

Abstract
Cisplatin treatment is effective against several types of carcinomas. However, it frequently leads to kidney injury, which warrants effective prevention methods. Sodium valproic acid is a prophylactic drug candidate with a high potential for clinical application against cisplatin‐induced kidney injury. Therefore, in this study, we aimed to elucidate the mechanism underlying the prophylactic effect of valproic acid on cisplatin‐induced kidney injury in a mouse model and HK2 and PODO cells with cisplatin‐induced toxicity. In the mouse model of cisplatin‐induced kidney injury, various renal function parameters and tubular damage scores were worsened by cisplatin, but they were significantly improved upon combination with valproic acid. No difference was observed in cisplatin accumulation between the cisplatin‐treated and valproic acid‐treated groups in whole blood and the kidneys. The mRNA expression levels of proximal tubular damage markers, apoptosis markers, and inflammatory cytokines significantly increased in the cisplatin group 72 h after cisplatin administration but significantly decreased upon combination with valproic acid. In HK2 cells, a human proximal tubular cell line, the cisplatin‐induced decrease in cell viability was significantly suppressed by co‐treatment with valproic acid. Valproic acid may inhibit cisplatin‐induced kidney injury by suppressing apoptosis, inflammatory responses, and glomerular damage throughout the kidneys by suppressing proximal tubular cell damage. However, prospective controlled trials need to evaluate these findings before their practical application.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Current clinical management practices do not completely alleviate cisplatin‐induced kidney damage, and more substantial supportive measures need to be developed.
WHAT QUESTION DID THIS STUDY ADDRESS?
Numerous pharmaceuticals and substances have demonstrated efficacy in mitigating cisplatin‐induced kidney injury through preclinical in vitro and in vivo investigations; however, their clinical application remains limited. In this research, we used large‐scale medical data analysis to identify potential preventive agents with significant prospects for clinical implementation.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Valproic acid reduced cisplatin‐induced kidney injury by suppressing the apoptosis of proximal tubular cells, thereby preventing damage to the entire kidney. These findings indicate that valproic acid can be a promising preventive agent for mitigating cisplatin‐induced kidney injury in a clinical setting.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Based on these findings, clinical trials can verify that sodium valproate may be a promising prophylactic agent to reduce cisplatin‐induced renal injury in clinical practice.
INTRODUCTION
Cisplatin is a prominent platinum‐based chemotherapy drug used in various cancers, particularly solid tumors, such as lung, bladder, and head and neck malignancies. 1 However, its adverse effects are often severe and include renal and hearing impairment, nausea, and vomiting. 1 Approximately 30% of patients receiving cisplatin develop acute kidney injury, 2 an important dose‐limiting toxicity. Cisplatin‐induced cell death and inflammation are predominantly observed in the proximal tubule segment, coinciding with the location of maximal cisplatin accumulation. 3 , 4 , 5 Acute kidney injury can result in chronic tubulointerstitial fibrosis or irreversible chronic tubulopathy, potentially leading to renal failure. 6 However, cisplatin continues to be the predominant drug used in clinical settings. To alleviate the nephrotoxicity caused by cisplatin, it is advisable to implement measures such as adequate fluid intake, including infusion of saline solution (>3 L/day), and diuretic medication administration during anticancer treatment. 7 Nevertheless, it is not possible to avert renal damage entirely, and additional supportive measures are necessary.
To develop a cisplatin‐induced kidney injury prophylaxis with high potential for clinical application, certain requirements must be met. In addition to ensuring pharmacodynamic safety, drugs used to treat cisplatin‐induced renal injury should demonstrate compatibility with cisplatin and other concurrent anticancer medications administered without compromising their anticancer effectiveness. Here, we focused on valproic acid (VPA) as a candidate agent for preventing cisplatin‐induced renal injury. VPA increases brain gamma‐aminobutyric acid concentrations and is used to treat seizures and other neuropsychiatric conditions. It is effective in reducing kidney damage; it may also exhibit anticancer properties. In renal models of tubulointerstitial fibrosis and lupus, VPA treatment has been shown to prevent renal injury and improve laboratory serum and urine analysis results. 8 , 9 , 10 , 11 , 12 , 13 VPA also decreases inflammatory cytokine expression in ischemia–reperfusion‐induced renal injury. 14 , 15 Furthermore, VPA treatment exerted anticancer effects, such as inhibiting the growth of pancreatic cancer cells 16 and primary tumors and their metastasis to the lungs, in a mouse breast cancer model. 17 Furthermore, our preliminary study using VigiBase showed that patients taking VPA reported less kidney damage as an adverse effect than those not taking VPA, suggesting that VPA may be effective as a preventive agent for kidney injury. Here, we aimed to evaluate the efficacy of VPA treatment in mitigating or preventing cisplatin‐induced renal injury in cells and animals.
METHODS
Data analysis
From January 1968 to December 2021, there were 28,617,525 voluntary reports documenting adverse events were recorded on VigiBase (https://who‐umc.org/). Duplicate data were excluded per recommendations, and the remaining 27,994,584 reports were used. SQLite was used for data processing and R (version 3.2.1; R Foundation for Statistical Computing) for statistical analyses. Acute renal failure was defined using 47 terms of “acute renal failure (SMQ 20000003),” excluding neonatal and pediatric diseases (Table S1), according to the Medical Dictionary for Regulatory Activities (MedDRA) version 23.1.
Adverse event risk was assessed using reporting odds ratio (ROR) and 95% confidence interval (CI). Patients who received cisplatin were classified into the following groups: (A) patients who used drug A and reported acute renal failure; (B) those who used drug A and did not report acute renal failure; (C) those who did not use drug A and reported acute renal failure; and (D) those who did not use drug A and did not report acute renal failure. ROR and 95% CI were calculated using the following equations:
All tests were two‐tailed; results with p < 0.05 were considered significant.
