

Abstract
The plant kingdom is constantly challenged by a battery of evolving pathogens. New species or races of pathogens are discovered on crops that were initially bred for disease resistance, and globalization is facilitating the movement of exotic pests. Among these pests, obligate biotrophic parasites make up some of the most damaging groups and have been particularly challenging to study. Here we demonstrate the utility of kinetic PCR (kPCR) (real-time PCR, quantitative PCR) to assess the growth of poplar rust, caused by Melampsora species, by quantification of pathogen DNA. kPCR allowed the construction of reliable growth curves from inoculation through the final stages of uredinial maturation, as well as pathogen monitoring before symptoms become visible. Growth parameters, such as latency period, generation time in logarithmic growth, and the increase in DNA mass at saturation, were compared in compatible, incompatible, and nonhost interactions. Pathogen growth was monitored in different applications dealing with plant pathology, such as host and pathogen diversity and transgenic crop improvement. Finally, the capacity of kPCR to differentiate pathogens in the same sample has broad molecular ecology applications for dynamically monitoring the growth of fungi in their environments or in mixed populations or to measure the efficacy of pest control strategies.
Leaf rust is the most important disease of hybrid poplars worldwide (28). In eastern North America, Melampsora medusae f. sp. deltoidae is the main poplar rust causal agent, whereas Melampsora larici-populina is the most important species in Europe. Melampsora infections appear as yellow-orange pustules on the lower leaf surface of poplar. The disease builds up rapidly and causes defoliation of susceptible poplar clones at the end of summer. Growth losses caused by Melampsora can reach 65% in planted areas (34). Melampsora rust fungi overwinter on fallen leaves as telia and complete their life cycle by producing basidiospores in the spring that infect an alternate coniferous host. Mating occurs on the alternate host to produce aeciospores that spread back to the poplar host (36).
Plant-pathogen interactions involving Melampsora and poplar have been described both visually and microscopically (12, 27, 31). Quantitative disease assessments have also been produced, but these have relied mainly on the appearance and/or quantification of symptoms, fruiting bodies, and spores (23). As for most biotrophic pathogens, there are few quantitative data concerning Melampsora disease progression from germination through spore production.
Several techniques have been developed to monitor fungal growth from diverse environmental samples. Immunological techniques, such as enzyme-linked immunosorbent assays, have been used, but they require the production of an epitope-specific antiserum. Consequently, it is very difficult to develop enzyme-linked immunosorbent assay procedures to differentiate closely related species, a problem which also applies to chemical quantification of fungus-specific macromolecules (7). A molecular approach relying on the quantification of constitutively expressed gene transcripts by Northern blotting was also developed (17). This procedure requires several days and demonstration that the expression of the chosen gene does not vary throughout growth; this demonstration becomes especially challenging when the fungus faces growth antagonists, such as plant defense reactions or antibiosis. Recently, tobacco pathogens and the biocontrol fungus Trichoderma atroviride were genetically engineered to express green fluorescent protein, and fluorescence was used to monitor their growth (3, 16). Although promising, this technique requires the fungus of interest to be transformed, which in itself limits the applicability.
On the other hand, PCR is widely used in plant pathology for specific identification and detection of pathogens (32). Recently, several groups have reported the use of kinetic PCR (kPCR) to detect and quantify fungal pathogens from different biological samples (5, 29, 30). However, in these studies the workers quantified fungi in an endpoint assay by kPCR from biological samples without interference of plant material, which does not fully exploit the quantitative capabilities of kPCR to monitor the dynamics of pathogen colonization in planta. The ability to accurately quantify pathogen DNA provided by kPCR allows construction of growth curves that provide details of pathogen infection that were previously unattainable. The objective of this study was to use kPCR to monitor the growth of biotrophic pathogens in planta at various times following host infection.
MATERIALS AND METHODS
Plant material and fungal inoculation procedure.
