

Summary
Asparagus bean (Vigna unguiculata ssp. sesquipedialis), a subspecies of V. unguiculata, is a vital legume crop widely cultivated in Asia for its tender pods consumed as vegetables. However, the existing asparagus bean assemblies still contain numerous gaps and unanchored sequences, which presents challenges to functional genomics research. Here, we present an improved reference genome sequence of an elite asparagus bean variety, Fengchan 6, achieved through the integration of nanopore ultra‐long reads, PacBio high‐fidelity reads, and Hi‐C technology. The improved assembly is 521.3 Mb in length and demonstrates several enhancements, including a higher N50 length (46.4 Mb), an anchor ratio of 99.8%, and the presence of only one gap. Furthermore, we successfully assembled 14 telomeres and all 11 centromeres, including four telomere‐to‐telomere chromosomes. Remarkably, the centromeric regions cover a total length of 38.1 Mb, providing valuable insights into the complex architecture of centromeres. Among the 30 594 predicted protein‐coding genes, we identified 2356 genes that are tandemly duplicated in segmental duplication regions. These findings have implications for defence responses and may contribute to evolutionary processes. By utilizing the reference genome, we were able to effectively identify the presence of the gene VuMYB114, which regulates the accumulation of anthocyanins, thereby controlling the purple coloration of the pods. This discovery holds significant implications for understanding the underlying mechanisms of color determination and the breeding process. Overall, the highly improved reference genome serves as crucial resource and lays a solid foundation for asparagus bean genomic studies and genetic improvement efforts.
Keywords: asparagus bean, reference genome, segmental and tandem duplication, anthocyanin biosynthesis
Introduction
Asparagus bean (Vigna unguiculata ssp. sesquipedialis), a warm‐season and drought‐tolerant subspecies of V. unguiculata, is primarily cultivated for its tender pods that measure 50–100 cm in length (Xia et al., 2019). It is widely cultivated in East/Southeast Asia and is regarded as one of the top ten Asian cultivated vegetables. The popularity of asparagus bean stems from its exceptional tolerance to heat and drought, making it an ideal crop for challenging environmental conditions. Additionally, asparagus bean is highly valued for its nutritional richness, containing significant amounts of proteins, vitamins, and minerals. Due to its nutritional profile and versatility, asparagus bean plays a crucial role in addressing malnutrition and food insecurity, providing a valuable resource for combating these challenges (Xia et al., 2019; Xu et al., 2017).
Decoding the complete genome sequence is vital for understanding genome structure and further facilitating the genetic improvement of critical agronomic traits (Zhang et al., 2023). The efforts to sequence the V. unguiculata genome have been underway for many years. In 2019, assemblies of two subspecies of V. unguiculata, namely, the asparagus bean variety Xiabao II and the common cowpea (V. unguiculata ssp. unguiculata) variety IT97K‐499‐35, were released (Lonardi et al., 2019; Xia et al., 2019). These assemblies were generated through de novo assembled using Illumina short reads and Pacific Biosciences (PacBio) continuous long reads (CLRs), respectively, followed by scaffold construction employing high‐density genetic maps. However, all current genomes contain numerous gaps (34 161 gaps for Xiabao II) and unanchored contigs (46.0 Mb for IT97K‐499‐35), which pose challenges to functional genomics research. Therefore, it is crucial to assemble a genome of higher‐quality genome, preferably a complete genome encompassing both centromeric and telomeric regions. Recently, the assembly of a telomere‐to‐telomere (T2T) genome using third‐generation sequencing technologies, including PacBio high‐fidelity (HiFi) long reads, ultra‐long reads from Oxford Nanopore Technologies (ONT), and high‐through chromosome conformation capture (Hi‐C) technology, has provided a more comprehensive assembly with reduced or no gaps (Nurk et al., 2022). This type of genome assembly enables the identification of variants and genes in regions previously known as ‘dark matter’, which encompass centromeres and segmental duplications (SDs). T2T assemblies have been accomplished in various plant species, such as Arabidopsis (Naish et al., 2021), rice (Li et al., 2021; Song et al., 2021), watermelon (Deng et al., 2022) and banana (Belser et al., 2021). However, to our knowledge, no T2T genome/chromosome has been reported in legume crops. Therefore, it is crucial to de novo assemble a high‐quality, preferably near‐complete genome of asparagus bean and analyse the features, structure, and distribution of these regions. This comprehensive genome will make a significant contribution to genetic and genomic studies in asparagus bean, facilitating a better understanding of its genetic composition and potential for future improvements.
As a fruit vegetable, the color of tender pods is a crucial consumer‐related trait for asparagus bean, significantly influencing consumer preference. Purple pods, in particular, are in high demand due to their coloration, which is attributed to anthocyanin accumulation (Li et al., 2020). Anthocyanins, which are water‐soluble pigments, play a crucial role in plant growth and development, providing protection against biotic or abiotic stress (Zheng et al., 2019). Furthermore, they play a substantial role in safeguarding against age‐related degenerative diseases, including their capacity to inhibit tumor cell growth (Mazewski et al., 2018). Breeding new asparagus bean varieties with high anthocyanin content in the pods is believed to enhance both the nutritional value and visual appeal of the crop. Previous metabolomics and transcriptomic analyses have shed light on the underlying mechanisms governing purple pod color, identifying multiple transcription factors (TFs) that regulate anthocyanin accumulation in these pods (Li et al., 2020). However, the specific candidate genes responsible for regulating purple pod color remain unidentified, posing a challenge to genetic improvement and breeding efforts in this regard. Further research is necessary to reveal the genetic factors involved and enable targeted breeding strategies for asparagus bean varieties exhibiting desirable purple pod characteristics.
In this study, we employed a comprehensive approach by harnessing the benefits of ONT ultra‐long read and PacBio HiFi read sequencing, along with Hi‐C data, to de novo assemble an improved asparagus bean reference genome using the elite cultivar Fengchan 6. The resulting high‐quality assembly exhibited exceptional continuity, completeness and accuracy, thereby offering a valuable reference genome for a wide range of genetic and genomic inquiries. Notably, this resource is highly relevant for investigating the genetic basis of purple pods in asparagus bean. Through the integration of genomic, transcriptomic and forward genetic analyses, we successfully identified candidate gene and potential causal variant associated with the development of purple pods. Overall, the significantly enhanced assembly of asparagus bean genome, presented in this study, serves as a crucial resource and establishes a strong foundation for future genomic studies and genetic improvement.
Results
Improved assembly of the asparagus bean genome
In this study, we employed a multi‐platform approach, utilizing various sequencing data types, to de novo assemble a high‐quality genome of the elite asparagus bean inbred line Fengchan 6. Fengchan 6 exhibits important agronomic traits, such as long straight pod, sprawling habit, good quality, high yield and wider environmental adaptability (Figure 1a). Overall, we obtained 258.9 gigabases (Gb) of high‐quality reads after quality control (Table S1), consisting of 72.1 Gb ONT ultra‐long reads, 30.7 Gb PacBio HiFi reads, 50.1 Gb Illumina paired‐end (PE) reads for the whole‐genome, and 50.9 Gb of Hi‐C reads. The genome size was estimated to be 500.1 and 529.6 megabases (Mb) using Illumina and PacBio HiFi reads, respectively (Figure S1). The GenomeScope model estimated the heterozygosity level of the genome to be approximately 0.03%–0.06%, which was lower than the heterozygosity level of the Arabidopsis genome (0.08%) (Wang et al., 2021). The remarkably low heterozygosity of this inbred suggested that it was suitable for assembling the genome without phasing the two haplotypes. To achieve a high‐quality assembly, we developed a pipeline that utilizes the length of ONT ultra‐long reads (N50 length 57.4 Kb), the accuracy of PacBio HiFi reads (99.8%) (Hon et al., 2020) and the scaffold building of Hi‐C data (Figure 1b). Initially, contigs were assembled using ONT ultra‐long and PacBio HiFi reads, resulting in two assemblies with total lengths of 518.7 Mb and 584.9 Mb, respectively (Table S2). Although the contig N50 lengths were similar (24.6 Mb for ONT and 23.9 Mb for PacBio), the contigs derived from PacBio HiFi reads exhibited relatively poor continuity due to a substantial number of small contigs (Figure S2). Consequently, we used the contigs derived from ONT ultra‐long reads as the backbone and substituted the syntenic regions with more accurate contigs from PacBio HiFi reads (Figure S3). To achieve a higher‐continuity assembly at the chromosomal level, we utilized Hi‐C data to order and orient the contigs, taking advantage of the generation of chromosomal interaction maps. Subsequently, we aligned ONT and PacBio reads to the scaffolds to fill the remaining gaps and properly orient them using insertion sequences and translocation information between the current scaffolds, respectively. Additionally, we polished the scaffolds using PacBio HiFi reads and further refined them using Illumina reads, with a particular focus on the bases at the boundaries of synteny regions. Ultimately, the genome assembly of Fengchan 6 encompassed a total size of 521.3 Mb, consisting of 17 scaffolds with a scaffold N50 length of 46.4 Mb (Table S3). Among them, 11 scaffolds with a cumulative length of 520.3 Mb corresponded to 11 chromosomes (Lonardi et al., 2019), accounting for 99.8% of the assembled genome sequence (Figure 1c, Table 1 and Table S3). Notably, we successfully assembled 14 telomeres with telomeric repeat sequences consisting of seven bases (CCCTAAA at the 5′ end and TTTAGGG at the 3′ end). The copy number of telomere repeats exhibited wide variation, ranging from 284 to 4805, with an average number of 1642 (Table S4). The assembled genome consisted of four telomere‐to‐telomere (T2T) chromosomes: Vu03, Vu04, Vu07 and Vu09.
Figure 1.
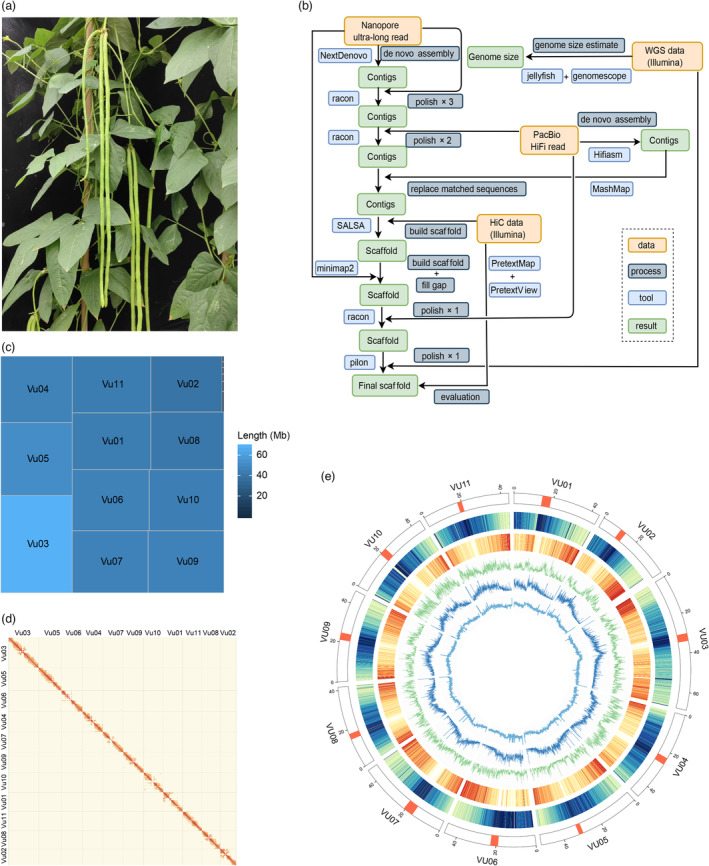
Telomere‐to‐telomere assembly of the Fengchan 6 genome. (a) Image of the leaves and pods of V. unguiculata variety ‘Fengchan 6’. (b) Workflow of de novo assembly by integrating multiple types of sequencing data. (c) Tree map of assembled scaffolds. (d) Hi‐C chromatin interaction map of the Fengchan 6 assembly. (e) Chromosomes characterization of the Fengchan 6 genome. The outer layer of blocks represents 11 chromosomes, and the red regions indicate the centromeres. The tracks from outside to inside are shown as follows: TE density, gene density, SNP and InDel density, methylation density, and GC content. The window size for these features is 50 Kb.
