

Abstract
On one side, brain dysfunction is a poorly explored complication of sepsis. On the other side, brain dysfunction may actively contribute to the pathogenesis of sepsis. The current review aimed at summarizing the current knowledge about the reciprocal interaction between the immune and central nervous systems during sepsis. The immune-brain cross talk takes part in circumventricular organs that, being free from blood-brain-barrier, interface between brain and bloodstream, in autonomic nuclei including the vagus nerve, and finally through the damaged endothelium. Recent observations have confirmed that sepsis is associated with excessive brain inflammation and neuronal apoptosis which clinical relevance remains to be explored. In parallel, damage within autonomic nervous and neuroendocrine systems may contribute to sepsis induced organ dysfunction.
Keywords: apoptosis, autonomic nervous system, central nervous system, hormones, inflammation, neuromediators
Introduction
It is clear that septic shock can be associated with a spectrum of cerebral damage and dysfunction [1-3]. Reciprocal interactions between the immune and central nervous systems are now considered to be major components of the host response in septic shock. This is the case even though the brain is often thought of as a privileged organ – one that is anatomically sequestered from the immune system by the blood–brain barrier (BBB), lacking a lymphatic system and with low expression of histocompatibility complex antigens on its parenchymal cells. Because the central nervous system controls a wide range of physiological functions that are crucial to maintaining homeostasis and orchestrating the host response at behavioural, neuroendocrine and autonomic levels [4-7], disturbances in any of these adaptive functions may deleteriously influence the course of septic shock. For example, they may perpetuate immune-inflammatory responses and haemodynamic failure. Here we review the areas of the brain that are involved in the response to infection, the pathways and mechanisms of immune–brain interaction during septic shock, and clinical aspects of cerebral dysfunction in human septic shock.
Neuroanatomy of the brain response to infection
The systemic response to infection, an example of the response to noxious stress that was first described nearly 70 years ago by Seyle [8], involves a complex, organized and coherent interaction between immune, autonomic, neuroendocrine and behavioural systems [4,7,9]. The brain structures involved in this response are, in roughly ascending order (Fig. 1), as follows:
Figure 1.
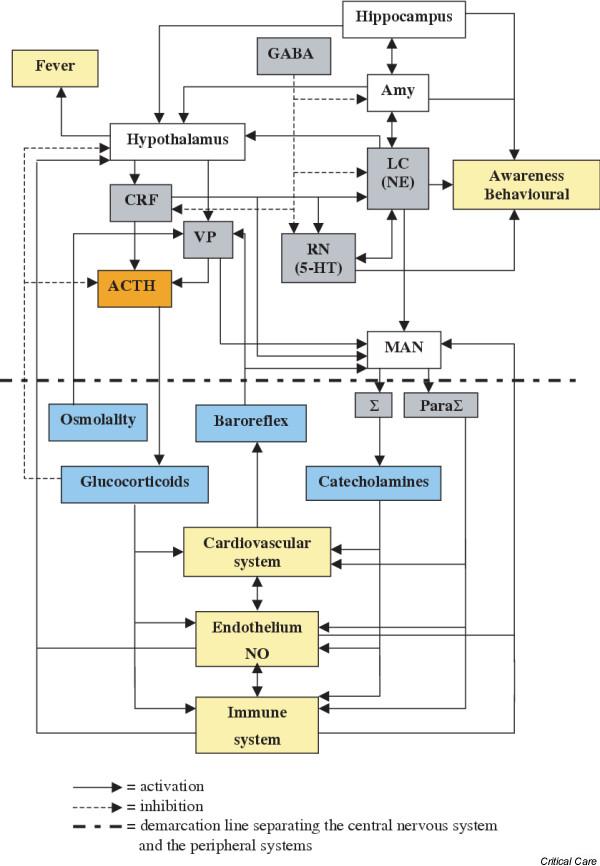
Main cerebral networks involved in the response to stress. ACTH, adrenocorticotrophic hormone; Amy, amygdala; CRF, corticotrophin-releasing factor; 5-HT, serotonin (5-hydroxytryptamine); LC, locus coeruleus; MAN, medullary autonomic nuclei; NE, norepinephrine (noradrenaline); NO, nitric oxide; ParaΣ, parasympathetic system; Σ, sympathetic system; RN, raphe nuclei; VP, vasopressin.
1. The medullary autonomic nuclei (i.e. solitary tract nuclei, the dorsal motor nucleus of the vagus and the ambiguus nuclei), which control parasympathetic output directly and sympathetic activity indirectly, through the intermediolateral cell column in the thoracic spinal cord.
2. The parabrachial nuclei, A5 cell group and the area postrema, which are located in the brainstem and control the medullary autonomic nuclei.
3 The midbrain raphe nuclei, which are the source of serotonergic fibre systems, and the reticular formation.
4 The locus coeruleus, which is both localized in the pons and the core of the noradrenergic network.
5. The hypothalamic paraventricular and supraoptic nuclei, which synthesize and release corticotrophin-releasing factor (CRF) and vasopressin.
6. The amygdala, which is located within the hippocampus and connected to the limbic system.
In addition to their neuroendocrine functions, CRF and vasopressin are both neurotransmitters with receptors that are expressed in the medullary autonomic nuclei and locus coeruleus [9]. All of these structures are interconnected, notably the paraventricular nucleus, locus coeruleus and nuclei of solitary tract, which have reciprocal projections [9].
The CRF, vasopressin and noradrenergic networks (termed CRF/VP and LC-NA systems) are coactivated during the response to stress and modulate each other [7]. They are also influenced by cerebral facilitatory (serotonergic and cholinergic networks) and inhibitory (γ-aminobutyric acid and opioid networks) systems, as well as by peripheral feedback mechanisms such as circulating inflammatory mediators, baroreflex afferents (vasopressin and autonomic nuclei), plasma corticosteroid level (adrenocorticotrophic hormone [ACTH] and CRF) and plasma osmolality (vasopressin).
There is an additional level of complexity, namely the interactive cellular organization of the brain, which includes endothelial cells, glial cells (astrocytes and microglia) and neurones. For example, astrocytes play a protective role in regulating local blood flow, transporting energy substrates from microvessels to neurones, preserving BBB properties, destroying pathogens, removing debris and promoting tissue repair [1,10]. However, activated glial cells acquire neurotoxic properties, notably by releasing nitric oxide (NO) and glutamate [10,11], in circumstances that include cerebral trauma, inflammation and infection.
Neuropathology of human septic shock
Neuropathological studies of human septic shock are scant, most of them being retrospective or performed in few patients [12,13]. In a recent prospective autopsy study of 23 patients who had died from septic shock, we found ischaemic lesions in all cases, haemorrhage in 26%, hypercoagulability syndrome in 9%, microabscesses in 9%, and multifocal necrotizing leucoencephalopathy in 9%, which was associated with both local expression and high circulating levels of proinflammatory cytokines [2]. This latter finding is of great interest because it shows that the brain can be damaged through purely inflammatory processes, as distinct from hypoperfusion or coagulation disturbances, during septic shock [14]. However, the incidence and features of brain lesions in the ante mortem period and in patients surviving septic shock remain to be assessed.