Poplar clones 717 (Populus tremula × Populus alba 717 I-B4), NM6 (Populus nigra × Populus maximowiczii NM6), and Jackii (Populus balsamifera L. × Populus deltoides Marsh.) and the chitinase-overexpressing line CH-11 (NM6 background) were grown in a greenhouse prior to inoculation. CH-11 expresses the endochitinase gene ech42 from the biocontrol fungus Trichoderma harzianum under the control of a cauliflower mosaic virus double 35S promoter, alfalfa mosaic virus leader (1). Leaf disks were used in all experiments because they were previously shown to reflect field infections (8). The fifth and sixth leaves from the index leaf were taken from at least five different trees and surface sterilized in a 1% household bleach solution for 5 min and then washed twice in sterile water. Disks (diameter, 2 cm) were inoculated on the abaxial side with M. medusae f. sp. deltoidae or M. larici-populina at a density of 1,000 to 3,000 spores (in 0.01% Tween) per cm2 by using a Crown spray tool (North American Professional Products, Vaughan, Ontario, Canada). After inoculation, the disks were kept on wet paper in large Parafilm-sealed petri dishes and incubated in a growth chamber at 18°C with a long photoperiod. Control disks were sprayed with 0.01% Tween. After 2 days the Parafilm was removed. This inoculation procedure was also used to amplify and maintain the fungal strains. Melampsora strains were amplified from field isolates that were genotyped on the basis of the internal transcribed spacer (ITS) sequence (9).
Staining and microscopy.
Leaf disks were stained with bromophenol blue and chlorazol black by using the procedure of Conner (4), with the clearing and staining time reduced to 45 min. Leaf disks were mounted in 50% glycerol and examined with a Zeiss Axioskop microscope.
Genomic DNA isolation and real-time PCR.
Disks were taken at specified intervals and frozen in liquid nitrogen in a 2-ml cylindrical tube. A large ceramic bead and 150 mg of glass beads (diameter, 0.5 mm) were added to each tube along with 400 μl of AP1 buffer (Plant DNeasy kit; QIAGEN, Chatsworth, Calif.) and 4 μl of RNase (supplied with the Plant DNeasy kit). The disks were homogenized in a FastPrep FP120 (Qbiogene, Carlsbad, Calif.) for 45 s at speed 6.0; a second homogenization was performed after a 3-min break. The remainder of the total genomic DNA preparation procedure was performed by using the instructions supplied with the Plant DNeasy kit (QIAGEN). Ten nanograms of total DNA was used in each kPCR. Amplifications were performed in 1× QuantiTect SYBR Green mixture (QIAGEN) with 0.3 μM 5′ oligonucleotides and 0.3 μM 3′ oligonucleotides (Table 1). Amplifications were carried out with an Opticon2 DNA engine (MJ Research, Waltham, Mass.). After an initial 15-min activation step at 95°C, 45 cycles (94°C for 15 s, 57°C for 1 min, and 72°C for 30 s) were performed, and a single fluorescent reading was obtained after each cycle immediately following the elongation at 72°C. Annealing was performed at 54°C for tubulin amplification. A melting curve was determined at the end of cycling to ensure that there was single amplification. Cycle threshold (Ct) values were determined with the Opticon Monitor 2 software supplied with the instrument at a manually set fluorescent threshold of 0.0160. ΔCt curves were generated by subtracting the raw Ct values (Fig. 1A and C) from the average Ct at day zero. To ensure a proper dose of DNA, kPCR was also performed with a poplar-specific primer pair (Fig. 1B and D). Each point on the curves represented the mean of four biological samples (infected disks) assayed by kPCR in triplicate (n = 12).
TABLE 1.
Oligonucleotides used in this study
| Amplicon | 5′ Oligonucleotide | 3′ Oligonucleotide | Amplicon size (bp) | Primer pair efficiency (%) |
|---|---|---|---|---|
| M. medusae ITS | TGAGTTGCTAAATGCATTC | TAAGCCAAAGTTGCCTTTGTC | 119 | 97.4 |
| M. larici-populina ITS | TGAGCGACTTTAATGTGACTC | ATGTAAATCAAAGTTGCCTTTGCG | 123 | 91.3 |
| Poplar | GAAGTAGCACTTGCTGCATAG | GTCAAAATCTTGGGAGTCAAAG | 122 | 100 |
| Tubulina | TGGGAAGTGGTGTCTGATGA | GATCGAACGGAATCCATTGT | 370/381b | NDc |
| Nested tubulin | ||||
| M. medusae | ACACTGATTCGTTCGAGTTG | 186 | 84.4 | |
| M. larici-populina | ACGCTGATTCGCTCGAGTTG | 184 | 84.8 |
The same primer pair was used for both M. medusae and M. larici-populina.
M. medusae amplicon size/M. larici-populina amplicon size.
ND, not determined.
FIG. 1.

Raw kPCR data. (A and C) M. medusae (A) and M. larici-populina (C) raw kPCR growth curve data for four different poplar host genotypes. (B and D) Raw kPCR growth curve data for poplar genomic DNA used as a control for equal starting material for M. medusae and M. larici-populina growth curves, respectively. The values are the means ± standard deviations for three individual kPCR runs with four different biological samples (n = 12). ▴, data obtained with infected clone Jackii; ▪, data obtained with infected clone NM6; •, data obtained with infected clone 717; ×, data obtained with infected clone CH-11.
