

Abstract
Intravenous remdesivir (RDV) is US Food and Drug Administration–approved for hospitalized and nonhospitalized individuals with coronavirus disease 2019. RDV undergoes intracellular metabolic activation to form the active triphosphate, GS‐443902, and other metabolites. Alternative administration routes, including localized pulmonary delivery, can lower systemic exposure and maximize exposure at the site of action. This study evaluated the pharmacokinetics (PK) and safety of inhaled RDV in healthy adults. This phase Ia, randomized, placebo‐controlled study evaluated inhaled RDV in healthy participants randomized 4:1 to receive RDV or placebo as single doses (4 cohorts) or multiple once‐daily doses (3 cohorts). Doses in cohorts 1–6 were administered as an aerosolized solution for inhalation through a sealed facemask; doses in cohort 7 were administered as an aerosolized solution for inhalation through a mouthpiece. Safety was assessed throughout the study. Seventy‐two participants were enrolled (inhaled RDV, n = 58 and placebo, n = 14). Following single RDV doses, RDV, GS‐704277, and GS‐441524 plasma PK parameters indicated dose‐proportional increases in area under the concentration‐time curve (AUC) extrapolated to infinite time, AUC from time zero to last quantifiable concentration, and maximum observed concentration. Analyte plasma concentrations after multiple RDV doses were consistent with those for single‐dose RDV. Analyte plasma exposures were lower when RDV was administered with a mouthpiece versus a sealed facemask. The most common adverse events included nausea, dizziness, and cough. Single‐ and multiple‐dose inhaled RDV exhibited linear and dose‐proportional plasma PK. Administration of RDV via inhalation was generally safe and well‐tolerated.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Intravenous remdesivir (RDV) is currently approved by the US Food and Drug Administration for the treatment of coronavirus disease 2019 (COVID‐19) in hospitalized adult and pediatric patients, as well as nonhospitalized patients with mild‐to‐moderate COVID‐19 who are at high risk for progression to severe COVID‐19. Identifying viable alternative routes of administration, such as localized inhalation, is important to expand the accessibility of RDV to a broader outpatient population.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study aimed to evaluate the pharmacokinetics (PK), safety, and tolerability of single and multiple ascending doses of RDV administered via inhalation to healthy adult participants.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
RDV administered via inhalation exhibited linear and dose‐proportional plasma PK, with multiple‐dose RDV PK results consistent with those of single‐dose RDV. Administration of inhaled RDV was generally safe and well‐tolerated.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Administering RDV via inhalation delivers medication directly to the primary site of infection in the respiratory tract, reducing overall systemic exposure.
INTRODUCTION
Intravenous (i.v.) remdesivir (RDV) is approved by the US Food and Drug Administration for the treatment of coronavirus disease 2019 (COVID‐19), which primarily affects the respiratory tract in hospitalized adult and pediatric patients. RDV is also approved for nonhospitalized patients with mild‐to‐moderate COVID‐19 who are at high risk for progression to severe COVID‐19, including hospitalization or death. 1 RDV is an adenosine nucleotide prodrug that undergoes intracellular activation to form GS‐443902, an analog of adenosine triphosphate, which inhibits viral RNA polymerases, thereby inhibiting viral replication. GS‐443902 can also inhibit viral replication via incorporation into viral RNA as a result of viral polymerase read‐through that may occur at higher nucleotide concentrations. 2 , 3 , 4 Intracellular activation of RDV in the pulmonary parenchyma begins with hydrolase cleavage via carboxylesterases (cathepsin A [CatA] and carboxylesterase 1 [CES1]) to form the intermediate metabolite, GS‐704277. 3 , 5 Subsequently, the phosphoramidate bond of GS‐704277 is cleaved by histidine triad nucleotide‐binding protein 1 (HINT1) to form the nucleoside analog monophosphate, which is then further phosphorylated to the pharmacologically active nucleoside triphosphate, GS‐443902. Each of these key enzymes for RDV metabolism (CatA, CES1, and HINT1) are highly expressed in the human lung. 3 Dephosphorylation of the nucleoside analog monophosphate results in the formation of the nucleoside analog, GS‐441524, which is not efficiently rephosphorylated. RDV and its metabolites (GS‐441524 and GS‐704277) are detectable in plasma. The antiviral efficacy of RDV is determined by the intracellular lung concentration of the active triphosphate metabolite, GS‐443902. 6 , 7 , 8 In current clinical studies, peripheral blood mononuclear cells (PBMCs) are used as a clinical surrogate for pulmonary parenchyma to assess GS‐443902 concentrations. 2
In two phase I studies evaluating single and multiple ascending doses of RDV in healthy participants, RDV administered via i.v. infusion exhibited dose‐proportional pharmacokinetics (PK). High concentrations of GS‐443902 were achieved in PBMCs, following either a 30‐min or a 2‐h infusion (220‐ to 370‐fold above the in vitro half‐maximal effective concentration against severe acute respiratory syndrome coronavirus 2 [SARS‐CoV‐2; 9.9 nM]). 2 Following multiple doses of 150 mg RDV once daily across 7 to 14 days, the metabolite GS‐441524 accumulated ~1.9‐fold and reached steady‐state by day 4 (consistent with a half‐life of ~24.5 h). Overall, RDV demonstrated favorable safety and PK profiles supporting once‐daily dosing recommendations. 2
As an i.v. infusion, RDV use is limited to certain clinical settings. Identifying viable alternative routes of administration is important to expand the accessibility of RDV to a broader nonhospitalized population. Localized pulmonary delivery could also provide rapid and direct antiviral activity in the respiratory tract, which is the target of SARS‐CoV‐2 infection, while achieving lower systemic drug exposure. The PK evaluations in nonhuman primates (NHPs) showed that 20‐fold lower inhaled RDV doses produced similar concentrations of the active triphosphate (GS‐443902) in pulmonary parenchyma as the equivalent clinical i.v. RDV doses. 4 The systemic and PBMC RDV exposures in NHPs with doses administered via inhalation that matched the GS‐443902 levels observed in the lungs were ~50‐fold lower than with i.v. administration. Moreover, repeated dosing of inhaled RDV in an African green monkey model of SARS‐CoV‐2 infection demonstrated reductions in viral replication in bronchoalveolar lavage fluid and respiratory tract tissues compared with placebo. The aim of the current clinical study, initiated early in the COVID‐19 pandemic (June 2020), was to evaluate the PK, safety, and tolerability of single and multiple ascending doses of RDV administered via inhalation to healthy adult participants. As guided by the nonclinical evaluations, estimated lung doses of up to 10 mg RDV of either the i.v. formulation (RDV solution for injection) or the new inhalation formulation (RDV solution for inhalation) were assessed in this study.
