

Abstract
A quantitative fingerprinting method, called the real-time terminal restriction fragment length polymorphism (real-time-t-RFLP) assay, was developed for simultaneous determination of microbial diversity and abundance within a complex community. The real-time-t-RFLP assay was developed by incorporating the quantitative feature of real-time PCR and the fingerprinting feature of t-RFLP analysis. The assay was validated by using a model microbial community containing three pure strains, an Escherichia coli strain (gram negative), a Pseudomonas fluorescens strain (gram negative), and a Bacillus thuringiensis strain (gram positive). Subsequently, the real-time-t-RFLP assay was applied to and proven to be useful for environmental samples; the richness and abundance of species in microbial communities (expressed as the number of 16S rRNA gene copies of each ribotype per milliliter) of wastewater and estrogen-degrading consortia (enriched with 17α-estradiol, 17β-estradiol, or estrone) were successfully characterized. The results of this study strongly suggested that the real-time-t-RFLP assay can be a powerful molecular tool for gaining insight into microbial communities in various engineered systems and natural habitats.
Terminal restriction fragment length polymorphism (t-RFLP) analysis is a powerful comparative fingerprinting molecular technique that is widely used to describe microbial community structure for different temporal and spatial variations, as well as geochemical alterations caused by natural and/or engineered interventions (1, 8, 13, 32, 38-40, 46). In t-RFLP analysis, the phylogenetic signatures (such as that of the gene encoding 16S rRNA) of total community DNA are first PCR amplified with fluorescence-labeled primers, and the PCR products are subsequently digested with a restriction enzyme to generate a series of peaks corresponding to various fluorescence-labeled terminal restriction fragment (T-RF) lengths. The profile of a series of T-RFs provides an estimate of the number of phylotypes in a microbial community, and the fluorescence intensity of each peak reflects the relative abundance of each phylotype (theoretically) in the microbial community (39). Because many phylogenetically similar species can contribute to the same T-RF, T-RFs are commonly referred to as operational taxonomic units. Nevertheless, the t-RFLP assay is still considered a useful fingerprinting method, particularly for comparative analysis of microbial communities.
Real-time PCR analysis has been accepted as a very sensitive quantitative molecular method that has applications ranging from clinical microbiology (4, 5, 24, 44) to molecular ecology (25, 53, 54) and environmental microbiology (3, 10, 18, 22, 28, 33, 34, 37). Unlike other quantitative molecular techniques, real-time PCR is a fluorescence-based kinetic PCR method, and it allows rapid quantification of the initial number of copies of the target gene (29). In TaqMan real-time PCR, an extra fluorescence-labeled probe complementary to the target DNA is initially bound to the target DNA. As PCR progresses, the fluorescent label is cleaved to release a fluorescent signal that is recorded at the end of each PCR cycle (23, 31). The detected fluorescent signals are compared to a set of standard curves for quantification of the target gene in the samples. In addition to its sensitivity for quantification, the real-time PCR assay can be designed for a specific strain, a phylogenetic group, or even a group of microorganisms exhibiting a similar function (49).
Currently, researchers are able to quantify a specific species of interest by using real-time PCR or are able to acquire qualitative information for microbial community structure by using molecular techniques, like t-RFLP analysis. However, integrating results obtained from various molecular assays in order to describe microbial diversity and the absolute abundance of species populations in a given community is quite a challenge and may not even be possible. Therefore, a single quantitative molecular method that allows effective fingerprinting of a given microbial community will be particularly valuable in microbe-related studies.
In this study, a single molecular assay, the real-time-t-RFLP assay, which allowed simultaneous quantification of all microbial ribotypes in a microbial community, was developed. This assay was applied to environmental samples (wastewater) and enrichment cultures (estrogen-degrading consortia). The estrogen-degrading consortia enriched from activated sludge were capable of degrading three different estrogens, 17α-estradiol, 17β-estradiol, and estrone. The limitations and potential applications of the real-time-t-RFLP assay are also discussed.
Method development and rationale.
The development of the real-time-t-RFLP assay was based on unique features of two novel molecular methods, real-time PCR and t-RFLP analysis. Figure 1 illustrates the rationale behind the real-time-t-RFLP assay. The use of a fluorescence-labeled primer in conjunction with a TaqMan probe is critical to the success of the real-time- t-RFLP assay because the labeled primer and probe are a link between the real-time PCR and t-RFLP techniques. The first step of the real-time-t-RFLP assay is to amplify and quantify 16S rRNA genes from total genomic DNA by using a 5′-end fluorescence-labeled primer and a fluorescence-labeled probe during real-time PCR. During the amplification, the fluorescence-labeled probe is cleaved from a complementary region of the target gene and emits a fluorescent signal for rapid quantification of the total copy of the initial DNA. Meanwhile, the fluorescence-labeled primer is incorporated into the amplified PCR products. In the second step, the fluorescence-labeled PCR products are harvested, digested with restriction enzymes, and analyzed with an automatic sequencer in order to generate a profile consisting of a series of T-RFs (t-RFLP analysis). The profile is used to provide information about the relative abundance of each ribotype (T-RF) in a microbial community. By combining two sets of information, one set collected from real-time PCR (i.e., the absolute numbers of copies of total 16S rRNA genes) and the other set collected from t-RFLP analysis (i.e., the relative distribution of each ribotype), the richness and abundance of microbial strains in a given community can be quickly assessed and presented as a profile of the 16S rRNA gene copies of each species.
FIG. 1.

Rationale behind the real-time-t-RFLP assay for quantitative fingerprinting of microbial communities.
MATERIALS AND METHODS
Chemicals.
Three natural estrogens (17α-estradiol, 17β-estradiol, and estrone) were used in this study. 17β-Estradiol (>99% pure), 17α-estradiol (>98% pure), and estrone (>98% pure) were purchased from Sigma-Aldrich Inc. (St. Louis, Mo.). Stock solutions of estrogens were prepared in acetone. Dimethylformamide and N,O-bis(trimethylsilyl)trifluoroacetamide were purchased from Pierce Chemicals, Dallas, Tex. Hi-Di formamide and the GeneScan 500 ROX size standard were purchased from Applied Biosystems, Warrington, United Kingdom.
Bacterial cultures.
Three bacterial strains were used in this study. Escherichia coli (gram negative), Pseudomonas fluorescens (gram negative), and Bacillus thuringiensis (gram positive) were obtained from the culture collection at the Center for Environmental Biotechnology, University of Tennessee, Knoxville. The 16S rRNA gene sequences of E. coli, P. fluorescens, and B. thuringiensis exhibit 99% identity to the sequences deposited under GenBank accession numbers AF233451, AF228366, and AF155954, respectively. The bacterial strains were grown in Luria-Bertani medium at 30°C overnight before they were harvested by centrifugation for experimental use.
Estrogen-degrading consortia.
