

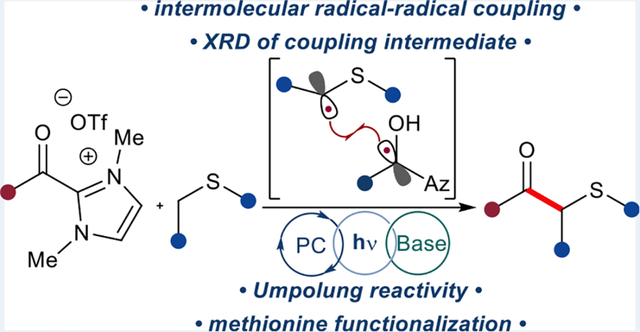
Abstract
α-Heteroatom functionalization is a key strategy for C–C bond formation in organic synthesis, as exemplified by the addition of a nucleophile to electrophilic functional groups, such as iminium ions; oxocarbenium ions; and their sulfur analogues, sulfenium ions. We envisioned a photoredox-enabled radical Pummerer-type reaction realized through the single-electron oxidation of a sulfide. Following this oxidative event, α-deprotonation would afford α-thio radicals that participate in radical–radical coupling reactions with azolium-bound ketyl radicals, thereby accessing a commonly proposed mechanistic intermediate of the radical–radical coupling en route to functionalized additive Pummerer products. This system provides a complementary synthetic approach to highly functionalized sulfurous products, including modification of methionine residues in peptides, and beckons further exploration in C–C bond formations previously limited in the standard two-electron process.
Keywords: azolium, sulfide, photocatalysis, Pummerer, thioether
Graphical Abstract
The Pummerer reaction and its modifications have had significant utility in organic synthesis since their discovery.1 These reactions transform a sulfoxide bearing an α-proton into a substituted sulfide via a sulfenium intermediate.2 The general approach involves oxidation of a sulfide followed by electrophilic activation of the resultant sulfoxide for elimination into a sulfenium species (Figure 1A). These electrophilic sulfenium ions are known to accept a range of soft nucleophiles in both intra- and intermolecular addition reactions to form carbon–carbon or carbon–heteroatom bonds. These nucleophiles include enolate equivalents,3 enol silanes,4 ene-donors,5 silanes,6 electron-rich arenes,7 and turbo Grignards.8 Traditional syntheses of these β-keto sulfides include the reaction of mercaptans with α-haloketones or sulfenyl halides with enolates.9 An alternate route could employ umpolung reactivity, i.e., polarity inversion, to access an acyl anion equivalent as a carbon-based nucleophile for addition into the sulfenium (Figure 1B). However, this approach remains unreported to the best of our knowledge. In this work, we present an intermolecular radical coupling approach to this umpolung bond formation by utilizing acyl and sulfenium radical equivalents (Figure 1C).
Figure 1.
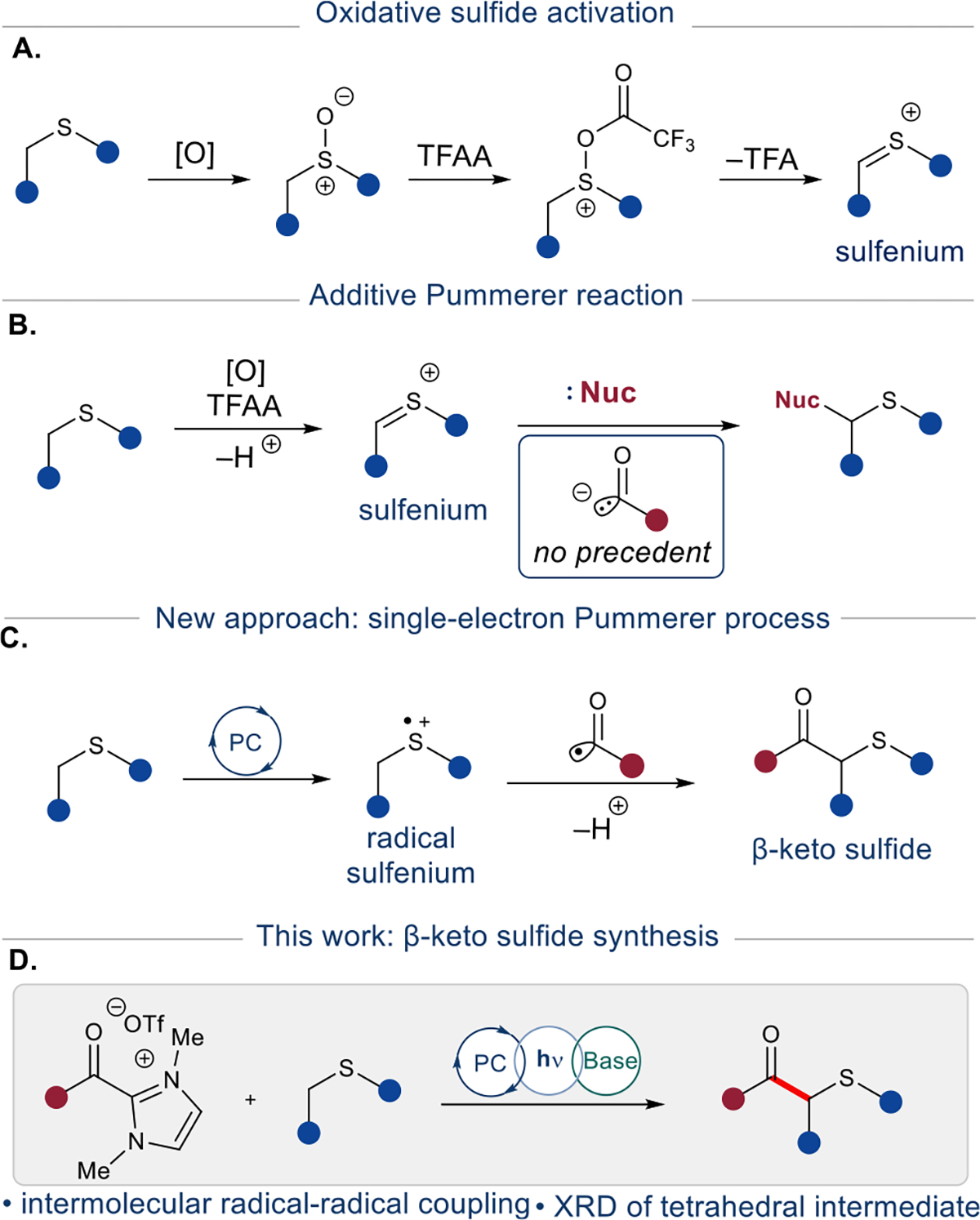
Reaction plan overview.