METHODS
Study design
This was a phase Ia, randomized, placebo‐controlled study with staggered dose escalation and adaptive dose selection to evaluate the safety and tolerability of RDV administered via inhalation in healthy participants from June 25, 2020 (first participant first visit) to March 12, 2021 (last participant last visit). Ascending single and multiple doses of inhaled RDV were administered to cohorts 1 to 5, followed by assessment of an additional formulation (cohort 6) and device configuration (cohort 7).
Following screening and admission assessments, participants were randomized 4:1 to receive either RDV or placebo as single doses (cohorts 1, 2, 4, and 7) or multiple once‐daily doses (cohorts 3, 5, and 6). Dosing in each single‐dose cohort was staggered, with two participants (one active and one placebo) dosed 24 h before additional participants were dosed. Cohorts 1 to 5 received the RDV for injection formulation, whereas cohorts 6 and 7 received the RDV for inhalation formulation. Doses in cohorts 1 to 6 were administered as an aerosolized solution for inhalation using a sealed facemask coupled with a PARI LC Sprint valved jet nebulizer (PARI Respiratory Equipment, Inc.) with a single inlet/outlet and exhaust filter. Doses in cohort 7 were administered as an aerosolized solution for inhalation using a mouthpiece coupled with an LC Sprint nebulizer. The sulfobutylether‐β‐cyclodextrin sodium content was 7.5% after reconstitution in the RDV for inhalation formulation and 15% after reconstitution in the RDV for injection formulation. Single ascending dose escalation from cohort 1 to cohort 2 occurred upon safety evaluation through at least day 4 from cohort 1. Commencement of cohort 4 occurred upon safety evaluation through at least day 8 from cohort 3. Multiple ascending dose escalation from cohort 2 to cohort 3 and from cohort 4 to cohort 5 occurred upon safety evaluation through at least day 4 from cohorts 2 and 4, respectively. Commencement of cohorts 6 and 7 was determined upon completion of previous cohorts.
Study treatments, overall delivery efficiency, and deposited and nominal doses within each cohort are presented in Table 1. Safety and tolerability were assessed through incidences of adverse events (AEs), clinical laboratory abnormalities, and evaluations of electrocardiograms (ECGs), vital signs, and forced expiratory volume in 1 s (FEV1). A safety review team was established to assess safety, make decisions on dose escalation, and define the maximum tolerated dose, if applicable. Eligible participants were healthy, nonsmoking participants aged 18 to 45 years (inclusive), with a body mass index (BMI) greater than or equal to 19.0 and less than or equal to 30.0 kg/m2, a creatinine clearance greater than or equal to 90 mL/min, an FEV1 greater than 80% of predicted, and an FEV1/forced vital capacity (FVC) ratio greater than 70%. Exclusion criteria included abnormal vital signs at screening, defined as systolic blood pressure less than 90 mmHg or greater than or equal to 140 mmHg, diastolic blood pressure less than 50 mmHg or greater than or equal to 90 mmHg, or heart rate less than 50 or greater than 90 beats per minute. Pregnant or breastfeeding participants were excluded.
TABLE 1.
Mode of administration and device efficiency by cohort.
| Test product | Device | Cohort | Nominal dose (estimated lung dose, overall delivery efficiency) | Mode of administration |
|---|---|---|---|---|
| RDV for injection formulation (containing 15% SBECD after reconstitution) | Sealed facemask coupled with an LC Sprint nebulizer with a single inlet/outlet and exhaust filter | 1 | 12.5 mg (2 mg, 16.1%) | Inhalation for ~7 min in the morning for 1 day |
| 2 | 31 mg (5 mg, 16.1%) | Inhalation for ~17 min in the morning for 1 day | ||
| 3 | 31 mg (5 mg, 16.1%) | Inhalation for ~17 min once daily in the morning for 5 days | ||
| 4 | 62 mg (10 mg, 16.1%) | Inhalation for ~34 min in the morning for 1 day | ||
| 5 | 62 mg (10 mg, 16.1%) | Inhalation for ~34 min once daily in the morning for 5 days | ||
| RDV for inhalation formulation (containing 7.5% SBECD after reconstitution) | Sealed facemask coupled with an LC Sprint nebulizer with a single inlet/outlet and exhaust filter | 6 | 60 mg (10.4 mg, 17%) | Inhalation for ~24 min once daily in the morning for 5 days |
| Mouthpiece coupled with an LC Sprint nebulizer | 7 | 39 mg (10 mg, 25.6%) | Inhalation for ~19 min in the morning for 1 day |
Abbreviations: RDV, remdesivir; SBECD, sulfobutylether β‐cyclodextrin sodium.
The protocol, consent forms, study participant information sheets, and advertisements were submitted by the investigator to a duly constituted institutional review board for review and approval before study initiation. The study was conducted under a US investigational new drug application and in accordance with recognized international scientific and ethical standards including, but not limited to, the International Council for Harmonization guideline for Good Clinical Practice and the original principles embodied in the Declaration of Helsinki.
Dose selection
Based on the results of a 7‐day rat non–Good Laboratory Practice local tolerance study, the no observed adverse effect level for the RDV for injection reconstituted solution administered by inhalation was 10.6 mg/kg presented dose (assuming 10% deposition), which corresponds to a deposited lung dose of 1.06 mg/kg of body weight or 0.154 mg/g of lung weight, estimated from actual body weight and lung weight derived from the 7‐day rat study. The average vehicle mass median aerodynamic diameter of the generated aerosol in that study was 3.04 μm, with an average geometric standard deviation (SD) of 1.67. The starting dose for the RDV for injection formulation evaluation in cohort 1 of this study was a single inhaled nominal dose of 12.5 mg (2 mg estimated lung dose given the 16.1% delivery efficiency of an LC Sprint equipped with a sealed facemask). Assuming 100% deposition of the nominal dose in the human lung, this dose would result in an estimated lung dose of 0.0125 mg/g of lung weight (assuming a 70 kg human with 1000 g lung weight), corresponding to a 12‐fold safety margin relative to the no observed adverse effect level (NOAEL). Doses of 31 mg, administered as a single dose or as 5‐day multiple doses, were evaluated in cohorts 2 and 3, respectively, and were expected to result in a safety margin of 5.0‐fold. Escalation to a nominal dose of 62 mg (10 mg estimated lung dose given an overall efficiency of 16.1% for the LC Sprint equipped with a sealed facemask; cohorts 4 and 5) was supported by results from the 14‐day Good Laboratory Practice toxicology studies in rats, which demonstrated a lack of AEs with a safety margin of 2.6‐fold. The doses evaluated in cohorts 6 and 7 were 60 and 39 mg nominal doses (10.4 and 10 mg estimated lung doses), respectively.