Immune–brain pathways
The immune system can be thought of as a diffuse sensory system that signals the presence of micro-organism constituents to the brain through three main mechanisms [15]. First are the circumventricular organs, which are composed of specialized tissue and located at a strategic position in the midline ventricular system. Because they are not protected by the BBB they can function as communicating structures between the brain and bloodstream. They encompass the pineal body, the subcommissural organ and the subfornical organ, but especially the organum vasculosum, the median eminence and the neurohypohysis; these are, respectively, part of the hypothalamic and pituitary centres and the area postrema, which is close to the medullar autonomic nuclei. The vagus nerve, by sensing peripheral inflammation (presumably through cytokine receptors on the nerve surface), conveys immune-related information to the medulla [16] and then suppresses the inflammatory response at the site of infection (through nicotinic acetylcholine receptors on monocytes) [17-19]. The third signalling pathway is via endothelial activation and leakage, which leads to release or passive diffusion of inflammatory and neurotoxic mediators.
Blood–brain barrier during infection
Diffuse endothelial activation, also termed panendothelitis, is considered to be the hallmark of septic shock. Both lipopolysaccharide (LPS) and proinflammatory cytokines induce the expression of CD40, vascular adhesion molecule-1 or intercellular adhesion molecule-1, and E-selectin on human brain microvessel endothelial cells [20-24]. They also cause transcriptional activation of the gene that encodes cyclooxygenase 2 and stimulation of the IκB-α /nuclear factor-κB (NF-κB) pathway [25-27]. Although brain endothelial cells do not express surface CD14, LPS also triggers the mitogen-activated protein kinase cascade through soluble CD14 [28]. LPS-activated brain endothelial cells exhibit IL-1 and tumour necrosis factor (TNF)-α receptors [29,30]; produce IL-1β, TNF-α and IL-6 [31-33]; and exhibit endothelial and inducible nitric oxide synthase (NOS) [34-37]. These mediators are able to interact with surrounding brain cells, relaying into the brain inflammatory response. This endothelial activation may result in alteration in the BBB [38-41]. Indeed, it has been shown that the BBB is rendered permeable in experimental models of septic shock [42-44], an effect that is attenuated by glial cells, dexamethasone, or NOS inhibition [42,45,46]. This endothelial activation may also result in cerebrovascular dysfunction. However, although a number of studies have assessed cerebral blood flow, endothelial reactivity and oxygen consumption during sepsis both in animal and human shock, they have yielded contradictory results, some showing impairment [47-49] and others not [50-53].
Cerebral immune system in infection
A coherent neuro–immune interaction requires that the brain can detect inflammatory mediators. Components of the innate and adaptive immune systems are expressed in the brain during experimental endotoxin shock [54]. Remarkably, their expression spreads from circumventricular organs to the deeper brain areas that control neuroendocrine and autonomic functions – a 'migratory' pattern of brain activation. Thus, LPS receptor CD14 is expressed sequentially, first in the circumventricular organs and then in hypothalamic and medullary autonomic nuclei during the very acute phase of experimental septic shock [55].
Toll-like receptor (TLR)2, TLR4 and TLR9 have been detected both in resting and LPS-activated animal or human glial cells (microglia, astrocytes and oligodendrocytes) [54,56,57], as may be expected because they are bone marrow derived monocytes. The issue of whether TLRs are expressed in neurones remains controversial, Lehnardt and coworkers [58] having recently shown that neuronal TLR remained undetectable after in vitro LPS stimulation. TLR4, which interacts with LPS-bound CD14, is constitutively expressed in circumventricular organs but also in the hypothalamus and medulla; in contrast to CD14, however, there is a downregulation of TLR4 mRNA in the brains of rats challenged by LPS [59]. There is also a strong and transient expression of the gene encoding TLR2 in the brains of LPS-3 challenged mice [60]. Microglial cells also express TLR9 mRNA, and its ligand has been found to activate these cells both in vitro and in animal models [61]. CD14 and TLR both trigger cellular transcription of proinflammatory molecules through the NF-κB pathway. Thus, IκB mRNA follows a CD14 migratory-like transcription pattern in the brain of rats following intraperitoneal LPS administration [62].
It has been established that LPS stimulation induces NO synthesis [63,64], and the release of proinflammatory and anti-inflammatory cytokines and their receptors from neurones, astrocytes and microglial cells both in vitro [65-68] and in vivo [31,34-36,69-72]. The coexpression of proinflammatory and anti-inflammatory cytokines suggests the existence of a highly organized immune counter-regulation within the brain [73].
Prostaglandins are key mediators in the brain response to inflammatory stimuli, their role in fever having been extensively investigated. Thus, following LPS stimulation astrocytes release significant amount of prostaglandin E [74], whereas microglia express prostaglandin receptors [75] and express cyclooxygenase 2 [76]. Finally, a number of other mediators are involved in the cerebral brain response to immune challenge including, among others, chemokines, macrophage migrating inhibitory factor, platelet activating factor, superoxide radicals and carbon monoxide.
Consequences of cerebral immune activation
There is a body of evidence that NO, cytokines and prostaglandins modulate brain neurotransmission [77-82], especially the β-adrenergic system, the production and release of CRF, ACTH and vasopressin, as well as medullary autonomic centre output [83,84]. Inversely, neurotransmitters and neurohormones also modulate cerebral expression of inflammatory mediators [85,86]. These effects have been described elsewhere [66,87]. The final neuroendocrine and autonomic response is variable because it depends on a highly complex and spatiotemporally changing process that involves both stimulatory and inhibitory factors, which themselves depend on interactions between glial, endothelial and neuronal cells. Disturbances in these relationships may lead to maladaptive responses, as illustrated by a recent experimental study [88] that showed that heart failure associated sympathetic hyperactivity was linked to decreased NO production in the paraventricular nucleus. The opposite phenomenon may occur in septic shock, which is associated with reduced sympathetic output [89].
At an intracellular level, various phenomena have been reported, including activation or inhibition of mitochondrial respiration [10,90], activation of mitogen-activated protein kinase and NF-κB pathways [91] and release of cytotoxic agents such as calcium and reactive oxygen species [92,93], as well as protective ones such as heat shock proteins [94]. However, although sepsis-related mitochondrial dysfunction has been extensively assessed in various human organs [95], it remains to be documented in the human brain, but it is of course the case that genetic mitochondrial diseases are well described causes of brain dysfunction in humans.
Clearly, an important aspect of cerebral dysfunction is brain cell apoptosis, which occurs as a consequence of multiple factors that are in play during septic shock, including ischaemia, glial cell activation, TNF-α, IL-1β. interferon-γ and NO [96-99]. LPS challenge is associated with either glial or neuronal apoptosis [99,100] and it appears that NO is the main apoptotic mediator, although the TLR4 pathway may also be involved [101]. On the other hand, recent experimental studies have suggested that IL-10 and cyclooxygenase inhibition attenuate LPS-induced apoptosis [97,102,103]. We recently found apoptotic microglial and neuronal cells in the hypothalamus and cardiovascular autonomic centres in the brains of patients who had died from septic shock [3]. Of note is that, in that study, neuronal apoptosis was closely correlated with endothelial cell inducible NOS expression [3].
Encephalopathy, neuroendocrine and autonomic dysfunction in septic shock
Septic encephalopathy
The prevalence of encephalopathy in severe sepsis varies from 9% to 71%, depending on the definition, which can be based on clinical criteria [1,104-106], electroencephalographic criteria [107,108], or, more recently, on sensory evoked potentials [109,110]. An important advantage of the latter technique is that it is not influenced by sedation [109,110]. The severity of encephalopathy has been found to correlate with the global severity of illness, as assessed by Acute Physiology and Chronic Health Evaluation II score or organ failure scores, and with mortality [104-106]. As described above, the pathophysiology of encephalopathy is multifactorial, including the following: cerebral endothelial dysfunction, with BBB disruption and cerebral blood flow impairment, fostering translocation of neurotoxic molecules and brain hypoperfusion/ischaemia, respectively [1]; neurotoxic amino acids (such as ammonium, tyrosine, tryptophan and phenylalanine), whose plasma levels are increased in sepsis because of muscle proteolysis and reduced hepatic clearance [1,77,111-114]; and endotoxin and inflammatory mediators, which alter glial and neuronal metabolism, as was described previously [1]. Renal and hepatic failure, metabolic disturbances and neurotoxic drugs may also contribute to the development of brain dysfunction. Finally, neurone-specific enolase, a marker of brain injury, may be a predictor of death in septic shock patients [115].
