

Abstract
Pseudomonas putida strain MnB1, a biofilm-forming bacterial culture, was used as a model for the study of bacterial Mn oxidation in freshwater and soil environments. The oxidation of aqueous Mn+2 [Mn+2(aq)] by P. putida was characterized by spatially and temporally resolving the oxidation state of Mn in the presence of a bacterial biofilm, using scanning transmission X-ray microscopy (STXM) combined with near-edge X-ray absorption fine structure (NEXAFS) spectroscopy at the Mn L2,3 absorption edges. Subsamples were collected from growth flasks containing 0.1 and 1 mM total Mn at 16, 24, 36, and 48 h after inoculation. Immediately after collection, the unprocessed hydrated subsamples were imaged at a 40-nm resolution. Manganese NEXAFS spectra were extracted from X-ray energy sequences of STXM images (stacks) and fit with linear combinations of well-characterized reference spectra to obtain quantitative relative abundances of Mn(II), Mn(III), and Mn(IV). Careful consideration was given to uncertainty in the normalization of the reference spectra, choice of reference compounds, and chemical changes due to radiation damage. The STXM results confirm that Mn+2(aq) was removed from solution by P. putida and was concentrated as Mn(III) and Mn(IV) immediately adjacent to the bacterial cells. The Mn precipitates were completely enveloped by bacterial biofilm material. The distribution of Mn oxidation states was spatially heterogeneous within and between the clusters of bacterial cells. Scanning transmission X-ray microscopy is a promising tool for advancing the study of hydrated interfaces between minerals and bacteria, particularly in cases where the structure of bacterial biofilms needs to be maintained.
The chemistry of many aqueous environments is dictated by reactions that occur at the interface between the aqueous solution phase and the solid mineral and organic phases (43). It is at this interface that microorganisms also accumulate (4, 14). Microorganisms are known to form layered communities (biofilms) characterized by production of extracellular polymers, in which the physical and chemical conditions may differ greatly from those of the bulk solution. Within the biofilm setting, microorganisms carry out dissolution and precipitation of minerals, exerting direct influence on the cycling of trace and contaminant metals alike (18, 51).
In particular, the precipitation of manganese (Mn) in soils, sediments, and water columns is primarily driven by microbial activity (11, 18, 33). Although conditions are thermodynamically favorable for the oxidation of aqueous Mn+2 [Mn+2(aq)] in the presence of dissolved oxygen, the homogeneous (reaction involving only solution-phase reactants) kinetics of oxidation are slow (32). Rates of Mn oxidation observed in natural waters are often too high to be attributed to homogeneous or surface-mediated reactions (32). Microbial processes are thought to control the precipitation and dissolution of Mn oxides in many natural systems, and Mn-oxidizing microorganisms have been isolated from a great diversity of environments (46).
Manganese oxide and oxyhydroxide minerals play an important role in the mobility and bioavailability of metals and organic compounds. It is therefore important to characterize the products of Mn oxidation to understand better biogeochemical cycles of elements such as carbon, iron, sulfur, and arsenic (32). Numerous laboratory studies have characterized Mn oxides as strong environmental oxidants influencing the speciation of redox-sensitive metals and metalloids, such as Cr (3) and As (41), as well as the processing of natural organic material (24). Studies of natural Mn oxides have confirmed their important role in regulating the speciation of trace metals, such as Zn, Ni, and Cu, in soils and sediments (23, 29).
In this study, Pseudomonas putida strain MnB1 was investigated as a model Mn-oxidizing bacterium to facilitate the study of Mn oxide chemistry in soil and freshwater settings. P. putida strain MnB1 was isolated from Mn-encrusted water pipes in Trier, Germany (46). The Mn-oxidizing factor expressed in P. putida GB-1, a bacterium with Mn-oxidizing characteristics similar to those of strain MnB1, was identified as an outer membrane bound protein (CumA) in the multicopper oxidase family (9). The mechanism by which P. putida transfers electrons from Mn(II) to Mn(IV) is currently unknown.
The presence of a Mn(III) species during Mn oxidation by Bacillus sp. strain SG-1 spores has been reported in a scanning transmission X-ray microscopy (STXM) study (35) but was not detected using unfocused X-ray absorption near-edge structure (XANES) spectroscopy (6). Manganese oxidation by Bacillus sp. strain SG-1 has also been studied in the presence of a strong Mn(III) complexing agent; these studies suggest that Mn oxidation by Bacillus sp. strain SG-1 consists of two one-electron transfers (45). The studies of Bacillus sp. strain SG-1 imply that Mn(III) is a possible biogenic intermediate species in Mn oxidation that may be spatially concentrated, avoiding detection with unfocused XANES spectroscopy. It is not currently known whether an Mn(III) reaction intermediate exists in oxidizing cultures of P. putida.
An understanding of the physical and chemical properties of the biogenic Mn oxide produced by P. putida is essential to careful studies of the environmental chemistry mediated by the Mn oxides. The present research is part of our ongoing study of the properties of biogenic Mn oxides. In previous work, we characterized the structure and oxidation state of Mn with X-ray diffraction (XRD) and X-ray absorption spectroscopic analyses, respectively (50). The biogenic Mn oxide was characterized after the biofilm and cellular material had been removed by oxidation with hypochlorite treatment. The clean biogenic Mn oxide had the layer structure of an Mn(III,IV) oxide with hexagonal sheet symmetry (birnessite), as determined by XRD. The average oxidation number of Mn in the clean biogenic oxide was 3.90 ± 0.05, as determined by titration, which was in agreement with the unfocused Mn XANES spectroscopy results for both the clean and untreated biogenic Mn oxide.
Manganese cycling in the environment is spatially and temporally heterogeneous, and its study presents significant challenges with respect to both chemical analysis and spatial resolution. The oxidation state of Mn is closely linked to numerous abiotic and biotic oxidation-reduction processes in which chemical gradients exist over minute spatial scales. As a result, samples are prone to chemical changes due to standard analysis procedures, such as extractions, sectioning, dehydration, and storage. Dehydration in particular, damages the structure of hydrated biofilms. STXM combined with near-edge X-ray absorption fine structure (NEXAFS) spectroscopy allows the Mn oxidation state to be measured in situ and at the submicron spatial scale of heterogeneity while minimizing sample degradation. This approach was applied recently to the study of Mn oxidation by the marine Bacillus species strain SG-1 (35) and formation of the Fe mineral akaganeite by microbially produced organic polymers under hydrated conditions (10).
A time-resolved, in situ, microspectroscopic investigation of Mn oxidation by P. putida was conducted. The spatial relationships between the bacterial cells, biogenic Mn oxides, and biofilm were determined in fully hydrated samples at a spatial resolution of 40 nm. Spatially resolved Mn L-edge NEXAFS spectra were used to measure quantitatively the relative abundances of Mn(II), Mn(III), and Mn(IV) as a function of time. The presented research will allow us to use the P. putida culture as a model for Mn-oxidizing bacteria in future studies of Mn oxide surface reactivity in the presence of the bacterial cells and biofilm. This study expands on the valuable research conducted by Pecher et al. (35) by studying a time series of Mn oxidation by a biofilm-forming bacterium at lower total Mn concentrations. The present study also differs from that of Pecher et al. (35) in the collection and processing of reference spectra as well as providing an estimation of radiation-induced damage to the sample.
MATERIALS AND METHODS
Bacterial growth.
