44 ± 2 |
| 26 4-CH3 | 46 ± 2 | 22 ± 1 | 42 ± 4 | 57 ± 1 | 34 ± 4 | 40 ± 8 | 61 ± 12 | 36 ± 2 | 72 ± 28 | 38 ± 3 | 67 ± 2 |
| 27 4-CH2CH3 | 64 ± 6 | <0 | 30 ± 10 | 59 ± 4 | 32 ± 7 | 16 ± 1 | 42 ± 3 | 36 ± 6 | 29 ± 6 | 38 ± 9 | 67 ± 5 |
| 28 4-F | 22 ± 0 | 16 ± 5 | 27 ± 6 | 50 ± 7 | 18 ± 4 | 28 ± 1 | 35 ± 4 | 38 ± 8 | 67 ± 33 | 35 ± 5 | 41 ± 2 |
| 29 4-Br | 58 ± 9 | 4 ± 6 | 28 ± 8 | 46 ± 2 | 19 ± 4 | 16 ± 3 | 54 ± 4 | 39 ± 3 | 48 ± 12 | 34 ± 9 | 58 ± 2 |
| 30 3-Cl | 42 ± 8 | 1 ± 2 | 31 ± 7 | 49 ± 2 | 18 ± 1 | 2 ± 4 | 24 ± 3 | 27 ± 2 | 33 ± 8 | 32 ± 7 | 43 ± 4 |
| 31 4-CF3 | 59 ± 3 | 16 ± 5 | 42 ± 5 | 67 ± 9 | 30 ± 3 | 31 ± 6 | 52 ± 4 | 41 ± 1 | 42 ± 7 | 43 ± 3 | 73 ± 9 |
| 32 4-NO2 | 50 ± 7 | 27 ± 3 | 47 ± 4 | 72 ± 7 | 40 ± 6 | 33 ± 7 | 48 ± 6 | 42 ± 1 | 37 ± 7 | 40 ± 3 | 69 ± 2 |
Colon.
Glioblastoma.
Breast.
Ovarian.
Lung.
Skin.
Prostate.
Neuroblastoma.
Pancreas.
Normal breast.
Scheme 2. Synthesis of Library 2 (25–32), Library 3 (33–42) and Library 4 (43–50) with piperazine analogues (13b–i) synthesised as per Scheme 1. Reagents and conditions: (i) CH2Cl2, NaOH, RT, 1–3 h. See tables for details.

Percentage growth inhibition evaluation (25 μM, CCK-8 assay, n = 3) of Library 1 compounds 2 and 14–24, relative to an untreated control, in an expanded pancreatic cell line panel. Higher values indicate higher compound potency.
| |||||||
|---|---|---|---|---|---|---|---|
| R1 | BxPC3 | MiaPaCa2 | HPDEa | Comp. | BxPC3 | MiaPaCa2 | HPDEa |
| 2 CH3CONH | 23.9 ± 11.1 | 30.3 ± 7.1 | 36.1 ± 4.8 | 19 F | 15.1 ± 4.2 | <10 | 40.8 ± 9.7 |
| 14 H | <10 | <10 | 15.8 ± 7.2 | 20 Cl | 22.9 ± 7.3 | <10 | 54.3 ± 7.3 |
| 15 CH3 | <10 | <10 | 45.0 ± 3.4 | 21 Br | 10.7 ± 8.3 | <10 | 32.6 ± 4.4 |
| 16 (CH3)2CH | <10 | 11.4 ± 7.3 | 48.8 ± 6.8 | 22 CF3 | <10 | <10 | 29.0 ± 8.8 |
| 17 (CH3)3C | <10 | <10 | 38.2 ± 12.4 | 23 NO2 | 21.1 ± 10.2 | <10 | 46.5 ± 2.5 |
| 18 OCH3 | <10 | 20.3 ± 12.4 | 33.9 ± 3.6 | 24 CH3CO | 22.8 ± 5.8 | <10 | 30.1 ± 9.7 |
Normal pancreatic cells.
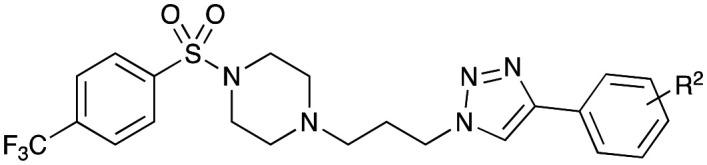
Library 3 synthesis saw the introduction of the 4-CF3 phenyl sulfonamide moiety on reaction of 4-trifluoromethylbenzene sulfonyl chloride (8j) and selected piperazines (13b–i). As with the other Libraries in this study, all Library 3 compounds (33–42) were afforded in moderate (45%) to good yields (79%) (Scheme 2).
Library 4 was developed to leverage the SAR from Libraries 1–3 and explore the position of the –CF3 substituent on the aromatic sulfonamide. Thus Library 4 was synthesised from 2-CF3 and 3-CF3 benzene sulfonyl chloride (8m and 8n) to give analogues 43–50 (Scheme 2). The yields were moderate (27%) to good (68%), again in keeping with that observed with the other focused libraries developed herein.
Two final analogues were explored with 4-CF3 substituted 51 (4-OCH3) and 52 (2-OCH3) derived as per Schemes 1 and 2 with 4- and 2-ethynylanisole.
Biology
Initial cytotoxicity evaluation was conducted on a broad-spectrum cancer cell line panel that comprised: HT29 (colon), U87 and SJ-G2 (glioblastoma), MCF-7 (breast), A2780 (ovarian), H460 (lung), A431 (skin), Du145 (prostate), BE2-C (neuroblastoma) MiaPaCa2 (pancreatic) and the normal cell line, MCF-10A (normal breast). Initial cell growth inhibition screening (MTT assay) was conducted with Library 1 at a single 25 μM compound concentration (Table S1; ESI†) and the percentage growth inhibition was determined. Of the Library 1 analogues, 14 (H), 15 (4-CH3), 18 (4-OCH3) and 21–23 (4-Br, 4-CF3 and 4-NO2, respectively) were deemed sufficiently active in at least one cell line to proceed to a full dose response analysis (Table 1). Moderate broad-spectrum activity was observed with 21 (4-Br; GI50 14 (MCF-7) to >50 μM (MiaPaCa-2)). All Library 1 analogues were active against the HT29 (colon) and A2780 (ovarian), and all except 23 (4-NO2) were active against the MCF-7 (breast) cancer cell line. No notable activity was observed against the U87 glioblastoma and MiaPaCa2 pancreatic cancer cell lines (GI50 > 50 μM). Library 1 modification failed to afford a significant cytotoxicity enhancement relative to lead 2, particularly against the MiaPaCa-2 cell line.
Growth inhibition evaluation (GI50, μM, MTT assay, n = 3) of Library 1 compounds 14, 15, 18 and 21–23 in a panel of cancer cell lines and MCF10A cells. GI50 is the concentration that inhibits cell growth by 50% relative to an untreated control, the lower the value the greater the growth inhibition.
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| R1 | HT29a | U87b | MCF-7c | A2780d | H460e | A431f | Du145g | BE2-Cb | SJ-G2h | MiaPaCa-2i | MCF10Aj |
| 14 H | 28 ± 2 | >50 | 35 ± 5 | 40 ± 1 | >50 | >50 | >50 | >50 | 42 ± 2 | >50 | >50 |
| 15 CH3 | 22 ± 1 | >50 | 33 ± 2 | 29 ± 4 | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
| 18 OCH3 | 30 ± 0 | >50 | 38 ± 1 | 27 ± 2 | >50 | >50 | >50 | >50 | 32 ± 2 | >50 | >50 |
| 21 Br | 18 ± 2 | >50 | 14 ± 1 | 20 ± 2 | 35 ± 3 | 40 ± 8 | 25 ± 1 | 20 ± 1 | 23 ± 3 | >50 | 29 ± 3 |
| 22 CF3 | 23 ± 3 | >50 | 28 ± 4 | 21 ± 3 | >50 | >50 | >50 | >50 | >50 | >50 | 39 ± 3 |
| 23 NO2 | 41 ± 4 | >50 | >50 | 27 ± 2 | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
Colon.
Glioblastoma.
Breast.
Ovarian.
Lung.
Skin.
Prostate.
Neuroblastoma.
Pancreas.
Normal breast.
Within this program we have developed a particular focus on targeting pancreatic cancer and thus established additional screening capabilities using a related CCK-8 assay. Both MTT and CCK-8 detect dehydrogenase activity and are considered equally valid to determine cell death, CCK-8 produces a more soluble product and by some is considered to be more sensitive.41 With the increased cost of the CCK-8 assay, we limit its use to circumstances were our preliminary MTT-screening on various cancerous cell lines warrant this. In this instance, it was used in evaluating efficacy against pancreatic cancer cell lines. Thus the effect of this library was examined against the BxPC3, MiaPaCa2 pancreatic cancer cell lines. Again, no Library 1 analogue displayed potency enhancement relative to 2 (Table 2). The highest levels of activity were noted against the normal pancreatic cell line, HPDE, with up to 54% inhibition at 25 μM compound concentration (20; 4-Cl) (Table 2).
In general, compounds in Library 2 displayed higher level growth inhibitory at 25 μM compound concentration effects than Library 1 compounds (Table S1; ESI†) against our target pancreatic cell line, MiaPaCa-2 cell line, with inhibition values ranging between 30–43%. Of the Library 2 analogues, 26 (4-CH3), 27 (4-CH2CH3), 31 (4-CF3) and 32 (4-NO2) showed slightly higher inhibition, with growth inhibition of 38%, 43%, 38% and 40% respectively compared with other library analogues. Analogues, 26 (4-CH3; Du145, SJ-G2, MCF-10A), 27 (4-CH2CH3; HT29, MCF10A), 28 (4-F; SJ-G2), 31 (4-CF3; A2780, MCF10A) and 32 (4-NO2; A2780, MCF10A) exhibited growth inhibition values of >60% against the cited cell lines. No analogue was deemed sufficiently active to proceed to full dose response evaluation.
Library 3 analogues at 25 μM compound concentration (Table S2; ESI†) revealed good levels of growth inhibition for 34 (4-CH3), 35 (4-CH2CH3), 36 (4-C(CH3)3), 38 (4-Br), and 40 (4-CF3) which proceed to full dose response evaluation (Table 4). Of these analogues 34 (4-CH3) and 35 (4-CH2CH3) gave the highest levels of broad-spectrum cytotoxicity with GI50 values of 3.1 (MCF-7) to 11 (U87) and 3.2 (MCF-7) to 19 (U87) μM, respectively.
Growth inhibition evaluation (GI50 μM, MTT assay, n = 3) of Library 3 compounds 34–36, 38 and 40, relative to an untreated control, in a panel of cancer cell lines and MCF10A cells.
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| R2 | HT29a | U87b | MCF-7c | A2780d | H460e | A431f | Du145g | BE2-Cb | SJ-G2h | MiaPaCa-2i | MCF10Aj |
| 34 4-CH3 | 3.4 ± 0.32 | 11 ± 3.5 | 3.1 ± 0.38 | 4.0 ± 0.19 | 4.4 ± 0.75 | 3.5 ± 0.32 | 3.9 ± 0.69 | 4.0 ± 0.17 | 3.2 ± 0.25 | 3.9 ± 0.07 | 3.6 ± 0.34 |
| 35 4-CH2CH3 | 3.5 ± 0.20 | 19 ± 3.2 | 3.2 ± 0.17 | 4.8 ± 0.17 | 5.9 ± 0.25 | 5.2 ± 1.3 | 6.7 ± 1.7 | 4.8 ± 0.10 | 4.8 ± 0.65 | 4.3 ± 0.28 | 4.5 ± 0.29 |
| 36 4-C(CH3)3 | 15 ± 0.67 | 27 ± 2.1 | 18 ± 0.88 | 13 ± 0.88 | 16 ± 0.58 | 19 ± 0.17 | 20 ± 2.2 | 15 ± 2.1 | 20 ± 1.2 | 17 ± 1.6 | 15 ± 0.67 |
| 38 4-Br | 4.7 ± 1.2 | 18 ± 0.88 | 5.5 ± 2.5 | 4.9 ± 0.61 | 4.0 ± 1.0 | 7.6 ± 2.0 | 7.1 ± 1.9 | 6.5 ± 1.8 | 5.6 ± 0.83 | 8.3 ± 1.9 | 5.8 ± 1.0 |
| 40 4-CF3 | 11 ± 3.2 | 17 ± 1.5 | 5.8 ± 0.68 | 6.1 ± 0.70 | 6.4 ± 1.4 | 10 ± 1.8 | 10 ± 4.4 | 8.8 ± 2.0 | 7.8 ± 1.6 | 15 ± 2.2 | 11 ± 1.6 |
Colon.
Glioblastoma.
Breast.
Ovarian.
Lung.
Skin.
Prostate.
Neuroblastoma.
Pancreas.
Normal breast.
Compounds 33–42 were also examined in an extended PC cell line panel BxPC-3, AsPC-1, Capan-2, PANC-1 and HPAC at 25 μM of drug concentration using the MTT assay (Table S3; ESI†). The potency of this Library justified the full dose response analysis of analogues 34 (4-CH3), 35 (4-CH2CH3), 38 (4-Br) and 40 (4-CF3) (Table 5). These analogues displayed potent inhibitory activity against the four shown PC cell lines with GI50 values 3.7–18 μM, with all but 40 (4-CF3) returning GI50 values <10 μM.
Growth inhibition evaluation (GI50 μM, CCK assay, n = 3) of Library 3 compounds 34, 35, 38 and 40, relative to an untreated control, in an expanded panel of five human PC cell lines.
| |||||
|---|---|---|---|---|---|
| BxPC-3 | AsPC-1 | Capan-2 | PANC-1 | HPAC | |
| 34 4-CH3 | 4.0 ± 0.35 | 7.3 ± 0.46 | 5.0 ± 0.67 | 6.1 ± 0.29 | 5.0 ± 0.47 |
| 35 4-CH2CH3 | 3.7 ± 0.15 | 8.0 ± 0.67 | 6.1 ± 0.86 | 6.1 ± 0.32 | 5.8 ± 0.21 |
| 38 4-Br | 8.0 ± 3.1 | 8.3 ± 1.2 | 7.7 ± 3.3 | 6.8 ± 0.13 | 9.6 ± 3.2 |
| 40 4-CF3 | 14 ± 2.7 | 18 ± 1.5 | 16 ± 1.5 | 16 ± 3.0 | 15 ± 1.5 |
Analysis of Library 4 analogues, 43–50 based on the percentage growth inhibition response saw only 45 (4-Br) advanced to full dose response evaluation (Table 6). While 45 (2-CF3; 4-Br) was active against HT29, MCF-7, A2780, and BE2-C cell lines with GI50 values 9–38 μM (and against the MCF10A normal breast cell line; GI50 = 12 μM), activity was only observed against the Capan-2 pancreatic cancer cell line (GI50 = 23 μM) (Table 6).
Percentage growth inhibition evaluation (at 25 μM, MTT assay, n = 3) of compounds in Library 4, 43–50, relative to an untreated control, in a panel of cancer cell lines and MCF10A cells. GI50 value (μM) are detailed in parentheses and italics for 45.
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| R | HT29a | MCF-7b | A2780c | BE2-Cd | MiaPaCa-2e | BxPC-3e | AsPC-1e | Capan-2e | PANC-1e | HPACe | MCF10Af |
| 43 4-CH3 | 86 ± 5 | 79 ± 3 | 75 ± 2 | 35 ± 9 | 38 ± 4 | 11 ± 6 | 57 ± 3 | 42 ± 6 | 48 ± 3 | 41 ± 13 | 61 ± 4 |
| 44 4-CH2CH3 | 79 ± 3 | 82 ± 5 | 75 ± 1 | 52 ± 9 | 39 ± 2 | 16 ± 5 | 59 ± 3 | 25 ± 2 | 48 ± 0 | 67 ± 5 | 63 ± 8 |
| 45 4-Br | 92 ± 2 (10 ± 2.0) | 87 ± 4 (9.2 ± 1.7) | 68 ± 5 (14 ± 5.4) | 58 ± 8 (38 ± 13) | 70 ± 13 (>50) | <10 (>50) | 51 ± 6 (>50) | 65 ± 8 (23 ± 0.33) | 53 ± 4 (>50) | 42 ± 7 (>50) | 72 ± 2 (12 ± 1.7) |
| 46 4-CF3 | 69 ± 4 | 75 ± 3 | 80 ± 4 | 62 ± 8 | 45 ± 5 | 19 ± 16 | 50 ± 4 | 42 ± 5 | 54 ± 4 | 29 ± 7 | 64 ± 5 |
Colon.
Breast.
Ovarian.
Neuroblastoma.
Pancreas.
Normal breast.
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 47 4-CH3 | 63 ± 5 | 86 ± 5 | 83 ± 3 | 60 ± 7 | 48 ± 3 | 32 ± 2 | 60 ± 4 | 54 ± 11 | 57 ± 2 | 40 ± 5 | 79 ± 4 |
| 48 4-CH2CH3 | 65 ± 5 | 86 ± 3 | 86 ± 3 | 61 ± 8 | 50 ± 4 | 39 ± 9 | 60 ± 5 | 58 ± 10 | 59 ± 2 | 41 ± 7 | 77 ± 4 |
| 49 4-Br | 46 ± 4 | 56 ± 3 | 72 ± 3 | 40 ± 7 | 35 ± 6 | 22 ± 10 | 53 ± 4 | 39 ± 5 | 41 ± 1 | <10 | 74 ± 5 |
| 50 4-CF3 | 50 ± 11 | 86 ± 14 | 65 ± 11 | 61 ± 6 | 58 ± 9 | 55 ± 13 | 50 ± 8 | 67 ± 16 | 60 ± 1 | 74 ± 8 | 59 ± 19 |
Compound 51 showed good levels of broad-spectrum cytotoxicity, as well as high levels of activity against all the pancreatic cancer cell lines examined spanning GI50 values of 5.2 (PANC-1) to 14 (AsPC-1) μM. The corresponding 2-OCH3 analogue, 52, returned an increase in potency with sub-micro molar potency across all cell lines examined. Analogue 52 was 10-fold more active than any analogue from Libraries 1–4 with GI50 values of 0.36 (HT29) to 0.80 (A2780) μM. Most significantly, 52 was sub-micromolar potent against the pancreatic cancer cell lines examined, GI50 0.48 (BxPC-3) to 0.68 (PANC-1) μM (Table 7).