Neuroendocrine dysfunction and autonomic failure
The endocrine response to sepsis is complex, and in this review we focus only on the hypothalamic–pituitary–adrenal axis and on vasopressin. Briefly, disruption of the hypothalamic–pituitary–adrenal axis is a common feature in severe sepsis and may be unmasked by a short Synacten test, when cortisol level increases by less than 9 μg/dl after an intravenous bolus of 250 μg corticotrophin [116]. It is now recognized that, in sepsis, adrenal insufficiency partly accounts for reduced vascular sensitivity to vasopressors [117] and an increased risk for death [116]. Moreover, in septic shock, correcting this disorder by cortisol replacement therapy improves haemodynamic status and survival [118].
Septic shock may also be associated with a relative vasopressin deficiency, a concept that is worthy of clarification. Indeed, it is one rationale for treating septic shock with vasopressin infusion, the optimal start, duration and target plasma vasopressin concentration of which remain unresolved [119-121]. First, deficiency implicitly suggests that plasma vasopressin levels are abnormally reduced. Landry and coworkers [122] originally reported significantly lower plasma vasopressin levels in late septic shock than in cardiogenic shock (3.1 ± 1.0 versus 22.7 ± 2.2 pg/ml). The latter observation, together with the demonstration of high vasopressin levels in experimental early endotoxic shock [123,124], suggests that circulating vasopressin levels wane as the course of septic shock progresses. Indeed, this pattern was confirmed in patients with septic shock [125].
Second, 'inappropriately low' means that the observed plasma vasopressin level does not match the expected value for a given level of plasma osmolality or a given degree of hypotension. It is highly difficult to apply such a criterion in septic shock. For instance, circulating vasopressin levels were inappropriately low in a third of patients with septic shock, mainly after the 36 hours from the onset of shock [125]. Vasopressin levels were thought to be inappropriate when they were 3.6 pg/ml or less (the upper limit for normonatraemic and normotensive healthy individuals) and sodium concentration was 145 mmol/l or more, or systolic blood pressure was less than 100 mmHg. One may argue that using the upper limit observed in hypernatraemic or hypotensive healthy individual or in cardiogenic shock as a reference would have resulted in a higher rate of inappropriate vasopressin levels. The latter issue concerns the limits of natraemia and systolic blood pressure to which one should refer. In such a life-threatening and complex condition as septic shock, it is conceivable that the osmo- and baro-thresholds of vasopressin secretion are respectively shifted to an upper level of natraemia and a lower level of systolic blood pressure, simply because vasopressin reserve must be preserved or vasopressin concentrations are appropriate for other physiological factors.
Keeping this in mind, it is noteworthy that, in patients with septic shock and adrenal insufficiency, plasma vasopressin levels were significantly higher in nonsurvivors [125]. It is therefore plausible that secretion of vasopressin, which is known to modulate ACTH release and to be regulated by circulating cortisol [7], was adapted to adrenal function. In addition, this observation may also suggest that plasma vasopressin deficiency is not associated with poorer outcomes. So, why should plasma vasopressin be normalized?
In an opposing and provocative view, one may argue that vasopressin secretion should be limited in some patients, particularly those with adrenal insufficiency. However, vasopressin infusion, if not beneficial in normalizing vasopressin deficiency, might be useful because of its haemodynamic properties [119-121]. The various mechanisms underlying inappropriately low circulating vasopressin levels may include increased vasopressin clearance from plasma, depleted vasopressin stores after the initial release, impaired baroreflex or osmoreceptor sensitivity, cytokines, or NO-induced decreased vasopressin synthesis or release [126-129]. We found normal vasopressinase activity, empty vasopressin neurohypophyseal stores on magnetic resonance imaging [130] and impaired baroreflex activity in some patients [125]. However, interpretation of baroreflex sensitivity is difficult because it is directly influenced, through the medullar V1b receptor, by plasma vasopressin level [131].
Autonomic failure was initially described in endotoxin challenged animals before it was documented in patients with septic shock, particularly by using spectral analysis of heart rate variability [89,132]. Impaired autonomic function is associated with an increased risk for death from critical illness [133,134].
Conclusion
Septic shock is often complicated by encephalopathy, neuroendocrine dysfunction and cardiovascular autonomic failure, all of which worsen patient outcomes. The mechanisms of these dysfunctions are highly complex and involve inappropriate immune–brain signalling, which results in brain cell activation; deleterious production of NO; dysfunction of intracellular metabolism; and cell death. Areas of the brain that are responsible for cardiovascular homeostasis appear to be specifically vulnerable during sepsis, creating a vicious cycle. The central role played by NO suggests that inhibition of inducible NOS expression would be beneficial but this needs to be demonstrated experimentally, especially because inhibition of endothelial NOS might worsen brain ischaemia. It may prove difficult to manipulate the complex and inter-related processes involved.
Competing interests
None declared.
Abbreviations
ACTH = adrenocorticotrophic hormone; BBB = blood–brain barrier; CRF = corticotrophin-releasing factor; IL = interleukin; LPS = lipopolysaccharide; NF-κB = nuclear factor-κB; NO = nitric oxide; NOS = nitric oxide synthase; TLR = Toll-like receptor; TNF = tumour necrosis factor.