The recipe for Leptothrix growth medium and the P. putida strain MnB1 culture were provided by Bradley Tebo (Scripps Institution of Oceanography) and are described in the work of Villalobos et al. (50). The Leptothrix growth medium consisted of 0.5 g of yeast extract liter−1, 0.5 g of Casamino Acids liter−1, 1 g of glucose liter−1, 2.38 g of HEPES buffer liter−1, 2 × 10−3 M CaCl2, 3.3 × 10−3 M MgSO4, 1.0 × 10−4 to 1.2 × 10−3 M MnCl2, 3.7 × 10−6 M FeCl3 and 1 ml of trace element solution liter−1. The trace element solution was prepared with the following metal salts (concentrations in mg liter−1 are given in parentheses): (10) CuSO4 · 5H2O, (44) ZnSO4 · 7H2O, (20) CoCl2 · 6H2O, and (13) Na2MoO4 · 2H2O. The growth medium and source solutions were prepared in MilliPore Milli-Q 18.2-MΩ/cm water (MQ water). The bacterial cultures were incubated at 27 to 30°C with 150-rpm orbital mixing.
Growth curves in liquid Leptothrix medium were prepared by spread plating and counting CFU. At 0, 12, 20, and 24 h, 1 ml of cell suspension was removed from three separate flasks and diluted. Several of the dilutions were then applied to agar plates (75 μl per plate) and incubated at 27°C, after which the colonies were counted. Attempts to use cell protein content as a proxy for cell number were not successful due to the culture's resistance to lysis via multiple freeze-thaw cycles, sodium dodecyl sulfate digestion, and bead beating.
Chemical analyses.
The amount of polysaccharides produced during growth was quantified with a colorimetric assay (13). The cells were grown in Leptothrix medium as described above. Subsamples were obtained 12, 24, 36, 48, and 72 h after inoculation. Eleven milliliters of cell suspension was centrifuged (9,400 relative centrifugal force, 4°C, 10 min) and resuspended in filter-sterilized MQ water three times. One milliliter of the rinsed cell suspension was digested in 2 ml of 10 M sulfuric acid at 120°C for 30 min in a fume hood (13, 39). The digest was passed through a glass fiber filter and rinsed with boiling MQ water. Two milliliters of undiluted filtrate was placed in acid-washed screw-top test tubes, followed by 0.05 ml of 80% (wt/wt) phenol-MQ and 5 ml of concentrated sulfuric acid. The tubes were capped and mixed gently, since the ensuing reaction is exothermic. The tubes were then placed in a 30°C water bath for 10 min to allow the solution color to develop. The resulting sample solution was added to a disposable cuvette (path length, 1 cm), and the absorbance at 490 nm was recorded (Shimadzu UV-160 spectrophotometer). The reference beam for the samples was a sulfuric acid-phenol solution. Total glucose content of the digested polysaccharides was measured, and glucose standards were processed and analyzed with each set of samples.
Total dissolved Mn was measured by inductively coupled plasma-atomic emission spectrometry (IRIS Thermo Jarrell Ash). All samples analyzed by inductively coupled plasma-atomic emission spectrometry were passed through a microfilter (pore diameter, 0.2 μm; polyethersulfone) and acidified with 10 μl of concentrated hydrochloric acid. Scandium was used as an instrumental internal standard. Calibration curves were constructed with gravimetrically prepared plasma standards (VHG Labs, Manchester, N.H.). Linear least-squares regressions were prepared from plots of the instrument response versus the standard concentrations.
Mn mass balance.
P. putida was grown in Leptothrix medium as described above. A mass balance calculation for Mn was performed 3 days after inoculation of the bacterial culture. The operational compartments for Mn were as follows: (i) Mn remaining in solution, (ii) Mn sorbed to biomaterial, and (iii) Mn removed from solution by oxidation and precipitation. The solution and sorbed Mn fractions were assumed to be Mn(II). The suspension containing bacterial cells, biofilm, and biogenic Mn oxide was centrifuged and resuspended in 40 ml of 0.02 M NaCl three times, these rinse cycles being 3, 24, and 48 h long. The residual media and subsequent rinses were analyzed for soluble Mn. The thoroughly rinsed cell oxide pellet was then resuspended in 20 ml of a solution containing 0.02 M NaCl and 260 μM CuCl2 · H2O. Since Cu+2 has a stronger affinity for Mn oxide surface sorption sites than Mn+2, it displaces sorbed Mn+2, allowing the amount of sorbed Mn+2 to be calculated (49). The Cu+2 may also replace Mn+2 from the bacterial cells and biofilm.
Electron microscopy.
All electron micrographs were collected at the Electron Microscopy Laboratory at the University of California, Berkeley. P. putida and the associated biogenic Mn oxide, grown in Leptothrix medium, were imaged by scanning electron microscopy (SEM). The sample was fixed with glutaraldehyde, postfixed with 1% osmium tetraoxide, and dehydrated with a series of ethanol-water solutions. After critical point drying, the sample was mounted on double-sided carbon tape, carbon coated, and imaged with a Hitachi S-5000 scanning electron microscope. P. putida, grown on solid agar, was imaged by transmission electron microscopy (TEM). The sample was fixed with glutaraldehyde, postfixed with 1% osmium tetraoxide, stained with 0.5% uranyl acetate, and dehydrated with acetone. The samples were embedded in resin, thin sectioned with a glass knife on a microtome, and imaged with a JEOL 100CX transmission electron microscope.
STXM. (i) Sample preparation.
As Mn oxidation proceeded, subsamples were collected from the growth flasks and immediately measured with STXM. A droplet (∼1 μl) of aqueous suspension was deposited onto a silicon nitride (Si3N4) window. A second window was then placed on top to form a “sandwich” that enclosed the sample suspension. The Si3N4 windows consist of a 500-μm2 by 100-nm membrane supported by a 5-mm2 by 200-μm silicon wafer (Silson Ltd). The two wafers were sealed at the edges with a thin layer of epoxy resin to prevent water evaporation; the epoxy layer does not come into contact with the sample solution. Thus sealed, the samples could be kept hydrated for several hours. Since absorption cross sections in the soft X-ray region are very high, very thin windows (100 nm) allowing 75% of the X-ray to be transmitted at the Mn L2,3 edges were used to image the samples. The 1/e attenuation lengths for water and Mn are about 800 and 300 nm, respectively, around the Mn-L3 edge. The attenuation lengths limit the effective sample thickness to a few micrometers for accurate spectroscopic measurement in transmission mode. The fully hydrated samples were analyzed with the microscope under He at atmospheric pressure to minimize the X-ray absorption and reduce potential absorption artifacts from O2 and N2. All reference compounds (except MnCl2) were in powder form. The Mn standards were finely ground by using an agate mortar. They were then suspended in MQ water, applied to a Si3N4 window by use of a micropipette, and air dried. Great care was taken to avoid saturation effects, such as those observed for spectra with optical densities above 2.
(ii) The STXM technique.
The soft X-ray spectromicroscopy measurements were performed with the same scanning transmission X-ray microscope at beamlines 7.0.1 and 11.0.2 of the Advanced Light Source at Lawrence Berkeley National Laboratory (26). The STXM uses zone plate optics to focus a monochromatic X-ray beam onto a 40-nm full-width-at-half-maximum spot. The microscope energy was calibrated by using the Mn-L3 absorption fine structure of MnCl2. The sample is raster scanned through the focused X-ray beam, and the intensity of transmitted photons is detected to form a two-dimensional image of the variation in photoelectric absorption signal through a column of material. A sequence of images, also referred to as a stack, is acquired at finely spaced incident monochromatic energy intervals in the Mn 2p region. Typical image sequences were obtained with a 10-μm2 field of view using a 70-nm2 pixel size with 40-nm spatial resolution and 0.1-eV energy resolution. The image sequences were acquired in approximately 50 min each. The images in each sequence were aligned via a spatial cross-correlation analysis using aXis2000 software (22). The absorption spectra could then be extracted from any pixel or group of pixels of the stack.