Bioanalytical procedures
The bioanalytical method for determining RDV and GS‐441524 in formic acid–treated human plasma was validated at QPS, LLC. This method involved protein precipitation extraction of RDV and GS‐441524 and stable isotype‐labeled internal standards GS‐829143 ([13C3]‐RDV) and GS‐828840 ([13C3]‐GS‐441524) from human plasma, followed by liquid chromatography–tandem mass spectrometry (LC–MS/MS). The calibration range was 0.2 to 200 ng/mL for RDV and 0.4 to 400 ng/mL for GS‐441524. The bioanalytical method for determination of GS‐704277 in formic acid–treated human plasma was also validated at QPS, LLC. Similarly, this method involved protein precipitation extraction of GS‐704277 and internal standard GS‐829466 ([13C3]‐GS‐704277) from human plasma, followed by LC–MS/MS with a calibration range of 2 to 2000 ng/mL. All samples were analyzed within the time frame supported by long‐term storage stability data.
PK analyses
Plasma concentrations and PK parameters (area under the concentration‐time curve [AUC] extrapolated to infinite time [AUCinf; single dose], AUC from time zero to the last quantifiable concentration [AUClast], apparent oral clearance [single dose], apparent oral clearance at steady‐state [multiple dose], terminal elimination half‐life, apparent volume of distribution, maximum observed concentration [C max], time [observed timepoint] of maximum observed concentration [T max], last observed quantifiable concentration [C last], time [observed timepoint] of C last, AUC over the dosing interval [AUCtau; multiple dose], AUC from time zero to 24 h [multiple dose], and observed concentration at the end of the dosing interval [C tau; multiple dose]) were summarized by cohort for all analytes using descriptive statistics.
Dose proportionality was evaluated based on AUClast, AUCinf, and C max values obtained on day 1 (single‐dose cohorts 1, 2, and 4) using both a power model and an analysis of variance (ANOVA) model. PK parameters of RDV and its metabolites using the RDV for injection and RDV for inhalation formulations were compared using an ANOVA model. PK parameters from the RDV for injection formulation were used as a reference; the test‐to‐reference ratio and the 90% confidence interval (CI) of the PK parameters between cohorts 5 and 6 were provided. An ANOVA model was also used to compare PK parameters of RDV and its metabolites achieved between the facemask and the mouthpiece; the test‐to‐reference ratio and the 90% CI of the PK parameters were compared between cohorts 4 and 7.
Safety and tolerability
Safety and tolerability were assessed throughout the study and included treatment‐emergent AEs (TEAEs), serious AEs (SAEs), vital signs, ECG, clinical laboratory analyses, and spirometry. TEAEs and SAEs were coded according to the Medical Dictionary for Regulatory Activities, version 24.0. The severity of TEAEs was graded using the Division of AIDS Table for Grading the Severity of Adult and Pediatric Adverse Events, version 2.1. Spirometry assessments were performed according to American Thoracic Society standardization. 9 Participants who developed a postdose decline in FEV1 of greater than 15% compared with the same‐day predose baseline values were monitored until their spirometry results returned to the predose baseline and all signs/symptoms resolved. Any participant who had a predose FEV1 value less than 80% of predicted did not receive study treatment on that day.
RESULTS
Participant disposition and demographics
Among 72 participants enrolled in the study, 58 (cohorts 1–6, n = 8 each and cohort 7, n = 10) received RDV via inhalation and 14 received placebo (Table 2). Across all cohorts, the mean (SD) age was 27 (7.1) years. The majority of participants were men (51.4%); 90.3% of participants were White, and 84.7% were not Hispanic or Latino. The mean (SD) BMI at baseline was 24.1 (2.9) kg/m2.
TABLE 2.
Participant disposition and baseline demographics.
| Characteristic | Cohort 1 2 mg RDV SAD (n = 8) | Cohort 2 5 mg RDV SAD (n = 8) | Cohort 3 5 mg RDV MAD (n = 8) | Cohort 4 10 mg RDV SAD (n = 8) | Cohort 5 10 mg RDV MAD (n = 8) | Cohort 6 10.4 mg RDV MAD inhalation formulation (n = 8) | Cohort 7 10 mg RDV SAD mouthpiece (n = 10) | Placebo (n = 14) | Total (N = 72) |
|---|---|---|---|---|---|---|---|---|---|
| Age, years | |||||||||
| Mean (SD) | 30 (6.9) | 26 (7.8) | 28 (6.9) | 26 (5.4) | 28 (6.6) | 30 (8.8) | 25 (6.4) | 27 (8.3) | 27 (7.1) |
| Sex at birth, n (%) | |||||||||
| Male | 5 (62.5) | 3 (37.5) | 4 (50.0) | 4 (50.0) | 5 (62.5) | 5 (62.5) | 7 (70.0) | 4 (28.6) | 37 (51.4) |
| Female | 3 (37.5) | 5 (62.5) | 4 (50.0) | 4 (50.0) | 3 (37.5) | 3 (37.5) | 3 (30.0) | 10 (71.4) | 35 (48.6) |
| Race, n (%) | |||||||||
| American Indian or Alaska Native | 0 | 0 | 0 | 0 | 1 (12.5) | 0 | 0 | 0 | 1 (1.4) |
| Asian | 0 | 0 | 0 | 1 (12.5) | 0 | 0 | 0 | 0 | 1 (1.4) |
| Black | 0 | 0 | 0 | 0 | 1 (12.5) | 1 (12.5) | 0 | 0 | 2 (2.8) |
| White | 8 (100) | 8 (100) | 8 (100) | 7 (87.5) | 6 (75.0) | 6 (75.0) | 8 (80.0) | 14 (100) | 65 (90.3) |
| Other | 0 | 0 | 0 | 0 | 0 | 1 (12.5) | 2 (20.0) | 0 | 3 (4.2) |
| Ethnicity, n (%) | |||||||||
| Hispanic or Latino | 0 | 1 (12.5) | 2 (25.0) | 2 (25.0) | 2 (25.0) | 1 (12.5) | 1 (10.0) | 2 (14.3) | 11 (15.3) |
| Not Hispanic or Latino | 8 (100) | 7 (87.5) | 6 (75.0) | 6 (75.0) | 6 (75.0) | 7 (87.5) | 9 (90.0) | 12 (85.7) | 61 (84.7) |
| BMI, kg/m2, mean (SD) | 25.2 (2.4) | 24.1 (3.5) | 26.2 (2.5) | 24.4 (3.0) | 23.2 (3.1) | 23.5 (2.8) | 24.0 (3.5) | 23.0 (1.8) | 24.1 (2.9) |
| FEV1/FVC, mean (SD) | 82.0 (5.3) | 86.1 (2.9) | 83.3 (5.2) | 82.2 (4.9) | 82.6 (6.5) | 78.9 (8.6) | 84.2 (6.3) | 80.8 (6.1) | NA |
Abbreviations: BMI, body mass index; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; MAD, multiple ascending dose; NA, not available; RDV, remdesivir; SAD, single ascending dose.