Determination of latency period, growth rates, and ITS copy number.
The latency period for a microorganism is the period between inoculation and the beginning of growth. Therefore, the latency period ended at the first day that significant growth was detected by kPCR. Growth rates were calculated after a linear regression was performed for data obtained from day 2 to day 5. The linear regression coefficients were between 0.9882 and 0.9984. The growth rate was then calculated with the following equation:
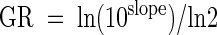 |
(1) |
where GR is the growth rate (in number of cell divisions per day). The ITS copy number was calculated by comparing the Ct value obtained for a single-copy gene (tubulin) with the Ct value of the ITS produced by a given sample. This evaluation could be performed only if the fluorescent threshold was kept constant, so that the DNA mass at the threshold was identical from run to run (25). The mass at threshold (Mt) could be calculated with equation 2:
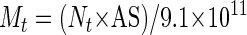 |
(2) |
where Nt is the number of amplicon molecules at the threshold, AS is the amplicon size (in base pairs), and 9.1 × 1011 is the number of single-base-pair molecules per nanogram (25). In turn, Nt could be calculated with the basic PCR equation:
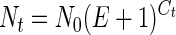 |
(3) |
where N0 is the initial number of molecules and E is the primer pair efficiency (25). The initial ITS copy number can be represented by Z × N0 compared with a single-copy gene in a given DNA mass. By inserting equation 3 into equation 2 and considering equal masses at the threshold, Z is determined as follows:
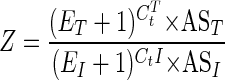 |
(4) |
where ET and EI are the primer pair efficiencies, CtT and CtI are the threshold cycles, and AST and ASI are the amplicon sizes of the tubulin and ITS primer pairs, respectively. The ITS copy number was determined for at least two pathogen DNA concentrations in four different runs, and the results were then averaged (n > 8).
RESULTS AND DISCUSSION
Interactions between M. medusae and poplar clones.
In this study, three poplar clones were chosen for their various levels of resistance to M. medusae and were characterized by visual observation (Fig. 2A) and staining (Fig. 2B and C). First, poplar clone Jackii was susceptible to M. medusae, which resulted in numerous uredinial pustules in 10 days (Fig. 2A and Table 2). Second, poplar clone NM6 showed resistance to M. medusae that looked like gene resistance because of the appearance of necrotic lesions (Fig. 2A) characteristic of the hypersensitive response (11). Finally, clone 717, although resistant to M. medusae, did not produce necrotic lesions (Fig. 2A), and the resistance could therefore be considered type 1 nonhost resistance (20). Although uredinial growth (Fig. 2B) or urediniospore germination (Fig. 2C) was observed after staining, it was not possible to derive growth parameters that are generally provided by growth curves.
FIG. 2.

M. medusae growth on three different poplar hosts. (A) Three poplar clones had different responses to M. medusae inoculation. Susceptible clone Jackii had numerous uredinial pustules 10 days after inoculation. Resistant clone NM6 had necrotic lesions characteristic of the hypersensitive response due to a gene for gene interaction. No symptoms were found on nonhost clone 717. (B) M. medusae grew on both clone Jackii and clone NM6. Microscopic observations showed that the uredinial pustule size increased dramatically from 3 to 10 days after inoculation on clone Jackii. The pustule size also increased on clone NM6 but did not reach the size seen on clone Jackii. The brown color on NM6 after 10 days was due to visualization of the uredinial pustule in a necrotic lesion. Magnification, ×100. (C) M. medusae germinated on clone 717. Germination was observed on clone 717 3 days after inoculation, but no further progress was observed after 10 days. Magnification, ×400.
TABLE 2.
Description of the plant-pathogen interactions
| Poplar clone |
M. medusae
|
M. larici-populina
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. of uredinial pustulesa | No. of necrotic lesionsa | Latency period (days) | Generation time (h)b | Increase in DNA mass (fold)c | No. of uredinial pustulesa | No. of necrotic lesionsa | Latency period (days) | Generation time (h)b | Increase in DNA mass (fold)c | |
| Jackii | 51 ± 10 | 0 | 3 | 8.0 | 2,728 | 88 ± 5 | 0 | 1 | 10.1 | 2,408 |
| NM6 | 6 ± 4 | 26 ± 5 | 4 | NDd | 28 | 79 ± 6 | 0 | 2 | 10.8 | 3,068 |
| 717 | 0 | 0 | >14 | ND | 0.45 | 0 | 0 | >14 | ND | 0.05 |
| CH-11 | 0 | 0 | >14 | ND | 0.24 | 35 ± 5 | 0 | 3 | 10.7 | 1,530 |
Counts were determined 10 days postinoculation (n = 16).