Recently, the rapidly expanding field of single-electron chemistry has offered new applications of oxidized sulfides. In 2020, a photoredox/weak Brønsted base dual catalytic system was reported. This method employs a photocatalyst to oxidize a sulfide to the corresponding sulfur radical cation, which is then deprotonated at the α-carbon.10 The resulting carbon-centered α-thio radical has been employed in radical conjugate addition reactions. More recently, this α-C(sp3)–H activation strategy has been employed in heteroarylation and radical coupling with isatins.11 Previous methods of α-thio radical generation include C–H abstraction, single-electron oxidation, and heterolytic cleavage of silyl, trifluoroborate, or carboxyl leaving groups.12
Inspired by this novel means of generating α-thio radicals, we envisioned a new variant of the Pummerer reaction wherein single-electron oxidation of a sulfide affords a radical sulfenium, which subsequently undergoes α-deprotonation and radical–radical coupling to yield functionalized products. Toward this end, we present the radical–radical coupling of α-thio radicals utilizing bench-stable acyl azolium reagents as acyl radical equivalents in photoredox catalysis (Figure 1D).
N-Heterocyclic carbenes, NHCs, are common acyl transfer catalysts in biological systems and have been employed in organocatalysis as umpolung operators to generate acyl anion equivalents.13 Our group initially reported the redox activity of the Breslow intermediate, an NHC–aldehyde adduct. This MnO2-enabled oxidation provides access to an acyl azolium. Facile displacement of the azolium by alcohols affords the ester product.14 Following this report, organic, catalytic transition metal, and electrochemical oxidants have been employed in the tandem oxidation process.15
In building on these recent developments, the single-electron transformation of these NHC–acyl adducts typically proceeds through oxidation of the strongly reducing Breslow intermediate or reduction of an electron-deficient acyl azolium. These connected approaches generate ketyl radicals with significant synthetic potential.16 In 2019, Ohmiya and co-workers reported the coupling of Breslow intermediate-derived ketyl- and carbon-centered radicals in the NHC-catalyzed decarboxylative alkylation of aldehydes.17 Our growing interest in SET (single-electron transfer) via photoredox catalysis motivated the development of a complementary radical–radical coupling of a reductively generated azolium radical intermediate. This unique approach involves a photocatalytic single-electron reduction of acyl azoliums to access a ketyl radical for application in radical–radical coupling reactions and, later, the adaptation to radical-relay methods.18 A unique aspect of these zwitterionic ketyl intermediates is presumably the advantageous delocalization into the aromatic azolium structure. These stabilized captodative radicals are more persistent relative to traditional acyl radicals, thus facilitating cross-coupling with transient radical coupling partners.16g,19
Recently, Wang and co-workers employed this ketyl radical in the coupling with anilines to form α-amino ketones.20 Expansion of the acyl azolium–photoredox system to the single-electron Pummerer process enables the synthesis of β-keto sulfides 3a–ae. Taking into account the differences in redox potentials between nitrogen and sulfur atoms, Wang and co-workers’ findings and our previous reports on acyl azolium chemistries presented a unique opportunity. Thioethers, in combination with the higher oxidation states of S(IV) (sulfoxides) and S(VI) (sulfones, sulfoximines, and others) are structurally integral to pharmaceuticals, natural products, materials, and agrochemicals.21 This system offers the coupling of α-heteroatom carbon-centered radicals, including the aliphatic peptide methionine, with azolium-derived ketyl radicals, as well as structural confirmation of a commonly proposed mechanistic intermediate 4d.
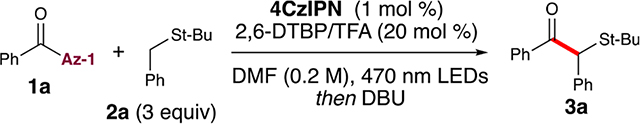
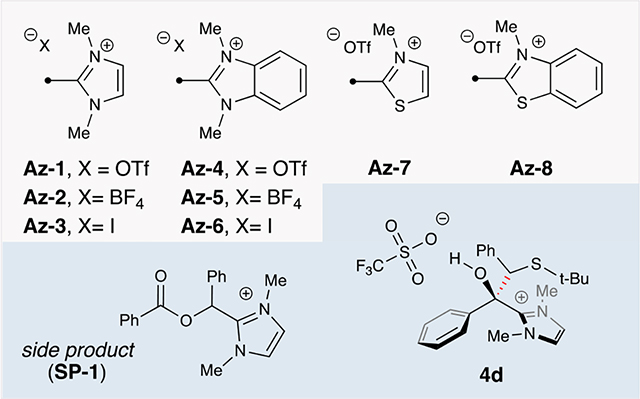
Our rational approach to develop a set of generalized conditions was guided by a broad screen of acyl azolium species and various reaction conditions (Table 1). Variables considered for optimization included acyl azoliums, sulfides, Brønsted bases, photocatalysts, and solvents. As part of this work, various isolated azoliums recently developed for cross-coupling were evaluated in the reaction with thioanisole. While we initially investigated conditions that were catalytic in N-heterocyclic carbene (results not shown), those reactions provided low yields, most likely because of low turnover numbers. Consequently, we turned quickly to using acyl azolium species as reagents given the ease of their preparation and storage. Interestingly, while the electron-rich thiazoliums (Az-7 and Az-8) and the highly conjugated benzimidazoliums (Az-5 to Az-6) all failed to yield the product 3a, the imidazolium Az-1 was the only species that resulted in isolable product in the qualitative LC-MS screen. Also, the triflate Az-4 provided a mass corresponding to trace product. This stands in contrast to previous work reported with Az-3 and Az-6 and suggests that iodide is incompatible with benzyl and aryl sulfide substrates under photocatalytic conditions, or that the triflate may participate in the reaction.18a Further development utilized Az-1 as the standard azolium for the screening of 13 photocatalysts, 6 Brønsted bases, and 6 Lewis acids (Supporting Information (SI), S5). A review of the preliminary screening established the photocatalyst 4CzIPN [2,4,5,6-Tetrakis(9H-carbazol-9-yl) isophthalonitrile] and a weak base (NaCO2CF3) as the optimal platform.10 Additionally, Lewis acids were determined to be deleterious. In our initial foray into this chemistry, GC-MS yields surprisingly failed to translate to isolated yields. Further investigations by LC-MS analysis revealed a mass corresponding to two potential intermediates. We hypothesized that heating transformed the intermediates to the desired ketone product during analysis. Thus, we performed a brief screen of workup conditions following the photochemical reaction and determined that addition of DBU [1,8-diazabicyclo(5.4.0)undec-7-ene] led to the disappearance of one intermediate (see 4d) and produced comparable yields with the high-throughput GC-MS screen.
Table 1.