Cell growth inhibition evaluation (GI50 μM, MTT assay, n = 3) of compounds 51 and 52 in Library 4 against various cancer cells and MCF10A cells.
| ||
|---|---|---|
| 51 R = 4-OCH3 | 52 R = 2-OCH3 | |
| HT29a | 8.6 ± 1.9 | 0.47 ± 0.02 |
| MCF-7ab | 6.1 ± 2.2 | 0.36 ± 0.03 |
| A2780c | 6.2 ± 1.3 | 0.80 ± 0.05 |
| BE2-Cd | 6.4 ± 1.1 | 0.59 ± 0.08 |
| MiaPaCa-2e | 7.8 ± 1.3 | 0.64 ± 0.01 |
| BxPC-3e | 12 ± 1.0 | 0.48 ± 0.08 |
| AsPC-1e | 14 ± 0.58 | 0.61 ± 0.06 |
| Capan-2e | 8.4 ± 1.9 | 0.56 ± 0.03 |
| PANC-1 | 5.2 ± 0.33 | 0.68 ± 0.09 |
| HPACe | 16 ± 1.7 | 0.43 ± 0.08 |
| MCF10Af | 4.9 ± 0.77 | 1.0 ± 0.18 |
Colon.
Breast.
Ovarian.
Neuroblastoma.
Pancreas.
Normal breast.
Molecular docking studies
The design parameters of this work were predicated on leads 1 and 2 blocking the interaction between S100A2 and p53, with these compounds occupying the S100A2–p53 binding groove and thus preventing the protein–protein interaction. Examination of the docked poses of selected Library 1 compounds 2 (CH3CONH), 21 (Br) and 22 (CF3) in the p53-groove of the S100A2–p53 binding pocket suggested that these analogues failed to engage with two regions of the p53-groove. The triazolophenyl moieties do not occupy the same region as p53 (yellow oval, Fig. 2B), nor does the benzene sulfonamide region of 2 (green oval, Fig. 2B). The intent of these studies to provide potential correlations between the observed SAR and a potential target. Currently the activity of these compounds, at sub-micromolar, does not yet warrant a full examination of the potential target as there remains a high probability of identifying off-targets that might confound later compound development. Thus far the modelling of these compounds and their modes of postulated interaction within the S100A2–p53 binding groove are consistent with this hypothesis. Enhanced activity will allow us, and other to more efficiently resolve the ultimate binding partners for analogues of this nature.
Fig. 2. The best predicted docking poses for, A. compound 2 (CH3CONH); and B. an overlay of 2 (CH3CONH), 15 (CH3), 18 (OCH3) and 24 (CH3CO); and the p53 peptide (red); all compounds are docked in the p53 groove of S100A2; compounds are coloured by atom type; protein surface coloured: purple: hydrophilic area; green: hydrophobic area. Red ribbon is the C-terminal fragment of p53 known to bind S100A2. Yellow oval – deviation from p53-groove by the triazolophenyl substituents in Library 1, green oval – deviation from the p53 groove by the benzene sulfonamide region of 2.

Molecular modelling suggested that 27 (4-CH2CH3) (Fig. 3A), 25 (4-H) and 29 (4-Br) (Fig. 3B) would effect a subtle change in the adopted conformation such that they would better align with the p53 groove. Specifically, the right-hand side of these docking poses better align with the p53 docking pose (Fig. 3B). Consistent with this, modelling showed a good alignment of the triazolophenyl moiety of 25, 27 and 29 with the p53 groove, but the benzene sulfonamide moiety remains partially misaligned (green oval, Fig. 3B).
Fig. 3. A. The best predicted docking poses for, A. compound 27 (4-CH2CH3); and B. overlay of 25 (H), 27 (4-CH2CH3), and 29 (4-Br) from Library 2, and the p53 peptide (red); all compounds are docked in the p53 groove of S100A2; compounds are coloured by atom type; protein surface coloured: purple: hydrophilic area; green: hydrophobic area. Red ribbon is the C-terminal fragment of p53 known to bind S100A2. Yellow oval – deviation from p53-groove by the benzene sulfonamide substituents in Library 2.

Building from Library 2, modelling supported an –NHCOCH3 (Fig. 4A and C) to –CF3 interchange to enable better engagement with the p53-groove, specifically by introduction of favourable –CF3 interactions with PheA89 (Fig. 4B and D).
Fig. 4. The interaction plots of A. compound 27 (4-CH2CH3) in the p53-groove with the image showing the adjacent amino acids, as generated by MOE; B. as for A with compound 35 (4-CH2CH3) in the p53-groove (A and B in codes of amino acids: sequence of S100A2 protein); C. the best predicted docking pose of compound 27; D. the best predicted docking pose of compound 35. All compounds are docked in the S100A2–p53 binding groove. Compounds are coloured by atom type; protein surface coloured: purple: hydrophilic.

As for compounds in Library 3, modelling analysis suggested that the inclusion of terminal tert-butyl substituent (36) would resulted in diminished S100A2–p53 binding groove engagement (with a lower predicted binding score (not shown)), and an anticipated steric clash within the S100A2–p53-groove (Fig. 5A). The result of this would be a shift in conformation of 36, with a resulting loss of good alignment with the benzene sulfonamide region and the p53 peptide. Meanwhile, the smaller triazolophenyl substituents (exemplified by 35 (4-CH3), Fig. 5B) align well with the p53-peptide.
Fig. 5. A. The docking pose of compound 36 (4-C(CH3)3) in the p53-groove of the S100A2–p53 heterodimer; B. as for A, with compound 34 (4-CH3). All compounds are docked in the p53 groove of S100A2; compounds are coloured by atom type; protein surface coloured: purple: hydrophilic area; green: hydrophobic area. Red ribbon is the C-terminal fragment of p53 known to bind S100A2.

Docking of 51 (4-OCH3) and 52 (2-OCH3) in the p53-binding groove, revealed these analogues aligns well with Library 2 and Library 3 analogues. MOE calculated binding scores for each pose are also consistent with the observed biological activity with 52 (−6.61 kcal.mol−1) returned a more negative value than 51 (−6.18 kcal.mol−1), indicating improved binding. Compound 51 was predicted to interact with the p53 groove via mainly hydrophobic interactions (Fig. 6A), while 52 was predicted to make key interactions with the p53 groove via a hydrogen bond with the sulfonamide and SerA86, and an arene–H interaction between the triazolophenyl ring and PheA89 (Fig. 6B). Importantly, when overlaid with the p53 peptide, compound 51 was not well aligned in the S100A2–p53 binding groove, with the triazolophenyl ring deviating from the binding groove (yellow oval, Fig. 6C). Compound 52, however, aligned well between the S100A2 and p53 peptide (Fig. 6D). On first examination, the improvement in activity noted with 52, albeit with a higher predicted binding interaction, is at odds with the apparent binding interactions. However, on docking both 51 and 52 in the same site, significant binding interaction differences are noted (Fig. 6E). Transposition of the OCH3 moiety from the 4- to 2-position results in a considerable change in the minimum energy conformation resulting in altered positioning in the S100A2–p53 groove, the terminal aromatic moiety adopts a significantly different conformation (cf. yellow oval and red oval Fig. 6E). This change to the 2-OCH3 moiety with 52 results in additional interactions with Thr14-OCH3, Phe89-π–π, Asp88-triazoloyl ring, Ser86 and Lys49 interactions with the 4-CF3Ph moiety.
Fig. 6. The interaction plots of A. compound 51 (4-OCH3) in the p53-groove with the image showing the adjacent amino acids, as generated by MOE; B. as for A with compound 52 (2-OCH3) in the p53-groove (A and B in codes of amino acids: sequence of S100A2 protein); C. the best predicted docking pose of compound 27; D. the best predicted docking pose of compound 35. All compounds are docked in the S100A2–p53 binding groove. Compounds are coloured by atom type; protein surface coloured: purple: hydrophilic. E. Compounds 51 (pale blue) and 52 (green) docked in the S100A2–p53 groove (atoms coloured by heteroatom type). Yellow and red ovals highlight the positioning of the CH3OPh substituents, a notable change in docked orientation. Key interacting amino acids interacting with 52 are highlighted (T14-OCH3, F89-π–π, D88-triazoloyl ring, S86 and K49 interactions with the 4CF3Ph moiety).

Finally, molecular dynamics studies were conducted for compound 52, to determine the stability of the predicted binding pose. After 500 ns, the pose remained mostly static (initial pose in pink, final pose in green, Fig. 7A). Importantly, the interaction that were observed initially between the sulfonamide and SerA86 was also maintained after the dynamics were performed (Fig. 7B): and whilst the arene–H interaction between the triazolophenyl ring and PheA89 was no longer observed, a new hydrogen bond between the triazole ring hydrogen and LysA49, due to a small conformational shift, was noted: confirming that the pose was quite stable.
Fig. 7. A. Compound 52 (2-OCH3) before (pink) and after (green) MD studies; B. the final pose for compound 52. All compounds are docked in the S100A2–p53 binding groove. Compounds are coloured by atom type; protein surface coloured: purple: hydrophilic.

Discussion
The inhibition of S100A2 is a poorly explored pancreatic cancer drug target. An in-house developed S100A2–p53 homology model based on the related S100B-heptamine protein ligand complex enabled the identification of the S100A2–p53 binding grove as a protein–protein interaction hot-spot.42 This pocket, predominately lined with hydrophobic amino acids (Val11, Thr15, Leu12, Glu41, Leu79, Val77, and Ala76) showed good, predicted binding of the p53 peptide (Fig. S1; ESI†). From this, and subsequent in silico screening we identified and developed the sulfonamides 1 and 2 to target this interaction resulting in the synthesis of analogues of 3 and 4 (Fig. 1).
Compound libraries were readily accessed via established synthesis approaches as described herein. Each library was specifically designed to interrogate key binding features predicted to block S100A2–p53 protein–protein interaction. Compounds were designed to occupy this S100A2–p53 binding groove, thus preventing p53 engagement.
Of the Library 1 analogues, 14 (H), 15 (4-CH3), 18 (4-OCH3) and 21–23 (4-Br, 4-NO2 and 4-CH3CO, respectively) were deemed sufficiently active in at least one cell line to proceed to a full dose response analysis (Table 1). In an expanded panel of pancreatic cancer cell lines, Library 1 analogues displayed no significant enhancement of potency relative to 2 (Table 2). Modelling analysis of the Library 1 screening outcomes with analogues 2 (4-CH3CONH), 21 (4-Br) and 22 (4-CF3) suggested that the sulfonamide moieties do not position in the same region as p53 (yellow oval, Fig. 2B), nor do the triazolophenyl moieties (green oval, Fig. 2B). This was in keeping with no observed cytotoxicity enhancement on modification of the sulfonamide moiety. This data was consistent with our previous findings with similar piperazine substituted analogues.16,17 Enhancing engagement in these two areas was hypothesized to result in higher levels of cytotoxicity in our target population of pancreatic cancer cells.
Modelling evaluation of proposed Library 2 analogues suggested that 31 (4-CF3) (Fig. 3A), 26 (4-CH2CH3) and 28 (4-F) (Fig. 3B) would effect a subtle change in the adopted conformation such that they would better align with the p53 groove. Specifically, the right-hand side of these docking poses better align with the p53 docking pose (Fig. 3B). The pendent bulky phenyl ring substituents provide a more compete block of the interaction between p53 and the binding pocket. This was borne out by the consistently higher cytotoxicity of this Library, both as broad spectrum active and against the MiaPaCa-2 pancreatic cell line (Table 3). Both electron withdrawing and donating substituents showed increased activity, with an increase in substituent size the common feature. This suggested that steric effects in this region dominated. Modelling analysis supported good p53-groove alignment of the terminal triazolophenyl moiety, but a misalignment of the sulfonamide.
To enhance sulfonamide engagement with the p53 groove, a CF3 group was predicted to make additional interactions with PheA89 (Fig. 4). Library 3, with the added 4-CF3 moiety, showed good levels of cytotoxicity supporting the S100A2–p53 binding model. Of particular note was the enhanced activity of the aliphatic substituted 34 (4-CH3) and 35 (4-CH2CH3), with 36 (4-C(CH3)3) exhibiting lower activity. This activity reduction with 36 was explained on re-examination of its binding pose noting that the t-butyl moity results in a steric clash at the right-hand side of the S100A2–p53 binding groove. The smaller 38 (4-Br), and 40 (4-CF3) align well with the p53-peptide as do the simple alkyl substituted analogues 34 (4-CH3) and 35 (4-CH2CH3) (Fig. 5B and C). The activity of this series of compounds is comparable with leads 3 and 4. These analogues also displayed higher levels of PC cell line toxicity (34 and 35; GI50 values of 4.0–7.3 and 3.7–8.0 μM). In a CCK-8 assay against the inherently drug resistant CFPAC1 pancreatic cancer cell line, 35 and 36 returned GI50 values of 16 and 56 μM, respectively.
Investigations of the most favourable CF3 substitution pattern through modelling (not shown) suggested that the 4-position was optimal with poorer binding predicted with 2-CF3 and 3-CF3 analogues. Both these moieties are predicted to cause a twisting of the sulfonamide aromatic ring, reducing binding site engagement. This was conformed on the synthesis and screening of Library 4 analogues with only 45 (2-CF3, 4-Br) proceeding to full dose response evaluation.
Two final analogues were explored with 4-CF3 substituted 51 (4-OCH3) and 52 (2-OCH3). It was anticipated that the 2-OCH3 (52) moiety would result in a conformational twist in the triazolo-aromatic substituent and adversely affect the observed cytotoxicity. Compound 51 showed good levels of broad-spectrum cytotoxicity, as well as high levels of activity against all the pancreatic cancer cell lines examined spanning GI50 values of 5.2 (PANC-1) to 14 (AsPC-1) μM. The corresponding 2-OCH3 analogue, 52, showed an increase in potency and sub-micro molar potent across all cell lines examined. Analogue 52 was 10-fold more active than any analogue from Libraries 1–4 with GI50 values of 0.36 (HT29) to 0.80 (A2780) μM. Most significantly, 52 was sub-micromolar potent against the pancreatic cancer cell lines examined, GI50 0.48 (BxPC-3) to 0.68 (PANC-1) μM (Table 7). Also of note, both the HT29 and MCF-7a cell lines are known to be S100A2 positive, providing data consistent with, but not defining, that these compounds (e.g.52) target S100A2 as proposed.43,44
The observed cytotoxicity of Library 1 to Library 4 compounds was consistent with the S100A2–p53 binding model used in the design of these compounds.
Conclusions
Four libraries of novel piperazine-1,2,3-triazoles, 45 compounds total, have been designed, synthesised and subjected to cell line growth inhibitory screening against various panels of human cancer cell lines. These panels included numerous PC cell lines in addition to the normal cell populations (HPDE and MCF10A). Building on lead 1, Library 1 retained the 3-OCH3 substituent of 2's triazolophenyl moiety. No Library 1 analogue showed enhanced growth inhibition relative to 1. The SAR data associated with Library 2 revealed a preference for steric bulk associated with the triazolophenyl moiety with the 4-CH3 (26), 4-CH2CH3 (27), 4-CF3 (31) and 4-NO2 (32) analogues displaying slightly higher efficacy. The introduction of a 4-CF3 sulfonamide substituent removed predicted unfavourable interactions in the S100A2–p53-binding groove associated with the acetamide moiety of lead 2 (and Library 2), with enhanced cytotoxicity consistent with the binding poses of these analogues. The corresponding 2-CF3 and 3-CF3 sulfonamide moiety were essentially inactive relative to the 4-CF3 analogues.
Analogues possessing an alkyl substituent displayed higher levels of PC cell line toxicity (34 and 35; GI50 values of 4.0–7.3 and 3.7–8.0 μM), and 35 and 36 retuned GI50 values of 16 and 56 μM, respectively, against the drug resistant CFPAC1 pancreatic cancer cell line. From the data presented in Table 6 it is apparent that a 2-CF3 (43–46) or a 3-CF3 (47–50) moiety is detrimental to observed cytotoxicity. Both these moieties are predicted to cause a twisting of the sulfonamide aromatic ring, reducing binding site engagement.
Repositioning of the –OCH3 moiety from 4-OCH3 (51) to 2-OCH3 (52) resulted in a 10-fold increase in potency against all cell lines examined, with sub-micromolar activity noted against the PC cell lines, MiaPaCa-2, BxPC-3, AsPC-1, Capan-2 and PANC-1. In each of these cases S100A2 is known to be upregulated. The data presented herein strongly supports the further development of these analogues. Based on these findings, 52, in particular represents and interesting lead compound towards the development of S100A2 targeted therapies and a possible treatment for pancreatic cancer.
Experimental
Chemistry
General methods
All reactions were performed using standard laboratory equipment and glassware, except for the flow synthesis of Bo-protected piperazines which used the Syrris FRX-100 flow system. Solvents and reagents were purchased from Sigma Aldrich, Alfa Aesar or AK Scientific and used as received. Organic solvents were of bulk quality and distilled from glass prior to use. Organic solvent extracts were dried over magnesium sulfate (MgSO4), and the solvent removed under reduced pressure with either Büchi or Heidolph rotary evaporators. Melting points were recorded in open capillaries on a Büchi 565 Melting Point Apparatus. Where available, literature values are provided and appropriately referenced. Electrospray mass spectra were recorded using 10% DMSO/H2O or HPLC-grade methanol or acetonitrile as carrier solvents on an Agilent Technologies 1260 Infinity UPLC system with a 6120 Quadrupole LC/MS in electrospray ionization (ESI†) positive and negative modes. TLC was performed on Merck silica gel 60 F254 precoated aluminium plates with a thickness of 0.2 mm. Nuclear magnetic resonance (NMR) spectroscopy was performed on a Brüker Avance III 400 MHz spectrometer, where proton NMR (1H NMR) spectra and carbon NMR (13C NMR) spectra were acquired at 400 and 100 MHz respectively, or a Brüker Avance III 600 MHz spectrometer, where proton NMR (1H NMR) spectra and carbon NMR (13C NMR) spectra were acquired at 600 and 150 MHz respectively. All spectra were recorded in deuterated dimethyl sulfoxide (DMSO-d6) obtained from Cambridge Isotope Laboratories Inc. Chemical shifts (d) were measured in parts per million (ppm) and referenced against the internal reference peaks. Coupling constants (J) were measured in Hertz (Hz). NMR assignments were determined through the interpretation of one- and two-dimensional spectra. Multiplicities are denoted as singlet (s), broad singlet (bs), doublet (d), doublet of doublets (dd), triplet (t), quartet (q), triplet of doublets (td), doublet of triplets (dt) and multiplet (m). Peaks are listed in decreasing chemical shift in the following format: chemical shift integration (1H), multiplicity (1H), coupling constant (1H).