Pathogen generation times were determined only for susceptible clones in the exponential growth phase.
Measured after 14 days of growth.
ND, not determined.
Construction of growth curves.
Since DNA replication is intimately linked to cell division, we used kPCR technology to quantify over time pathogen DNA in leaf tissue and to generate growth curves. Figure 1 shows the raw kPCR data for pathogen growth over time. Reductions in the Ct value were observed every time that pathogen growth was detected, meaning that there was an increase in pathogen DNA mass (Fig. 1A,C). The Ct values for poplar DNA mass did not change significantly over time, confirming that there was equal loading of kPCR mixtures (Fig. 1B and D). In order to present our data in a more conventional way, when pathogen growth actually resulted in an increased value, two methods for construction of growth curves were examined: a relative approach based upon differences in the Ct, in which the Ct determined for each sample (Fig. 1) was subtracted from the average Ct at day zero (ΔCt method) (Fig. 3A), and an absolute approach, in which Ct values were converted to pathogen DNA mass. The latter approach required the generation of a standard curve for pathogen DNA plotted against the Ct, from which the amount of pathogen DNA was calculated (Fig. 3B). The resulting growth curves were nearly identical (compare Fig. 3A and C). The ΔCt method (Fig. 3A), which did not require generation of standard curves, brought all curves to the same starting point; it therefore was insensitive to differences in inocula. This method was the most convenient way to graphically present the data and to describe the timing of events throughout growth, such as the initial latency period, the logarithmic growth, and the saturation phase. It was also perfectly suitable for describing pathogen growth for different host backgrounds. However, the DNA mass approach (Fig. 3C) was essential to appreciate the extent of the infection or to derive growth parameters; therefore, the two methods were complementary.
FIG. 3.

kPCR-derived Melampsora growth curves for different poplar host backgrounds. Poplar clones Jackii (▴), NM6 (▪), and 717 (•) were chosen for their different levels of resistance to M. medusae. (A) M. medusae growth curves for three different poplar clones with different levels of resistance. (B) Standard curve used for conversion of Ct values to pathogen DNA mass. The equation resulting from linear regression of the standard curve is shown. (C) M. medusae DNA mass growth curves for three different poplar clones. (D) M. larici-populina growth curves for three different poplar clones. (E and F) M. medusae (E) and M. larici-populina (F) growth curves for the chitinase-overexpressing line CH-11 (□) and the wild-type background clone NM6 (▪). The values are the means ± standard deviations for four different biological samples assayed in triplicate (n = 12).
Analysis of the M. medusae-clone Jackii compatible interaction.
Analysis of M. medusae growth on susceptible clone Jackii revealed an apparent exponential growth phase from day 2 to day 5 (Fig. 3A and C). During this period, M. medusae had a steady growth rate (three cell divisions per day, for a generation time of 8 h). At saturation (14 days), the pathogen DNA mass reached 20% of the total DNA mass (including the mass of plant and fungal DNAs). Although exponential growth is common for unicellular organisms, it is presumed that filamentous fungi, which grow primarily by hyphal tip extension, should not grow exponentially. However, modeling of fungal growth in batch cultivation has provided several examples in which exponential growth can be achieved. The most probable scenario for the exponential growth observed here is the free mycelium model proposed by Lejeune and Biron (13), who described fungal growth by hyphal tip extension while new tips were created by branching. Free mycelium is thought to occur only in the early growth of filamentous fungi, as observed here, where the number of branches or tips and the hyphal length increase exponentially and at the same specific rate. Recently, branching was found to be closely linked to nuclear division (6), reinforcing the utility of kPCR for studying fungal growth in planta.
Effects of different host backgrounds on the growth of M. medusae.
In order to test how different host backgrounds influence pathogen growth, clones with different levels of resistance were compared. The pathogen DNA mass increased 57-fold with resistant clone NM6 before growth was blocked at day 5, and it decreased 28-fold by day 14. In contrast, susceptible clone Jackii supported about 100 times greater accumulation of pathogen DNA over the same 14-day period (Table 2). A slight reduction in M. medusae DNA mass was observed on nonhost clone 717 (Fig. 3A). The outcome of the infection could also be assessed prior to the appearance of symptoms, before day 5 in the case of M. medusae (Fig. 3). This demonstrated the effectiveness of kPCR for monitoring the in planta progress of an obligate biotroph.