When incident X-rays of intensity I0 pass through a column of material, they are attenuated to an intensity I given by the Lambert-Beer law,
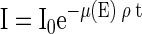 |
(1) |
where μ is the absorption coefficient (square centimeters per gram) and is a function of energy (E), ρ is the density of the element, and t is the thickness of the sample (centimeters). Since the sample thickness is not known, one can rewrite equation 1 as
 |
(2) |
where m = ρ t (grams per square centimeter) gives the sample mass per area. This substitution in equation 2 allows for the characterization of the sample in terms of a mass density per area, eliminating the sample thickness term. The coefficient μ is proportional to the total atomic absorption cross-section of Mn in the region of interest and can be calculated from Henke's optical values (21) for photon energies above about 30 eV and far from absorption edges, where the cross-section does not depend on the probability of populating unoccupied d-state molecular orbitals. In the image sequences, the regions used for normalization (I0) are free of Mn precipitates and are chosen such that the X-ray absorption can be assumed to be constant over a small energy interval. The regions of interest (I) are those containing Mn precipitates.
(iii) NEXAFS spectroscopy at Mn L2,3 absorption edges.
When an incident X-ray has an energy tuned at or above the binding energy of an inner shell electron, the electron can be completely removed from the atom and the absorption cross-section makes a “jump”. Near the absorption edge, the electron can be promoted to unoccupied molecular orbitals, yielding near-edge absorption fine structures (42). The absorption structures can be used as “fingerprints” of the available final states for every 3dn initial state and to probe the 3d orbital electronic occupancy, i.e., oxidation state (1, 17). When comparing 3d5 Mn(II), 3d4 Mn(III), and 3d3 Mn(IV), a shift towards higher energy with higher valency is observed, and the spectral shape changes significantly. Consequently, Mn L2,3 spectroscopy, when performed at high-energy resolution, can give information on the oxidation state, coordination environment, and spin state of the absorbing Mn ion (12). There is a detection limit of a few percent relative atomic abundance of Mn, corresponding to about a 0.1 mM concentration of homogeneously distributed Mn.
(iv) Characterization of Mn reference and test compounds.
The source or synthesis recipe and properties of the Mn reference and test materials utilized in this study are summarized in Table 1 (see Results). The hausmannite (Mn3O4) and bixbyite (Mn2O3) were purchased from Sigma Corp. The manganite, acid birnessite, triclinic Na-birnessite, c-disordered H-birnessite, and δ-Mn(IV)O2 were synthesized in the laboratory (28, 31, 50). The reference and test materials were characterized with powder XRD, N2 Brunauer-Emmett-Teller specific surface area determination, and chemical titration to determine the average Mn oxidation number. Powder XRD patterns were measured on a Siemens Kristalloflex Diffraktometer at 40 kV and 30 mA with Cu K-α radiation. The procedure for determining the average Mn oxidation number by chemical titration of Mn oxides is described in detail in the work of Villalobos et al. (50). Briefly, the average oxidation number of the Mn in oxide and oxyhydroxide reference materials was determined by using an oxalate titration method. Fifty milligrams of Mn solid was added to a sulfuric acid and oxalic acid (standardized) solution, and the suspension was stirred until Mn solid dissolution. The dissolved Mn sample solution was then heated to just below 100°C and was titrated with standardized potassium permanganate solution until the appearance of pink color. The average oxidation number of Mn in the sample was determined in triplicate. The stoichiometries of the layer-type Mn oxides, acid birnessite, triclinic Na-birnessite, and δ-Mn(IV)O2, are presented in Table 1 (Villalobos et al., submitted).
TABLE 1.
Summary of characteristics of Mn reference and test materialsa
| Name | Formula | Surface area (m2 g−1)e | Avg oxid. no.f | XRDg | NEXAFSh |
|---|---|---|---|---|---|
| — | MnCl2b | — | 2 | — | x |
| Bixbyiteb | Mn2O3 | — | 3.00 | 0 | x |
| Hausmanniteb | Mn3O4 | — | 2.68 | 0 | x |
| Manganitec | γ-MnOOH | 46 | 3.01 | 0 | x |
| Acid birnessited | H0.06K0.18H2O0.54Mn(III)0.08[Mn(IV)0.88,vac0.12]O2 | 36 | 3.96 | 0 | x |
| Triclinic Na-birnessited | Na0.26H2Ox[Mn(IV)0.74,Mn(III)0.26]O2 | 39 | 3.57i | 0 | 0 |
| “C-dis” H-birnessited | HaNabH2OcMn(III)tc[Mn(IV)d,Mn(III)e,vacf]O2 | 154 | 3.70 | 0 | 0 |
| δ-MnO2d | Na0.24H2O0.72[Mn(IV)0.94,vac0.06]O2 | 121 | 4.02 | 0 | x |
The formulas of the Mn(III, IV) layer type Mn oxides, acid birnessite, triclinic Na-birnessite, c-dis H-birnessite, and δ-MnO2 are complicated by the high proportions of Mn vacancy sites, and the specific stoichiometry of the c-disordered H-birnessite is not known (M. Villalobos, B. Lanson, A. Manceau, B. Toner, and G. Sposito, submitted for publication). Symbols: x, the data were collected and are displayed here; —, the data were not collected or are not applicable; 0, the data were collected but are not displayed.
Sigma Corporation.
Prepared by Evan Wong.
Synthesis, XRD pattern and average oxidation number of Mn in reference 50.
Courtesy of Sabine Goldberg, George E. Brown Salinity Laboratory, ±10 m2 g−1.
Except in the case of MnCl2, these values were determined by chemical titration; values ± 0.02. Oxid., oxidation.
XRD collected in this study and others. Hausmannite and bixbyite XRD patterns collected by Jasquelin Pena.
Sample containing 23 mol % manganite.
(v) Radiation damage.
An image of the stack (see “The STXM technique” for a definition) region was collected (at one energy) before and after the stack was recorded. These “before” and “after” images were then compared to one another to check for sample damage during the stack collection. Although direct evidence of beam damage was not observed when collecting a stack on the biogenic Mn oxides samples, its potential impact was explored. For this purpose, the Mn oxides were exposed to X-rays for an extended period in the presence of organic material (growth medium). A well-characterized Mn oxide, acid birnessite, was suspended in sterile Leptothrix medium or MQ water. The growth medium was chosen to provide a rich organic medium that closely resembled the experimental conditions. Three successive stacks were then recorded, using parameters identical to the ones used to acquire stacks on the incubated microbial samples.
(vi) Data analysis. (a) Reference and test compounds.
Four well-characterized single-valence reference compounds, MnCl2, γ-MnOOH, Mn2O3, and δ-Mn(IV)O2 (representing Mn oxidation states II, III, III, and IV, respectively), were employed to fit the biogenic Mn oxide spectra. The Mn L-edge NEXAFS spectra of reference and test compounds were measured by using STXM in a configuration identical to that used to measure the incubated microbial samples. The spectra of the reference and test compounds were normalized to correspond to a known amount of Mn (grams per square centimeter). This was accomplished by measuring the spectra far above the absorption edges at 690 eV, where photoelectric absorption gives rise to an unbound electron and the resulting spectrum does not depend on the probability of populating unoccupied 3d molecular orbitals. The reference spectra were normalized against the Mn atomic absorption computed by using tabulated optical constants (21). The tabulated optical constants provide the atomic absorption of 1 μg of Mn cm−2. The measured reference spectra were normalized to the computed atomic absorption value at both 675 and 690 eV to estimate the normalization uncertainty.
(b) Biogenic Mn oxide samples.
