

Abstract
Background
Glioblastomas (GBMs) display striking dysregulation of metabolism to promote tumor growth. Glioblastoma stem cells (GSCs) adapt to regions of heterogeneous nutrient availability, yet display dependency on de novo cholesterol biosynthesis. The transcription factor Sterol Regulatory Element-Binding Protein 2 (SREBP2) regulates cholesterol biosynthesis enzymes and uptake receptors. Here, we investigate adaptive behavior of GSCs under different cholesterol supplies.
Methods
In silico analysis of patient tumors demonstrated enrichment of cholesterol synthesis associated with decreased angiogenesis. Comparative gene expression of cholesterol biosynthesis enzymes in paired GBM specimens and GSCs were performed. In vitro and in vivo loss-of-function genetic and pharmacologic assays were conducted to evaluate the effect of SREBP2 on GBM cholesterol biosynthesis, proliferation, and self-renewal. Chromatin immunoprecipitation quantitative real-time PCR was leveraged to map the regulation of SREBP2 to cholesterol biosynthesis enzymes and uptake receptors in GSCs.
Results
Cholesterol biosynthetic enzymes were expressed at higher levels in GBM tumor cores than in invasive margins. SREBP2 promoted cholesterol biosynthesis in GSCs, especially under starvation, as well as proliferation, self-renewal, and tumor growth. SREBP2 governed the balance between cholesterol biosynthesis and uptake in different nutrient conditions.
Conclusions
SREBP2 displays context-specific regulation of cholesterol biology based on its availability in the microenvironment with induction of cholesterol biosynthesis in the tumor core and uptake in the margin, informing a novel treatment strategy for GBM.
Keywords: cholesterol biosynthesis, cholesterol, glioblastoma, glioblastoma stem cell, SREBP2
Key Points.
In GBM, cholesterol biosynthesis is spatially regulated with greater activity in the tumor core.
SREBP2 promotes GSC maintenance and in vivo tumor growth.
SREBP2 balances cholesterol levels based on local availability with differential biosynthesis or uptake.
Importance of the Study.
Here, we demonstrate that stem-like tumor cells display spatial heterogeneity in cholesterol availability with adaptation through switching from cholesterol biosynthesis to cholesterol uptake. SREBP2 serves as a master regulator of cholesterol synthesis and regulation, serving as a rheostat in tumor cell interactions with the microenvironment. Genetic and pharmacologic targeting of SREBP2 attenuated GSC proliferation, stemness, and in vivo tumor growth. As spatial variation in nutrient availability promotes adaptive tumor metabolism and growth, SREBP2 represents a central signaling node amenable to therapeutic targeting for glioblastoma therapy.
Glioblastoma (GBM) is the most common and aggressive type of human primary brain tumor, residing in the most cholesterol-rich organ of the body.1–3 Despite extensive molecular characterization, the prognosis of GBM is extremely poor.4,5 GBM remains essentially fatal with median survival of all patients of no more than 1 year, even under standard-of-care therapy consisting of maximal surgical resection, radiotherapy, and adjuvant chemotherapy.6–8 Intra-tumoral heterogeneity derived from interactions between tumor genetics, epigenetics, and microenvironmental interactions contributes to therapeutic failure and malignancy of GBM. GBM displays a cellular hierarchy of differentiation with glioblastoma stem cells (GSCs) at the apex.9–12 GSCs are functionally defined through self-renewal, sustained proliferation, and tumor initiation.13–16
Dysregulated cell growth requires cellular adaption to nutrient availability for tumor initiation and growth. Within the tumor microenvironment, nutrient availability is determined by the accumulation of myriad factors, including cell type and density, proximity to vasculature, etc. Metabolic states are often dichotomized between oxidative phosphorylation and glycolysis, but GSCs display striking plasticity to adapt to regions of abundant or insufficient nutrients.17–19 GBM tumor margins where tumor cells invade into normal brain and coopt existing vasculature enjoy relative abundance of oxygen and nutrients. Other regions are highly vascular, but the vasculature is often inefficient and delivery can be impaired. Finally, the tumor core is often nutrient-deficient and can include necrotic regions with hypoxia and inflammation.20 We previously reported that GBM commonly has dysregulated cholesterol metabolism associated with epigenetic induction of the cholesterol synthetic pathway.21 Cholesterol levels are determined by a balance between biosynthesis and uptake to increase intracellular levels with export and esterification reducing available cholesterol.22 The Sterol Regulatory Element-Binding Protein 2 (SREBP2) is a master regulator of the cholesterol biosynthetic pathway through transcriptional regulation, including the rate-limiting enzymes of the biosynthetic pathway.23 Cholesterol circulates in low-density lipoproteins and is taken up by binding the low-density lipoprotein receptor (LDLR), which is also regulated by SREBP2.23–25 Thus, SREBP2 collectively promotes cholesterol biosynthesis and enhances the uptake of extracellular cholesterol.26 However, the expression and role of SREBP2 in glioblastoma cells, particularly in GSCs are poorly understood.
Here, we demonstrate that the cholesterol pathway is spatially distinct in glioblastomas, depending on the extracellular cholesterol availability. Furthermore, patient-derived GSCs activated cholesterol biosynthesis to a greater degree than differentiated glioblastoma cells (DGCs) in an SREBP2-dependent manner. SREBP2 regulated GSCs proliferation, self-renewal, and GBM tumor growth. Taken together, these data suggest an important role of SREBP2 for cholesterol biosynthesis and uptake in GBM, and reveal a potential target for glioblastoma therapy in the metabolic regulation strategy.
Materials and Methods
In Silico Data Curation
Gene expression data were acquired via GlioVis (https://gliovis.shinyapps.io/GlioVis/). RNA-seq data of GSC were from the Rich laboratory. RNA-seq and ChIP-seq data of GSC and DGC were accessed from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibusdatabase at GSE54792.27 Kyoto Encyclopedia of Genes and Genomes analysis gene sets were downloaded from the kyoto encyclopedia of genes and genomes pathway database (https://www.genome.jp/kegg/pathway.html). Gene sets for Gene Set Enrichment Analysis were from the Molecular Signatures Database (https://www.gsea-msigdb.org/gsea).
Human Glioblastoma Specimens
Five human glioblastoma specimens were obtained from surgical resection specimens of glioma patients at the First Affiliated Hospital of Nanjing Medical University (Nanjing, China). The study was approved by the Research Ethics Committee of Nanjing Medical University (Nanjing, Jiangsu, China). Written informed consent was obtained from all patients (2021-SR-076).
Culture of GSCs
Glioblastoma tissues of GSCs were obtained from excess surgical resection samples from patients who underwent treatment at Case Western Reserve University after reviewing by a neuropathologist with appropriate consent and following an Institutional Review Board-approved protocol (090401). All patient studies were conducted in accordance with the Declaration of Helsinki. GSCs were cultured as neurospheres in Neurobasal medium supplemented with B27, L-glutamine, sodium pyruvate, basic fibroblast growth factor, and epidermal growth factor. See Supplementary Material for details.