Estrogen-degrading consortia were enriched from activated sludge from two wastewater treatment plants (WWTPs) (designated WWTP1 and WWTP2) near Knoxville, Tenn. WWTP1 treats 1.9 × 104 m3 of wastewater per day and consists of an oxidation ditch activated sludge system. Wastewater samples were collected from the following three locations: primary treatment (i.e., after the primary treatment serving as the influent for the ditch), the center of the ditch, and the clarifier. WWPT2 treats 1.1 × 105 m3 of wastewater per day and consists of a one-sludge nitrification system. A sludge sample was collected from the nitrified activated sludge tank of WWTP2.
Enrichment of the estrogen-degradation consortia was performed in 40-ml glass vials containing 10 ml of nitrate mineral salts medium (14), 1 ml of activated sludge as the inoculum, and 1 mg of estrogen (17α-estradiol, 17β-estradiol, or estrone) per liter. The inoculated vials and abiotic controls (without activated sludge) were incubated at 30°C at 150 rpm. The abiotic loss of estrogen was less than 5%. The experiment was performed in duplicate. Weekly subculturing was performed by transferring 1 ml of the liquid suspension to a new vial containing nitrate mineral salts medium with an estrogenic compound added as described above. After five transfers, enrichment cultures were collected for microbial community structure analysis by the real-time-t-RFLP assay. The microbial community structure after enrichment with three different estrogenic compounds was compared to the structure in the original activated sludge samples.
Analytical methods. (i) DNA extraction.
Genomic DNA of each bacterium was extracted with a FastDNA kit (Q-Biogene, Carlsbad, Calif.) in accordance with the manufacturer's instructions. For environmental samples (wastewater and activated sludge) and enrichment cultures, a FastDNA SPIN kit for soil (Q-Biogene) was used with the following minor modifications: the silica binding matrix-DNA complex was washed twice with 80% (vol/vol) ethanol after the recommended salt-ethanol wash step (18). DNA concentrations were determined with a Hoefer DyNa Quant 200 fluorometer (Pharmacia Biotech, San Francisco, Calif.).
(ii) Real-time-t-RFLP assay.
A region of the 16S rRNA gene sequence (length, 352 bp) was selected for development of the real-time-t-RFLP assay in this study, because primers (16S1055f and 16S1392r) and a TaqMan probe (16STaq1115f) for this region have been designed and successfully tested with wastewater samples (28). With no base pair mismatches, the primers and probe capture more than 9,000 bacterial 16S rRNA gene sequences in the GenBank database (28) and generate a longer PCR amplicon (352 bp versus 263 bp) than the other primer set and probe for total bacterial 16S rRNA genes (2). The target region in extracted DNA was amplified by using the primers and the TaqMan probe, with the following modifications. The PCR was performed with fluorescence-labeled forward primer 16SHex1055f (5′-hexachlorofluorescein-ATGGCTGTCGTCAGCT-3′), reverse primer 16S1392r (ACGGGCGGTGTGTAC-3′), and TaqMan probe 16STaq1115f (5′-[6-carboxyfluorescein]-CAACGAGCGCAACCC-[6-carboxytetramethylrhodamine]-3′). The PCR mixture (total volume, 25 μl) contained 2.5 μl of 10× PCR buffer A, 1.25 U of Taq polymerase (Fisher Scientific, Fair Lawn, N.J.), 0.5 μg of bovine serum albumin, each of four deoxynucleoside triphosphates at a concentration of 200 μM, 4.5 mM MgCl2, each primer at a concentration of 600 nM, 250 nM TaqMan probe 16STaq1115f, and 10 ng of DNA template. PCR amplification was performed with a DNA Engine Opticon continuous fluorescence detection system (MJ Research, Waltham, Mass.). The PCR conditions were as follows: initial denaturation at 95°C for 10 min, followed by 30 cycles of 95°C for 30 s, 54°C for 1 min, a plate read step, and 72°C for 2 min, and a final incubation at 4°C. Standard curves for quantifying 16S rRNA gene copies in samples were constructed by using plasmid #931, which carries a Nitrospira partial 16S rRNA gene (GenBank accession number AF420301) (18, 28). Standard curves based on amounts ranging from 2.3 × 103 to 2.3 × 108 copies of the 16S rRNA gene were generated parallel to the sample analysis.
PCR products were separated on a 1.5% agarose gel in 1× Tris-acetate-EDTA buffer, and the DNA bands of the expected size (352 bp) were excised. The PCR products were then recovered and purified by using MicroSpin columns (Amersham Biosciences, Piscataway, N.J.), followed by ethanol precipitation and two desalinization steps with 80% ethanol.
Purified PCR products were then digested with restriction enzyme MspI at 37°C for 8 h following 20 min of incubation at 70°C. The digestion reaction mixture (30 μl) contained 60 to 90 ng of the purified PCR products and 10 U of MspI (or HhaI). Digested PCR products were precipitated with ethanol and resuspended in 40 μl of high-performance liquid chromatography water.
The lengths of T-RFs of digested PCR products were determined with an ABI Prism 310 genetic analyzer (Applied Biosystems, Foster City, Calif.) in GeneScan mode. Each sample was prepared by adding 5 μl of desalted digested PCR product, 10 μl of Hi-Di formamide, and 0.5 μl of Genescan ROX 500 size standards. Samples were denatured at 95°C for 5 min, followed by rapid chilling on ice. By using an injection time of 10 s, a final DNA concentration in the range from 0.05 to 0.7 ng/μl was introduced electrokinetically into a capillary containing polymer (GS STR POP4). Electrophoresis was performed at 15 kV at 60°C for 25 min. The lengths of T-RFs were automatically determined by comparison with the internal standards by using the GeneScan software, version 3.1.
(iii) Estrogen analysis.
Concentrations of estrogens in the growth medium were determined as described by Raman et al. (50). The estrogen in a liquid suspension was extracted with an equal volume of ethyl ether (pesticide grade; Fisher Scientific). The ether layer (500 μl) was carefully transferred to 2-ml autosampling gas chromatography vials and evaporated under a gentle stream of nitrogen. The dried extracts were resuspended with 450 μl of dimethylformamide and then derivatized with 50 μl of N,O-bis(trimethylsilyl)trifluoroacetamide. The derivatized samples (1 μl) were injected into an HP 6890 series gas chromatograph equipped with a DB-5MS capillary column (length, 30 m; film thickness, 0.25 μm; J&W Scientific) and an HP 5973 mass selective detector. The analysis was performed in the selective ion monitoring mode. Standard curves for estrogens, containing at least five data points ranging from 0.001 to 5 mg/liter, were used for calculation of estrogen concentrations.
Data acquisition and analysis.