A semiempirical approach was used to calculate the relative amounts of Mn(II), Mn(III), and Mn(IV) from the experimental Mn L-edge NEXAFS spectra using single-valence reference compounds. A linear least-squares fitting, adjusted empirically to best reproduce the fine structures, was performed by using a linear fitting routine with the software Igor (WaveMetrics). A linear least-squares fitting was also performed with SixPack v 2.3.1 and aXis2000 softwares for comparison to the results obtained with Igor. The experimental spectra were fit using a linear combination of normalized single-valence reference spectra by optimization of equation 3,
 |
(3) |
The variables A, B, and C represent the normalized Mn(II), Mn(III), and Mn(IV) reference spectra, f is a normalization parameter, and a, b, and c are the relative amounts of the Mn oxidation states (with a + b + c = 1). The spectral background, bkg, is a function of a constant background, bg, a normalization parameter, g, and E, the photon energy in eV,
 |
(4) |
The fits to the experimental spectra resulted in quantitative relative abundances of Mn(II), Mn(III), and Mn(IV), expressed as percentages of total Mn present. The ability to distinguish Mn(II), Mn(III), and Mn(IV) spectroscopically depends on how different the L-edge fine structures are for each Mn oxidation state and if those differences are generated by the charge state of Mn rather than by other factors, such as local bonding structure. In the case when the spectral features are dominated by the charge state rather than the structure, it is reasonable to expect that errors in the quantitative analyses caused by choosing the “wrong” Mn reference material will be small. To test this assumption, two different Mn(III) reference compounds, bixbyite and manganite, were used along with MnCl2 and δ-Mn(IV)O2 for spectral fitting. Manganite yielded the best fits to the measured biogenic Mn oxide spectra, and all reported relative abundances were obtained by using this Mn(III) reference compound. The bixbyite spectrum was used to estimate the error introduced by the choice of the Mn(III) compound. In total, the errors associated with (i) the uncertainty of normalizing the references with the computed Mn atomic absorption, (ii) the effects of radiation damage, and (iii) the choice of the Mn(III) intermediate were added in quadrature.
RESULTS
Bacterial growth and Mn oxidation.
When grown in liquid medium, P. putida cells aggregate and produce a biofilm during the exponential and stationary phase of growth (Fig. 1C and 2A and B). The cells enter the stationary phase of growth 20 h after inoculation (Fig. 1A). In the early stationary phase of growth, the culture removed soluble Mn+2(aq) from solution and a precipitate was observed (Fig. 1B and 2C and D). The stringy rope features observed in the SEM images (Fig. 2A and B) indicate the presence of a biofilm that has been desiccated during sample preparation (25). The TEM results suggest that the Mn precipitate is closely associated with the cells and is composed of small, wafer-shaped particles (Fig. 2D). However, the location of the biofilm relative to the bacterial cells and biogenic Mn oxide was not clearly resolved with either SEM or TEM.
FIG. 1.

(A) Growth of P. putida in Leptothrix medium. The cells enter the stationary phase of growth at ∼20 h. (B) Removal of Mn+2(aq) from growth medium occurs after P. putida enters the stationary phase of growth. (C) The production of extracellular polysaccharides (biofilm) during growth. The arrow indicates for each graph where the cells enter the stationary phase of growth. susp., suspension.
FIG. 2.

Electron micrographs of P. putida collected during the stationary phase of growth. (A and B) SEM images show the cell oxide aggregates (∼10 by 30 μm) and the associated desiccated biofilm (extracellular polysaccharide material), which is seen as strings or ropes. (C and D) TEM images show the biogenic Mn oxide particles (electron-dense wafers) surrounding the bacterial cell. Scale bar = 2 μm (A), 0.5 μm (B), 0.2 μm (C), or 0.1 μm (D).
The growth curve, presented in Fig. 1A, exhibits substantial variability. This may be caused by the aggregation of the cells during the exponential phase of growth. Counting CFU was not an ideal method for determining the cell concentration. Enumeration of bacteria by counting CFU assumes that one colony on the agar plate was produced by one bacterium suspended in solution. Since the bacterial cells clump together, one colony is not equivalent to one bacterium in solution.
The extent of Mn oxidation was variable in flasks prepared and incubated identically. The average amount of Mn+2(aq) remaining unoxidized in solution in eight replicate flasks after 72 h was 1.8% ± 3.9% of initial MnCl2 added, with the values ranging from 0.1 to 11.2%. The average amount of Mn+2(aq) sorbed to biogenic surfaces (biogenic Mn oxide, cells, and biofilm) was 7.4% ± 6.8% of initial MnCl2 added, with the values ranging from 0.1 to 18.8%.
Radiation damage.
The results of the radiation damage experiment are presented in Fig. 3. The NEXAFS Mn-L edge spectrum extracted from the first stack on the acid birnessite (suspended in MQ water) was considered the baseline spectrum (Fig. 3A, Stack 1). Acid birnessite was chosen because it has a structure similar to that of the biogenic Mn oxide produced by P. putida, as determined by XRD, and the average oxidation number of Mn is known (50). In MQ water, small spectral differences can be seen when comparing the first stack to the second and third, specifically a slight decrease in the Mn-L3 peak intensity and a growing shoulder on the low-energy side of the peak. The second and third stacks collected from the acid birnessite, suspended in organic growth medium, exhibited a clear degradation of the shape of the Mn-L3 peak as well as a shift towards lower energy (i.e., lower valency) (Fig. 3B). By comparing the first stacks collected on both acid birnessite suspended in MQ water and acid birnessite suspended in organic medium, a 5% reduction in total Mn(IV) was estimated using the linear fitting routine. The radiation-induced Mn reduction was incorporated into the error estimates for the calculated relative proportions of Mn(II), Mn(III), and Mn(IV). In practice, a short dwell time per pixel (0.5 ms), a reasonably big pixel size (70 nm2), and a slightly defocused sample were used to record all of the image sequences in order to minimize the potential for radiation damage.
FIG. 3.

Mn L3edge NEXAFS spectra extracted from three successive stacks, recorded from 620 to 690 eV, on the same spatial region of hydrated acid birnessite. The extent of the chemical changes induced by soft X-rays can be observed in (A) acid birnessite suspended in MQ water and to a much greater extent in (B) acid birnessite suspended in organic growth medium. In each panel the first, second, and third stacks collected are presented from bottom to top.
Mn reference materials.
The manganite diffraction pattern was compared to experimental and calculated XRD patterns; no contaminant peaks were present (37). The XRD pattern collected for hausmannite reveals a 10 to 15% manganite contaminant; the proportion of the contaminant was estimated by comparing the peak intensities of hausmannite and manganite. The average oxidation number of Mn in the hausmannite reference material is 2.68 ± 0.02 as determined by titration. A 10% manganite contaminant in the hausmannite reference material is within the error estimate for the average oxidation number of Mn in the hausmannite. The bixbyite XRD pattern has a single unidentified peak not attributable to any Mn mineral in the available XRD database. The average oxidation number of Mn in the bixbyite (measured by titration) was 3.00 ± 0.02, agreeing with the theoretical value. The XRD patterns of the acid birnessite, triclinic Na-birnessite, c-disordered H-birnessite, and δ-Mn(IV)O2 are presented in the work of Villalobos et al. (50). The normalized NEXAFS spectra of the reference compounds extracted from STXM image sequences or stacks are displayed in Fig. 4. The reference spectra are presented with the calculated atomic absorption of one atom of Mn. When comparing the spectra of Mn(II), Mn(III), and Mn(IV) compounds, a shift towards higher energy with higher oxidation state is clearly observed, and the spectral shape changes significantly. The Mn L-edge spectra reported here are consistent with previously measured spectra (15, 35).
FIG. 4.

(Top) STXM images of the Mn reference materials, MnCl2, Mn2O3 (bixbyite), γ-MnOOH (manganite), and δ-MnIVO2, were collected at the Mn-L3 absorption edge. The spectra were extracted from regions of interest (I), which are encircled by white lines, and normalized by the I0 region, encircled by black lines. (Bottom) The corresponding extracted NEXAFS spectra for the Mn reference materials are presented with the computed atomic absorption used for normalization.
Test of data analysis procedure.
