

Abstract
Age-associated diseases are becoming progressively more prevalent, reflecting the increased lifespan of the world’s population. However, the fundamental mechanisms of physiologic aging are poorly understood, and in particular, the molecular pathways that mediate cardiac aging and its associated dysfunction are unclear. Here, we focus on certain ion flux abnormalities of the mitochondria that may contribute to cardiac aging and age-related heart failure. Using oxidative phosphorylation, mitochondria pump protons from the matrix to the intermembrane space to generate a proton gradient across the inner membrane. The protons are returned to the matrix by the ATPase complex within the membrane to generate ATP. However, a portion of protons leak back to the matrix and do not drive ATP production, and this event is called proton leak or uncoupling. Accumulating evidence suggests that mitochondrial proton leak is increased in the cardiac myocytes of aged hearts. In this mini-review, we discuss the measurement methods and major sites of mitochondrial proton leak with an emphasis on the adenine nucleotide transporter 1 (ANT1), and explore the possibility of inhibiting augmented mitochondrial proton leak as a therapeutic intervention to mitigate cardiac aging.
Keywords: Mitochondria, Proton leak, Cardiac aging, Heart failure, Adenine nucleotide transporter 1, Pleiotropic role
Introduction
Mitochondria use oxidative phosphorylation to pump protons from the matrix to the intermembrane space to generate a proton gradient that is responsible for the proton electrochemical potential (ΔμH+), which is the major contributor to the mitochondrial membrane potential (ΔΨm). Subsequently, the protons are driven by the ΔμH+ to re-enter the matrix through ATP synthase, which generates the ATP molecules that support the life of the organism. However, a portion of protons leak back to the matrix but do not drive ATP production, an event called proton leak or uncoupling (Fig. 1A). This event also is referred to as futile proton leak [1].
Fig. 1.
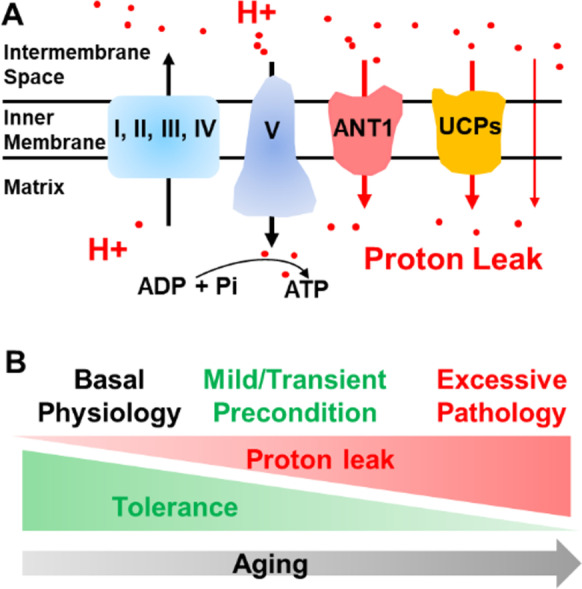
Mitochondrial proton leak and its role in cardiac aging. A Mitochondria pump protons through complexes I, III and IV to generate a proton gradient. Protons return to the matrix through complex V (ATPase, ATP synthase) to drive ATP production. Protons leak back to matrix through ANT1, UCPs and the mitochondrial inner membrane. B Aging increases the proton leak and concurrently reduces the tolerance to the potentially detrimental effects of proton leak. Basal proton leak is needed to maintain thermogenesis and other physiological processes. Mild increases in proton leak to mimic caloric restriction or exert a preconditioning effect may be beneficial. However, excessive proton leak may contribute to the pathogenesis of aging in the heart and contribute to pathologies in other organs
Mitochondrial proton leak has been reported in many organs and cell types including the liver [2, 3], kidney [3], brain [4], thymus [5], skeletal muscle [6], heart [3], and pancreatic β cells [7] of mammals, ectotherms [8, 9], and invertebrates [10]. In this mini-review, we briefly introduce several methods used to measure mitochondrial proton leak, and then showcase major sites of mitochondrial proton leak with the focus on the role of adenine nucleotide transporter 1 (ANT1). Finally, we discuss the functional impact of the increased mitochondrial proton leak on the aging heart and explore the therapeutic potential of inhibiting excessive proton leak to mitigate age-related diseases of the heart.
Measurement of mitochondrial proton leak (see summary in Table 1)
Table 1.
Measurement of mitochondrial proton leak
| Methods | Mechanism | Advantages | Limitations | Ref. |
|---|---|---|---|---|
| Clark-type oxygen electrode | [O2] change | Can measure phosphorylation related O2 consumption in one experiment. |
One sample each time. Needs isolated mitochondria or permeabilized cells. Requires ATPase blocker. Cannot exclude “proton slip”. |
[12] |
| Seahorse assay | [O2] change |
Can measure phosphorylation related O2 consumption, maximal respiration in one experiment. Good for isolated mitochondria or intact cells. Suitable for high throughput (24- or 96-well plates). Measure ECAR simultaneously. |
Requires ATPase blocker. Cannot exclude “proton slip”. |
https://www.agilent.com/en/product/cell-analysis/how-to-run-an-assay. |
| Mitoplast patch-clamp | Proton electricity current |
Direct Accurate |
Requires mitoplast. Requires high skills. |
[15, 16] |
| pH indicator-based measurement | pH indicator fluorescence change |
Direct No substrate. Reflects the purely proton leak. Suitable for high throughput. |
Requires a genetically encoded pH indicator. | [20] |
Oxygen consumption rate-based measurements
Clark-type oxygen electrode
The Clark-type oxygen electrode is named after Dr. Leland Clark, who invented the oxygen electrode in the 1950s to measure the pressure of oxygen (pO2) dissolved in solution [11]. The Clark-type oxygen probe uses a platinum electrode as the cathode, which directly contacts an oxygen-permeable Teflon membrane to allow oxygen to reach the cathode, the anode (a silver electrode) and a reference electrode Table 1. The electron flow between the electrodes generates a current, which is directly proportional to the level of pO2.