Animal model of cisplatin‐induced nephrotoxicity
Animal studies were conducted following the ARRIVE guidelines and regulations set by the Animal Research Committee of Tokushima University Graduate School. The experimental protocol was approved by the Institutional Review Board of Tokushima University Graduate School for Animal Protection (Permit Number: T30‐85, Approval Date: October 1, 2018). Male C57BL/6N mice, aged 9–10 weeks (Nippon CLEA) and weighing 23–28 g, were acquired and placed under unrestricted access to NMF‐type food (Oriental Yeast) and water. Male mice were used because the degree of renal dysfunction in the acute renal failure model is clearly more severe in male mice than in female mice, and the expression of OCT2, which is involved in cisplatin uptake, is higher in male mice. The relative humidity and temperature of the breeding room were 50% ± 10%, and 26°C ± 1°C, respectively, following a 12‐h light–dark cycle (on at 8:00 h, off at 20:00 h). To establish a mouse model of cisplatin‐induced renal injury, we used C57BL/6N male mice following established protocols described previously. 18 , 19
Mice were randomly allocated to six groups (n = 5–8 mice/group): group 1, vehicle‐injected; group 2, VPA‐treated; group 3, cisplatin‐injected; and groups 4–6, cisplatin‐injected with add‐on VPA at 10, 30, and 100 mg/kg, respectively. Mice were intraperitoneally injected with cisplatin (groups 3–6, 15 mg/kg) or vehicle (groups 1 and 2, saline) once. Twenty‐four h before being injected with cisplatin, the mice were orally administered VPA (groups 3–6) or vehicle (groups 1 and 2, saline) once daily for 2 or 4 consecutive days, 24 or 72 h after cisplatin treatment. The experimental mice were anesthetized and euthanized, and blood, serum, urine, and kidney tissue samples were obtained for further analyses. Anesthesia was induced by administering isoflurane via inhalation in a vaporizer set at 4% dilution (vaporized in oxygen at a flow rate of 1 L/min) and maintained using 2% isoflurane in the vaporizer. Isoflurane was administered via a small facial mask.
Measurement of serum and urine creatinine levels as well as blood urea nitrogen levels
Levels of serum creatinine (Cr), urine Cr, blood urea nitrogen (BUN), serum albumin (ALB), urine ALB, aspartate aminotransferase (AST), and alanine aminotransferase in the collected serum and urine samples at 72 h after cisplatin administration were determined by Oriental Yeast Industries (Shiga, Japan). Creatinine clearance (Ccr) was calculated using the following formula:
Histological analysis
Renal tubular injury was evaluated according to an established protocol. 20 Kidney tissue samples were fixed in 4% paraformaldehyde solution and embedded in paraffin. The paraffin‐embedded samples were sectioned to 4‐μm‐thick slices and stained with hematoxylin and eosin. Tubular damage was performed by greater than or equal to three independent experts who were not involved in the study. Experts evaluated the extent of damage based on various criteria, including tubular necrosis, loss of brush border, cast formation, tubular dilation, and tubular degeneration. Damage was scored on a scale of 0–4: 0 for normal, 1 for less than 25%, 2 for 25%–50%, 3 for 50%–75%, and 4 for greater than 75%. Ten random microscopic fields were examined per kidney section using a BX53 microscope (Olympus) for quantification.
Immunostaining of kidneys
Formaldehyde‐fixed kidney samples were embedded in paraffin and cut into 4‐μm‐thick sections. For deparaffinization, paraffin‐embedded sections of the kidneys were washed in Heme‐De (FALMA), 90% ethanol, 80% ethanol, and 70% ethanol for 5 min each, and in phosphate‐buffered saline (PBS) using 50 μL Triton/PBS (0.1%) and 50 μL bovine serum albumin/PBS at 4°C for 20 min each. The sections were incubated overnight at 4°C with the Rat Anti‐Mouse primary antibody F4/80 (1:100; MCA497G; Bio‐Rad). The sections were then washed in PBS and incubated with a secondary antibody, Alexa Fluor 488 goat anti‐rat IgG (1:1000; A11006; Thermo Fisher Scientific) for 1 h at 20°C, followed by the use of ProLong Gold antifade reagent with DAPI (P36935; Thermo Fisher Scientific). The sections were observed under a confocal laser scanning microscope (LSM700; Carl Zeiss).
For each group of immunostained sections, five randomly selected non‐overlapping digital images were captured for subsequent quantification. Quantitative immunostaining analysis was performed by counting the F4/80‐positive cells using the measurement function of Image‐Pro Plus software (Media Cybernetics). The result is expressed as the number of F4/80‐positive cells to the total area evaluated. Error bars indicate standard error of the mean.
Cell culture
HK2 (ATCC CRL‐2190; American Type Culture Collection) and PODO/TERT256 cells (CHT‐033‐0256; EVERCYTE) were used to examine the effects of VPA treatment on cisplatin‐induced cell death. The potential antitumor properties of VPA were evaluated using human lung cancer (A549) and gastric cancer (MKN‐1) cells. The cells were cultured and passaged in Roswell Park Memorial Institute medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. Culture conditions were maintained at 37°C in a humidified atmosphere of 95% air and 5% CO2. Subculturing was performed when the cells reached ~80% confluence, and experiments were conducted using cells at passages 10–20.
Cell viability assay
Cell viability was determined using the Cell Counting Kit‐8 (CCK‐8) assay (Dojindo Laboratories), following the manufacturer's instructions. The cells were plated in 96‐well plates at 5 × 104 cells/well density and incubated at 37°C for 24 h. The cells were then exposed to culture media containing or without 10 μM cisplatin for 24 h. VPA (100, 300, and 1000 μM) was then used to treat these cells (either cells cultured in media with cisplatin or those cultured in media without it). Cell viability was determined by quantifying the optical density of WST‐8 formazan at 450 nm using a microplate reader (Model 680; Bio‐Rad).
Real‐time polymerase chain reaction
Kidney samples and HK2 cells were obtained from the mice with cisplatin‐induced kidney injury. RNA extraction was performed using an RNA extraction solution (NIPPON GENE) following the manufacturer's instructions. Reverse transcription of RNA into cDNA was performed using the PrimeScript RT Reagent Kit (Takara Bio) and a polymerase chain reaction (PCR) Thermal Cycler Dice (Takara Bio). Each cDNA sample was combined with forward and reverse primers and a THUNDERBIRD SYBR qPCR mix (Toyobo), according to the manufacturer's protocol. The PCR mixture consisted of 1 μL cDNA, 5 μL Thunderbird SYBR qPCR mix, 0.2 μL PCR primers, and 3.6 μL RNase‐free water. PCR was performed using the Applied Biosystems StepOnePlus system. Briefly, there were 45 cycles of denaturation at 95°C for 15 s, followed by annealing and extension at 60°C for 1 min. Initial analysis was performed using StepOnePlus version 2.3 (Applied Biosystems). Relative fold changes in gene expression compared with those in the control group were determined using mouse GAPDH as the internal reference. PCR was performed using the primer sets listed in Table S2. Each experiment was conducted in triplicate.
Statistical analysis
Statistical analysis was conducted using Student's t‐test for group comparisons. A one‐way analysis of variance was performed for comparisons involving three or more groups. Post hoc analysis was performed using Tukey's test. Statistical analyses were performed using R version 3.2.1 for Windows. Results with p < 0.05 (two‐tailed) were considered significant.
RESULTS
VigiBase analysis
Among the 28,617,525 spontaneous adverse event reports submitted between January 1968 and December 2021, there were 124,685 cases that involved cisplatin administration. The ROR for acute renal failure due to VPA administration was examined. To confirm the accuracy of data analysis, we used ondansetron and palonosetron. The ROR for the VPA and cisplatin combination was 0.57 (95% CI: 0.08–4.12), that for the ondansetron and cisplatin combination was 1.33 (95% CI: 1.10–1.61), and that for palonosetron was 0.45 (95% CI: 0.32–0.61; Table 1).