Synthesis of Library 1 – based on 3-ethynylanisole compounds 2, 14–24
Tert-butylpiperazine-1-carboxylate ESI† material
Boc2O (5.5 g, 25 mmol) in CH2Cl2 (115 mL) was added to piperazine (2 eq., 4.3 g, 50 mmol) in CH2Cl2 (40 mL) at 0 °C using the FRX flow system at a flow rate of 0.2 mL min−1. The resultant mixture was allowed to stir for an additional 2 h and allowed to warm to RT. The solid was collected and the filtrate evaporated in vacuo. The residue was taken up in H2O (50 mL), saturated with K2CO3, and extracted with EtOAc (3 × 100 mL). The organic extracts were washed with H2O (50 mL), saturated brine (50 mL), dried over Mg2SO4, and the solvent removed in vacuo. The desired product was obtained as a white solid, 6.0 g (65%), m.p. dec. >137 °C; TLC (CH3OH/DCM: 10 : 1, Rf: 0.30). 1H NMR (400 MHz, CDCl3) δ 3.39 (t, J = 12.8 Hz, 4H), 2.85 (s, 1H), 2.80 (t, J = 12.2 Hz, 4H), 1.46 (s, 9H); 13C NMR (101 MHz, DMSO-d6) δ 155.0 (C O), 79.7 (2C), 47.2, 46.0 (2C), 38.6 (3C); IR υmax/cm−1: 2978 (N–H), 1684(C O), 1160 (C–O), 1102 (C–N).
Tert-butyl 4-(3-hydroxypropyl)piperazine-1-carboxylate (10) ESI† material
Batch methodology
Boc-piperazine (1.9 g, 10 mmol) and K2CO3 (2 eq., 2.8 g, 20 mmol) in CH3CN (80 mL) was added 3-bromo-1-propanol (1.5 eq., 6.68 g, 15 mmol), and heated to reflux for 3 h. After filtration, the solvent was removed in vacuo and the residue taken up in water (50 mL), washed with EtOAc three times (3 × 100 mL) and the combined organic extracts evaporated in vacuo. The residue was triturated with cold diethyl ether (3 × 50 mL). The desired product was obtained as a white crystalline solid, 1.1 g, (47%), m.p. 68–70 °C; TLC (CH3OH/DCM: 10 : 1, Rf: 0.56).
Flow methodology
A solution of Boc2O (1.0 eq., 5.456 g, 25 mmol) in CH2Cl2 (115 mL) was added to a stirred solution of anhydrous piperazine (2.0 eq., 4.307 g, 50 mmol) in CH2Cl2 (40 mL) at 0 °C using the FRX flow system at a flow rate of 0.2 mL min−1. The obtained mixture was allowed to stir for an additional 2 h and warmed to RT, monitored by TLC analysis (CH3OH/DCM: 10 : 1, Rf: 0.30). The obtained suspension was filtered, evaporated to dryness in vacuo. The residue was dissolved in H2O (50 mL), saturated with K2CO3 and extracted with EtOAc (3 × 100 mL). The combined organic extracts were washed with H2O (50 mL), saturated brine (50 mL) and dried over Mg2SO4. Evaporation of the solvent afforded Boc-piperazine as a white solid.44,4 1H NMR (400 MHz, CDCl3) δ 3.81 (t, J = 5.3 Hz, 2H), 3.43 (t, J = 5.3 Hz, 4H), 3.39 (s, 1H), 2.62 (t, J = 5.8 Hz, 2H), 2.47 (t, J = 4.9 Hz, 4H), 1.76–1,71 (m, 2H), 1.46 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 154.8, 80.2, 80.0, 64.7 (2C), 59.0 (2C), 53.3, 28.5 (3C), 27.2; IR υmax/cm−1: 3179 (O–H), 1688 (C O), 1164 (C–O), 1125 (C–O), 1098 (C–N).
3-(Piperazin-1-yl)propylmethanesulfonate (11) ESI† material
To a solution of tert-butyl-4-(3-hydroxypropyl)piperazine-1-carboxylate (10) (10 mmol, 2.4 g) and K2CO3 (2 eq., 2.8 g, 20 mmol) in dry CH2Cl2 (40 mL) was added MsCl (1.3 eq., 1.5 g, 13 mmol). The resulting solution was stirred at RT for 3 h. The mixture was filtered, and the solvent removed under a stream of N2. The resulting residue was washed with cold diethyl ether (3 × 20 mL) to afford the desired product as a white solid, 2.1 g (66%), m.p. dec. >159 °C; TLC (CH3OH/DCM: 10 : 1, Rf: 0.56). 1H NMR (400 MHz, DMSO-d6) δ 10.85 (s, 1H), 4.31 (t, J = 6.0 Hz, 2H), 4.00 (d, J = 13.2 Hz, 2H), 3.47 (d, J = 11.3 Hz, 2H), 3.23–3.16 (m, 7H), 3.02–2.94 (m, 2H), 2.33 (s, 1H), 2.19–2.12 (m, 2H), 1.42 (s, 9H); 13C NMR (101 MHz, DMSO-d6) δ 153.4, 79.9, 67.6, 53.4, 52.4, 50.7 (d, J = 5.0 Hz, 1C), 42.3, 39.8, 36.7, 28.0 (3C), 26.3, 23.3; IR υmax/cm−1: 2976 (N–H), 1625 (C C), 1277 (S O), 1157 (S O), 1142 (C–F), 1108 (C–N).
Tert-butyl 4-(3-azidopropyl)piperazine-1-carboxylate (12) ESI† material
To a solution of tert-butyl 4-(3-((methylsulfonyl)oxy)propyl)piperazine-1-carboxylate (11) (0.32 g, 1 mmol) in CH3CN (100 mL) was added NaN3 (2.5 eq., 0.16 g, 2.5 mmol) and heated to reflux for 3 h. The solvent was removed under a stream of N2, 10 mL CHCl3 was added to the residue and the precipitate collected. The filtrate was evaporated under a stream of N2 to yield the desired product as white solid, 0.23 g, (86%), m.p. 117–120 °C; TLC (CH3OH/DCM: 10 : 1, Rf: 0.59). 1H NMR (400 MHz, CDCl3) δ 3.60 (t, J = 6.5 Hz, 1H), 3.42 (t, J = 4.6 Hz, 4H), 3.35 (t, J = 6.7 Hz, 1H), 2.49 (t, J = 7.0 Hz, 1H), 2.43 (t, J = 7.1 Hz, 1H), 2.39–2.36 (m, 4H), 1.98–1.90 (m, 1H), 1.80–1.73 (m, 1H), 1.46 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 154.9, 79.8, 55.5, 55.4, 53.1(d, J = 7.7 Hz, 1C), 49.6, 43.2, 29.9, 28.6 (3C), 26.3; IR υmax/cm−1: 2094 (N N N), 1691 (C O), 1168 (C–O), 1124 (C–O), 1241 (C–N).
1-(3-(4-(3-Methoxyphenyl)-1H-1,2,3-triazol-1-yl)propyl)piperazine (13a) ESI† material
To a solution of tert-butyl-4-(3-azidopropyl)piperazine-1-carboxylate (12) (1 eq., 1.3 g, 5 mmol) in acetone (20 mL), sodium ascorbate (0.2 eq., 0.20 g, 1 mmol) in water (1 mL) and CuSO4·5H2O (0.1 eq., 0.12 g, 0.5 mmol) in H2O (0.5 mL), 3-ethynylanisole (8a) (1.2 eq., 0.79 g, 6 mmol) was added. The mixture was stirred at RT for 24 h, monitored by TLC (CH3OH/DCM: 10 : 1, Rf: 0.58). After completion, the mixture was filtered, and the solvent was removed in vacuo. The residue was de-protected in a solution of ethyl acetate (10 mL) and concentrated HCl (5 mL) (volume: 2 : 1) for 30 min. The mixture was dried under a stream of compressed air, and the residue was washed with acetone to afford the desired product as a yellow solid (1.5 g, 86%, purity >99%, UPLC-MS). The solid was used without further purification. These intermediates are highly hygroscopic and used as soon as prepared.
N-(4-((4-(3-(4-(3-Methoxyphenyl)-1H-1,2,3-triazol-1-yl)propyl)piperazin-1-yl)-sulfonyl)phenyl)acetamide (2)
A solution of 1-(3-(4-(3-methoxyphenyl)-1H-1,2,3-triazol-1-yl)propyl)piperazine hydrochloride (13a) (0.25 g, 0.75 mmol) and NaOH (2.5 eq., 0.08 g, 1.9 mmol) in CH2Cl2 (20 mL), was stirred for 15–20 min, to this 4-acetamidobenzenesulfonyl chloride (8a) (1.5 eq., 0.26 g, 1.1 mmol) in CH2Cl2 (5 mL) was added. The mixture was stirred for 40 min at RT, and when complete by UPLC-MS analysis, the mixture was filtered, the solvent was removed under a stream of compressed air and the residue was dissolved in methanol (5 mL). Trituration with cold diethyl ether (3 × 5 mL) afforded the desired compound 2 as an off-white solid. Yield: 0.29 g, 78%, m.p.: dec. >140 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.38 (s, 1H), 8.53 (s, 1H),7.82 (d, J = 8.8 Hz, 2H), 7.66 (d, J = 8.8 Hz), 7.39–7.32 (m, 3H), 6.90–6.87 (m, 1H), 4.34 (t, 2H, J = 7.0 Hz), 3.80 (s, 3H), 2.86 (s, 4H), 2.42 (s, 4H), 2.29 (t, 2H, J = 6.7 Hz), 2.09 (s, 3H), 2.00–1.93 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 169.1, 159.6, 146.1, 143.5, 132.2, 130.0 (2C), 128.8 (2C), 128.2, 121.6, 118.6, 117.4, 113.5, 110.3, 55.1, 53.8, 51.5 (2C), 47.6, 45.9 (2C), 26.8, 24.1; IR υmax/cm−1: 3392 (N–H), 1693 (C O), 1592 (C C), 1348 (S O), 1244 (CO), 1160 (S O), 1096 (CN), 1035 (CO). LRMS (ESI†) m/z: 499.3 [M + H]; HRMS calculated for C24H30N6O4S [M + H]: 499.2100; found: 499.2122.
1-(3-(4-(3-Methoxyphenyl)-1H-1,2,3-triazol-1-yl)propyl)-4-(phenylsulfonyl)-piperazine benzene sulfonate (14)
Synthesized using the general procedure as for 2, from 1-(3-(4-(3-methoxyphenyl)-1H-1,2,3-triazol-1-yl)propyl)piperazine hydrochloride (13a) (1 eq., 0.67 g, 2 mmol) and benzene sulfonyl chloride (8b) (1.1 eq., 0.39 g, 2.2 mmol). The desired product (as the benzenesulfonate salt) 14 as an off-white solid, 0.16 g (27%); m.p. 141–144 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.73 (s), 8.59 (s, 1H),7.78–7.71 (m, 5H), 7.58 (s, 2H), 7.39–7.31 (m, 6H), 6.91 (d, J = 8.0, 1H), 4.47 (s, 2H), 3.80 (s, 5H), 3.55 (s, 2H), 3.15 (s, 4H), 2.58 (s, 2H), 2.24 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 159.7, 148.2, 146.3, 133.9, 132.0, 130.1, 130.0, 128.5 (4C), 127.7 (2C), 125.5 (2C), 121.8 (2C), 117.5, 113.6, 110.4, 55.2, 52.9, 50.4 (2C), 46.8, 43.2 (2C), 24.2; IR υmax/cm−1: 1584 (C C), 1349 (S O), 1229 (C–O), 1162.1 (S O), 1126 (C–N), 1042 (C–O); LRMS (ESI†) m/z: 442.2 [M–C6H5SO3H + H]; HRMS calculated for C28H33N5O6S6 [M–C6H5SO3H + H]: 442.1898; found: 442.1907.
1-(3-(4-(3-Methoxyphenyl)-1H-1,2,3-triazol-1-yl)propyl)-4-tosylpiperazine (15)
Synthesized using the general procedure as for 2, from 1-(3-(4-(3-methoxyphenyl)-1H-1,2,3-triazol-1-yl)propyl)piperazine hydrochloride (13a) (1 eq., 0.17 g, 0.5 mmol) and 4-methylbenzene sulfonyl chloride (8c) (1 eq., 0.10 g, 0.5 mmol). When complete by UPLC-MS analysis, the mixture was filtered, the solvent was removed under a stream of compressed air and the residue was dissolved in methanol (5 mL). Trituration with cold diethyl ether (3 × 5 mL) afforded 11 as an off-white solid. Yield: 0.08 g, 35%, m.p.: 118–120 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.55 (s, 1H), 7.62 (d, J = 8.0 Hz, 2H), 7.45 (d, J = 8.0 Hz, 2H), 7.40–7.33 (m, 3H), 6.89 (s, J = 8.4 Hz, 1H), 4.36 (s, 2H), 3.81 (s, 3H), 2.85 (s, 4H), 2.42 (s, 7H), 2.28 (s, 2H), 1.97 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 159.7, 146.1, 143.7, 132.2, 131.8, 130.0, 129.9, 128.0, 127.6, 125.5, 121.6, 117.4, 113.4, 110.3, 55.1, 53.7, 51.4 (2C), 47.5, 45.9 (2C), 26.7, 21.0; IR υmax/cm−1: 1594 (C C), 1345 (S O), 1243 (CO), 1160 (S O), 1094 (C–N), 1033 (CO); LRMS (ESI†) m/z: 456.2 [M + H]; HRMS calculated for C23H29N5O3S [M + H]: 456.2061; found: 456.2064.
1-(4-Isopropylphenylsulfonyl)-4-(3-(4-(3-methoxyphenyl)-1H-1,2,3-triazol-1-yl)-propyl) piperazine hydrochloride (16)
Synthesized using the general procedure as for 2, from 1-(3-(4-(3-methoxyphenyl)-1H-1,2,3-triazol-1-yl)propyl)piperazine hydrochloride (13a) (1 eq., 0.25 g, 0.75 mmol) and 4-iso-propylbenzene sulfonyl chloride (8d) (1 eq., 0.16 g, 0.75 mmol). When complete by UPLC-MS analysis, the mixture was filtered, the solvent was removed under a stream of compressed air. The residue was added acetone 4 mL and 10% HCl 10 mL and filtered to afford a white solid and the solid was dissolved in acetone (5 mL). Trituration with cold diethyl ether (3 × 5 mL) afforded compound 12 hydrochloride as an off-white solid. Yield: 0.11 g, 51%, m.p.: 124–126 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.85 (s, 1H), 8.62 (s, 1H), 7.70 (d, J = 8.0 Hz, 2H), 7.56 (d, J = 8.0 Hz, 2H), 7.41–7.34 (m, 3H), 6.91 (d, J = 8.2 Hz, 1H), 4.49 (t, 2H, J = 6.1 Hz), 3.81 (s, 3H), 3.76 (s, 2H), 3.56–3.53 (m, 2H), 3.14–2.99 (m, 5H), 2.75–2.69 (m, 2H), 2.35–2.26. (m, 2H), 1.24 (s, 3H), 1.23 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 159.7, 154.6, 146.3, 132.0, 131.8, 130.0, 127.9 (2C), 127.6 (2C), 121.8, 117.5, 113.5, 110.4, 55.1, 52.8, 50.2 (2C), 46.8, 43.0 (2C), 33.4, 23.9, 23.4 (2C); IR υmax/cm−1: 2968 (N–H), 1595 (C C), 1339 (S O), 1250 (CO), 1157 (S O), 1096 (C–N), 1056 (CO); LRMS (ESI†) m/z: 484.3 [M–HCl + H]; HRMS calculated for C25H34ClN5O3S [M–HCl + H]: 484.2365; found: 484.2377.
1-((4-(tert-Butyl)phenyl)sulfonyl)-4-(3-(4-(3-methoxyphenyl)-1H-1,2,3-triazol-1-yl)propyl)piperazine hydrochloride (17)
Synthesized using the general procedure as for 2, from 1-(3-(4-(3-methoxyphenyl)-1H-1,2,3-triazol-1-yl)propyl)piperazine hydrochloride (13a) (1 eq., 0.17 g, 0. 5 mmol) and 4-tert-butylbenzene sulfonyl chloride (8e) (1.1 eq., 0.13 g, 0.55 mmol). When complete by UPLC-MS analysis, the mixture was filtered, the solvent was removed under a stream of compressed air. The residue was added acetone 4 mL and 10% HCl 10 mL and filtered to afford a white solid and the solid was dissolved in acetone (5 mL). Trituration with cold diethyl ether (3 × 5 mL) afforded compound 17 hydrochloride as an off-white solid. Yield: 0.16 g, 60%, m.p.: dec. >186 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.94 (s, 1H), 8.63 (s, 1H), 7.71 (s, 4H), 7.39–7.34 (m, 3H), 6.91 (d, J = 6.8 Hz, 1H), 4.49 (s, 2H), 3.81–3.74 (s, 5H), 3.54 (s, 2H), 3.14 (s, 4H), 2.77–2.71 (m, 2H), 2.30 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 159.7, 156.8, 146.3, 132.0, 131.6, 130.0, 127.6 (2C), 126.5 (2C), 121.8, 117.5, 113.6, 110.4, 55.1, 52.8, 50.2 (2C), 46.9, 43.0 (2C), 35.0, 30.7 (3C), 23.9; IR υmax/cm−1: 2968 (N–H), 1592 (C C), 1328 (S O), 1244 (C–O), 1172 (S O), 1090 (C–N), 1030 (C–O); LRMS (ESI†) m/z: 498.3 [M–HCl + H]; HRMS calculated for C26H36ClN5O3S [M–HCl + H]: 498.2525; found: 498.2533.