The number of 16S rRNA gene copies of the ith T-RF per unit volume (Ci) in a microbial community can be determined as follows:
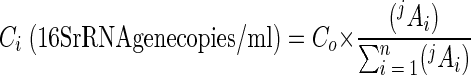 |
where Co is the initial number of gene copies in the sample, expressed in number of 16S rRNA gene copies per milliliter; jAi is the peak area of the ith T-RF of j length (in bases) that is measured from the electropherogram; and n is the total number of T-RFs in the electropherogram. Co was determined by comparing the threshold cycle (Ct) mesured from samples to standard curves (28). The Ct was defined as the number of PCR cycles when the generated fluorescence exceeded 10 times the standard deviation of background fluorescence. The Ct value was determined automatically with computer software (Opticon Monitor, version 1.4) installed in a DNA Engine Opticon continuous fluorescence detection system (MJ Research). Standard curves were constructed by plotting the Ct values over a range of known numbers of gene copies per unit volume. The ratio of each peak area to the total peak area, 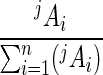 , can be considered the relative abundance of the ith T-RF in a microbial community.
, can be considered the relative abundance of the ith T-RF in a microbial community.
Since T-RFs less than 30 bp long were observed in all samples and their intensities were independent of the input DNA, the peaks of these T-RFs were excluded from the data analyses. A threshold peak area value of 1% has been used in other studies (11, 43), and use of a 5% relative peak area threshold has been shown to minimize the effects of the amount of labeled products (one of the significant factors) affecting t-RFLP profiles (51). In this study, peak area that contributed less than 1% of the total area was regarded as background noise and was excluded from the analysis.
RESULTS
Development and validation of the real-time-t-RFLP assay.
Two sets of experiments with pure strains and constructed communities were conducted to develop and validate the real-time-t-RFLP assay to provide quantitative information for the number of 16S rRNA gene copies of each ribotype (expressed as T-RF) in a microbial community profile.
The first set of experiments was conducted to determine (i) whether a specific region of the 16S rRNA gene of three known strains could be amplified with a fluorescence-labeled forward primer (16SHex1055f), a nonlabeled reverse primer (16S1392r), and a fluorescence-labeled probe (16STaq1115f) and (ii) whether the amplified PCR products generated a consistent microbial community profile after restriction digestion in the t-RFLP analysis. Figure 2A shows the sizes of the measured T-RFs of three strains, as follows: 82 bp for E. coli, 103 bp for B. thuringiensis, and 105 bp for P. fluorescens. The measured T-RFs were 0 to 3 bp shorter than the predicted lengths based on TAP analysis (a t-RFLP analysis program) of Ribosomal Database Project II entries (41). These discrepancies have been noted by other investigators and have yet to be explained (15, 38, 39, 48). The results of the real-time-t-RFLP analysis of these three strains were plotted as the numbers of 16S rRNA gene copies per milliliter (y axis) versus the corresponding T-RFs (x axis) (Fig. 2B). Experiments with different amounts of 16S rRNA gene template per PCR mixture (range, 0.5 to 25 ng/PCR reaction) were conducted to test the reliability of the real-time-t-RFLP assay. Linear correlation coefficients (r2) of >0.99 were obtained for the number of 16S rRNA gene copies and the amount of DNA template per PCR reaction (Fig. 2B, insets), suggesting that the amplification efficiency with the labeled primers and probe was quite constant.
FIG. 2.

Real-time-t-RFLP analysis of three known strains (E. coli, B. thuringiensis, and P. fluorescens). A 10-ng DNA template was used in each real-time PCR. (A) T-RF peaks for E. coli, B. thuringiensis, and P. fluorescens at 82, 103, and 105 bp, respectively. The sizes (in base pairs) of T-RFs of the three strains predicted from TAP analysis of Ribosomal Database Project II (41) are indicated in parentheses. (B) 16S rRNA gene copies of the three strains (expressed as T-RFs) were quantified by the real-time-t-RFLP assay. The error bars indicatethe range of results of duplicate experiments. The insets show good linear correlations between the measured number of 16S rRNA gene copies and the amount of DNA template per PCR reaction (range, 0.5 to 25 ng/PCR reaction). (C) Comparison of results of real-time PCR assays performed with labeled forward primer and nonlabeled forward primer.
An additional set of experiments was conducted to examine if the labeled primer affects the quantification of 16S rRNA gene copies in samples during real-time PCR amplification. For direct comparison, real-time PCR assays were conducted with labeled and nonlabeled forward primer by using plasmid #931, three pure strains, and the model microbial community (10 ng of template DNA). The results of real-time PCR assays with plasmid #931 are shown in Fig. 2C. A high correlation between the threshold cycle and the log copy number was obtained with both labeled forward primer and nonlabeled forward primer (r2 > 0.99 in each case). In addition, the difference between the slopes and intercepts of two standard curves when labeled and nonlabeled primers were used was very small (0.5%). t tests were also used to determine whether the gene copy values obtained with the labeled forward primer in real-time PCR assays were significantly different from the values obtained by using the nonlabeled forward primer. As shown in Fig. 2C, the differences between the values obtained with the labeled forward primer and the values obtained with the nonlabeled forward primer are not statistically significant (P ≤ 0.05). These results (Fig. 2) demonstrated that the labeled forward primer could be easily incorporated into the PCR product without interfering with real-time PCR detection and that the fluorescence of the PCR products after digestion was stable and sufficient to generate a consistent microbial community profile.
The second set of experiments was conducted to examine whether the real-time-t-RFLP assay is capable of quantifying the 16S rRNA gene copies of each strain in a model microbial community. A model community was constructed by mixing equal amounts of genomic DNA from three strains. For experiments in which 10 ng of community DNA was used as the template, the real-time-t-RFLP assay successfully quantified the 16S rRNA gene copies of E. coli, B. thuringiensis, and P. fluorescence, expressed as three distinct T-RFs (82, 103, and 105 bp, respectively) (Fig. 3). Consistent results were also obtained in duplicate experiments when we used amounts of community DNA template ranging from 0.5 to 25 ng per PCR reaction (Table 1). Over this template range, variations ranging from 10 to 20% in the ratios of the areas measured under T-RFs were observed. The variation was most likely a result of instrument errors, including errors in the values obtained with the fluorometer used for DNA concentration measurement and with the PCR machine used for real-time PCR amplification. Experimental errors and preferred PCR amplification for different species (i.e., slightly different slopes [10% variation] for the three strains shown in the insets in Fig. 2B) were also possible. Overall, the results of the experiments performed with community DNA suggested that the real-time-t-RFLP assay could qualitatively and quantitatively characterize the microbial community structure.
FIG. 3.

Real-time-t-RFLP analysis of a constructed microbial community containing E. coli, B. thuringiensis, and P. fluorescens. (A) T-RF peaks for E. coli, B. thuringiensis, and P. fluorescens at 82, 103, and 105 bp, respectively. (B) Numbers of 16S rRNA gene copies in all three strains (expressed as T-RFs) in the constructed community quantified by real-time-t-RFLP analysis.
TABLE 1.