TABLE 1.
Effect of drugs on the occurrence of cisplatin‐induced ARF based on VigiBase analysis.
| Drug | ARF (%) without the drug | ARF (%) with the drug | ROR (95% CI) | pvalue |
|---|---|---|---|---|
| Ondansetron | 2.28 (2758/121,000) | 3.01 (111/3685) | 1.33 (1.10–1.61) | <0.01 |
| Palonosetron | 2.34 (2828/120,823) | 1.06 (41/3862) | 0.45 (0.32–0.61) | <0.01 |
| VPA | 2.30 (2868/124,610) | 1.33 (1/75) | 0.57 (0.08–4.12) | 1 |
Note: Statistical analysis was performed using Fisher's exact test.
Abbreviations: ARF, acute renal failure; CI, confidence interval; ROR, reporting odds ratio; VPA, valproic acid.
Effects of VPA treatment on cisplatin‐induced kidney injury in mice
Body weight was lower for mice 72 h after cisplatin treatment than for mice treated with a vehicle, but there was no significant change in kidney weight (Table 2). However, at 24 h after cisplatin treatment, body weight remained unchanged (Table S3). At 72 h after cisplatin treatment, renal damage significantly increased (p < 0.01), and the levels of Kim‐1 and Lcn‐2, which serve as indicators of proximal tubular injury, increased (p < 0.01). BUN, urine ALB, urine CRE ratio, and AST levels increased (p < 0.01), whereas urine volume and Ccr decreased (p < 0.05) compared with those in the vehicle mice (Table 2; Figure 1a,b).
TABLE 2.
Body weight, kidney weight, renal function, and liver function in vehicle‐treated mice and 72‐h cisplatin‐treated mice with or without VPA treatment.
| Vehicle | VPA (100) | Cisplatin | Cisplatin‐VPA (10) | Cisplatin‐VPA (30) | Cisplatin‐VPA (100) | |
|---|---|---|---|---|---|---|
| Initial body weight (g) | 26.4 ± 0.6 | 25.1 ± 0.4 | 26.8 ± 0.5 | 25.6 ± 0.6 | 26.8 ± 0.3 | 26.8 ± 0.6 |
| Final body weight (g) | 25.2 ± 0.6 | 23.8 ± 0.3 | 20.8 ± 0.3 ** | 20.3 ± 0.6 | 21.6 ± 0.6 | 21.6 ± 0.6 |
| Kidney weight (mg) | 187.2 ± 5.2 | 167.6 ± 9.5 | 164.0 ± 9.6 * | 150.3 ± 7.7 | 165.6 ± 3.2 | 157.9 ± 3.3 |
| Kidney weight/body weight (mg/g) | 7.57 ± 0.23 | 7.06 ± 0.22 | 7.89 ± 0.17 | 7.42 ± 0.17 | 7.68 ± 0.18 | 7.28 ± 0.19 |
| Urine volume (mL) | 1.18 ± 0.13 | 0.90 ± 0.19 | 0.64 ± 0.11 * | 0.80 ± 0.13 | 0.96 ± 0.21 | 1.02 ± 0.13 |
| BUN (mg/dL) | 21.3 ± 1.3 | 20.4 ± 0.7 | 72.0 ± 5.9 ** | 55.3 ± 4.1 | 59.1 ± 6.4 | 54.8 ± 8.5 |
| Ccr (mL/min/kg) | 6.60 ± 0.58 | 5.77 ± 0.73 | 2.41 ± 0.33 * | 3.84 ± 0.48 | 4.69 ± 0.78 | 7.17 ± 1.49†† |
| Serum ALB (g/dL) | 2.87 ± 0.05 | 2.85 ± 0.06 | 2.94 ± 0.04 | 3.01 ± 0.08 | 3.13 ± 0.03 | 2.91 ± 0.03 |
| Urine ALB (μg/mL) | 21.6 ± 2.6 | 22.5 ± 0.3 | 3289.2 ± 710.3** | 1170.9 ± 583.6† | 592.9 ± 113.8†† | 808.6 ± 221.2†† |
|
Urine ALB Urine Cr−1 ratio |
0.97 ± 0.1 | 1.07 ± 0.1 | 95.96 ± 20.1** | 39.86 ± 19.8 | 20 ± 3.7†† | 24.21 ± 6.7†† |
| AST (IU/L) | 142.3 ± 9.5 | 152.0 ± 25.1 | 234.5 ± 10.2** | 209.6 ± 8.6 | 229.9 ± 15.9 | 207.9 ± 11.1 |
| ALT (IU/L) | 38.8 ± 8.8 | 33.8 ± 8.9 | 50.0 ± 5.4 | 41 ± 2.7 | 51.5 ± 6.1 | 43.9 ± 2.6 |
Note: Data are presented as mean ± standard error of the mean.
Abbreviations: ALB, serum albumin; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BUN, blood urea nitrogen; Ccr, creatinine clearance; Cr, creatinine.
*p < 0.05 and **p < 0.01 versus vehicle, † p < 0.05 and †† p < 0.01 vs. cisplatin, n = 4–8 in each group.
FIGURE 1.
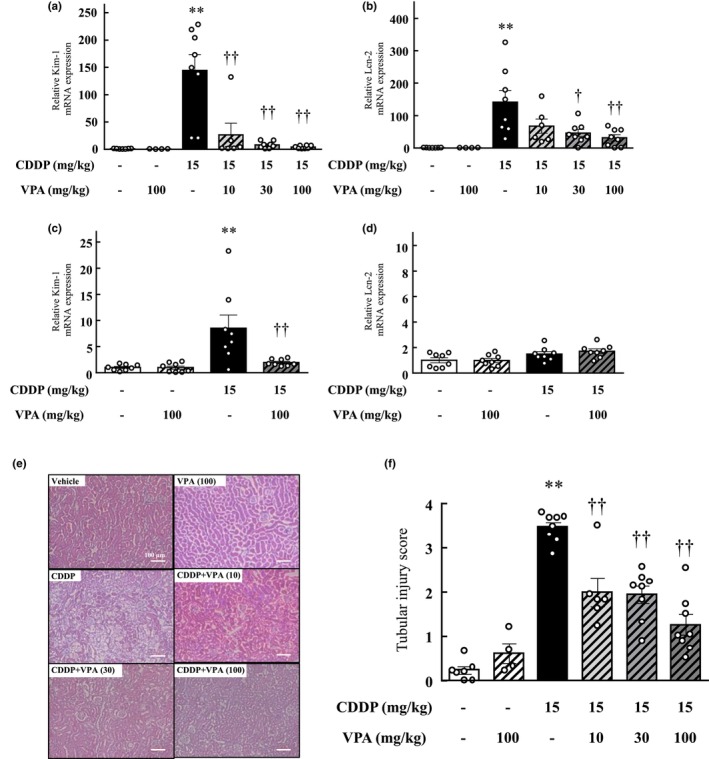
Effect of valproic acid treatment on cisplatin‐induced toxicity in the mouse kidneys. (a–d) The mRNA expression levels of the kidney injury markers Kim‐1 (a, c) and Lcn‐2 (b, d) were assessed in the renal tissue of mice from each experimental group. The measurements were conducted at two different timepoints: (a, b) 72 h after cisplatin treatment and (c, d) 24 h following cisplatin treatment. (e, f) Histological evaluation of the renal tubules in mice. (e) Representative hematoxylin and eosin staining of kidney sections from vehicle‐ and cisplatin‐treated mice with or without valproic acid treatment. Scale bar indicates 100 μm. (f) Quantitative analysis of renal damage scores. Values are expressed as mean ± standard error of the mean. **p < 0.01 versus vehicle, † p < 0.05, †† p < 0.01 versus cisplatin, n = 4–8 in each group. CDDP, cisplatin; VPA, valproic acid.