Table 1.
A comparison between Fengchan 6 genome and other published V. unguiculata genomes
| Genome feature | Fengchan 6 | Xiabao II | IT97K‐499‐35 |
|---|---|---|---|
| Assembled genome size (Mb) | 521.3 | 597.5 | 519.4 |
| Unplaced sequence (Mb) | 1.0 | 0 | 46.0 |
| No. of gap | 1 | 34 161 | 68 |
| GC content (%) | 33.0 | 28.8 | 33.0 |
| Contig N50 (Kb) | 24 485 | 15.2 | 10 912 |
| Contig number | 42 | – | 765 |
| Scaffold N50 (Mb) | 46.4 | 2.7 | 16.4 |
| Scaffold number | 17 | 11 | 686 |
| Assembled telomere number | 14 | 0 | 0 |
| Repeat content (%) | 51.8 | 46.5 | 49.5 |
| Protein‐coding genes | 30 594 | 42 609 | 29 773 |
| Missing BUSCOs (%) | 0.7% | 6.8% | 4.1% |
| No. of total BUSCOs | 1614 | 1440 | 1440 |
| Reference | This study | Xia et al. (2019) | Lonardi et al. (2019) |
Evaluation and annotation of the genome assembly
The quality of the Fengchan 6 assembly was assessed using multiple methods. The genome structure and quality were validated by mapping the Hi‐C data, revealing no noticeable instances of misalignment intra‐ or inter‐chromosomes (Figure 1d). Aligning the PacBio HiFi reads to the assembly, we observed 1.4 Mb gaps distributed across multiple chromosomes, such as the end of Vu03 (Figure S4), indicating a primarily contribution of ONT ultra‐long reads to the contiguity. To evaluate the accuracy of the genome at the single‐base level, we aligned the Illumina whole‐genome short reads to the assembly. Several findings provide evidence for the accuracy of the assembled genome. Firstly, 98.1% of the Illumina PE reads generated in this study mapped correctly to the assembly, surpassing the mapping rate of IT97K‐499‐35 (87.0%) using its own Illumina PE reads. Secondly, based on the mapping, we identified 7586 homozygous single nucleotide polymorphisms (SNPs) and 6485 homozygous short insertions and deletions (InDels), indicating a nucleotide accuracy rate of 99.993% (Table S5). Additionally, we detected 98 134 heterozygous SNPs and 54 241 heterozygous InDels, indicating a heterozygosity rate of 0.029% and confirmed the very low heterozygosity of the Fengchan 6 genome. Thirdly, we assessed the base accuracy of the genomes using k‐mer‐based quality estimation (k = 19 bp). The quality value (QV) increased from 39.0 to 44.5 compared to IT97K‐499‐35. Furthermore, to evaluate the completeness of the gene regions of the assembled genome, we utilized benchmarking universal single‐copy orthologs (BUSCO) by searching the single‐copy orthologs identified in the embryophyta lineage. Overall, out of the 1614 conserved BUSCO genes, we identified 1598 (99.0%) complete and 5 (0.3%) fragmented genes, representing the highest numbers reported among the assembled V. unguiculata genomes (93.2% for Xiabao II and 95.9% for IT97K‐499‐35, Table 1). These results collectively demonstrate the high contiguity, completeness and accuracy of the Fengchan 6 assembly.
To analyse the repetitive sequences in the Fengchan 6 assembly, we utilized RepeatMasker with the RepeatModeler result and the Dfam database as the libraries. Finally, we annotated a total of 269.8 Mb, accounting for 51.8% of the genome, as repetitive elements (Table S6 and Figure 1e). The repetitive sequence content was higher compared to previous reports for Xiabao II (46.5%) and IT97K‐499‐35 (49.5%), confirming the distinct advantages of our strategy in assembling the repetitive regions. To predict the gene structure, we integrated RNA data from six tissues (root, stem, leaf, flower, pod and seed) and utilized the Fengchan 6 genome after masking repetitive sequence. Using MAKER, we predicted 30 594 protein‐coding genes in our assembly, with a tendency for these genes to be located at the ends of chromosomes (Figure 1e). The predicted protein‐coding genes in the Fengchan 6 assembly displayed an average gene length of 3942 bp (ranging from 152 to 166 973 bp), with an average coding sequence (CDS) length of 1241 bp (ranging from 148 to 48 621). Among the predicted protein‐coding genes, 29 787 (97.4%) were functionally annotated in at least one of the NR (nonredundant, NCBI), SwissProt, InterPro, Pfam, KEGG and GO databases (Table S7), highlighting the reliability of the predicted genes. Additionally, we identified various non‐coding RNA genes throughout the Fengchan 6 genome, including 1969 microRNAs, 1764 transfer RNAs, 2372 ribosomal RNAs and 773 small nucleolar RNAs (Table S8).
Sequence architecture of the centromere
To determine the locations of the centromeres within our assembly, we conducted a search using centromere‐specific 455 bp (CEN455) satellite sequences, which are predominantly found in centromere‐specific histone H3 (CENH3)‐bound DNAs of at least seven chromosomes in V. unguiculata (Ishii et al., 2020). The remaining four chromosomes were reported to contain the major centromere components, namely 721 bp (CEN721) and 1600 bp (CEN1600) satellite sequences (Ishii et al., 2020). We successfully identified core centromeric regions ranging from 2.2 Mb to 5.1 Mb across the chromosomes, with an average length of 3.5 Mb (Table S9 and Figure S5). In total, we identified 38.1 Mb of sequences covering the core centromeres, surpassing the coverage in the previously assembled IT97K‐499‐35 genome (20.2 Mb) using PacBio CLR data (Lonardi et al., 2019). Our result indicated that the combination of the ONT ultra‐long and PacBio HiFi datasets used in this study provided an advantage in successfully assembling in centromere regions compared to the assembly from PacBio CLR data. We identified a large number of CEN455 on seven of the 11 chromosomes, with copy numbers ranging from 2369 to 6977 (Table S10), which is in consistent with previous studies. CEN1600 satellites were detected in the remaining four chromosomes, namely, Vu02, Vu03, Vu09 and Vu11, with copy numbers ranging from 557 to 1185. Overall, we identified 30 622 and 4073 complete copies for CEN455 and CEN1600 satellites, respectively. Furthermore, we observed that the majority of the centromere‐specific satellites within chromosomes exhibited at least 90% sequence identity (Figure 2a and Figure S5). Using the sequences of these two satellites as the repeat library, we identified a total of 25.9 Mb using RepeatMasker, accounting for 68.0% of the centromeric sequences. Consistent with previous studies that demonstrated the enrichment of repetitive sequences (Pootakham et al., 2021), we found that centromere‐specific satellites arrays were surrounded by retroelements, which accounted for 26.7% of the centromeric sequences (Table S11 and Figure S6). Among them, long terminal repeat (LTR) retrotransposons represented the largest portion (20.8% of the centromeric sequences), with the Gypsy superfamily (19.4%) being dominant cluster (Figure 2b, Figure S6 and Table S11). Small amounts of the Copia superfamily (1.1%) were also detected in this region. Additionally, rDNA sequences were found to cluster in the flanking regions of centromeres, such as those on chromosome Vu01 (Figure 2b). Notably, the PacBio HiFi reads struggled to cover the rDNA sequence region in Vu01 (13 575–13 645 Kb), resulting in a 49.6 Kb gap (Figure 2b). However, the ONT ultra‐long reads completely covered this region, confirming the advantage of ONT data in assembling low‐struggled to cover sequence regions. In addition, we utilized the ONT ultra‐long reads to profile DNA methylation, detecting a total of 4.9 million CG methylations throughout the genome. We observed that the CG methylation levels of the satellite arrays within the centromeric regions were lower compared to the retroelements‐enriched flanking regions (Figure 2b), aligning with the findings reported in Arabidopsis (Wlodzimierz et al., 2023). However, the median percentage of CG methylation levels in the centromeric regions (Table S9) was significantly higher than that in the non‐centromeric regions (0.90 vs. 0.59, two‐tailed t test, P = 4.9 × 10−12, Figure S7). This finding was consistent with previous studies (Song et al., 2021; Wlodzimierz et al., 2023), suggesting that DNA methylation may contribute to the maintenance of chromosomal stability at the centromere level (Scelfo and Fachinetti, 2019).
Figure 2.
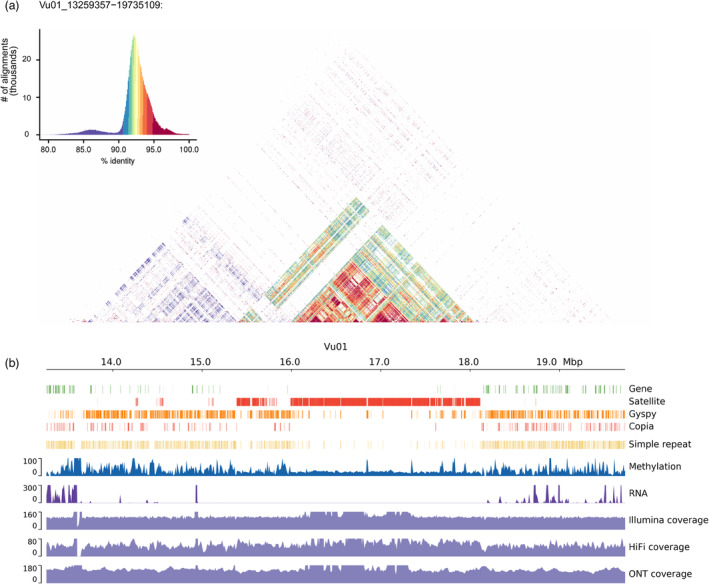
Characterization of the Vu01 centromeric region. (a) Sequence identity of the Vu01 centromeric region (14 259–18 735 Kb) and flanking regions with a distance of 1 Mb. (b) Track displaying density of genes, satellites, Gypsy elements, Copia elements, simple sequence repeats and rDNA, followed by the methylation level, gene expression and coverages of Illumina short reads, PacBio HiFi reads and ONT ultra‐long reads.
Although the satellite arrays and the retroelements occupied most of the centromeres, we still detected 127 predicted protein‐coding genes located in the centromeric regions, with 37 (29.1%) genes expressed in at least one tissue. The lower transcription ratio of gene in centromeric regions compared to the entire genome might be attributed to heterochromatin repression or the presence of non‐expressed pseudogenes. Out of these genes, 22 exhibited transcription in all the six tissues of the plant, indicating their continuous gene functionality.
Comparison of the V. unguiculata assemblies
Compared with the findings of the previously published report on the common cowpea assembly IT97K‐499‐35, our assembly of Fengchan 6 exhibited significant enhancements in continuity, completeness and accuracy. Notably, our assembly has greatly improved the contiguity of the V. unguiculata genome compared to Xiabao II (scaffold N50 = 2.7 Mb) (Xia et al., 2019) and IT97K‐499‐35 (scaffold N50 = 16.4 Mb, Table 1) (Lonardi et al., 2019). Although the total assembly length of IT97K‐499‐35 reaches 519.4 Mb, only 473.5 Mb of sequences (91.1%) can be successfully anchored to the 11 chromosomes, which is considerably smaller than our anchored assembly (520.7 Mb). Collinear alignment of the genomes revealed a substantial deficiency of sequences, particularly in the centromeric regions, in IT97K‐499‐35 (Figure 3a). Furthermore, we detected abundant genetic variations, particularly structural variations (SVs), between these two subspecies. The cleaned PacBio CLR data of IT97K‐499‐35, totalling 154.8 Gb, were mapped to the Fengchan 6 genome at a mapping rate of 80.8%, which was lower than that of Fengchan 6 data mapped to its own genome (99.3% for ONT reads and 99.8% for PacBio HiFi reads), indicating the existence of numerous variants between the two genomes. Following the mapping process, we detected a total of 24 854 SVs distributed throughout the genome, predominantly comprised of deletions (DELs) and insertions (INSs), which accounted for 12 823 and 11 862 events, respectively (Table S12). The numbers of duplications (DUPs) and inversion (INVs) were 77 and 92, respectively. It is worth noting that there was no significant correlation between the number of SVs and chromosome length (Pearson correlation test, r = 0.278, P = 0.407, Figure 3b). These SVs were unevenly distributed across the genome, with 23 hotspots identified on 10 chromosomes, particularly on chromosomes Vu04, Vu10 and Vu11 (Table S13), indicating regions of concentrated variation. Among these SVs, there were 18 large INVs (>100 Kb) observed between the two genomes, including the previously reported 4.2 Mb INV (Lonardi et al., 2019). Additionally, we discovered a 4.8 Mb INV with well‐defined breakpoints (Vu10:13 237 623–18 079 870, Table S12 and Figure 3c) between Fengchan 6 and IT97K‐499‐35, which was validated through comparing analysis of the two assemblies (Figure 3a). We observed that 2372 DELs intersected with 3838 genes, and 1744 INSs were located in 1338 genes. Overall, a total of 4661 distinct genes were potentially impacted by these DELs and INSs, thereby significantly contributing to the differentiation and adaptation of these two subspecies.