References
- Papadopoulos MC, Davies DC, Moss RF, Tighe D, Bennett ED. Pathophysiology of septic encephalopathy: a review. Crit Care Med. 2000;28:3019–3024. doi: 10.1097/00003246-200008000-00057. [DOI] [PubMed] [Google Scholar]
- Sharshar T, Annane D, de la Grandmaison G, Brouland JP, Hopkinson NS, Gray F. The neuropathology of septic shock. Brain Pathol. 2004;14:21–33. doi: 10.1111/j.1750-3639.2004.tb00494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharshar T, Gray F, Lorin de la Grandmaison G, Hopkinson NS, Ross E, Dorandeu A, Orlikowski D, Raphael J-C, Gajdos P, Annane D. Apoptosis of neurons in cardiovascular autonomic centres triggered by inducible nitric oxide synthase after death from septic shock. Lancet. 2003;362:1799–1805. doi: 10.1016/S0140-6736(03)14899-4. [DOI] [PubMed] [Google Scholar]
- Spyer KM. Neural mechanisms involved in cardiovascular control during affective behaviour. Trends Neurosci. 1989;12:506–513. doi: 10.1016/0166-2236(89)90111-2. [DOI] [PubMed] [Google Scholar]
- Saper CB, Breder CD. The neurologic basis of fever. N Engl J Med. 1994;330:1880–1886. doi: 10.1056/NEJM199406303302609. [DOI] [PubMed] [Google Scholar]
- Chrousos GP, Gold PW. The concepts of stress and stress system disorders. Overview of physical and behavorial homeostasis. JAMA. 1992;267:1244–1252. doi: 10.1001/jama.267.9.1244. [DOI] [PubMed] [Google Scholar]
- Chrousos GP. The hypothalamic-pituitary-adrenal-axis and immune-mediated inflammation. N Engl J Med. 1995;332:1351–1362. doi: 10.1056/NEJM199505183322008. [DOI] [PubMed] [Google Scholar]
- Seyle HA. A syndrome produced by diverse nocuous agents. Nature. 1936;138:32. doi: 10.1176/jnp.10.2.230a. [DOI] [PubMed] [Google Scholar]
- Carrasco GA, Van de Kar LD. Neuroendocrine pharmacology of stress. Eur J Phamarcol. 2003;463:235–272. doi: 10.1016/S0014-2999(03)01285-8. [DOI] [PubMed] [Google Scholar]
- Bal-Price A, Brown GC. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci. 2001;21:6480–6491. doi: 10.1523/JNEUROSCI.21-17-06480.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS. Regional difference in susceptibility to lipopolysaccharide neurotoxicity in the rat brain: role of microglia. J Neurosci. 2000;20:6309–6316. doi: 10.1523/JNEUROSCI.20-16-06309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AC, Gilbert JJ, Young GB, Bolton CF. The encephalopathy of sepsis. Can J Neurol. 1985;12:303–307. doi: 10.1017/s0317167100035381. [DOI] [PubMed] [Google Scholar]
- Bleck TP, Smith MC, Pierre-Louis SJ, Jares JJ, Murray J, Hansen CA. Neurologic complications of critical medical illnesses. Crit Care Med. 1993;21:98–103. doi: 10.1097/00003246-199301000-00019. [DOI] [PubMed] [Google Scholar]
- Sharshar T, Gray F, Poron F, Raphael JC, Gajdos P, Annane D. Multifocal necrotizing leukoencephalopathy in septic shock. Crit Care Med. 2002;30:2371–2375. doi: 10.1097/00003246-200210000-00031. [DOI] [PubMed] [Google Scholar]
- Goehler LE, Gaykema RP, Hansen MK, Anderson K, Maier SF, Watkins LR. Vagal immune-to-brain communication: a visceral chemosensory pathway. Auton Neurosci. 2000;85:49–59. doi: 10.1016/S1566-0702(00)00219-8. [DOI] [PubMed] [Google Scholar]
- Maier SF, Goehler LE, Fleshner M, Watkins LR. The role of the vagus nerve in cytokine-to-brain communication. Ann N Y Acad Sci. 1998;840:289–300. doi: 10.1111/j.1749-6632.1998.tb09569.x. [DOI] [PubMed] [Google Scholar]
- Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–462. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Yang H, Ulloa L, Al-Abed Y, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- Goehler LE, Gaykema RP, Hammack SE, Maier SF, Watkins LR. Interleukin-1 induces c-fos immunoreactivity in primary afferent neurons of the vagus nerve. Brain Res. 1998;804:306–310. doi: 10.1016/S0006-8993(98)00685-4. [DOI] [PubMed] [Google Scholar]
- Omari KM, Dorovini-Zis K. CD40 expressed by human brain endothelial cells regulate CD4+ T cell adhesion to endothelium. J Neuroimmunol. 2003;134:166–178. doi: 10.1016/S0165-5728(02)00423-X. [DOI] [PubMed] [Google Scholar]
- Wong D, Dorovini-Zis K. Expression of vascular cell adhesion molecule-1 (VCAM-1) by human brain microvessel endothelial cells in primary culture. Microvasc Res. 1995;49:325–339. doi: 10.1006/mvre.1995.1028. [DOI] [PubMed] [Google Scholar]
- Hess DC, Thompson Y, Sprinkle A, Carroll J, Smith J. E-selectin on human brain microvascular endothelial cells. Neurosci Lett. 1996;213:37–40. doi: 10.1016/0304-3940(96)12837-8. [DOI] [PubMed] [Google Scholar]
- Rieckmann P, Michel U, Albrecht M, Bruck W, Wockel L, Felgenhauer K. Cerebral endothelial cells are a major source for soluble intercellular adhesion molecule-1 in the human central nervous system. Neurosci Lett. 1995;186:61–64. doi: 10.1016/0304-3940(95)11282-2. [DOI] [PubMed] [Google Scholar]
- Hess DC, Bhutawala T, Sheppard JC, Zhao W, Smith J. ICAM-1 expression on human brain microvascular endothelial cells. Neurosci Lett. 1994;168:201–204. doi: 10.1016/0304-3940(94)90450-2. [DOI] [PubMed] [Google Scholar]
- Lacroix S, Rivest S. Effect of acute systemic inflammatory response and cytokines on the transcription genes encoding cyclooxigenase enzimes (COX-1 and COX-2) in the rat brain. J Neurochem. 1998;70:452–466. doi: 10.1046/j.1471-4159.1998.70020452.x. [DOI] [PubMed] [Google Scholar]
- Cao C, Matsumura K, Ozaki M, Watanabe Y. Lipopolysaccharide injected into the cerebral ventricle evokes fever through induction of cyclooxygenase-2 in brain endothelial cells. J Neurosci. 1998;19:716–725. doi: 10.1523/JNEUROSCI.19-02-00716.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura K, Cao C, Ozaki M, Morii H, Nakadate K, Watanabe Y. Brain endothelial cells express cyclooxygenase-2 during lipopolysaccharide-induced fever: light and electron microscopic immunocytochemical studies. J Neurosci. 1998;18:6279–6289. doi: 10.1523/JNEUROSCI.18-16-06279.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arditi M, Zhou J, Torres M, Durden DL, Stins M, Kim KS. Lipopolysaccharide stimulates the tyrosine phosphorylation of mitogen-activated kinases p44, p42, and p41 in vascular endothelial cells in a soluble CD14-dependent manner. Role of protein tyrosine phosphorylation in lipopolysaccharide-induced stimulation of endothelial cells. J Immunol. 1995;155:3994–4003. [PubMed] [Google Scholar]
- Konsman JP, Vigues S, Mackerlova L, Bristow A, Blomqvist A. Rat brain vascular distribution of interleukin-1 type-1 receptor immunoreactivity: relastionship to patterns of inducible cyclooxigenase expression by peripheral inflammatory stimuli. J Comp Neurol. 2004;472:113–129. doi: 10.1002/cne.20052. [DOI] [PubMed] [Google Scholar]
