

Key Points
-
•
Acquired BAX mutations represent a novel mechanism of adaptive resistance to venetoclax-based therapy for patients with AML.
-
•
Defective BAX enhances resistance to BH3-mimetics targeting diverse BCL-2 family prosurvival proteins in AML.
Visual Abstract
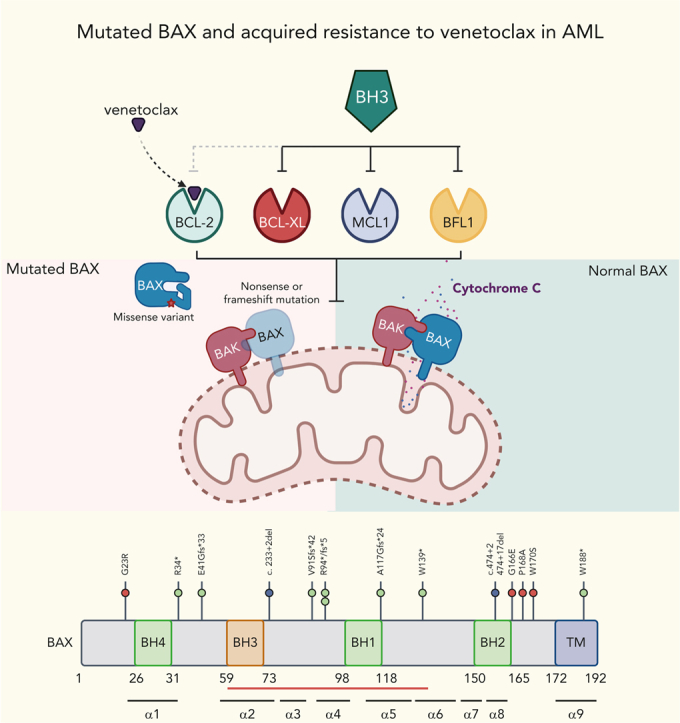
Abstract
Randomized trials in acute myeloid leukemia (AML) have demonstrated improved survival by the BCL-2 inhibitor venetoclax combined with azacitidine in older patients, and clinical trials are actively exploring the role of venetoclax in combination with intensive chemotherapy in fitter patients with AML. As most patients still develop recurrent disease, improved understanding of relapse mechanisms is needed. We find that 17% of patients relapsing after venetoclax-based therapy for AML have acquired inactivating missense or frameshift/nonsense mutations in the apoptosis effector gene BAX. In contrast, such variants were rare after genotoxic chemotherapy. BAX variants arose within either leukemic or preleukemic compartments, with multiple mutations observed in some patients. In vitro, AML cells with mutated BAX were competitively selected during prolonged exposure to BCL-2 antagonists. In model systems, AML cells rendered deficient for BAX, but not its close relative BAK, displayed resistance to BCL-2 targeting, whereas sensitivity to conventional chemotherapy was variable. Acquired mutations in BAX during venetoclax-based therapy represent a novel mechanism of resistance to BH3-mimetics and a potential barrier to the long-term efficacy of drugs targeting BCL-2 in AML.
The BH3 mimetic venetoclax improves outcomes for older, unfit patients with acute myeloid leukemia (AML) when combined with azacytidine or low-dose cytosine arabinoside, but relapse is common. Moujalled et al identified loss-of-function BAX mutations in 17% of AML samples from patients relapsing after venetoclax, providing a novel explanation for resistance that also applies in vitro to other BH3 mimetics. In contrast, these mutations are uncommon after intensive chemotherapy, reinforcing the case for exploration of venetoclax–chemotherapy combinations.
Introduction
The treatment landscape for patients with acute myeloid leukemia (AML) has witnessed remarkable change, with Food and Drug Administration approval of 9 new drugs since 2017, including the BH3-mimetic venetoclax to target prosurvival BCL-2 in patients with AML unfit for intensive chemotherapy.1 BCL-2 and related prosurvival family proteins, for example, MCL1, repress activation of apoptosis effectors BAX and BAK, until sufficiently neutralized by apoptosis-initiating BH3-only proteins.2 Although the clinical activity of venetoclax monotherapy in AML was modest, clinical responses and survival improved significantly when venetoclax was combined with azacitidine or low-dose cytarabine.3, 4, 5 High response rates have also been observed among fitter patients with newly diagnosed or relapsed/refractory AML receiving venetoclax in combination with intensive chemotherapy.6,7 Nevertheless, despite promising clinical progress, relapse remains an important cause of treatment failure.