Figure 3.
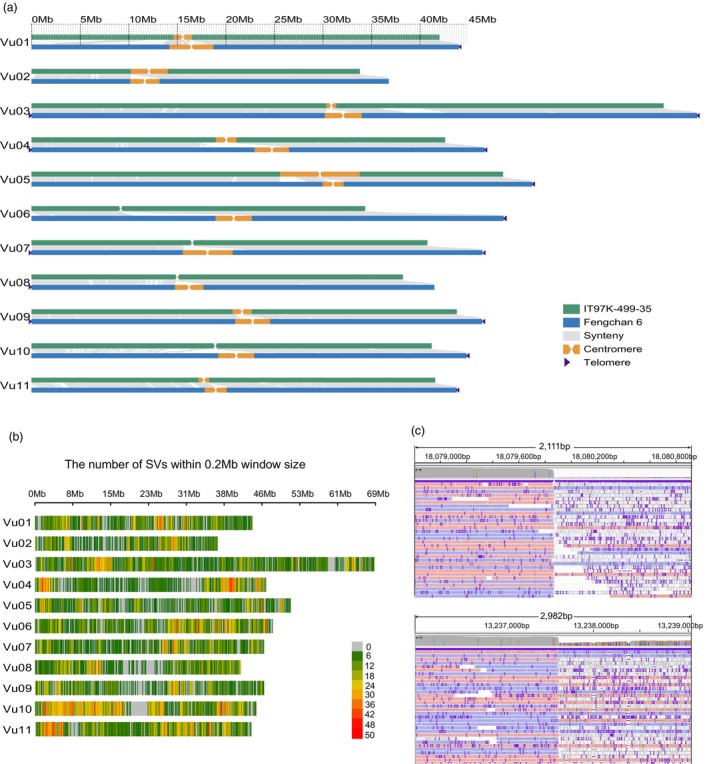
Comparison of between the Fengchan 6 genome with the previously published IT97K‐499‐35 genome. (a) Collinearity between the Fengchan 6 and IT97K‐499‐35 genomes. The collinear regions are represented by grey lines. Orange and yellow regions indicate the centromeric regions and INVs, respectively. Black triangles indicate the presence of telomere sequence repeats. (b) SV density between Fengchan 6 and IT97K‐499‐35 across the chromosomes. (c) IGV screenshot of the breakpoints of the 4.8 Mb INV (Vu10:13 237 623–18 079 870) between Fengchan 6 and IT97K‐499‐35.
Segmentally and tandemly duplicated genes
Gene duplication is a crucial evolutionary mechanism that contributes to the emergence of novel functions (Belser et al., 2021). Throughout the genome, duplicated sequences with a length of at least 1 Kb and a sequence identity of 90% or higher are referred to as SDs (Bailey et al., 2001), which play a significant role in gene evolution as the major sources of duplicated genes (Vollger et al., 2022a). The high‐quality genome assembly of Fengchan 6 provides a valuable opportunity to robustly and accurately characterize SDs. In total, we identified 24 164 SDs, spanning a combined length of 79.6 Mb, which accounting for 15.3% of the genome (Table S14). This value falls within a moderate range compared to other surveyed genomes, such as 7.0% for humans and 24.2% for rice (Li et al., 2021; Vollger et al., 2022a). The distribution of SDs across the genome is non‐random (Figure 4a). Notably, chromosomes Vu04 and Vu06 contained 10.7 Mb and 14.5 Mb SDs, respectively, which is approximately twice the number found in the other chromosomes (Table S14). This finding strongly indicates that these two chromosomes played a substantial role in the evolutionary process of V. unguiculata through extensive duplication events. Additionally, we observed a decrease in the number of SDs as the increasing fragment size increased (Figure 4b). The 1–2 Kb fragments contributed the most, accounting for 66.0% of the total number of SDs. Although the number of SD tended to decrease with increasing sequence identity, the percentage variation was relatively small, ranging from 6.0% to 12.7% with an average of 10.0% (Figure 4c). Gene ontology (GO) enrichment analysis revealed that 3338 genes in the SD regions were enriched in metabolic process (GO:0008152), cellular response to heat (GO:0034605), defence response to bacterium (GO:0009816), induced systemic resistance (GO:0009682) and maintenance of inflorescence meristem identity (GO:0010077) (Figure 4d). This indicates that SDs have contributed to the expansion of the functional genes related to environmental adaptability.
Figure 4.
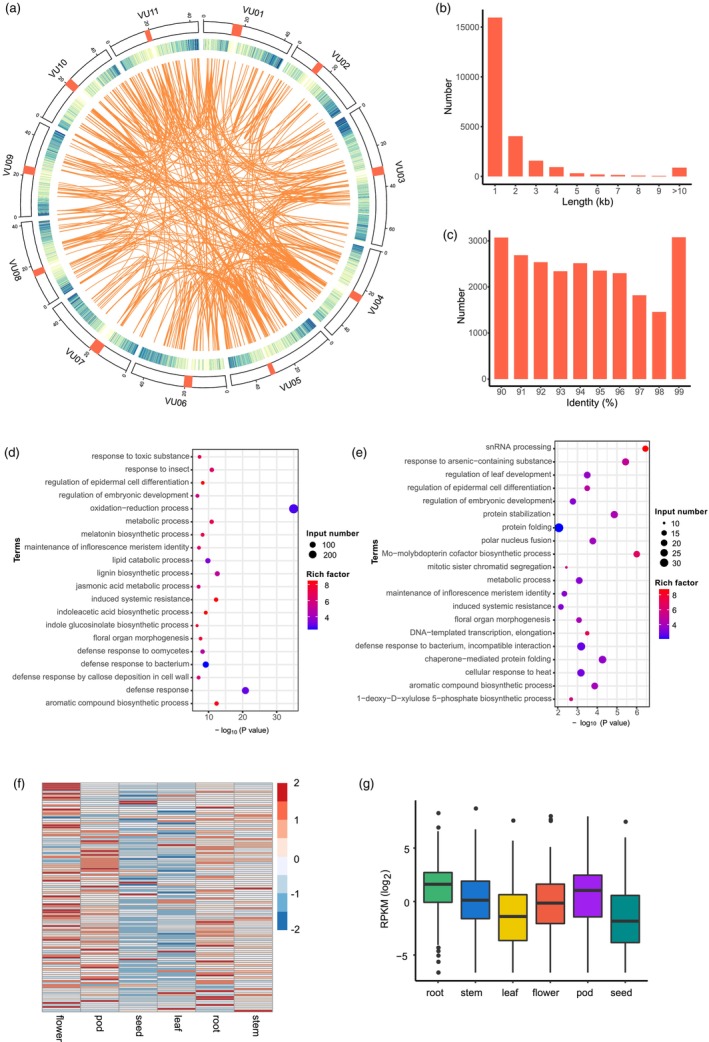
Segmentally and tandemly duplicated genes in Fengchan 6. (a) Heatmap in the circos plot indicates the gene density. The linked regions indicate the SD regions between chromosomes. (b) Length distribution of SD regions. (c) Number distribution of SD sequence identity. (d) GO (biological process) enrichment analysis of genes in SD regions. The P values were corrected using the Benjamini–Hochberg method. (e) GO (biological process) enrichment analysis of tandemly duplicated genes. The P values were corrected using the Benjamini–Hochberg method. (f) Heatmap displays the expression levels of the PLCP gene family across six tissues: root, stem, leaf, flower, pod and seed. (g) Expression levels of NBS‐LRR genes across six tissues.
In addition to the expansion in SDs, we also detected 914 clusters of tandemly duplicated genes, comprising a total of 2356 genes (Table S15). These genes exhibited a predominant enrichment in defence response (GO:0006952, GO:0009625, GO:0042742, GO:0002229 and GO:0009636) and biosynthetic process (GO:0009809, GO:0019438, GO:0009684, GO:0030187 and GO:0009759), enhancing resistance to biotic and abiotic stress (Figure 4e). Among the tandemly duplicated genes, 654 clusters consisted of two genes and 158 clusters (17.3) consisted of three genes. The largest cluster comprised 18 genes that encod patatin‐like cysteine proteases (PLCPs). PLCPs constitute a large gene family widely present in plants and are involved in plant defence against biotic and abiotic stress (La Camera et al., 2009; Matos et al., 2000). The Fengchan 6 genome contained a total of 302 PLCP annotated genes of which 116 genes expressed in at least two tissues. Their expression levels displayed clear tissue‐specific patterns (Figure 4f), suggesting functional differentiation of these genes across different tissues.
Nucleotide‐binding site leucine‐rich repeat (NBS‐LRR) genes, the largest group of disease resistance genes, are crucial components of the plant's defence system against diseases (Wang et al., 2022). We identified 181 NBS‐LRR genes in the Fengchan 6 genome (Table S16), which is a greater number compared to chickpea (121) (Sagi et al., 2017). We observed that 23 NBS‐LRR genes were located in SD regions with no enrichment (Fisher's exact test, odds ratio (OR) = 1.2, P = 0.48). In contrast, 77 NBS‐LRR genes form clusters and exhibited significant enrichment among tandemly duplicated genes (Fisher's exact test, OR = 5.5, P = 1.9 × 10−27). Additionally, we found that tissue‐specific expression patterns for 181 NBS‐LRR genes. Among the six tissues analysed in this study, the roots exhibited the highest median expression of NBS‐LRR genes in 2.9 RPKM (Figure 4). This finding suggests that roots may exhibit an enhanced response to microbial exposures, potentially indicating a higher susceptibility to infection.
Metabolome and transcriptome profiles in anthocyanin biosynthesis
As the main edible part, the color of tender pod is an important appearance and quality characteristic for asparagus bean, with a wide range of diversity obsevered across various varieties, including white, green, purple and brindle. To investigate the differences in the biosynthesis pathway of different pod color pigments, we conducted a metabolite analysis of fresh pods from two cultivars, PRS (purple pod) and WSS1 (light greed pod) (Figure 5a). Our investigation unveiled a substantial presence of metabolites related to anthocyanin biosynthesis, along with the upstream phenylpropanoid and flavonoid biosynthesis pathways. Notably, out of the 73 different flavonoid metabolites, 67 exhibited significantly higher levels in PRS when compared to WSS1, including key compounds such as naringenin chalcone, naringenin and dihydroquercetin (Table S17). Additionally, six anthocyanins were detected in the pods, with the purple pod variety showing significant elevations in their concentrations (Table S17).
Figure 5.