Comparison of Melampsora species.
The aggressiveness of closely related pathogens was also examined. The same poplar clones were inoculated with the European poplar rust, M. larici-populina. Reflective of the nonhost type, the M. larici-populina DNA mass decreased over time on clone 717 (Fig. 3D). Although M. larici-populina grew on clone Jackii, its latency period (1 day) was significantly shorter that that of M. medusae (3 days) (Fig. 3A and D). Clone NM6, which was resistant to M. medusae, was clearly susceptible to M. larici-populina, as shown by a 3,068-fold increase in the DNA mass (Table 2). The growth of M. larici-populina was similar on both susceptible clones; the latency period on clone Jackii (1 day) was significantly shorter than that on clone NM6 (2 days) (Fig. 3D). Furthermore, the generation time of M. larici-populina during the exponential growth phase was slightly greater on NM6 (10.8 h) than on Jackii (10 h), which translated into earlier saturation of pathogen growth on Jackii (Fig. 3D and Table 2). M. larici-populina was recently discovered in eastern North America (9), where the native fungus M. medusae was the only previously reported poplar rust. Growth curves can be an important tool for monitoring the interaction between native and exotic pests occupying the same niche.
Species-specific detection of pathogens in mixed samples.
In order to test the capacity of kPCR to differentiate two pathogens in the same sample, we reconstituted DNA samples containing different quantities of pathogens. Increasing amounts of DNA extracted from either M. medusae- or M. larici-populina-infected clone Jackii leaves (10 days) were mixed together and subjected to kPCR with species-specific primers, and the resulting Ct values are shown in Tables 3 and 4. We found that the M. medusae (Table 3) and M. larici-populina (Table 4) primer pairs were species specific because no amplification was detected when the respective DNAs were omitted from the DNA mixtures. Tables 3 and 4 also show that pathogen detection was not significantly altered by other DNAs since the Ct values for a specific pathogen were nearly identical for all DNA mixtures containing the same amount of the pathogen. These results demonstrate the ability of kPCR with species-specific primers to differentiate pathogens in the same sample, reinforcing its utility for studying pathogen population dynamics.
TABLE 3.
M. medusae primer pair specificity evaluated by comparing Ct values in mixed DNA samples
| Amt of M. medusae DNAa (ng) | Amt of M. larici-populina DNAa (ng)
|
Avgb | |||
|---|---|---|---|---|---|
| 0 | 0.05 | 0.5 | 5 | ||
| 0 | Nonec | None | None | None | None |
| 0.05 | 20.7 ± 0.3 | 20.7 ± 0.4 | 20.8 ± 0.1 | 20.9 ± 0.1 | 20.8 ± 0.2 |
| 0.5 | 17.4 ± 0.3 | 17.5 ± 0.4 | 17.5 ± 0.2 | 17.7 ± 0.3 | 17.5 ± 0.3 |
| 5 | 14.0 ± 0.2 | 13.9 ± 0.1 | 14.0 ± 0.5 | 13.9 ± 0.3 | 14.0 ± 0.3 |
DNA was extracted from infected clone Jackii leaves at 10 days postinfection. The mixtures contained 0, 0.05, 0.5, or 5 ng of DNA isolated from M. medusae- or M. larici-populina-infected leaves. Amplification was carried out in triplicate with the M. medusae ITS primer pair.
Average for all 12 runs.
None, no amplification detected.
TABLE 4.
M. larici-populina primer pair specificity evauated by comparing Ct values in mixed DNA samples
| Amt of M. medusae DNAa (ng) | Amt of M. larici-populina DNAa (ng)
|
|||
|---|---|---|---|---|
| 0 | 0.05 | 0.5 | 5 | |
| 0 | Noneb | 21.4 ± 0.3 | 18.2 ± 0.8 | 14.3 ± 0.4 |
| 0.05 | None | 21.7 ± 0.4 | 18.1 ± 0.5 | 14.5 ± 0.7 |
| 0.5 | None | 21.2 ± 0.3 | 18.0 ± 0.4 | 14.4 ± 0.4 |
| 5 | None | 21.5 ± 0.4 | 18.1 ± 0.4 | 14.5 ± 0.5 |
| Avgc | None | 21.4 ± 0.4 | 18.1 ± 0.5 | 14.4 ± 0.4 |
DNA was extracted from infected clone Jackii leaves at 10 days postinfection. The mixtures contained 0, 0.05, 0.5, or 5 ng of DNA isolated from M. medusae- or M. larici-populina-infected leaves. Amplification was carried out in triplicate with the M. larici-populina ITS primer pair.