Organoid Culture
Our methods for establishing GSC organoids were originally based on protocols from the Rich laboratory.28 A total of 5000 GSC 387 cells were embedded in matrigel per organoid and cultured in Neurobasal medium for 6 weeks. The organoids were treated with low-density lipoprotein (LDL) for 7 days before being harvested.
Quantitative Real-Time PCR
Trizol reagent (Takara) was used for isolating total cellular RNA, followed by reverse transcription into cDNA using the qScript cDNA Synthesis Kit (Takara). Real-time polymerase chain reaction (PCR) was performed using Applied Biosystems 7900HT cycler using SYBR-Green PCR Master Mix (Thermo Fisher Scientific). Primer sequences are shown in Supplementary Table 1.
Plasmids and Lentiviral Transduction
See Supplementary Material for details on plasmids and lentiviral transduction. All shRNAs and sgRNAs sequences are listed in Supplementary Table 2.
Proliferation and Neurosphere Formation Assay
Cell proliferation was measured using CellTiter-Glo (Promega, Madison, WI, USA). All data were normalized to day 1 and presented as mean ± standard error of the mean (SEM). Neurosphere formation was measured by in vitro limiting dilution. Briefly, decreasing numbers of cells per well (50, 20, 10, 5, and 1) were plated into 96-well plates. Seven days after plating, the presence and number of neurospheres in each well were recorded. Extreme limiting dilution analysis was performed using software available at http://bioinf.wehi.edu.au/software/elda.
Western Blotting
See Supplementary Material for details on western blotting. The antibodies used are listed in Supplementary Table 3.
Immunofluorescence
See Supplementary Material for details on immunofluorescence assay. The antibodies used are listed in Supplementary Table 3.
Flow Cytometry of Cell Cycle Progression
Cells were stained with propidium iodide and measured through a BD flow cytometer. See Supplementary Material for details.
Flow Cytometry Analysis of Apoptosis
Cells were stained with propidium iodide and annexin V-Alexa Fluor647 and measured through a BD flow cytometer. See the Supplementary Material for details.
Chromatin Immunoprecipitation Quantitative Real-Time PCR
ChIP was performed using a ChIP Assay Kit (Cat# 17-295, Merck Millipore) with antibodies against SREBP2. Primer sequences are shown in Supplementary Table 1. See Supplementary Material for details.
Filipin Staining
Cells were fixed, permeabilized, and stained under light-protected conditions for 2 hours at room temperature with 0.1 mg/mL filipin solution and cell nuclei were stained with 7-AAD.
In Vivo Xenograft Model
Four-week-old, female BALB/c nude mice were transplanted with patient-derived GSCs into the right cerebral cortex at a depth of 3.5 mm. Intracranial tumor growth was measured by bioluminescence imaging. The mice were humanely euthanized 2 to 10 weeks after implantation, and their brains were harvested, paraffin-embedded, stained with H&E to confirm the presence of tumor, and subjected to immunofluorescence staining. For animal survival analysis, mice were maintained until the manifestation of neurological symptoms (ie, hunched back, loss of body weight, reduced food consumption, and inactivity) from tumor burden developed or until 100 days after injection. All experiments used for animal experiments were approved by the Institutional Ethics Committee of Nanjing Medical University, and the Institutional Animal Care and Use Committee (IACUC-2006033) at Nanjing Medical University in accordance with NIH and institutional guidelines. See the Supplementary Material for details.
Statistical Analysis
Data are represented as mean ± SEM of biological triplicates. Significance was calculated by a 2-tailed Student’s t-test for multiple comparisons using GraphPad Prism 9.0 software. A log-rank (Mantel–Cox) analysis was used to determine the statistical significance of Kaplan–Meier survival curves. For all statistical tests, P-values ≤.05 were considered significant.
Results
The Cholesterol Biosynthesis Landscape in GBM
Tumors display striking variation regionally in vasculature, cellular variation, and oncotic pressure that manifests in spatial variation of nutrient availability. Therefore, we interrogated metabolic pathway gene sets in bulk GBM tumor specimens in the The Cancer Genome Atlas (TCGA) dataset, revealing inverse expression of cholesterol biosynthesis and angiogenesis (Figure 1A–C). Cholesterol biosynthesis enzyme messenger ribonucleic acid (mRNA) levels were higher in GBM tumor cores than those in the tumor margin in 3 paired glioblastoma specimens (Figure 1D). As noted above, SREBP2 is a master transcriptional regulator that controls enzymes in cholesterol biosynthesis and uptake receptor.29,30 Across 528 GBM tumor samples analyzed, SREBP2 expression positively correlated with cholesterol biosynthesis-related enzymes and LDLR (Figure 1E). SQLE (Squalene monooxygenase) catalyzes the stereospecific oxidation of squalene to (S)-2,3-epoxysqualene, and is considered to be a rate-limiting enzyme in steroid biosynthesis.31,32 The correlation coefficient between SREBP2 and SQLE was 0.49, supporting the coordinated regulation of genes involved in the cholesterol biosynthesis pathway in GBM. In the Chinese Glioma Genome Atlas (CGGA) and Rembrandt (REpository for Molecular BRAin Neoplasia DaTa), genes related to tumor stemness and cholesterol biosynthesis were also positively correlated (Figure 1F). Given the differences between the tumor core and margin microenvironment, we considered the potential impact of differential extracellular cholesterol availability in tumor cell growth. However, LDL treatment of patient-derived GSCs (4121 and 387) did not alter GSC viability (Figure 1G). We, therefore, examined mRNA expression levels of cholesterol biosynthesis pathway genes (SQLE, LSS, CYP51A1, SC5D, and DHCR7) in GSCs treated with LDL, revealing that LDL induced a reduction in gene expression consistent with a negative feedback loop (Figure 1H, I). In contrast, withdrawal of LDL increased the mRNA expression levels of those cholesterol biosynthesis enzymes, validating the compensatory activation of cholesterol biosynthesis in cholesterol-poor environment (Figure 1J). Collectively, these results suggest that the GBMs activate cholesterol biosynthesis in response to nutrient deprivation.
Figure 1.

Cholesterol biosynthesis in GBM. (A) Heatmap showing metabolic pathway associated with angiogenesis. The top 10 pathways from each group were selected and ordered based on the fold change difference of pathway intensity. Data were from TCGA GBM HG-U133A. (B) Metabolic pathway enrichment analysis of angiogenesis intensity low versus high in GBM. Data were from TCGA GBM HG-U133A. (C) Correlation between angiogenesis signature and cholesterol biosynthesis signature. Analysis of gene expression in TCGA GBM HG-U133A datasets. (D) qRT-PCR quantification of mRNA levels of cholesterol biosynthesis enzymes (SQLE, LSS, SC5D, and 7-dehydrocholesterol reductase) in 3 paired GBM margins and core specimens. Data are presented as the mean ± SEM of 3 independent experiments. Significant results are presented as *P < .05, **P < .01. (E) Pairwise correlation analysis of 6 representative genes in cholesterol pathway. Data were from TCGA GBM HG-U133A. Correlation coefficient (R) values are shown. (F) Correlation between stemness signature and cholesterol biosynthesis signature. Analysis of gene expression in CGGA, GBM (left), and Rembrandt GBM (right). (G) Cell growth of GSC 4121 and 387 cells treated with LDL or not was measured by CellTiter-Glo assay. Data are presented as the mean ± SEM of 6 independent experiments. (H–J) qRT-PCR quantification of mRNA levels of cholesterol biosynthesis enzymes (SQLE, LSS, CYP51A1, SC5D, and DHCR7) in Glioblastoma stem cells (GSCs) treated with LDL over a concentration course in 24 hours (H), GSCs treated with 5 µg/ml LDL over a time course (I), and GSCs treated with 5 µg/ml LDL and withdraw LDL in a time course (J). GSCs derived from 2 glioblastoma patient-derived xenografts (4121 and 387). Data are presented as the mean ± SEM of 3 independent experiments. Significant results are presented as *P < .05, **P < .01.