PK of single ascending doses of RDV, GS‐704277, and GS‐441524
Overall, the plasma PK profiles of RDV, GS‐704277, and GS‐441524 showed similar terminal elimination phases across the studied dose range. Following single RDV estimated lung doses of 2, 5, and 10 mg, participants had detectable RDV concentrations up to 1.1, 1.8, and 2.6 h postdose, respectively (Figure 1). Participants had detectable GS‐704277 concentrations up to 2.0, 2.3, and 2.6 h postdose and detectable GS‐441524 concentrations up to 24, 48, and 72 h (last PK sampling timepoint) postdose, respectively.
FIGURE 1.
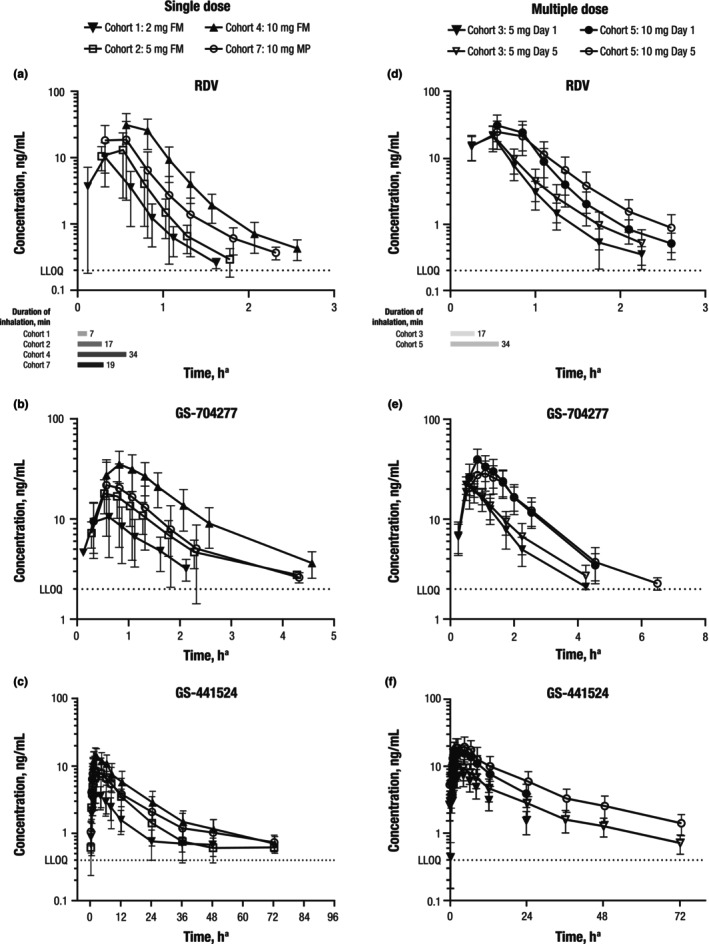
Mean (SD) plasma concentration versus time for (a) RDV, (b) GS‐704277, and (c) GS‐441524 for single‐dose RDV and (d) RDV, (e) GS‐704277, and (f) GS‐441524 for multiple‐dose RDV administered via inhalation. FM, facemask; LLOQ, lower limit of quantification; MP, mouthpiece; RDV, remdesivir. Error bars denote standard deviations. aTime from start of nebulization.
Plasma PK parameters for RDV, GS‐704277, and GS‐441524 are presented in Table 3. Results of ANOVA and power model analyses indicated approximately dose‐proportional increases in AUCinf, AUClast, and C max of RDV, GS‐704277, and GS‐441524 across the studied dose range (Tables S1 and S2). The median T max was between 0.28 and 0.58 h for RDV, 0.50 and 0.84 h for GS‐704277, and 2.08 and 4.24 h for GS‐441524.
TABLE 3.
PK parameters of RDV, GS‐704277, and GS‐441524 after single‐ or multiple‐dose RDV administered by inhalation.