Optimization of Reaction Conditions
| ||
|---|---|---|
| entry | deviation from standard | yield (%) |
| 1 | none | 63 |
| 2 | DCM instead of DMF | 47 |
| 3 | NaCO2CF3, DCM | 37 |
| 4 | NaCO2CF3, DCM, sulfide (1 equiv) | 28 |
| 5 | no TFA | 47 |
| 6 | no 2,6-di-tert-butylpyridine | 49 |
| 7 | no 2,6-di-tert-butylpyridine/TFA | 32 |
| 8 | no 4CzIPN | 0 |
| 9 | no light | 0 |
| 10 | no 4CzIPN/370 nm | 14 |
| ||
Formal optimization of the reaction was performed with azolium 1a and the sulfide 2a to afford the coupled product 3a. When the reaction was conducted with NaCO2CF3, a yield of 28% was observed (Table 1, entry 4). This improved further by increasing the equivalency of the sulfide 2a, which achieved a 37% yield (Table 1, entry 3). However, undesirable byproducts persisted in the reaction mixture. The selection of base proved crucial to the success of this radical coupling process. For example, the absence of a Brønsted acid–base pair resulted in a 32% yield (Table 1, entry 7), thereby illustrating its key role in generating the α-thio radical 4b for the overall transformation. Previous kinetic isotope effect (KIE) studies of photo-activatived sulfides by Alfonzo and Hande suggest that deprotonation is the rate-limiting step in the formation of α-thio radicals.11b Informed by this work, TFA in combination with 2,6-di-tert-butylpyridine (2,6-DTBP) enabled in situ generation of the weak base trifluoroacetate (Table 1, entry 2), which provided a 47% yield. The significant improvement may be attributed to a proton shuttle process, thus enabling rapid protonation of the ketyl radical anion and mitigation of undesired azolium side products. During the LC-MS analysis, a byproduct (SP-1) was detected in the reaction. Its structure was determined to result from either transesterification of the ketyl radical anion or over-reduction to the secondary alcohol on the basis of analysis of MS data and NMR spectroscopy. Considering the recognized reactivity of acyl azoliums as acylating agents in the presence of alcohols, the presence of this byproduct was expected. However, it remains crucial to inhibit this pathway to ensure the desired outcome of the reaction. This observation also supports a possible proton-coupled electron transfer (PCET) or proton-mediated mechanism with the conjugate acid of 2,6-DTBP.22 A brief solvent screen, indeed, demonstrated preference for high dielectric solvents: using DMF instead of DCM (Table 1, entry 1) resulted in intermediate 4d, which was transformed to the product 3a in a satisfactory yield of 63%. Independent yields for 2,6-di-tert-butylpyridine (Table 1, entry 5) or TFA (Table 1, entry 6) were 47% and 49%, respectively.
With these optimized conditions, we surveyed of the scope of acyl azoliums amenable to radical–radical cross coupling (Table 2). A range of electron-withdrawing and electron-donating aromatic substituents were tolerated on the acyl subunit of the dimethyl imidazolium. Naphthyl ketone 3b was isolated in high yield, which suggested the importance of additional resonance stabilization in radical persistence. Sterically encumbered substrate 3c and the biphenyl substrate 3d also achieved good yields. Notably, the heteroaromatic acyl furan product 3e could be obtained in 16% yield. Halogenated meta- and para-substituted acyl azoliums gave products 3f–j in good yield. Electron-rich alkoxy-substituted substrate 3k demonstrated a slower rate of reaction but ultimately a higher overall yield after 120 h. The strongly electron-donating 3,4-methylenedioxy substrate 31 proceeded very slowly and stalled at a 26% overall yield. Also, the nitrile 3m was successfully isolated, albeit in reduced yields. Additionally, an azido-functionalized product 3n was prepared in moderate yield, thereby offering a functional handle for application in click reactions. Gratifyingly, the aliphatic substrates 3o, 3p, and 3q could be obtained in up to 34% yield, despite the challenge posed by aliphatic substrates in previous reports.16d,18b,c
Table 2.
β-Keto Sulfide Scopec
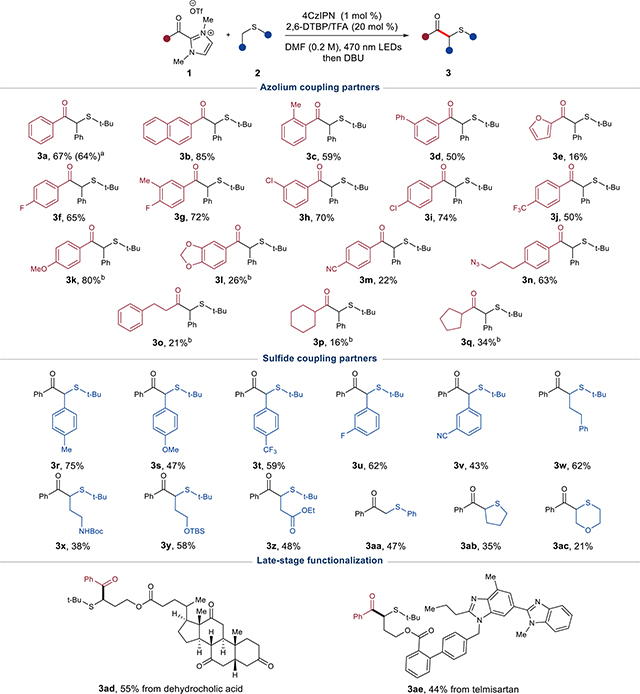
|
Gram-scale preparation (1.034 g, 64%) utilizing 456 nm Kessil lamp.
Extended reaction time of 120 h.
Standard reaction conditions: azolium (1–3 equiv), sulfide (1–3 equiv), 2,6-ditertbutyl pyridine (20 mol %), TFA (20 mol %), 4CzIPN (1 mol %), DMF (0.2 M), 470 nm, 18–24 h, then DBU (1 equiv).
A variety of benzylic sulfides were initially examined to investigate the alkyl sulfide scope in the coupling reaction. A methyl-substituted sulfide was well tolerated and provided product 3r with improved yields. However, stronger electron-donating groups, such as methoxy, led to moderate yields (see ketone 3s). Electron-withdrawing substituents at the meta and para positions provided ketones 3t and 3u in yields consistent with the unsubstituted benzyl tert-butyl sulfide. Product 3v bearing a meta-substituted nitrile was accessed in moderate yield, thereby offering a functional handle for further diversification. In general, secondary sulfides with a variety of functional groups performed well in the reaction. Ketones derived from an alkyl sulfide 3w, carbamate-protected amine 3x, silyl-protected alcohol 3y, and ethyl ester 3z were isolated in moderate yields. Additionally, coupling of the arene-stabilized sulfide thioanisole to yield product 3aa proceeded under these conditions. Products 3ab and 3ac containing sulfur heterocycles were isolated in reduced yields. Late-stage functionalization was performed on esterified derivatives of the pharmaceutical drugs dehydrocholic acid and telmisartan to afford 3ad and 3ae.
In line with our optimization, we present a mechanism dependent on the photocatalyst (Table 1, entry 8) and light (Table 1, entry 9). To determine the role of the photocatalyst, Stern–Volmer quenching studies revealed fluorescence quenching of 4CzIPN* [E1/2 (4CzIPN•+/4CzIPN*) (V) = −1.18 V] by the acyl azolium 1a (−1.13 V, see SI, S163).18a,23 This leads to the proposal of an oxidative quenching cycle, whereby the excitation by visible light affords the excited-state photocatalyst 4CzIPN*, and proceeds by the reduction of azolium 1a to generate the oxidized photocatalyst 4CzIPN•+ and the π-stabilized azolium radical intermediate 4c, a persistent radical species.24 The photocatalyst is turned over [E1/2 (4CzIPN•+/4CzIPN) (V) = 1.49 V] via oxidation of the corresponding dialkyl sulfide 2a, a class of sulfides with known reduction potentials of 1.06–1.50 V (Figure 2A).25 The resulting sulfur radical cation 4a is then deprotonated by trifluoroacetate at the α-position to afford the α-thio radical 4b.