SREBP2 Induces Cholesterol Biosynthesis in GSCs
GSCs display striking plasticity in response to their microenvironment and metabolic demands.33 In both the TCGA and CGGA GBM datasets, SREBP2 positively correlated with markers of tumor stemness (Figure 2A and Supplementary Figure 1A). Gene set enrichment analysis of GBM RNA-seq revealed that SREBP2 was positively correlated with cholesterol homeostasis and negatively correlated with angiogenesis in GSC (Figure 2B, C). In silico comparative RNA-seq data from matched GSCs and DGCs identified selective upregulation of cholesterol biosynthesis pathway genes and stemness markers in GSCs (Figure 2D). Chromatin analysis of cholesterol biosynthetic genes across 3 matched pairs of GSCs and differentiated GBM cells (DGCs) derived from 3 independent GBM patients27 revealed increased H3K27ac peak levels in GSCs, consistent with increased gene expression (Figure 2E). In direct validation experiments, we induced differentiation in 2 patient-derived GSCs (4121 and 387) and found that differentiation was associated with reduced mRNA levels of cholesterol biosynthesis genes (Figure 2F). SREBP2 preferentially marked SRY-box transcription factor 2 (SOX2)-positive tumor cells in clinical GBM tissues (Supplementary Figure 1B), and was higher in GSCs compared with matched DGCs (Figure 2G); however, GSCs were resistant to LDL-induced changes in SREBP2, both in cell and organoid culture (Figure 2H and Supplementary Figure 1C). Collectively, these results support the preferential upregulation of the cholesterol biosynthesis pathway in GSCs.
Figure 2.

Sterol Regulatory Element-Binding Protein 2 (SREBP2) activated cholesterol biosynthesis in Glioblastoma stem cells (GSCs). (A) Correlation between stemness signature and SREBP2 expression. Analysis of gene expression in the CGGA GBM dataset. (B–C) gene set enrichment analysis plot of SREBP2 and cholesterol homeostasis (B) or angiogenesis (C) in GSC. The normalized enrichment score and P value are indicated. RNA-seq data were from the Rich laboratory. (D) Heatmap showing gene expression based on global mRNA levels from RNA-seq data derived from paired GSC and DGC samples. A total of 21 genes from the cholesterol biosynthesis pathway were selected. RNA-seq data were downloaded from NCBI Gene Expression Omnibus GSE54792. (E) H3K27ac ChIP-seq enrichment plot centered at the gene locus for SREBP2, SQLE, LSS, CYP51A1, SC5D, and DHCR7. Enrichment is shown for 3 matched pairs of GSCs and differentiated glioblastoma cells (DGCs) from patient-derived glioblastoma specimens. H3K27ac ChIP-seq data were downloaded from NCBI Gene Expression Omnibus GSE54792. (F) qRT-PCR quantification of mRNA levels of cholesterol biosynthesis enzymes in GSCs and DGCs. GSCs were treated with serum to induce differentiation over a time course (2, 4, and 6 days). Data are presented as the mean ± SEM of 3 independent experiments. Significant results are presented as **P < .01. (G) Protein levels for SREBP2 were assessed by immunoblotting in 3 matched pairs of GSCs and DGCs derived from patient-derived glioblastoma models (4121, 387, and GSC23). Tubulin was used as a loading control. (H) Immunoblot assessment of SREBP2 protein levels. GSCs (4121, 387, and GSC23) were treated with LDL (5ug/ml) over a time course (12, 24, 36, and 48 hours). Tubulin was used as a loading control.
SREBP2 is Essential for GSC Maintenance and Tumor Growth
Based on the hypothesis that SREBP2 regulates GSC proliferation and self-renewal, we performed loss-of-function and overexpression studies. Silencing SREBP2 using 2 nonoverlapping short hairpin RNAs (shRNA) in 4121 GSCs and 387 GSCs reduced the mRNA and protein levels in GSCs, as expected (Figure 3A, B). Knockdown of SREBP2 decreased GSC proliferation, cell viability, and self-renewal (Figure 3C–G), but was dispensable in DGCs and normal neural stem cells (Supplementary Figure 2A, B). In addition, we identified that overexpression of SREBP2 in GSCs promoted GSC growth (Figure 3H, I).
Figure 3.

Sterol Regulatory Element-Binding Protein 2 (SREBP2) maintains GSC proliferation and self-renewal. (A) SREBP2 mRNA levels were assayed by real-time PCR quantification in GSC 4121 and 387 cells with SREBP2 knockdown using 2 independent shRNAs. Data are presented as the mean ± SEM of 3 independent experiments. Significance results are presented as **P < .01. (B) The protein levels of SREBP2 after transduction in GSC 4121 and 387 cells with 2 independent shRNAs. A non-targeting shRNA (shCONT) as the control. Tubulin was used as a loading control. (C–D) Two independent shRNAs targeting SREBP2 decreased the growth of GSC 4121 (top) and 387 (bottom) cells compared with shCONT, as measured by cell number count (C) and a CellTiter-Glo assay (D). Data are presented as the mean ± SEM of 3 (C) or 6 (D) independent experiments, respectively. Significant results are presented as **P < .01. (E) The extreme limiting dilution assays (ELDA) reveal that knockdown of SREBP2 in GSC 4121 (top) and 387 (bottom) cells decreased the sphere formation. Data are presented as the mean ± SEM of 3 independent experiments. (F) Quantification of the number of spheres formed by GSC 4121 (top) and 387 (bottom) cells in 2 independent shRNAs targeting SREBP2 and shCONT. Data are presented as the mean ± SEM of 6 independent experiments. Significant results are presented as **P < .01. (G) Representative images of neurospheres of GSC 4121 (left) and 387(right) cells expressing shCONT, shSREBP2-1, or shSREBP2-2. Scale bar, 100μm. (H) The protein levels of SREBP2 after transduction in GSC 4121 and 387 cells with empty vector or SREBP2 overexpression vector. Tubulin was used as a loading control. (I) Cell viability of GSC 4121 and 387 cells transduced with empty vector or SREBP2 overexpression vector, as measured by CellTiter-Glo assay. Data are presented as mean ± SEM from 6 independent experiments. Significant results are presented as *P < .05, **P < .01.