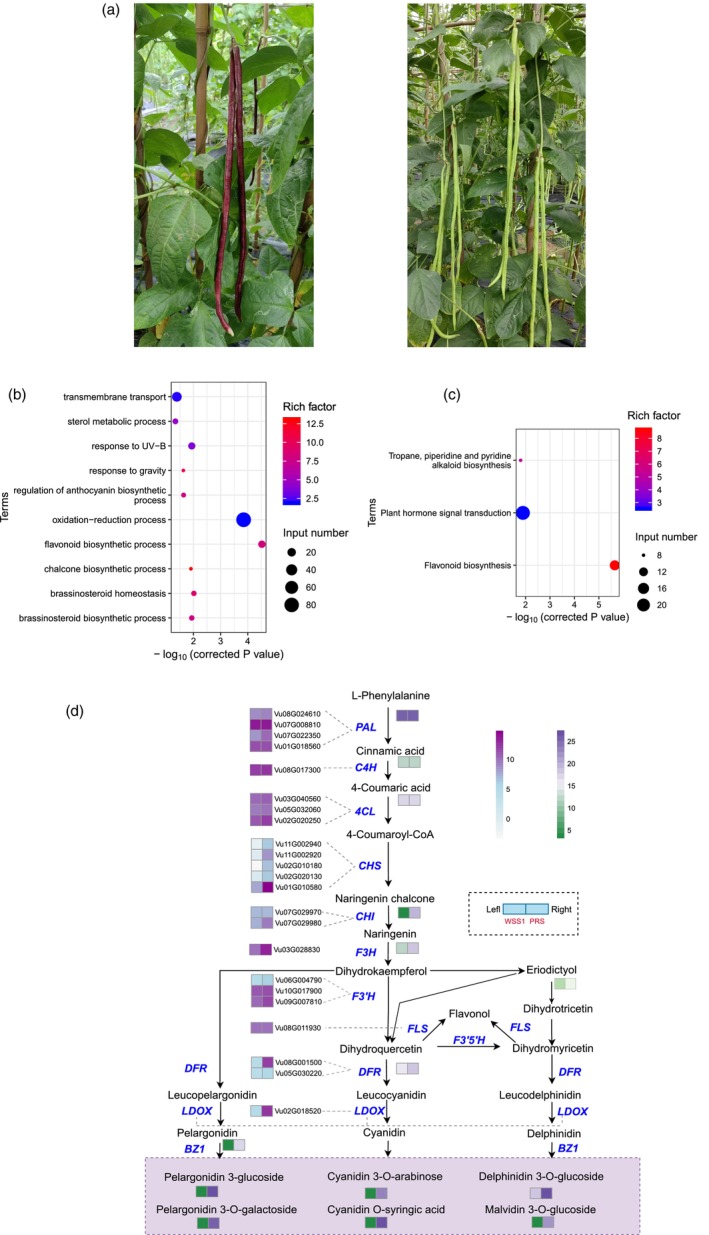
Genes and metabolites involved in anthocyanin biosynthesis. (a) Image of the leaves and pods of V. unguiculata varieties PRS (purple pods) and WSS1 (light green pods). (b) GO enrichment analysis of DEGs between PRS and WSS1. (c) KEGG enrichment analysis of DEGs between PRS and WSS1. (d) Profiles of gene and metabolites related to flavonoid and anthocyanin biosynthesis for PRS and WSS1.
To gain further insight into the molecular mechanism underlying purple pod formation, we performed transcriptome sequencing on similar tissues utilized in the metabolomic analysis. The analysis of differentially expressed genes (DEGs) showed a significant enrichment in the GO term associated with the flavonoid biosynthetic process (GO:0009813, Fisher's exact test, OR = 8.0, corrected P = 3.0 × 10−5, Figure 5b), as well as in Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways associated with flavonoid biosynthesis (map00941, Fisher's exact test, OR = 8.7, corrected P = 2.2 × 10−6, Figure 5c). Notably, gene expression analysis demonstrated upregulation of flavonoid and anthocyanin‐related genes, including PAL, F3H, DFR and LDOX, in the purple pod variety (PRS) compared to the light green pod variety (WSS1) (Figure 5d). These findings indicate that the upregulation of genes involved in flavonoid and anthocyanin biosynthesis pathways contribute to the purple coloration of pods in asparagus bean. Through the integration of metabolomic and transcriptomic approaches, our study highlights the crucial role of abundant metabolites and highly expressed genes involved in the flavonoid and anthocyanin biosynthesis pathways contributing to the formation of purple pods and elucidating the underlying mechanism of color determination in asparagus bean.
Gene identification of purple pods using the Fengchan 6 genome
To determine the genes associated with purple pods, we conducted a genetic analysis through reciprocal hybridization between the PRS and WSS1 varieties (Figure 5a). A total of 304 F2 individuals were obtained, consisting of 220 and 84 individuals with purple and light green pods, respectively. Based on the phenotype of F1 generation (purple pods) and the segregation ratio of 3:1 among F2 individuals (Chi‐square = 1.12, P > 0.05), we determined that the purple color of the pods was controlled by a single dominant gene. We sequenced the two parents as well as two bulked pools with extreme phenotypes, and the bulk segregant analysis (BSA) identified a locus on chromosome Vu05 (Figure 6a) using the Fengchan 6 genome as a reference. By investigating the variant number with Δ(SNP‐index) >0.9 in a 250‐Kb window with a step size of 50 Kb, we identified a genomic region spanning 0.55 Mb in length (3.10–3.65 Mb), which contained 54 genes (Figure 6b and Table S18).
Figure 6.
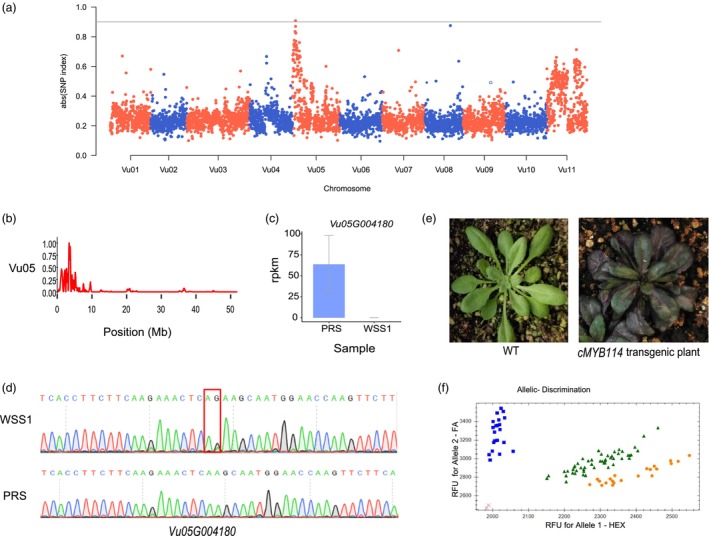
Identification of genes associated with purple pods. (a) Manhattan plot of the absolute value of Δ(SNP‐index) for two bulks of individuals with purple and white pods. The x‐axis represents the physical position across the chromosomes, and each dot indicates the average absolute value of SNP_index within a 100‐Kb window. The grey line indicates the significance threshold (0.9). (b) Number of SNPs with an absolute value of Δ(SNP‐index) larger than 0.9 in a 250‐Kb window with a step size of 50 Kb. The interval of 0.8 times the maximum number was regarded as the candidate region (3.10–3.65 Mb). (c) Expression levels of two DEGs in the candidate region. The grey line indicates the standard deviation of three repeats. (d) Sanger sequencing of the two‐base variation in the coding region of Vu05G004180 between the two parental complementary chains. (e) Phenotypes of wild‐type Arabidopsis thaliana and the transgenic positive strain. (f) Validation of KASP marker VuP‐KASP1 on the 94 F2 plants derived from WSS1 and PRS. Orange, green and blue color clusters represent homozygous purple pod allele classes: −/−, heterozygous purple pod allele classes AG/− and green pod allele classes AG/AG, respectively. The red cross represents the non‐template control.
Furthermore, transcriptome analysis of the pods in PRS and WSS1 identified two DEGs within the candidate region: Vu05G004180 and Vu05G004220 (Table S18 and Figure 6c). Comparative analysis revealed a two‐base (CT) insertion in the exon of Vu05G004180 in WSS1 compared to PRS, resulting in the loss of function of the gene due to a premature stop codon (Figure 6d). Vu05G004180, named as VuMYB114, shares homology with AT1G66380 (AtMYB114) in Arabidopsis, which is a member of the MYB family of TFs involved in anthocyanin biosynthesis regulation (Heppel et al., 2013; Yan et al., 2021). Therefore, Vu05G004180 (VuMYB114) was considered the candidate gene controlling purple pods trait.
To validate the candidacy of the gene, we opted to conduct the validation in Arabidopsis, primarily because the transgenic system in asparagus bean is still in early stages of development. The CDSs of VuMYB114 (cVuMYB114) were successfully transformed into Arabidopsis, resulting in the emergence of transgenic lines displaying varying levels of purple coloration (Figure 6e), thus confirming the function of VuMYB114. Additionally, a kompetitive allele‐specific polymerase chain reaction (KASP) marker named VuP1‐KASP1 was designed based on the two‐base variation and utilized for genotyping 94 F2 plants derived from WSS1 and PRS. The genotypes exhibited distinct clustering into three categories, which cosegregated with pod color phenotypes (Figure 6f), further supporting the role of the TF VuMYB114 in the regulation of anthocyanin metabolism.
Discussion
A high‐quality genome assembly is crucial for comprehending the genetic basis of economically important traits and enabling molecular breeding in plants (Xu et al., 2023). In recent years, two V. unguiculata genomes have been assembled, however, numerous sequences remained unassembled or unplaced (Lonardi et al., 2019; Xia et al., 2019). In this study, we integrated multiple sequencing platforms and assembled a high‐quality reference genome, Fengchan 6. Our assembly outperforms the previous genomes by successfully assembling all 11 centromeres, 14 of 22 telomeres, and four T2T chromosomes utilizing PacBio HiFi and ONT ultra‐long reads. We observed only one gap and four unanchored contigs, which comprised just 1 Mb and 1% of the assembly. This is a significant improvement compared to XiaBao II (which had 34 161 gaps) (Xia et al., 2019) and IT97K‐499‐35 (which had 8.9% unanchored sequences and 68 gaps) (Lonardi et al., 2019). Moreover, our assembly exhibits higher N50 lengths for both contigs and scaffolds, indicating enhanced continuity. Additionally, our assembly achieved a higher QV compared to IT97K‐499‐35 (44.5 vs. 39.0), demonstrating improved base accuracy.
The utilization of PacBio HiFi and ONT ultra‐long reads greatly facilitated the assembly and characterization of telomere sequences and centromeric regions. These genomic regions are recognized for containing abundant satellite arrays and retroelements, which have demonstrated evolutionary implications and functional importance in centromeres (Talbert and Henikoff, 2022). The previous released genome (IT97K‐499‐35) assembled a 20.2‐Mb centromeric sequence using PacBio CLR reads, which was far from complete compared with our assembly (38.1 Mb). In the Fengchan 6 assembly, we identified abundant CEN455 and CEN1600 arrays, which were distributed across centromeric regions of different chromosomes but did not detect the CEN180 sequence enriched in Arabidopsis and rice centromeres (Naish et al., 2021; Song et al., 2021), suggesting species‐specific centromere‐enriched satellites. Furthermore, we observed the presence of retroelements, such as Gypsy elements, similar to ATHILA elements found in Arabidopsis (Naish et al., 2021). The species‐specific satellites and retroelements predominantly occupy centromeric sequences, which exhibit variation in monomer composition among different chromosomes. Consequently, our findings enable the investigation of centromere evolution within and between species, in comparison to genomes that successfully assemble centromeric sequences in the Vigna genus.
The advent of long‐read sequencing technologies, such as ONT ultra‐long read and PacBio HiFi read sequencing, has demonstrated their effectiveness in generating high‐quality genome assemblies. This is exemplified by the complete sequencing of a haploid human genome (Nurk et al., 2022) and two rice genomes (Li et al., 2021; Song et al., 2021). Despite our efforts to perform de novo assembly of the genome by integrating multiple sequencing data types, achieving a complete genome assembly without any gap remains challenging. This challenge may arise from the relatively large genome size of asparagus bean (521 Mb) compared to rice (392–398 Mb) (Li et al., 2021; Song et al., 2021) and watermelon (369 Mb) (Deng et al., 2022). Additionally, the presence of low‐complexity and specific sequences, such as those in SDs, rDNAs or telomeres, which might extend beyond the read length of third generation sequencing (TGS), further hampers the assembly of the complete genome, as observed in Arabidopsis (Naish et al., 2021). Although Hi‐C data were employed to construct the scaffolds, its utility was limited due to the short‐read lengths, making it challenging to span low‐complexity regions. To addressed this, future efforts could explore the application of BAC‐long sequencing (>150 Kb) on the ONT platform or Pore‐C, a method capable of capturing multiway chromatin contacts using long‐read sequencing. These approaches have the potential to enhance the assembly by generating more contiguous contigs or anchoring the remaining contigs to the chromosomes.