Summary of real-time-t-RFLP analysis of a three-member bacterial community
| No. of DNA templates per PCR mixture, ng/PCR reaction (82A/103A/105A ratio)a | No. of 16S rRNA gene copies/mlb
|
||
|---|---|---|---|
| E. coli | B. thuringiensis | P. fluorescens | |
| 0.5 (1.1:1.1:1) | (0.96 ± 0.04) × 106 | (1.18 ± 0.05) × 106 | (0.79 ± 0.15) × 106 |
| 5 (1.0:1.1:1) | (1.28 ± 0.15) × 107 | (1.32 ± 0.52) × 107 | (0.82 ± 0.06) × 107 |
| 10 (0.9:0.9:1) | (2.87 ± 0.46) × 107 | (2.76 ± 0.42) × 107 | (2.26 ± 0.45) × 107 |
| 25 (0.9:0.8:1) | (7.41 ± 1) × 107 | (6.59 ± 0.18) × 107 | (6.00 ± 0.08) × 107 |
82A, area under 82-bp T-RF, representing E. coli; 103A, area under 103-bp T-RF, representing B. thuringiensis; 105A, area under 105-bp T-RF, representing P. fluorescens.
The values were calculated by performing data analysis as described in Materials and Methods and indicate the ranges for duplicate experiments.
Application of the real-time-t-RFLP assay to wastewater.
Experiments were also conducted to determine whether the real-time-t-RFLP assay can be applied to environmental samples, particularly samples containing complex microbial communities, like wastewater. To address this question, wastewater samples were collected from primary effluent, the center of an oxidation ditch, and a clarifier of WWTP1, and the microbial communities were analyzed by the real-time-t-RFLP assay (Fig. 4). A comparison of the t-RFLP electropherograms for these three microbial communities showed little variation (Fig. 4A), suggesting that the microbial communities in the samples were similar. These results were unexpected since (i) higher nutrient levels were present in the primary effluent and lower nutrient levels were present in the clarifier, (ii) biomass was recycled back from the bottom of the clarifier to the oxidation ditch, and (iii) less microbial biomass and diversity were expected in the upper region of the clarifier (where the sample was collected) due to settlement. Unlike the electropherograms in Fig. 4A, the results of a real-time-t-RFLP assay shown in Fig. 4B revealed a 100-fold-greater number of 16S rRNA gene copies of each corresponding T-RF in the oxidation ditch than in the primary effluent and clarifier. This quantitative information is particularly useful because it provides a link between microbial community function, such as nitrification-denitrification and bioremediation processes, and the abundance of specific ribotypes. Such quantitative data can be rapidly obtained only through real-time-t-RFLP analysis.
FIG. 4.

Changes in the microbial ecology of wastewater as treatment proceeds. (A) Diagram of WWTP1 and t-RFLP profiles of the microbial communities from the primary effluent, oxidation ditch, and clarifier. HRT, hydraulic retention time; SRT, solid retention time; BOD, biological oxygen demand; DO, dissolved oxygen. An asterisk indicates a sampling location. (B) Real-time-t-RFLP assay revealed the variation in the number of 16S rRNA gene copies per milliliter for each ribotype in the three wastewater samples examined.
The real-time-t-RFLP assay was also applied to activated sludge samples collected from three WWTPs in which different biological treatment processes were used (Fig. 5). WWTP1 consists of an oxidation ditch, WWTP2 consists of a nitrified completely mixed activated sludge system, and WWTP3 consists of a plug flow conventional activated sludge reactor with even aeration. The oxidation ditch is considered a plug flow reactor in which the nutrient levels (dissolved oxygen, biological oxygen demand, nitrogen, and phosphate contents) change over time as wastewater travels through the reactor. Unlike the completely mixed bioreactor used in WWTP2, the nutrient levels are considered uniform throughout the reactor. The nutrient levels influence the microbial community structure and function in the reactor; subsequently, the changes in microbial community structure and function modify the nutrient levels in the reactor. As shown in Fig. 5, different patterns of microbial communities were observed in the three activated sludge samples. For the WWTP1 sample the levels of 7 of 14 T-RFs (84, 89, 98, 103, 105, 315, and 328 bp) were in the range from 2.8 × 108 to 1.2 × 109 16S rRNA gene copies/ml, accounting for 80% of the 5.0 × 109 16S rRNA gene copies/ml. For WWTP2 the levels of 7 of 16 T-RFs (83, 98, 103, 105, 187, 327, and 329 bp) were in the range from 4.0 × 108 to 1.4 × 109 16S rRNA gene copies/ml, accounting for 84% of the total copies of the 16S rRNA gene. And for the WWTP3 sample the levels of 6 of the 14 T-RFs (84, 98, 103, 104, 105, 108, and 327 bp) were in the range from 1.5 × 109 to 3.8 × 109 16S rRNA gene copies/ml, accounting for 88% of total 16S rRNA gene copies measured (1.5 × 1010 16S rRNA gene copies/ml).
FIG. 5.

Real-time-t-RFLP analysis of microbial communities in activated sludge collected at three different WWTPs from an oxidation ditch, nitrified completely mixed activated sludge, and conventional plug flow activated sludge. An asterisk indicates a sampling location.
Application of real-time-t-RFLP to estrogen-degrading consortia.
Estrogen-degrading consortia enriched from activated sludge collected from WWTP1 and WWTP2 were capable of degrading three different estrogens (17α-estradiol, 17β-estradiol, and estrone). After five successive enrichments, the consortia showed enhanced degradation ability; the time necessary for complete degradation of 17α-estradiol, 17β-estradiol, or estrone (1 mg/liter) decreased from 7 to 3 days.
In response to the presence of different estrogens, the microbial community structures in the enrichment consortia were expected to change. Not surprisingly, shifts in diversity and abundance of ribotypes were observed in all three enrichment consortia (Fig. 6). For 17α-estradiol-enriched consortia, the number of ribotypes (expressed as T-RFs) decreased from 16 to 12. For 17β-estradiol- and estrone-enriched consortia, the number of ribotypes decreased to 10. Meanwhile, the abundance of certain ribotypes either increased or decreased after the enrichment. Interestingly, the 91-, 98-, 103-, 105-, 114-, 324-, and 327-bp T-RFs were observed in all consortia, regardless of the type of estrogen, suggesting that these ribotypes were directly or indirectly related to complete degradation of all three estrogens. The 72-bp T-RF was observed only in 17β-estradiol-enriched consortia, implying that this ribotype might be related to 17β-estradiol degradation but not to 17α-estradiol or estrone degradation. Similarly, some T-RFs were observed only in 17α-estradiol-enriched consortia, suggesting that there was a unique growth requirement for these T-RFs.
FIG. 6.

Microbial communities of estrogen-degrading consortia based on real-time-t-RFLP assays. (A) Community profiles of consortia before and after the fifth enrichment with the estrogens 17α-estradiol, 17β-estradiol, and estrone. Activated sludge from WWTP2 was used as the inoculum. (B) Real-time-t-RFLP analysis of estrogen-degrading microbial consortia.