Betulin is a pharmacologic SREBP inhibitor with antitumor and chemo-preventive activities.34–36 Betulin attenuated GSC proliferation at submicromolar concentrations (Figure 4A). SREBP2 targeting using lentiviral shRNA transduction decreased the protein levels of the GSC markers, SOX2 and oligodendrocyte transcription factor 2 (OLIG2) (Figure 4B) and mRNA expression of SOX2, OLIG2, and MYC (Figure 4C). In orthogonal CRISPR-based targeting, sgRNAs against SREBP2 reduced its protein levels (Supplementary Figure 3A) and reduced mRNA expression of SOX2, OLIG2, and MYC (Figure 4D). Overexpression of SREBP2 promoted the protein levels of SOX2 and OLIG2 (Supplementary Figure 3B). Knockdown of SREBP2 induced GSC apoptosis, as demonstrated by increased Annexin V-staining and cleaved poly (ADP-ribose) polymerase (PARP) and caspase-3 (Figure 4E, F); meanwhile, immunofluorescent showing that knockdown of SREBP2 decreased the protein levels of stemness marker, SOX2, and increased the protein levels of cleaved caspase-3 (Supplementary Figure 3C). Loss of SREBP2 increased G1 and G2/M with reduced S phase fraction in GSCs (Supplementary Figure 3D).
Figure 4.
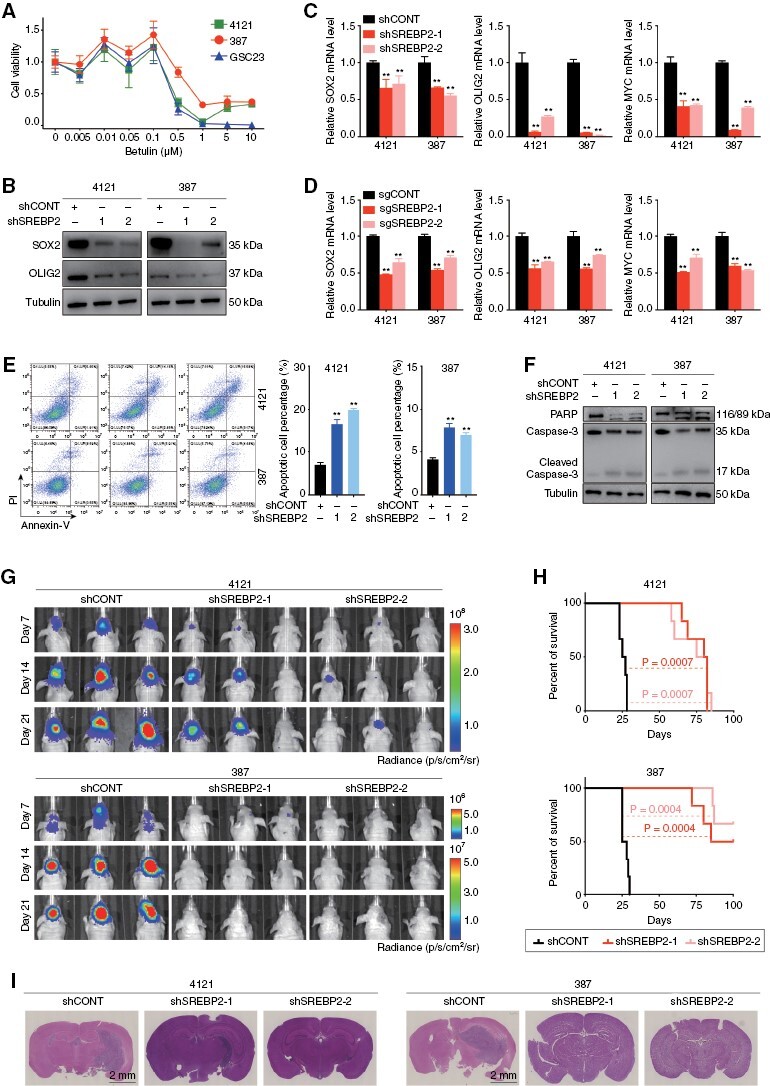
Sterol Regulatory Element-Binding Protein 2 (SREBP2) regulated GSC survival and tumor growth. (A) Sensitivity of 3 patient-derived GSC models (4121, 387, and GSC23) to the SREBP2 inhibitor betulin. (B) Immunoblot showing knockdown of SREBP2 in GSC 4121 and 387 cells decreased the protein levels of stemness markers SOX2 and OLIG2. Tubulin was used as a loading control. (C–D) qRT-PCR quantification showing that GSC 4121 and 387 cells transduced with shSREBP2 (C) and sgSREBP2 (D) decreased the stemness markers, including SOX2, OLIG2, and MYC proto-oncogene (MYC). Data are presented as the mean ± SEM of 3 independent experiments. Significant results are presented as **P < .01. (E) Representative images of flow cytometry analysis of apoptosis show that knockdown of SREBP2 induced the apoptosis of GSC 4121 and 387 cells (left). Quantification of the percentage of apoptosis cells (right). Data are presented as the mean ± SEM of 3 independent experiments. Significant results are presented as **P < .01. (F) The protein levels of PARP and caspase-3 were detected by immunoblotting in GSC 4121 and 387 cells after transduction with shCONT, shSREBP2-1, and shSREBP2-2. Tubulin was used as a loading control. (G) In vivo bioluminescent imaging of tumor growth was performed in nude mice bearing glioblastoma xenografts derived from 105 GSCs (4121 and 387) transduced with shCONT, shSREBP2-1, or shSREBP2-2 on days 7, 14, and 21. (H) Kaplan–Meier survival curves of immunodeficiency mice with intracranial GSC 4121 or 387 cells expressing shCONT, shSREBP2-1, and shSREBP2-2. (I) Representative images of H&E staining of mouse brains collected on day 21 after transplantation of GSC 4121 or 387 cells expressing shCONT, shSREBP2-1, or shSREBP2-2. Scale bar, 2 mm. Each image is representative of at least 3 similar experiments.
In vivo tumor initiation is the gold standard of cancer stem cells. To validate the in vivo function of SREBP2 in GBM growth, we transduced 2 patient-derived GSCs (4121 and 387) with either 1 of the 2 shSREBP2s or non-targeting control short hairpin ribonucleic acid (shCONT), then implanted the GSCs into the brains of immunocompromised mice. Loss of SREBP2 prolonged survival of tumor-bearing mice compared with mice bearing GSCs transduced with a non-targeting control shRNA (Figure 4G–I). Consistently, we identified the preferential expression of SREBP2 in SOX2-positive cells and increased expression of cleaved PARP in the core region of mouse tumors (Supplementary Figure 4A, B). Furthermore, mice transplanted with GSCs overexpressing SREBP2 portended poorer overall survival with increased tumor volume as compared to those with GSCs expressing empty vector (Supplementary Figure 4C–E). And the in vivo study targeting SREBP2 using botulin strengthened the point that SREBP2 is critical for GSC maintenance and tumor growth (Supplementary Figure 4F–H).