1-(3-(4-(3-Methoxyphenyl)-1H-1,2,3-triazol-1-yl)propyl)-4-((4-methoxyphenyl)-sulfonyl)piperazine 4-methoxy benzenesulfonate (18)
Synthesized using the general procedure as for 2, from 1-(3-(4-(3-methoxyphenyl)-1H-1,2,3-triazol-1-yl)propyl)piperazine hydrochloride (13a) (1 eq., 0.17 g, 0.5 mmol) and 4-methoxybenzene sulfonyl chloride (8f) (1.1 eq., 0.11 g, 0.55 mmol). When complete by UPLC-MS analysis, the mixture was filtered, the solvent was removed under a stream of compressed air and the residue was dissolved in methanol (5 mL). Trituration with cold diethyl ether (3 × 5 mL) afforded compound 18 as an off-white solid. Yield: 0.11 g, 34%, m.p.: 161–164 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.44 (s, 1H), 8.62 (s, 1H), 7.72 (d, J = 8.4 Hz, 2H), 7.51 (d, J = 8.4 Hz, 1H), 7.40–7.34 (m, 5H), 7.21 (d, J = 8.4 Hz, 2H), 6.91 (d, J = 7.6 Hz, 1H), 6.85 (d, J = 8.4 Hz, 1H), 4.48–4.47 (m, 2H), 3.87–3.71 (m, 11H), 3.54 (d, J = 8.9 Hz, 2H), 3.14 (s, 4H,), 2.67 (t, 2H, J = 10.4 Hz), 2.28 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 163.7, 160.1, 159.7, 146.6, 141.3, 132.5, 130.5, 130.4 (2C), 127.5 (2C), 126.0, 122.3, 118.0 (2C), 115.3 (2C), 114.0, 113.2, 110.9, 56.3, 55.6 (2C), 53.3, 50.7 (2C), 47.3, 43.6 (2C), 24.5; IR υmax/cm−1: 2975 (N–H), 1594 (C C), 1335 (S O), 1259 (C–O), 1154 (S O), 1096 (CN), 1025 (CO); LRMS (ESI†) m/z: 472.3 [M–C7H7SO3H + H]; HRMS calculated for C30H37N5O8S2 [M–C7H7SO3H + H]: 472.2006; found: 472.2013.
1-((4-Fluorophenyl)sulfonyl)-4-(3-(4-(3-methoxyphenyl)-1H-1,2,3-triazol-1-yl)-propyl)piperazine hydrochloride (19)
Synthesized using the general procedure as for 2, from 1-(3-(4-(3-methoxyphenyl)-1H-1,2,3-triazol-1-yl)propyl)piperazine hydrochloride (13a) (1 eq., 0.17 g, 0.5 mmol) and 4-fluorobenzene sulfonyl chloride (8g) (1.1 eq., 0.11 g, 0.55 mmol). When complete by UPLC-MS analysis, the mixture was filtered, the solvent was removed under a stream of compressed air. The residue was added acetone 4 mL and 10% HCl 10 mL and filtered to afford a white solid and the solid was dissolved in acetone (5 mL). Trituration with cold diethyl ether (3 × 5 mL) afforded compound 19 hydrochloride as an off-white solid. Yield: 0.14 g, 57%, m.p.: dec. >174 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.96 (s, 1H), 8.62 (s, 1H), 7.87 (dd, J = 8.6, 5.1 Hz, 2H), 7.55 (t, J = 8.7 Hz, 2H), 7.41–7.34 (m, 3H), 6.91 (d, J = 8.2 Hz, 1H), 4.49 (t, J = 6.5 Hz, 2H), 3.81–3.54 (s, 5H), 3.54 (s, 2H), 3.13 (s, 4H), 2.80–2.74 (m, 2H), 2.30 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 164.9 (d, J = 252.7), 159.7, 146.3, 132.0, 130.9 (d, J = 9.7 Hz), 130.8 (d, J = 2.8 Hz, 2C), 130.1, 121.8, 117.5, 117.0 (d, J = 22.8 Hz, 2C), 113.6, 110.4, 55.1, 52.7, 50.1 (2C), 46.9, 42.9 (2C), 23.9; 19F NMR (376 MHz, DMSO-d6) δ −101.7; IR υmax/cm−1: 2980 (N–H), 1586 (C C), 1357 (S O), 1230 (C–O), 1171.5 (S O), 1157 (C–F), 1090 (C–N), 1041 (C–O), 763 (C–Cl); LRMS (ESI†) m/z: 460.2 [M–HCl + H]; HRMS calculated for C22H27ClFN5O3S [M–HCl + H]: 460.1802; found: 460.1813.
1-((4-Chlorophenyl)sulfonyl)-4-(3-(4-(3-methoxyphenyl)-1H-1,2,3-triazol-1-yl)-propyl)piperazine (20)
Synthesized using the general procedure as for 2, from 1-(3-(4-(3-methoxyphenyl)-1H-1,2,3-triazol-1-yl)propyl)piperazine hydrochloride (13a) (1 eq., 0.17 g, 0.5 mmol) and 4-chlorobenzene sulfonyl chloride (8h) (1 eq., 0.12 g, 0.55 mmol). When complete by UPLC-MS analysis, the mixture was filtered, the solvent was removed under a stream of compressed air and the residue was dissolved in methanol (5 mL). Trituration with cold diethyl ether (3 × 5 mL) afforded compound 20 as an off-white solid. Yield: 012 g, 51%, m.p.: 124–126 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.44 (s, 1H), 8.62 (s, 1H), 7.72 (d, J = 8.4 Hz, 2H), 7.51 (d, J = 8.4 Hz, 1H), 7.40–7.34 (m, 5H), 7.21 (d, J = 8.4 Hz, 2H), 6.91 (d, J = 7.6 Hz, 1H), 6.85 (d, J = 8.4 Hz, 1H), 4.48–4.47 (m, 2H), 3.87–3.71 (m, 11H), 3.54 (d, J = 8.9 Hz, 2H), 3.14 (s, 4H,), 2.67 (t, 2H, J = 10.4 Hz), 2.28 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 159.6, 146.0, 138.3, 133.8, 132.2130.0, 129.6 (2C), 129.4 (2C), 121.6, 117.4, 113.4, 110.3, 55.1, 53.8, 51.5 (2C), 47.6, 45.9 (2C), 26.7; IR υmax/cm−1: 1583 (C C), 1345 (S O), 1230 (CO), 1169 (S O), 1093 (CN), 1041 (CO), 763 (C–Cl); LRMS (ESI†) m/z: 476.2 [M + H, 35Cl, 100%]/478.2 [M + H, 37Cl, 35%]; HRMS calculated for C22H26ClN5O3S [M + H, 35Cl]: 476.1508/[M + H, 37Cl]: 478.1479; found: 476.1518/478.1518.
1-((4-Bromophenyl)sulfonyl)-4-(3-(4-(3-methoxyphenyl)-1H-1,2,3-triazol-1-yl) propyl)piperazine (21)
Synthesized using the general procedure for 2, from 1-(3-(4-(3-methoxyphenyl)-1H-1,2,3-triazol-1-yl)propyl)piperazine hydrochloride (13a) (0.17 g, 0.5 mmol) and 4-bromobenzene sulfonyl chloride (8i) (1 eq., 0.13 g, 0.5 mmol). When complete by UPLC-MS analysis, the mixture was filtered, the solvent was removed under a stream of compressed air and the residue was dissolved in methanol (5 mL). Trituration with cold diethyl ether (3 × 5 mL) afforded compound 21 as an off-white solid. Yield: 0.13 g, 50%, m.p.: 131–133 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.54 (s, 1H), 7.87 (d, J = 8.4 Hz), 7.66 (d, J = 8.4 Hz), 7.38–7.33 (m, 3H), 6.90 (d, J = 8.0 Hz), 4.36 (t, J = 6.8 Hz, 2H), 3.81 (s, 3H), 2.90 (s, 4H), 2.43 (s, 4H), 2.30 (s, 2H), 1.99–1.96 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 159.6, 146.1, 134.2, 132.5 (2C), 132.2, 130.0, 129.5 (2C), 127.3, 121.6, 117.4, 113.4, 110.3, 55.1, 53.8 (2C), 51.5, 47.6 (2C), 45.9, 26.7; IR υmax/cm−1: 1584 (C C), 1347 (S O), 1168 (C–O) and (S O), 1096 (C–N), 1041 (CO); LRMS (ESI†) m/z: 520.1 [M + H, 79Br, 90%]/522.1 [M + H, 81Br, 100%]; HRMS calculated for C22H26BrN5O3S [M + H, 79Br]: 520.1008/[M + H, 81Br]: 522.0981; found: 520.1012/522.1012.
1-(3-(4-(3-Methoxyphenyl)-1H-1,2,3-triazol-1-yl)propyl)-4-((4-(trifluoromethyl)-phenyl)sulfonyl) piperazine (22)
Synthesized using the general procedure as for 2, from 1-(3-(4-(3-methoxyphenyl)-1H-1,2,3-triazol-1-yl)propyl)piperazine hydrochloride (13a) (1 eq., 0.17 g, 0.5 mmol) and 4-trifluoromethylbenzene sulfonyl chloride (8j) (1.1 eq., 0.13 g, 0.55 mmol). When complete by UPLC-MS analysis, the mixture was filtered, the solvent was removed under a stream of compressed air and the residue was dissolved in acetone (5 mL). Trituration with cold diethyl ether (3 × 5 mL) afforded compound 22 as an off-white solid. Yield: 0.15 g, 59%, m.p. 132–135 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.53 (s, 1H), 8.00 (dd, J = 31.5, 7.2 Hz, 4H), 7.36–7.30 (m, 3H), 6.89 (d, J = 7.0 Hz, 1H), 4.35 (s, 2H), 3.80 (s, 3H), 2.95 (s, 4H), 2.43 (s, 4H), 2.29 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 159.7, 146.1, 139.0, 132.9 (q, J = 32.5 Hz,), 132.2, 130.0 (2C), 128.5 (2C), 126.7 (q, J = 3.6 Hz, aryl C), 123.4 (q, J = 271.3 Hz, CF3), 121.6 (2C), 117.4, 113.5, 110.3, 55.1, 53.8 (2C), 51.5, 47.6 (2C), 45.8, 26.7; 19F NMR Analysis: 19F NMR (376 MHz, DMSO-d6) δ −58.6; IR υmax/cm−1: 1586 (C C), 1348 (S O), 1322 (C–F), 1247 (C–O), 1172 (S O), 1090 (C–N), 1061 (C–O); LRMS (ESI†) m/z: 510.2 [M + H]; HRMS calculated for C23H26F3N5O3S [M + H]: 510.1773; found: 510.1781.
1-(3-(4-(3-Methoxyphenyl)-1H-1,2,3-triazol-1-yl)propyl)-4-((4-nitrophenyl)-sulfonyl)piperazine hydrochloride (23)
Synthesized using the general procedure as for 2, from 1-(3-(4-(3-methoxyphenyl)-1H-1,2,3-triazol-1-yl)propyl)piperazine hydrochloride (13a) (1 eq., 0.17 g, 0. 5 mmol) and 4-nitrobenzene sulfonyl chloride (8k) (0.9 eq., 0.10 g, 0.45 mmol). When complete by UPLC-MS analysis, the mixture was filtered, the solvent was removed under a stream of compressed air. The residue was added acetone 4 mL and 10% HCl 10 mL and filtered to afford a white solid and the solid was dissolved in methanol (5 mL). Trituration with cold diethyl ether (3 × 5 mL) afforded compound 23 hydrochloride as an off-white solid. Yield: 0.23 g, 89%, m.p.: 219–223 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.21 (s, 1H), 8.62 (s, 1H),8.49 (d, J = 8.4 Hz, 2H), 8.08 (d, J =8.4 Hz, 2H), 7.41–7.34 (m, 3H), 6.92–6.89 (m, 1H), 4.50 (t, 2H, J = 6.8 Hz), 3.84 (s, 2H), 3.81 (s, 3H), 3.57 (s, 2H), 3.13(s, 4H), 2.95–2.89 (m, 2H), 2.35–2.27 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 159.6, 150.4, 146.3, 140.3, 132.0, 130.1, 129.3 (2C), 124.9 (2C), 121.8, 117.5, 113.5, 110.4, 55.1, 52.7, 50.1 (2C), 46.9, 42.8 (2C), 23.9; IR υmax/cm−1: 2951 (N–H), 1583 (C C), 1351 (S O), 1166 (C–O), (S O), 1096 (C–N) and 1044 (CO); LRMS (ESI†) m/z: 487.2 [M–HCl + H]; HRMS calculated for C22H27Cl2N6O5S [M–HCl + H]: 487.1760; found: 487.1758.
1-(4-((4-(3-(4-(3-Methoxyphenyl)-1H-1,2,3-triazol-1-yl)propyl)piperazin-1-yl)-sulfonyl)phenyl)ethanone hydrochloride (24)
Synthesized using the general procedure as for 2, from 1-(3-(4-(3-methoxyphenyl)-1H-1,2,3-triazol-1-yl)propyl)piperazine hydrochloride (13a) (1 eq., 0.17 g, 0.5 mmol) and 4-acetylbenzene sulfonyl chloride (8l) (0.9 eq., 0.10 g, 0.45 mmol). When complete by UPLC-MS analysis, the mixture was filtered, the solvent was removed under a stream of compressed air. The residue was added acetone 4 mL and 10% HCl 10 mL and filtered to afford a white solid and the solid was dissolved in methanol (5 mL). Trituration with cold diethyl ether (3 × 5 mL) afforded compound 24 hydrochloride as an off-white solid. Yield: 0.15 g, 58%, m.p.: 188–191 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.97 (s, 1H), 8.61 (s, 1H), 8.21 (d, J = 8.4 Hz, 2H), 7.93 (d, J = 8.4 Hz, 2H), 7.39–7.36 (m, 3H), 6.91 (s, J = 8.2 Hz, 1H), 4.49 (t, 2H, J = 6.7 Hz), 3.80 (s, 5H), 3.54 (s, 2H), 3.14 (s, 4H), 2.84–2.77 (m, 2H), 2.67 (s, 3H), 2.30 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 197.3, 159.7, 146.3, 140.4, 138.1, 132.0, 130.0, 129.3 (2C), 128.0 (2C), 121.8, 117.5, 113.5, 110.4, 55.1, 52.7, 50.1 (2C), 46.8, 42.9 (2C), 27.1, 23.9; IR υmax/cm−1: 2986 (N–H), 1687 (C O), 1586 (C C), 1354 (S O), 1267 (CO), 1169 (S O), 1099 (C–N), 1047 (CO); LRMS (ESI†) m/z: 484.2 [M–HCl + H]; HRMS calculated for C24H30ClN5O4S [M–HCl + H]: 484.2003; found: 484.2013.
Synthesis of Library 2 – based on 4-acetylaminobenzenesulfonyl chloride compounds 25–32
N-(4-{4-[3-(4-Phenyl-[1,2,3]triazol-1-yl)-propyl]-piperazine-1-sulfonyl}-phenyl)-acetamide hydrochloride (25)
Synthesized using the general procedure as for 2, from tert-butyl-4-(3-azidopropyl)piperazine-1-carboxylate (12) (1 eq., 0.20 g, 0.74 mmol) and 1-ethynylbenzene (4b) (1.3 eq., 0.10 g, 0.96 mmol) to afford 1-[3-(4-phenyl-[1,2,3]triazol-1-yl)-propyl]-piperazine hydrochloride (13b) (1 eq., 0.11 g, 0.36 mmol), then reacting with 4-acetylaminobenzene sulfonyl chloride (8a) (1.2 eq., 0.10 g, 0.43 mmol) (monitored by UPLC-MS analysis) to afford compound 25 hydrochloride as a pale pink solid. Yield: 0.09 g, 56%, m.p.: 198–202 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.63 (s, 1H), 10.51 (s, 1H), 8.59 (s, 1H), 7.93–7.78 (m, 4H), 7.71 (d, J = 8.8 Hz, 2H), 7.45 (t, J = 7.6 Hz, 2H), 7.34 (t, J = 7.4 Hz, 1H), 4.49 (t, J = 6.7 Hz, 2H), 3.71 (d, J = 7.0 Hz, 2H), 3.54 (d, J = 9.7 Hz, 2H), 3.13 (s, 4H), 2.68 (t, J = 11.7 Hz, 2H), 2.28 (s, 2H), 2.10 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 169.2, 146.4, 144.0, 130.7, 130.0 (2C), 128.9 (2C), 127.9, 127.2, 125.1 (2C), 121.5, 118.8 (2C), 52.8, 50.2 (2C), 46.8, 43.1 (2C), 24.2, 24.0; IR υmax/cm−1: 2985 (N–H), 1681.8 (C O), 1589.9 (C C), 1333.7 (S O), 1159.6 (S O), 1095.4 (C–N); LRMS (ESI†) m/z: 469.3 [M–HCl + H]; HRMS calculated for C23H29ClN6O3S [M–HCl + H]: 469.2011; found: 469.2016.
N-(4-{4-[3-(4-p-Tolyl-[1,2,3]triazol-1-yl)-propyl]-piperazine-1-sulfonyl}-phenyl)-acetamide hydrochloride (26)
Synthesized using the general procedure as for 2, from tert-butyl-4-(3-azidopropyl)piperazine-1-carboxylate (12) (1 eq., 0.20 g, 0.74 mmol) and 4-methyl-1-ethynylbenzene (4c) (1.3 eq., 0.11 g, 0.96 mmol) to afford 1-[3-(4-(4-tolyl)-[1,2,3]triazol-1-yl)-propyl]-piperazine hydrochloride (13c) (1 eq., 0.21 g, 0.65 mmol), then reacting with 4-acetylaminobenzene sulfonyl chloride (8a) (1.5 eq., 0.23 g, 0.98 mmol) (monitored by UPLC-MS analysis) to afford compound 26 hydrochloride as an off-white solid. Yield: 0.16 g, 52%, m.p.: dec. >156 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.68 (s, 1H), 10.52 (s, 1H), 8.53 (s, 1H), 7.87 (d, J = 8.8 Hz, 2H), 7.71 (d, J = 8.5 Hz, 4H), 7.26 (d, J = 8.0 Hz, 2H), 4.47 (t, J = 6.6 Hz, 2H), 3.76–3.65 (m, 2H), 3.54 (d, J = 10.3 Hz, 2H), 3.12 (s, 4H), 2.68 (t, J = 11.6 Hz, 2H), 2.33 (s, 3H), 2.28 (s, 3H), 2.10 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 169.2, 146.4, 144.0, 137.2, 129.5 (2C), 129.0 (2C), 127.9, 127.3, 125.1 (2C), 121.1, 118.8 (2C), 52.8, 50.2 (2C), 46.8, 43.1 (2C), 24.2, 24.0, 20.8; IR υmax/cm−1: 3028 (N–H), 1686 (C O), 1589 (C C), 1351 (S O), 1158 (S O), 1096 (C–N); LRMS (ESI†) m/z: 483.3 [M–HCl + H]; HRMS calculated for C24H31Cl2N6O3S [M–HCl + H]: 483.2166; found: 483.2173.