None, no amplification detected.
Average for all 12 runs.
Effects of ectopic endochitinase gene expression on M. medusae and M. larici-populina growth.
The ability to determine the efficacy of disease resistance in transgenic crops by using growth curves was investigated. We characterized the growth of both M. medusae and M. larici-populina on transgenic NM6 poplar trees ectopically expressing an endochitinase gene. This endochitinase gene was introduced into apple trees to improve scab resistance (1). No significant pathogen growth was detected on the transgenic line when it was inoculated with M. medusae, which resulted in a symptomless infection similar to the one observed on clone 717 (Fig. 3E and Table 2). This contrasts with the 28-fold increase in M. medusae DNA mass observed on wild-type clone NM6 (Fig. 3E and Table 2). However, expression of the endochitinase gene affected only the initial stages of M. larici-populina infection, resulting in a 1-day increase in the latency period (Fig. 3F). Once the pathogen was established, its generation time in logarithmic growth was almost unaltered (Table 2). These results clearly demonstrated that there were different pathogen responses to the same transgenic poplar line. The transgenic strategy was appropriate for controlling M. medusae, for which a complete block of pathogen progress was observed (Fig. 3E), while it was unsuitable for M. larici-populina. Nevertheless, a 50% reduction in fruiting bodies was observed with M. larici-populina, indicating that there was slightly improved resistance (Table 2). Chitinases have the ability to enhance the antifungal effects of nonenzymatic compounds, microorganisms, and plant defense mechanisms (14, 15). It seemed that the apparent gene for resistance of clone NM6 to M. medusae (Fig. 2A and 3A and Table 2) might show some synergism with the endochitinase overexpression to completely block pathogen growth. No such disease response occurred with M. larici-populina (Fig. 3D and Table 2), and this might be the reason why endochitinase overexpression failed to block pathogen growth. In addition, chitinases have been shown to have a substantial inhibitory effect on spore germination and hyphal elongation (10). Since the chitinase effect was perceived in the initial stages of infection, the in vivo data presented here support the hypothesis that chitinases are more efficient with newly synthesized chitin. These observations reinforce the importance of generating growth curves to understand the pathogen response to transgenic crop improvement.
ITSs provide increased sensitivity.
The ITS is widely used as a target for diagnostic purposes, largely because it is present in multiple copies in tandem repeats (2, 33). However, variation in the number of copies among different organisms can be problematic for quantification. When increases in DNA mass are measured or when the ΔCt approach is used, quantification becomes independent of copy number, thus allowing users to retain the sensitivity of the ITS-based diagnostic and the quantification accuracy provided by kPCR. In this study, pathogen detection was linear for between 5 ng and 5 fg of M. medusae or M. larici-populina DNA (Fig. 3B and data not shown), which is 3 orders of magnitude more sensitive than the results previously reported for detection of fungal pathogens with kPCR with single-copy genes (18, 30). Moreover, by comparing Ct values obtained from a single-copy gene (the tubulin gene) in the present study with the values obtained with the ITS primers, we determined that the numbers of rRNA gene repeats were 233 ± 40 and 175 ± 55 per haploid genome for M. medusae and M. larici-populina, respectively. These numbers reflect the 200-fold increase in sensitivity observed here compared with previous studies.
Conclusions.
kPCR offers all the advantages of PCR, which are maximal sensitivity and specificity, and a wide dynamic range of quantification in addition to high throughput capability. Therefore, kPCR is the method of choice for generating growth curves for obligate biotrophic pathogens. Since the majority of microorganisms are not yet known, mostly due to our inability to cultivate them (21), we can use kPCR in conjunction with molecular phylogeny approaches (19, 22, 26) to study microbial population dynamics in the environment. kPCR could also be used to evaluate in-host competition of diseases (24) or the host diversity impact of multihost pathogens (35). kPCR provides a means for monitoring the growth of microbes in their environment, whether it be in planta, in soil, or even during infection of mammals.
Acknowledgments
We are grateful to R. G. Rutledge for critical reading and valuable comments on the manuscript and to I. Lamarre for editorial work. We also thank M. J. Bergeron for access to the tubulin gene sequences prior to GenBank deposition.
This research was supported by Canadian Biotechnology Strategy grants to A.S. and R.C.H.