SREBP2 Regulates Cholesterol Biosynthesis and Uptake in GBM
Cholesterol biosynthesis is frequently dysregulated in multiple cancers. In GBM, SREBP2 levels correlate with hypoxia.37 To identify the downstream targets of SREBP2 in GBM, we examined mRNA and protein levels of cholesterol biosynthesis enzymes and the uptake receptor using genetic (shRNA or sgRNA) or pharmacologic approaches. The mRNA and protein levels of all relevant genes were consistently reduced after shSREBP2 transduction (Figure 5A and Supplementary Figure 5A), sgSREBP2 transduction (Figure 5B and Supplementary Figure 5B), and pharmacological inhibition of SREBP2 (Figure 5C and Supplementary Figure 5C). Consistently, protein levels of all relevant enzymes and the uptake receptor were increased after SREBP2 overexpression (Supplementary Figure 5D). Notably, knockdown of SREBP2 in GSCs induced the protein levels of SQLE, a key rate-limiting enzymes in cholesterol biosynthesis (Figure 5D). To determine if extracellular cholesterol rescued the effect of SREBP2 knockdown on GSC, we treated GSCs transfected with shSREBP2 with LDL. Exogenous LDL administration failed to reverse the effects of targeting SREBP2 on GSC cell viability, self-renewal, and intracellular cholesterol level (Figure 5E–G and Supplementary Figure 5E), suggesting that SREBP2 regulates not only cholesterol biosynthesis but also cholesterol uptake.
Figure 5.
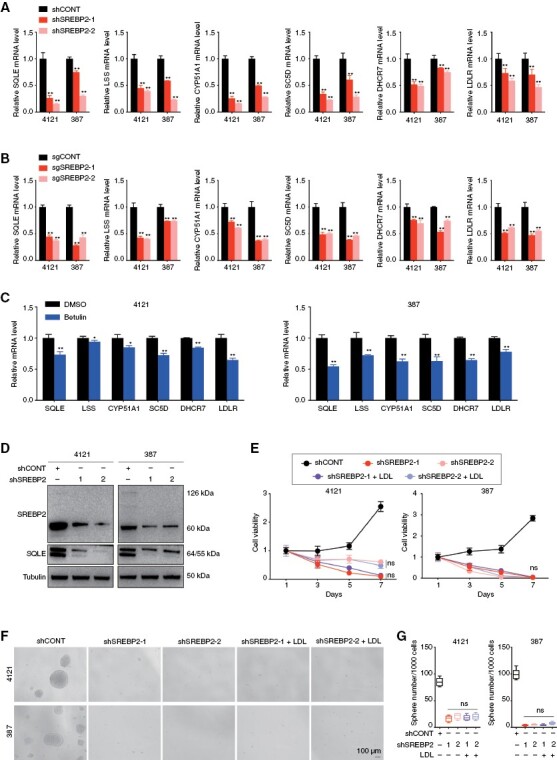
Sterol Regulatory Element-Binding Protein 2 (SREBP2) regulated both cholesterol biosynthesis and uptake in GBM. (A–C) qRT-PCR quantification of mRNA levels of cholesterol biosynthesis enzymes and low-density lipoprotein receptor in GSC 4121 and 387 cells transduced with shSREBP2 (A), sgSREBP2 (B) and treated with SREBP2 inhibitor betulin for 48 hours (C). Data are presented as the mean ± SEM of 3 independent experiments. Significant results are presented as *P < .05, **P < .01. (D) Immunoblot showing knockdown of SREBP2 in GSC 4121 and 387 cells decreased the protein levels of SQLE. Tubulin was used as a loading control. (E) Cell growth of GSC 4121 and 387 cells transduced with shCONT, shSREBP2-1, shSREBP2-2, shSREBP2-1 treated with LDL, and shSREBP2-2 treated with LDL was measured by CellTiter-Glo assay. Data are presented as the mean ± SEM of 3 independent experiments. (F) Representative images of neurospheres of GSC 4121 (top) and 387 (bottom) cells expressing shCONT, shSREBP2-1, shSREBP2-2, shSREBP2-1 treated with LDL, and shSREBP2-2 treated with LDL. Scale bar, 100μm. (G) Quantification of the number of spheres formed by GSC 4121 (left) and 387 (right) cells expressing shCONT, shSREBP2-1, shSREBP2-2, shSREBP2-1 treated with LDL, and shSREBP2-2 treated with LDL. Data are presented as the mean ± SEM of 6 independent experiments.
Impaired Intracellular Biosynthesis Sensitizes GSCs to Cholesterol Uptake
To confirm the direct binding of SREBP2 to the relevant gene promoters given its role as a transcription factor, we performed chromatin immunoprecipitation followed by PCR for 2 cholesterol biosynthesis genes, SQLE and LSS, and the cholesterol uptake receptor gene, LDLR, revealing enrichment of SREBP2 binding compared to the IgG control (Figure 6A). As SQLE is a key rate-limiting enzymes in downstream cholesterol biosynthesis,31 correlates tightly with SREBP2 levels (Figure 1E), and is regulated by SREBP2, we sought to determine the role of SQLE in GSCs to isolate the dependency of GSCs on the roles of cholesterol biosynthesis from uptake. We transduced GSCs with 2 nonoverlapping shRNA targeting SQLE, which reduced SQLE mRNA and protein levels in GSCs (Figure 6B and Supplementary Figure 6A). Knockdown of SQLE decreased GSC cell proliferation, viability, and self-renewal (Figure 6C–G). To identify the impact when GSCs lose intracellular cholesterol biosynthesis, we silenced SQLE in GSCs and provided exogenous LDL, which should permit cellular recovery if cholesterol uptake is unaffected by SQLE targeting. Exogenous LDL in the culture medium partially rescued the effects of SQLE knockdown on GSC cell viability, self-renewal, and intracellular cholesterol level (Figure 6H–J and Supplementary Figure 6B), suggesting that GSCs coopt extracellular cholesterol uptake to compensate for impaired cholesterol biosynthesis. Menin inhibitor 2 was developed as a menin inhibitor for leukemia treatment and acts by disrupting the interaction between menin and its binding partner mixed lineage leukemia 1 but in other cells is a cholesterol biosynthesis enzyme inhibitor directly inhibiting lanosterol synthase (LSS, also known as oxidosqualene cyclase).38,39 Menin inhibitor 2 inhibited GSC proliferation (Supplementary Figure 6C), but was less effective than the SREBP2 inhibitor, betulin. Collectively, these results demonstrate that SREBP2 regulates SQLE to activate cholesterol biosynthesis, with SQLE independent of cholesterol uptake to maintain GSC proliferation and self-renewal. GSCs rely on SREBP2-dependent cholesterol uptake when intracellular cholesterol biosynthesis was impaired.
Figure 6.