At 24 h after cisplatin treatment, the mice showed renal damage with increased expression of Kim‐1 (p < 0.01) but presented no change in Lcn‐2 expression (Figure 1c,d). The cisplatin and VPA combination resulted in a significant dose‐dependent alleviation of cisplatin‐induced renal dysfunction (Table 2, Figure 1).
Histological evaluation
At the 72‐h timepoint after cisplatin treatment, mice displayed evident signs of renal tissue degeneration and damage, such as nuclear loss, atrophy of proximal tubular cells, and tubular enlargement. However, the VPA and cisplatin combination resulted in reduced tubular dilation and necrotic tubular cell proportion compared to cisplatin (Figure 1e). The groups administered the VPA and cisplatin combination demonstrated lower scores than the group administered cisplatin alone (p < 0.01), and this effect was dose‐dependent (Figure 1f).
Effect of VPA on platinum concentration in whole‐blood and kidney tissue samples
Eight h after cisplatin administration, renal platinum and whole‐blood platinum concentrations did not change with VPA treatment (Figure S1), indicating that VPA does not affect platinum accumulation in the kidneys or whole blood.
Influence on inflammatory cytokines in the kidneys
Quantitative real‐time PCR confirmed that the mRNA expression of the inflammatory cytokines TNF‐α, IL‐1β, and IL‐6 in the kidneys increased 72 h after cisplatin treatment (IL‐1β, p < 0.05; TNF‐α, and IL‐6, p < 0.01); however, TNF‐α, IL‐1β, and IL‐6 expression significantly reduced in the kidneys of the cisplatin and VPA combination group compared with that in the kidneys of the cisplatin‐alone group (TNF‐α and IL‐1β, p < 0.05; IL‐6, p < 0.01). This decrease in expression was dose‐dependent (Figure 2a–c).
FIGURE 2.
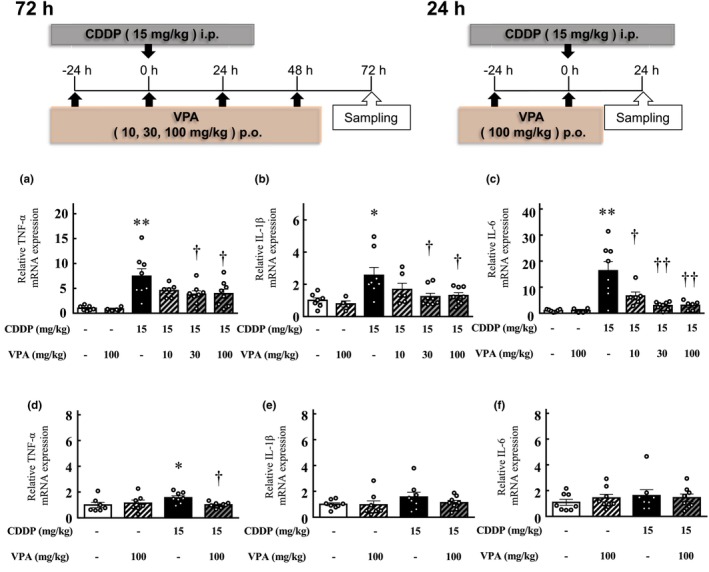
Impact of valproic acid administration on the expression of inflammatory cytokines in the renal tissues of mice. The mRNA levels of TNF‐α (a, d), IL‐1β (b, e), and IL‐6 (c, f), which are inflammatory cytokines, were measured in the kidneys of mice in each experimental group. (a–c) Mice at 72 h after cisplatin treatment. (d–f) Mice 24 h after cisplatin treatment. Values are expressed as mean ± standard error of the mean. *p < 0.05, **p < 0.01 versus vehicle, † p < 0.05, †† p < 0.01 versus cisplatin, n = 4–8 in each group. CDDP, cisplatin; VPA, valproic acid.
In contrast, quantitative real‐time PCR confirmed that TNF‐α expression in the kidneys increased 24 h after cisplatin treatment (p < 0.05); however, its expression reduced in the kidneys of the cisplatin and VPA combination group compared with that in the cisplatin‐alone group (p < 0.05; Figure 2d). No significant differences were observed in IL‐1β and IL‐6 expression in the kidneys of mice treated with cisplatin for 24 h and vehicle‐treated mice (Figure 2e,f).
Effect of VPA treatment on cisplatin‐induced inflammatory cell infiltration
Figure 3a shows a typical image of a kidney from each group that was immunostained for F4/80. At 72 h after cisplatin treatment, the number of F4/80 cells significantly increased (p < 0.01; Figure 3b). Mice treated with VPA had fewer F4/80 cells than those treated with cisplatin alone (p < 0.01; Figure 3b).
FIGURE 3.
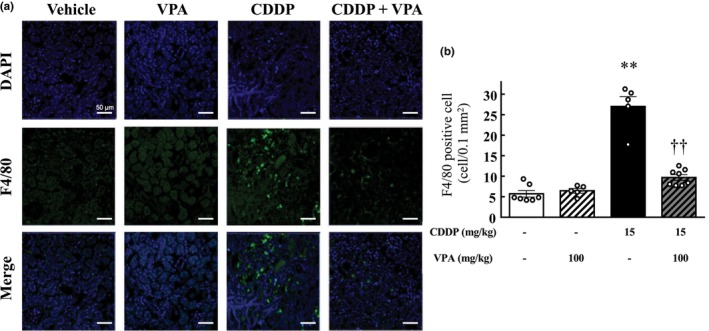
Effect of valproic acid on inflammatory cell infiltration in mouse kidneys. (a) Representative confocal images of immunostaining of F4/80 (green), DAPI nuclear staining (blue), merged images of F4/80 immunostaining (green), and DAPI nuclear staining (blue) of kidney sections of vehicle‐ and cisplatin‐treated mice with or without valproic acid treatment. Scale bar indicates 50 μm. (b) Quantification of cells that stained positive for F4/80. Values are expressed as mean ± standard error of the mean. **p < 0.01 versus vehicle, †† p < 0.01 versus cisplatin, n = 5–8 in each group. CDDP, cisplatin; VPA, valproic acid.