N-[4-(4-{3-[4-(4-Ethylphenyl)-[1,2,3]triazol-1-yl]-propyl}-piperazine-1-sulfonyl)-phenyl]-acetamide hydrochloride (27)
Synthesized using the general procedure as for 1, from tert-butyl-4-(3-azidopropyl)piperazine-1-carboxylate (12) (1 eq., 0.25 g, 0.93 mmol) and 4-ethyl-1-ethynylbenzene (4d) (1.3 eq., 0.16 g, 1.21 mmol) to afford 1-{3-[4-(4-ethylphenyl)-[1,2,3]triazol-1-yl]-propyl}-piperazine hydrochloride (13d) (1 eq., 0.17 g, 0.51 mmol), then reacting with 4-acetylaminobenzene sulfonyl chloride (8a) (1.5 eq., 0.18 g, 0.76 mmol) (monitored by UPLC-MS analysis) to afford compound 27 hydrochloride as a yellow solid. Yield: 0.13 g, 48%, m. p.: dec. >153 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.79 (s, 1H), 10.53 (s, 1H), 8.54 (s, 1H), 7.87 (d, J = 8.8 Hz, 2H), 7.72 (t, J = 8.3 Hz, 4H), 7.29 (d, J = 8.1 Hz, 2H), 4.48 (t, J = 6.7 Hz, 2H), 3.72–3.69 (m, 2H), 3.12 (s, 4H), 2.72–2.60 (m, 4H), 2.32–2.25 (m, 2H), 2.10 (s, 3H), 1.20 (t, J = 7.6 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 169.2, 146.5, 144.0, 143.5, 129.0 (2C), 128.3 (2C), 128.2, 127.3, 125.2 (2C), 121.1, 118.8 (2C), 52.8, 50.2 (2C), 46.8, 43.0 (2C), 27.9, 24.2, 24.0, 15.5; IR υmax/cm−1: 2970 (N–H), 1689 (C O), 1589 (C C), 1315 (S O), 1158 (S O), 1094 (C–N); LRMS (ESI†) m/z: 497.3 [M–HCl + H]; HRMS calculated for C25H33ClN6O3S [M–HCl + H]: 497.2324; found: 497.2329.
N-[4-(4-{3-[4-(4-Fluorophenyl)-[1,2,3]triazol-1-yl]-propyl}-piperazine-1-sulfonyl)-phenyl]-acetamide hydrochloride (28)
Synthesized using the general procedure as for 2, from tert-butyl-4-(3-azidopropyl)piperazine-1-carboxylate (12) (1 eq., 0.25 g, 0.93 mmol) and 4-fluoro-1-ethynylbenzene (4e) (2 eq., 0.22 g, 1.86 mmol) to afford 1-{3-[4-(4-fluorophenyl)-[1,2,3]triazol-1-yl]-propyl}-piperazine hydrochloride (13e) (1 eq., 0.17 g, 0.52 mmol), then reacting with 4-acetylaminobenzene sulfonyl chloride (8a) (1.5 eq., 0.18 g, 0.78 mmol) (monitored by UPLC-MS analysis) to afford compound 28 hydrochloride as an off-white solid. Yield: 0.18 g, 69%, m.p.: 226–229 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.75 (s, 1H), 10.53 (s, 1H), 8.59 (s, 1H), 7.88–7.86 (m, 4H), 7.72 (d, J = 6.8 Hz, 2H), 7.30 (dd, J = 8.7, 6.6 Hz, 2H), 4.49 (s, 2H), 3.70 (d, J = 28.2 Hz, 2H), 3.54 (d, J = 26.7, 2H), 3.12 (s, 4H), 2.69 (s, 2H), 2.29 (s, 2H), 2.11 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 169.2, 161.8 (d, J = 242.9 Hz), 145.5 (2 overlapping signals, unconfirmed by 2D NMR.), 140.0, 129.0 (2C), 127.3 (d, J = 2.9 Hz), 127.1 (d, J = 8.2 Hz, 2C), 121.5, 118.8 (2C), 115.9 (d, J = 21.6 Hz, 2C), 52.7, 50.2 (2C), 46.9, 43.0 (2C), 24.2, 24.0; 19F NMR (376 MHz, DMSO-d6) δ −110.9; IR υmax/cm−1: 3046 (N–H), 1691 (C O), 1591 (C C), 1332 (S O), 1267 (C–F), 1159 (S O), 1096 (C–N); LRMS (ESI†) m/z: 487.3 [M–HCl + H]; HRMS calculated for C23H28ClFN6O3S [M–HCl + H]: 487.1916; found: 487.1922.
N-[4-(4-{3-[4-(4-Bromophenyl)-[1,2,3]triazol-1-yl]-propyl}-piperazine-1-sulfonyl)-phenyl]-acetamide hydrochloride (29)
Synthesized using the general procedure as for 2, from tert-butyl-4-(3-azidopropyl) piperazine-1-carboxylate (12) (1 eq., 0.20 g, 0.74 mmol) and 4-bromo-1-ethynylbenzene (4f) (1.3 eq., 0.17 g, 0.96 mmol) to afford 1-{3-[4-(4-bromophenyl)-[1,2,3]triazol-1-yl]-propyl}piperazine hydrochloride (13f) (1 eq., 0.18 g, 0.46 mmol), then reacting with 4-acetylaminobenzene sulfonyl chloride (8a) (1.5 eq., 0.16 g, 0.69 mmol) (monitored by UPC-MS analysis) to afford compound 29 hydrochloride as an off-white solid. Yield: 0.10 g, 27%, m.p.: 207–211 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.65 (s, 1H), 10.51 (s, 1H), 8.64 (s, 1H), 7.87 (d, J = 8.6 Hz, 2H), 7.79 (d, J = 8.2 Hz, 2H), 7.71 (d, J = 8.5 Hz, 2H), 7.65 (d, J = 8.2 Hz, 2H), 4.49 (s, 2H), 3.70 (s, 2H), 3.53 (s, 2H), 3.12 (s, 4H), 2.68 (s, 2H), 2.27 (s, 2H), 2.10 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 169.2, 145.3, 144.0, 131.9 (2C), 130.0, 129.0 (2C), 127.3, 127.1 (2C), 121.9, 120.9, 118.8 (2C), 52.8, 50.2 (2C), 46.9, 43.1 (2C), 24.2, 24.0; IR υmax/cm−1: 2981 (N–H), 1687 (C O), 1590 (C C), 1334 (S O), 1157 (S O), 1096 (C–N), 551 (C–Br); LRMS (ESI†) m/z: 547.1 [M–HCl + H, 79Br, 90%]/549.1 [M–HCl + H, 81Br, 100%]; HRMS calculated for C23H28BrClN6O3S [M–HCl + H, 79Br]: 547.1118/[M–HCl + H, 81Br]: 549.1095; found: 547.1122/549.1122.
N-[4-(4-{3-[4-(3-Chloro-phenyl)-[1,2,3]triazol-1-yl]-propyl}-piperazine-1-sulfonyl)-phenyl]-acetamide hydrochloride (30)
Synthesized using the general procedure as for 2, from tert-butyl-4-(3-azidopropyl)piperazine-1-carboxylate (12) (1 eq., 0.25 g, 0.93 mmol) and 3-chloro-1-ethynylbenzene (4g) (1.3 eq., 0.17 g, 1.21 mmol) to afford 1-{3-[4-(3-chlorophenyl)-[1,2,3]triazol-1-yl]-propyl}-piperazine hydrochloride (13g) (1 eq., 0.17 g, 0.50 mmol), then reacting with 4-acetylaminobenzene sulfonyl chloride (8a) (1 eq., 0.11 g, 0.50 mmol) (monitored by UPLC-MS analysis) to afford compound 30 hydrochloride as a pale yellow solid. Yield: 0.07 g, 28%, m.p.: 188–191 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 10.53 (s, 1H), 8.71 (s, 1H), 7.88–7.86 (m, 3H), 7.83–7.79 (m, 1H), 7.71 (d, J = 8.8 Hz, 2H), 7.49 (t, J = 7.9 Hz, 1H), 7.42–7.39 (m, 1H), 4.50 (t, J = 6.7 Hz, 2H), 3.72–3.69 (m, 2H), 3.57–3.52 (m, 2H), 3.12 (s, 4H), 2.70 (t, J = 11.4 Hz, 2H), 2.29 (s, 2H), 2.10 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 169.2, 145.0, 144.0, 133.7, 132.8, 130.9, 129.0 (2C), 127.7, 127.3, 124.7, 123.7, 122.3, 118.8 (2C), 52.7, 50.2 (2C), 47.0, 43.0 (2C), 24.2, 23.9; IR υmax/cm−1: 3032 (N–H), 1685 (C O), 1589 (C C), 1350 (S O), 1157 (S O), 1095 (C–N), 738 (C–Cl); LRMS (ESI†) m/z: 503.2 [M–HCl + H]; HRMS calculated for C23H28Cl2N6O3S [M–HCl + H, 35Cl]: 503.1625/[M–HCl + H, 37Cl]: 505.1594; found: 503.1627/505.1627.
N-[4-(4-{3-[4-(4-Trifluoromethylphenyl)-[1,2,3]triazol-1-yl]-propyl}-piperazine-1-sulfonyl)-phenyl]-acetamide hydrochloride (31)
Synthesized using the general procedure as for 2, from tert-butyl-4-(3-azidopropyl)piperazine-1-carboxylate (12) (1 eq., 0.25 g, 0.93 mmol) and 4-trifluoromethyl-1-ethynylbenzene (4h) (1.6 eq., 0.25 g, 1.48 mmol) to afford 1-{3-[4-(4-trifluoromethylphenyl)-[1,2,3]triazol-1-yl]-propyl}-piperazine hydrochloride (13h) (1 eq., 0.19 g, 0.50 mmol), then reacting with 4-acetylaminobenzene sulfonyl chloride (8a) (1.5 eq., 0.17 g, 0.75 mmol) (monitored by UPLC-MS analysis) to afford compound 31 hydrochloride as a pale yellow solid. Yield: 0.17 g, 61%, m. p.: dec. >145 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.68 (s, 1H), 10.52 (s, 1H), 8.78 (s, 1H), 8.05 (d, J = 8.1 Hz, 2H), 7.85 (dd, J = 17.4, 8.6 Hz, 4H), 7.71 (d, J = 8.8 Hz, 2H), 4.52 (t, J = 6.6 Hz, 2H), 3.71 (d, J = 6.5 Hz, 2H), 3.54 (d, J = 9.0 Hz, 2H), 3.13 (s, 4H), 2.68 (t, J = 11.9 Hz, 2H), 2.30 (s, 2H), 2.10 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 169.2, 145.0, 144.0, 134.6, 128.9 (2C), 128.0 (q, J = 31.7 Hz, aryl C), 127.2, 125.9 (q, J = 3.8 Hz, aryl C, 2C), 125.6 (2C), 124.2 (q, J = 271.4 Hz, CF3), 122.8, 118.8 (2C), 52.7, 50.2 (2C), 47.0, 43.0 (2C), 24.2, 24.0; 19F NMR (376 MHz, DMSO-d6) δ −57.6; IR υmax/cm−1: 2931 (N–H), 1689 (C O), 1589 (C C), 1326 (S O), 1164 (S O), 1065 (C–F),1096 (C–N); LRMS (ESI†) m/z: 537.3 [M–HCl + H]; HRMS calculated for C24H28ClF3N6O3S [M–HCl + H]: 537.1884; found: 537.1890.
N-[4-(4-{3-[4-(4-Nitrophenyl)-[1,2,3]triazol-1-yl]-propyl}-piperazine-1-sulfonyl)-phenyl]-acetamide hydrochloride (32)
Synthesized using the general procedure as for 2, from tert-butyl-4-(3-azidopropyl)piperazine-1-carboxylate (12) (1 eq., 0.20 g, 0.74 mmol) and 4-nitro-1-ethynylbenzene (4i) (1.3 eq., 0.14 g, 1.96 mmol) to afford 1-{3-[4-(4-nitrophenyl)-[1,2,3]triazol-1-yl]-propyl}-piperazine hydrochloride (13i) (1 eq., 0.16 g, 0.47 mmol), then reacting with 4-acetylaminobenzene sulfonyl chloride (8a) (0.9 eq., 0.10 g, 0.42 mmol) (monitored by UPLC-MS analysis) to afford 32 hydrochloride as an off-white solid. Yield: 0.12 g, 48%, m.p.: 232–236 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.78 (s, 1H), 10.52 (s, 1H), 8.87 (s, 1H), 8.38–8.28 (m, 2H), 8.17–8.07 (m, 2H), 7.87 (d, J = 8.8 Hz, 2H), 7.71 (d, J = 8.8 Hz, 2H), 4.54 (t, J = 6.6 Hz, 2H), 3.71 (d, J = 6.4 Hz, 2H), 3.53 (s, 2H), 3.13 (s, 4H), 2.70 (t, J = 11.1 Hz, 2H), 2.31 (s, 2H), 2.10 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 169.2, 146.6, 144.5, 144.0, 137.1, 129.0 (2C), 127.3, 125.9 (2C), 124.4 (2C), 123.7, 118.8 (2C), 52.7, 50.2 (2C), 47.1, 43.1 (2C), 24.2, 23.9; IR υmax/cm−1: 3046 (N–H), 1689 (C O), 1592 (C C), 1516 (N–O), 1332 (S O), 1164 (S O), 1097 (C–N); LRMS (ESI†) m/z: 514.3 [M–HCl + H]; HRMS calculated for C23H28ClN7O5S [M–HCl + H]: 514.1860; found: 514.1867.
Synthesis of Library 3 – based on 4-trifluoromethylbenzenesulfonyl chloride compounds 33–42
1-(3-(4-Phenyl-1H-1,2,3-triazol-1-yl)propyl)-4-((4-(trifluoromethyl)phenyl)-sulfonyl)piperazine hydrochloride (33)
As for 2, 1-(3-(4-phenyl-1H-1,2,3-triazol-1-yl)propyl)piperazine hydrochloride (13b) (1 eq., 0.15 g, 0.5 mmol) was reacted with 4-trifluoromethylbenzenesulfonyl chloride (8j) (1 eq., 0.12 g, 0.5 mmol) (monitored by UPLC-MS) to afford 33 as a hydrochloride salt, off-white solid. Yield: 0.18 g, 70%, m.p.: 227–229 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.38 (s, 1H), 8.62 (s, 1H), 8.06 (dd, J = 26.2, 8.4 Hz, 4H), 7.84–7.82 (m, 2H), 7.45 (t, J = 7.6 Hz, 2H), 7.34 (t, J = 7.4 Hz, 1H), 4.51 (t, J = 6.8 Hz, 2H), 3.80 (d, J = 12.2 Hz, 2H), 3.55 (d, J = 11.5 Hz, 2H), 3.14 (d, J = 6.2 Hz, 4H), 2.89 (t, J = 11.9 Hz, 2H), 2.34–2.31 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 146.4, 138.5, 133.3 (q, J = 31.3 Hz, aryl C), 130.8, 129.0, 128.7 (2C), 128.0, 127.0 (unresolved quartet, J = 3.7 Hz, aryl C, 2C), 125.2 (2C), 123.4 (q, J = 271.5 Hz, CF3), 121.6 (2C), 52.7, 50.1 (2C),46.9, 42.9 (2C), 23.9; 19F NMR (376 MHz, DMSO-d6) δ −58.6; IR υmax/cm−1: 1609 (C C), 1321 (S O), 1167 (S O), 1133 (C–F), 1097 (C–N); LRMS (ESI†) m/z: 480.2 [M–HCl + H]; HRMS calculated for C22H25ClF3N5O2S [M–HCl + H]: 480.1668; found: 480.1676.
1-(3-(4-(p-Tolyl)-1H-1,2,3-triazol-1-yl)propyl)-4-((4-(trifluoromethyl)phenyl)-sulfonyl)piperazine hydrochloride (34)
As for 2, 1-(3-(4-(p-tolyl)-1H-1,2,3-triazol-1-yl)propyl)piperazine hydrochloride (13c) (1 eq., 0.16 g, 0.5 mmol) was reacted with 4-trifluoromethylbenzene sulfonyl chloride (8j) (1 eq., 0.12 g, 0.5 mmol) (monitored by UPLC-MS) to afford 34 as a hydrochloride salt, off-white solid. Yield: 0.12 g, 45%, m.p.: 213–217 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.88 (s, 1H), 8.53 (s, 1H), 8.06 (dd, J = 28.2, 8.2 Hz, 4H), 7.71 (d, J = 7.9 Hz, 2H), 7.26 (d, J = 7.9 Hz, 2H), 4.48 (s, 2H), 3.81 (d, J = 10.5 Hz, 2H), 3.55 (d, J = 12.0 Hz, 2H), 3.15 (s, 4H), 2.82 (t, J = 11.1 Hz, 2H), 2.33 (s, 5H); 13C NMR (101 MHz, DMSO-d6) δ 146.5, 138.5, 137.3, 133.4 (q, J = 32.1 Hz, aryl C), 129.6, 128.8 (2C), 128.0, 137.0 (d, J = 3.1 Hz, aryl C, 2C), 125.2 (2C), 123.1 (q, J = 271.3 Hz, CF3), 121.1 (2C), 52.8, 50.2 (2C),46.8, 43.0 (2C), 24.0, 20.8; 19F NMR (376 MHz, DMSO-d6) δ −58.5; IR υmax/cm−1: 1641 (C C), 1321 (S O), 1170 (S O), 1136 (C–F), 1096 (C–N); LRMS (ESI†) m/z: 494.2 [M–HCl + H]; HRMS calculated for C23H27ClF3N5O2S [M–HCl + H]: 494.1821; found: 494.1832.