SQLE-depleted Glioblastoma stem cells (GSCs) rely on extracellular cholesterol uptake. (A) Sterol Regulatory Element-Binding Protein 2 (SREBP2) binds directly to promoter regions of SQLE, LSS and low-density lipoprotein receptor (LDLR). Cross-linked chromatin was prepared from GSC 4121 and 387 cells, and then immunoprecipitated using an anti-SREBP2 antibody or goat IgG control followed by real-time PCR using primers specific to promoter regions of SQLE, LSS and LDLR. Data are presented as mean ± SEM from 3 independent experiments. **, P < .01. (B) qRT-PCR quantification of mRNA levels of SQLE in GSC 4121 and 387 cells with SREBP2 knockdown. Data are presented as the mean ± SEM of 3 independent experiments. Significant results are presented as **P < .01. (C–D) Two independent shRNAs targeting SQLE decreased the growth of GSC 4121 (top) and 387 (bottom) cells compared shCONT, as measured by cell number count (C) and a CellTiter-Glo assay (D). Data are presented as the mean ± SEM of 3 (C) or 6 (D) independent experiments, respectively. Significant results are presented as **P < .01. (E) The extreme limiting dilution assays (ELDA) reveal that knockdown of SQLE in GSC 4121 (top) and 387 (bottom) cells decreased the sphere formation. (F) Quantification of the number of spheres formed by GSC 4121 (top) and 387 (bottom) cells in 2 independent shRNAs targeting SQLE and shCONT. Data are presented as the mean ± SEM of 6 independent experiments. Significant results are presented as **P < .01. (G) Representative images of neurospheres of GSC 4121 (left) and 387(right) cells expressing shCONT, shSQLE-1, or shSQLE-2. Scale bar, 100μm. (H) Cell growth of GSC 4121 and 387 cells transduced with shCONT, shSQLE-1, and shSQLE-1 treated with LDL was measured by CellTiter-Glo assay. Data are presented as the mean ± SEM of 6 independent experiments. Significant results are presented as **P < .01. (I) Quantification of the number of spheres formed by GSC 4121 (left) and 387 (right) cells expressing shCONT, shSQLE-1, and shSQLE-1 treated with LDL. Data are presented as the mean ± SEM of 6 independent experiments. Significant results are presented as **P < .01. (J) Representative images of neurospheres of GSC 4121 (top) and 387 (bottom) cells expressing shCONT, shSQLE-1, and shSQLE-1 treated with LDL. Scale bar, 100μm.
Discussion
GBM was previously designated as glioblastoma multiforme, based on its striking intra-tumoral morphologic heterogeneity, which is reflected in cellular diversity in which different tumor cell populations present various nutrient availability and therapeutic response. Here, we focus on cholesterol dependence in GBM. Free cholesterol is essential for cellular viability. Michikawa et al. reported that inhibition of cholesterol production induces neuronal cell death.40 The intracellular availability of cholesterol is regulated by the LDLR through the uptake of LDL. The transcription of LDLR is responsive to the cholesterol levels in cells.30 In GBM, the tumor core and margin display extreme differences in tumor vasculature and nutrient availability. Tumor cells in the margin have access to uptake cholesterol. On the other hand, the cells located in the hypoxic necrotic area of the tumor core face challenges to obtain abundant cholesterol in nutrient-deficient microenvironment. Hence, in our study, we found the significant enrichment of cholesterol biosynthesis in GBM associated with decreased angiogenesis among metabolic pathways, and focused on the activation level of cholesterol biosynthesis and uptake in the tumor core or margin, aiming to figure out the cholesterol source switching under different supplies of cholesterol in the tumor stem cell microenvironment, which is not restricted in tumor grades.41 We found that mRNA levels of several enzymes for cholesterol biosynthesis were significantly increased in the core compared with the margin samples in paired GBM specimens (Figure 1D). Therefore, we hypothesize that the sources of cholesterol for tumor cells present in the tumor core and margin regions are potentially different. In the core region, tumor cells largely rely on cholesterol biosynthesis, whereas in the marginal region, cholesterol tend to be absorbed from the tumor microenvironment because of its easy availability in vascular microenvironment.
GSCs, a small subpopulation of GBM cells that have self-renewal and tumor-initiating activity, contribute to glioblastoma development and recurrence.15,42 GSCs closely resemble neural precursor cells, express stem cell or precursor cell markers, and generate spheres in serum-free medium, which possess potent tumorigenic ability in vivo.43,44 GSCs have the capacity to adapt to regions of abundant or insufficient nutrients, and play a crucial role in tumor growth and response to therapy. In our study, GSCs could adapt to the culture conditions with or without LDL, which encouraged us to figure out the cholesterol homeostasis behavior of GSCs.
The SREBPs are a family of membrane-bound transcription factors that regulate cholesterol homeostasis and activate the expression of genes involved in the biosynthesis of cholesterol.45 We focused on the specific roles of SREBP2 under different supplies of cholesterol. Our results showed that, SREBP2, serving as a transcription factor, regulates cholesterol biosynthesis genes, including SQLE and LSS, and cholesterol uptake receptor, LDLR. SREBP2 mainly activates cholesterol biosynthesis genes in the cholesterol-deficient core region, while cholesterol uptake-related genes are in the marginal region, where cholesterol is in abundance. SREBP2 is indispensable for GSC proliferation, self-renewal, and tumor growth. Further, we found that knocking down SQLE in GSCs can effectively weaken the cell viability and self-renewal. Supplying LDL to SQLE-depleted GSCs could partially reverse loss of proliferation, showing the adaptability of GSCs to different supplies of cholesterol.
Taken together, our study demonstrated the distinct requirement of cholesterol biosynthesis and uptake for the proliferation and self-renewal of GSCs, an intra-tumoral population difficult to eradicate with current treatment strategies. SREBP2 plays a vital role in maintaining tumor growth in both cholesterol-rich and starved areas of tumor, and can serve as novel therapeutic targets for GBM.
Supplementary Material
Contributor Information
Danling Gu, National Health Commission Key Laboratory of Antibody Techniques, Department of Cell Biology, Jiangsu Provincial Key Laboratory of Human Functional Genomics, School of Basic Medical Sciences, Nanjing Medical University, Nanjing, Jiangsu, China.
Fengqi Zhou, Institute for Brain Tumors, Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Personalized Cancer Medicine, Nanjing Medical University, Nanjing, Jiangsu, China; Department of Neurosurgery, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, China.
Hao You, National Health Commission Key Laboratory of Antibody Techniques, Department of Cell Biology, Jiangsu Provincial Key Laboratory of Human Functional Genomics, School of Basic Medical Sciences, Nanjing Medical University, Nanjing, Jiangsu, China; Institute for Brain Tumors, Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Personalized Cancer Medicine, Nanjing Medical University, Nanjing, Jiangsu, China.
Jiancheng Gao, National Health Commission Key Laboratory of Antibody Techniques, Department of Cell Biology, Jiangsu Provincial Key Laboratory of Human Functional Genomics, School of Basic Medical Sciences, Nanjing Medical University, Nanjing, Jiangsu, China; Institute for Brain Tumors, Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Personalized Cancer Medicine, Nanjing Medical University, Nanjing, Jiangsu, China; Department of Neurosurgery, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, China.
Tao Kang, National Health Commission Key Laboratory of Antibody Techniques, Department of Cell Biology, Jiangsu Provincial Key Laboratory of Human Functional Genomics, School of Basic Medical Sciences, Nanjing Medical University, Nanjing, Jiangsu, China; Institute for Brain Tumors, Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Personalized Cancer Medicine, Nanjing Medical University, Nanjing, Jiangsu, China.
Deobrat Dixit, Department of Medicine, Division of Regenerative Medicine, University of California, San Diego, La Jolla, California, United States.
Qiulian Wu, University of Pittsburgh Medical Center Hillman Cancer Center, Pittsburgh, Pennsylvania, United States.
Kailin Yang, Department of Radiation Oncology, Taussig Cancer Center, Cleveland Clinic, Cleveland, Ohio, United States.