The Clark-type oxygen electrode measures the oxygen consumption rate (OCR), which in turn, reflects mitochondrial respiration. With this method, the proton leak can be determined indirectly by measuring the OCR under non-phosphorylating conditions after addition of the ATPase inhibitor oligomycin. Mitochondrial membrane potential is measured simultaneously by a triphenylmethylphosphonium (TPMP+) sensing probe. In isolated mitochondria or permeabilized cells, mitochondrial respiration is initiated either with malate and glutamate to provide complex I substrates or with succinate as a complex II substrate to establish a steady-state membrane potential. Then the kinetics of proton leak are determined by plotting respiration rates against their corresponding membrane potentials. For details of the method, see reference [12].
Seahorse Assay
Since 2009, Seahorse XF technology [13] has been widely used to measure OCR. In addition to evaluating the OCR in isolated mitochondria and permeabilized cells, the Seahorse Assay can measure the OCR in intact cells. Using the Mitochondrial Stress Assay, the basal oxygen consumption rate is recorded initially before the ATPase inhibitor oligomycin is injected to prevent oxygen consumption dependent on mitochondrial ATP. Then, the proton ionophore cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP) is added to trigger maximal respiration. Finally, the complex I inhibitor rotenone and complex III inhibitor antimycin are injected simultaneously to block mitochondria-related respiration; the residual OCR is non-mitochondrial respiration. The proton leak is equivalent to the OCR recorded after oligomycin injection minus the non-mitochondrial OCR. For further methodological details, see the Agilent Seahorse website https://www.agilent.com/en/product/cell-analysis/how-to-run-an-assay.
The Seahorse Assay outgrew the popularity of the Clark-type electrode because (1) it can measure the mitochondrial respiration in intact cells; (2) it requires a very small amount of sample and thereby can detect the OCR in hundreds to thousands of cells plated on a 24-well or 96-well plate; and (3) it can measure the extracellular acidification rate (ECAR) simultaneously. However, both the traditional Clark-type oxygen electrode and Seahorse Assay measurements of proton leak are based on indirect calculation of OCR and rely on adding mitochondrial ATPase blockers and substrates. Moreover, these OCR-based proton leak measurements cannot exclude “proton slip,” which is defined as mitochondrial respiratory chain activity that consumes oxygen and transfers electrons without extruding protons from the matrix to the intermembrane space.
Mitoplast patch-clamp
Patch-clamp is a very powerful approach to directly measure the mitochondrial proton leak across the inner mitochondrial membrane (mitoplast). This method requires fine preparation of the mitoplast and elegant patch-clamp skills. The mitoplasts with fully preserved integrity are usually prepared mechanically either with a French Press or with passive osmotic swelling to disrupt the outer mitochondrial membrane but leave the mitochondrial inner membrane and matrix intact. The whole-mitoplast patch-clamp pipettes are similar to those used for standard whole-cell recordings except the pipette tip is smaller in size (~2–5 μm), and the tip resistance of the pipettes is higher (between 25 and 35 MΩ). In 2012, Dr. Kirichok’s laboratory initially published this challenging method in which they recorded H+ current mediated by the uncoupling protein 1 (UCP1) transporter in mitoplasts prepared from brown adipose tissue [14]. Recently, using the mitoplast patch-clamp method, the same group reported that proton transport is another integral function of ANT1 in the heart [15]. For methodological details, see reference [16].
Application of the patch-clamp method to mitoplasts enables measurement of the mitochondrial proton leak directly and accurately with very high time (< 1 ms) and amplitude (< 1 pA) resolutions [16]. However, this method requires a highly skilled operator and does not lend itself to performing high throughput testing.
pH indicator-based measurement
In response to the challenges mentioned above, our group established a direct method for measuring proton leak using a mitochondrial-targeted pH-sensitive indicator mt-cpYFP [17–20]. Mitochondrial-targeted pH indicators have been used extensively to measure changes in mitochondrial pH [21–26]. Based on this technology, we subsequently adapted the pH indicator protocols to pioneer the direct estimation of mitochondrial proton leak in cardiac myocytes [20]. There are two excitation wavelengths for mt-cpYFP: 488 nm, which is sensitive to pH change, and 405 nm, which is pH-insensitive [20]. Taking advantage of this property, we used saponin to permeabilize cardiomyocytes over-expressing mt-cpYFP and exposed the mitochondria to a pH gradient stress. The pH of the solution was then progressively lowered from pH 7.5, in 8-min steps by addition of HCl in quantities previously titrated, to deliver a final pH of 7.3, 6.9, 5.3, and 4.5. The drop in mt-cpYFP fluorescence 488/405 ratio in response to stepwise lower pH is due to the proton leak through the mitochondrial inner membrane into the mitochondrial matrix. For detailed methods, see reference [20].