DISCUSSION
In studies performed with other fingerprinting methods, such as t-RFLP analysis and denaturing gradient gel electrophoresis, workers observed fallacious microbial community structure due to the presence of artificial peaks that commonly resulted from incomplete digestion of PCR products (15, 19, 38, 45, 48). For real-time-t-RFLP analysis, complete digestion of the PCR product is essential for obtaining accurate quantification of each ribotype since only complete digestion can result in a correct percentage of each ribotype (T-RF) and the percentage is subsequently used to calculate the number of 16S rRNA gene copies for each ribotype. To optimize the real-time-t-RFLP assay for complete digestion of PCR products, different mass ratios of PCR products to restriction enzyme (MspI) were examined by using two different digestion procedures: a single-step procedure (MspI was added at the beginning) and a two-step procedure (equal doses of MspI were added in the beginning and 6 h after the first dose). PCR products were digested under recommended digestion temperature and duration conditions. It was found that a single 10-U dose of MspI was sufficient to completely digest 60- to 90-ng PCR products in a 30-μl digestion reaction mixture (results not shown), as described in Materials and Methods.
In addition to incomplete digestion, many other factors (including nonspecific PCR products) can result in artificial peaks in t-RFLP profiles (15). In this study, a factor that caused artificial peaks that has not been reported previously was noted. When a digested DNA sample was overloaded during capillary electrophoresis, artificial peaks occurred at sizes 6 to 10% shorter than the sizes of measured T-RFs. As shown in Fig. 7D, an artificial peak at 48 bp was observed along with the measured T-RF (51 bp) of HhaI-digested PCR products when plasmid #931 was used as a template. When the loaded sample sizes were decreased proportionally (2, 1, 0.5, and 0.25 ng/μl), the area under the T-RF peak at 48 bp decreased rapidly but not proportionally. As expected, the area under the T-RF peak at 51 bp decreased in proportion to the loaded sample size. Furthermore, the area ratios of the 48-bp T-RF peak to the 51-bp T-RF peak did not remain constant, indicating that the 48-bp T-RF peak was an artificial peak. To avoid generation of artificial peaks, digested samples were loaded at concentrations of less than 0.5 ng/μl. Nevertheless, it is also recommended that each sample be run with at least two different dilutions during capillary electrophoresis to avoid overestimating the diversity and/or underestimating the abundance of ribotypes in a microbial community.
FIG. 7.

Observation of artificial peaks due to overloaded DNA sample during capillary electrophoresis.
Selection of a proper amplicon size for a real-time-t-RFLP assay can be a challenge due to the inherent limitations of the real-time PCR and t-RFLP techniques. For TaqMan real-time PCR, the recommended amplicon length is less than 400 bp because shorter amplicons allow effective binding of probes and primers to the complementary sequences (12). For t-RFLP analysis, the recommended size is between 400 and 700 bp, an amplicon size that is long enough to include sequence divergence among microorganisms and at least one restriction site (38). Based on both criteria, the real-time-t-RFLP assay with an amplicon size of 352 bp was successfully developed. According to TAP analysis of the 16S rRNA gene sequences in the Ribosomal Database Project database (allowing at most no mismatch in the last four nucleotides from the 3′ end of the forward primer and three mismatches in any other nucleotide), this amplicon should generate 188 unique T-RFs when a sample is digested with MspI and only 157 of 11,274 primed sequences (1.4%) showed no MspI restriction sites in this 352-bp amplicon. In this study, the 352-bp T-RF was observed in only two digested samples, the clarifier sample and the 17α-estradiol-degrading consortium enriched from the clarifier sample from WWTP1 (<5% of the total area under T-RF peaks), suggesting that there was a lack of restriction sites for MspI in amplicons of these two samples. As the amplicon used in this study was not located in variable region I in the 16S rRNA gene and the first 500 bp of variable region I provides better resolution for t-RFLP analysis (39), designing different amplicons (such as amplicons targeting a different region of the 16S rRNA gene) and using different restriction enzymes to digest amplified PCR products should improve the resolution and avoid undigested ampilcons.
Although the real-time-t-RFLP assay is subject to systematic biases like any PCR-based method (56) and has the same limitations as t-RFLP analysis (39), the application of the real-time-t-RFLP assay to environmental samples was considered successful. The microbial ecology of activated sludge is very complex and has not yet been fully illustrated. Many predominant bacterial groups in activated sludge have been identified by using a wide range of molecular techniques, including clonal library analysis (7), in situ hybridization (36, 52), denaturing gradient gel electrophoresis (47), and t-RFLP analysis (21, 57). The predominant bacterial phyla are the Proteobacteria, Planctomycetes, Bacteroidetes, Firmicutes, and Actinobacteria. As shown in Table 2, the microbial ecology in wastewater and activated sludge analyzed by the real-time-t-RFLP assay (Fig. 4 and 5) was consistent with the microbial ecology determined in other studies. For example, the measured T-RFs are consistent with the expected T-RFs of a wide range of known microorganisms, including foam-causing, filamentous, nitrifying-denitrifying, and presumptive phosphorus-accumulating bacteria. By developing different combinations of amplicons and restriction enzymes, workers may be able to quantify specific functional groups of microorganisms by the real-time-t-RFLP assay. Quantitative information pertaining to specific functional groups of microorganisms not only provides interesting insights into these functional communities but may also provide a fundamental basis for fine-tuning operational parameters to prevent foaming and sludge bulking, as well as to enhance the nitrogen and phosphorus removal efficiency in treatment units. The quantitative feature of the real-time-t-RFLP assay is a powerful tool for understanding the microbial ecology underlying microbially meditated processes.
TABLE 2.