- Nadeau S, Rivest S. Effects of circulating tumor necrosis on the neuronal activity of the genes encoding the tumor necrosis factors receptors (p55 and p75) in the rat brain: a view from the blood–brain barrier. Neuroscience. 1999;93:1449–1464. doi: 10.1016/S0306-4522(99)00225-0. [DOI] [PubMed] [Google Scholar]
- Breder CD, Hazuka C, Ghayur T, Klug C, Huginin M, Yasuda K, Teng M, Saper CB. Regional induction of tumor necrosis factor alpha expression in the mouse brain after systemic lipopolysaccharide administration. Proc Natl Acad Sci USA. 1994;91:11393–11397. doi: 10.1073/pnas.91.24.11393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes TM, Fabry Z, Coe CL. Brain endothelial cell production of neuroprotective cytokine, interleukin-6, in response to noxious stimuli. Brain Res. 1999;1999:215–220. doi: 10.1016/S0006-8993(99)02189-7. [DOI] [PubMed] [Google Scholar]
- Fabry Z, Fitzsimmons KM, Herlein JA, Moninger TO, Dobbs MB, Hart MN. Production of cytokines interleukin-1 and 6 by murine brain microvessel endothelium and smooth muscle pericytes. J Neuroimmunol. 1993;47:23–34. doi: 10.1016/0165-5728(93)90281-3. [DOI] [PubMed] [Google Scholar]
- Iwase K, Miyanaka K, Shimizu A, Nagasaki A, Gotoh T, Mori M, Takiguchi M. Induction of endothelial nitric-oxyde syjthase in rat brain astrocytes by systemic lipopolysaccharide treatment. J Biol Chem. 2000;275:11929–11933. doi: 10.1074/jbc.275.16.11929. [DOI] [PubMed] [Google Scholar]
- Wong ML, Bongiorno PB, al-Shekhlee A, Esposito A, P PK, Licinio J. IL-1 beta, IL-1 receptor type I and iNOS gene expression in rat brain vasculature and perivascular areas. NeuroReport. 1996;7:2445–2448. doi: 10.1097/00001756-199611040-00008. [DOI] [PubMed] [Google Scholar]
- Wong ML, Rettori V, Shekhlee Aa, Bongiorno PB, Canteros G, McCann SM, Gold PW, Licinio J. Inducible nitric oxide synthase gene expression in the brain during systemic inflammation. Nat Med. 1996;2:581–584. doi: 10.1038/nm0596-581. [DOI] [PubMed] [Google Scholar]
- Freyer D, Manz R, Ziegenhorn A, M Weih MA, Docke WD, Meisel A, Schumann RR, Dirnagl U, Weber JR. Cerebral endothelial cells release TNF-alpha after stimulation with cell walls of Streptococcus pneumoniae and regulate inducible nitric oxide synthase and ICAM-1 expression via autocrine loops. J Immunol. 1999;1:4308–4314. [PubMed] [Google Scholar]
- Clawson CC, Hartmann JF, Vernier RL. Electron microscopy of the effect of gram-negative endotoxin on the blood–brain barrier. J Comp Neurol. 1966;127:183–198. doi: 10.1002/cne.901270204. [DOI] [PubMed] [Google Scholar]
- Jeppsson B, Freund HR, Z G, James JH, von Meyenfeldt MF, Fischer JE. Blood–brain barrier derangement in sepsis: cause of septic encephalopathy? Am J Surg. 1981;141:136–142. doi: 10.1016/0002-9610(81)90026-X. [DOI] [PubMed] [Google Scholar]
- Ring A, Weiser JN, Tuomanen EI. Pneumoccocal trafficking accross the blood-brain barrier : molecular analysis of a novel bidirectionnal pathway. J Clin Invest. 1998;102:347. doi: 10.1172/JCI2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos MC, Lamb FJ, Moss RF, Davies DC, Tighe D, Bennett ED. Faecal peritonitis causes oedema and neuronal injury in pig cerebral cortex. Clin Sci (Lond) 1999;96:461–466. doi: 10.1042/CS19980327. [DOI] [PubMed] [Google Scholar]
- Descamps L, Coisne C, Dehouck B, Cecchelli R, Torpier G. Protective effect of glial cells against lipopolysaccharide-medietd blood-brain barrier injury. Glia. 2003;42:46–58. doi: 10.1002/glia.10205. [DOI] [PubMed] [Google Scholar]
- Xaio H, Banks WA, Niehoff ML, Morley JE. Effect of LPS on the permeability of the blood-brain barrier to insulin. Brain Res. 2001;896:1–2. doi: 10.1016/S0006-8993(00)03247-9. [DOI] [PubMed] [Google Scholar]
- Mayhan G. Effect of lipopolysaccharide on the permeability and reactivity of the cerebral microcirculation: role of inducible nitric oxide synthase. Brain Res. 1998;792:353–357. doi: 10.1016/S0006-8993(98)00259-5. [DOI] [PubMed] [Google Scholar]
- Boje KM. Inhibition of nitric oxide synthase attenuates blood–brain barrier disruption during experimental meningitis. Brain Res. 1996;720:75–83. doi: 10.1016/0006-8993(96)00142-4. [DOI] [PubMed] [Google Scholar]
- Gaillard PJ, de Boer AB, Breimer DD. Pharmalogical investigations on lipopolysaccharide-induced changes in the blood brain barrier in vitro. Microvasc Res. 2003;65:24–31. doi: 10.1016/S0026-2862(02)00009-2. [DOI] [PubMed] [Google Scholar]
- Bowton DL, Bertels NH, Prough DS, Stump DA. Cerebral blood flow is reduced in patients with sepsis syndrome. Crit Care Med. 1989;17:399–403. doi: 10.1097/00003246-198905000-00004. [DOI] [PubMed] [Google Scholar]
- Maekawa T, Fujii Y, Sadamitsu D, Yokota K, Soejima Y, Ishikawa T, Miyauchi Y, Takeshita H. Cerebral circulation and metabolism in patients with septic encephalopathy. Am J Emerg Med. 1991;9:139–143. doi: 10.1016/0735-6757(91)90175-J. [DOI] [PubMed] [Google Scholar]
- Terborg C, Schummer W, Albrecht M, Reinhart K, Weiller C, Rother J. Dysfunction of vasomotor reactivity in severe sepsis and septic shock. Intensive Care Med. 2001;27:1231–1234. doi: 10.1007/s001340101005. [DOI] [PubMed] [Google Scholar]
- Christenson JT, Kuikka JT, Owunwanne A, Al-Sarraf AA. Cerebral circulation during endotoxin shock with special emphasis on the regional cerebral blood flow in vivo. Nucl Med Commun. 1986;7:531–540. doi: 10.1097/00006231-198607000-00007. [DOI] [PubMed] [Google Scholar]
- Ekstrom-Jodal B, Larsson LE. Effects of dopamine of cerebral circulation and oxygen metabolism in endotoxic shock: an experimental study in dogs. Crit Care Med. 1982;10:375–377. doi: 10.1097/00003246-198206000-00008. [DOI] [PubMed] [Google Scholar]
- Matta BF, Stow P. Sepsis-induced vasoparalysis does not involve the cerebral vasculature: indirect evidence from autoregulation and carbon dioxide reactivity studies. Br J Anaesth. 1996;76:790–794. doi: 10.1093/bja/76.6.790. [DOI] [PubMed] [Google Scholar]
- Pollard V, Prough DS, Deyo DJ, Conroy B, Uchida T, Daye A, Traber LD, Traber DL. Cerebral blood flow during experimental endotoxemia in volunteers. Crit Care Med. 1997;25:1700–1706. doi: 10.1097/00003246-199710000-00020. [DOI] [PubMed] [Google Scholar]
- Rivest S. Molecular insights on the cerebral innate immune system. Brain Behav Immun. 2003;17:13–19. doi: 10.1016/S0889-1591(02)00055-7. [DOI] [PubMed] [Google Scholar]