An advantage of the pH indicator-based method to measure proton leak is that the buffers lack metabolic substrates, ATP and ADP, which makes the cells energetically inactive and thereby allows separation of the physical property of the mitochondrial inner membrane (i.e., proton leak) from energetically active processes such as ion pumping and proton slipping. Thus, the pH indicator-based proton leak allows mechanistic insights that can be gained from evaluating the leak as a purely biophysical aspect of the mitochondrial inner membrane. Moreover, using cells with stable expression of pH indicators makes it feasible to use high throughput screening to identify drugs that regulate the proton leak.
The sites of mitochondrial proton leak
There are two general classifications of mitochondrial proton leak: constitutive proton leak and regulated proton leak [27]. The constitutive mitochondrial proton leak is composed of (1) proton leak through the mitochondrial inner membrane phospholipid bilayer and (2) ANT1-mediated proton leak. Considering that proton flux directly through the lipid bilayer accounts for ~5% of total proton leak in intact mitochondria [28] and that the phospholipid fatty acid composition has no correlation with it [29], there has been little research on the regulation of this aspect of proton leak. More studies have focused on ANT1, which mediates the majority of constitutive proton leak and will be discussed in more detail below. The regulated or inducible proton leak is catalyzed by uncoupled protein (UCP) transporters, including UCP1, 2, and 3 [27]. The regulated proton leak mediated by UCPs, especially UCP1, has been extensively investigated and summarized. For example, see references [10, 30–32].
ANT1 is a major transport protein of the mitochondrial inner membrane that controls ATP production by exporting ATP to the cytosol while importing ADP to the mitochondrial matrix. ANT1 has two configurations (states) in which it either faces the mitochondrial matrix or the cytosol. Two well-defined ANT1 specific inhibitors can stabilize ANT1 in one of these two configurations: bongkrekic acid (BKA) locks ANT1 facing the matrix [33] and carboxyatractyloside (CAT) locks it facing the cytosol [34].
Although ANT1 mediates the majority of constitutive proton leak, the molecular determinants of the leak and regulatory mechanisms are poorly understood. Using the Clark-type oxygen probe, Brand et al. [35] reported that the abundance of ANT1 positively correlated with basal mitochondrial proton leak in Drosophila in which ANT1 was under-expressed, over-expressed or expressed in wild-type levels. In this preparation, ANT1 contributed one-half to two-thirds of the basal mitochondrial proton leak in skeletal muscle [35]. Patch-clamp measurements of proton leak later revealed that ANT1-mediated mitochondrial proton leak requires fatty acids and that CAT inhibits it [15]. ANT1 switches between the ADP/ATP exchange mode and the distinct proton leak mode, and purine nucleotides negatively regulate the ANT1-mediated proton leak [15]. Using pH indicator-based proton leak measurements, we found that both BKA and CAT suppress ANT1-mediated proton leak in aged cardiomyocytes. This evidence suggests that constraining the conformational state in either configuration or directly blocking the proton pore can reduce ANT1-mediated proton leak. However, which amino acid residues in the ANT1 protein structure are essential for the proton leak remains unclear and, similarly, which post-translational protein modifications may modify the function of ANT1 to alter proton leak requires clarification.
Interestingly, it appears that the site of proton leak is highly heterogeneous between tissues. For example, ANT1 is the major site of proton leak in heart [15] and skeletal muscle [15, 35], whereas UCP1 is the dominant uncoupling protein responsible for proton leak in brown and beige fat [14, 36, 37]. In contrast, UCP2 primarily mediates proton leak in pancreatic β cells [7]. The differential expression of proton leak mediators between cell types raises the possibility of synthesizing therapeutic molecules that could modulate mitochondrial proton leak in a tissue-specific manner. In this regard, an intriguing patch-clamp study in mitoplasts recently revealed that two commonly used proton ionophores, 2,4-dinitrophenol (DNP), and FCCP, which act as “protonophores” by enabling protons to leak across the mitochondrial inner membrane and thereby bypass ATP synthase to uncouple mitochondrial oxidative phosphorylation, actually bind to ANT1 and UCP1 to induce the proton leak [38]. Accordingly, deletion or pharmacologic inhibition of ANT1 dramatically reduces the DNP- or FCCP-induced proton leak in mitoplasts prepared from the heart [38]. Similarly, deletion or inhibition of UCP1 suppresses DNP- and FCCP-induced proton leak in brown fat tissue [38]. The discovery that synthetic protonophores can bind to and activate ANT1 and UCP1 has reshaped our understanding of these commonly used reagents and strengthened the idea that therapeutic molecules can be designed to selectively modulate mitochondrial proton leak.
Augmented mitochondrial proton leak and its role in aged heart
Increased mitochondrial proton leak associated with aging has been reported during the past two decades in multiple tissues and cell types, including heart [3, 39], liver [3, 40], kidney [3], skeletal muscle [41–43], testis [44], and brain microvessels [45] in rodents and invertebrates [46]. We also detected increased proton leak in intact cardiomyocytes from aged mice by Seahorse Assay [20] and in permeabilized aged rat cardiomyocytes by pH indicator mt-cpYFP-based measurements [20].