The high‐quality genome assembly is a valuable resource for studying important traits and aiding genetic breeding. By utilizing this assembly, we were able to narrow down the potential genomic region to 0.55 Mb through BSA analysis between the light green and purple pod pools. By analysing differential expression and gene annotation, we identified Vu05G004180 (VuMYB114) as a potential candidate gene for purple pods. Through alignment to the genome, we found a two‐base variation, confirmed by Sanger sequencing, suggesting its role as a causal variation. The gene with this variation was validated in transgenic Arabidopsis, establishing the two‐base variation as a functional marker for future genetic improvement. The high‐quality genome has enabled us to explore SDs across the entire genome, which play a pivotal role in driving genome evolution. They result from the replication and insertion of genomic segments, leading to the presence of multiple copies of genetic material in the same genome. This has profound implications for genetic diversity and innovation, including the expansion of gene families and the acquisition of new functions through duplication and subfunctionalization. In our study, we identified 914 clusters of tandemly duplicated genes in SD regions, enriched in biosynthetic and defence response genes like PLCP genes. Notably, these tandemly duplicated genes display tissue‐specific expression patterns, highlighting their functional differentiation across various tissues. This emphasizes the importance of SDs in shaping the adaptive potential of genomes and their involvement in tissue‐specific processes.
High‐quality genomes play a significant role in comparative genome analysis by facilitating precise alignment and comparison of sequences across different species. This facilitates the identification of conserved regions, evolutionary changes and genetic variations. For instance, previous studies investigating synteny blocks in the Vigna genus revealed truncated MYB26 genes in V. angularis and V. unguiculata, which could be responsible for non‐shattering phenotypes, suggesting parallel domestication pathways (Takahashi et al., 2020). Similarly, conserved genes like PSAT1 have been identified as potential targets for domestication efforts across Vigna genus (Takahashi et al., 2023). Comparative genome analysis using high‐quality genomes also detects SVs and functional elements, providing insights into evolutionary relationships, genomic organization and the genetic basis of phenotypic variation. This enhances our understanding of genome evolution and species diversification.
In this study, we observed that different pod colors are associated with the presence of distinct metabolites, particularly flavonoids, which serve as substrates for anthocyanin synthesis and contribute to varying levels of anthocyanins, resulting in different shades of purple in the pods. Transcriptome analysis revealed significant upregulation of almost all endogenous structural genes involved in the general flavonoid pathway and subsequent anthocyanin biosynthesis pathway. Among the candidate genes, Vu05G004180 (VuMYB114) was identified as a key regulator associated with purple pods. MYB114 is a homologous gene that encodes an R2R3‐MYB TF, which is recognized for its role in regulating genes involved in the flavonoid biosynthesis pathway and downstream anthocyanin biosynthetic pathway in various plant species (Xie et al., 2020; Xu et al., 2015). It is worth noting that R2R3‐MYB TFs from different species exhibit distinct regulatory mechanisms and have the ability to activate different target genes (Lai et al., 2013). Although the regulatory mechanisms of VuMYB114 are not fully understood, the successful development and validation of a functional marker associated with purple pods offers a valuable tool for molecular breeding programmes aimed at improving the nutritional value of asparagus bean varieties.
Experimental procedures
Plant materials
The elite inbred line Fengchan 6 of asparagus bean, which has been self‐pollination for several generations, was used to de novo assemble the genome. Genomic DNA (gDNA) was extracted from the young leaves for constructing DNA sequencing libraries, and RNAs were separately extracted from root, stem, leaf, flower, pod and seed for RNA library construction. In addition, the varieties PRS (purple pod) and WSS1 (green light pod) were self‐pollination for at least three generations. PRS and WSS1 were crossed to obtain the F1 hybrids, having purple pods; A total of 304 F2 individuals were obtained from selfing of the F1 hybrids. Among them, the individuals with purple and light green pods were 220 and 84, respectively. Thirty individuals were selected for each color and pooled into two separate bulks for subsequent analysis. Simultaneously, RNAs were extracted from pods of both PRS and WSS1, and the pods of these two varieties were also used for the detection of metabolic components. Transcriptome and metabolome were performed with three biological replicates for each material.
Library preparation and sequencing
For the ONT ultra‐long library, approximately 8–10 μg of high‐molecular‐weight genomic DNA (gDNA) was size‐selected (>50 Kb) using SageHLS HMW library system (Sage Science), and the Ligation sequencing 1D kit (SQK‐LSK109, Oxford Nanopore Technologies, UK) was then used to process the DNA library according to the manufacturer's instructions. About 800 ng DNA library was constructed and subsequently sequenced on the PromethION (Oxford Nanopore Technologies, UK) at the Genome Center of Grandomics (Wuhan, China). In addition, a SMRTbell target size library was constructed for sequencing according to PacBio's standard protocol (Pacific Biosciences, CA) using 15‐Kb preparation solutions. The SMRTbell library was then purified using AMPure PB beads, and the size of library fragments was assessed using an Agilent 2100 Bioanalyzer (Agilent technologies). Sequencing was performed on a PacBio Sequel II instrument with Sequencing Primer V2 and Sequel II Binding Kit 2.0 in Grandomics.
For Illumina short‐read sequencing, a total of 1.5 μg of DNA was used as input material for DNA sample preparation. Sequencing libraries were generated using the Truseq Nano DNA HT Sample Kit (Illumina) following the manufacturer's recommendations, and index codes were added to attribute sequences to each sample. Briefly, the DNA sample was fragmented by sonication to a size of 350 bp, and then DNA fragments were end polished, A‐tailed, and ligated with the full‐length adapter for Illumina sequencing with further polymerase chain reaction (PCR) amplification. At last, the PCR products were purified (AMPure XP system), and libraries were analysed for size distribution by Agilent2100 Bioanalyzer and quantified using real‐time PCR. These constructed libraries were sequenced on Illumina NovaSeq platform, and 150 bp PE reads were generated.
According to Hi‐C procedure, nuclear DNA from the leaves was cross‐linked and lysed using the restriction enzyme DpnII, leaving pairs of distally located but physically interacting DNA molecules attached to one another. The sticky ends of these digested fragments were then biotinylated and ligated to each other to form chimeric circles. The biotinylated circles, which are chimeras of the physically associated DNA molecules from the initial original cross‐linking, were enriched, sheared and sequenced using the Illumina NovaSeq platform with 150 bp PE reads.
RNA was isolated using Plant RNA Kit (R6827, OMEGA), and first strand cDNA was synthesized using random primers. 1 μg of RNA per sample was used as input material for RNA‐seq. RNA‐seq libraries were prepared using Illumina mRNA‐seq Library Preparation kit and sequenced by the Illumina NovaSeq, generating 150 bp PE reads.
Quality control of sequencing data
In order to obtain high‐quality sequencing data, the quality control was performed to remove sequencing adapters and bases with low quality. For Illumina short‐read sequencing data, we used fastp (Chen et al., 2018) v0.20.1 with the following parameters: ‘‐‐length_required 80 ‐‐qualified_quality_phred 15 ‐‐unqualified_percent_limit 30 ‐‐cut_front ‐‐cut_tail ‐‐cut_window_size 1 ‐‐cut_mean_quality 20’. For ONT ultra‐long reads, we detected base quality at both ends of reads using NanoQC (De Coster et al., 2018) v0.8.1 and trimmed 40 bases of start and 30 bases of end for the raw reads using NanoFilt (De Coster et al., 2018) v2.2.0 due to their lower quality. And we kept the reads with length >1 Kb and mean quality score >7 for subsequent analysis.
Genome assembly
The genome size was estimated by employing jellyfish (Marçais and Kingsford, 2011) v2.2.10 based on k‐mer distribution analysis of whole‐genome Illumina short‐read sequencing data. Subsequently, the heterozygosity rate of the genome was estimated using GenomeScope (Ranallo‐Benavidez et al., 2020) v2.0, which guided the subsequent genome assembly. To achieve a more contiguous assembly, de novo genome assembly was performed using NextDenovo (https://github.com/Nextomics/NextDenovo) v2.4.0 with default parameters based on 72.1 Gb ONT ultra‐long reads, whose maximal and N50 lengths were 381.4 Kb and 57.4 Kb, respectively. The assembly was corrected with the ONT reads using racon (https://github.com/isovic/racon) v1.4.13 with three rounds. We further improved the accuracy of the assembly by employing polish with 30.7 Gb PacBio HiFi reads using racon by two rounds. To obtain high‐accuracy assembly, the PacBio HiFi reads was used to de novo assemble the genome using Hifiasm (Cheng et al., 2021) v0.15.5‐r350 with parameter ‘‐10’ to disable duplication purging due to the very low heterozygosity rate of the gnome. In order to obtain the syntenic region between the assemblies from ONT ultra‐long and PacBio HiFi reads, we used MashMap (Jain et al., 2018) v2.0 to perform comparisons between two genomes with parameters ‘‐‐perc_identity 95 ‐‐segLength 50000 ‐‐filter_mode one‐to‐one’, and the dot plot was generated by generateDotPlot in MashMap. The contigs assembled from PacBio HiFi reads were used to link the break ends of some contigs from ONT data and then filled the gaps. In order to get the assembly with high continuity and high accuracy, the assembly sequences from ONT data with matched length >100 Kb were replaced with contig sequences from PacBio HiFi data. Next, we build the scaffold using SALSA (Ghurye et al., 2017) v2.3 with 50.9 Gb Hi‐C data. In short, we aligned the Hi‐C data to the replaced contigs using BWA (Li, 2013) v0.7.17‐r1188 with default parameter. Then, the SALSA was used to clyster the contigs into scaffold. To further improve the scaffold building and fill the gaps, we aligned the ONT and PacBio data to the above scaffold with parameters ‘‐‐MD ‐ax map‐ont’ and ‘‐‐MD ‐ax asm20’, respectively. Sniffles (Sedlazeck et al., 2018) v1.0.11 was used to call SVs for BAM file from ONT data using parameters ‘‐‐min_support 10 ‐‐min_length 50 ‐‐minmapping_qual 20 ‐‐num_reads_report ‐1 ‐‐min_seq_size 500 ‐‐genotype ‐‐report_BND ‐‐report‐seq’ and plus ‘‐‐ccs_reads’ for BAM file from PacBio HiFi data. The unanchored contigs were aligned to scaffold based on translocation (TRA) information, and gaps are filled using the sequences of INS. To obtain the final accurate assembly, the scaffold was polished with PacBio HiFi reads using racon, followed by polishing by pilon (Walker et al., 2014) v1.23 using Illumina data. To get the chromosome information of assembled scaffolds, we aligned the assembly to previously published genome of different subspecies variety IT97K‐499‐35 (Lonardi et al., 2019) using MashMap.
Quality evaluation of the assembly
To validate the final chromosome‐level genome assembly, the Hi‐C data were aligned to the genome and then processed with PretextMap (https://github.com/wtsi‐hpag/PretextMap) v0.1.6 to visually evaluate the chromosomes as a contact map in PretextView (https://github.com/wtsi‐hpag/PretextView) v0.2.5. To further evaluate the accuracy of the genome assembly, cleaned Illumina short reads were aligned on the Fengchan 6 genome using BWA with default parameters. Picard (https://broadinstitute.github.io/picard/) v2.18.29 was used to identify and remove duplicated reads caused by PCR in the process of library construction. Small variation calling was performed according to the Genome Analysis Toolkit (GATK) (Van der Auwera et al., 2013) v4.0.6.0 with parameters ‘QD < 2.0 || MQ < 40.0 || FS > 60.0 || ReadPosRankSum < ‐8.0’ applied to SNPs, and ‘QD < 2.0 || FS > 200.0 || ReadPosRankSum < ‐20.0’ applied to InDels. The completeness of gene regions was evaluated using BUSCO (Simao et al., 2015) v4.1.4 by searching the 1614 conserved protein models in the BUSCO embryophyta_odb10 dataset against Fengchan 6 genome with default parameters. In addition, we assessed the QV of genome assembly using Merqury (Rhie et al., 2020) v1.3 based on 19‐mer database from Illumina short reads. The QV was estimated for both the assemblies of Fengchan 6 and IT97K‐499‐35 whose genome sequences and annotation information were downloaded from the Legume Information System (https://data.legumeinfo.org/Vigna/unguiculata/genomes/IT97K‐499‐35.gnm1.QnBW/) (Lonardi et al., 2019). Illumina short reads of IT97K‐499‐35 were downloaded from NCBI with project accession PRJNA836573, and run ID is SRR19215719 (https://www.ncbi.nlm.nih.gov/sra/SRR19215719). The Xiabao II assembly (NCBI accession GCA_003958685) was also downloaded to evaluate the quality.