Influence of VPA treatment on apoptosis marker expression in the kidneys
Quantitative real‐time PCR analysis demonstrated upregulated expression of the apoptosis markers Bax/Bcl‐2, FOXO3, and augmented renal clearance (ARC) in the kidneys at 72 h after cisplatin treatment (FOXO3 and ARC, p < 0.05; Bax/Bcl‐2, p < 0.01); however, their expression decreased in the kidneys of the cisplatin and VPA combination group compared with that in the cisplatin‐alone group (p < 0.05; Figure 4a–c).
FIGURE 4.
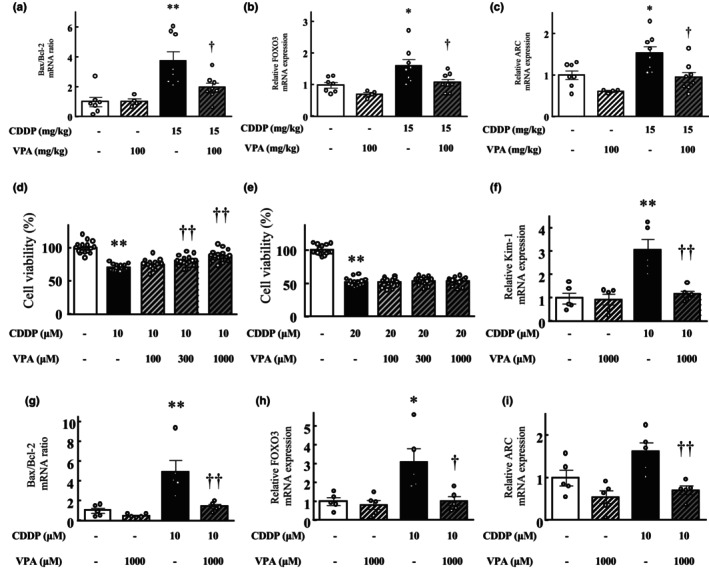
Effect of valproic acid treatment on the apoptosis‐related gene ratio or expression in mouse kidneys 72 h after cisplatin treatment and on cisplatin‐induced toxicity in kidney cells. (a) The ratio of Bax/Bcl‐2 mRNA levels. mRNA expression levels of apoptosis markers FOXO3 (b) and ARC (c) in each group. Values are expressed as mean ± standard error of the mean. *p < 0.05, **p < 0.01 versus vehicle, †† p < 0.01 versus cisplatin, n = 4–8 in each group. (d, e) Preventive effect of valproic acid treatment against cisplatin‐induced toxicity in HK2 (d) and PODO (e) kidney cells. Viability of cells after 24 h of incubation in a medium with or without cisplatin treatment (10 or 20 μM) was calculated as 100% for vehicle‐treated cells. Valproic acid and cisplatin were simultaneously administered. Values are expressed as mean ± standard error of the mean. **p < 0.01 versus vehicle‐treated cells, †† p < 0.01 versus cisplatin‐treated cells, n = 16 in each group. (f–i) The effect of valproic acid treatment on cisplatin‐induced toxicity in HK cells. (f) The mRNA expression level of Kim‐1. (g) The ratio of Bax/Bcl‐2 mRNA levels. mRNA expression levels of FOXO3 (h) and ARC (i) in each group. Values are expressed as mean ± standard error of the mean. *p < 0.05, **p < 0.01 versus vehicle‐treated cells, † p < 0.05, †† p < 0.01 versus cisplatin‐treated cells, n = 5 in each group. CDDP, cisplatin; VPA, valproic acid.
Effect of VPA treatment on cytotoxicity induced by cisplatin
After 24 h of cisplatin treatment (10 or 20 μM), the viability of cisplatin‐treated HK2 and PODO cells markedly reduced compared with that of vehicle‐treated cells (p < 0.01; Figure 4d,e). The 1000‐μM VPA add‐on treatment increased HK2 cell viability compared with that in the cisplatin‐only treatment group (p < 0.01; Figure 4d). However, treatment with VPA at any dose did not alter the viability of PODO cells compared with that in the cisplatin‐only treatment group (Figure 4e).
Effects of VPA treatment on cisplatin‐induced kidney injury and apoptosis in HK2 cells
Quantitative real‐time PCR analysis validated the upregulation in Kim‐1, Bax/Bcl‐2, FOXO3, and ARC expression in HK2 cells upon treatment with cisplatin alone (FOXO3, p < 0.05; Kim‐1 and Bax/Bcl‐2, p < 0.01); however, these expressions were lower in the cisplatin and VPA combination‐treated cells than in the cisplatin‐treated cells (FOXO3, p < 0.05; Kim‐1, Bax/Bcl‐2, and ARC, p < 0.01; Figure 4f–i).
Anticancer effect of VPA with cisplatin
The cytotoxicity of the VPA and cisplatin combination was evaluated in A549 and MKN‐1 cells, which are representative human lung and stomach cancer cell lines, respectively. After 24 h of treatment with cisplatin (25 μM), the viability of both A549 and MKN‐1 cells markedly reduced compared with that of cells cultured in cisplatin‐free medium (p < 0.01; Figure 5). Adding VPA did not alter the viability of A549 cells compared with that of cells cultured in media containing cisplatin but without VPA (Figure 5a). The viability of MKN‐1 cells treated with cisplatin and VPA (300 and 1000 μM) was significantly lower than that of cells treated with cisplatin alone (Figure 5b). In HT29, Caco2 human colon cancer cells, Colon26 mouse colon cancer cells, and LLC mouse lung cancer cells, VPA in combination with cisplatin did not affect the antitumor effect of cisplatin (data not shown).
FIGURE 5.
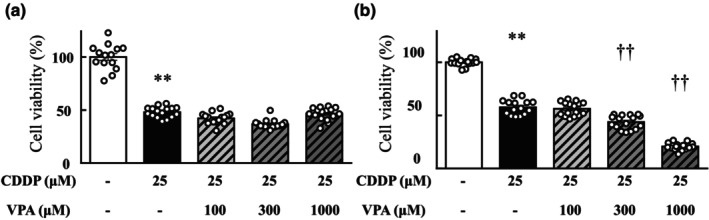
Anticancer effects of valproic acid with cisplatin on tumor cells. Viability of A549 (a) and MKN‐1 (b) cells. Viability of cells after 24 h of incubation in a medium with or without 25 μM cisplatin treatment was calculated as 100% for vehicle treatment. Valproic acid and cisplatin were simultaneously administered. Values are expressed as mean ± standard error of the mean. **p < 0.01 versus vehicle‐treated cells, †† p < 0.01 versus cisplatin‐treated cells, n = 8–16 in each group. CDDP, cisplatin; VPA, valproic acid.