As discussed in “The sites of mitochondrial proton leak”, ANT1 is the major site of proton leak in the heart [15]. Both ANT1 inhibitors, BKA and CAT, inhibit the proton inward current in heart mitoplasts [15]. However, there are very few reports on the particular site and mechanism of the increased mitochondrial proton leak in the aged heart. Measured by a Clark-type oxygen probe in cardiac mitochondria isolated from aged rats, Bellanti et al. [39] reported that CAT fails to prevent the excessive proton leak, but GDP (an UCP inhibitor) suppresses it. The same authors also found that protein levels of UCP2 (the dominant UCP isoform in the heart) and UCP3 are increased in the aged rat heart, which may contribute to the increased proton leak [39]. Inconsistent with this report, however, the UCP2 mRNA level is comparable in aged and young rat heart [47]. Similarly, we found that the UCP2 protein level shows no significant change in the aged mouse heart [20]. Moreover, we used the Seahorse Assay and mt-cpYFP-based measurement of proton leak to show that the ANT1 inhibitors BKA and CAT inhibit the excessive proton leak in aged cardiac mitochondria, whereas inhibition of UCP2 by genipin had no effect on it [20]. More comprehensively, we used the ATP synthase inhibitor, oligomycin A, to examine the ATPase contribution to the excessive passive proton leak in the aged heart and found no evidence for a contribution, although ATP generation consumes protons [20]. The inconsistent findings between earlier and recent reports may relate to different proton leak measure systems, animal preparations, or inhibitors used. Collectively, however, there may be reason to believe that ANT1 is an under-appreciated mediator of excessive proton leak in the aged mitochondria from heart.
Another controversial topic is whether the increased proton leak is a “friend or foe” in the pathogenesis of aging. Collective reports suggest that the augmented mitochondrial proton leak is a double-edged sword. There is a dogma that promoting proton leak, i.e., “uncoupling for survival” [48], is advantageous, as reducing the mitochondrial membrane potential presumably reduces the leak of electrons and protons across the gradient. This is supported by positive correlations between increased skeletal muscle proton uncoupling and mouse lifespan [49], and the observation that overexpression of UCP2 extends fly lifespan [50]. Moreover, increased UCP2 activity leads to improved resistance to high glucose stress in endothelial cells [51]. Also, mild proton leak achieved by low concentrations of DNP shows beneficial effects in several disease models [52–54] and promotes energy metabolism, redox balance, and prolongs mouse life span [55].
Although there is supportive evidence for the notion that proton leak is advantageous, this concept is challenged by reports that overexpression of UCP3 shortens fly lifespan [56] and overexpression of UCP2 alone [57], or UCP2 plus UCP3 [58], fails to extend mouse lifespan. Moreover, the increased proton leak induced by higher doses of DNP sold to the public as an oral weight loss medication at the start of the 20th century was associated with acute toxicity and fatalities, and it was subsequently banned by the FDA in 1938 [59]. An interesting finding is that a small increase in proton leak mediated by UCP2 and UCP3 during cardiac ischemic preconditioning (three 5-min episodes of ischemia, interspersed with 5 min of reperfusion) is protective, whereas a major increase in proton leak mediated by ANT1 in ischemia reperfusion (25 min of global ischemia followed by 30 min of reperfusion) is detrimental [60].
These collective findings suggest that a chronic excessive mitochondrial proton leak is detrimental. When there is an excessive proton leak, it burdens the mitochondrial workload, resulting in a decline in respiratory efficiency, decreasing ATP output, and increasing the electron leak, which in turn exacerbates superoxide generation, especially in the energetically active cardiomyocyte. Therefore, decreasing the pathological proton leak may represent a novel strategy to reduce mitochondrial workload and restore normal mitochondrial function and cell biology, ultimately helping to attenuate or reverse cardiac dysfunction in aging. In support of this concept, we found that SS-31 (aka elamipretide, a four amino acid peptide) binds to ANT1 [20, 61] and prevents excessive mitochondrial proton leak in aged cardiomyocytes [20], and 8 weeks of systemic treatment with SS-31 protects the heart from age-related dysfunction [62]. However, due to the pleiotropic effects of SS-31, we still lack direct proof that prevention of excessive proton leak protects against cardiac dysfunction. In this regard, Dr. Wojtovich and colleagues recently used a novel mitochondrial-targeted light-activated proton pump to pump protons from the mitochondrial matrix into the intermembrane space, thereby by-passing the electron transport chain to increase mitochondrial membrane potential. This intervention extended the lifespan of Caenorhabditis elegans by 30 to 40% [63], thereby providing direct causal evidence that rescuing the age-related decline in mitochondrial membrane potential (ΔΨm), of which the proton electrochemical potential (ΔμH+) is the major component, is sufficient to slow the rate of aging and extend lifespan in this organism.
Thus, a mild proton leak may decrease ROS production by reducing the mitochondrial potential gradient, while an excessive proton leak may increase ROS production. However, the exact conditions that separate these two domains are as of yet unclear.