REFERENCES
- 1.Bolar, J. P., J. L. Norelli, K.-W. Wong, C. K. Hayes, G. E. Harman, and H. S. Aldwinckle. 2000. Expression of endochitinase from Trichoderma harzianum in transgenic apple increases resistance to apple scab and reduces vigor. Phytopathology 90:72-77. [DOI] [PubMed] [Google Scholar]
- 2.Borneman, J., and R. J. Hartin. 2000. PCR primers that amplify fungal rRNA genes from environmental samples. Appl. Environ. Microbiol. 66:4356-4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen, N., T. Hsiang, and P. H. Goodwin. 2003. Use of green fluorescent protein to quantify the growth of Colletotrichum during infection of tobacco. J. Microbiol. Methods 53:113-122. [DOI] [PubMed] [Google Scholar]
- 4.Conner, P. J. 2002. A detached leaf technique for studying race-specific resistance to Cladosporium caryigenum in pecan. J. Am. Soc. Hortic. Sci. 127:781-785. [Google Scholar]
- 5.Cullen, D. W., A. K. Lees, I. K. Toth, and J. M. Duncan. 2001. Conventional PCR and real-time quantitative PCR detection of Helminthosporium solani in soil and on potato tubers. Eur. J. Plant Pathol. 107:387-398. [Google Scholar]
- 6.Dynesen, J., and J. Nielsen. 2003. Branching is coordinated with mitosis in growing hyphae of Aspergillus nidulans. Fungal Genet. Biol. 40:15-24. [DOI] [PubMed] [Google Scholar]
- 7.Gessner, M. O., and A. L. Schmitt. 1996. Use of solid-phase extraction to determine ergosterol concentrations in plant tissue colonized by fungi. Appl. Environ. Microbiol. 62:415-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamelin, R. C., R. S. Ferriss, L. Shain, and B. A. Thielges. 1994. Prediction of poplar leaf rust epidemics from a leaf-disk assay. Can. J. For. Res. 24:2085-2088. [Google Scholar]
- 9.Innes, L., L. Marchand, P. Frey, M. Bourassa, and R. C. Hamelin. 2004. First report of Melampsora larici-populina on Populus spp. in eastern North America. Plant Dis. 88:85. [DOI] [PubMed] [Google Scholar]
- 10.Kubicek, C. P., R. L. Mach, C. K. Peterbauer, and M. Lorito. 2001. Trichoderma: from genes to biocontrol. J. Plant Pathol. 83:11-23. [Google Scholar]
- 11.Lam, E., N. Kato, and M. Lawton. 2001. Programmed cell death, mitochondria and the plant hypersensitive response. Nature 411:848-853. [DOI] [PubMed] [Google Scholar]
- 12.Laurans, F., and G. Pilate. 1999. Histological aspects of a hypersensitive response in poplar to Melampsora larici-populina. Phytopathology 89:233-238. [DOI] [PubMed] [Google Scholar]
- 13.Lejeune, R., and G. V. Baron. 1998. Modeling the exponential growth of filamentous fungi during batch cultivation. Biotechnol. Bioeng. 60:169-179. [DOI] [PubMed] [Google Scholar]
- 14.Lorito, M., S.-L. Woo, M. D'Ambrosio, G.-E. Harman, C.-K. Hayes, C.-P. Kubicek, and F. Scala. 1996. Synergistic interaction between cell wall degrading enzymes and membrane affecting compounds. Mol. Plant-Microbe Interact. 9:206-213. [Google Scholar]
- 15.Lorito, M., S. L. Woo, I. G. Fernandez, G. Colucci, G. E. Harman, J. A. Pintor-Toro, E. Filippone, S. Muccifora, C. B. Lawrence, A. Zoina, S. Tuzun, and F. Scala. 1998. Genes from mycoparasitic fungi as a source for improving plant resistance to fungal pathogens. Proc. Natl. Acad. Sci. USA 95:7860-7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu, Z., R. Tombolini, S. Woo, S. Zeilinger, M. Lorito, and J. K. Jansson. 2004. In vivo study of Trichoderma-pathogen-plant interactions, using constitutive and inducible green fluorescent protein reporter systems. Appl. Environ. Microbiol. 70:3073-3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahe, A., J. Grisvard, and M. Dron. 1992. Fungal- and plant-specific gene markers to follow the bean anthracnose infection process and normalize a bean chitinase mRNA induction. Mol. Plant-Microbe Interact. 5:242-248. [Google Scholar]