| PK parameter a | Cohort 1 2 mg RDV SAD (n = 8) | Cohort 2 5 mg RDV SAD (n = 8) | Cohort 4 10 mg RDV SAD (n = 8) | Cohort 3 5 mg RDV MAD (n = 8) | Cohort 5 10 mg RDV MAD (n = 8) | Cohort 6 10.4 mg RDV MAD (n = 8) | |||
|---|---|---|---|---|---|---|---|---|---|
| Day 1 | Day 5 | Day 1 | Day 5 | Day 1 | Day 5 | ||||
| RDV | |||||||||
| AUCinf, h·ng/mL | 4.4 (59.7) | 7.4 (58.9) | 23.4 (49.8) | – | – | – | – | – | – |
| AUClast, h·ng/mL | 4.3 (62.0) | 7.2 (59.6) | 23.2 (49.9) | – | – | – | – | 23.2 (31.2) | 30.2 (57.5) |
| AUC, h·ng/mL b | – | – | – | 12.7 (41.3) | 15.5 (38.6) | 23.9 (44.2) | 23.0 (48.4) | 23.5 (31.3) | 30.5 (57.1) |
| C max, ng/mL | 10.2 (64.4) | 14.4 (69.7) | 31.1 (48.6) | 21.7 (41.6) | 23.3 (39.1) | 31.9 (43.4) | 25.6 (45.7) | 33.6 (29.2) | 35.0 (40.4) |
| T max, h | 0.35 (0.34, 0.36) | 0.28 (0.25, 0.50) | 0.58 (0.54, 0.61) | 0.50 (0.48, 0.53) | 0.50 (0.48, 0.55) | 0.59 (0.57, 0.63) | 0.61 (0.52, 0.71) | 0.60 (0.43, 0.67) | 0.57 (0.43, 0.75) |
| T last, h | 1.12 (1.10, 1.59) | 1.75 (1.28, 1.75) | 2.58 (2.29, 2.61) | 2.02 (1.74, 2.28) | 2.23 (2.23, 2.30) | 2.58 (2.54, 2.60) | 2.56 (2.49, 2.61) | – | – |
| C last, ng/mL | 0.4 (34.7) | 0.4 (41.4) | 0.4 (35.9) | 0.3 (28.0) | 0.5 (59.6) | 0.5 (40.0) | 0.9 (58.2) | – | – |
| t 1/2, h | 0.23 (0.19, 0.24) | 0.28 (0.23, 0.29) | 0.41 (0.35, 0.44) | 0.30 (0.26, 0.42) | 0.41 (0.38, 0.45) | 0.48 (0.45, 0.52) | 0.47 (0.46, 0.50) | 0.46 (0.38, 0.48) | 0.42 (0.37, 0.60) |
| CL/F, mL/h | 586,000 (51.3) | 822,000 (37.2) | 595,000 (71.7) | – | – | – | – | – | – |
| CLss/F, mL/h | – | – | – | – | 364,000 (38.1) | – | 575,000 (66.2) | – | – |
| V z/F, mL | 226,000 (86.0) | 335,000 (55.1) | 327,000 (67.8) | – | 219,000 (42.6) | – | 407,000 (72.9) | – | – |
| GS‐704277 | |||||||||
| AUCinf, h·ng/mL | 17.2 (36.4) c | 28.6 (36.6) | 63.5 (42.2) | – | – | – | – | – | – |
| AUClast, h·ng/mL | 12.0 (56.4) | 23.4 (44.4) | 56.7 (45.9) | – | – | – | – | 69.3 (35.1) | 77.3 (55.8) |
| AUC, h·ng/mL b | – | – | – | 32.2 (29.6) | 38.5 (26.3) | 76.0 (26.6) | 68.6 (33.1) | 74.4 (35.4) | 82.7 (52.0) |
| C max, ng/mL | 10.6 (59.5) | 18.0 (36.6) | 35.3 (35.0) | 22.1 (19.9) | 20.4 (15.5) | 39.7 (26.7) | 30.0 (32.5) | 41.9 (40.2) | 37.7 (44.4) |
| T max, h | 0.60 (0.59, 0.61) | 0.50 (0.50, 0.53) | 0.84 (0.81, 0.88) | 0.50 (0.48, 0.53) | 0.73 (0.50, 0.78) | 0.84 (0.82, 0.88) | 0.99 (0.86, 1.08) | 0.67 (0.62, 0.68) | 0.93 (0.68, 1.05) |
| T last, h | 2.09 (1.84, 2.10) | 2.25 (2.25, 2.28) | 2.63 (2.56, 4.56) | 2.27 (2.24, 3.28) | 2.47 (2.23, 4.28) | 4.59 (4.57, 4.63) | 4.61 (4.56, 5.58) | – | – |
| C last, ng/mL | 3.1 (22.3) | 4.1 (24.2) | 5.6 (52.8) | 3.8 (40.6) | 4.3 (45.3) | 3.5 (30.3) | 3.0 (31.1) | – | – |
| t 1/2, h | 0.87 (0.63, 1.12) c | 0.88 (0.82, 1.05) | 0.80 (0.75, 0.98) | 0.77 (0.72, 0.91) | 1.13 (0.95, 1.14) | 1.12 (1.04, 1.16) | 1.06 (0.97, 1.21) | 1.08 (1.02, 1.15) | 1.14 (1.00, 1.23) |
| GS‐441524 | |||||||||
| AUCinf, h·ng/mL | 58.7 (42.4) | 129 (33.6) | 249 (40.8) | – | – | – | – | – | – |
| AUClast, h·ng/mL | 49.6 (46.9) | 119 (32.4) | 225 (41.6) | – | – | – | – | 183.3 (24.6) | 413.6 (29.6) |
| AUC, h·ng/mL b | – | – | – | 90.8 (30.4) | 141 (29.3) | 211 (31.5) | 273 (38.8) | 183.5 (24.6) | 277.0 (25.3) |
| C max, ng/mL | 3.9 (38.0) | 7.9 (19.5) | 15.3 (31.8) | 7.5 (28.4) | 10.7 (30.3) | 16.2 (28.7) | 20.2 (37.6) | 15.4 (29.0) | 20.2 (22.0) |
| T max, h | 3.11 (1.84, 4.10) | 4.24 (2.27, 4.25) | 2.08 (1.75, 2.33) | 3.25 (1.99, 4.27) | 2.30 (2.20, 4.23) | 2.29 (1.47, 3.63) | 3.53 (2.33, 4.57) | 1.98 (1.35, 2.42) | 4.37 (2.55, 4.43) |
| T last, h | 24.1 (24.1, 30.1) | 48.3 (42.2, 48.3) | 72.6 (48.6, 72.6) | 23.9 (23.9, 23.9) | 72.3 (72.2, 72.4) | 23.9 (23.9, 23.9) | 72.6 (72.5, 72.7) | – | – |
| C last, ng/mL | 0.6 (18.5) | 0.5 (12.8) | 0.7 (17.8) | 1.5 (37.6) | 0.7 (31.9) | 3.9 (37.9) | 1.4 (34.9) | – | – |
| t 1/2, h | 9.65 (8.51, 11.6) | 11.5 (10.1, 14.5) | 21.5 (16.8, 27.7) | – | 27.4 (25.0, 29.7) | – | 28.6 (24.6, 30.5) | – | 27.5 (19.9, 29.8) |
Abbreviations: %CV, percentage of coefficient of variation; AUC, area under the concentration‐time curve; AUC0‐24, AUC from time zero to 24 h; AUCinf, AUC extrapolated to infinite time; AUClast, AUC from time zero to the last quantifiable concentration; AUCtau, AUC over the dosing interval; CL/F, apparent oral clearance; C last, last observed quantifiable concentration; CLss/F, apparent oral clearance at steady‐state; C max, maximum observed concentration; MAD, multiple ascending dose; PK, pharmacokinetic; Q, quartile; RDV, remdesivir; SAD, single ascending dose; t 1/2, terminal elimination half‐life; T last, time (observed timepoint) of last observed quantifiable concentration; T max, time (observed timepoint) of maximum observed concentration; V z/F, apparent volume of distribution.