In an attempt to integrate diverse observations in a unifying framework and reconcile the disparities, we conceptualized a model of antagonistic pleiotropy [64] to explain the role of increased proton leak (Fig. 1B). The first tenet of this model is that the degree and duration of the mitochondrial proton leak play a key role in determining its beneficial or detrimental balance. It appears that basal proton leak is required for maintaining thermogenesis. A mild increase in proton leak can mimic the situation of caloric restriction as the most effective way to extend life span [55]. Alternatively, a short-term increase in proton leak may be beneficial because it transiently or partially reduces the ΔΨm, resulting in a preconditioning-type protection from ischemic injury [60]. However, excessive proton leak can raise the mitochondrial workload by accelerating electron transport and the concomitant electron leak, ultimately exacerbating superoxide (oxygen free radical) generation to promote pathogenic events and cell injury.
The second tenet of this model is that the ability to tolerate and/or compensate for the excessive proton leak is weakened in the aged heart. It is known that the organization of mitochondrial supercomplexes is disrupted in the aged heart [65], which can result in functional defects in electron transport. Additionally, aging results in lowered activities of the mitochondrial complexes I [66], III [67] and IV [68], which are all major proton pump sites. Finally, ATP synthase activity may decrease by ~40% in the aged heart [69, 70]. All of these structural and functional declines impair the mitochondrial capacity to pump more protons to restore the ΔμH+ or generate ATP that supports many physiological processes. Thus, it is possible that the same level of proton leak that is beneficial or tolerable in the young heart may be detrimental to the old heart, which already may be challenged by the higher proton leak associated with aging (Fig. 1). This would be an example of antagonistic pleiotropy in aging [64].
Perspective
In summary, the mitochondrial proton leak is a ubiquitous phenomenon that contributes to the cellular metabolic rate in all eukaryotic cells. It appears to play a pleiotropic role depending on the health of the cell or organism, leak severity, and duration; either conferring protection or exacerbating injury caused by biological insults including the aging process.
Although the mitochondrial proton leak has been documented for more than a half century, many questions remain to be answered. For example, what is the relationship between mitochondrial proton leak and production of reactive oxygen species (ROS)? Does excessive proton leak actually cause cardiac dysfunction and will therapeutic prevention of excessive proton leak benefit the aged heart? Finally, what is the molecular basis of mitochondrial proton leak with regard to the structural aspects of the ANT1 molecule that mediates much of the proton leak, and which signaling pathways enable its regulation?
We anticipate that these questions and others will be addressed by the energized research teams rapidly advancing our knowledge of mitochondrial proton leak in different tissues and by the implementation of improved measurement techniques. One can envision a new treatment paradigm in which pharmacological modulators of mitochondrial proton leak are implemented as medications to combat aging and other diseases of mitochondrial dysfunction.
Funding
The authors received support from the following sources: NIH P01 AG001751 (P. S. R); American Heart Association 19 CDA 34660311 (H. Z.); NIH UW CTMR pilot award under P30 AR074990 pilot award (H. Z.); University of Arkansas for Medical Sciences (UAMS) Bronson Foundation Award (H. Z.); NSF 2217757 (X. Q.); NIH TL1 TR003109 (N. J. R.); NIH U54 TR002732 (N. J .R.); NIH R01 HL146713 (S. M.); and UAMS Bronson Foundation Award (S. M.)
Declarations
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Peter S. Rabinovitch, Email: petersr@uw.edu
Huiliang Zhang, Email: hzhang2@uams.edu.
References
- 1.Brand MD, et al. The causes and functions of mitochondrial proton leak. Biochim Biophys Acta. 1994;1187(2):132–139. doi: 10.1016/0005-2728(94)90099-x. [DOI] [PubMed] [Google Scholar]
- 2.Brand MD. The efficiency and plasticity of mitochondrial energy transduction. Biochem Soc Trans. 2005;33(Pt 5):897–904. doi: 10.1042/BST0330897. [DOI] [PubMed] [Google Scholar]
- 3.Serviddio G, et al. Bioenergetics in aging: mitochondrial proton leak in aging rat liver, kidney and heart. Redox Rep. 2007;12(1):91–95. doi: 10.1179/135100007X162112. [DOI] [PubMed] [Google Scholar]
- 4.Jekabsons MB, Nicholls DG. In situ respiration and bioenergetic status of mitochondria in primary cerebellar granule neuronal cultures exposed continuously to glutamate. J Biol Chem. 2004;279(31):32989–33000. doi: 10.1074/jbc.M401540200. [DOI] [PubMed] [Google Scholar]