DISCUSSION
Here, we assessed the effect of VPA treatment on cisplatin‐induced renal injury using VigiBase, animal models, and cell lines. Our findings indicate that VPA has the potential to serve as a preventive medication against cisplatin‐induced renal injury.
Although several candidates have been identified for managing cisplatin‐induced renal injury, none have been commercialized. 21 Drugs that are effective in basic experiments are not necessarily useful in clinical settings. Irrespective of their preclinical safety and effectiveness, ~90% of the drugs are discontinued during the clinical development phase. 22 Hence, we used VigiBase to examine the potential of VPA as an inhibitor of cisplatin‐induced kidney injury. We compared the reported incidence of kidney injury in patients receiving cisplatin alone versus in those receiving cisplatin in combination with candidate drugs. A previous study using a large medical information database and hospital medical records found an increased incidence of cisplatin‐induced kidney injury in patients administered ondansetron and a decreased incidence in those administered palonosetron. 18 Our results are similar to those of the previous study, thus confirming the accuracy of the present analysis. VPA exhibited an ROR of less than 1, indicating reduced occurrence of cisplatin‐induced renal injury (Table 1). These findings strongly support the clinical potential of VPA as a preventive agent for cisplatin‐induced renal injury.
To elucidate the renal protective effect and mechanism of action of VPA, we used a cisplatin‐induced renal injury mouse model. VPA dose was established by referring to the doses used in previous mouse studies. 23 , 24 The half‐maximal effective concentration of VPA, an index of anti‐epileptic effect in mice, is 400–500 mg/kg. The dose of valproic acid at which renal damage was prevented in this study was 100 mg/kg, which is lower than that used for achieving anti‐epileptic effects. Therefore, there is a high possibility that the same dose currently used clinically in humans will also be effective in preventing renal damage. We demonstrated that VPA is a safe drug to treat cisplatin‐induced kidney injury, as it did not induce any overt adverse tissue responses (Table 2). VPA‐treated kidneys appeared similar to vehicle‐treated kidneys when imaged. Evaluation at 72 h after cisplatin administration showed that renal function significantly decreased because of cisplatin treatment, whereas co‐administration of VPA led to a dose‐dependent improvement. The levels of markers of proximal tubular injury, Kim‐1 and Lcn2, 7 , 25 as well as inflammatory markers IL‐6, IL‐1β, and TNF‐α, significantly increased following cisplatin administration, but their levels were significantly reduced when VPA was co‐administered. IL‐6, IL‐1β, and TNF‐α are cytokines that promote inflammation. TNF‐α is particularly critical in the progression of cisplatin‐induced kidney damage and contributes to the release of other pro‐inflammatory cytokines. The production of TNF‐α in kidney cells, rather than in immune cells, exacerbates renal injury. 26 , 27 Inflammatory cell infiltration was also decreased with VPA co‐treatment (Figure 3). A previous study reported that VPA reduces conjunctival inflammation in a mouse model of conjunctival scarring. VPA has been suggested as a potential drug for therapeutic targeting of macrophages. 28 Furthermore, a marker of glomerular injury, the urine ALB/CRE ratio, significantly increased with cisplatin treatment, but VPA co‐administration attenuated this effect. Until now, cisplatin‐induced renal injury has been mainly associated with acute proximal tubular injury. 29 , 30 However, our study revealed, for the first time, that glomerular injury is also induced by cisplatin. Interestingly, VPA significantly suppressed cisplatin‐induced glomerular injury as well, providing important evidence that glomerular injury inhibition may have implications for preventing renal failure progression.
To clarify the action mechanism of VPA, we analyzed the early pathophysiology of cisplatin‐induced renal injury. At 24 h after cisplatin administration, renal dysfunction and elevated inflammatory markers were not detected, but a significant increase in Kim‐1 expression indicated early proximal tubular cell injury, which was effectively suppressed by VPA co‐administration. Kim‐1 expression level may be a promising marker for the early detection of renal damage. 15 , 31 , 32 Our results are consistent with those of previous studies. Here, VPA did not demonstrate a protective effect in PODO cells against cisplatin‐induced cell damage, but it showed a protective effect in HK2 cells.
An important factor in the development of cisplatin‐induced renal injury is the amount of cisplatin accumulated in the kidneys. 4 Our previous study suggested that differences in cisplatin accumulation at 4 h after cisplatin administration affect the extent of cisplatin‐induced renal injury. 18 To demonstrate that VPA is not related to cisplatin levels, renal platinum concentrations were determined to examine the effect of VPA on cisplatin accumulation in the kidneys. Our findings revealed no alterations in platinum levels in the kidneys or whole blood (Figure S1). Based on these results, we propose that cisplatin‐induced renal injury may initiate from proximal tubular injury, followed by a subsequent inflammatory response leading to renal dysfunction and glomerular injury. VPA may exert its protective effect by inhibiting early proximal tubular injury after cisplatin administration, thereby attenuating subsequent inflammatory responses and glomerular damage.
We focused on FOXO3 as an upstream factor targeted by VPA. FOXO activation promotes apoptosis. 33 , 34 , 35 , 36 Cisplatin covalently binds to purine bases, induces the dephosphorylation and nuclear translocation of FOXO3, suppresses downstream gene expression, and induces apoptosis. 37 Upon FOXO3 activation, gene expression in the mitochondria is suppressed, leading to cell death processes. 38 FOXO expression was significantly downregulated by the VPA add‐on compared with that by cisplatin only in the kidneys and HK2 cells. This finding suggests that, in normal cells, VPA may suppress FOXO expression and inhibit apoptosis. Previous studies have reported that histone deacetylase (HDAC) inhibitors inhibit FOXO3 expression and exhibit cell‐protective effects in the heart and liver. 39 , 40 VPA induces neuroprotective effects by inhibiting HDAC. 41 , 42 , 43 We believe that a similar mechanism was involved in the inhibition of proximal tubular cell damage observed in the present study. Recently, it has been reported that VPA inhibits the decrease in mitochondrial antioxidant enzyme expression in the kidneys caused by cisplatin through its HDAC inhibitory effect, which supports our findings. 44 Contrarily, in various cancer cells, HDAC inhibitors, such as VPA, induce FOXO expression and promote cell death. 34 , 35 , 38 , 45 , 46 We also confirmed that VPA, as a single agent, exerts antitumor effects in various cancer cells (Figure S2). Experiments using A549 and MKN‐1 cells showed that the anticancer effect of cisplatin was not suppressed by VPA, suggesting that VPA does not impede the therapeutic efficacy of cisplatin.