1-(3-(4-(4-Ethylphenyl)-1H-1,2,3-triazol-1-yl)propyl)-4-((4-(trifluoromethyl)phenyl)-sulfonyl)piperazine hydrochloride (35)
As for 2, 1-(3-(4-(p-ethylphenyl)-1H-1,2,3-triazol-1-yl)propyl)piperazine hydrochloride (13d) (1 eq., 0.17 g, 0.5 mmol) was reacted with 4-trifluoromethylbenzene sulfonyl chloride (8j) (1 eq., 0.12 g, 0.5 mmol) (monitored by UPLC-MS) to afford 35 as a hydrochloride salt, off-white solid. Yield: 0.21 g, 77%, m.p.: dec. >210 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.28 (s, 1H), 8.55 (s, 1H), 8.06 (dd, J = 26.8, 8.4 Hz, 4H), 7.74 (d, J = 8.1 Hz, 2H), 7.29 (d, J = 8.1 Hz, 2H), 4.49 (t, J = 6.8 Hz, 2H), 3.80 (d, J = 12.4 Hz, 2H), 3.55 (d, J = 12.1 Hz, 2H), 3.14 (d, J = 6.8 Hz, 4H), 2.88 (t, J = 12.1 Hz, 2H), 2.63 (q, J = 7.6 Hz, 2H), 2.37–2.25 (m, 2H), 1.20 (t, J = 7.6 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 146.5, 143.5, 138.5, 133.3 (q, J = 32.3 Hz, aryl C), 128.7(2C), 128.3 (2C), 128.2, 126.9 (q, J = 3.7 Hz, aryl C, 2C), 125.2 (2C) 124.9 (q, J = 271.4 Hz, CF3), 121.2, 52.7, 50.1 (2C), 46.8, 42.9 (2C), 27.9, 23.9, 15.5; 19F NMR (376 MHz, DMSO-d6) δ −58.4; IR υmax/cm−1: 1635 (C C), 1322 (S O), 1170 (S O), 1134 (C–F), 1092 (C–N); LRMS (ESI†) m/z: 508.3 [M–HCl + H]; HRMS calculated for C24H29ClF3N5O2S [M–HCl + H]: 508.1992; found: 508.1989.
1-(3-(4-(4-(tert-Butyl)phenyl)-1H-1,2,3-triazol-1-yl)propyl)-4-((4-(trifluoromethyl)-phenyl)sulfonyl) piperazine hydrochloride (36)
As for 2, 1-(3-(4-(4-(tert-butyl)phenyl)-1H-1,2,3-triazol-1-yl)propyl)piperazine hydrochloride (13j) (1 eq., 0.18 g, 0.5 mmol) was reacted with 4-trifluoromethylbenzene sulfonyl chloride (8j) (1 eq., 0.12 g, 0.5 mmol) (monitored by UPLC-MS) to afford 36 as a hydrochloride salt, off-white solid. Yield: 0.18 g, 63%, m.p.: dec. >203 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.87 (s, 1H), 8.54 (s, 1H), 8.09 (d, J = 8.4 Hz, 2H), 8.02 (d, J = 8.3 Hz, 2H), 7.75–7.72 (m, 2H), 7.48–7.44 (m, 2H), 4.48 (t, J = 6.6 Hz, 2H), 3.80 (d, J = 12.0 Hz, 2H), 3.55 (d, J = 10.4 Hz, 2H), 3.14 (s, 4H), 2.81 (t, J = 10.9 Hz, 2H), 2.29 (s, 2H), 1.30 (s, 9H); 13C NMR (101 MHz, DMSO-d6) δ 150.4, 146.4, 138.5, 133.3 (q, J = 32.2 Hz, aryl C), 128.7 (2C), 128.0, 126.9 (q, J = 3.6 Hz, aryl C, 2C), 125.6 (2C), 124.9 (2C), 123.4 (q, J = 271.3 Hz, CF3), 121.2, 52.7, 50.1 (2C), 46.8, 42.9 (2C), 34.4, 31.1 (3C), 23.9; 19F NMR (376 MHz, DMSO-d6) δ −58.3; IR υmax/cm−1: 2971 (N–H), 1609 (C C), 1323 (S O), 1166 (S O), 1136 (C–F), 1099 (C–N); LRMS (ESI†) m/z: 536.3 [M–HCl + H]; HRMS calculated for C26H33ClF3N5O2S [M–HCl + H]: 536.2290; found: 536.2302.
1-(3-(4-(4-Fluorophenyl)-1H-1,2,3-triazol-1-yl)propyl)-4-((4-(trifluoromethyl)-phenyl)sulfonylpiperazine hydrochloride (37)
As for 2, 1-(3-(4-(4-fluorophenyl)-1H-1,2,3-triazol-1-yl)propyl)piperazine hydrochloride (13e) (1 eq., 0.16 g, 0.5 mmol) was then reacted with 4-trifluoromethylbenzene sulfonyl chloride (8j) (1 eq., 0.12 g, 0.5 mmol) (monitored by UPLC-MS) to afford 37 as a hydrochloride salt, off-white solid. Yield: 0.21 g, 79%, m.p.: 207–210 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.31 (s, 1H), 8.61 (s, 1H), 8.06 (dd, J = 27.0, 8.4 Hz, 4H), 7.89–7.85 (m, 2H), 7.31–7.26 (m, 2H), 4.50 (t, J = 6.8 Hz, 2H), 3.80 (d, J = 12.2 Hz, 2H), 3.55 (d, J = 12.8 Hz, 2H), 3.17–3.13 (m, 4H), 2.88 (t, J = 11.8 Hz, 2H), 2.35–2.28 (m, 2H). The piperazine peaks was not well resolved; 13C NMR (101 MHz, DMSO-d6) δ 161.9 (d, J = 242.9), 145.7, 138.6, 133.5 (q, J = 32.2 Hz, aryl C), 128.8, 127.4, 127.3 (unresolved quartet, J = 4.3 Hz, 2C), 127.1 (q, J = 3.6 Hz, aryl C, 2C), 123.5 (q, J = 271.4 Hz, CF3), 121.6 (2C), 116.0 (unresolved quartet, J = 21.5 Hz, 2C), 51.9, 50.3 (2C), 47.0, 43.0 (2C), 24.1; 19F NMR (376 MHz, DMSO-d6) δ −58.4, −110.8; IR υmax/cm−1: 1612 (C C), 1330 (S O), 1167 (S O), 1147 (C–F), 1097 (C–N); LRMS (ESI†) m/z: 498.2 [M–HCl + H]; HRMS calculated for C22H24ClF4N5O2S [M–HCl + H]: 498.1572; found: 498.1581.
1-(3-(4-(4-Bromophenyl)-1H-1,2,3-triazol-1-yl)propyl)-4-((4-trifluoromethyl)-phenyl)sulfonyl piperazine hydrochloride (38)
As for 2, 1-(3-(4-(4-bromophenyl)-1H-1,2,3-triazol-1-yl)propyl)piperazine hydrochloride (13k) (1 eq., 0.19 g, 0.5 mmol) was reacted with 4-trifluoromethylbenzene sulfonyl chloride (8j) (1 eq., 0.12 g, 0.5 mmol) (monitored by UPLC-MS) to afford 38 as a hydrochloride salt, off-white solid. Yield: 0.16 g, 54%, m.p.: 212–225 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.98 (s, 1H), 8.65 (s, 1H), 8.06 (dd, J = 28.2, 8.4 Hz, 4H), 7.79 (d, J = 8.4 Hz, 2H), 7.66 (d, J = 8.4 Hz, 2H), 4.50 (t, J = 6.5 Hz, 2H), 3.81 (d, J =12.0 Hz, 2H), 3.54 (s, 2H), 3.14 (s, 4H), 2.83 (t, J = 12.0 Hz, 2H), 2.30 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 145.3, 138.5, 133.3 (q, J = 32.3 Hz, aryl C), 131.9 (2C), 130.0, 128.7 (2C), 127.1 (2C), 126.9 (q, J = 3.6 Hz, aryl C, 2C), 123.4 (q, J = 271.3 Hz, CF3), 122.0, 120.9, 52.7, 50.1 (2C), 46.9, 42.9 (2C), 23.9; 19F NMR (376 MHz, DMSO-d6) δ −58.3; IR υmax/cm−1: 1608 (C C), 1330 (S O), 1167 (S O), 1147 (C–F), 1096 (C–N), 617 (C–Br); LRMS (ESI†) m/z: 558.1 [M–HCl + H, 79Br]/560.1 [M–HCl + H, 81Br]; HRMS calculated for C23H24ClF6N5O2S [M–HCl + H, 79Br]: 558.0768/[M–HCl + H, 81Br]: 560.0768; found: 558.0781/560.0781.
1-(3-(4-(3-Chlorophenyl)-1H-1,2,3-triazol-1-yl)propyl)-4-((4-(trifluoromethyl)-phenyl)sulfonyl)piperazine hydrochloride (39)
As for 2, 1-(3-(4-(3-chlorophenyl)-1H-1,2,3-triazol-1-yl)propyl)piperazine hydrochloride (13g) (1 eq., 0.17 g, 0.5 mmol) was reacted with 4-trifluoromethylbenzene sulfonyl chloride (8j) (1 eq., 0.12 g, 0.5 mmol) (monitored by UPLC-MS) to afford 39 as a hydrochloride salt, off-white solid. Yield: 0.19 g, 69%, m.p.: dec. >215 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.01 (s, 1H), 8.71 (s, 1H), 8.09 (d, J = 8.4 Hz, 2H), 8.02 (d, J = 8.3 Hz, 2H), 7.88 (t, J = 1.7 Hz, 1H), 7.81 (d, J = 7.8 Hz, 1H), 7.49 (t, J = 7.9 Hz, 1H), 7.40 (dd, J = 8.0, 1.1 Hz, 1H), 4.51 (t, J = 6.7 Hz, 2H), 3.81 (d, J = 10.4 Hz, 2H), 3.55 (d, J = 11.5 Hz, 2H), 3.14 (s, 4H), 2.84 (t, J = 12.1 Hz, 2H), 2.29 (d, J = 6.4 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 145.1, 138.4, 133.7, 133.2 (q, J = 32.2 Hz, aryl C), 132.8, 130.9, 128.7, 127.7, 126.9 (unresolved quartet, J = 3.4 Hz, aryl C, 2C), 124.7, 123.7, 123.4 (q, J = 271.4 Hz, CF3), 122.3 (unresolved quartet, J = 10.9 Hz, aryl C, 2C), 52.7, 50.2 (2C), 47.0, 43.0 (2C), 24.0; 19F NMR (376 MHz, DMSO-d6) δ −58.4; IR υmax/cm−1: 1611 (C C), 1321 (S O), 1173 (S O), 1143 (C–F), 1096 (C–N), 726 (C–Cl); LRMS (ESI†) m/z: 514.1 [M–HCl + H]; HRMS calculated for C22H24Cl2F3N5O2S [M–HCl + H]: 514.1276/516.1376; found: 514.1286/516.1286.
1-(4-Trifluoromethylbenzenesulfonyl)-4-{3-[4-(4-trifluoromethylphenyl)-[1,2,3]-triazol-1-yl]-propyl}-piperazine hydrochloride (40)
As for 2, 1-(3-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)propyl) piperazine hydrochloride (13h) (1 eq., 0.19 g, 0.5 mmol) was reacted with 4-trifluoromethylbenzenesulfonyl chloride (8j) (1 eq., 0.12 g, 0.5 mmol) (monitored by UPLC-MS) to afford 40 as a hydrochloride salt, off-white solid. Yield: 0.22 g, 75%, m.p.: dec. >220 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.34 (s, 1H), 8.80 (s, 1H), 8.06 (dt, J = 13.3, 8.4 Hz, 6H), 7.83 (d, J = 8.3 Hz, 2H), 4.54 (t, J = 6.8 Hz, 2H), 3.81 (d, J = 12.0 Hz, 2H), 3.56 (d, J = 11.6 Hz, 2H), 3.15 (s, 4H), 2.89 (t, J = 11.6 Hz, 2H), 2.37–2.30 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 145.0, 138.5, 134.7, 133.3 (q, J = 32.2 Hz, aryl C), 128.7(2C), 128.0 (q, J = 31.7 Hz, aryl C), 126.9 (q, J = 3.7 Hz, aryl C, 2C), 126.1 (q, J = 271.4 Hz, CF3), 125.6 (2C), 125.9 (q, J = 3.8 Hz, aryl C, 2C), 123.4 (q, J = 271.4 Hz, CF3), 120.2, 52.6, 50.1 (2C), 47.0, 42.9 (2C), 23.8; 19F NMR (376 MHz, DMSO-d6) δ −58.2, −58.9; IR υmax/cm−1: 1623 (C C), 1330 (S O), 1175 (S O), 1120 (C–F), 1082 (C–N); LRMS (ESI†) m/z: 548.2 [M–HCl + H]; HRMS calculated for C23H24ClF6N5O2S [M–HCl + H]: 548.1535; found: 548.1549.
1-(3-(4-(4-Nitrophenyl)-1H-1,2,3-triazol-1-yl)propyl)-4-((4-(trifluoromethyl)-phenyl)sulfonyl)piperazine hydrochloride (41)
As for 2, 1-(3-(4-(4-nitrophenyl)-1H-1,2,3-triazol-1-yl)propyl)piperazine hydrochloride (13i) (1 eq., 0.18 g, 0.5 mmol) was reacted with 4-trifluoromethylbenzene sulfonyl chloride (8j) (1 eq., 0.12 g, 0.5 mmol) (monitored by UPLC-MS) to afford 41 as a hydrochloride salt, off-white solid. Yield: 0.18 g, 64%, m.p.: dec. >214 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.10 (s, 1H), 8.88 (s, 1H), 8.33 (d, J = 8.8 Hz, 2H), 8.10 (t, J = 8.0 Hz,4H), 8.02 (d, J = 8.0 Hz, 2H), 4.55 (t, J = 6.6 Hz, 2H), 3.81 (d, J = 11.1 Hz, 2H), 3.56 (d, J = 9.9 Hz, 2H), 3.15 (s, 4H), 2.85 (t, J = 11.6 Hz, 2H), 2.33 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 146.8, 144.6, 138.4, 137.2, 133.5 (q, J = 32.3 Hz, aryl C), 128.8, 127.1 (q, J = 3.6 Hz, aryl C, 2C), 126.1(2C), 124.6 (2C), 123.8 (2C), 123.5 (q, J = 271.4 Hz, CF3), 52.9, 50.4 (2C), 47.2, 43.2 (2C), 24.1; 19F NMR (376 MHz, DMSO-d6) δ −58.5; IR υmax/cm−1: 1607 (C C), 1319 (S O), 1174 (S O), 1149 (C–F), 1100 (C–N); LRMS (ESI†) m/z: 525.2 [M–HCl + H]; HRMS calculated for C22H24ClF3N6O4S [M–HCl + H]: 525.1516; found: 525.1526.
N,N-Dimethyl-4-(1-(3-(4-((4-(trifluoromethyl)phenyl)sulfonyl)piperazin-1-yl)-propyl)-1H-1,2,3-triazol-4-yl)aniline hydrochloride (42)
As for 2, N,N-dimethyl-4-(1-(3-(piperazin-1-yl)propyl)-1H-1,2,3-triazol-4-yl) aniline hydrochloride (13l) (1 eq., 0.18 g, 0.5 mmol) was reacted with 4-trifluoromethylbenzene sulfonyl chloride (8j) (1 eq., 0.12 g, 0.5 mmol) (monitored by UPLC-MS) to afford 42 as a hydrochloride salt, pale brown solid. Yield: 0.13 g, 46%, m.p.: dec. >187 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.51 (s, 1H), 8.57 (s, 1H), 8.08 (d, J = 8.4 Hz, 2H), 8.02 (d, J = 8.3 Hz, 2H), 7.82 (d, J = 7.8 Hz, 2H), 7.36 (s, 2H), 4.49 (t, J = 6.7 Hz, 2H), 3.80 (d, d, J = 12.6 Hz, 2H), 3.55 (d, J = 11.6 Hz, 2H), 3.20–3.11 (m, 4H), 3.03 (s, 6H), 2.91 (t, J = 11.8 Hz, 2H), 2.36–2.28 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 145.7, 144.6, 138.7 (2C), 133.4 (q, J = 32.2 Hz, aryl C), 128.8 (2C), 127.0 (q, J = 3.5 Hz, aryl C, 2C), 126.6 (2C), 123.5 (q, J = 271.3 Hz, CF3), 122.0 (2C), 119.6, 52.9, 50.3 (2C), 47.1, 44.4 (2C), 43.0 (2C), 24.1; 19F NMR (376 MHz, DMSO-d6) δ −58.3; IR υmax/cm−1: 1609 (C C), 1323 (S O), 1159 (S O), 1129 (C–N), 1052 (C–F); LRMS (ESI†) m/z: 523.3 [M–HCl + H]; HRMS calculated for C24H30ClF3N6O2S [M–HCl + H]: 523.2087; found: 523.2098.
Synthesis of Library 4 – based on 2- and 3-trifluoromethylbenzenesulfonyl chloride compounds 43–50
1-[3-(4-p-Tolyl-[1,2,3]triazol-1-yl)-propyl]-4-(2-trifluoromethylbenzenesulfonyl)-piperazine hydrochloride (43)
As for 2, 1-[3-(4-(4-tolyl)-[1,2,3]triazol-1-yl)-propyl]-piperazine hydrochloride (13c) (1 eq., 0.14 g, 0.43 mmol) was reacted with 2-trifluoromethylbenzene sulfonyl chloride (8m) (1.1 eq., 0.12 g, 0.47 mmol) (monitored by UPLC-MS) to afford 43 as a hydrochloride salt, off-white solid. Yield: 0.10 g, 44%, m.p.: 216–218 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.19 (s, 1H), 8.54 (s, 1H), 8.13–8.07 (m, 2H), 7.98–7.92 (m, 2H), 7.72 (d, J = 8.1 Hz, 2H), 7.26 (d, J = 7.9 Hz, 2H), 4.50 (t, J = 6.7 Hz, 2H), 3.81 (s, 2H), 3.56 (s, 2H), 3.26–3.10 (m, 6H), 2.36–2.29 (m, 5H); 13C NMR (101 MHz, DMSO-d6) δ 146.4, 137.2, 135.9, 134.1, 133.7, 131.6, 129.4 (2C), 128.9 (q, J = 6.4 Hz, aryl C), 128.0, 126.3 (q, J = 32.4 Hz, aryl C), 125.1 (2C), 122.5 (q, J = 272.2 Hz, CF3), 121.1, 52.7, 50.4 (2C), 46.9, 42.4 (2C), 23.9, 20.8; 19F NMR (376 MHz, DMSO-d6) δ −53.2; IR υmax/cm−1: 2980 (N–H), 1621 (C C), 1307 (S O), 1172 (S O), 1149 (C–F), 1118 (C–N); LRMS (ESI†) m/z: 494.2 [M–HCl + H]; HRMS calculated for C23H27ClF3N5O2S [M–HCl + H]: 494.1822; found: 494.1832.