- 5.Buttgereit F, et al. The effects of methylprednisolone on oxidative phosphorylation in Concanavalin-A-stimulated thymocytes. Top-down elasticity analysis and control analysis. Eur J Biochem. 1994;223(2):513–519. doi: 10.1111/j.1432-1033.1994.tb19020.x. [DOI] [PubMed] [Google Scholar]
- 6.Rolfe DF, Brand MD. Contribution of mitochondrial proton leak to skeletal muscle respiration and to standard metabolic rate. Am J Physiol. 1996;271(4 Pt 1):C1380–C1389. doi: 10.1152/ajpcell.1996.271.4.C1380. [DOI] [PubMed] [Google Scholar]
- 7.Affourtit C, Brand MD. Uncoupling protein-2 contributes significantly to high mitochondrial proton leak in INS-1E insulinoma cells and attenuates glucose-stimulated insulin secretion. Biochem J. 2008;409(1):199–204. doi: 10.1042/BJ20070954. [DOI] [PubMed] [Google Scholar]
- 8.Savina MV, Gamper NL. Respiration and adenine nucleotides of Baltic lamprey (Lampetra fluviatilis l.) hepatocytes during spawning migration. Comp Biochem Physiol B, Biochem. 1998;120(2):375–383. [Google Scholar]
- 9.Brand MD, et al. Evolution of energy metabolism. Proton permeability of the inner membrane of liver mitochondria is greater in a mammal than in a reptile. Biochem J. 1991;275(Pt 1):81–86. doi: 10.1042/bj2750081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stuart JA, et al. Mitochondrial proton leak and the uncoupling protein 1 homologues. Biochim Biophys Acta. 2001;1504(1):144–158. doi: 10.1016/s0005-2728(00)00243-7. [DOI] [PubMed] [Google Scholar]
- 11.Clark LC, Jr, et al. Continuous recording of blood oxygen tensions by polarography. J Appl Physiol. 1953;6(3):189–193. doi: 10.1152/jappl.1953.6.3.189. [DOI] [PubMed] [Google Scholar]
- 12.Affourtit C, Quinlan CL, Brand MD. Measurement of proton leak and electron leak in isolated mitochondria. Methods Mol Biol. 2012;810:165–182. doi: 10.1007/978-1-61779-382-0_11. [DOI] [PubMed] [Google Scholar]
- 13.Liu S, et al. Insulin signaling regulates mitochondrial function in pancreatic beta-cells. PLoS One. 2009;4(11):e7983. doi: 10.1371/journal.pone.0007983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fedorenko A, Lishko PV, Kirichok Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell. 2012;151(2):400–413. doi: 10.1016/j.cell.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bertholet AM, et al. H(+) transport is an integral function of the mitochondrial ADP/ATP carrier. Nature. 2019;571(7766):515–520. doi: 10.1038/s41586-019-1400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bertholet AM, Kirichok Y. Patchclamp analysis of the mitochondrial H(+) leak in brown and beige fat. Front Physiol. 2020;11:326. doi: 10.3389/fphys.2020.00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei-LaPierre L, et al. Respective contribution of mitochondrial superoxide and pH to mitochondria-targeted circularly permuted yellow fluorescent protein (mt-cpYFP) flash activity. J Biol Chem. 2013;288(15):10567–10577. doi: 10.1074/jbc.M113.455709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Demaurex N, Schwarzlander M. Mitochondrial flashes: dump superoxide and dance with protons now. Antioxid Redox Signal. 2016;25(9):550–551. doi: 10.1089/ars.2016.6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang W, et al. Mitochondrial Flash: integrative reactive oxygen species and pH signals in cell and organelle biology. Antioxid Redox Signal. 2016;25(9):534–549. doi: 10.1089/ars.2016.6739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang H, et al. Reduction of elevated proton leak rejuvenates mitochondria in the aged cardiomyocyte. Elife. 2020:9. [DOI] [PMC free article] [PubMed]
- 21.Llopis J, et al. Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc Natl Acad Sci U S A. 1998;95(12):6803–6808. doi: 10.1073/pnas.95.12.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han J, Burgess K. Fluorescent indicators for intracellular pH. Chem Rev. 2010;110(5):2709–2728. doi: 10.1021/cr900249z. [DOI] [PubMed] [Google Scholar]
- 23.Cao L, et al. In vivo observation of the pH alternation in mitochondria for various external stimuli. Chem Commun (Camb) 2015;51(97):17324–17327. doi: 10.1039/c5cc07118f. [DOI] [PubMed] [Google Scholar]
- 24.Lee MH, et al. Mitochondria-immobilized pH-sensitive off-on fluorescent probe. J Am Chem Soc. 2014;136(40):14136–14142. doi: 10.1021/ja506301n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abad MF, et al. Mitochondrial pH monitored by a new engineered green fluorescent protein mutant. J Biol Chem. 2004;279(12):11521–11529. doi: 10.1074/jbc.M306766200. [DOI] [PubMed] [Google Scholar]
- 26.Casey JR, Grinstein S, Orlowski J. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol. 2010;11(1):50–61. doi: 10.1038/nrm2820. [DOI] [PubMed] [Google Scholar]
- 27.Divakaruni AS, Brand MD. The regulation and physiology of mitochondrial proton leak. Physiology (Bethesda) 2011;26(3):192–205. doi: 10.1152/physiol.00046.2010. [DOI] [PubMed] [Google Scholar]