Figure 2.
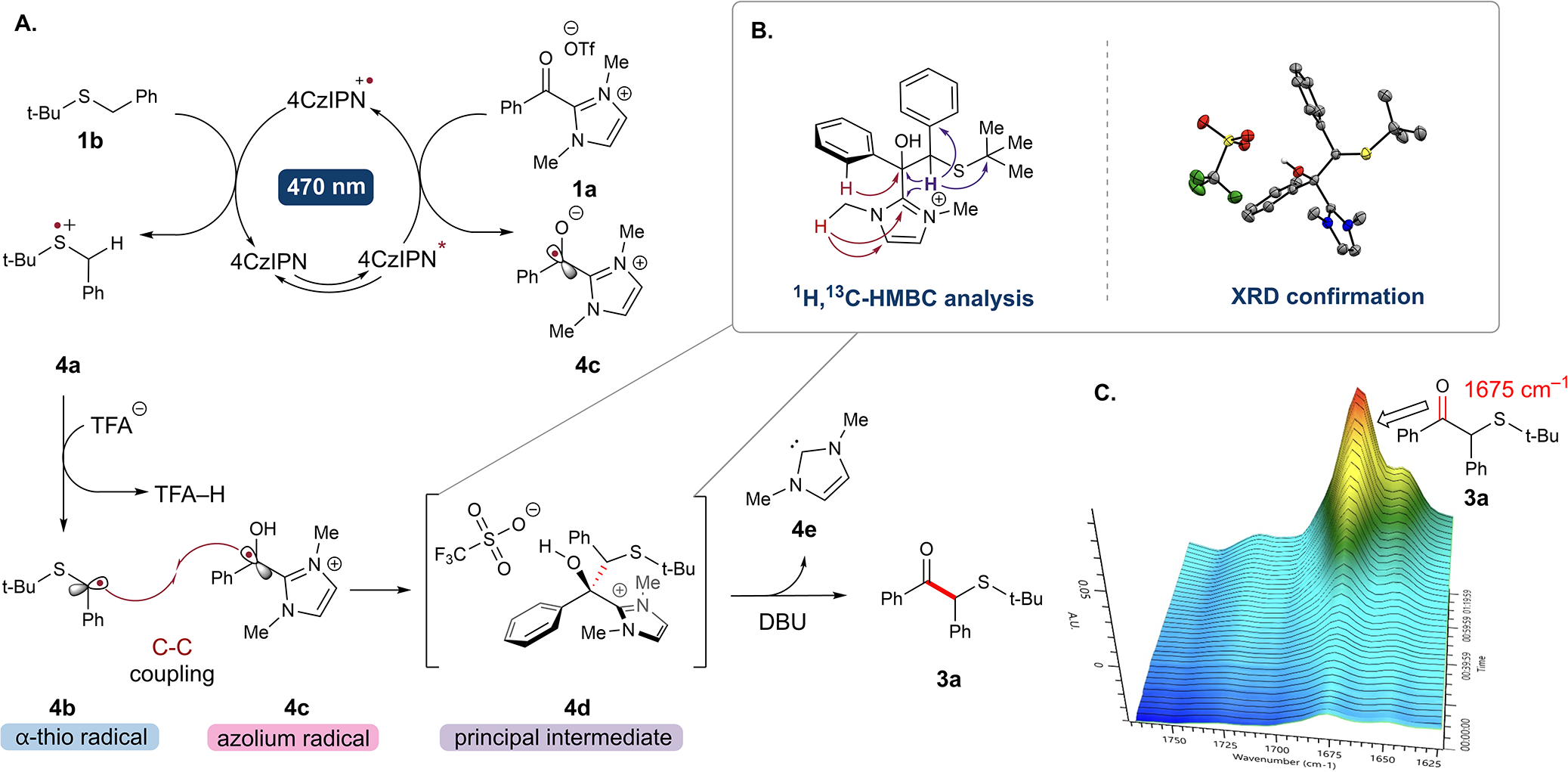
(A) Proposed photoredox mechanism for the coupling of acyl azolium 1a and sulfide 2a. (B) Intermediate 4d characterization. (C) In situ infrared spectroscopy displaying the transformation of intermediate 4d to the product 3a and monitored formation of the respective ketone peak 1675 cm−1. Reaction conditions: DBU (0.2 equiv), MeCN (0.05 M), −20 °C, 1.2 h.
We propose that the radical–radical cross-coupling between the α-thio radical 4b and the azolium radical 4c proceeds via a mechanism analogous with previously reported DFT calculations.18d,26 The cross coupled product under mildly acidic reaction conditions results in a tetrahedral tertiary alcohol intermediate 4d that was characterized by single-crystal X-ray diffraction (Figure 2B). The transformation of this principal intermediate to the product in the presence of DBU was monitored by in situ infrared spectroscopy (Figure 2C), thereby liberating the neutral azolium 4e and the desired β-keto sulfide product 3a.
In light of our recent research on utilizing the triplet states of acyl azoliums in hydrogen atom transfer (HAT) and radical recombination reactions, we conducted an experiment involving a photocatalyst-free reaction at 370 nm (Table 1, entry 10).27 The resulting yield was 14%. Mass spectrometry analysis confirmed the presence of the common intermediate 4d, which indicates that the triplet pathway shares a common route to the product. Interestingly, 4CzIPN is a known energy transfer catalyst that is particularly effective in a DMF solvent.28 Stern–Volmer experiments have demonstrated the quenching of 4CzIPN by 1a. This quenching may occur through a single electron transfer (SET) process or an energy transfer (EnT) process with a photocatalyst triplet excitation energy of (4CzIPN*, 58.3 kcal/mol).29 Both pathways are plausible and are supported by the experimentally determined reduction potential and calculated triplet excitation energy of the azolium (1a, 57.5 kcal/mol).27 To elucidate the overall process further, including the mechanism of 4CzIPN quenching investigations, the involvment of transition absorption spectroscopy would be needed, which is currently beyond the scope of this study. Regardless, the overall transformation does require light-promoted excitation and produces useful compounds.
We also sought to apply our methodology to the functionalization of the amino acid methionine and related tripeptides (Table 3). A limited number of methods have reported site-selective modification at methionine via sulfonium and sulfinimine intermediates.30 The continued development of direct bioconjugation methods targeting methionine residues could have significant utility in chemistry and the biological sciences.30a,b Motivated by these opportunities, the MacMillan group and others have applied photocatalytic α-thio radical generation to methionine in Giese-type coupling reactions but have not engaged the corresponding radicals in radical–radical coupling reactions.31 We were able to apply our method to the acylation of N-Boc-methyl methionate 5a in 61% yield with 2:3 (1°/2°) regioselectivity. Building upon these results, tripeptides bearing a C-terminal and internal methionine residue were successfully functionalized, thereby providing ketones 5b and 5c in 53% and 57% yields, respectively.30
Table 3.