- Lacroix S, Feinstein D, Rivest The bacterial endotoxin lipopolysaccharide has the ability to target the brain in upregulating its membrane CD14 receptor within specific cellular populations. Brain Pathol. 1998;8:625–640. doi: 10.1111/j.1750-3639.1998.tb00189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman CC, Rasley A, Tranguch SL, Marriott I. Cultured astrocytes express toll-like receptors for bacterial products. Glia. 2003;43:281–291. doi: 10.1002/glia.10256. [DOI] [PubMed] [Google Scholar]
- Bsibi M, Ravid R, Gveric D, van Noort JM. Broad expression of toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- Lehnardt S, Lachance C, Patrizi S, Lefebvre S, Follett PL, Jensen FE, Rosenberg PA, Volpe JJ, Vartanian T. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. Neuroscience. 2002;22:2478–2486. doi: 10.1523/JNEUROSCI.22-07-02478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laflamme N, Rivest S. Toll-like receptor 4: the missing link of the cerebral innate immune response triggered by circulating gram-negative bacterial cell components. FASEB J. 2001;15:155–163. doi: 10.1096/fj.00-0339com. [DOI] [PubMed] [Google Scholar]
- Laflamme N, Souci G, Rivest S. Circulating cell wall components derived from Gram-negative and not Gram-positive bacteria cause of a profound transcriptionnal activation of the gene Toll-like receptor 2 in the CNS. J Neurochem. 2001;70:648–657. doi: 10.1046/j.1471-4159.2001.00603.x. [DOI] [PubMed] [Google Scholar]
- Dalpke AH, Schafer MK, Frey M, Zimmerman S, Tebbe J, Weihe E, Hegg K. Immunostimulatory CpG-DNA activates murine microglia. J Immunol. 2002;168:4854–4863. doi: 10.4049/jimmunol.168.10.4854. [DOI] [PubMed] [Google Scholar]
- Quan N, Whiteside MB, Kim H, Herkenham M. Induction of inhibitory factor kappaBalpha mRNA in the central nervous system after peripheral lipopolysaccharide administration: an in situ hybridization histochemistry study in the rat. Proc Natl Acad Sci USA. 1997;94:10985–10990. doi: 10.1073/pnas.94.20.10985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minc-Golomb D, Tsarfaty I, Schwartz JP. Expression of inducible nitric oxide synthase by neurones following exposure to endotoxin and cytokine. Br J Pharmacol. 1994;112:720–722. doi: 10.1111/j.1476-5381.1994.tb13136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galea E, Reis DJ, Feinstein DL. Cloning and expression of inducible nitric oxide synthase from rat astrocytes. J Neuroscience Res. 1994;37:406–414. doi: 10.1002/jnr.490370313. [DOI] [PubMed] [Google Scholar]
- Aloisi F, Penna G, Cerase J, Menendez Iglesias B, Adorini L. IL-12 production by central nervous system microglia is inhibited by astrocytes. J Immunol. 1997;159:1604–1612. [PubMed] [Google Scholar]
- Turnbull AW, Rivier CL. Regulation of the hypothalamic–pituitary–adrenal axis cytokines: actions and mechanisms of action. Physiol Rev. 1999;79:1–71. doi: 10.1152/physrev.1999.79.1.1. [DOI] [PubMed] [Google Scholar]
- Lee YB, Nagai A, Kim SU. Cytokines, chemokines, and cytokine receptors in human microglia. J Neurosci Res. 2002;69:94–103. doi: 10.1002/jnr.10253. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Breve JJ, Wierinckx A, van der Jagt S, Bristow AF, Leyson JE, Tilders FJ, Van Dam AM. Expression and regulation of interleukin-10 and interleukin-10 receptor in rat astroglial and microglial cells. Eur J Neurosci. 2002;16:1175–1185. doi: 10.1046/j.1460-9568.2002.02200.x. [DOI] [PubMed] [Google Scholar]
- Konsman JP, Kelley K, Dantzer R. Temporal and spatial relationships between lipopolysaccharide-induced expression of Fos, interleukin-1 beta and inducible nitric oxide synthase in rat brain. Neuroscience. 1999;89:535–548. doi: 10.1016/S0306-4522(98)00368-6. [DOI] [PubMed] [Google Scholar]
- Quan N, Stern EL, Whiteside MB, Herkenham M. Induction of proinflammatory cytokine mRNAs in the brain after peripheral injection of subseptic doses of lipopolysaccharide in the rat. J Neuroimmunol. 1999;93:72–80. doi: 10.1016/S0165-5728(98)00193-3. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Hwang PM, Snyder SH. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature. 1990;347:768–770. doi: 10.1038/347768a0. [DOI] [PubMed] [Google Scholar]
- Wong ML, Bongiorno PB, Rettori V, McCann SM, Licinio J. Interleukin (IL)-1β, IL-1 receptor antagonist, IL-10, and IL-13 gene expression in the central nervous system during systemic inflammation: pathophysiological implications. Proc Natl Acad Sci USA. 1997;94:227–232. doi: 10.1073/pnas.94.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyen JR, Ye S, Finck BN, Johnson RW. Interleukin(IL)-10 inhibits IL-6 production in microglia by preventing activation of NF-kappaB. Brain Res Mol Brain Res. 2000;77:138–147. doi: 10.1016/S0169-328X(00)00042-5. [DOI] [PubMed] [Google Scholar]
- Fontana A, Kristensen F, Dubs R, Gemsa D, Weber E. Production of prostaglandin E and an interleukin-1 like factor by cultured astrocytes and C6 glioma cells. J Immunol. 1982;129:2413–2419. [PubMed] [Google Scholar]
- Caggiano AO, Kraig RP. Prostaglandin E receptor subtypes in cultured rat microglia and their role in reducing lipopolysaccharide-induced interleukin-1beta production. J Neurochem. 1999;72:565–575. doi: 10.1046/j.1471-4159.1999.0720565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmquist JK, Scammell TE, Saper CB. Mechansims of CNS resposne to systemic immune challenge: the febrile response. Trends Neurosci. 1997;20:565–570. doi: 10.1016/S0166-2236(97)01138-7. [DOI] [PubMed] [Google Scholar]
- Kadoi Y, Saito S. An alteration in the gamma-aminobutyric acid receptor system in experimentally induced septic shock in rats. Crit Care Med. 1996;24:298–305. doi: 10.1097/00003246-199602000-00020. [DOI] [PubMed] [Google Scholar]
- Kadoi Y, Saito S, Kunimoto F, Imai T, Fujita T. Impairment of the brain beta-adrenergic system during experimental endotoxemia. J Surg Res. 1996;61:496–502. doi: 10.1006/jsre.1996.0153. [DOI] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci USA. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn AJ. Endotoxin-induced activation of cerebral catecholamine and serotonin metabolism: comparison with interleukin-1. J Pharmacol Exp Ther. 1992;261:964–969. [PubMed] [Google Scholar]
- Dunn AJ. Cytokine activation of the HPA axis. Ann N Y Acad Sci. 2000;917:608–617. doi: 10.1111/j.1749-6632.2000.tb05426.x. [DOI] [PubMed] [Google Scholar]