- 28.Brookes PS, Rolfe DF, Brand MD. The proton permeability of liposomes made from mitochondrial inner membrane phospholipids: comparison with isolated mitochondria. J Membr Biol. 1997;155(2):167–174. doi: 10.1007/s002329900168. [DOI] [PubMed] [Google Scholar]
- 29.Brookes PS, Hulbert AJ, Brand MD. The proton permeability of liposomes made from mitochondrial inner membrane phospholipids: no effect of fatty acid composition. Biochim Biophys Acta. 1997;1330(2):157–164. doi: 10.1016/s0005-2736(97)00160-0. [DOI] [PubMed] [Google Scholar]
- 30.Ricquier D, Bouillaud F. The uncoupling protein homologues: UCP1, UCP2, UCP3, StUCP and AtUCP. Biochem J. 2000;345(Pt 2):161–179. [PMC free article] [PubMed] [Google Scholar]
- 31.Stuart JA, et al. Mitochondrial proton leak and the uncoupling proteins. J Bioenerg Biomembr. 1999;31(5):517–525. doi: 10.1023/a:1005456725549. [DOI] [PubMed] [Google Scholar]
- 32.Nicholls DG. Mitochondrial proton leaks and uncoupling proteins. Biochim Biophys Acta Bioenerg. 2021;1862(7):148428. doi: 10.1016/j.bbabio.2021.148428. [DOI] [PubMed] [Google Scholar]
- 33.Ruprecht JJ, et al. The molecular mechanism of transport by the mitochondrial ADP/ATP carrier. Cell. 2019;176(3):435–447. doi: 10.1016/j.cell.2018.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pebay-Peyroula E, et al. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature. 2003;426(6962):39–44. doi: 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]
- 35.Brand MD, et al. The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem J. 2005;392(Pt 2):353–362. doi: 10.1042/BJ20050890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parker N, et al. Uncoupling protein-1 (UCP1) contributes to the basal proton conductance of brown adipose tissue mitochondria. J Bioenerg Biomembr. 2009;41(4):335–342. doi: 10.1007/s10863-009-9232-8. [DOI] [PubMed] [Google Scholar]
- 37.Monemdjou S, Kozak LP, Harper ME. Mitochondrial proton leak in brown adipose tissue mitochondria of Ucp1-deficient mice is GDP insensitive. Am J Physiol. 1999;276(6):E1073–E1082. doi: 10.1152/ajpendo.1999.276.6.E1073. [DOI] [PubMed] [Google Scholar]
- 38.Bertholet AM, et al. Mitochondrial uncouplers induce proton leak by activating AAC and UCP1. Nature. 2022;606(7912):180–187. doi: 10.1038/s41586-022-04747-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bellanti F, et al. Many faces of mitochondrial uncoupling during age: damage or defense? J Gerontol - Biol Sci. 2013;68(8):892–902. doi: 10.1093/gerona/gls332. [DOI] [PubMed] [Google Scholar]
- 40.Harper ME, et al. Age-related increase in mitochondrial proton leak and decrease in ATP turnover reactions in mouse hepatocytes. Am J Physiol. 1998;275(2 Pt 1):E197–E206. doi: 10.1152/ajpendo.1998.275.2.E197. [DOI] [PubMed] [Google Scholar]
- 41.Asami DK, et al. Effect of aging, caloric restriction, and uncoupling protein 3 (UCP3) on mitochondrial proton leak in mice. Exp Gerontol. 2008;43(12):1069–1076. doi: 10.1016/j.exger.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crescenzo R, et al. Alterations in proton leak, oxidative status and uncoupling protein 3 content in skeletal muscle subsarcolemmal and intermyofibrillar mitochondria in old rats. BMC Geriatr. 2014;14:79. doi: 10.1186/1471-2318-14-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lal SB, et al. Effects of caloric restriction on skeletal muscle mitochondrial proton leak in aging rats. J Gerontol A Biol Sci Med Sci. 2001;56(3):B116–B122. doi: 10.1093/gerona/56.3.b116. [DOI] [PubMed] [Google Scholar]
- 44.Amaral S, et al. Testicular aging involves mitochondrial dysfunction as well as an increase in UCP2 levels and proton leak. FEBS Lett. 2008;582(30):4191–4196. doi: 10.1016/j.febslet.2008.11.020. [DOI] [PubMed] [Google Scholar]
- 45.Sakamuri SS, et al. Aging related impairment of brain microvascular bioenergetics involves oxidative phosphorylation and glycolytic pathways. J Cereb Blood Flow Metab. 2022;42(8):1410–1424. doi: 10.1177/0271678X211069266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Philipp E, Portner HO, Abele D. Mitochondrial ageing of a polar and a temperate mud clam. Mech Ageing Dev. 2005;126(5):610–619. doi: 10.1016/j.mad.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 47.Barazzoni R, Nair KS. Changes in uncoupling protein-2 and -3 expression in aging rat skeletal muscle, liver, and heart. Am J Physiol Endocrinol Metab. 2001;280(3):E413–E419. doi: 10.1152/ajpendo.2001.280.3.E413. [DOI] [PubMed] [Google Scholar]
- 48.Brand MD. Uncoupling to survive? The role of mitochondrial inefficiency in ageing. Exp Gerontol. 2000;35(6-7):811–820. doi: 10.1016/s0531-5565(00)00135-2. [DOI] [PubMed] [Google Scholar]
- 49.Speakman JR, et al. Uncoupled and surviving: individual mice with high metabolism have greater mitochondrial uncoupling and live longer. Aging Cell. 2004;3(3):87–95. doi: 10.1111/j.1474-9728.2004.00097.x. [DOI] [PubMed] [Google Scholar]