Methionine Functionalizationa
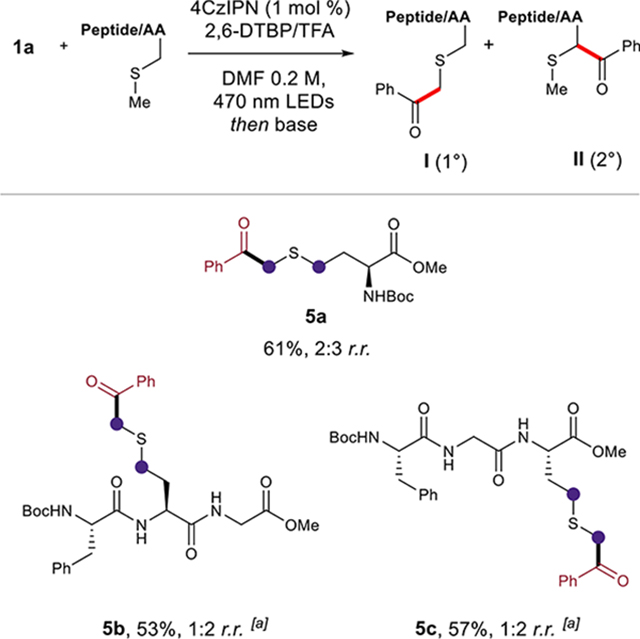
|
Regioselectivity ratio (r.r.) reported as (1°/2°).
Substrates containing tert-butyl sulfide moieties avoid the complications of regioselectivity in this transformation and offer further utility through functional group interconversion. For example, the tert-butyl sulfide undergoes a facile Lewis acid-mediated conversion to the thioester product 6a in 89% yield (Scheme 1). The thioester protecting group has been shown to provide the free thiol under mild conditions in the presence of various functional groups while mitigating oxidation to the undesired disulfides.32 These β-keto sulfide products 3 are also useful precursors in the synthesis of a known class of ligands.33 Toward this end, a zinc borohydride reduction of the standard substrate 3a afforded the respective 1,2-thio alcohols 6b and 6c in 13:1 d.r. and excellent yield. The corresponding alcohol 6c underwent further transformation to afford the phthalimide 6d and the benzyl ester 6e.
Scheme 1.
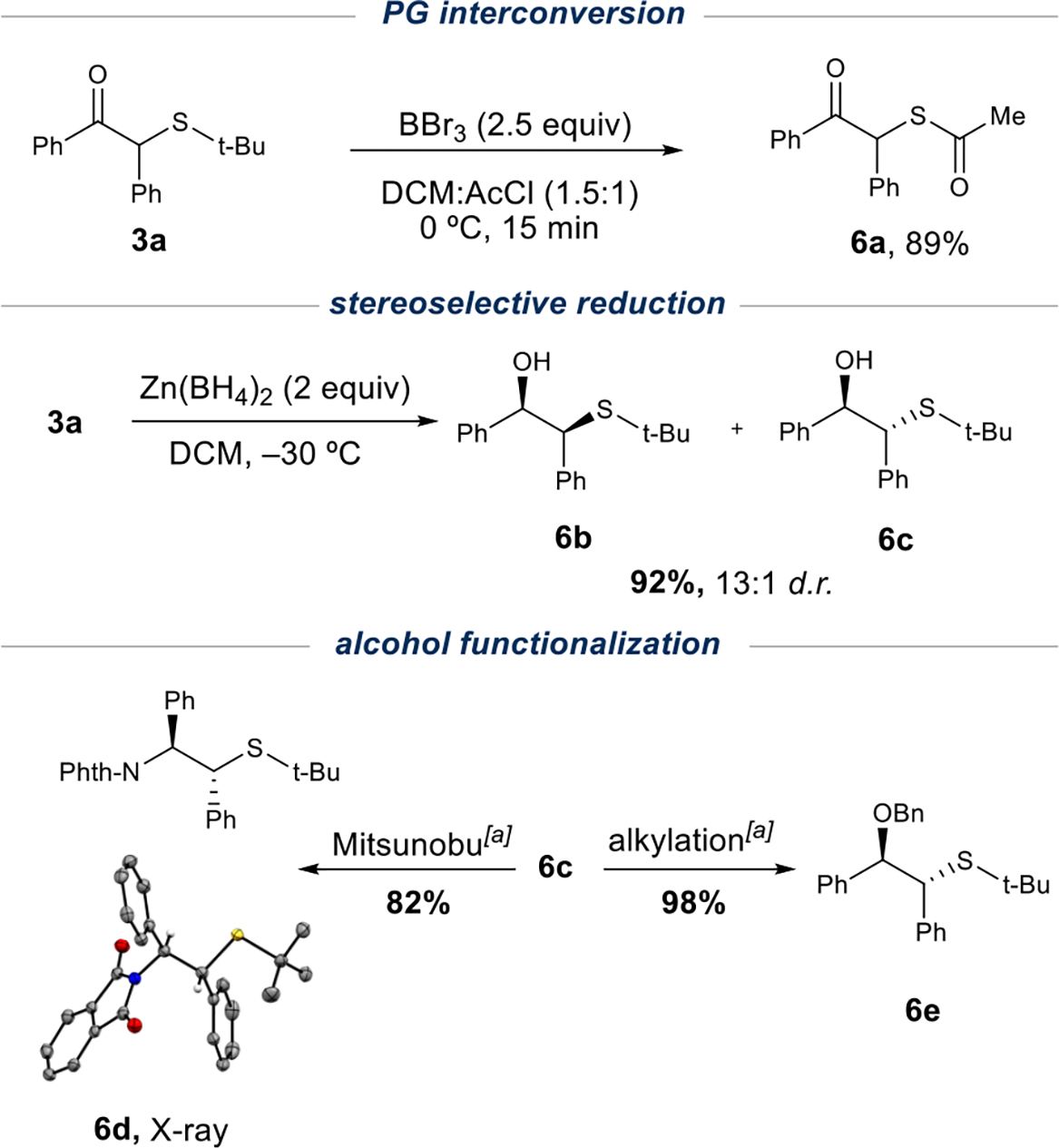
Application of Standard Substratea
aSee SI for details.
The work herein details the development of a convergent and intermolecular photoredox-catalyzed coupling of sulfides with acyl azoliums to access β-keto sulfides. This advancement in single-electron acyl azolium chemistry offers a mild umpolung strategy for α-heteroatom functionalization. Notably, the azolium component allows for facile access to ketyl radicals that then couple effectively with α-thio radicals generated in situ. The resulting tetrahedral intermediates after coupling were observed and characterized by NMR spectroscopy and single-crystal X-ray analysis. This method enables access to a wide variety of β-keto sulfides with applications in small molecule synthesis, late-stage functionalization, and methionine conjugation. Ongoing investigations include the application of this technology to larger biomolecules in pursuit of an acyl azolium and photoredox-enabled bioconjugation platform.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dalton R. Kim (NU) and Dr. Saman Shafaie (NU) for assistance with HRMS. We also thank Charlotte Stern (NU) for assistance with X-ray crystallography and Dr. Zhiquan Lei for assistance with NMR spectroscopy. We thank Joshua L. Zhu for assistance with cyclic voltammetry. The authors thank Northwestern University (NU) and the National Institute of General Medical Sciences (R35 GM136440) for support of this work.