The linear fitting procedure was tested with the NEXAFS spectrum of hausmannite (Mn3O4). The hausmannite spectrum was fit with two combinations of Mn(II) and Mn(III) spectra: (i) reference spectra MnCl2 and manganite (γ-MnOOH) and (ii) reference spectra MnCl2 and bixbyite (Mn2O3). The fitted results were compared with the chemically determined value. The chemically determined average oxidation number of Mn in hausmannite is 2.68 ± 0.02 (Table 1). The fits to the hausmannite spectrum were 0.140 Mn(II) and 0.860 Mn(III) with bixbyite as the Mn(III) reference and 0.195 Mn(II) and 0.805 Mn(III) with manganite as the Mn(III) reference. The fitted percentages of Mn(II) and Mn(III), with error estimates included, were 19.5 ± 5.7 and 80.5 ± 7.9, respectively. The NEXAFS fitting predicts that the average oxidation number of Mn in hausmannite is 2.5 to 3.1. This value is in agreement with the theoretical and measured values of the average oxidation number of Mn in hausmannite based on the chemical formula, the XRD pattern and manganite contaminant, and the results of the chemical titration. Of the estimated errors caused by normalization (<5%), radiation damage (<5%), and the choice of Mn(III) reference material (1 to 25%), the choice of Mn(III) reference material introduced the greatest error to the calculated value of the proportions of Mn(II), Mn(III), and Mn(IV).
The sensitivity of the NEXAFS spectra to small changes in average oxidation number of Mn between 3.5 and 4.0 was characterized by collecting Mn L-edge spectra for numerous Mn layer-type oxides with average oxidation numbers of Mn ranging from 3.57 to 4.02 ± 0.02 (Table 1). The spectral features, peak position and shape, are very similar among the layer-type Mn oxides with these Mn average oxidation numbers. This result indicates that Mn L-edge NEXAFS spectroscopic analysis cannot differentiate among Mn layer-type oxides with average oxidation numbers of Mn above approximately 3.5 to 4.0.
Chemical and spatial distribution of Mn as function of time.
Manganese oxidation experiments in liquid growth medium were conducted at both 0.1 and 1 mM total initial Mn concentrations. A summary of the collected STXM images is presented in Fig. 5. The Mn L-edge NEXAFS spectra obtained from the encircled areas on the sample images are also presented, along with linear best fits (Fig. 6).
FIG. 5.

STXM image sequences or stacks collected during growth of P. putida and Mn oxidation. Samples were collected at t = 16, 24, 34, and 48 h in the 1 mM Mn reaction series (left) and at t = 23, 35, and 47 h in the 0.1 mM Mn reaction series (right). The areas encircled by black lines were used to normalize (I0), and the areas encircled by white lines represent the regions of interest (I).
FIG. 6.

Mn L-edge NEXAFS spectra obtained from time-resolved Mn oxidation experiments. The spectra were extracted from the STXM stacks presented in Fig. 5 from areas encircled by the white lines representing I (sample transmitted) and normalized by areas representing I0 (background transmitted) encircled by the black lines. The spectra (bold line) were fit (light line) with linear combinations of Mn(II)Cl2, γ-Mn(III)OOH, and δ-Mn(IV)O2. The numerical results of the spectral fits are presented in Fig. 7.
The STXM images indicate that the cells aggregate during the exponential phase of growth; this timing agrees with the observed increases in production of biofilm (Fig. 1C). Slight accumulation of Mn(II) in the biofilm but not in the bacterial cells was observed in the 1 mM Mn reaction series (Fig. 5, panel for 16 h, area 1). Manganese(III) was detected as a reaction intermediate in the bacterially mediated oxidation of Mn+2(aq) in both the 0.1 and 1 mM reaction series. The progression in the NEXAFS spectral features from pure Mn+2(aq) to combinations of Mn(II) and Mn(III) to combinations of Mn(III) and Mn(IV) is clearly visible when experimental spectra are compared to the three Mn reference spectra (Fig. 4 and 6).
Spatial heterogeneity of Mn oxidation states within bacterial aggregates of the same age was observed in both reaction series and can be seen by comparing the spectra collected from different areas of the stack at the same sampling time (Fig. 6). In the 0.1 mM Mn reaction series at t = 23 h, the sample had regions where Mn(III) was 61.5% ± 6.7% (Fig. 5, 23 h, area 2) and 16.5% ± 5.7% (Fig. 5, 23 h, area 1) of the total Mn present. Although the progression of Mn oxidation states is similar in the 0.1 mM and 1 mM reaction series, the amount of Mn(III) remaining in the aggregates after 48 h is greater in the 1 mM series (Fig. 7). In the 0.1 mM Mn reaction series at t = 47 h, the selected areas were 90.0% ± 4.5% Mn(IV) and 10.0% ± 0.5% Mn(II). At t = 48 h for the 1 mM Mn reaction series, there was 64.0% ± 4.4% Mn(IV), 29.0% ± 5.1% Mn(III), and 7.0% ± 1.4% Mn(II) (Fig. 5, 48 h, area 1) and 35.0% ± 12.5% Mn(IV), 60.0% ± 13.6% Mn(III), and 5.0% ± 1.0% Mn(II) (Fig. 5, 48 h, area 2).
FIG. 7.

Manganese oxidation by P. putida as a function of time with 0.1 and 1 mM initial MnCl2. The relative abundances of Mn(II), Mn(III), and Mn(IV) are calculated by linear fitting of experimental Mn L-edge spectra with reference spectra. The error bars represent uncertainty in the normalization of reference spectra, the choice of reference compounds, and chemical changes due to radiation damage. Values for duplicate sampling areas at time points ≥24 h are displayed and indicate spatial heterogeneity within the samples.
DISCUSSION
Bacterial growth and Mn oxidation.
The observed timing of the onset of Mn oxidation in the stationary phase of growth by the culture is in agreement with previous research conducted on this Pseudomonas strain (46). Although the production of a biofilm has not been reported in previous studies of P. putida strain MnB1, it is consistent with numerous studies of other Pseudomonas cultures, including P. putida strain DMP-1 (16), Pseudomonas sp. soil isolate (39), Pseudomonas putida G7 (25), and Pseudomonas putida mt-2 (2). The STXM images confirm that the biogenic Mn oxide particles are precipitated and retained within the biofilm. The effect of the biofilm on the reactivity of the biogenic Mn oxide is not known and could be an important issue regarding the reactivity of natural Mn oxides.
Radiation damage.
Manganese oxides are prone to reduction of the oxidation state of Mn through exposure to hard X-rays, particularly in the presence of organic matter; the Mn(II) contribution to unfocused Mn XANES spectra increased after repeated scans were collected from soils, indicating Mn reduction via beam damage (40). In particular, with the focused soft X-ray beam of STXM, the likelihood of radiation damage has to be considered because small portions of the sample are subjected to a large photon flux (7).
This research involved measurement of the relative amounts of single-valence species [Mn(II), Mn(III), and Mn(IV)] in hydrated, carbon-rich samples. The radiation-induced damage is clearly accelerated by the presence of reduced organic material (Fig. 3). The observed spectral changes in the Mn oxidation state of the acid birnessite in the irradiated region are interpreted as X-ray-induced photoreduction with the organic carbon present in the sample providing the electrons (40). The production of free-radical species as incident X-ray energy is absorbed by the dissolved organic compounds may cause the observed photoreduction.
Mn reference materials.
Determination of the relative quantitative amounts of Mn(II), Mn(III), and Mn(IV) in the biogenic Mn oxide samples depends greatly on two factors: (i) the choice and quality of the reference materials and (ii) the normalization of reference compound spectra. Although Mn has seven possible oxidation states, Mn(II), Mn(III), and Mn(IV) are the environmentally relevant oxidation states of Mn (32, 33). The Mn reference materials, MnCl2, γ-MnOOH, and δ-Mn(IV)O2, were chosen to provide as pure as possible single-oxidation-state references. MnCl2 and δ-Mn(IV)O2 are specifically relevant to the experimental conditions. Manganese chloride was the Mn compound introduced to the growth medium initially, and δ-Mn(IV)O2 has properties similar to those of the Mn oxide final product produced by P. putida under similar conditions and is a pure Mn(IV) oxide (50). The reference and test compounds acid birnessite, hausmannite, and bixbyite are also consistent with those chosen for other studies of bacterially produced Mn oxides (6, 35) and are considered environmentally relevant forms of Mn (38, 44).