Data are presented as mean (%CV), except for T max, T last, and t 1/2, which are presented as median (Q1, Q3).
AUC0‐24 is presented for day 1, and AUCtau is presented for day 5.
There were seven participants; PK parameters are not reportable for one participant.
Systemic exposures of RDV administered via inhalation were substantially (~7‐ to 10‐fold) lower compared with i.v. RDV administration at the same dose level. An estimated lung dose of 10 mg RDV (cohort 4) in the current study yielded an RDV AUCinf of ~23 h*ng/mL compared with an RDV AUCinf of ~230 h*ng/mL for a 10 mg RDV dose administered i.v. in a prior study. 10 This observed decrease in systemic exposure of RDV when administered via inhalation suggests substantial extraction and/or first‐pass metabolism of RDV in the lungs. The AUCinf values of RDV metabolites were comparable between the i.v. and inhalation routes of administration, as the enzymes responsible for metabolism of RDV are known to be expressed in the lungs. The plasma exposures to RDV and metabolites were consistent with the nonclinical PK data. 3 The AUCinf of GS‐704277 following a 10 mg i.v. administration of RDV was ~30 h*ng/mL10 compared with an AUCinf of ~64 h*ng/mL following an estimated lung dose of 10 mg RDV (cohort 4) in the current study. The AUCinf of GS‐441524 following a 10 mg i.v. administration of RDV was ~264 h*ng/mL10 compared with an AUCinf of ~249 h*ng/mL following an estimated lung dose of 10 mg RDV (cohort 4) in the current study.
PK of multiple ascending doses of RDV, GS‐704277, and GS‐441524
Overall, the plasma concentrations of RDV, GS‐704277, and GS‐441524 after administration of multiple ascending doses of RDV by inhalation were consistent with those expected based on PK after single‐dose RDV (Figure 1; Figure S1). In all participants, RDV, GS‐704277, and GS‐441524 were detectable for 2.0 to 2.6, 2.3 to 4.6, and 23.9 to 72.6 h, respectively (Table 3). Consistent with the single‐dose half‐life of 0.23 to 0.41 h for RDV and 0.80 to 0.88 h for GS‐704277, no accumulation of either analyte was observed following 5 days of dosing. Modest accumulation of GS‐441524 was observed based on ratios for geometric least‐squares mean values following 5 days of dosing (1.3‐ to 1.5‐fold on AUC, 1.2‐ to 1.4‐fold on C max, and 1.5‐ to 1.9‐fold on C tau), consistent with the single‐dose half‐life of 9.65 to 21.5 h. Although the study was not powered to assess bioequivalence, a statistical comparison showed RDV, GS‐704277, and GS‐441524 exposures (AUCtau and C max) were similar regardless of the RDV formulation (Figure S1; Table S3).
Comparison of PK exposures between mouthpiece and sealed facemask administration
Statistical comparisons indicated that RDV, GS‐704277, and GS‐441524 exposures (AUCinf and C max) were lower when RDV was administered through a mouthpiece (cohort 7) versus a sealed facemask (cohort 4; Table 4). Higher variability in PK parameters was also observed when RDV was administered through a mouthpiece compared with a sealed facemask.
TABLE 4.
Inhaled single‐dose plasma PK parameters for RDV, GS‐704277, and GS‐441524 in cohort 7 (mouthpiece) compared with cohort 4 (facemask).
| Analyte PK parameter a | Cohort 7 10 mg RDV SAD, mouthpiece (n = 10) b | Cohort 4 10 mg RDV SAD, facemask (n = 8) | %GLSM ratio (cohort 7 [test] vs. cohort 4 [reference]; 90% CI) |
|---|---|---|---|
| RDV | |||
| AUCinf, h·ng/mL | 13.9 (74.8) | 23.4 (49.8) | 46.63 (20.95, 103.76) |
| AUClast, h·ng/mL | 13.8 (75.1) | 23.2 (49.9) | – |
| C max, ng/mL | 22.7 (72.7) | 31.1 (48.6) | 57.94 (26.38, 127.26) |
| T max, h | 0.43 (0.34, 0.58) | 0.58 (0.54, 0.61) | – |
| t 1/2, h | 0.32 (0.20, 0.37) | 0.41 (0.35, 0.44) | – |
| GS‐704277 | |||
| AUCinf, h·ng/mL | 39.7 (61.3) c | 63.5 (42.2) | 58.67 (35.35, 97.38) |
| AUClast, h·ng/mL | 31.9 (78.3) | 56.7 (45.9) | – |
| C max, ng/mL | 24.7 (65.8) | 35.3 (35.0) | 57.97 (32.04, 104.87) |
| T max, h | 0.60 (0.58, 0.67) | 0.84 (0.81, 0.88) | – |
| t 1/2, h | 0.72 (0.54, 0.92) c | 0.80 (0.75, 0.98) | – |
| GS‐441524 | |||
| AUCinf, h·ng/mL | 181.8 (71.6) | 248.7 (40.8) | 55.04 (25.50, 118.83) |
| AUClast, h·ng/mL | 163.7 (72.7) | 225.4 (41.6) | – |
| C max, ng/mL | 9.8 (64.3) | 15.3 (31.8) | 49.36 (24.70, 98.64) |
| T max, h | 1.81 (1.08, 2.09) | 2.08 (1.75, 2.33) | – |
| t 1/2, h | 18.00 (13.35, 26.42) | 21.48 (16.77, 27.68) | – |
Abbreviations: %GLSM, percentage of geometric least‐squares mean; AUCinf, area under the concentration‐time curve extrapolated to infinite time; AUClast, area under the concentration‐time curve from time zero to the last quantifiable concentration; CI, confidence interval; C max, maximum observed concentration; PK, pharmacokinetic; Q, quartile; RDV, remdesivir; SAD, single ascending dose; t 1/2, terminal elimination half‐life; T max, time (observed timepoint) of maximum observed concentration.