Genome annotation
For annotating the repetitive sequences of Fengchan 6 genome, the de novo repeat library was created using RepeatModeler (https://www.repeatmasker.org/RepeatModeler/) v1.0.3 with default parameters. The repetitive elements of the genome were predicted and masked by RepeartMasker (https://www.repeatmasker.org/) v4.1.0 using homologous identification by running against the Dfam database v3.1 and repeat library is created by RepeatModeler.
For gene structure annotation of the genome, we downloaded 256 277 protein sequences from genus Vigna in NCBI (https://www.ncbi.nlm.nih.gov/protein?term=txid3913[Organism]) v20220111. The redundant protein sequences were removed using CD‐HIT (Li and Godzik, 2006) v4.8.1 with parameters ‘cd‐hit ‐i protein.fa ‐o protein_cdhit.fa ‐c 0.9 ‐n 5 ‐d 0 ‐M 0 ‐T 24’. In this study, a total of 55.1 Gb clean RNA data from six tissues, namely root, stem, leaf, flower, pod and seed, were obtained, ranging from 8.3 to 10.0 Gb. The RNA data for each tissue were independently aligned to the genome using Hisat2 (Kim et al., 2015) v2.2.1with parameters ‘‐q ‐‐sensitive’. The BAM files were merged to perform reference‐based assembly of the transcripts using Trinity (Grabherr et al., 2011) v2.9.1 with parameters ‘‐‐genome_guided_bam merged.bam ‐‐genome_guided_max_intron 10000 ‐‐max_memory 100G’. The non‐redundant transcripts were obtained using CD‐HIT with parameters ‘cd‐hit‐est ‐i transcript.fa ‐o transcipt.cdhit.fa ‐c 0.9 ‐n 8 ‐d 0 ‐M 0 ‐T 24’. Finally, gene predictions based on ab initio approaches and transcript and protein evidence were integrated using the annotation pipeline of MAKER (Yandell, 2011) v3.01.03.
The function information of the predicted protein‐coding genes was annotated through homology searching against the public databases including NCBI non‐redundant (NR) proteins (v20211225), Swiss‐Prot and TrEMBL (v20211117) (Boeckmann, 2003), using Diamond (Buchfink et al., 2015) v2.0.11 (e‐value <1E‐5). Domains were identified by searching against Pfam database (Finn et al., 2016) v35.0 using HMMER (Mistry et al., 2013) v3.1b2. The terms of KEGG and GO were obtained based on homologous alignment against the genes of Arabidopsis (TAIR10).
For non‐coding RNA (ncRNA) annotation, tRNAscan‐SE (Chan et al., 2021) v2.0.7 was used to identify transfer RNAs (tRNAs), and Infernal (Nawrocki and Eddy, 2013) v1.1.4 was used to identify microRNA (miRNA), ribosome RNA (rRNA) and small nuclear RNA (snRNA) by searching against Rfam database (Griffiths‐Jones et al., 2003) (v20210517).
Identification of centromeric and telomeric sequences
The centromeric regions were defined based on the 455 bp centromere‐specific satellite sequence that was previously identified in V. unguiculata by fluorescence in situ hybridization (FISH) (Iwata‐Otsubo et al., 2016). In addition, we also used the tandem repeats 721 bp (DDBJ ID: LC490941) and 1600 bp (DDBJ ID: LC490942) identified by (Ishii et al., 2020). We aligned the above sequences to the assembly genome using BLAST with e‐value <1E‐5 and sequence coverage >80%. The heatmap of sequence identity in centromere was displayed using StainedGlass (Vollger et al., 2022b) v0.4. The tracks of satellite and transposon elements around the centromeres were plotted by pyGenomeTracks (Lopez‐Delisle et al., 2021) v3.3.
Structural variation (SV) analysis
We downloaded the previously public PacBio CLR data (154.8 Gb) of IT97K‐499‐35 via NCBI project accession PRJNA325510. After quality control, we aligned the clean reads to the Fengchan 6 genome using minimap2 with parameters ‘‐‐MD ‐a ‐x map‐pb’. Afterwards, we used sniffles (Sedlazeck et al., 2018) v1.0.12 to call four type SVs, namely deletion (DEL), INS, duplication (DUP) and INV, with length >50 bp Using parameters ‘‐‐min_support 10 ‐‐min_length 50 ‐‐minmapping_qual 20 ‐‐num_reads_report ‐1 ‐‐min_seq_size 500 ‐‐genotype ‐‐report_BND ‐‐report‐seq’.
Segmental duplication and tandemly duplicated genes
Segmental duplications were identified using SEDEF (Numanagic et al., 2018) v1.1 with default parameters after repeat masking with RepeatMasker. so that only regions with homology outside of common repeat elements would be identified by SEDEF.
Tandemly duplicated genes were detected through an all‐against‐all comparison of the genome proteins using Diamond (Buchfink et al., 2015) v2.0.11. The output was filtered according to the following parameters: an e‐value <10E−20 and a coverage of the smallest protein greater than 80% (Belser et al., 2021). Genes were considered as tandemly duplicated if they were co‐localized on the same chromosome and located within a distance of no more than 10 genes for each gene pair. Sequentially, sequential detected tandem genes were grouped into clusters.
Identification of NBS‐LRR genes and gene families involved in anthocyanin biosynthesis
To identify NBS‐LRR genes, predicted protein sequences of 30 594 genes were initially scanned for the hidden Markov model (HMM) profile of NBS/NB‐ARC domain (pfam00931) in HMMER (Mistry et al., 2013) v3.1b2 using hmmsearch with e‐value <1E‐10 and bit score >50.
Based on a previous study (Li et al., 2020), we constructed the anthocyanin biosynthesis pathway and compiled the gene families associated with the pathway. Comparative analysis with homologous genes in Arabidopsis allowed us to identify genes encoding the enzymes involved in anthocyanin biosynthesis in V. unguiculata. The protein‐coding genes in Arabidopsis involved in anthocyanin biosynthesis were aligned against the genome‐wide gene set of Fengchan 6 using BLASTP with an e‐value <1E‐10 and identity >50%.
Methylation analysis for ONT ultra‐long reads
The ONT ultra‐long reads were aligned to the assembly in this study using minimap2. BAM files were filtered for primary alignments with SAMtools (Danecek et al., 2021) v1.12. Then we used Nanopolish to measure CpG methylation. The methylation calls were filtered using the nanopore_methylation_utilities tool (https://github.com/timplab/nanopore‐methylation‐utilities) using the threshold of a log‐likelihood ratio of 1.5. CpG sites with log‐likelihood ratios greater than 1.5 (methylated) were considered high quality and included in the analysis (Gershman et al., 2022).
Bulked segregant analysis
Genomic DNAs from 30 individuals were mixed to create bulked pools of purple and light green pods, respectively. And the genomic DNAs of two bulks and two parents were extracted from young leaves and used to constructed PE libraries for sequencing. To identify SNPs and InDels, the cleaned reads were mapped to the assemble genome Fengchan 6 using BWA (Li, 2013) v0.7.17‐r1188 with parameter ‘mem ‐M’. Variant calling was performed using GATK with aforementioned parameters. And all variants were annotated using SnpEff (Cingolani et al., 2012) v5.0. Association analysis was performed using the SNP‐index (Abe et al., 2012) and the Δ(SNP‐Index) (Takagi et al., 2013).
Differentially expressed genes analysis
A total of six RNA libraries were constructed, representing three replicates each of varieties PRS and WSS1. The libraries were sequenced using Illumina to obtain 2 × 150 bp PE reads. After quality control, approximately 7.4 Gb of clean data per repeat were obtained (ranging from 6.1 to 9.4 Gb). Afterwards, the cleaned reads were aligned to the assembly genome in this study using Hisat2 (Kim et al., 2015) v2.2.1. The feature Counts (Liao et al., 2014) v1.6.4 was used to count the reads for exons of the genes, and then the reads per kilobase per million mapped reads (RPKM) were calculated for each replicates. The DEGs between the varieties was detected using edgeR (Robinson et al., 2010) v3.36.0, with the criterion fold‐change >2 and FDR <0.01. The GO and KEGG terms of the genes were obtained through aligning then to the genes of Arabidopsis. GO and KEGG enrichments were analysed by Fisher's exact test, and corrected P values were calculated using Benjamini–Hochberg method.
Metabolome detection and analysis
The freeze‐dried leaf was crushed using a mixer mill (MM 400, Retsch) with a zirconia bead for 1.5 min at 30 Hz. The 100 mg powder was weighed and extracted overnight at 4 °C with 0.6 mL 70% of aqueous methanol. Then the extracts were centrifuged at 10 000 g for 10 min and filtrated (SCAA‐104, 0.22 μm pore size) before UPLC‐MS/MS analysis. The sample extracts were analysed using an UPLC‐ESI‐MS/MS system (UPLC, Shim‐pack UFLC SHIMADZU CBM30A system; MS, Applied Biosystems 4500 QTRAP) equipped with a Waters ACQUITY UPLC HSS T3 C18 column (1.8 μm, 2.1 mm × 100 mm). The mobile phase was consisted of solvent A (pure water with 0.04% acetic acid) and solvent B (acetonitrile with 0.04% acetic acid). The gradient was set followed previous study (Zhang et al., 2021). The column oven was set 40 °C, and the injection volume was 4 μL. The effluent was connected alternately to an ESI‐triple quadrupole‐linear ion trap (QTRAP)‐MS. The MS parameters were set as follows: ion spray voltage with 5500 V, electrospray ionization temperature was set to 55 °C, and curtain gas was set to 25.0 psi (Zhang et al., 2021). Data processing was performed using Analyst 1.6.3 software (AB Sciex). Significantly regulated metabolites between groups were determined based on a VIP ≥ 1 and absolute Log2FC (fold change) ≥ 1. VIP values were extracted from the OPLS‐DA result, which also contain score plots and permutation plots and was generated using R package MetaboAnalystR (Chong and Xia, 2018) v2.0.
Plasmid construction and plant genetic transformation
The CDS of the candidate gene was cloned and then transferred into a pEGOEP35S‐H‐ binary vector. The detail of the vector construction as described according to In‐fusion technology (https://www.takarabio.com/). The constructed vector was transferred into Agrobacterium tumefaciens strain GV3101 and then transformed into the wild‐type Arabidopsis Columbia line.
KASP marker development and validation
In order to develop KASP markers for VuMYB114, sequences containing the two‐base variation were converted into KASP marker. The KASP primer pair, VuP‐KASP1, which consisting of forward primer (Primer_AlleleFAM and Primer_AlleleHEX) and reverse primers (Primer_Common) (Table S19), were synthesized. KASP PCR reactions were performed in 96‐well microplates in 10 μL reaction volumes. Each reaction contained approximately 1.0 μL of DNA (10.0 ng/μL), 5.0 μL of KASP 2× Master Mix, 0.1 μL of primer mixture (10 ng/μL) and 5 μL distilled deionized water. The PCR amplification protocol was as follows: 95 °C for 10 min, followed by 10 cycles of 95 °C for 15 s and touchdown from 61 °C to 55 °C for 1 min, and then 28 cycles of 95 °C for 15 s and 55 °C for 1 min. The KASP data were visualized and exported using the Bio‐Rad CFX Manager 3.1 software package (Bio‐Rad, Inc.).
Accession numbers
All data supporting the findings of this study are available in the paper and the supplemental information files. All the sequencing data with fastq format have been deposited in Genome Sequence Archive (GSA) the National Genomics Data Center (NGDC) under accession CRA007957 (https://ngdc.cncb.ac.cn/gsa/browse/CRA007957). The assembly genome sequence of Fengchan 6 has been deposited to in the Genome Warehouse of NGDC under accession GWHCBIQ00000000 (https://ngdc.cncb.ac.cn/gwh/Assembly/37824/show).