An important issue to consider is the potential spectral differences between Mn L-edge spectra from different compounds that have the same Mn 3d-shell occupancy or Mn oxidation state. For the purposes of this analysis, it was assumed that any differences between different compounds with the same nominal oxidation state contribute small errors to the quantitative analysis. This assumption was scrutinized by using two different Mn(III) reference compounds, manganite (γ-MnOOH) and bixbyite (Mn2O3). The Mn(III) spectra (Fig. 4) are indeed similar and are strikingly different from the spectra of the Mn(II) and Mn(IV) compounds.
Chemical and spatial distribution of Mn as function of time.
The sampling time points reflect the general series of events in Mn+2(aq) oxidation by P. putida under the experimental conditions. At t > 0 but < 24 h, Mn oxidation has not yet begun, and all the Mn in the sample is in the form of Mn+2(aq). At t ≥ 24 h, the culture is in early stationary phase, and small amounts of Mn precipitation are normally visible. At t ≥ 36 and 48 h, the growth flasks are visibly dense with dark-brown Mn precipitates, and the concentration of Mn+2(aq) in solution has decreased (Fig. 1B and 7).
Previous research with Bacillus sp. strain SG-1 has determined that in the presence of high Mn+2(aq) concentrations (e.g., 10 mM Mn), a variety of Mn(III) solids are formed: hausmannite (Mn3O4), feitknechtite (β-MnOOH), and manganite (γ-MnOOH) (30). However, in the presence of Mn+2(aq) concentrations of 0.1 to 1.0 mM, Mn(IV) minerals are the dominant product of bacterial oxidation (6, 30). Taken together, the Bacillus sp. strain SG-1 studies suggest that the production of Mn(III) oxides at higher Mn+2(aq) concentrations is the result of favorable abiotic surface-catalyzed reactions and that the product of Mn oxidation is a Mn(IV) oxide (5). In the present study, with Mn+2(aq) concentrations 10 to 100 times lower than 10 mM, a Mn(III) reaction intermediate was clearly observed in the Mn oxidation sequence catalyzed by P. putida.
Manganese(III) was also detected, with STXM, during Mn+2(aq) oxidation by marine Bacillus sp. strain SG-1, where oxygen was supplied by diffusion and the initial Mn concentration was 10 mM (35). The presence of Mn(III) in the Bacillus sp. strain SG-1 study was interpreted as evidence for a one-electron transfer mechanism. The mechanism by which bacterial proteins oxidize Mn is currently unknown. Two mechanisms have been proposed (46). The protein may mediate two separate one-electron transfers with a Mn(III) complex as an intermediate, or the protein may mediate a two-electron transfer in a single step (46). The results presented here, with the appearance of Mn(III) as a reaction intermediate, may be considered consistent with either model. According to the first mechanism, Mn+2(aq) may be oxidized to Mn(III), which either precipitates or remains complexed near the bacterial outer membrane. The Mn(III) intermediate then either disproportionates into Mn(II) and Mn(IV) (20) or is oxidized to Mn(IV) in a second step. According to the second proposed mechanism, Mn(II) is oxidized directly to Mn(IV), which forms a precipitate. The Mn(IV) precipitate then reacts with the excess solution-phase Mn+2(aq) to form Mn(III) in an abiotic reaction (6, 36). If the Mn(III) product in this case precipitates, the transition from Mn(III) to Mn(IV) may be via disproportionation or dissolution as the solution Mn+2(aq) quickly decreases. In either scenario, a detectable Mn(III) intermediate is possible.
The Mn L-edge NEXAFS spectra do not provide a structural identification of the Mn(III) or Mn(IV) species found in the samples. However, the NEXAFS spectra do provide general clues about the Mn(III) intermediate. The detection limit of the instrument for homogeneously distributed Mn, e.g., Mn+2(aq) in solution, is approximately 0.1 mM. No Mn L-edge spectrum was obtained for the t < 24 h sample in the 0.1 mM Mn series, since the concentration was too low for detection with the transmission technique being used. However, at t ≥ 23 h, Mn(II) and Mn(III) were detectable. This suggests that Mn is accumulating on or between cells. Therefore, a solid precipitate or perhaps a membrane-bound Mn(III) intermediate is more likely than a solution-phase Mn(III) complex. In addition, a solution-phase Mn(III) complex is less likely than a bound Mn(III) complex, since P. putida, when grown on solid medium, is not known to produce a diffusible Mn-oxidizing factor. When the culture and solid growth medium were separated by a 0.2-μm-pore-size membrane filter, Mn oxidation occurred only next to the colonies; therefore, Mn+2(aq) was diffusing through the membrane filter and being oxidized within the colonies rather than Mn-oxidizing factor(s) or a Mn(III) complex diffusing into the solid medium.
The first appearance of Mn oxidation, or the detection of Mn(III), was well within the cellular aggregates. Within the center of a bacterial aggregate, consisting of aerobic heterotrophs in the late exponential phase and early stationary phase of growth, the chemical conditions present are expected to be out of equilibrium with the bulk solution. In particular, the dissolved oxygen concentrations in the interstitial solution may be low. P. putida thus catalyzes the oxidation of Mn+2(aq) in areas where oxygen concentrations are potentially the lowest, even though bacterial Mn-oxidizing proteins remove electrons from Mn+2(aq) and likely transfer them to molecular oxygen (46). This result is consistent with observations from studies of Mn+2(aq) oxidation in the environment. In depth profiles of oxygen, sulfide, and Mn particulates in the Baltic Sea and Black Sea, the peak concentration of Mn particulates was observed in the region adjacent to the sulfide interface, where dissolved oxygen concentrations are very low (34, 47). Bacterial Mn+2(aq) oxidation appears to be a microaerobic process in certain circumstances.
Although the Mn oxidation reaction in the 1 mM series did not proceed entirely to Mn(IV), from a bulk solution perspective this culture behaved as expected; the cells grew and filled the flask with dark-brown precipitates at the appropriate time, and 48 h after inoculation, less than 10% of the initial Mn+2(aq) remained in solution. However, the NEXAFS spectroscopy measurements reveal that the oxidation process in this experiment was complex. It is important to note that in experiments identical to the 1 mM series presented here, complete oxidation of Mn to Mn(IV) was observed. Variability in the extent of Mn oxidation in flasks prepared and incubated identically was observed in the bulk solution and at the nano-to-micrometer scale. We speculate that the observed variability may result from differences in the rate and extent of cell aggregation during incubation and cell growth. The physical and chemical conditions in each flask are the same; however, as the growth curve presented in Fig. 1 suggests, there is considerable variability in the extent of aggregation.
Conclusions
Scanning transmission X-ray microscopy was used to examine Mn oxidation by the biofilm-forming bacterium P. putida strain MnB1 as a function of time. The fully hydrated samples were analyzed in situ, at atmospheric pressure and a spatial resolution of 40 nm. The quantitative relative proportions of Mn(II), Mn(III), and Mn(IV) were calculated with linear fitting of Mn L-edge NEXAFS spectra. The bacterial cells oxidized Mn+2(aq) to Mn(III) and Mn(IV) during the stationary phase of growth. Manganese(III) and Mn(IV) were detected only within the cellular biofilm flocs and were adjacent to and filling the spaces between bacterial cells. Manganese(III) was detected in both the 0.1 mM and 1 mM total Mn reaction series. The substantial proportion of Mn present as Mn(III) in certain locations during oxidation is an important finding, because Mn(III) is an oxidant and its function in natural waters is largely unknown (27). The standard reduction potential (E0) of the Mn(III) + e− → Mn(II) couple varies considerably depending on the Mn(III) species present; as examples, MnOOH(s), +1.50 (46); Mn(III)-EDTA(aq), +0.823 (19); and Mn(III)-porphyrin(aq), range from −0.37 to +0.01 (8). The presence of Mn(III) in these experiments and its potential role in environmental chemistry is even more compelling when the dynamic redox cycling of Mn in natural waters and sediments is considered (48). In these experiments, Mn(III) is an intermediate species; however, its presence further supports the hypothesis that Mn(III) may be a significant environmental species, especially in regions of active Mn cycling. As a microscopic and spectroscopic technique, STXM was uniquely suited for this study of metal-microbe interactions. The application of STXM to future studies of microbe-metal-mineral interactions, particularly in the case of redox active metals and metalloids, such as Fe, Cr, and As, has great potential to yield important insights into geomicrobiological processes.