Comparison of observed T-RFs to the T-RFs of known bacteria in activated sludge
| Phylum | Class | Species and/or strain(s) (size of T-RF [bp])a | T-RF size (bp)b |
|---|---|---|---|
| Proteobacteria | α | Rhodobacter sphaeroides (42),cRhodobacter blasticus (42),cParacoccus denitrificans (42)c,d | 39 |
| Sphingomonas capsulata (106),eMesorhizobium loti (106),cHyphomicrobium aestuarii (106),cHyphomicrobium vulgare (106),cNitrobacter winogradskyi (106),fHyphomicrobium zavarzinii (106)d | 103 | ||
| Beijerinckia indica (328)c,g | 325 | ||
| β | Eikelboom type 1863 (87)h | 84 | |
| Aquabacterium commune (101),gIdeonella dechloratans (101),cBrachymonas denitrificans (101),cLeptothrix cholodnii (101),c,hComamonas denitrificans (101),dLeptothrix mobilis (101),hSphaerotilus natans (101),hLeptothrix discophora (101)h | 98 | ||
| Comamonas testosteroni (105),eRhodoferax fermentans (105),eVariovorax paradoxus (105)e | 102 | ||
| Rhodocyclus tenuis (106),gThauera aromatica, (106),gDunganella zoologleoides (106),gZoogloea ramigera (106),cAzoarcus evansii (106),cNitrosococcus mobilis (106),fNitrosomonas marina (106),fRhodocyclus sp. strain R6 (106)i | 103 | ||
| Nitrosomonas europaea (107),fNitrosomonas eutropha (107)f | 104 | ||
| Leucothrix mucor (108)h | 105 | ||
| γ | Acinetobacter johnsonii (87)e | 84 | |
| Aeromonas hydrophila (108),ePseudomonas stutzeri (108)d | 105 | ||
| Acinetobacter calcoaceticus (249)e | 246 | ||
| ɛ | Arcobacter cryaerophilus (329)e | 326 | |
| Nitrospira | Nitrospira sp. strain RC14 (77),gNitrospira moscoviensis (77)c,f | 74 | |
| Bacteroidetes | Haliscomenobacter hydrossis (106)h | 103 | |
| Verrucomicrobia | Prosthecobacter dejongeii FC1 (77)g | 74 | |
| Actinobacteria | Gordona amarae (87),h,jTetrasphaera japonica (87)i | 84 | |
| Nocardia asteroides (108),hRhodococcus rhodochrous (108),hCandidatus “Nostocoida limicola” (108),h,iTetrasphaera australienesis (108),iTetrasphaera elongata (108)i | 105 | ||
| Firmicutes | Streptococcus bovis (106),eErysipelothrix rhusiopathiae (106),eEubacterium bioforme (106),eLactosphaera pasteurii (106)h | 103 | |
| Clostridium sticklandii (281)c | 278 |
The predicted sizes of T-RFs were based on a TAP analysis (41). MspI was used as the restriction enzyme.
Size of the most likely T-RF measured by the real-time-t-RFLP assay, assuming that the measured T-RFs are 3 bases shorter than the predicted T-RFs.
16S rRNA gene clone library from activated sludge (35). The most closely related microorganisms (sequence similarity, >95%) are listed.
Microorganism identified by fluorescence in situ hybridization in activated sludge (52).
Microorganism identified by cloning and sequencing in a conventional anaerobic-aerobic sequencing batch reactor (16).
Known filamentous bacterium in WWTP (57).
Known foam-causing bacterium in WWTP (17).
Biodegradation of estrogen in activated sludge has been strongly suggested in previous studies; however, the diversity and abundance of estrogen-degrading cultures in activated sludge remain unclear. The changes in the microbial diversity and quantities in response to addition of three different types of estrogens (Fig. 6) indicated the potential presence of estrogen-degrading cultures in activated sludge. Studies which focus on isolation and characterization of estrogen-degrading cultures in activated sludge should be useful for the development of optimal operating conditions for better estrogen removal from wastewater.
The strategy used in real-time-t-RFLP assay development can be extended from eubacteria to archaea, a specific phylogenetic group, and/or functionally similar groups. In this study, the 16S rRNA gene was used to target all eubacteria. Similarly, some functional genes can be used for development of a method for quantifying a specific group of functional microorganisms. For example, the amoA genes have shown higher diversity than the 16S rRNA gene in ammonia-oxidizing bacteria (49) and can be further adapted into a real-time- t-RFLP assay for studying ammonia-oxidizing bacteria. For studies designed to increase the T-RF resolution at the species level, the 16S-23S intergenic region that has significant heterogeneity in both length and nucleotides (20) can be considered during design of the amplicon.
The real-time-t-RFLP assay will be particularly useful for in situ or ex situ bioremediation in which the microbial activity changes in response to increases or decreases in specific microbial populations. The information obtained should allow better assessment of the treatment duration required for achieving target clean-up goals. In addition, this assay should also be useful for studying microbial ecology in soils and water in which the microbial communities vary due to spatial or temporal differences, pH variation, nutrient availability, and many engineered biological treatment processes (such as processes in bioreactors) in which microbial populations shift in response to environmental toxicants and/or stimulants.
Acknowledgments
We thank Alice Layton and Hebe Dionisi for their technical advice during the development of the real-time-t-RFLP assay. We also thank John Sanseverino, Steve Ripp, Jack McPherson, and Jim Fleming for helpful comments about the manuscript.
REFERENCES
- 1.Avaniss-Aghajani, E., K. Jones, D. Chapman, and C. Brunk. 1994. A molecular technique for identification of bacteria using small subunit ribosomal RNA sequences. BioTechniques 17:144-146. [PubMed] [Google Scholar]
- 2.Bach, H.-J., J. Tomanova, M. Schloter, and J. C. Munch. 2002. Enumeration of total bacteria and bacteria with genes for proteolytic activity in pure cultures and in environmental samples by quantitative PCR mediated amplification. J. Microbiol. Methods 49:235-245. [DOI] [PubMed] [Google Scholar]
- 3.Beller, H. R., S. R. Kane, T. C. Legler, and P. J. J. Alvarez. 2002. A real-time polymerase chain reaction method for monitoring anaerobic, hydrocarbon-degrading bacteria based on a catabolic gene. Environ. Sci. Technol. 36:3977-3984. [DOI] [PubMed] [Google Scholar]
- 4.Berger, C., P. Day, G. Meier, W. Zingg, W. Bossart, and D. Nadal. 2001. Dynamics of Epstein-Barr virus DNA levels in serum during EBV-associated disease. J. Med. Virol. 64:505-512. [PubMed] [Google Scholar]
- 5.Bieche, I., P. Onody, I. Laurendeau, M. Olivi, D. Vidaud, R. Lidereau, and M. Vidaud. 1999. Real-time reverse transcription-PCR assay for future management of ERBB2-based clinical applications. Clin. Chem. 45:1148-1156. [PubMed] [Google Scholar]
- 6.Blackall, L. L., E. M. Seviour, D. Bradford, S. Rossetti, V. Tandoi, and R. J. Seviour. 2000. ‘Candidatus Nostocoida limicola,’ a filamentous bacterium from activated sludge. Int. J. Syst. Evol. Microbiol. 50:703-709. [DOI] [PubMed] [Google Scholar]