- 50.Fridell YW, et al. Targeted expression of the human uncoupling protein 2 (hUCP2) to adult neurons extends life span in the fly. Cell Metab. 2005;1(2):145–152. doi: 10.1016/j.cmet.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 51.Koziel A, Sobieraj I, Jarmuszkiewicz W. Increased activity of mitochondrial uncoupling protein 2 improves stress resistance in cultured endothelial cells exposed in vitro to high glucose levels. Am J Physiol Heart Circ Physiol. 2015;309(1):H147–H156. doi: 10.1152/ajpheart.00759.2014. [DOI] [PubMed] [Google Scholar]
- 52.Wu B, et al. 2,4 DNP improves motor function, preserves medium spiny neuronal identity, and reduces oxidative stress in a mouse model of Huntington's disease. Exp Neurol. 2017;293:83–90. doi: 10.1016/j.expneurol.2017.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dorighello GG, et al. Mild mitochondrial uncoupling decreases experimental atherosclerosis, a proof of concept. J Atheroscler Thromb. 2022;29(6):825–838. doi: 10.5551/jat.62796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goldgof M, et al. The chemical uncoupler 2,4-dinitrophenol (DNP) protects against diet-induced obesity and improves energy homeostasis in mice at thermoneutrality. J Biol Chem. 2014;289(28):19341–19350. doi: 10.1074/jbc.M114.568204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caldeira da Silva CC, et al. Mild mitochondrial uncoupling in mice affects energy metabolism, redox balance and longevity. Aging Cell. 2008;7(4):552–560. doi: 10.1111/j.1474-9726.2008.00407.x. [DOI] [PubMed] [Google Scholar]
- 56.Humphrey DM, et al. Expression of human uncoupling protein-3 in Drosophila insulin-producing cells increases insulin-like peptide (DILP) levels and shortens lifespan. Exp Gerontol. 2009;44(5):316–327. doi: 10.1016/j.exger.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Andrews ZB, Horvath TL. Uncoupling protein-2 regulates lifespan in mice. Am J Physiol Endocrinol Metab. 2009;296(4):E621–E627. doi: 10.1152/ajpendo.90903.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McDonald RB, et al. Characterization of survival and phenotype throughout the life span in UCP2/UCP3 genetically altered mice. Exp Gerontol. 2008;43(12):1061–1068. doi: 10.1016/j.exger.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 59.Grundlingh J, et al. 2,4-dinitrophenol (DNP): a weight loss agent with significant acute toxicity and risk of death. J Med Toxicol. 2011;7(3):205–212. doi: 10.1007/s13181-011-0162-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nadtochiy SM, Tompkins AJ, Brookes PS. Different mechanisms of mitochondrial proton leak in ischaemia/reperfusion injury and preconditioning: implications for pathology and cardioprotection. Biochem J. 2006;395(3):611–618. doi: 10.1042/BJ20051927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chavez JD, et al. Mitochondrial protein interaction landscape of SS-31. Proc Natl Acad Sci U S A. 2020;117(26):15363–15373. doi: 10.1073/pnas.2002250117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chiao YA, et al. Late-life restoration of mitochondrial function reverses cardiac dysfunction in old mice. Elife. 2020;9:e55513. doi: 10.7554/eLife.55513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Berry BJ, Vodičková A, Müller-Eigner A, Meng C, Ludwig C, Kaeberlein M, Peleg S, Wojtovich AP. Optogenetic rejuvenation of mitochondrial membrane potential extends C. elegans lifespan. Nat Aging. 2022;3:157–161. doi: 10.1038/s43587-022-00340-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mitteldorf J. What is antagonistic pleiotropy? Biochemistry (Mosc) 2019;84(12):1458–1468. doi: 10.1134/S0006297919120058. [DOI] [PubMed] [Google Scholar]
- 65.Gomez LA, et al. Supercomplexes of the mitochondrial electron transport chain decline in the aging rat heart. Arch Biochem Biophys. 2009;490(1):30–35. doi: 10.1016/j.abb.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lenaz G, et al. Mitochondrial complex I defects in aging. Mol Cell Biochem. 1997;174(1-2):329–333. [PubMed] [Google Scholar]
- 67.Lesnefsky EJ, et al. Aging decreases electron transport complex III activity in heart interfibrillar mitochondria by alteration of the cytochrome c binding site. J Mol Cell Cardiol. 2001;33(1):37–47. doi: 10.1006/jmcc.2000.1273. [DOI] [PubMed] [Google Scholar]
- 68.Paradies G, et al. Age-dependent decline in the cytochrome c oxidase activity in rat heart mitochondria: role of cardiolipin. FEBS Lett. 1997;406(1-2):136–138. doi: 10.1016/s0014-5793(97)00264-0. [DOI] [PubMed] [Google Scholar]
- 69.Guerrieri F, et al. Age-dependent changes in the mitochondrial F0F1 ATP synthase. Arch Gerontol Geriatr. 1992;14(3):299–308. doi: 10.1016/0167-4943(92)90029-4. [DOI] [PubMed] [Google Scholar]
- 70.Davies SM, et al. Measurements of protein carbonyls, ortho- and meta-tyrosine and oxidative phosphorylation complex activity in mitochondria from young and old rats. Free Radic Biol Med. 2001;31(2):181–190. doi: 10.1016/s0891-5849(01)00576-7. [DOI] [PubMed] [Google Scholar]