The codes of genome assembly pipeline in this study are publicly available via GitHub repository (https://github.com/ZhikunWu/T2T‐Assembly).
Conflict of interest
The authors declare no competing interests.
Author contributions
The authors confirm contribution to the paper as follows: Y.Z. conceived and supervised the work. Y.Z., Z.K.W. and Y.Y. designed the study. Y.Y. and Z.K.W. analysed the data. Y.Y., Z.X.W., T.Y.L., Z.S. and X.Z. conducted the experiments. Y.Y. and Z.K.W. prepared the manuscript. Z.K.W., Y.Y. and Y.Z. wrote the manuscript. G.J.L. and X.Y.W. revised the manuscript. All authors read and approved the manuscript.
Supporting information
Figure S1 Genome size estimate of Fengchan 6. (a) Genome size estimate based on Illumina short reads. (b) Genome size estimate based on PacBio HiFi reads.
Figure S2 Tree maps of assembled contigs. (a) Tree map of assembled contigs from ONT ultra‐long reads. (b) Tree map of assembled contigs from PacBio HiFi reads.
Figure S3 The syntenic regions of the assemblies. The illustration of dot plots showing syntenic regions between two assemblies. (a) Horizontal axis shows the coordinate of contigs from ONT ultra‐long reads, and the vertical axis shows the coordinate of contigs from PacBio HiFi reads. (b) Horizontal axis shows the coordinate of contigs from ONT data after linking by PacBio contigs, and the vertical axis shows the coordinate of contigs from PacBio HiFi reads. (c) Horizontal axis shows the coordinate of contigs from ONT data after replacing them with PacBio contigs for the synteny regions, and the vertical axis shows the coordinate of previously published assembly IT97K‐499‐35. (d) Horizontal axis shows the coordinate of the assembly derived from ONT contigs after scaffold building using Hi‐C data, and the vertical axis shows the coordinate of previously published assembly IT97K‐499‐35.
Figure S4 The read coverage for the region of chromosome V03. Distributions of Illumina short reads, PacBio HiFi reads and ONT ultra‐long reads for Fengchan 6 genome for region 2.0–4.5 Mb in chromosomes Vu03.
Figure S5 Sequences identities of the centromeric regions of 11 chromosomes.
Figure S6 Sequence characterization of centromeric regions. The features from top to bottom are densities of gene, satellite, Gyspy, Copia and CG methylation.
Figure S7 The percentages of CG methylation of sequences in centromeric and non‐centromeric regions. In the boxplots, the upper and lower hinges represent the first and third quartiles, respectively. The whiskers extended to the most extreme value within 1.5 times the interquartile range on both end of the distribution. The centre line represented the median.
Figure S8 Phenotype cVuMYB114 transgenic Arabidopsis.
Table S1 Summary of the cleaned sequencing data for Fengchan 6.
Table S2 The statistics of de novo assemblies using different methods.
Table S3 Summary of Fengchan 6 assembly.
Table S4 Telomere repeat abundances of chromosomes.
Table S5 Evaluation of base accuracy of Fengchan 6 assemblies.
Table S6 Statistics of repetitive elements in Fengchan 6 assembly.
Table S7. Statistics of functional annotation of predcted protein‐coding genes.
Table S8 Summary of predcted noncoding genes.
Table S9 Predicted core centromeric regions.
Table S10 The number of centromere‐specific satellites.
Table S11 Statistics of repetitive elements in core centromeric regions.
Table S12 Summary of structural variations between Fengchan 6 and IT97K‐499‐35 revealed by PacBio CLR reads.
Table S13 Hotspots of SVs between Fengchan 6 and IT97K‐499‐35.
Table S14 Unique segmental duplication regions.
Table S15 Summary of clusters of tandem duplicated genes.
Table S16 Summary of predicted NBS‐LRR genes.
Table S17 Different metabolites in flavonoid and anthocyanin biosynthesis between WSS1 and PRS.
Table S18 Genes and corresponding expressions of PRS and WSS1 in genomic region revealed by BSA.
Table S19 KASP marker VuP‐KASP1 developed from the VuMYB114 genome DNA sequence region primer information.
Acknowledgements
This study was supported by the Natural Science Foundation of Guangdong Province (Grant No. 2022A1515010281), Key Realm R & D Program of Guangdong Province (Grant No. 2020B020220002) and the Innovation Fund Projects of the Guangdong Academy of Agricultural Sciences (Grant No. 202107). We are deeply thankful to Dr. Mayank Gautam for editing the English version of the manuscript. We would like to extend our gratitude to Tong Li for the invaluable assistance and insightful suggestions during the data analysis process.
References
- Abe, A. , Kosugi, S. , Yoshida, K. , Natsume, S. , Takagi, H. , Kanzaki, H. , Matsumura, H. et al. (2012) Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 30, 174–178. [DOI] [PubMed] [Google Scholar]
- Bailey, J.A. , Yavor, A.M. , Massa, H.F. , Trask, B.J. and Eichler, E.E. (2001) Segmental duplications: organization and impact within the current human genome project assembly. Genome Res. 11, 1005–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belser, C. , Baurens, F.C. , Noel, B. , Martin, G. , Cruaud, C. , Istace, B. , Yahiaoui, N. et al. (2021) Telomere‐to‐telomere gapless chromosomes of banana using nanopore sequencing. Commun. Biol. 4, 1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckmann, B. (2003) The SWISS‐PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink, B. , Xie, C. and Huson, D.H. (2015) Fast and sensitive protein alignment using DIAMOND. Nat. Methods, 12, 59–60. [DOI] [PubMed] [Google Scholar]
- Chan, P.P. , Lin, B.Y. , Mak, A.J. and Lowe, T.M. (2021) tRNAscan‐SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, S. , Zhou, Y. , Chen, Y. and Gu, J. (2018) fastp: an ultra‐fast all‐in‐one FASTQ preprocessor. Bioinformatics, 34, i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, H. , Concepcion, G.T. , Feng, X. , Zhang, H. and Li, H. (2021) Haplotype‐resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods, 18, 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong, J. and Xia, J. (2018) MetaboAnalystR: an R package for flexible and reproducible analysis of metabolomics data. Bioinformatics, 34, 4313–4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani, P. , Platts, A. , Wang le, L. , Coon, M. , Nguyen, T. , Wang, L. , Land, S.J. et al. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly (Austin), 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek, P. , Bonfield, J.K. , Liddle, J. , Marshall, J. , Ohan, V. , Pollard, M.O. , Whitwham, A. et al. (2021) Twelve years of SAMtools and BCFtools. Gigascience, 10, giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Coster, W. , D'Hert, S. , Schultz, D.T. , Cruts, M. and Van Broeckhoven, C. (2018) NanoPack: visualizing and processing long‐read sequencing data. Bioinformatics, 34, 2666–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, Y. , Liu, S. , Zhang, Y. , Tan, J. , Li, X. , Chu, X. , Xu, B. et al. (2022) A telomere‐to‐telomere gap‐free reference genome of watermelon and its mutation library provide important resources for gene discovery and breeding. Mol. Plant, 15, 1268–1284. [DOI] [PubMed] [Google Scholar]
- Finn, R.D. , Coggill, P. , Eberhardt, R.Y. , Eddy, S.R. , Mistry, J. , Mitchell, A.L. , Potter, S.C. et al. (2016) The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershman, A. , Sauria, M.E.G. , Guitart, X. , Vollger, M.R. , Hook, P.W. , Hoyt, S.J. , Jain, M. et al. (2022) Epigenetic patterns in a complete human genome. Science, 376, eabj5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghurye, J. , Pop, M. , Koren, S. , Bickhart, D. and Chin, C.S. (2017) Scaffolding of long read assemblies using long range contact information. BMC Genomics, 18, 527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr, M.G. , Haas, B.J. , Yassour, M. , Levin, J.Z. , Thompson, D.A. , Amit, I. , Adiconis, X. et al. (2011) Full‐length transcriptome assembly from RNA‐Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths‐Jones, S. , Bateman, A. , Marshall, M. , Khanna, A. and Eddy, S.R. (2003) Rfam: an RNA family database. Nucleic Acids Res. 31, 439–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppel, S.C. , Jaffe, F.W. , Takos, A.M. , Schellmann, S. , Rausch, T. , Walker, A.R. and Bogs, J. (2013) Identification of key amino acids for the evolution of promoter target specificity of anthocyanin and proanthocyanidin regulating MYB factors. Plant Mol. Biol. 82, 457–471. [DOI] [PubMed] [Google Scholar]
- Hon, T. , Mars, K. , Young, G. , Tsai, Y.C. , Karalius, J.W. , Landolin, J.M. , Maurer, N. et al. (2020) Highly accurate long‐read HiFi sequencing data for five complex genomes. Sci. Data, 7, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii, T. , Juranic, M. , Maheshwari, S. , Bustamante, F.O. , Vogt, M. , Salinas‐Gamboa, R. , Dreissig, S. et al. (2020) Unequal contribution of two paralogous CENH3 variants in cowpea centromere function. Commun. Biol. 3, 775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata‐Otsubo, A. , Lin, J.Y. , Gill, N. and Jackson, S.A. (2016) Highly distinct chromosomal structures in cowpea (Vigna unguiculata), as revealed by molecular cytogenetic analysis. Chromosome Res. 24, 197–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain, C. , Koren, S. , Dilthey, A. , Phillippy, A.M. and Aluru, S. (2018) A fast adaptive algorithm for computing whole‐genome homology maps. Bioinformatics, 34, i748–i756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. , Langmead, B. and Salzberg, S.L. (2015) HISAT: a fast spliced aligner with low memory requirements. Nat. Methods, 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Camera, S. , Balagué, C. , Göbel, C. , Geoffroy, P. , Legrand, M. , Feussner, I. , Roby, D. et al. (2009) The Arabidopsis patatin‐like protein 2 (PLP2) plays an essential role in cell death execution and differentially affects biosynthesis of oxylipins and resistance to pathogens. MPMI, 22, 469–481. [DOI] [PubMed] [Google Scholar]
- Lai, Y. , Li, H. and Yamagishi, M. (2013) A review of target gene specificity of flavonoid R2R3‐MYB transcription factors and a discussion of factors contributing to the target gene selectivity. Front. Biol. 8, 577–598. [Google Scholar]
- Li, H. (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. arXiv .