Shusheng Ci, National Health Commission Key Laboratory of Antibody Techniques, Department of Cell Biology, Jiangsu Provincial Key Laboratory of Human Functional Genomics, School of Basic Medical Sciences, Nanjing Medical University, Nanjing, Jiangsu, China; Institute for Brain Tumors, Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Personalized Cancer Medicine, Nanjing Medical University, Nanjing, Jiangsu, China.
Danyang Shan, National Health Commission Key Laboratory of Antibody Techniques, Department of Cell Biology, Jiangsu Provincial Key Laboratory of Human Functional Genomics, School of Basic Medical Sciences, Nanjing Medical University, Nanjing, Jiangsu, China; Institute for Brain Tumors, Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Personalized Cancer Medicine, Nanjing Medical University, Nanjing, Jiangsu, China.
Xiao Fan, Institute for Brain Tumors, Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Personalized Cancer Medicine, Nanjing Medical University, Nanjing, Jiangsu, China; Department of Neurosurgery, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, China.
Wei Yuan, Department of Pathology, The Yancheng Clinical College of Xuzhou Medical University, The First people’s Hospital of Yancheng, Yancheng, Jiangsu, China; Department of Central Laboratory, Yancheng Medical Research Center of Nanjing University Medical School, Yancheng, Jiangsu, China.
Qian Zhang, National Health Commission Key Laboratory of Antibody Techniques, Department of Cell Biology, Jiangsu Provincial Key Laboratory of Human Functional Genomics, School of Basic Medical Sciences, Nanjing Medical University, Nanjing, Jiangsu, China; Institute for Brain Tumors, Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Personalized Cancer Medicine, Nanjing Medical University, Nanjing, Jiangsu, China.
Chenfei Lu, National Health Commission Key Laboratory of Antibody Techniques, Department of Cell Biology, Jiangsu Provincial Key Laboratory of Human Functional Genomics, School of Basic Medical Sciences, Nanjing Medical University, Nanjing, Jiangsu, China; Institute for Brain Tumors, Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Personalized Cancer Medicine, Nanjing Medical University, Nanjing, Jiangsu, China; Department of Neurosurgery, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, China.
Daqi Li, National Health Commission Key Laboratory of Antibody Techniques, Department of Cell Biology, Jiangsu Provincial Key Laboratory of Human Functional Genomics, School of Basic Medical Sciences, Nanjing Medical University, Nanjing, Jiangsu, China; Institute for Brain Tumors, Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Personalized Cancer Medicine, Nanjing Medical University, Nanjing, Jiangsu, China.
Ningwei Zhao, China Exposomics Institute, Shanghai, China.
Zhumei Shi, Department of Neurosurgery, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, China.
Wei Gao, National Health Commission Key Laboratory of Antibody Techniques, Department of Cell Biology, Jiangsu Provincial Key Laboratory of Human Functional Genomics, School of Basic Medical Sciences, Nanjing Medical University, Nanjing, Jiangsu, China; Institute for Brain Tumors, Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Personalized Cancer Medicine, Nanjing Medical University, Nanjing, Jiangsu, China.
Fan Lin, National Health Commission Key Laboratory of Antibody Techniques, Department of Cell Biology, Jiangsu Provincial Key Laboratory of Human Functional Genomics, School of Basic Medical Sciences, Nanjing Medical University, Nanjing, Jiangsu, China.
Jianghong Man, State Key Laboratory of Proteomics, National Center of Biomedical analysis, Beijing, China.
Qianghu Wang, Institute for Brain Tumors, Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Personalized Cancer Medicine, Nanjing Medical University, Nanjing, Jiangsu, China.
Xu Qian, Institute for Brain Tumors, Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Personalized Cancer Medicine, Nanjing Medical University, Nanjing, Jiangsu, China; Department of Nutrition and Food Hygiene, Center for Global Health, School of Public Health, Nanjing Medical University, Nanjing, Jiangsu, China.
Stephen C Mack, Division of Brain Tumor Research, Department of Developmental Neurobiology, St. Jude Children’s Research Hospital, Memphis, Tennessee, United States.
Weiwei Tao, College of Biomedicine and Health and College of Life Science and Technology, Huazhong Agricultural University, Wuhan, Hubei, China.
Sameer Agnihotri, Brain Tumor Biology and Therapy Lab, Department of Neurosurgery, University of Pittsburgh Medical Center, Pittsburgh, Pennsylvania, United States.
Nu Zhang, Department of Neurosurgery, The First Affiliated Hospital of Sun Yat-sen University, Guangdong Provincial Key Laboratory of Brain Function and Disease, Guangdong Translational Medicine Innovation Platform, Guangzhou, Guangdong, China.
Yongping You, Institute for Brain Tumors, Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Personalized Cancer Medicine, Nanjing Medical University, Nanjing, Jiangsu, China; Department of Neurosurgery, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, China.
Jeremy N Rich, University of Pittsburgh Medical Center Hillman Cancer Center, Pittsburgh, Pennsylvania, United States; Department of Neurology, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, United States.
Junxia Zhang, Institute for Brain Tumors, Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Personalized Cancer Medicine, Nanjing Medical University, Nanjing, Jiangsu, China; Department of Neurosurgery, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, China.
Xiuxing Wang, National Health Commission Key Laboratory of Antibody Techniques, Department of Cell Biology, Jiangsu Provincial Key Laboratory of Human Functional Genomics, School of Basic Medical Sciences, Nanjing Medical University, Nanjing, Jiangsu, China; Institute for Brain Tumors, Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Personalized Cancer Medicine, Nanjing Medical University, Nanjing, Jiangsu, China; Jiangsu Cancer Hospital, Affiliated Cancer Hospital of Nanjing Medical University, Nanjing, Jiangsu, China.
Funding
This work was supported by the National Natural Science Foundation of China (82072779 to X. Wang).
Conflict of interest statement
The authors declare that the manuscript, or any part of it, has not been previously published or submitted concurrently to any other journal.
Authorship
Conception and design: D.G., J.G. Collection and assembly of data: F.Z., H.Y. Data analysis and interpretation: D.G., J.G. Manuscript writing: D.G., F.Z., H.Y. Revision and final approval of manuscript: All authors. Accountable for all aspect of the study: All authors.