- Matsuoko Y, Furuyashiki T, Bito H, Ushikubi F, Tanaka Y, Kobayashi T, Muro S, Satoh N, Kayahara T, Higashi M, et al. Impaired adrenocorticotropic hormone response to bacterial endotoxin in mice deficient in prostaglandin E receptor EP1 and EP3 subtypes. Proc Natl Acad Sci USA. 2003;100:4132–4137. doi: 10.1073/pnas.0633341100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Forstermann U. Nitric oxide in the pathogenesis of vascular disease. J Pathol. 2000;190:244–254. doi: 10.1002/(SICI)1096-9896(200002)190:3<244::AID-PATH575>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Persson PB. Modulation of cardiovascular control mechanisms and their interaction. Physiol Rev. 1996;76:193–232. doi: 10.1152/physrev.1996.76.1.193. [DOI] [PubMed] [Google Scholar]
- Weidenfeld J, Kahbha K, Reches A, Shohami E. Role of the central adrenergic system in the regulation of prostaglandin biosynthesis in rat brain. J Neurochem. 1992;58:694–699. doi: 10.1111/j.1471-4159.1992.tb09773.x. [DOI] [PubMed] [Google Scholar]
- Garthwaite J. Glutamate, nitric oxide and cell-cell signalling in the nervous system. Trends Neurosci. 1991;14:60–67. doi: 10.1016/0166-2236(91)90022-M. [DOI] [PubMed] [Google Scholar]
- McCann SM, Kimura M, Karanth S, Yu WH, Mastronardi CA, Rettori V. The mechanism of action of cytokines to control the release of hypothalamic and pituitary hormones in infection. Ann N Y Acad Sci. 2000;917:14–18. doi: 10.1111/j.1749-6632.2000.tb05368.x. [DOI] [PubMed] [Google Scholar]
- Li Y-F, Patel KP. Paraventricular nucleus of the hypothalamus and elevated sympathetic activity in heart failuer: the altered inhibitory mechanisms. Acta Physiol Scand. 2003;177:17–26. doi: 10.1046/j.1365-201X.2003.01043.x. [DOI] [PubMed] [Google Scholar]
- Annane D, Trabold F, Sharshar T, Jarrin I, Blanc AS, Raphael JC, Gajdos P. Inappropriate sympathetic activation at onset of septic shock: a spectral analysis approach. Am J Respir Crit Care Med. 1999;160:458–465. doi: 10.1164/ajrccm.160.2.9810073. [DOI] [PubMed] [Google Scholar]
- Kramer BC, Yabut JA, Cheong J, JnoBaptiste R, Robakis T, Olanow CW, Mytilineou C. Lipopolysaccharide prevents cell death caused by glutathione depletion: possible mechanisms of protection. Neuroscience. 2002;114:361–372. doi: 10.1016/S0306-4522(02)00310-X. [DOI] [PubMed] [Google Scholar]
- Lohrer P, Gloddek J, Carbia Nagashima A, Korali Z, Hopfner U, Paez Pereda M, Arzt E, Stalla GK, Renner U. Lipopolysaccharide directly stimulates the intrapituitary interleukin-6 production by folliculostellate cells via specific receptors and the p38a mitogen-activated protein kinase/NF-kB pathways. Endocrinology. 2000;141:4457–4465. doi: 10.1210/en.141.12.4457. [DOI] [PubMed] [Google Scholar]
- Zhan RZ, Fujiwara N, Shimoji K. Regionally different elevation of intracellular free calcium in hippocampus of septic rat brain. Shock. 1996;6:293–297. doi: 10.1097/00024382-199610000-00012. [DOI] [PubMed] [Google Scholar]
- Wang T, Qin L, Liu B, Liu Y, Wilson B, Eling TE, Langenbach R, Taniura S, Hong JS. Role of reactive oxygen species in LPS-induced production of prostaglandin E2 in microglia. J Neurochem. 2004;88:939–947. doi: 10.1046/j.1471-4159.2003.02232.x. [DOI] [PubMed] [Google Scholar]
- Feinstein DL, Galea E, Aquino DA, Li GC, Xu H, Reis DJ. Heat shock protein 70 suppress astroglial-inducible nitric-oxide synthase expression by decreasing NFkappaB activation. J Biol Chem. 1996;271:17724–17732. doi: 10.1074/jbc.271.46.29489. [DOI] [PubMed] [Google Scholar]
- Brealey D, Brand M, Hargreaves I, Heales S, Land J, Smolenski R, Davies NA, Cooper CE, Singer M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360:219–223. doi: 10.1016/S0140-6736(02)09459-X. [DOI] [PubMed] [Google Scholar]
- Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- Lynch AM, Walsh C, Delaney A, Nolan Y, Campbell VA, Lynch MA. Lipopolysaccharide-induced increase in signalling hippocampus is abrogated by IL-10: a role for IL-1beta. J Neurochem. 2004;88:635–646. doi: 10.1046/j.1471-4159.2003.02157.x. [DOI] [PubMed] [Google Scholar]
- Lee J, Hur J, Lee P, Kim JY, Cho N, Kim SY, Kim H, Lee MS, Suk K. Dual role of inflammatory stimuli in activation-induced cell death of mouse microglial cells. Initiation of two separate apoptotic pathways via induction of interferon regulatory factor-1 and caspase-11. J Biol Chem. 2001;276:32956–32965. doi: 10.1074/jbc.M104700200. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Loschmann PA, Gleichmann M, Weller M, Schulz JB, Wullner U, Klockgether T. Induction of nitric oxide synthase and nitric oxide mediated apoptosis in neural PC12 cells after stimulation with tumor necrosis factor-alpha/lipopolysaccharide. J Neurochem. 1998;71:88–94. doi: 10.1046/j.1471-4159.1998.71010088.x. [DOI] [PubMed] [Google Scholar]
- Lee P, Lee J, Kim S, Lee MS, Yagita H, Kim SY, Kim H, Suk K. NO as an autocrine mediator in the apoptosis of activated microglial cells: correlation between activation and apoptosis of microglial cells. Brain Res. 2001;892:380–385. doi: 10.1016/S0006-8993(00)03257-1. [DOI] [PubMed] [Google Scholar]
- Lehnardt S, Massilllon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, Volpe JJ, Vartanian T. Activation of innate immunity in the CNS triggers neurodegeneration through a toll-like receptor 4-dependent pathway. Proc Natl Acad Sci USA. 2003;100:8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata H, Katsuki H, Nishikawa M, Kume T, Kaneko S, Akaike A. Lipopolysaccharide-induced dopaminergic cell death in rat midbrain slice cultures: role of inducible nitric oxide synthase and protection by indomethacin. J Neurochem. 2003;86:1201–1212. doi: 10.1046/j.1471-4159.2003.01929.x. [DOI] [PubMed] [Google Scholar]
- Monje ML, Toda H, Palmer TD. Inflammatory blockad restores adult hippocampal neurogenesis. Science. 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- Young GB, Bolton CF, Austin TW, Archibald YM, Gonder J, Wells GA. The encephalopathy associated with septic illness. Clin Invest Med. 1990;13:297–304. [PubMed] [Google Scholar]
- Sprung CL, Peduzzi PN, Shatney CH, Schein RMH, Wilson MF, Sheagren JN, Hinshaw LB, The Veterans Administration Systemic Sepsis Cooperative Study Group Impact of encephalopathy on mortality of sepsis. Crit Care Med. 1990;18:801–806. doi: 10.1097/00003246-199008000-00001. [DOI] [PubMed] [Google Scholar]
- Eidelman LA, Putterman D, Putterman C, Sprung CL. The spectrum of septic encephalopathy. Definitions, etiologies, and mortalities. JAMA. 1996;275:470–473. doi: 10.1001/jama.275.6.470. [DOI] [PubMed] [Google Scholar]
- Straver JS, Keunen RW, Stam CJ, Tavy DL, de Ruiter GR, Smith SJ, Thijs LG, Schellens RG, Gielen G. Nonlinear analysis of EEG in septic encephalopathy. Neurol Res. 1998;20:100–106. doi: 10.1080/01616412.1998.11740490. [DOI] [PubMed] [Google Scholar]
- Young GB, Bolton CF, Archibald YM, Austin TW, Wells GA. The electroencephalogram in sepsis-associated encephalopathy. J Clin Neurophysiol. 1992;9:145–152. doi: 10.1097/00004691-199201000-00016. [DOI] [PubMed] [Google Scholar]