- 7.Bond, P. L., P. Hugenholtz, J. Keller, and L. L. Blackall. 1995. Bacterial community structures of phosphate-removing and non-phosphate-removing activated sludges from sequencing batch reactors. Appl. Environ. Microbiol. 61:1910-1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braker, G., H. L. Ayala-del-Rio, A. H. Devol, A. Fesefeldt, and J. M. Tiedje. 2001. Community structure of denitrifiers, bacteria, and archaea along redox gradients in Pacific Northwest marine sediments by terminal restriction fragment length polymorphism analysis of amplified nitrite reductase (nirS) and 16S rRNA genes. Appl. Environ. Microbiol. 67:1893-1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braker, G., A. Fesefeldt, and K.-P. Witzel. 1998. Development of PCR primer systems for amplification of nitrite reductase genes (nirK and nirS) to detect denitrifying bacteria in environmental samples. Appl. Environ. Microbiol. 64:3769-3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brechtbuehl, K., S. A. Whalley, G. M. Dusheiko, and N. A. Saunders. 2001. A rapid real-time quantitative polymerase chain reaction for hepatitis B virus. J. Virol. Methods 93:105-113. [DOI] [PubMed] [Google Scholar]
- 11.Buchan, A., S. Y. Newell, J. I. L. Moreta, and M. A. Moran. 2002. Analysis of internal transcribed spacer (ITS) regions of rRNA genes in fungal communities in a southeastern U.S. salt marsh. Microb. Ecol. 43:329-340. [DOI] [PubMed] [Google Scholar]
- 12.Bustin, S. A. 2000. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 25:169-193. [DOI] [PubMed] [Google Scholar]
- 13.Chin, K.-J., T. Lukow, S. Stubner, and R. Conrad. 1999. Structure and function of the methanogenic archaeal community in stable cellulose-degrading enrichment cultures at two different temperatures (15 and 30°C). FEMS Microbiol. Ecol. 30:313-326. [DOI] [PubMed] [Google Scholar]
- 14.Chu, K. H., and L. Alvarez-Cohen. 1996. Trichloroethylene degradation by methane-oxidizing cultures grown with various nitrogen sources. Water Environ. Res. 68:76-82. [Google Scholar]
- 15.Clement, B. G., L. E. Kehl, K. L. DeBord, and C. L. Kitts. 1998. Terminal restriction fragment patterns (TRFPs), a rapid, PCR-based method for the comparison of complex bacterial communities. J. Microbiol. Methods 31:135-142. [Google Scholar]
- 16.Dabert, P., B. Sialve, J.-P. Delgenes, R. Moletta, and J.-J. Godon. 2001. Characterization of the microbial 16S rDNA diversity of an aerobic phosphorus-removal ecosystem and monitoring of its transition to nitrate respiration. Appl. Microbiol. Biotechnol. 55:500-509. [DOI] [PubMed] [Google Scholar]
- 17.de los Reyes, F. L., III, and L. Raskin. 2002. Role of filamentous microorganisms in activated sludge foaming: relationship of mycolata levels to foaming initiation and stability. Water Res. 36:445-459. [DOI] [PubMed] [Google Scholar]
- 18.Dionisi, H. M., A. C. Layton, G. Harmes, I. R. Gregory, K. G. Robinson, and G. S. Sayler. 2002. Quantification of Nitrosomonas oligotropha-like ammonia-oxidizing bacteria and Nitrospira spp. from full-scale wastewater treatment plants by competitive PCR. Appl. Environ. Microbiol. 68:245-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fey, A., and R. Conrad. 2000. Effect of temperature on carbon and electron flow and on the archaeal community in methanogenic rice field soil. Appl. Environ. Microbiol. 66:4790-4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fisher, M. M., and E. W. Triplett. 1999. Automated approach for ribosomal intergenic spacer analysis of microbial diversity and its application to freshwater bacterial communities. Appl. Environ. Microbiol. 65:4630-4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forney, L. J., W. T. Liu, J. B. Guckert, Y. Kumagai, E. Namkung, T. Nishihara, and R. J. Larson. 2001. Structure of microbial communities in activated sludge: potential implications for assessing the biodegradability of chemicals. Ecotoxicol. Environ. Saf. 49:40-53. [DOI] [PubMed] [Google Scholar]
- 22.Fortin, N. Y., A. Mulchandani, and W. Chen. 2001. Use of real-time polymerase chain reaction and molecular beacons for the detection of Escherichia coli O157:H7. Anal. Biochem. 289:281-288. [DOI] [PubMed] [Google Scholar]
- 23.Gibson, U. E., C. A. Heid, and P. M. Williams. 1996. A novel method for real time quantitative RT-PCR. Genome Res. 6:995-1001. [DOI] [PubMed] [Google Scholar]
- 24.Giovangrandi, Y., B. Parfait, M. Asheuer, M. Olivi, R. Lidereau, M. Vidaud, and I. Bieche. 2001. Analysis of the human CGB/LHB gene cluster in breast tumors by real-time quantitative RT-PCR assays. Cancer Lett. 168:93-100. [DOI] [PubMed] [Google Scholar]
- 25.Gruntzig, V., S. C. Nold, J. Z. Zhou, and J. M. Tiedje. 2001. Pseudomonas stutzeri nitrite reductase gene abundance in environmental samples measured by real-time PCR. Appl. Environ. Microbiol. 67:760-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gumaelius, L., G. Magnusson, B. Pettersson, and G. Dalhammar. 2001. Comamonas denitrificans sp. nov., an efficient denitrifying bacterium isolated from activated sludge. Int. J. Syst. Evol. Microbiol. 51:999-1006. [DOI] [PubMed] [Google Scholar]
- 27.Hanada, S., W.-T. Liu, T. Shintani, Y. Kamagata, and K. Nakamura. 2002. Tetrasphaera elongata sp. nov., a polyphosphate-accumulating bacterium isolated from activated sludge. Int. J. Syst. Evol. Microbiol. 52:883-887. [DOI] [PubMed] [Google Scholar]
- 28.Harms, G., A. C. Layton, H. M. Dionisi, I. R. Gregory, S. Hawkins, K. G. Robinson, and G. S. Sayler. 2003. Real-time PCR quantification of nitrifying bacteria in a municipal wastewater treatment plant. Environ. Sci. Technol. 37:343-351. [DOI] [PubMed] [Google Scholar]
- 29.Heid, C. A., J. Stevens, K. J. Livak, and P. M. Williams. 1996. Real time quantitative PCR. Genome Res. 6:986-994. [DOI] [PubMed] [Google Scholar]
- 30.Hesselmann, R. P. X., C. Werlen, D. Hahn, J. R. Van der Meer, and A. J. B. Zehnder. 1999. Enrichment, phylogenetic analysis, and detection of a bacterium that performs enhanced biological phosphate removal in activated sludge. Syst. Appl. Microbiol. 22:454-465. [DOI] [PubMed] [Google Scholar]
- 31.Higuchi, R., C. Fockler, C. Dollinger, and R. Watson. 1993. Kinetic PCR analysis: real time monitoring of DNA amplification reactions. Bio/Technology 11:1026-1030. [DOI] [PubMed] [Google Scholar]
- 32.Horz, H.-P., J.-H. Rotthauwe, T. Lukow, and W. Liesack. 2000. Identification of major subgroups of ammonia-oxidizing bacteria in environmental samples by T-RFLP analysis of amoA PCR products. J. Microbiol. Methods 39:197-204. [DOI] [PubMed] [Google Scholar]