Data are presented as mean (%CV), except for T max and t 1/2, which are presented as median (Q1, Q3).
There were eight participants; two participants in cohort 7 experienced issues during dosing (excessive foaming obstructing mouthpiece) that resulted in dosing interruption or incomplete dosing.
There were seven participants; PK parameters were not estimable for one additional participant.
Safety
In total, 31 (43.1%) participants experienced greater than or equal to one TEAE (Table 5). Of these participants, 23 (74.2%) received RDV and 8 (25.8%) received placebo. No participants in the study died, experienced a nonfatal treatment‐emergent SAE, experienced a grade 3 or 4 TEAE, had study treatment discontinued due to a TEAE, or discontinued the study due to a TEAE. The percentage of participants with TEAEs was lower in cohorts receiving single doses of the RDV for injection formulation through a sealed facemask (cohorts 1 and 2, 1 [12.5%] participant each; cohort 4, 3 [37.5%] participants) compared with the cohort receiving a single dose of the RDV for inhalation formulation through a mouthpiece (cohort 7, 5 [50.0%] participants). In the cohorts receiving multiple doses of the RDV for injection formulation through a sealed facemask, the percentage of participants with TEAEs was the same for cohorts 3 and 5 (4 [50.0%] participants each) but slightly higher in cohort 6 (5 [62.5%] participants). The most common TEAEs that occurred in greater than or equal to two participants receiving RDV in any cohort were nausea (2/8 [25.0%] participants in cohort 3), dizziness (2/8 [25.0%] participants each in cohorts 4 and 5), and cough (2/10 [20.0%] participants in cohort 7). The only events that occurred in greater than or equal to two participants receiving placebo were dizziness and upper respiratory tract congestion (2/14 [14.3%] participants each).
TABLE 5.
Overall summary of AEs.
| n (%) | Cohort 1 2 mg RDV SAD (n = 8) | Cohort 2 5 mg RDV SAD (n = 8) | Cohort 3 5 mg RDV MAD (n = 8) | Cohort 4 10 mg RDV SAD (n = 8) | Cohort 5 10 mg RDV MAD (n = 8) | Cohort 6 10.4 mg RDV MAD inhalation formulation (n = 8) | Cohort 7 10 mg RDV SAD mouthpiece (n = 10) | Placebo (n = 14) | Total (N = 72) |
|---|---|---|---|---|---|---|---|---|---|
| TEAE a | 1 (12.5) | 1 (12.5) | 4 (50.0) | 3 (37.5) | 4 (50.0) | 5 (62.5) | 5 (50.0) | 8 (57.1) | 31 (43.1) |
| TEAE grade ≥2 b | 0 | 0 | 1 (12.5) | 1 (12.5) | 0 | 2 (25.0) | 0 | 0 | 4 (5.6) |
| TEAE related to study treatment | 0 | 1 (12.5) | 2 (25.0) | 3 (37.5) | 3 (37.5) | 4 (50.0) | 2 (20.0) | 7 (50.0) | 22 (30.6) |
| TEAE related to study procedure | 1 (12.5) | 1 (12.5) | 3 (37.5) | 3 (37.5) | 4 (50.0) | 3 (37.5) | 4 (40.0) | 8 (57.1) | 27 (37.5) |
| TE SAE | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| TEAE leading to study treatment discontinuation | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| TEAE leading to study discontinuation | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Abbreviations: AE, adverse event; MAD, multiple ascending dose; RDV, remdesivir; SAD, single ascending dose; SAE, serious adverse event; TE, treatment‐emergent; TEAE, treatment‐emergent adverse event.
TEAEs were those that began after the study treatment start date up to 30 days after permanent discontinuation of study treatment.
Severity grades were defined by the Division of AIDS Table for Grading the Severity of Adult and Pediatric Adverse Events, version 2.1.
Overall, 28 of 72 (38.9%) participants had greater than or equal to one graded, treatment‐emergent abnormal laboratory finding reported during the study, most of which were grade 1. No notable changes from predose to any postdose timepoint were observed in systolic blood pressure, diastolic blood pressure, pulse rate, body temperature, respiration rate, or oxygen saturation. Similarly, no notable changes from predose to any postdose timepoint were observed in FEV1, FVC, or FEV1/FVC. Although several participants had abnormal ECG findings during the study, most of these were present prior to administration of study treatment and none were considered to be clinically significant.
DISCUSSION
Results from the current analysis demonstrate that following administration of RDV via inhalation, RDV, GS‐704277, and GS‐441524 exhibited linear and dose‐proportional plasma PK, with multiple‐dose PK showing results consistent with those of single‐dose PK. RDV, GS‐704277, and GS‐441524 plasma exposures were similar for both RDV formulations used for nebulization. Systemic exposures of the metabolites GS‐704277 and GS‐441524 were comparable with those observed after i.v. administration at the equivalent dose, highlighting the feasibility and efficiency of RDV administration via inhalation. In this study, sufficient FEV1 was one of the inclusion criteria as a safety precaution. Furthermore, FEV1 was tested prior to dosing throughout the study to ensure that poor FEV1 would not interfere with RDV delivery. Further PK evaluations in participants with a range of FEV1 would be required to fully understand the respective implications and potential need for dose adjustment.
RDV was generally safe and well‐tolerated when administered as a single ascending dose or multiple ascending doses via inhalation. No participants experienced grade 3 or 4 TEAEs or SAEs or discontinued due to TEAEs, reflecting the low systemic exposures. Additionally, there were no changes in spirometry parameters, indicating no detriment to pulmonary function when RDV is delivered locally to the lungs. These findings demonstrate that inhalation could be an alternative route for RDV delivery that would be especially useful for self‐administration in outpatient settings.
Along with two formulations of inhaled RDV, this study evaluated an LC Sprint valved jet nebulizer equipped with either a sealed facemask or a mouthpiece as the patient interface. The mouthpiece was hypothesized to be a more user‐friendly patient interface for at‐home administration. It also showed a higher overall delivery efficiency based on in vitro testing (25.6% for the mouthpiece vs. 16.1%–17% for the facemask). However, in this study, plasma RDV exposures were lower and more variable with the mouthpiece configuration compared with the facemask. Although formal assessment of taste was not conducted in this trial, the unpleasant taste, as informally reported by some study participants, may have contributed to overall efficiency of nebulization. These results indicate that further evaluation of the device is required to enable efficient and convenient home use. Simple and easy‐to‐use devices are necessary to expand the accessibility of inhaled RDV to a wider outpatient demographic.