1-{3-[4-(4-Ethylphenyl)-[1,2,3]triazol-1-yl]-propyl}-4-(2-trifluoromethylbenzenesulfonyl)-piperazine hydrochloride (44)
As for 2, 1-{3-[4-(4-ethylphenyl)-[1,2,3]triazol-1-yl]-propyl}-piperazine hydrochloride (13d) (1 eq., 0.14 g, 0.41 mmol) was reacted with 2-trifluoromethylbenzene sulfonyl chloride (8m) (1.1 eq., 0.11 g, 0.46 mmol) (monitored by UPLC-MS) to afford 44 as a hydrochloride salt, off-white solid. Yield: 0.15 g, 67%, m.p.: 187–190 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.48 (s, 1H), 8.56 (s, 1H), 8.14–8.07 (m, 2H), 7.99–7.92 (m, 2H), 7.74 (d, J = 8.2 Hz, 2H), 7.29 (d, J = 8.3 Hz, 2H), 4.51 (t, J = 6.8 Hz, 2H), 3.83 (d, J = 12.9 Hz, 2H), 3.57 (d, J = 12.1 Hz, 2H), 3.30–3.09 (m, 6H), 2.63 (q, J = 7.5 Hz, 2H), 2.38–2.31 (m, 2H), 1.20 (t, J = 7.6 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 146.5, 143.5, 135.9, 134.1, 133.7, 131.7, 128.9 (q, J = 6.5 Hz, aryl C), 128.3 (2C), 128.2, 126.3 (q, J = 32.5 Hz, aryl C), 125.2 (2C), 122.6 (q, J = 269.3 Hz, CF3), 121.2, 52.7, 50.4 (2C), 46.9, 42.3 (2C), 27.9, 23.9, 15.5; 19F NMR (376 MHz, DMSO-d6) δ −53.2; IR υmax/cm−1: 2971 (N–H), 1622 (C C), 1310 (S O), 1168 (S O), 1141 (C–F), 1111 (C–N); LRMS (ESI†) m/z: 508.3 [M–HCl + H]; HRMS calculated for C24H29ClF3N5O2S [M–HCl + H]: 508.1989; found: 508.1989.
1-{3-[4-(4-Bromophenyl)-[1,2,3]triazol-1-yl]-propyl}-4-(2-trifluoromethylbenzene sulfonyl)-piperazine hydrochloride (45)
As for 2, 1-{3-[4-(4-bromophenyl)-[1,2,3]triazol-1-yl]-propyl}-piperazine hydrochloride (13f) (1 eq., 0.16 g, 0.42 mmol) was reacted with 2-trifluoromethylbenzene sulfonyl chloride (8m) (1.1 eq., 0.11 g, 0.46 mmol) (monitored by UPLC-MS) to afford 45 as a hydrochloride salt, off-white solid. Yield: 0.14 g, 57%, m.p.: 200–203 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.11 (s, 1H), 8.66 (s, 1H), 8.13–8.07 (m, 2H), 7.98–7.92 (m, 2H), 7.79 (d, J = 8.5 Hz, 2H), 7.66 (d, J = 8.5 Hz, 2H), 4.52 (t, J = 6.4 Hz, 2H), 3.84 (d, J = 11.1 Hz, 2H), 3.57 (d, J = 11.1 Hz, 2H), 3.25–3.09 (m, 6H), 2.33 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 145.3, 135.9, 134.1, 133.7, 131.9 (2C), 131.6, 130.0, 128.9 (q, J = 6.4 Hz, aryl C), 127.1 (2C), 126.3 (q, J = 32.4 Hz, aryl C), 122.5 (q, J = 272.2 Hz, CF3), 121.9, 120.8, 52.7, 50.2 (2C), 47.0, 42.4 (2C), 23.9; 19F NMR (376 MHz, DMSO-d6) δ −53.1; IR υmax/cm−1: 2978 (N–H), 1607 (C C), 1306 (S O), 1168 (S O), 1147 (C–F), 1119 (C–N), 581 (C–Br); LRMS (ESI†) m/z: 558.1 [M–HCl + H, 79Br, 95%]/560.1 [M–HCl + H, 81Br, 100%]; HRMS calculated for C22H24BrClF3N5O2S [M–HCl + H]: 558.0784/[M–HCl + H]: 560.0757; found: 558.0781/560.0781.
1-(2-Trifluoromethylbenzenesulfonyl)-4-{3-[4-(4-trifluoromethylphenyl)-[1,2,3]-triazol-1-yl]-propyl}-piperazine hydrochloride (46)
As for 2, 1-{3-[4-(4-trifluorophenyl)-[1,2,3]triazol-1-yl]-propyl}-piperazine hydrochloride (13h) (1 eq., 0.16 g, 0.43 mmol) was reacted with 2-trifluoromethylbenzene sulfonyl chloride (8m) (1.1 eq., 0.11 g, 0.47 mmol) (monitored by UPLC-MS) to afford 46 as a hydrochloride salt, off-white solid. Yield: 0.12 g, 49%, m.p.: 228–232 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.24 (s, 1H), 8.79 (s, 1H), 8.13–8.05 (m, 4H), 7.99–7.92 (m, 2H), 7.83 (d, J = 8.2 Hz, 2H), 4.55 (t, J = 6.6 Hz, 2H), 3.84 (d, J = 11.4 Hz, 2H), 3.58 (d, J = 10.2 Hz,2H), 3.27–3.10 (m, 6H), 2.35 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 145.0, 135.9, 134.7, 134.1, 133.7, 131.6, 128.8 (q, J = 6.4 Hz, aryl C), 128.0 (q, J = 31.6 Hz, aryl C), 126.3 (q, J = 32.4 Hz, aryl C), 125.9 (q, J = 3.7 Hz, aryl 2C), 125.7 (2C), 124.2 (q, J = 272.1 Hz, CF3), 122.5 (q, J = 272.1 Hz, CF3), 122.9, 52.6, 50.4 (2C), 47.1, 42.4 (2C), 23.9; 19F NMR (376 MHz, DMSO-d6) δ −53.2, 58.2; IR υmax/cm−1: 2927 (N–H), 1624 (C C), 1330 (S O), 1309 (S O), 1154.4 (C–F), 1111 (C–N); LRMS (ESI†) m/z: 548.2 [M–HCl + H]; HRMS calculated for C23H24ClF6N5O2S [M–HCl + H]: 548.1550; found: 548.1549.
1-[3-(4-p-Tolyl-[1,2,3]triazol-1-yl)-propyl]-4-(3-trifluoromethylbenzenesulfonyl)-piperazine hydrochloride (47)
As for 2, 1-[3-(4-tolyl-[1,2,3]triazol-1-yl)-propyl]-piperazine hydrochloride (13c) (1 eq., 0.14 g, 0.43 mmol) was reacted with 3-trifluoromethylbenzene sulfonyl chloride (8n) (1 eq., 0.11 g, 0.43 mmol) (monitored by UPLC-MS) to afford 47 as a hydrochloride salt, off-white solid. Yield: 0.10 g, 43%, m.p.: dec. >165 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.91 (s, 1H), 8.53 (s, 1H), 8.19–7.97 (m, 4H), 7.71 (s, 2H), 7.26 (s, 2H), 4.48 (s, 2H), 3.83 (s, 2H), 3.56 (s, 2H), 3.14 (s, 4H), 2.81 (s, 2H), 2.32 (s, 5H); 13C NMR (101 MHz, DMSO-d6) δ 146.4, 137.2, 135.6, 131.7, 131.4, 130.6 (q, J = 3.4 Hz, aryl C), 130.3 (q, J = 32.7 Hz, aryl C), 129.5 (2C), 128.0, 125.1 (2C), 124.0 (q, J = 3.9 Hz, aryl C), 123.3 (q, J = 271.3 Hz, CF3), 121.1, 52.7, 50.0 (2C), 46.9, 43.0 (2C), 23.9, 20.8; 19F NMR (376 MHz, DMSO-d6) d −57.9; IR υmax/cm−1: 2972 (N–H), 1609 (C C), 1326 (S O), 1167 (S O), 1134 (C–F), 1106 (C–N); LRMS (ESI†) m/z: 494.2 [M–HCl + H]; HRMS calculated for C23H27ClF3N5O2S [M–HCl + H]: 494.1822; found: 494.1832.
1-{3-[4-(4-Ethylphenyl)-[1,2,3]triazol-1-yl]-propyl}-4-(3-trifluoromethylbenzenesulfonyl)-piperazine 3-trifluoromethy-benzenesulfonate (48)
As for 2, 1-{3-[4-(4-ethylphenyl)-[1,2,3]triazol-1-yl]-propyl}-piperazine hydrochloride (13k) (1 eq., 0.10 g, 0.41 mmol) was reacted with 3-trifluoromethylbenzene sulfonyl chloride (8n) (1 eq., 0.13 g, 0.41 mmol) (monitored by UPLC-MS) to afford 48 3-trifluoromethylbenzenesulfonate, as an off-white solid Yield: 0.08 g, 27%, m.p.: dec. >127 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.17 (s, 1H), 8.50 (s, 1H), 8.21 (d, J = 7.8 Hz, 1H), 8.14 (d, J = 7.9 Hz, 1H), 8.05 (s, 1H), 7.98 (t, J = 7.9 Hz, 1H), 7.89–7.85 (m, 2H), 7.74–7.68 (m, 3H), 7.59 (t, J = 7.7 Hz, 1H), 7.28 (d, J = 8.2 Hz, 2H), 4.45 (t, J = 6.5 Hz, 2H), 3.84 (s, 2H), 3.57 (s, 2H), 3.17 (s, 4H), 2.65–2.56 (m, 4H), 2.21 (s, 2H), 1.19 (t, J = 7.6 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 149.3, 146.5, 143.6, 135.3, 132.0, 131.5, 130.7 (q, J = 3.4 Hz, aryl C), 130.3 (q, J = 32.7 Hz, aryl C), 129.2, 129.5 (unresolved quartet, J = 1.0 Hz, aryl C), 128.5 (q, J = 31.5 Hz, aryl C), 128.3 (2C), 128.2, 125.1 (q, i = 3.8 Hz, aryl C), 125.2 (2C), 124.1 (q, J = 270.7 Hz, CF3), 124.1 (q, J = 3.9 Hz, aryl C), 123.3 (q, J = 271.2 Hz, CF3), 122.0 (q, J = 3.9 Hz, aryl C), 121.2, 53.0, 50.4 (2C), 46.7, 43.3 (2C), 27.9, 24.3, 15.5; 19F NMR (376 MHz, DMSO-d6) δ −58.79, 58.84; IR υmax/cm−1: 2971 (N–H), 1609 (C C), 1325 (S O), 11 634 (S O), 1122 (C–F), 1068 (C–N); LRMS (ESI†) m/z: 508.2 [M–C7H4F3O2SOH + H]; HRMS calculated for C31H33F6N5O5S2 [M–C7H4F3O2SOH + H]: 508.1989; found: 508.1989.
1-{3-[4-(4-Bromophenyl)-[1,2,3]triazol-1-yl]-propyl}-4-(3-trifluoromethylbenzenesulfonyl)-piperazine hydrochloride (49)
As for 2, 1-{3-[4-(4-bromophenyl)-[1,2,3]triazol-1-yl]-propyl}-piperazine hydrochloride (13f) (1 eq., 0.22 g, 0.58 mmol) was reacted with 3-(trifluoromethyl)benzene sulfonyl chloride (8n) (1 eq., 0.14 g, 0.58 mmol) (monitored by UPLC-MS) to afford 49 as a hydrochloride salt, off-white solid. Yield: 0.23 g, 68%, m.p.: dec. >180 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.76 (s, 1H), 8.64 (s, 1H), 8.19 (d, J = 7.7 Hz, 1H), 8.13 (d, J = 7.3 Hz, 1H), 8.04 (s, 1H), 7.96 (t, J = 7.6 Hz, 1H), 7.78 (d, J = 8.4 Hz, 2H), 7.65 (d, J = 8.4 Hz, 2H), 4.49 (s, 2H), 3.83 (s, 2H), 3.55 (s, 2H), 3.14 (s, 4H), 2.79 (s, 2H), 2.29 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 145.3, 135.9, 131.9 (2C), 131.7, 131.4, 130.6, 130.3 (q, J = 32.7 Hz, aryl C), 130.0, 127.1 (2C), 124.0 (q, J = 3.8 Hz, aryl C), 123.3 (q, J = 271.8 Hz, CF3), 121.9, 120.8, 52.8, 50.2 (2C), 47.0, 43.0 (2C), 23.9; 19F NMR (376 MHz, DMSO-d6) δ −58.7; IR υmax/cm−1: 2963 (N–H), 1609 (C C), 1325 (S O), 1168 (S O), 1129 (C–F), 1104 (C–N), 578 (C–Br); LRMS (ESI†) m/z: 558.1 [M–HCl + H, 79Br]/560.1 [M–HCl + H, 81Br]. HRMS calculated for C20H22ClBrF3N5O2S [M–HCl + H, 79Br]: 558.0784/[M–HCl + H, 81Br]: 560.0758; found: 558.0781/560.0781.
1-(3-Trifluoromethylbenzenesulfonyl)-4-{3-[4-(4-trifluoromethyl-phenyl)-[1,2,3]-triazol-1-yl]-propyl}-piperazine hydrochloride (50)
As for 2, 1-{3-[4-(4-trifluorophenyl)-[1,2,3]triazol-1-yl]-propyl}-piperazine hydrochloride (13h) (1 eq., 0.20 g, 0.54 mmol) was reacted with 3-trifluoromethylbenzene sulfonyl chloride (8n) (1 eq., 0.13 g, 0.54 mmol) (monitored by UPLC-MS) to afford 50 as a hydrochloride salt, off-white solid. Yield: 0.17 g, 54%, m.p.: dec. >156 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.82 (s, 1H), 8.77 (s, 1H), 8.19 (d, J = 7.5 Hz, 1H), 8.13 (d, J = 7.5 Hz, 1H), 8.05 (d, J = 8.1 Hz, 3H), 7.96 (t, J = 7.8 Hz, 1H), 7.82 (d, J = 8.3 Hz, 2H), 4.51 (s, 2H), 3.83 (s, 2H), 3.56 (s, 2H), 3.15 (s, 4H), 2.80 (s, 2H), 2.30 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 145.0, 135.9, 134.7, 131.7, 131.4, 130.5, 130.3 (q, J = 32.7 Hz, aryl C), 128.0 (q, J = 31.7 Hz, aryl C), 125.9 (q, J = 3.8 Hz, aryl C, 2C), 125.7 (2C), 124.3 (q, J = 270.4 Hz, CF3), 124.0 (q, J = 3.8 Hz, aryl C), 123.3 (q, J = 271.3 Hz, CF3), 122.9, 52.8, 50.2 (2C), 47.1, 43.1 (2C), 24.1; 19F NMR (376 MHz, DMSO-d6) δ −57.6, 58.0; IR υmax/cm−1: 2981.4 (N–H), 1623 (C C), 1325 (S O), 1171 (S O), 1129 (C–F), 1106 (C–N); LRMS (ESI†) m/z: 548.2 [M–HCl + H]; HRMS calculated for C23H24ClF6N5O2S [M–HCl + H]: 548.1550; found: 548.1549.
1-{3-[4-(4-Methoxyphenyl)-[1,2,3]triazol-1-yl]-propyl}-4-(4-trifluoromethylbenzenesulfonyl)-piperazine (51)
As for 2, 1-{3-[4-(4-methoxyphenyl)-[1,2,3]triazol-1-yl]-propyl}-piperazine hydrochloride (13m) (1 eq, 0.15 g, 0.43 mmol) was reacted with 4-trifluoromethylbenzene sulfonyl chloride (8j) (1.1 eq., 0.12 g, 0.47 mmol) (monitored by UPLC-MS) to afford 51 as an off-white solid. Yield: 0.10 g, 46%, m.p.: dec >270 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.39 (s, 1H), 8.04 (d, J = 8.4 Hz, 2H), 7.96 (d, J = 8.3 Hz, 2H), 7.71 (d, J = 8.8 Hz, 2H), 6.99 (d, J = 8.8 Hz, 2H), 4.33 (t, J = 6.9 Hz, 2H), 3.78 (s, 3H), 2.95 (s, 4H), 2.43 (t, J = 5.5 Hz, 4H), 2.29 (t, J = 6.7 Hz, 2H), 1.99–1.92 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 158.9, 146.1, 139.0, 132.8 (q, J = 32.1 Hz, aryl C), 128.5 (2C), 126.6 (d, J = 3.7 Hz, aryl C, 2C), 126.4 (2C), 123.5, 123.4 (q, J = 271.3 Hz, CF3), 120.4, 114.2 (2C), 55.1, 53.8, 51.5 (2C), 47.5, 45.9 (2C), 27.2; 19F NMR (376 MHz, DMSO-d6) δ −58.9; IR υmax/cm−1: 1616 (C C), 1328 (S O), 1169 (S O), 1141 (C–F), 1062 (C–N); LRMS (ESI†) m/z: 510.2 [M + H]; HRMS calculated for C23H26F3N5O3S [M + H]: 510.1772; found: 510.1781.
1-{3-[4-(2-Methoxyphenyl)-[1,2,3]triazol-1-yl]-propyl}-4-(4-trifluoromethylbenzenesulfonyl)-piperazine (52)
As for 2, 1-{3-[4-(2-methoxyphenyl)-[1,2,3]triazol-1-yl]-propyl}-piperazine hydrochloride (13n) (1 eq, 0.15 g, 0.43 mmol) was reacted with 4-trifluoromethylbenzene sulfonyl chloride (8j) (1.1 eq., 0.12 g, 0.47 mmol) (monitored by UPLC-MS) to afford 52 as an white solid. Yield: 0.11 g, 49%, m.p.: dec >271 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.30 (s, 1H), 8.10 (dd, J = 7.7, 1.7 Hz, 1H), 8.03 (d, J = 8.4 Hz, 2H), 7.96 (d, J = 8.4 Hz, 2H), 7.34–7.30 (m, 1H), 7.12–7.09 (m, 1H), 7.05–7.01 (m, 1H), 4.37 (t, J = 6.9 Hz, 2H), 3.87 (s, 3H), 2.96 (s, 4H), 2.43 (d, J = 4.3 Hz, 4H), 2.27 (t, J = 6.7 Hz, 2H), 1.99–1.93 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 155.2, 141.5, 139.0, 132.8 (q, J = 32.1 Hz, aryl C), 128.8, 128.5 (2C), 126.6 (d, J = 3.7 Hz, aryl C, 2C), 126.5 (2C), 124.0, 123.4 (q, J = 271.3 Hz, CF3), 119.2, 111.5, 55.4, 53.7, 51.5 (2C), 47.3, 45.8 (2C), 26.8; 19F NMR (376 MHz, DMSO-d6) δ −58.9; IR υmax/cm−1: 1607 (C C), 1322 (S O), 1170 (S O), 1132 (C–F), 1063 (C–N); LRMS (ESI†) m/z: 510.2 [M + H]; HRMS calculated for C23H26F3N5O3S [M + H]: 510.1770; found: 510.1781.