Acknowledgments
B.T., M.V., and G.S. are grateful for funding from the National Science Foundation, Collaborative Research Activities in Environmental Molecular Science program (CHE-0089208). The Advanced Light Source and work on BL7.0.1 and BL11.0.2 STXM are supported by the Office of Basic Energy Sciences, Division of Materials Sciences, Division of Chemical Sciences, Geosciences, and Biosciences of the DOE at Lawrence Berkeley National Laboratory under contract DOE-AC03-76SF00098.
B.T. and M.V. thank the Electron Microscopy Laboratory (UC Berkeley) for training and sample preparation assistance, Sabine Goldberg (George E. Brown Salinity Laboratory) for surface area determination, Evan Wong for synthesizing the manganite, and Jasquelin Pena for collecting hausmannite and bixbyite XRD patterns. S.F. thanks E. Rotenberg at BL7.0.1 and D. K. Shuh, T. Tyliszczak, and M. Gilles at BL11.0.2 for providing beam time.
REFERENCES
- 1.Abbate, M., F. M. F. deGroot, J. C. Fuggle, A. Fujimori, Y. Tokura, Y. Fujishima, O. Strebel, M. Domke, G. Kaindl, J. van Elp, B. T. Thole, G. A. Sawatsky, M. Sacchi, and N. Tsuda. 1991. Soft-x-ray absorption studies of the location of extra charges induced by substitution in controlled-valence materials. Phys. Rev. B Condens. Matter 44:419-5422. [DOI] [PubMed] [Google Scholar]
- 2.Auerbach, I., C. Sorensen, H. Hansma, and P. Holden. 2000. Physical morphology and surface properties of unsaturated Pseudomonas putida biofilms. J. Bacteriol. 182:3809-3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banerjee, D., and H. W. Nesbitt. 1999. Oxidation of aqueous Cr(III) at birnessite surfaces: constraints on reaction mechanism. Geochim. Cosmochim. Acta 63:1671-1687. [Google Scholar]
- 4.Banfield, J. F., and R. J. Hamers. 1997. Processes at minerals and surfaces with relevance to microorganisms and prebiotic synthesis, p. 81-122. In J. F. Banfield and K. H. Nealson (ed.), Geomicrobiology: interactions between microbes and minerals. Reviews in Mineralogy, vol. 35. Mineralogical Society of America, Washington, D.C.
- 5.Bargar, J. R., B. M. Tebo, U. Bergmann, S. M. Webb, P. Glatzel, V. Q. Chiu, and M. Villalobos. 2005. Biotic and abiotic products of Mn(II) oxidation by spores of the marine Bacillus sp. strain SG-1. Am. Mineral. 90:143-154.
- 6.Bargar, J. R., B. M. Tebo, and J. E. Villinski. 2000. In situ characterization of Mn(II) oxidation by spores of the marine Bacillus sp. strain SG-1. Geochim. Cosmochim. Acta 64:2775-2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beetz, T., and C. Jacobsen. 2003. Soft x-ray radiation-damage studies in PMMA using cryo-STXM. J. Synchr. Radiat. 10:280-283. [DOI] [PubMed] [Google Scholar]
- 8.Boucher, L. J. 1972. Manganese porphyrin complexes. Coordin. Chem. Rev. 7:289-329. [Google Scholar]
- 9.Brouwers, G. J., J. P. M. deVrind, P. L. A. M. Corstjens, P. Cornelis, C. Baysse, and E. W. deVrind-deJong. 1999. cumA, a gene encoding a multicopper oxidase, is involved in Mn +2 oxidation in Pseudomonas putida GB-1. Appl. Environ. Microbiol. 65:1762-1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan, C., G. DeStasio, S. Welch, M. Girasole, B. Frazer, M. Nesterova, S. Fakra, and J. Banfield. 2004. Microbial polysaccharides template assembly of nanocrystal fibers. Science 303:1656-1658. [DOI] [PubMed] [Google Scholar]
- 11.Chapnick, S., W. S. Moore, and K. H. Nealson. 1982. Microbially mediated manganese oxidation in a freshwater lake. Limnol. Oceanogr. 17:1004-1014. [Google Scholar]
- 12.Cramer, S. P., F. M. F. deGroot, Y. Ma, C. T. Chen, F. Sette, C. A. Kipke, D. M. Eichhorn, M. K. Chan, W. H. Armstrong, E. Libby, G. Christou, S. Brooker, V. McKee, O. C. Mullins, and J. C. Fuggle. 1991. Ligand field strengths and oxidation states from manganese L-edge spectroscopy. J. Am. Chem. Soc. 113:7937-7940. [Google Scholar]
- 13.Dubois, M., K. Gilles, J. Hamilton, P. Rebers, and F. Smith. 1956. Colorimetric method for determination of sugars and related substances. Anal. Chem. 28:350-356. [Google Scholar]
- 14.Fortin, D., F. G. Ferris, and T. J. Beveridge. 1997. Surface-mediated mineral development by bacteria, p. 161-180. In J. F. Banfield and K. H. Nealson (ed.), Geomicrobiology: interactions between microbes and minerals. Reviews in Mineralogy, vol. 35. Mineralogical Society of America, Washington, D.C.
- 15.Gilbert, B., B. H. Frazer, A. Belz, P. G. Conrad, K. H. Nealson, D. Haskel, J. C. Lang, G. Srajer, and G. D. Stasio. 2003. Multiple scattering calculations of bonding and x-ray absorption spectroscopy of manganese oxides. J. Phys. Chem. 107:2839-2847. [Google Scholar]
- 16.Gilbert, E. S., A. Khlebnikov, W. Meyer-Ilse, and J. D. Keasling. 1999. Use of soft x-ray microscopy for analysis of early-stage biofilm formation. Water Sci. Technol. 39:269-272. [Google Scholar]
- 17.Grush, M. M., J. Chen, T. L. Stemmler, S. J. George, C. Y. Ralston, R. T. Stibrany, A. Gelasco, G. Christou, S. M. Gorum, J. E. Penner-Hahn, and S. P. Cramer. 1996. Manganese L-edge x-ray absorption spectroscopy of manganese catalase from Lactobacillus plantarum and mixed valence manganese complexes. J. Am. Chem. Soc. 118:65-69. [Google Scholar]
- 18.Haack, E. A., and L. A. Warren. 2003. Biofilm hydrous manganese oxyhydroxides and metal dynamics in acid rock drainage. Environ. Sci. Technol. 37:4138-4147. [DOI] [PubMed] [Google Scholar]
- 19.Hamm, R. E., and M. A. Suwyn. 1967. Preparation and characterization of some aminopolycarboxylate complexes of manganese(III). Inorg. Chem. 6:139-142. [Google Scholar]
- 20.Hem, J. D., and C. J. Lind. 1983. Nonequilibrium models for predicting forms of precipitated manganese oxides. Geochim. Cosmochim. Acta 47:2037-2046. [Google Scholar]
- 21.Henke, B. L., E. M. Gullikson, and J. C. Davis. 1993. At. Nucl. Data Tables 54:181-342. [Google Scholar]
- 22.Hitchcock, A. P. 2000. aXis2000 is an IDL-based analytical package. http://unicorn.mcmaster.ca.