- Zauner C, Gendo A, Kramer L, Kranz A, Grimm G, Madl C. Metabolic encephalopathy in critically ill patients suffering from septic or nonseptic multiple organ failure. Crit Care Med. 2000;28:1310–1315. doi: 10.1097/00003246-200005000-00009. [DOI] [PubMed] [Google Scholar]
- Zauner C, Gendo A, Kramer L, Funk GC, Bauer E, Schenk P, Ratheipser K, Madl C. Impaired subcortical and cortical sensory evoked potential pathways in septic patients. Crit Care Med. 2002;30:1136–1139. doi: 10.1097/00003246-200205000-00030. [DOI] [PubMed] [Google Scholar]
- Freund HR, Muggia-Sullam M, Peiser J, Melamed E. Brain neurotransmitter profile is deranged during sepsis and septic encephalopathy in the rat. J Surg Res. 1985;38:267–271. doi: 10.1016/0022-4804(85)90037-X. [DOI] [PubMed] [Google Scholar]
- Freund HR, Muggia-Sullam M, LaFrance R, Holroyde J, Fischer JE. Regional brain amino acid and neurotransmitter derangements during abdominal sepsis and septic encephalopathy in the rat. The effect of amino acid infusion. Arch Surg. 1986;121:209–216. doi: 10.1001/archsurg.1986.01400020095011. [DOI] [PubMed] [Google Scholar]
- Winder TR, Minuk GY, Sargeant EJ, Seland TP. γ-Aminobutyric acid (GABA) and sepsis-related encephalopathy. Can J Neurol Sci. 1988;15:23–25. doi: 10.1017/s0317167100027128. [DOI] [PubMed] [Google Scholar]
- Sprung CL, Cerra FB, Freund HR, Schein RM, Konstantinides FN, Marcial EH, Pena M. Amino acid alterations and encephalopathy in the sepsis syndrome. Crit Care Med. 1991;19:753–757. doi: 10.1097/00003246-199106000-00004. [DOI] [PubMed] [Google Scholar]
- Weigand MA, Volkmann M, Scmidt H, Martin E, Bohrer H, Bardenheuer HU. Neuron-specific enolase as a marker of fatal outcome in patients with severe sepsis or septic shock. Anesthesiology. 2000;92:905–907. doi: 10.1097/00000542-200003000-00057. [DOI] [PubMed] [Google Scholar]
- Annane D, Sebille V, Troche G, Raphael JC, Gajdos P, Bellissant E. A 3-level prognostic classification in septic shock based on cortisol levels and cortisol response to corticotropin. JAMA. 2000;283:1038–1045. doi: 10.1001/jama.283.8.1038. [DOI] [PubMed] [Google Scholar]
- Annane D, Bellissant E, Sebille V, Lesieur O, Mathieu B, Raphael JC, Gajdos P. Impaired pressor sensitivity to noradrenaline in septic shock patients with and without impaired adrenal function reserve. Br J Clin Pharmacol. 1998;46:589–597. doi: 10.1046/j.1365-2125.1998.00833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annane D, Sebille V, Charpentier C, Bollaert PE, Francois B, Korach JM, Capellier G, Cohen Y, Azoulay E, Troche G, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002;288:862–871. doi: 10.1001/jama.288.7.862. [DOI] [PubMed] [Google Scholar]
- Landry DW, Levin HR, Gallant EM, Ashton RC, Seo S, D'alessandro D, Oz MC, Oliver JA. Vasopressin pressor hypersensitivity in vasodilatory septic shock. Crit Care Med. 1997;25:1279–1282. doi: 10.1097/00003246-199708000-00012. [DOI] [PubMed] [Google Scholar]
- Mutlu GM, Factor P. Role of vasopressin in the management of septic shock. Int Care Med. 2004;30:1276–1291. doi: 10.1007/s00134-004-2283-8. [DOI] [PubMed] [Google Scholar]
- Russell JA. Vasopressin in septic shock: clinical equipoise mandates a time for restraint. Crit Care Med. 2003;31:2707–2709. doi: 10.1097/01.CCM.0000092458.16716.EE. [DOI] [PubMed] [Google Scholar]
- Landry DW, Levin HR, Gallant EM, Ashton RC, Seo S, D'alessandro D, Oz MC, Oliver JA. Vasopressin deficiency contributes to the vasodilation of septic shock. Circulation. 1997;95:1122–1125. doi: 10.1161/01.cir.95.5.1122. [DOI] [PubMed] [Google Scholar]
- Wilson MF, Brackett DJ, Hinshaw LB, Tompkins P, Archer LT, Benjamin BA. Vasopressin release during sepsis and septic shock in baboons and dogs. Surg Gyn Obstet. 1981;153:869–872. [PubMed] [Google Scholar]
- Brackett DJ, Schaeffer CF, Tompkins P, Fagraeus L, Peters LJ, Wilson MF. Evaluation of cardiac output, total peripheral vascular resistance, and plasma concentrations of vasopressin in the conscious, unrestrained rat during endotoxemia. Circ Res. 1985;17:273–284. [PubMed] [Google Scholar]
- Sharshar T, Blanchard A, Paillard M, Raphael JC, Gajdos P, Annane D. Circulating vasopressin in septic shock. Crit Care Med. 2003;31:1752–1758. doi: 10.1097/01.CCM.0000063046.82359.4A. [DOI] [PubMed] [Google Scholar]
- Giusti-Paiva A, De Castro M, Antunes-Rodrigues J, Carnio EC. Inducible nitric oxide synthase pathway in the central nervous system and vasopressin release during experimental septic shock. Crit Care Med. 2002;30:1306–1310. doi: 10.1097/00003246-200206000-00025. [DOI] [PubMed] [Google Scholar]
- Kadekaro M, Liu H, Terrell ML, Gestl S, Bui V, Summy-Long JY. Role of NO on vasopressin and oxytocin release and blood pressure during osmotic stimulation in rats. Am J Physiol. 1997;273:R1024–R1030. doi: 10.1152/ajpregu.1997.273.3.R1024. [DOI] [PubMed] [Google Scholar]
- Landgraf R, Neumann I, Holsboer F, Pittman QJ. Interleukin-1β stimulates both central and peripheral release of vasopressin and oxytocin in the rat. Eur J Neurosci. 1995;7:592–598. doi: 10.1111/j.1460-9568.1995.tb00663.x. [DOI] [PubMed] [Google Scholar]
- Reid IA. Role of vasopressin deficiency in vasodilatation of septic shock. Crit Care Med. 1997;95:1108–1110. doi: 10.1161/01.cir.95.5.1108. [DOI] [PubMed] [Google Scholar]
- Sharshar T, Carlier R, Blanchard A, Feydy A, Gray F, Paillard M, Raphael JC, Gajdos P, Annane D. Depletion of neurohypophyseal content of vasopressin in septic shock. Crit Care Med. 2002;30:497–500. doi: 10.1097/00003246-200203000-00001. [DOI] [PubMed] [Google Scholar]
- Luk J. Role of V1 receptors in the action of vasopressin on the baroreflex control of heart rate. Am J Physiol. 1993;265:R524–R529. doi: 10.1152/ajpregu.1993.265.3.R524. [DOI] [PubMed] [Google Scholar]
- Goldstein B, Kempski MH, Stair D, Tipton RB, D DD, DeAsla R, Cox C, Lund N, Woollf PD. Autonomic modulation of heart rate variability during endotoxin shock in rabbits. Crit Care Med. 1995;23:1694–1702. doi: 10.1097/00003246-199510000-00014. [DOI] [PubMed] [Google Scholar]
- Winchell RJ, Hoyt DB. Spectral analysis of heart rate variability in the ICU: a measure of autonomic function. J Surg Res. 1996;63:11–16. doi: 10.1006/jsre.1996.0214. [DOI] [PubMed] [Google Scholar]
- Korach M, Sharshar T, Jarrin I, Fouillot JP, Raphael JC, Gajdos P, Annane D. Cardiac variability in critically ill adults: influence of sepsis. Crit Care Med. 2001;29:1380–1385. doi: 10.1097/00003246-200107000-00013. [DOI] [PubMed] [Google Scholar]