- 18.Mercado-Blanco, J., M. Collado-Romero, S. Parrilla-Araujo, D. Rodríguez-Jurado, and R. M. Jiménez-Díaz. 2003. Quantitative monitoring of colonization of olive genotypes by Verticillium dahliae pathotypes with real-time polymerase chain reaction. Physiol. Mol. Plant Pathol. 63:91-105. [Google Scholar]
- 19.Moon-van der Staay, S. Y., R. De Wachter, and D. Vaulot. 2001. Oceanic 18S rDNA sequences from picoplankton reveal unsuspected eukaryotic diversity. Nature 409:607-610. [DOI] [PubMed] [Google Scholar]
- 20.Mysore, K. S., and C.-M. Ryu. 2004. Nonhost resistance: how much do we know? Trends Plant Sci. 9:97-104. [DOI] [PubMed] [Google Scholar]
- 21.Pace, N. R. 1997. A molecular view of microbial diversity and the biosphere. Science 276:734-740. [DOI] [PubMed] [Google Scholar]
- 22.Pawlowska, T. E., and J. W. Taylor. 2004. Organization of genetic variation in individuals of arbuscular mycorrhizal fungi. Nature 427:733-737. [DOI] [PubMed] [Google Scholar]
- 23.Pei, M. H., C. Ruiz, J. Harris, and T. Hunter. 2003. Quantitative inoculations of poplars with Melampsora larici-populina. Eur. J. Plant Pathol. 109:269-276. [Google Scholar]
- 24.Read, A. F., and L. H. Taylor. 2001. The ecology of genetically diverse infections. Science 292:1099-1102. [DOI] [PubMed] [Google Scholar]
- 25.Rutledge, R. G., and C. Côté. 2003. Mathematics of quantitative kinetic PCR and the application of standard curves. Nucleic Acids Res. 31:e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schadt, C. W., A. P. Martin, D. A. Lipson, and S. K. Schmidt. 2003. Seasonal dynamics of previously unknown fungal lineages in tundra soils. Science 301:1359-1361. [DOI] [PubMed] [Google Scholar]
- 27.Sharma, S., and R. Sharma. 2003. Influence of host genotype and pathogen isolate in the development of poplar leaf rust. J. Plant Dis. Prot. 110:359-365. [Google Scholar]
- 28.Steenackers, J., M. Steenackers, V. Steenackers, and M. Stevens. 1996. Poplar diseases, consequences on growth and wood quality. Biomass Bioenerg. 10:267-274. [Google Scholar]
- 29.van de Graaf, P., A. K. Lees, D. W. Cullen, and J. M. Duncan. 2003. Detection and quantification of Spongospora subterranea in soil, water and plant tissue samples using real-time PCR. Eur. J. Plant Pathol. 109:589-597. [Google Scholar]
- 30.Vandemark, G. J., and B. M. Barker. 2003. Quantifying Phytophthora medicaginis in susceptible and resistant alfalfa with a real-time fluorescent PCR assay. J. Phytopathol. 151:577-583. [Google Scholar]
- 31.Villar, M., F. Lefèvre, H. D. Bradshaw, Jr., and E. Teissier du Cros. 1996. Molecular genetics of rust resistance in poplars (Melampsora larici-populina Kleb/Populus sp.) by bulked segregant analysis in a 2 × 2 factorial mating design. Genetics 143:531-536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ward, E., A. Tahiri-Alaoui, and J. F. Antoniw. 1998. Applications of PCR in fungal-plant interactions, p. 289-307. In P. D. Bridge, D. K. Arora, C. A. Reddy, and R. P. Elander (ed.), Applications of PCR in mycology. CAB International, Wallingford, United Kingdom.
- 33.White, T. J., T. Bruns, S. Lee, and J. Taylor. 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics, p. 315-322. In M. A. Innis, D. H. Gelfand, J. J. Sninsky, and T. J. White (ed.), PCR protocols: a guide to methods and applications. Academic Press, San Diego, Calif.
- 34.Widin, K. D., and A. L. Schipper, Jr. 1981. Effect of Melampsora medusae leaf rust infection on yield of hybrid poplars in the north-central United States. Eur. J. For. Pathol. 11:438-448. [Google Scholar]
- 35.Woolhouse, M. E. J., L. H. Taylor, and D. T. Haydon. 2001. Population biology of multihost pathogens. Science 292:1109-1112. [DOI] [PubMed] [Google Scholar]
- 36.Ziller, W. G. 1974. The tree rusts of western Canada. Publication no. 1329. Canadian Forestry Service, Victoria, Canada.