- 23.Jackson, T. A., and T. Bistricki. 1995. Selective scavenging of copper, zinc, lead, and arsenic by iron and manganese oxyhydroxide coatings on plankton in lakes polluted with mine and smelter wastes: results of energy dispersive X-ray micro-analysis. J. Geochem. Explor. 52:97-125. [Google Scholar]
- 24.Jokic, A., A. I. Frenkel, M. A. Vairavamurthy, and P. M. Huang. 2001. Birnessite catalysis of the Maillard reaction: its significance in natural humification. Geophys. Res. Lett. 28:3899-3902. [Google Scholar]
- 25.Kachlany, S., S. Levery, J. Kim, B. Reuhs, L. Lion, and W. Ghiorse. 2001. Structure and carbohydrate analysis of the exopolysaccharide capsule of Pseudomonas putida G7. Environ. Microbiol. 3:774-784. [DOI] [PubMed] [Google Scholar]
- 26.Kilcoyne, A. L. D., T. Tyliszczak, W. F. Steele, S. Fakra, P. Hitchcock, K. Franck, E. Anderson, B. Harteneck, E. G. Rightor, G. E. Mitchell, A. P. Hitchcock, L. Yang, T. Warwick, and H. Ade. 2003. Interferometer-controlled scanning transmission x-ray microscopes at the Advanced Light Source. J. Synchr. Radiat. 10:125-136. [DOI] [PubMed] [Google Scholar]
- 27.Kostka, J. E., G. W. Luther III, and K. H. Nealson. 1995. Chemical and biological reduction of Mn(III)-pyrophosphate complexes: potential importance of dissolved Mn(III) as an environmental oxidant. Geochim. Cosmochim. Acta 59:885-894. [Google Scholar]
- 28.Larsen, I., B. Little, K. H. Nealson, R. Ray, A. Stone, and J. Tian. 1998. Manganite reduction by Shewanella putrefaciens MR-4. Am. Mineral 83:1564-1572. [Google Scholar]
- 29.Manceau, A., N. Tamura, R. S. Celestre, A. A. Macdowell, N. Geoffroy, G. Sposito, and H. A. Padmore. 2003. Molecular-scale speciation of Zn and Ni in soil ferromanganese nodules from loess soils of the Mississippi Basin. Environ. Sci. Technol. 37:75-80. [DOI] [PubMed] [Google Scholar]
- 30.Mandernack, K. W., J. Post, and B. M. Tebo. 1995. Manganese mineral formation by bacterial spores of the marine Bacillus, strain SG-1: evidence for the direct oxidation of Mn(II) to Mn(IV). Geochim. Cosmochim. Acta 59:4393-4408. [Google Scholar]
- 31.McKenzie, R. M. 1971. The synthesis of birnessite, cryptomelane, and some other oxides and hydroxides of manganese. Mineral Mag. 38:493-502. [Google Scholar]
- 32.Morgan, J. J. 2000. Manganese in natural waters and Earth's crust: its availability to organisms. .In A. S. A. H. Sigel (ed.), Manganese and its role in biological processes, vol. 37. Marcel Dekker, Inc., New York, N.Y. [PubMed]
- 33.Nealson, K., B. Tebo, and R. Rosson. 1988. Occurrence and mechanisms of microbial oxidation of manganese. Adv. Appl. Microbiol. 33:279-317. [Google Scholar]
- 34.Neretin, L., C. Pohl, G. Jost, T. Leipe, and F. Pollehne. 2003. Manganese cycling in the Gotland Deep, Baltic Sea. Mar. Chem. 82:125-143. [Google Scholar]
- 35.Pecher, K., D. McCubbery, E. Kneedler, J. Rothe, J. Bargar, G. Meigs, L. Cox, K. Nealson, and B. Tonner. 2003. Quantitative charge state analysis of manganese biominerals in aqueous suspension using scanning transmission X-ray microscopy (STXM). Geochim. Cosmochim. Acta 67:1089-1098. [Google Scholar]
- 36.Pérez-Benito, J. 2002. Reduction of colloidal manganese dioxide by manganese(II). J. Colloid Interface Sci. 248:130-135. [DOI] [PubMed] [Google Scholar]
- 37.Post, J. E. 1992. Crystal structures of manganese oxide minerals, p. 51-73. In H. C. W. Skinner and R. W. Fitzpatrick (ed.), Biomineralization processes. Catena Verlag, Cremlingen-Destedt, Germany.
- 38.Post, J. E. 1999. Manganese oxide minerals: crystal structures and economic and environmental significance. Proc. Natl. Acad. Sci. USA 96:3447-3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roberson, E., and M. Firestone. 1992. Relationship between desiccation and exopolysaccharide production in a soil Pseudomonas sp. Appl. Environ. Microbiol. 58:1284-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ross, D., H. Hales, G. Shea-McCarthy, and A. Lanzirotti. 2001. Sensitivity of soil manganese oxides: XANES spectroscopy may cause reduction. Soil Sci. Soc. Am. J. 65:744-752. [Google Scholar]
- 41.Scott, M. J., and J. J. Morgan. 1995. Reactions at oxide surfaces. 1. Oxidation of As(III) by synthetic birnessite. Environ. Sci. Technol. 29:1898-1904. [DOI] [PubMed] [Google Scholar]
- 42.Stohr, J. 1992. NEXAFS spectroscopy. Springer-Verlag, Berlin, Germany.
- 43.Stumm, W. 1997. Catalysis of redox processes by hydrous oxide surfaces. Croat. Chem. Acta 70:71-93. [Google Scholar]
- 44.Stumm, W., and R. Giovanoli. 1976. On the nature of particulate manganese in simulated lake waters. Chimia 30:423-425. [Google Scholar]
- 45.Tebo, B. M., J. R. Bargar, B. G. Clement, G. J. Dick, K. J. Murray, D. Parker, R. Verity, and S. M. Webb. 2004. Biogenic manganese oxides: properties and mechanisms of formation. Annu. Rev. Earth Planet Sci 32:287-328. [Google Scholar]
- 46.Tebo, B. M., W. C. Ghiorse, L. G. V. Waasbergen, P. L. Siering, and R. Caspi. 1997. Bacterially-mediated mineral formation: insights into manganese(II) oxidation from molecular genetic and biochemical studies, p. 225-266. In J. F. Banfield and K. H. Nealson (ed.), Geomicrobiology: interactions between microbes and minerals, vol. 35. Mineralogical Society of America, Washington, D.C.
- 47.Tebo, B. M., K. H. Nealson, S. Emerson, and L. Jacobs. 1984. Microbial mediation of Mn(II) and Co(II) precipitation at the O2/H2S interfaces in two anoxic fjords. Limnol. Oceanogr. 29:1247-1258. [Google Scholar]
- 48.Thamdrup, B., R. N. Glud, and J. W. Hansen. 1994. Manganese oxidation and in situ fluxes from a coastal sediment. Geochim. Cosmochim. Acta 58:2563-2570. [Google Scholar]
- 49.Traina, S. J., and H. E. Doner. 1985. Copper-manganese(II) exchange on a chemically reduced birnessite. Soil Sci. Soc. Am. J. 49:307-313. [Google Scholar]
- 50.Villalobos, M., B. Toner, J. Bargar, and G. Sposito. 2003. Characterization of the manganese oxide produced by Pseudomonas putida strain MnB1. Geochim. Cosmochim. Acta 67:2649-2662. [Google Scholar]
- 51.Wilson, A. R., L. W. Lion, Y. M. Nelson, M. L. Shuler, and W. C. Ghiorse. 2001. The effects of pH and surface composition on Pb adsorption to natural freshwater biofilms. Environ. Sci. Technol. 35:3182-3189. [DOI] [PubMed] [Google Scholar]