Data Availability
All data accessed from external sources and prior publications have been referenced in the text and corresponding figure legends. Additional data used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- 1. Gusyatiner O, Hegi ME.. Glioma epigenetics: From subclassification to novel treatment options. Semin Cancer Biol. 2018;51:50–58. [DOI] [PubMed] [Google Scholar]
- 2. Björkhem I, Meaney S.. Brain cholesterol: Long secret life behind a barrier. Arterioscler Thromb Vasc Biol. 2004; 24(5):806–815. [DOI] [PubMed] [Google Scholar]
- 3. Ostrom QT, Gittleman H, de Blank PM, et al. American Brain Tumor Association adolescent and young adult primary brain and central nervous system tumors diagnosed in the United States in 2008-2012. Neuro Oncol. 2016; 18(suppl 1):i1–i50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nicholson JG, Fine HA.. Diffuse glioma heterogeneity and its therapeutic implicationsdiffuse glioma heterogeneity and its therapeutic implications. Cancer Discov. 2021;11(3):575–590. [DOI] [PubMed] [Google Scholar]
- 5. Wen PY, Packer RJ.. The 2021 WHO Classification of Tumors of the Central Nervous System: Clinical Implications. Vol. 23. US: Oxford University Press; 2021:1215–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stupp R, Hegi ME, Mason WP, et al. ; European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. [DOI] [PubMed] [Google Scholar]
- 7. Omuro A, DeAngelis LM.. Glioblastoma and other malignant gliomas: A clinical review. JAMA. 2013;310(17):1842–1850. [DOI] [PubMed] [Google Scholar]
- 8. Tanaka S, Louis DN, Curry WT, Batchelor TT, Dietrich J.. Diagnostic and therapeutic avenues for glioblastoma: No longer a dead end? Nat Rev Clin Oncol. 2013;10(1):14–26. [DOI] [PubMed] [Google Scholar]
- 9. Wen PY, Kesari S.. Malignant gliomas in adults. N Engl J Med. 2008;359(5):492–507. [DOI] [PubMed] [Google Scholar]
- 10. Stupp R, Mason WP, van den Bent MJ, et al. ; European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. [DOI] [PubMed] [Google Scholar]
- 11. Garnier D, Meehan B, Kislinger T, et al. Divergent evolution of temozolomide resistance in glioblastoma stem cells is reflected in extracellular vesicles and coupled with radiosensitization. Neuro Oncol. 2018;20(2):236–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. [DOI] [PubMed] [Google Scholar]
- 13. Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. [DOI] [PubMed] [Google Scholar]
- 14. Goodwin CR, Laterra JN.. unmasking the multiforme in glioblastoma. Nat Rev Neurol. 2010;6(6):304–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Venere M, Fine HA, Dirks PB, Rich JN.. Cancer stem cells in gliomas: Identifying and understanding the apex cell in cancer’s hierarchy. Glia. 2011;59(8):1148–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bhat KPL, Balasubramaniyan V, Vaillant B, et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell. 2013;24(3):331–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang X, Yang K, Xie Q, et al. Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nat Neurosci. 2017;20(5):661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mashimo T, Pichumani K, Vemireddy V, et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell. 2014;159(7):1603–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li X, Yu W, Qian X, et al. Nucleus-translocated ACSS2 promotes gene transcription for lysosomal biogenesis and autophagy. Mol Cell. 2017;66(5):684–697.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gimple RC, Bhargava S, Dixit D, Rich JN.. Glioblastoma stem cells: Lessons from the tumor hierarchy in a lethal cancer. Genes Develop. 2019;33(11–12):591–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Villa GR, Hulce JJ, Zanca C, et al. An LXR-cholesterol axis creates a metabolic co-dependency for brain cancers. Cancer Cell. 2016;30(5):683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Silvente-Poirot S, Poirot M.. Cholesterol and cancer, in the balance. Science. 2014;343(6178):1445–1446. [DOI] [PubMed] [Google Scholar]
- 23. Luo J, Yang H, Song B-L.. Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol. 2020;21(4):225–245. [DOI] [PubMed] [Google Scholar]
- 24. Goldstein JL, DeBose-Boyd RA, Brown MS.. Protein sensors for membrane sterols. Cell. 2006;124(1):35–46. [DOI] [PubMed] [Google Scholar]
- 25. Yokoyama C, Wang X, Briggs MR, et al. SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell. 1993;75(1):187–197. [PubMed] [Google Scholar]
- 26. Brown MS, Goldstein JL.. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89(3):331–340. [DOI] [PubMed] [Google Scholar]
- 27. Suva ML, Rheinbay E, Gillespie SM, et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell. 2014;157(3):580–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hubert CG, Rivera M, Spangler LC, et al. A three-dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found in vivo. Cancer Res. 2016;76(8):2465–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eberlé D, Hegarty B, Bossard P, Ferré P, Foufelle F.. SREBP transcription factors: Master regulators of lipid homeostasis. Biochimie. 2004;86(11):839–848. [DOI] [PubMed] [Google Scholar]
- 30. Horton JD, Goldstein JL, Brown MS.. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109(9):1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Coates HW, Chua NK, Brown AJ.. Consulting prostate cancer cohort data uncovers transcriptional control: Regulation of the MARCH6 gene. Biochim Biophys Acta Mol Cell Biol Lipids. 2019;1864(11):1656–1668. [DOI] [PubMed] [Google Scholar]
- 32. Howe V, Sharpe LJ, Prabhu AV, Brown AJ.. New insights into cellular cholesterol acquisition: Promoter analysis of human HMGCR and SQLE, two key control enzymes in cholesterol synthesis. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862(7):647–657. [DOI] [PubMed] [Google Scholar]
- 33. Dirkse A, Golebiewska A, Buder T, et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat Commun. 2019;10(1):1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Król SK, Kiełbus M, Rivero-Müller A, Stepulak A.. Comprehensive review on betulin as a potent anticancer agent. Biomed Res Int. 2015;2015:584189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tang J-J, Li J-G, Qi W, et al. Inhibition of SREBP by a small molecule, betulin, improves hyperlipidemia and insulin resistance and reduces atherosclerotic plaques. Cell Metab. 2011;13(1):44–56. [DOI] [PubMed] [Google Scholar]
- 36. Xue L, Qi H, Zhang H, et al. Targeting SREBP-2-regulated mevalonate metabolism for cancer therapy. Front Oncol. 2020;10:1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lewis CA, Brault C, Peck B, et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene. 2015;34(40):5128–5140. [DOI] [PubMed] [Google Scholar]
- 38. Nes WD. Biosynthesis of cholesterol and other sterols. Chem Rev. 2011;111(10):6423–6451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Phillips RE, Yang Y, Smith RC, et al. Target identification reveals lanosterol synthase as a vulnerability in glioma. Proc Natl Acad Sci USA. 2019;116(16):7957–7962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Michikawa M, Yanagisawa K.. Inhibition of cholesterol production but not of nonsterol isoprenoid products induces neuronal cell death. J Neurochem. 1999;72(6):2278–2285. [DOI] [PubMed] [Google Scholar]
- 41. Li D, Li S, Xue AZ, Smith Callahan LA, Liu Y.. Expression of SREBP2 and cholesterol metabolism related genes in TCGA glioma cohorts. Medicine. 2020;99(12):e18815e18815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Clara JA, Monge C, Yang Y, Takebe N.. Targeting signalling pathways and the immune microenvironment of cancer stem cells - a clinical update. Nat Rev Clin Oncol. 2020;17(4):204–232. [DOI] [PubMed] [Google Scholar]
- 43. Chen J, Li Y, Yu TS, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488(7412):522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cheng L, Huang Z, Zhou W, et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell. 2013;153(1):139–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang Y, Muneton S, Sjövall J, Jovanovic JN, Griffiths WJ.. The effect of 24S-hydroxycholesterol on cholesterol homeostasis in neurons: Quantitative changes to the cortical neuron proteome. J Proteome Res. 2008;7(4):1606–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data accessed from external sources and prior publications have been referenced in the text and corresponding figure legends. Additional data used and/or analyzed during the current study are available from the corresponding author on reasonable request.