This study has certain limitations. First, the VigiBase database comprises voluntary reports, which could be influenced by biases, such as under‐reporting and over‐reporting. Moreover, VigiBase included only a small number of patients with acute renal failure treated with cisplatin in combination with VPA. Unfortunately, this study did not provide data regarding certain factors that may contribute to cisplatin‐induced renal injury, such as other nephrotoxic substances and hydration status. Second, this study relied solely on mouse and cell models to demonstrate the potential mitigation of cisplatin‐induced kidney injury by VPA. Therefore, it is important to exercise caution when extrapolating these findings to humans, as clinical trials are necessary to validate the clinical application of our results. Prospective controlled trials are needed to further investigate the efficacy and safety of VPA in this context.
Nevertheless, the study suggests the potential of VPA as a promising preventive agent for mitigating cisplatin‐induced kidney injury. To our knowledge, this is the first study to elucidate that VPA can effectively suppress proximal tubular cell apoptosis through the inhibition of FOXO3 expression, thereby inhibiting the progression of cisplatin‐induced renal dysfunction and glomerular injury. The observed prophylactic effect of VPA against cisplatin‐induced renal injury may be partially attributed to its ability to inhibit HDAC activity. Clinical trials are warranted to verify our findings in humans.
AUTHOR CONTRIBUTIONS
M.G. and T.Y. designed the research. T.Y., S.I., Y.S., K.M., and H.H. performed the experiments. H.H., M.K., F.A., K.Y., Y.I.I., K.M., T.N., T.S., Y.Z., and K.I. analyzed the data. T.Y. and M.G. wrote the manuscript.
FUNDING INFORMATION
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI [grant numbers 20K07132, 20H05799, and JP 22H04922 (AdAMS)]. The funders had no involvement in the design of the study; collection, analysis, and interpretation of data; writing of the report; or decision to submit the manuscript for publication.
CONFLICT OF INTEREST STATEMENT
The authors declared no competing interests for this work. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
The authors thank Editage (Tokyo, Japan) for English language editing.
Yoshioka T, Goda M, Kanda M, et al. Valproic acid treatment attenuates cisplatin‐induced kidney injury by suppressing proximal tubular cell damage. Clin Transl Sci. 2023;16:2369‐2381. doi: 10.1111/cts.13638
DATA AVAILABILITY STATEMENT
The data and codes that support the findings of this study are available from the corresponding author on request.
REFERENCES
- 1. Chovanec M, Abu Zaid M, Hanna N, el‐Kouri N, Einhorn LH, Albany C. Long‐term toxicity of cisplatin in germ‐cell tumor survivors. Ann Oncol. 2017;28:2670‐2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Isnard‐Bagnis C, Moulin B, Launay‐Vacher V, Izzedine H, Tostivint I, Deray G. [Anticancer drug‐induced nephrotoxicity]. Nephrol Ther. 2005;1:101‐114. [DOI] [PubMed] [Google Scholar]
- 3. Okada K, Ma D, Warabi E, et al. Amelioration of cisplatin‐induced nephrotoxicity in peroxiredoxin I‐deficient mice. Cancer Chemother Pharmacol. 2013;71:503‐509. [DOI] [PubMed] [Google Scholar]
- 4. Yokoo S, Yonezawa A, Masuda S, Fukatsu A, Katsura T, Inui KI. Differential contribution of organic cation transporters, OCT2 and MATE1, in platinum agent‐induced nephrotoxicity. Biochem Pharmacol. 2007;74:477‐487. [DOI] [PubMed] [Google Scholar]
- 5. Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int. 2008;73:994‐1007. [DOI] [PubMed] [Google Scholar]
- 6. Latcha S, Jaimes EA, Patil S, Glezerman IG, Mehta S, Flombaum CD. Long‐term renal outcomes after cisplatin treatment. Clin J Am Soc Nephrol. 2016;11:1173‐1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Karasawa T, Steyger PS. An integrated view of cisplatin‐induced nephrotoxicity and ototoxicity. Toxicol Lett. 2015;237:219‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cao Y, Semanchik N, Lee SH, et al. Chemical modifier screen identifies HDAC inhibitors as suppressors of PKD models. Proc Natl Acad Sci USA. 2009;106:21819‐21824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Marumo T, Hishikawa K, Yoshikawa M, Hirahashi J, Kawachi S, Fujita T. Histone deacetylase modulates the proinflammatory and ‐fibrotic changes in tubulointerstitial injury. Am J Physiol Renal Physiol. 2010;298:F133‐F141. [DOI] [PubMed] [Google Scholar]
- 10. Noh H, Oh EY, Seo JY, et al. Histone deacetylase‐2 is a key regulator of diabetes‐ and transforming growth factor‐beta1‐induced renal injury. Am J Physiol Renal Physiol. 2009;297:F729‐F739. [DOI] [PubMed] [Google Scholar]
- 11. Pang M, Kothapally J, Mao H, et al. Inhibition of histone deacetylase activity attenuates renal fibroblast activation and interstitial fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol. 2009;297:F996‐F1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sayyed SG, Gaikwad AB, Lichtnekert J, et al. Progressive glomerulosclerosis in type 2 diabetes is associated with renal histone H3K9 and H3K23 acetylation, H3K4 dimethylation and phosphorylation at serine 10. Nephrol Dial Transplant. 2010;25:1811‐1817. [DOI] [PubMed] [Google Scholar]
- 13. Van Beneden K, Geers C, Pauwels M, et al. Valproic acid attenuates proteinuria and kidney injury. J Am Soc Nephrol. 2011;22:1863‐1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Amirzargar MA, Yaghubi F, Hosseinipanah M, et al. Anti‐inflammatory effects of valproic acid in a rat model of renal ischemia/reperfusion injury: alteration in cytokine profile. Inflammation. 2017;40:1310‐1318. [DOI] [PubMed] [Google Scholar]
- 15. Speir RW, Stallings JD, Andrews JM, Gelnett MS, Brand TC, Salgar SK. Effects of valproic acid and dexamethasone administration on early bio‐markers and gene expression profile in acute kidney ischemia‐reperfusion injury in the rat. PLoS One. 2015;10:e0126622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lin T, Ren Q, Zuo W, et al. Valproic acid exhibits anti‐tumor activity selectively against EGFR/ErbB2/ErbB3‐coexpressing pancreatic cancer via induction of ErbB family members‐targeting microRNAs. J Exp Clin Cancer Res. 2019;38:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gottlicher M, Minucci S, Zhu P, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20:6969‐6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goda M, Kanda M, Yoshioka T, et al. Effects of 5‐HT((3)) receptor antagonists on cisplatin‐induced kidney injury. Clin Transl Sci. 2021;14:1906‐1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kanda M, Goda M, Maegawa A, et al. Discovery of preventive drugs for cisplatin‐induced acute kidney injury using big data analysis. Clin Transl Sci. 2022;15:1664‐1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tanaka H, Ishikawa E, Teshima S, Shimizu E. Histopathological study of human cisplatin nephropathy. Toxicol Pathol. 1986;14:247‐257. [DOI] [PubMed] [Google Scholar]