Footnotes
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acscatal.3c01558
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.3c01558.
Experimental details and characterization data, computational data, and crystallographic files [CCDC 2195732 (4d) and 2223444 (6d)] (PDF)
Contributor Information
Michael J. Rourke, Department of Chemistry, Northwestern University, Evanston, Illinois 60208, United States
Charles T. Wang, Department of Chemistry, Northwestern University, Evanston, Illinois 60208, United States
Cullen R. Schull, Department of Chemistry, Northwestern University, Evanston, Illinois 60208, United States
Karl A. Scheidt, Department of Chemistry, Northwestern University, Evanston, Illinois 60208, United States
REFERENCES
- (1).(a) Bur SK; Padwa A The Pummerer Reaction: Methodology and Strategy for the Synthesis of Heterocyclic Compounds. Chem. Rev. 2004, 104, 2401–2432. [DOI] [PubMed] [Google Scholar]; (b) Feldman KS Modern Pummerer-type reactions. Tetrahedron 2006, 62, 5003–5034. [Google Scholar]; (c) Akai S; Kita Y Recent Advances in Pummerer Reactions. In Sulfur-Mediated Rearrangements I; Schaumann E, Ed.; Springer: Berlin, Heidelberg, Germany, 2007; pp 35–76. [Google Scholar]
- (2).Pummerer R Über Phenyl-sulfoxyessigsäure. Chem. Ber. 1909, 42, 2282–2291. [Google Scholar]
- (3).(a) Peng B; Geerdink D; Farés C; Maulide N Chemoselective Intermolecular α-Arylation of Amides. Angew. Chem., Int. Ed. 2014, 53, 5462–5466. [DOI] [PubMed] [Google Scholar]; (b) Shang L; Chang Y; Luo F; He J-N; Huang X; Zhang L; Kong L; Li K; Peng B Redox-Neutral α-Arylation of Alkyl Nitriles with Aryl Sulfoxides: A Rapid Electrophilic Rearrangement. J. Am. Chem. Soc. 2017, 139, 4211–4217. [DOI] [PubMed] [Google Scholar]
- (4).Feldman KS; Vidulova DB; Karatjas AG Extending Pummerer Reaction Chemistry. Development of a Strategy for the Regio- and Stereoselective Oxidative Cyclization of 3-(ω-Nucleophile)-Tethered Indoles. J. Org. Chem. 2005, 70, 6429–6440. [DOI] [PubMed] [Google Scholar]
- (5).Tamura Y; Maeda H; Choi HD; Ishibashi H Ene Reaction with Pummerer Reaction Intermediates of a-(Methylsulfinyl)-acetamides: A Novel Synthesis of Pellitorine. Synthesis 1982, 1982, 56–57. [Google Scholar]
- (6).(a) Eberhart AJ; Imbriglio JE; Procter DJ Nucleophilic Ortho Allylation of Aryl and Heteroaryl Sulfoxides. Org. Lett. 2011, 13, 5882–5885. [DOI] [PubMed] [Google Scholar]; (b) Eberhart AJ; Procter DJ Nucleophilic ortho-Propargylation of Aryl Sulfoxides: An Interrupted Pummerer/Allenyl Thio-Claisen Rearrangement Sequence. Angew. Chem., Int. Ed. 2013, 52, 4008–4011. [DOI] [PubMed] [Google Scholar]
- (7).(a) Kobatake T; Fujino D; Yoshida S; Yorimitsu H; Oshima K Synthesis of 3-Trifluoromethylbenzo[b]furans from Phenols via Direct Ortho Functionalization by Extended Pummerer Reaction. J. Am. Chem. Soc. 2010, 132, 11838–11840. [DOI] [PubMed] [Google Scholar]; (b) Yanagi T; Otsuka S; Kasuga Y; Fujimoto K; Murakami K; Nogi K; Yorimitsu H; Osuka A Metal-Free Approach to Biaryls from Phenols and Aryl Sulfoxides by Temporarily Sulfur-Tethered Regioselective C–H/C–H Coupling. J. Am. Chem. Soc. 2016, 138, 14582–14585. [DOI] [PubMed] [Google Scholar]; (c) Shrives HJ; Fernández-Salas JA; Hedtke C; Pulis AP; Procter DJ Regioselective synthesis of C3 alkylated and arylated benzothio-phenes. Nat. Commun. 2017, 8, 14801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Colas K; Martín-Montero R; Mendoza A Intermolecular Pummerer Coupling with Carbon Nucleophiles in Non-Electrophilic Media. Angew. Chem., Int. Ed. 2017, 56, 16042–16046. [DOI] [PubMed] [Google Scholar]
- (9).(a) Marigo M; Bachmann S; Halland N; Braunton A; Jørgensen KA Highly Enantioselective Direct Organocatalytic α-Chlorination of Ketones. Angew. Chem., Int. Ed. 2004, 43, 5507–5510. [DOI] [PubMed] [Google Scholar]; (b) Yadav JS; Subba Reddy BV; Jain R; Baishya G N-Chlorosuccinimide as a versatile reagent for the sulfenylation of ketones: a facile synthesis of α-ketothioethers. Tetrahedron Lett. 2008, 49, 3015–3018. [Google Scholar]; (c) Younai A; Fettinger JC; Shaw JT Influence of chiral thiols on the diastereoselective synthesis of γ-lactams from cyclic anhydrides. Tetrahedron 2012, 68, 4320–4327. [Google Scholar]; (d) Kearney AM; Murphy L; Murphy CC; Eccles KS; Lawrence SE; Collins SG; Maguire AR Synthesis and reactivity of α-sulfenyl-β-chloroenones, including oxidation and Stille cross-coupling to form chalcone derivatives. Tetrahedron 2021, 88, 132091. [Google Scholar]
- (10).Alfonzo E; Hande SM Photoredox and Weak Brønsted Base Dual Catalysis: Alkylation of α-Thio Alkyl Radicals. ACS Catal. 2020, 10, 12590–12595. [Google Scholar]
- (11).(a) Tan Z; Zhu S; Liu Y; Feng X Photoinduced Chemo-, Site- and Stereoselective α-C(sp3)-H Functionalization of Sulfides. Angew. Chem., Int. Ed. 2022, 61, No. e202203374. [DOI] [PubMed] [Google Scholar]; (b) Alfonzo E; Hande SM α-Heteroarylation of Thioethers via Photoredox and Weak Brønsted Base Catalysis. Org. Lett. 2021, 23, 6115–6120. [DOI] [PubMed] [Google Scholar]
- (12).(a) Hasegawa E; Brumfield MA; Mariano PS; Yoon UC Photoadditions of ethers, thioethers, and amines to 9,10-dicyanoanthracene by electron transfer pathways. J. Org. Chem. 1988, 53, 5435–5442. [Google Scholar]; (b) Baciocchi E; Del Giacco T; Elisei F; Lapi A Sulfur Radical Cations. Kinetic and Product Study of the Photoinduced Fragmentation Reactions of (Phenylsulfanylalkyl)trimethylsilanes and Phenylsulfanylacetic Acid Radical Cations. J. Org. Chem. 2006, 71, 853–860. [DOI] [PubMed] [Google Scholar]; (c) Li Y; Miyazawa K; Koike T; Akita M Alkyl- and aryl-thioalkylation of olefins with organotrifluoroborates by photoredox catalysis. Org. Chem. Front. 2015, 2, 319–323. [Google Scholar]; (d) Le C; Liang Y; Evans RW; Li X; MacMillan DWC Selective sp(3) C-H alkylation via polarity-match-based cross-coupling. Nature 2017, 547, 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Dutta S; Erchinger JE; Schäfers F; Das A; Daniliuc CG; Glorius F Chromium/Photoredox Dual-Catalyzed Synthesis of α-Benzylic Alcohols, Isochromanones, 1,2-Oxy Alcohols and 1,2-Thio Alcohols. Angew. Chem., Int. Ed. 2022, 61, No. e202212136. [DOI] [PubMed] [Google Scholar]