- Li, K. , Jiang, W. , Hui, Y. , Kong, M. , Feng, L.Y. , Gao, L.Z. , Li, P. et al. (2021) Gapless indica rice genome reveals synergistic contributions of active transposable elements and segmental duplications to rice genome evolution. Mol. Plant, 14, 1745–1756. [DOI] [PubMed] [Google Scholar]
- Li, W. and Godzik, A. (2006) Cd‐hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics, 22, 1658–1659. [DOI] [PubMed] [Google Scholar]
- Li, Y. , Chen, Q. , Xie, X. , Cai, Y. , Li, J. , Feng, Y. and Zhang, Y. (2020) Integrated metabolomics and transcriptomics analyses reveal the molecular mechanisms underlying the accumulation of anthocyanins and other flavonoids in cowpea pod (Vigna unguiculata L.). J. Agric. Food Chem. 68, 9260–9275. [DOI] [PubMed] [Google Scholar]
- Liao, Y. , Smyth, G.K. and Shi, W. (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics, 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lonardi, S. , Munoz‐Amatriain, M. , Liang, Q. , Shu, S. , Wanamaker, S.I. , Lo, S. , Tanskanen, J. et al. (2019) The genome of cowpea (Vigna unguiculata [L.] Walp.). Plant J. 98, 767–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Delisle, L. , Rabbani, L. , Wolff, J. , Bhardwaj, V. , Backofen, R. , Gruning, B. , Ramirez, F. et al. (2021) pyGenomeTracks: reproducible plots for multivariate genomic datasets. Bioinformatics, 37, 422–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marçais, G. and Kingsford, C. (2011) A fast, lock‐free approach for efficient parallel counting of occurrences of k‐mers. Bioinformatics, 27, 764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos, A.R. , d'Arcy‐Lameta, A. , França, M. , Zuily‐Fodil, Y. and Pham‐Thi, A.T. (2000) A patatin‐like protein with galactolipase activity is induced by drought stress in Vigna unguiculata leaves. Biochem. Soc. Trans. 28, 779–781. [PubMed] [Google Scholar]
- Mazewski, C. , Liang, K. and Gonzalez de Mejia, E. (2018) Comparison of the effect of chemical composition of anthocyanin‐rich plant extracts on colon cancer cell proliferation and their potential mechanism of action using in vitro, in silico, and biochemical assays. Food Chem. 242, 378–388. [DOI] [PubMed] [Google Scholar]
- Mistry, J. , Finn, R.D. , Eddy, S.R. , Bateman, A. and Punta, M. (2013) Challenges in homology search: HMMER3 and convergent evolution of coiled‐coil regions. Nucleic Acids Res. 41, e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naish, M. , Alonge, M. , Wlodzimierz, P. , Tock, A.J. , Abramson, B.W. , Schmucker, A. , Mandakova, T. et al. (2021) The genetic and epigenetic landscape of the Arabidopsis centromeres. Science, 374, eabi7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki, E.P. and Eddy, S.R. (2013) Infernal 1.1: 100‐fold faster RNA homology searches. Bioinformatics, 29, 2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numanagic, I. , Gokkaya, A.S. , Zhang, L. , Berger, B. , Alkan, C. and Hach, F. (2018) Fast characterization of segmental duplications in genome assemblies. Bioinformatics, 34, i706–i714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk, S. , Korean, S. , Rhie, A. , Rautiainen, M. , Bzikadze, A.V. , Mikheenko, A. , Vollger, M.R. et al. (2022) The complete sequence of a human genome. Science, 376, 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pootakham, W. , Nawae, W. , Naktang, C. , Sonthirod, C. , Yoocha, T. , Kongkachana, W. , Sangsrakru, D. et al. (2021) A chromosome‐scale assembly of the black gram (Vigna mungo) genome. Mol. Ecol. Resour. 21, 238–250. [DOI] [PubMed] [Google Scholar]
- Ranallo‐Benavidez, T.R. , Jaron, K.S. and Schatz, M.C. (2020) GenomeScope 2.0 and Smudgeplot for reference‐free profiling of polyploid genomes. Nat. Commun. 11, 1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhie, A. , Walenz, B.P. , Koren, S. and Phillippy, A.M. (2020) Merqury: reference‐free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, M.D. , McCarthy, D.J. and Smyth, G.K. (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagi, M.S. , Deokar, A.A. and Tar'an, B. (2017) Genetic analysis of NBS‐LRR gene family in chickpea and their expression profiles in response to ascochyta blight infection. Front. Plant Sci. 8, 838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scelfo, A. and Fachinetti, D. (2019) Keeping the centromere under control: a promising role for DNA methylation. Cell, 8, 912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlazeck, F.J. , Rescheneder, P. , Smolka, M. , Fang, H. , Nattestad, M. , von Haeseler, A. and Schatz, M.C. (2018) Accurate detection of complex structural variations using single‐molecule sequencing. Nat. Methods, 15, 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simao, F.A. , Waterhouse, R.M. , Ioannidis, P. , Kriventseva, E.V. and Zdobnov, E.M. (2015) BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Song, J.M. , Xie, W.Z. , Wang, S. , Guo, Y.X. , Koo, D.H. , Kudrna, D. , Gong, C. et al. (2021) Two gap‐free reference genomes and a global view of the centromere architecture in rice. Mol. Plant, 14, 1757–1767. [DOI] [PubMed] [Google Scholar]
- Takagi, H. , Abe, A. , Yoshida, K. , Kosugi, S. , Natsume, S. , Mitsuoka, C. , Uemura, A. et al. (2013) QTL‐seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J., 74, 174–183. [DOI] [PubMed] [Google Scholar]
- Takahashi, Y. , Kongjaimun, A. , Muto, C. , Kobayashi, Y. , Kumagai, M. , Sakai, H. , Satou, K. et al. (2020) Same locus for non‐shattering seed pod in two independently domesticated legumes, Vigna angularis and Vigna unguiculata . Front. Genet. 11, 748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, Y. , Sakai, H. , Ariga, H. , Teramoto, S. , Shimada, T.L. , Eun, H. , Muto, C. et al. (2023) Domesticating Vigna stipulacea: chromosome‐level genome assembly reveals VsPSAT1 as a candidate gene decreasing hard‐seededness. Front. Plant Sci. 14, 1119625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbert, P.B. and Henikoff, S. (2022) The genetics and epigenetics of satellite centromeres. Genome Res. 32, 608–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera, G.A. , Carneiro, M.O. , Hartl, C. , Poplin, R. , Del Angel, G. , Levy‐Moonshine, A. , Jordan, T. et al. (2013) From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics, 43, 11 10 11–11 10 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollger, M.R. , Guitart, X. , Dishuck, P.C. , Mercuri, L. , Harvey, W.T. , Gershman, A. , Diekhans, M. et al. (2022a) Segmental duplications and their variation in a complete human genome. Science, 376, eabj6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollger, M.R. , Kerpedjiev, P. , Phillippy, A.M. and Eichler, E.E. (2022b) StainedGlass: Interactive visualization of massive tandem repeat structures with identity heatmaps. Bioinformatics, 38, 2049–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, B.J. , Abeel, T. , Shea, T. , Priest, M. , Abouelliel, A. , Sakthikumar, S. , Cuomo, C.A. et al. (2014) Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One, 9, e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, B. , Yang, X. , Jia, Y. , Xu, Y. , Jia, P. , Dang, N. , Wang, S. et al. (2021) High‐quality Arabidopsis thaliana genome assembly with Nanopore and HiFi long reads. Genomics Proteomics Bioinformatics, 20, 4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. , Yang, C. , Wu, X. , Wang, Y. , Wang, B. , Wu, X. , Lu, Z. et al. (2022) Genome‐wide characterization of NBS‐LRR family genes and expression analysis under powdery mildew stress in Lagenaria siceraria . Physiol. Mol. Plant Pathol. 118, 101798. [Google Scholar]
- Wlodzimierz, P. , Rabanal, F.A. , Burns, R. , Naish, M. , Primetis, E. , Scott, A. , Mandakova, T. et al. (2023) Cycles of satellite and transposon evolution in Arabidopsis centromeres. Nature, 618, 557–565. [DOI] [PubMed] [Google Scholar]
- Xia, Q. , Pan, L. , Zhang, R. , Ni, X. , Wang, Y. , Dong, X. , Gao, Y. et al. (2019) The genome assembly of asparagus bean, Vigna unguiculata ssp. sesquipedialis. Sci. Data, 6, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, S. , Lei, Y. , Chen, H. , Li, J. , Chen, H. and Zhang, Z. (2020) R2R3‐MYB transcription factors regulate anthocyanin biosynthesis in grapevine vegetative tissues. Front. Plant Sci. 11, 527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, L. , Wang, Y. , Dong, J. , Zhang, W. , Tang, M. , Zhang, W. , Wang, K. et al. (2023) A chromosome‐level genome assembly of radish (Raphanus sativus L.) reveals insights into genome adaptation and differential bolting regulation. Plant Biotechnol. J. 21, 990–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, P. , Wu, X. , Munoz‐Amatriain, M. , Wang, B. , Wu, X. , Hu, Y. , Huynh, B.L. et al. (2017) Genomic regions, cellular components and gene regulatory basis underlying pod length variations in cowpea (V. unguiculata L. Walp). Plant Biotechnol. J. 15, 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, W. , Dubos, C. and Lepiniec, L. (2015) Transcriptional control of flavonoid biosynthesis by MYB‐bHLH‐WDR complexes. Trends Plant Sci. 20, 176–185. [DOI] [PubMed] [Google Scholar]
- Yan, H. , Pei, X. , Zhang, H. , Li, X. , Zhang, X. , Zhao, M. , Chiang, V.L. et al. (2021) MYB‐mediated regulation of anthocyanin biosynthesis. Int. J. Mol. Sci. 22, 3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yandell, C.H.M. (2011) MAKER2 an annotation pipeline and genome‐database management tool for second‐generation genome projects. BMC Bioinformatics, 12, 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Liang, J. , Chen, H. , Zhang, Z. , Wu, J. and Wang, X. (2023) A near‐complete genome assembly of Brassica rapa provides new insights into the evolution of centromeres. Plant Biotechnol. J. 21, 1022–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q. , Yang, W. , Liu, J. , Liu, H. , Lv, Z. , Zhang, C. , Chen, D. et al. (2021) Postharvest UV‐C irradiation increased the flavonoids and anthocyanins accumulation, phenylpropanoid pathway gene expression, and antioxidant activity in sweet cherries (Prunus avium L.). Postharvest Biol. Technol. 175, 111490. [Google Scholar]
- Zheng, J. , Wu, H. , Zhu, H. , Huang, C. , Liu, C. , Chang, Y. , Kong, Z. et al. (2019) Determining factors, regulation system, and domestication of anthocyanin biosynthesis in rice leaves. New Phytol. 223, 705–721. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Genome size estimate of Fengchan 6. (a) Genome size estimate based on Illumina short reads. (b) Genome size estimate based on PacBio HiFi reads.
Figure S2 Tree maps of assembled contigs. (a) Tree map of assembled contigs from ONT ultra‐long reads. (b) Tree map of assembled contigs from PacBio HiFi reads.
Figure S3 The syntenic regions of the assemblies. The illustration of dot plots showing syntenic regions between two assemblies. (a) Horizontal axis shows the coordinate of contigs from ONT ultra‐long reads, and the vertical axis shows the coordinate of contigs from PacBio HiFi reads. (b) Horizontal axis shows the coordinate of contigs from ONT data after linking by PacBio contigs, and the vertical axis shows the coordinate of contigs from PacBio HiFi reads. (c) Horizontal axis shows the coordinate of contigs from ONT data after replacing them with PacBio contigs for the synteny regions, and the vertical axis shows the coordinate of previously published assembly IT97K‐499‐35. (d) Horizontal axis shows the coordinate of the assembly derived from ONT contigs after scaffold building using Hi‐C data, and the vertical axis shows the coordinate of previously published assembly IT97K‐499‐35.
Figure S4 The read coverage for the region of chromosome V03. Distributions of Illumina short reads, PacBio HiFi reads and ONT ultra‐long reads for Fengchan 6 genome for region 2.0–4.5 Mb in chromosomes Vu03.
Figure S5 Sequences identities of the centromeric regions of 11 chromosomes.
Figure S6 Sequence characterization of centromeric regions. The features from top to bottom are densities of gene, satellite, Gyspy, Copia and CG methylation.
Figure S7 The percentages of CG methylation of sequences in centromeric and non‐centromeric regions. In the boxplots, the upper and lower hinges represent the first and third quartiles, respectively. The whiskers extended to the most extreme value within 1.5 times the interquartile range on both end of the distribution. The centre line represented the median.
Figure S8 Phenotype cVuMYB114 transgenic Arabidopsis.
Table S1 Summary of the cleaned sequencing data for Fengchan 6.
Table S2 The statistics of de novo assemblies using different methods.
Table S3 Summary of Fengchan 6 assembly.
Table S4 Telomere repeat abundances of chromosomes.
Table S5 Evaluation of base accuracy of Fengchan 6 assemblies.
Table S6 Statistics of repetitive elements in Fengchan 6 assembly.
Table S7. Statistics of functional annotation of predcted protein‐coding genes.
Table S8 Summary of predcted noncoding genes.
Table S9 Predicted core centromeric regions.
Table S10 The number of centromere‐specific satellites.
Table S11 Statistics of repetitive elements in core centromeric regions.
Table S12 Summary of structural variations between Fengchan 6 and IT97K‐499‐35 revealed by PacBio CLR reads.
Table S13 Hotspots of SVs between Fengchan 6 and IT97K‐499‐35.
Table S14 Unique segmental duplication regions.
Table S15 Summary of clusters of tandem duplicated genes.
Table S16 Summary of predicted NBS‐LRR genes.
Table S17 Different metabolites in flavonoid and anthocyanin biosynthesis between WSS1 and PRS.
Table S18 Genes and corresponding expressions of PRS and WSS1 in genomic region revealed by BSA.
Table S19 KASP marker VuP‐KASP1 developed from the VuMYB114 genome DNA sequence region primer information.