Biology
Cell culture and stock solutions
Stock solutions were prepared as follows and stored at −20 °C: drugs were stored as 40 mM solutions in DMSO. All cell lines were cultured in a humidified atmosphere 5% CO2 at 37 °C. The cancer cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM) (Trace Biosciences, Australia) supplemented with 10% foetal bovine serum, 10 mM sodium bicarbonate, penicillin (100 IU mL−1), streptomycin (100 μg mL−1), and glutamine (4 mM). The non-cancer MCF10A cell line was cultured in DMEM : F12 (1 : 1) cell culture media, 5% heat inactivated horse serum, supplemented with penicillin (50 IU mL−1), streptomycin (50 μg mL−1), 20 mM Hepes, l-glutamine (2 mM), epidermal growth factor (20 ng mL−1), hydrocortisone (500 ng mL−1), cholera toxin (100 ng mL−1), and insulin (10 μg mL−1).46
In vitro growth inhibition MTT assay
Cells in logarithmic growth were transferred to 96-well plates. Cytotoxicity was determined by plating cells in duplicate in 100 μL medium at a density of 2500–4000 cells per well. On day 0, (24 h after plating) when the cells were in logarithmic growth 100 μL medium with or without the test agent was added to each well. After 72 h drug exposure growth inhibitory effects were evaluated using the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazoliumbromide) assay and absorbance read at 540 nm. Percentage growth inhibition was determined at a fixed drug concentration of 25 μM. A value of 100% is indicative of complete cell growth inhibition. Those analogues showing appreciable percentage growth inhibition underwent further dose response analysis allowing for the calculation of a GI50 value. This value is the drug concentration at which cell growth is 50% inhibited based on the difference between the optical density values on day 0 and those at the end of drug exposure.45,46
In vitro growth inhibition CCK-8 assay
Human pancreatic cancer cell lines including MiaPaCa2, BxPC3, CFPAC-1 and a normal human pancreatic ductal epithelial cells (HPDE) were used for this assessment and maintained in a humidified atmosphere 5% CO2 at 37 °C. MiaPaCa2 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM), supplemented with 10% fetal bovine serum (FBS), 2.5% horse serum and l-glutamine (100 μg mL−1). BxPC3 and CFPAC-1 cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 and Iscove's Modified Dulbecco's Media (IMDM) supplemented with 10% FBS and l-glutamine (100 μg mL−1), respectively. HPDE cells were cultured in Keratinocyte Serum-Free Media (KSFM) supplemented with human Recombinant Epidermal Growth Factor (rEGF) and Bovine Pituitary Extract (BPE) (100 μg mL−1).
Cell viability was assessed using the Dojindo Cell Counting Kit-8 (CCK-8). 200 μL of suspended cells were seeded into a 96-well plate at 3 × 103 cells per well for MiaPaCa2, 7 × 103 cells per well for BxPC3 and CFPAC-1 and 1 × 104 cells per well for HPDE, and incubated for 24 h. Then, a range of concentrations of the compounds and gemcitabine (50 nM) or a vehicle control (0.5% DMSO) were added to the wells. After 72 h, 10 μL of CCK-8 solution was added to each well and incubated at 37 °C for 1 h. The absorbance was measured at 450 nm by a multi-plate reader (BIORAD Benchmark Plus™, Bio-Rad Laboratories Pty Ltd., Gladesville, NSW, Australia) and cell viability was calculated as a percentage compared to negative control (0.5% DMSO). To eliminate any background that occurs due to turbidity, the absorbance of the wells was also measured at 650 nm as a reference, and this value was subtracted from the absorbance of the same well measured at 450 nm. The IC50 values (concentration that inhibits cell growth by 50%) were determined based on a dose response curve (100–0.05 μM).45,46
Molecular docking
Molecular docking simulations were performed using the default settings of Molecular Operating Environment (MOE). The human S100A2 crystal structure (RCSB ID: 2RGI) was prepared in MOE using the protonate 3D function: the system was protonated, partial charges added, and minimization performed. The designed 3D structures were built using the software molecular builder and were subjected to conformational analysis using a stochastic search approach. Each conformational library was docked into the p53 binding groove of the S100A2 homodimer using “Triangle Matcher” as the placement method and “London dG” as the scoring function for the initial ligand placement. The top 20 poses were then refined with the GBVI/WSA scoring function. The highest ranked pose for each compound was relaxed in the binding groove using LigX energy minimisation. Analysis and visualisation of the docking output, such as identification of hydrogen bonds, steric clashes, hydrophobic interactions, or π–π interactions, were performed in MOE.46–48 Molecular dynamics was performed using the above crystal structure: the system was solvated with water and a 100 ps equilibration was conducted, followed by a 500 ps molecular dynamics simulation.
Author contributions
JS, JRB, PJC and CCR: chemical synthesis and molecular modelling studies; HNTP, CDG, and JAS: Biological screening; CJS, and JAS: project conception and supervision; AM, CJS and JAS: manuscript preparation.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
JS is the recipient of a joint scholarship of UON UPRS and Binzhou Medical University (China), and this project has received funding from the UON Priority Research Centre for Drug Development. AM is the recipient of funding from the Australian Cancer Research Foundation and the Ramaciotti Foundation.
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00289b
References
- Rahib L. Smith B. D. Aizenberg R. Rosenzweig A. B. Fleshman J. M. Matrisian L. M. Cancer Res. 2014;74:2913–2921. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- Siegel R. L. Miller K. D. Fuchs H. E. Jemal A. Ca-Cancer J. Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- Teague A. Lim K.-H. Wang-Gillam A. Ther. Adv. Med. Oncol. 2015;7:68–84. doi: 10.1177/1758834014564775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Initiative A. P. C. G. Bailey P. Chang D. K. Nones K. Johns A. L. Patch A.-M. Gingras M.-C. Miller D. K. Christ A. N. Bruxner T. J. C. Quinn M. C. Nourse C. Murtaugh L. C. Harliwong I. Idrisoglu S. Manning S. Nourbakhsh E. Wani S. Fink L. Holmes O. Chin V. Anderson M. J. Kazakoff S. Leonard C. Newell F. Waddell N. Wood S. Xu Q. Wilson P. J. Cloonan N. Kassahn K. S. Taylor D. Quek K. Robertson A. Pantano L. Mincarelli L. Sanchez L. N. Evers L. Wu J. Pinese M. Cowley M. J. Jones M. D. Colvin E. K. Nagrial A. M. Humphrey E. S. Chantrill L. A. Mawson A. Humphris J. Chou A. Pajic M. Scarlett C. J. Pinho A. V. Giry-Laterriere M. Rooman I. Samra J. S. Kench J. G. Lovell J. A. Merrett N. D. Toon C. W. Epari K. Nguyen N. Q. Barbour A. Zeps N. Moran-Jones K. Jamieson N. B. Graham J. S. Duthie F. Oien K. Hair J. Grützmann R. Maitra A. Iacobuzio-Donahue C. A. Wolfgang C. L. Morgan R. A. Lawlor R. T. Corbo V. Bassi C. Rusev B. Capelli P. Salvia R. Tortora G. Mukhopadhyay D. Petersen G. M. Munzy D. M. Fisher W. E. Karim S. A. Eshleman J. R. Hruban R. H. Pilarsky C. Morton J. P. Sansom O. J. Scarpa A. Musgrove E. A. Bailey U.-M. H. Hofmann O. Sutherland R. L. Wheeler D. A. Gill A. J. Gibbs R. A. Pearson J. V. Waddell N. Biankin A. V. Grimmond S. M. Nature. 2016;531:47–52. doi: 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- Fink D. M. Steele M. M. Hollingsworth M. A. Cancer Lett. 2016;381:217–236. doi: 10.1016/j.canlet.2015.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kota J. Hancock J. Kwon J. Korc M. Cancer Lett. 2017;391:38–49. doi: 10.1016/j.canlet.2016.12.035. [DOI] [PubMed] [Google Scholar]
- Arias-Pinilla G. A. Modjtahedi H. Cancers. 2021;13:1781. doi: 10.3390/cancers13081781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadha A. S. Khoo A. Aliru M. L. Arora H. K. Gunther J. R. Krishnan S. Semin. Radiat. Oncol. 2016;26:320–337. doi: 10.1016/j.semradonc.2016.05.002. [DOI] [PubMed] [Google Scholar]
- Taboada A. G. M. Lominchar P. L. Roman L. M. García-Alfonso P. Martin A. J. M. Rodriguez J. A. B. Pascual J. M. A. Ann. Hepatobiliary Pancreat. Surg. 2021;25:179–191. doi: 10.14701/ahbps.2021.25.2.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancemi P. Buttacavoli M. Cara G. D. Albanese N. N. Bivona S. Pucci-Minafra I. Feo S. Oncotarget. 2018;9:29064–29081. doi: 10.18632/oncotarget.25561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. Xu C. Jin Q. Liu Z. Am. J. Cancer Res. 2014;4:89–115. [PMC free article] [PubMed] [Google Scholar]
- Bresnick A. R. Weber D. J. Zimmer D. B. Nat. Rev. Cancer. 2015;15:96–109. doi: 10.1038/nrc3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Publish Ahead of Print, NA; ; Dreyer S. B. Pinese M. Jamieson N. B. Scarlett C. J. Colvin E. K. Pajic M. Johns A. L. Humphris J. L. Wu J. Cowley M. J. Chou A. Nagrial A. M. Chantrill L. Chin V. T. Jones M. D. Moran-Jones K. Carter C. R. Dickson E. J. Samra J. S. Merrett N. D. Gill A. J. Kench J. G. Duthie F. Miller D. K. Cooke S. Aust D. Knösel T. Rümmele P. Grützmann R. Pilarsky C. Nguyen N. Q. Musgrove E. A. Bailey P. J. McKay C. J. Biankin A. V. Chang D. K. Initiative A. P. C. G. Laboratory G. P. O. Ann. Surg. 2020;272:366–376. doi: 10.1097/SLA.0000000000003143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biankin A. V. Kench J. G. Colvin E. K. Segara D. Scarlett C. J. Nguyen N. Q. Chang D. K. Morey A. L. Lee C.-S. Pinese M. Kuo S. C. L. Susanto J. M. Cosman P. H. Lindeman G. J. Visvader J. E. Nguyen T. V. Merrett N. D. Warusavitarne J. Musgrove E. A. Henshall S. M. Sutherland R. L. Network N. P. C. Gastroenterology. 2009;137:558–568.e11. doi: 10.1053/j.gastro.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Tartaglione S. Mancini P. Viggiani V. Chirletti P. Angeloni A. Anastasi E. PLoS One. 2021;16:e0251656. doi: 10.1371/journal.pone.0251656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J. Baker J. R. Russell C. C. Cossar P. J. Pham H. N. T. Sakoff J. A. Scarlett C. J. McCluskey A. ChemMedChem. 2021;16:2851–2863. doi: 10.1002/cmdc.202000949. [DOI] [PubMed] [Google Scholar]
- Sun J. Ambrus J. I. Russell C. C. Baker J. R. Cossar P. J. Pirinen M. J. Sakoff J. A. Scarlett C. J. McCluskey A. ChemMedChem. 2021;16:2851–2863. doi: 10.1002/cmdc.202000949. [DOI] [PubMed] [Google Scholar]
- Zeng M.-L. Zhu X.-J. Liu J. Shi P.-C. Kang Y.-L. Lin Z. Cao Y.-P. BioMed Res. Int. 2020;2020:1–15. doi: 10.1155/2020/8206849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.-Y. Song H.-M. Zhou Q. Medicine. 2020;99:e22777. doi: 10.1097/MD.0000000000022777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S. Liu L. Xue T. Jing C. Xu X. Wu Y. Wang M. Xie X. Zhang B. Front. Genet. 2021;12:648156. doi: 10.3389/fgene.2021.648156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J. Ambrus J. I. Baker J. R. Russell C. C. Cossar P. J. Sakoff J. A. Scarlett C. J. McCluskey A. Bioorg. Med. Chem. Lett. 2022;61:128591. doi: 10.1016/j.bmcl.2022.128591. [DOI] [PubMed] [Google Scholar]
- Sedaghat F. Notopoulos A. Hippokratia. 2008;12:198–204. [PMC free article] [PubMed] [Google Scholar]
- Totti S. Vernardis S. I. Meira L. Pérez-Mancera P. A. Costello E. Greenhalf W. Palmer D. Neoptolemos J. Mantalaris A. Velliou E. G. Drug Discovery Today. 2017;22:690–701. doi: 10.1016/j.drudis.2017.01.012. [DOI] [PubMed] [Google Scholar]
- Marenholz I. Heizmann C. W. Fritz G. Biochem. Biophys. Res. Commun. 2004;322:1111–1122. doi: 10.1016/j.bbrc.2004.07.096. [DOI] [PubMed] [Google Scholar]
- Deshpande R. Woods T. L. Fu J. Zhang T. Stoll S. W. Elder J. T. J. Invest. Dermatol. 2000;115:477–485. doi: 10.1046/j.1523-1747.2000.00078.x. [DOI] [PubMed] [Google Scholar]
- Ilg E. C. Schäfer B. W. Heizmann C. W. Int. J. Cancer. 1996;68:325–332. doi: 10.1002/(SICI)1097-0215(19961104)68:3<325::AID-IJC10>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Koch M. Diez J. Fritz G. J. Mol. Biol. 2008;378:933–942. doi: 10.1016/j.jmb.2008.03.019. [DOI] [PubMed] [Google Scholar]
- Tan M. Heizmann C. W. Guan K. Schafer B. W. Sun Y. FEBS Lett. 1999;445:265–268. doi: 10.1016/S0014-5793(99)00135-0. [DOI] [PubMed] [Google Scholar]
- Ohuchida K. Mizumoto K. Ishikawa N. Fujii K. Konomi H. Nagai E. Yamaguchi K. Tsuneyoshi M. Tanaka M. Clin. Cancer Res. 2005;11:7785–7793. doi: 10.1158/1078-0432.CCR-05-0714. [DOI] [PubMed] [Google Scholar]
- Ohuchida K. Mizumoto K. Miyasaka Y. Yu J. Cui L. Yamaguchi H. Toma H. Takahata S. Sato N. Nagai E. Yamaguchi K. Tsuneyoshi M. Tanaka M. J. Pathol. 2007;213:275–282. doi: 10.1002/path.2250. [DOI] [PubMed] [Google Scholar]
- Mueller A. Schäfer B. W. Ferrari S. Weibel M. Makek M. Höchli M. Heizmann C. W. J. Biol. Chem. 2005;280:29186–29193. doi: 10.1074/jbc.M505000200. [DOI] [PubMed] [Google Scholar]
- Leclerc E. Vetter S. W. Biochim. Biophys. Acta, Mol. Basis Dis. 2015;1852:2706–2711. doi: 10.1016/j.bbadis.2015.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresnick A. R. Biophys. Rev. 2018;10:1617–1629. doi: 10.1007/s12551-018-0471-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilder P. T. Varney K. M. Weber D. J. Methods Mol. Biol. 2019;1929:291–310. doi: 10.1007/978-1-4939-9030-6_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalier M. C. Pierce A. D. Wilder P. T. Alasady M. J. Hartman K. G. Neau D. B. Foley T. L. Jadhav A. Maloney D. J. Simeonov A. Toth E. A. Weber D. J. Biochemistry. 2014;53:6628–6640. doi: 10.1021/bi5005552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camara R. Ogbeni D. Gerstmann L. Ostovar M. Hurer E. Scott M. Mahmoud N. G. Radon T. Crnogorac-Jurcevic T. Patel P. Mackenzie L. S. Chau D. Y. S. Kirton S. B. Rossiter S. Eur. J. Med. Chem. 2020;203:112621. doi: 10.1016/j.ejmech.2020.112621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaquiquzzaman M. Verma G. Marella A. Akhter M. Akhtar W. Khan M. F. Tasneem S. Alam M. M. Eur. J. Med. Chem. 2015;102:487–529. doi: 10.1016/j.ejmech.2015.07.026. [DOI] [PubMed] [Google Scholar]
- Rathi A. K. Syed R. Shin H.-S. Patel R. V. Expert Opin. Ther. Pat. 2016;26:777–797. doi: 10.1080/13543776.2016.1189902. [DOI] [PubMed] [Google Scholar]
- Lin A. J. S. Russell C. C. Baker J. R. Frailey S. L. Sakoff J. A. McCluskey A. Org. Biomol. Chem. 2016;14:8732–8742. doi: 10.1039/C6OB01153E. [DOI] [PubMed] [Google Scholar]
- Kolb H. C. Finn M. G. Sharpless K. B. Angew. Chem., Int. Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Failli A. Legitimo A. Orsini G. Castagna M. Spisni R. Miccoli P. Consolini R. J. Biol. Regul. Homeostatic Agents. 2013;27:275–284. [PubMed] [Google Scholar]
- Cossar P. J. Lewis P. J. McCluskey A. Med. Res. Rev. 2020;40:469–494. doi: 10.1002/med.21519. [DOI] [PubMed] [Google Scholar]
- Buckley N. E. D'Costa Z. Kamiska M. Mullan P. B. Cell Death Dis. 2014;5:e1070. doi: 10.1038/cddis.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. Chen Q. Zhou Y. Niu Y. Wang X. Li X. Zheng H. Wei T. Zhao L. Gao H. FASEB J. 2020;34:13333–13344. doi: 10.1096/fj.202000555R. [DOI] [PubMed] [Google Scholar]
- Baker J. R. Russell C. C. Gilbert J. McCluskey A. Sakoff J. A. RSC Med. Chem. 2021;12:929–942. doi: 10.1039/D1MD00021G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker J. R. Russell C. C. Gilbert J. Sakoff J. A. McCluskey A. ChemMedChem. 2020;15:490–505. doi: 10.1002/cmdc.201900643. [DOI] [PubMed] [Google Scholar]
- Abdel-Hamid M. K. McCluskey A. Molecules. 2014;19:6609–6622. doi: 10.3390/molecules19056609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Hamid M. K. Macgregor K. A. Odell L. R. Chau N. Mariana A. Whiting A. Robinson P. J. McCluskey A. Org. Biomol. Chem. 2015;13:8016–8028. doi: 10.1039/C5OB00751H. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.