- (13).(a) Patel MS; Nemeria NS; Furey W; Jordan F The pyruvate dehydrogenase complexes: structure-based function and regulation. J. Biol. Chem. 2014, 289, 16615–16623. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Flanigan DM; Romanov-Michailidis F; White NA; Rovis T Organocatalytic Reactions Enabled by N-Heterocyclic Carbenes. Chem. Rev. 2015, 115, 9307–9387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Maki BE; Scheidt KA N-Heterocyclic Carbene-Catalyzed Oxidation of Unactivated Aldehydes to Esters. Org. Lett. 2008, 10, 4331–4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).(a) Guin J; De Sarkar S; Grimme S; Studer A Biomimetic Carbene-Catalyzed Oxidations of Aldehydes Using TEMPO. Angew. Chem., Int. Ed. 2008, 47, 8727–8730. [DOI] [PubMed] [Google Scholar]; (b) Finney EE; Ogawa KA; Boydston AJ Organocatalyzed Anodic Oxidation of Aldehydes. J. Am. Chem. Soc. 2012, 134, 12374–12377. [DOI] [PubMed] [Google Scholar]; (c) Zhao J; Mück-Lichtenfeld C; Studer A. Cooperative N-Heterocyclic Carbene (NHC) and Ruthenium Redox Catalysis: Oxidative Esterification of Aldehydes with Air as the Terminal Oxidant. Adv. Synth. Catal. 2013, 355, 1098–1106. [Google Scholar]
- (16).(a) Yang W; Hu W; Dong X; Li X; Sun J N-Heterocyclic Carbene Catalyzed γ-Dihalomethylenation of Enals by Single-Electron Transfer. Angew. Chem., Int. Ed. 2016, 55, 15783–15786. [DOI] [PubMed] [Google Scholar]; (b) Liu J; Xing X-N; Huang J-H; Lu L-Q; Xiao W-J Light opens a new window for N-heterocyclic carbene catalysis. Chem. Sci. 2020, 11, 10605–10613. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mavroskoufis A; Jakob M; Hopkinson MN Light-Promoted Organocatalysis with N-Heterocyclic Carbenes. ChemPhotoChem. 2020, 4, 5147–5153. [Google Scholar]; (d) Ishii T; Nagao K; Ohmiya H Recent advances in N-heterocyclic carbene-based radical catalysis. Chem. Sci. 2020, 11, 5630–5636. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Dai L; Xu Y-Y; Xia Z-H; Ye S γ-Difluoroalkylation: Synthesis of γ-Difluoroalkyl-α,β-Unsaturated Esters via Photoredox NHC-Catalyzed Radical Reaction. Org. Lett. 2020, 22, 8173–8177. [DOI] [PubMed] [Google Scholar]; (f) Dai L; Ye S Recent advances in N-heterocyclic carbene-catalyzed radical reactions. Chin. Chem. Lett. 2021, 32, 660–667. [Google Scholar]; (g) Delfau L; Nichilo S; Molton F; Broggi J; Tomás-Mendivil E; Martin D Critical Assessment of the Reducing Ability of Breslow-type Derivatives and Implications for Carbene-Catalyzed Radical Reactions**. Angew. Chem., Int. Ed. 2021, 60, 26783–26789. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Chen K-Q; Sheng H; Liu Q; Shao P-L; Chen X-Y N-heterocyclic carbene-catalyzed radical reactions. Sci. China Chem. 2021, 64, 7–16. [Google Scholar]; (i) Bay AV; Scheidt KA Single-electron carbene catalysis in redox processes. Trends Chem. 2022, 4, 277–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ishii T; Kakeno Y; Nagao K; Ohmiya H N-Heterocyclic Carbene-Catalyzed Decarboxylative Alkylation of Aldehydes. J. Am. Chem. Soc. 2019, 141, 3854–3858. [DOI] [PubMed] [Google Scholar]
- (18).(a) Bayly AA; McDonald BR; Mrksich M; Scheidt KA High-throughput photocapture approach for reaction discovery. Proc. Natl. Acad. Sci. U.S.A 2020, 117, 13261–13266. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bay AV; Fitzpatrick KP; Betori RC; Scheidt KA Combined Photoredox and Carbene Catalysis for the Synthesis of Ketones from Carboxylic Acids. Angew. Chem., Int. Ed. 2020, 59, 9143–9148. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Meng Q-Y; Döben N; Studer A Cooperative NHC and Photoredox Catalysis for the Synthesis of β-Trifluoromethylated Alkyl Aryl Ketones. Angew. Chem., Int. Ed. 2020, 59, 19956–19960. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Bay AV; Fitzpatrick KP; González-Montiel GA; Farah AO; Cheong PH-Y; Scheidt KA Light-Driven Carbene Catalysis for the Synthesis of Aliphatic and α-Amino Ketones. Angew. Chem., Int. Ed. 2021, 60, 17925–17931. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhu JL; Scheidt KA Photocatalytic acyl azolium-promoted alkoxycarbonylation of trifluoroborates. Tetrahedron 2021, 92, 132288. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zuo Z; Daniliuc CG; Studer A Cooperative NHC/Photoredox Catalyzed Ring-Opening of Aryl Cyclopropanes to 1-Aroyloxylated-3-Acylated Alkanes. Angew. Chem., Int. Ed. 2021, 60, 25252–25257. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Meng Q-Y; Lezius L; Studer A Benzylic C-H acylation by cooperative NHC and photoredox catalysis. Nat. Commun. 2021, 12, 2068. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Liu K; Studer A Direct α-Acylation of Alkenes via N-Heterocyclic Carbene, Sulfinate, and Photoredox Cooperative Triple Catalysis. J. Am. Chem. Soc. 2021, 143, 4903–4909. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Wang P; Fitzpatrick KP; Scheidt KA Combined Photoredox and Carbene Catalysis for the Synthesis of γ-Aryloxy Ketones. Adv. Synth. Catal. 2022, 364, 518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Bay AV; Farnam EJ; Scheidt KA Synthesis of Cyclohexanones by a Tandem Photocatalyzed Annulation. J. Am. Chem. Soc. 2022, 144, 7030. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Ren S-C; Lv W-X; Yang X; Yan J-L; Xu J; Wang F-X; Hao L; Chai H; Jin Z; Chi YR Carbene-Catalyzed Alkylation of Carboxylic Esters via Direct Photoexcitation of Acyl Azolium Intermediates. ACS Catal. 2021, 11, 2925–2934. [Google Scholar]; (l) Sato Y; Goto Y; Nakamura K; Miyamoto Y; Sumida Y; Ohmiya H Light-Driven N-Heterocyclic Carbene Catalysis Using Alkylborates. ACS Catal. 2021, 11, 12886–12892. [Google Scholar]; (m) Ren S-C; Yang X; Mondal B; Mou C; Tian W; Jin Z; Chi YR Carbene and photocatalyst-catalyzed decarboxylative radical coupling of carboxylic acids and acyl imidazoles to form ketones. Nat. Commun. 2022, 13, 2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).(a) Leifert D; Studer A The Persistent Radical Effect in Organic Synthesis. Angew. Chem., Int. Ed. 2020, 59, 74–108. [DOI] [PubMed] [Google Scholar]; (b) Liu W; Vianna A; Zhang Z; Huang S; Huang L; Melaimi M; Bertrand G; Yan X Mesoionic carbene-Breslow intermediates as super electron donors: Application to the metal-free arylacylation of alkenes. Chem. Catalysis 2021, 1, 196–206. [Google Scholar]
- (20).Wang X; Zhu B; Liu Y; Wang Q Combined Photoredox and Carbene Catalysis for the Synthesis of α-Amino Ketones from Carboxylic Acids. ACS Catal. 2022, 12, 2522–2531. [Google Scholar]
- (21).(a) Boyd DA Sulfur and Its Role In Modern Materials Science. Angew. Chem., Int. Ed. 2016, 55, 15486–15502. [DOI] [PubMed] [Google Scholar]; (b) Devendar P; Yang G-F Sulfur-Containing Agrochemicals. Top. Curr. Chem. 2017, 375, 82. [DOI] [PubMed] [Google Scholar]; (c) Scott KA; Njardarson JT Analysis of US FDA-Approved Drugs Containing Sulfur Atoms. Top. Curr. Chem. 2018, 376, 5. [DOI] [PubMed] [Google Scholar]
- (22).(a) Tarantino KT; Liu P; Knowles RR Catalytic Ketyl-Olefin Cyclizations Enabled by Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2013, 135, 10022–10025. [DOI] [PubMed] [Google Scholar]; (b) Du J; Espelt LR; Guzei IA; Yoon TP Photocatalytic reductive cyclizations of enones: Divergent reactivity of photogenerated radical and radical anion intermediates. Chem. Sci. 2011, 2, 2115–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Speckmeier E; Fischer TG; Zeitler K A Toolbox Approach To Construct Broadly Applicable Metal-Free Catalysts for Photoredox Chemistry: Deliberate Tuning of Redox Potentials and Importance of Halogens in Donor–Acceptor Cyanoarenes. J. Am. Chem. Soc. 2018, 140, 15353–15365. [DOI] [PubMed] [Google Scholar]
- (24).Nakanishi I; Itoh S; Suenobu T; Inoue H; Fukuzumi S Redox Behavior of Active Aldehydes Derived from Thiamin Coenzyme Analogs. Chem. Lett. 1997, 26, 707–708. [Google Scholar]
- (25).Cottrell PT; Mann CK Electrochemical Oxidation of Aliphatic Sulfides under Nonaqueous Conditions. J. Electrochem. Soc. 1969, 116, 1499. [Google Scholar]
- (26).Jin S; Sui X; Haug GC; Nguyen VD; Dang HT; Arman HD; Larionov OV N-Heterocyclic Carbene-Photo-catalyzed Tricomponent Regioselective 1,2-Diacylation of Alkenes Illuminates the Mechanistic Details of the Electron Donor–Acceptor Complex-Mediated Radical Relay Processes. ACS Catal. 2022, 12, 285–294. [Google Scholar]
- (27).Zhu JL; Schull CR; Tam AT; Rentería-Gómez Á; Gogoi AR; Gutierrez O; Scheidt KA Photoinduced Acylations Via Azolium-Promoted Intermolecular Hydrogen Atom Transfer. J. Am. Chem. Soc. 2023, 145, 1535–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lu J; Pattengale B; Liu Q; Yang S; Shi W; Li S; Huang J; Zhang J Donor–Acceptor Fluorophores for Energy-Transfer-Mediated Photocatalysis. J. Am. Chem. Soc. 2018, 140, 13719–13725. [DOI] [PubMed] [Google Scholar]
- (29).Huang S; Zhang Q; Shiota Y; Nakagawa T; Kuwabara K; Yoshizawa K; Adachi C Computational Prediction for Singlet- and Triplet-Transition Energies of Charge-Transfer Compounds. J. Chem. Theory Comput. 2013, 9, 3872–3877. [DOI] [PubMed] [Google Scholar]
- (30).(a) Lin S; Yang X; Jia S; Weeks AM; Hornsby M; Lee PS; Nichiporuk RV; Iavarone AT; Wells JA; Toste FD; Chang CJ Redox-based reagents for chemoselective methionine bioconjugation. Science 2017, 355, 597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Taylor MT; Nelson JE; Suero MG; Gaunt MJ A protein functionalization platform based on selective reactions at methionine residues. Nature 2018, 562, 563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zang J; Chen Y; Zhu W; Lin S Chemoselective Methionine Bioconjugation on a Polypeptide, Protein, and Proteome. Biochemistry 2020, 59, 132–138. [DOI] [PubMed] [Google Scholar]
- (31).Kim J; Li BX; Huang RYC; Qiao JX; Ewing WR; MacMillan DWC Site-Selective Functionalization of Methionine Residues via Photoredox Catalysis. J. Am. Chem. Soc. 2020, 142, 21260–21266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).(a) Stuhr-Hansen N The tert -Butyl MoietyA Base Resistent Thiol Protecting Group Smoothly Replaced by the Labile Acetyl Moiety. Synth. Commun. 2003, 33, 641–646. [Google Scholar]; (b) Sørensen JK; Vestergaard M; Kadziola A; Kilså K; Nielsen MB Synthesis of Oligo(phenyleneethynylene)-Tetrathiafulvalene Cruciforms for Molecular Electronics. Org. Lett. 2006, 8, 1173–1176. [DOI] [PubMed] [Google Scholar]
- (33).(a) Evans DA; Campos KR; Tedrow JS; Michael FE; Gagné MR Chiral Mixed Phosphorus/Sulfur Ligands for Palladium-Catalyzed Allylic Alkylations and Aminations. J. Org. Chem. 1999, 64, 2994–2995. [DOI] [PubMed] [Google Scholar]; (b) Evans DA; Campos KR; Tedrow JS; Michael FE; Gagné MR Application of Chiral Mixed Phosphorus/Sulfur Ligands to Palladium-Catalyzed Allylic Substitutions. J. Am. Chem. Soc. 2000, 122, 7905–7920. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.