- 21. Ali BH, Al Moundhri MS. Agents ameliorating or augmenting the nephrotoxicity of cisplatin and other platinum compounds: a review of some recent research. Food Chem Toxicol. 2006;44:1173‐1183. [DOI] [PubMed] [Google Scholar]
- 22. Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3:711‐715. [DOI] [PubMed] [Google Scholar]
- 23. Leviel V, Naquet R. A study of the action of valproic acid on the kindling effect. Epilepsia. 1977;18:229‐234. [DOI] [PubMed] [Google Scholar]
- 24. Cao BJ, Peng NA. Magnesium valproate attenuates hyperactivity induced by dexamphetamine‐chlordiazepoxide mixture in rodents. Eur J Pharmacol. 1993;237:177‐181. [DOI] [PubMed] [Google Scholar]
- 25. Yu X, Meng X, Xu M, et al. Celastrol ameliorates cisplatin nephrotoxicity by inhibiting NF‐kappaB and improving mitochondrial function. EBioMedicine. 2018;36:266‐280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Faubel S, Lewis EC, Reznikov L, et al. Cisplatin‐induced acute renal failure is associated with an increase in the cytokines interleukin (IL)‐1beta, IL‐18, IL‐6, and neutrophil infiltration in the kidney. J Pharmacol Exp Ther. 2007;322:8‐15. [DOI] [PubMed] [Google Scholar]
- 27. Zhang B, Ramesh G, Norbury CC, Reeves WB. Cisplatin‐induced nephrotoxicity is mediated by tumor necrosis factor‐alpha produced by renal parenchymal cells. Kidney Int. 2007;72:37‐44. [DOI] [PubMed] [Google Scholar]
- 28. Seet LF, Toh LZ, Finger SN, Chu SWL, Wong TT. Valproic acid exerts specific cellular and molecular anti‐inflammatory effects in post‐operative conjunctiva. J Mol Med (Berl). 2019;97:63‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vickers AE, Rose K, Fisher R, et al. Kidney slices of human and rat to characterize cisplatin‐induced injury on cellular pathways and morphology. Toxicol Pathol. 2004;32:577‐590. [DOI] [PubMed] [Google Scholar]
- 30. Cornelison TL, Reed E. Nephrotoxicity and hydration management for cisplatin, carboplatin, and ormaplatin. Gynecol Oncol. 1993;50:147‐158. [DOI] [PubMed] [Google Scholar]
- 31. Huang JX, Kaeslin G, Ranall MV, et al. Evaluation of biomarkers for in vitro prediction of drug‐induced nephrotoxicity: comparison of HK‐2, immortalized human proximal tubule epithelial, and primary cultures of human proximal tubular cells. Pharmacol Res Perspect. 2015;3:e00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Abdelsalam M, Elmorsy E, Abdelwahab H, et al. Urinary biomarkers for early detection of platinum based drugs induced nephrotoxicity. BMC Nephrol. 2018;19:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reagan‐Shaw S, Ahmad N. RNA interference‐mediated depletion of phosphoinositide 3‐kinase activates forkhead box class O transcription factors and induces cell cycle arrest and apoptosis in breast carcinoma cells. Cancer Res. 2006;66:1062‐1069. [DOI] [PubMed] [Google Scholar]
- 34. Yang Y, Zhao Y, Liao W, et al. Acetylation of FoxO1 activates Bim expression to induce apoptosis in response to histone deacetylase inhibitor depsipeptide treatment. Neoplasia. 2009;11:313‐324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shats I, Gatza ML, Liu B, Angus SP, You L, Nevins JR. FOXO transcription factors control E2F1 transcriptional specificity and apoptotic function. Cancer Res. 2013;73:6056‐6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Prasad SB, Yadav SS, Das M, et al. Down regulation of FOXO1 promotes cell proliferation in cervical cancer. J Cancer. 2014;5:655‐662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fernandez de Mattos S, Villalonga P, Clardy J, Lam EW. FOXO3a mediates the cytotoxic effects of cisplatin in colon cancer cells. Mol Cancer Ther. 2008;7:3237‐3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhong S, Chen W, Wang B, et al. Energy stress modulation of AMPK/FoxO3 signaling inhibits mitochondria‐associated ferroptosis. Redox Biol. 2023;63:102760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Aune SE, Herr DJ, Mani SK, Menick DR. Selective inhibition of class I but not class IIb histone deacetylases exerts cardiac protection from ischemia reperfusion. J Mol Cell Cardiol. 2014;72:138‐145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mihaylova MM, Vasquez DS, Ravnskjaer K, et al. Class IIa histone deacetylases are hormone‐activated regulators of FOXO and mammalian glucose homeostasis. Cell. 2011;145:607‐621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Leng Y, Wang J, Wang Z, et al. Valproic acid and other HDAC inhibitors upregulate FGF21 gene expression and promote process elongation in glia by inhibiting HDAC2 and 3. Int J Neuropsychopharmacol. 2016;19:pyw035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Silva MR, Correia AO, Dos Santos GCA, et al. Neuroprotective effects of valproic acid on brain ischemia are related to its HDAC and GSK3 inhibitions. Pharmacol Biochem Behav. 2018;167:17‐28. [DOI] [PubMed] [Google Scholar]
- 43. Sinn DI, Kim SJ, Chu K, et al. Valproic acid‐mediated neuroprotection in intracerebral hemorrhage via histone deacetylase inhibition and transcriptional activation. Neurobiol Dis. 2007;26:464‐472. [DOI] [PubMed] [Google Scholar]
- 44. Li Y, Li K, Zhao W, et al. VPA improves ferroptosis in tubular epithelial cells after cisplatin‐induced acute kidney injury. Front Pharmacol. 2023;14:1147772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yamada T, Amann JM, Tanimoto A, et al. Histone deacetylase inhibition enhances the antitumor activity of a MEK inhibitor in lung cancer cells harboring RAS mutations. Mol Cancer Ther. 2018;17:17‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kiweler N, Brill B, Wirth M, et al. The histone deacetylases HDAC1 and HDAC2 are required for the growth and survival of renal carcinoma cells. Arch Toxicol. 2018;92:2227‐2243. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Data Availability Statement
The data and codes that support the findings of this study are available from the corresponding author on request.