- 33.Hristova, K. R., C. M. Lutenegger, and K. M. Scow. 2001. Detection and quantification of methyl tert-butyl ether-degrading strain PM1 by real-time TaqMan PCR. Appl. Environ. Microbiol. 67:5154-5160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ibekwe, A. M., and C. M. Grieve. 2003. Detection and quantification of Escherichia coli O157:H7 in environmental samples by real-time PCR. J. Appl. Microbiol. 94:421-431. [DOI] [PubMed] [Google Scholar]
- 35.Juretschko, S., A. Loy, A. Lehner, and M. Wagner. 2002. The microbial community composition of a nitrifying-denitrifying activated sludge from an industrial sewage treatment plant analyzed by the full-cycle rRNA approach. Syst. Appl. Microbiol. 25:84-99. [DOI] [PubMed] [Google Scholar]
- 36.Kampfer, P., R. Erhart, C. Beimfohr, J. Bohringer, M. Wagner, and R. Amann. 1996. Characterization of bacterial communities from activated sludge: culture-dependent numerical identification versus in situ identification using group- and genus-specific rRNA-targeted oligonucleotide probes. Microb. Ecol. 32:101-121. [DOI] [PubMed] [Google Scholar]
- 37.Kikuchi, T., K. Iwasaki, H. Nishihara, Y. Takamura, and O. Yagi. 2002. Quantitative and rapid detection of the trichloroethylene-degrading bacterium Methylocystis sp. M in groundwater by real-time PCR. Appl. Microbiol. Biotechnol. 59:731-736. [DOI] [PubMed] [Google Scholar]
- 38.Kitts, C. L. 2001. Terminal restriction fragment patterns: a tool for comparing microbial communities and assessing community dynamics. Curr. Issues Intest. Microbiol. 2:17-25. [PubMed] [Google Scholar]
- 39.Liu, W. T., T. L. Marsh, H. Cheng, and L. J. Forney. 1997. Characterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA. Appl. Environ. Microbiol. 63:4516-4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lukow, T., P. F. Dunfield, and W. Liesack. 2000. Use of the T-RFLP technique to assess spatial and temporal changes in the bacterial community structure within an agricultural soil planted with transgenic and non-transgenic potato plants. FEMS Microbiol. Ecol. 32:241-247. [DOI] [PubMed] [Google Scholar]
- 41.Marsh, T. L., P. Saxman, J. Cole, and J. Tiedje. 2000. Terminal restriction fragment length polymorphism analysis program, a web-based research tool for microbial community analysis. Appl. Environ. Microbiol. 66:3616-3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maszenan, A. M., R. J. Seviour, B. K. C. Patel, P. Schumann, J. Burghardt, Y. Tokiwa, and H. M. Stratton. 2000. Three isolates of novel polyphosphate-accumulating gram-positive cocci, obtained from activated sludge, belong to a new genus, Tetrasphaera gen. nov., and description of two new species, Tetrasphaera japonica sp. nov. and Tetrasphaera australiensis sp. nov. Int. J. Syst. Evol. Microbiol. 50:593-603. [DOI] [PubMed] [Google Scholar]
- 43.Matz, C., and K. Jurgens. 2003. Interaction of nutrient limitation and protozoan grazing determines the phenotypic structure of a bacterial community. Microb. Ecol. 45:384-398. [DOI] [PubMed] [Google Scholar]
- 44.Mercier, B., L. Burlot, and C. Ferec. 1999. Simultaneous screening for HBV DNA and HCV RNA genomes in blood donations using a novel TaqMan PCR assay. J. Virol. Methods 77:1-9. [DOI] [PubMed] [Google Scholar]
- 45.Mills, D. K., K. Fitzgerald, C. D. Litchfield, and P. M. Gillevet. 2003. A comparison of DNA profiling techniques for monitoring nutrient impact on microbial community composition during bioremediation of petroleum-contaminated soils. J. Microbiol. Methods 54:57-74. [DOI] [PubMed] [Google Scholar]
- 46.Moeseneder, M. M., C. Winter, J. M. Arrieta, and G. J. Herndl. 2001. Terminal-restriction fragment length polymorphism (T-RFLP) screening of a marine archaeal clone library to determine the different phylotypes. J. Microbiol. Methods 44:159-172. [DOI] [PubMed] [Google Scholar]
- 47.Nielsen, A. T., W.-T. Liu, C. Filipe, L. Grady, Jr., S. Molin, and D. A. Stahl. 1999. Identification of a novel group of bacteria in sludge from a deteriorated biological phosphorus removal reactor. Appl. Environ. Microbiol. 65:1251-1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Osborn, A. M., E. R. Moore, and K. N. Timmis. 2000. An evaluation of terminal-restriction fragment length polymorphism (T-RFLP) analysis for the study of microbial community structure and dynamics. Environ. Microbiol. 2:39-50. [DOI] [PubMed] [Google Scholar]
- 49.Purkhold, U., A. Pommerening-Roser, S. Juretschko, M. C. Schmid, H.-P. Koops, and M. Wagner. 2000. Phylogeny of all recognized species of ammonia oxidizers based on comparative 16S rRNA and amoA sequence analysis: implications for molecular diversity surveys. Appl. Environ. Microbiol. 66:5368-5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Raman, D. R., A. C. Layton, L. B. Moody, J. P. Easter, G. S. Sayler, R. T. Burns, and M. D. Mullen. 2001. Degradation of estrogens in dairy waste solids: effects of acidification and temperature. Trans. ASAE 44:1881-1888. [Google Scholar]
- 51.Sait, L., M. Galic, R. A. Strugnell, and P. H. Janssen. 2003. Secretory antibodies do not affect the composition of the bacterial microbiota in the terminal ileum of 10-week-old mice. Appl. Environ. Microbiol. 69:2100-2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Snaidr, J., R. Amann, I. Huber, W. Ludwig, and K.-H. Schleifer. 1997. Phylogenetic analysis and in situ identification of bacteria in activated sludge. Appl. Environ. Microbiol. 63:2884-2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suzuki, M. T., L. T. Taylor, and E. F. DeLong. 2000. Quantitative analysis of small-subunit rRNA genes in mixed microbial populations via 5′-nuclease assays. Appl. Environ. Microbiol. 66:4605-4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takai, K., D. P. Moser, M. DeFlaun, T. C. Onstott, and J. K. Fredrickson. 2001. Archaeal diversity in waters from deep South African gold mines. Appl. Environ. Microbiol. 67:5750-5760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Teske, A., E. Alm, J. M. Regan, S. Toze, B. E. Rittmann, and D. A. Stahl. 1994. Evolutionary relationships among ammonia- and nitrite-oxidizing bacteria. J. Bacteriol. 176:6623-6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.von Wintzingerode, F., U. B. Gobel, and E. Stackebrandt. 1997. Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiol. Rev. 21:213-229. [DOI] [PubMed] [Google Scholar]
- 57.Wagner, M., A. Loy, R. Nogueira, U. Purkhold, N. Lee, and H. Daims. 2002. Microbial community composition and function in wastewater treatment plants. Antonie Leeuwenhoek 81:665-680. [DOI] [PubMed] [Google Scholar]