This study was initiated early in the COVID‐19 pandemic (June 2020; Figure S2), a time when minimizing SARS‐CoV‐2 transmission and identifying effective treatments and vaccines against COVID‐19 were of paramount importance. Only a month prior to this study, i.v. RDV was shown to shorten time to recovery in adults hospitalized with COVID‐19. 11 Since then, RDV has been approved for the treatment of COVID‐19 in the United States, the European Union, Japan, and greater than 50 other countries worldwide at an initial dose of 200 mg i.v., followed by 100 mg i.v. for the next 4 or 9 days. 1
The advantage of administering RDV via inhalation is the direct delivery of the drug to the primary site of infection in the respiratory tract, thereby reducing overall systemic drug exposure. The disadvantage is that inhalation requires special equipment, longer administration time, and further optimization. There was no direct evaluation of active metabolite formation in human lungs (either through biopsy or bronchoalveolar lavage) following inhalation of RDV in this study, partially due to the invasive nature of such assessments. However, based on the preclinical data, the doses evaluated were expected to deliver comparable levels of the active triphosphate metabolite as the clinical regimen of 200/100 mg of i.v. RDV. 4 Using PBMCs as a surrogate for active metabolite formation was not feasible either, considering the low systemic exposures following inhalation.
At the time of the current study, multiple programs pursuing alternative routes of RDV administration, including subcutaneous injection, intramuscular injection, as well as the oral administration of novel nucleosides, were in development. In recent years, orally administered COVID‐19 therapeutics have shown promise in clinical trials due to the simplicity, convenience, and improved efficacy of oral compared with inhaled or i.v. routes of administration. 12 , 13
Overall, RDV administration via inhalation was generally safe and well‐tolerated regardless of the nebulization formulation used. Together, these data demonstrate that the inhaled route of RDV administration shows promise in the clinic, but exploration of alternative devices could offer more stable and predictable drug delivery to the lungs.
AUTHOR CONTRIBUTIONS
All authors wrote the manuscript. R.H., K.J., S.C., S.E., A.S., M.J., S.D., A.dZ., J.L., A.O., S.G., R.P., and M.D. designed the research. R.H., K.J., S.C., S.E., A.S., M.J., S.D., A.dZ., J.L., A.O., S.G., R.P., and M.D. performed the research. R.H., K.J., S.C., S.E., O.A., D.X., A.S., M.J., S.D., A.dZ., J.L., A.O., H.W., S.G., R.P., and M.D. analyzed the data.
FUNDING INFORMATION
This study was funded by Gilead Sciences, Inc.
CONFLICT OF INTEREST STATEMENT
R.H., S.C., S.E., O.A., D.X., A.S., S.D., J.L., A.O., H.W., S.G., R.P., and M.D. are employees and stockholders of Gilead Sciences, Inc. K.J. is a former employee and stockholder of Gilead Sciences, Inc. and a current employee of Corcept Therapeutics. M.J. is an employee of ICON plc. A.dZ. is a former employee and stockholder of Gilead Sciences, Inc. and a current employee of Atea Pharmaceuticals.
Supporting information
Data S1
ACKNOWLEDGMENTS
The authors would like to thank the participants of this study, their families, and the support staff. Medical writing support was provided by Melanie Chen, PharmD, of Lumanity Communications Inc., and was funded by Gilead Sciences, Inc.
Humeniuk R, Juneja K, Chen S, et al. Pharmacokinetics, safety, and tolerability of inhaled remdesivir in healthy participants. Clin Transl Sci. 2023;16:2276‐2288. doi: 10.1111/cts.13627
REFERENCES
- 1. VEKLURY® (remdesivir) injection, for intravenous use [package insert]. Gilead Sciences, Inc. 2022.
- 2. Humeniuk R, Mathias A, Kirby BJ, et al. Pharmacokinetic, pharmacodynamic, and drug‐interaction profile of remdesivir, a SARS‐CoV‐2 replication inhibitor. Clin Pharmacokinet. 2021;60(5):569‐583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li R, Liclican A, Xu Y, et al. Key metabolic enzymes involved in remdesivir activation in human lung cells. Antimicrob Agents Chemother. 2021;65:e0060221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vermillion MS, Murakami E, Ma B, et al. Inhaled remdesivir reduces viral burden in a nonhuman primate model of SARS‐CoV‐2 infection. Sci Transl Med. 2022;14(633):eabl8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Leegwater E, Moes D, Bosma LBE, et al. Population pharmacokinetics of remdesivir and GS‐441524 in hospitalized COVID‐19 patients. Antimicrob Agents Chemother. 2022;66:e0025422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. de Wit E, Feldmann F, Cronin J, et al. Prophylactic and therapeutic remdesivir (GS‐5734) treatment in the rhesus macaque model of MERS‐CoV infection. Proc Natl Acad Sci USA. 2020;117:6771‐6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Williamson BN, Feldmann F, Schwarz B, et al. Clinical benefit of remdesivir in rhesus macaques infected with SARS‐CoV‐2. Nature. 2020;585:273‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Warren TK, Jordan R, Lo MK, et al. Therapeutic efficacy of the small molecule GS‐5734 against Ebola virus in rhesus monkeys. Nature. 2016;531:381‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Graham BL, Steenbruggen I, Miller MR, et al. Standardization of spirometry 2019 update: an official American Thoracic Society and European Respiratory Society technical statement. Am J Respir Crit Care Med. 2019;200:e70‐e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Humeniuk R, Mathias A, Cao H, et al. Safety, tolerability, and pharmacokinetics of remdesivir, an antiviral for treatment of COVID‐19, in healthy subjects. Clin Transl Sci. 2020;13:896‐906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beigel JH, Tomashek KM, Dodd LE, et al. Remdesivir for the treatment of Covid‐19—final report. N Engl J Med. 2020;383:1813‐1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jayk Bernal A, Gomes da Silva MM, Musungaie DB, et al. Molnupiravir for oral treatment of Covid‐19 in nonhospitalized patients. N Engl J Med. 2022;386:509‐520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hammond J, Leister‐Tebbe H, Gardner A, et al. Oral nirmatrelvir for high‐risk, nonhospitalized adults with Covid‐19. N Engl J Med. 2022;386:1397‐1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1