Longitudinal analyses of paired diagnosis/relapse samples from patients receiving venetoclax-based therapy for AML have identified expansion or gain of clones harboring activated kinases (such as internal tandem duplications [ITDs] of FLT3) or disrupted TP53.7, 8, 9 Selection of AML populations with monocytic differentiation at relapse has also been reported, potentially related to higher levels of MCL1 expression inherent to cells of monocytic lineage.10
To investigate other causes of venetoclax resistance in AML models, two independent research groups used genome-wide CRISPR-Cas9 deletion screens and identified genes disrupting the apoptosis pathway, such as TP53, BAX, and NOXA.11,12 We have recently shown that deficient TP53 impairs activation of downstream BAX and BAK in response to sublethal concentrations of venetoclax, giving TP53-defective AML cells a competitive advantage and predisposing to earlier disease progression.13 More recently, in patients with chronic lymphocytic leukemia (CLL), expansion of BAX variants was identified in the myeloid compartment of patients with long-term exposure to venetoclax, suggesting that BAX defects favored outgrowth of preleukemic populations exposed to drugs targeting BCL-2.14
We now document the presence of BAX variants in 17% of patients relapsing after venetoclax-based therapy for AML, whereas such populations were rare in patients relapsing after standard chemotherapy. Observed BAX variants included frameshift abnormalities predicted to truncate the protein and/or cause nonsense-mediated decay of its messenger RNA, as well as missense variants in the COOH-terminal region that impair BAX proapoptotic function by retaining it in the cytosol. In some cases, evolution of pathogenic BAX variants at relapse was polyclonal, with a dominant BAX variant accompanied by additional minor BAX clones. We found that mutant BAX typically arose from a hematopoietic population ancestral to the primary AML clone dominant at diagnosis. Single-cell multiomic studies confirmed that mutant BAX could arise in either the leukemic or preleukemic bone marrow (BM) compartment. In model systems, we demonstrate that leukemic cells rendered deficient for BAX, but not for the closely related pro-apoptotic effector BAK, are resistant to BCL-2–targeted drugs administered either alone or in combination with other BH3-mimetics. Although deficient or functionally defective BAX was associated with resistance to inhibitors of BCL-2 in various AML models, cross resistance to other BH3-mimetics and cytotoxic drugs was variable. These findings highlight deleterious BAX variants as a prominent and therapy-related cause of resistance in patients receiving BH3-mimetics targeting prosurvival proteins in AML.
Methods
Primary AML specimens
BM samples were obtained from patients with AML after informed consent and in accordance with procedures approved by Alfred Health Human Research Ethics Committee.
Single-cell DNA and protein library preparation and sequencing
The Tapestri Platform (Mission Bio) and custom panel containing 144 amplicons covering BAX and 22 genes mutated in AML were used. Sequencing was performed on a NovaSeq 6000 (Illumina). Results were visualized using Tapestri Insights.
BAX-targeted next-generation sequencing
Bulk sequencing for BCL-2, BAX, and myeloid genes was performed using a custom QIAseq Targeted DNA Panel for error-corrected digital DNA sequencing.14
Drugs and cytotoxic agents
S55746 and S63845 were provided by Servier Laboratories. A1331852 was provided by Guillaume Lessene (Walter and Eliza Hall Institute). Venetoclax, idarubicin, cytarabine, and daunorubicin were purchased from Selleckchem.
Generation of OCI-AML3 cells with acquired resistance to BH3-mimetics
Induced resistance to BCL-2 and MCL1 inhibitors (BCL-2i and MCL1i, respectively) was achieved by progressive exposure of OCI-AML3 cells to increasing doses of BCL-2i, MCL1i, or combined BCL-2i and MCL1i over a period of 3 months. Once resistance was established, cells were cultured in the absence of drug for 6 weeks.
Generation of BAX or BAK knockout cell lines using CRISPR-Cas9 gene editing
Single-guide RNAs targeting human BAX or BAK were synthesized and cloned into FgH1tUTG (Addgene Plasmid, #70183), which permits doxycycline-inducible expression of the single-guide RNA and constitutive expression of a green fluorescent protein reporter. All lentiviruses were produced in HEK293T cells and cells were transduced using established protocols.
Assessing BAX integration by carbonate extraction
Cells were incubated with 500 nM venetoclax, permeabilized with 0.025% weight-to-volume ratio digitonin, and fractionated by centrifugation for immunoblotting assays.
Generating MOLM13 cells expressing BAX WT or BAXP168A by retroviral infection
BAX wild-type (WT) or P168A mutant constructs were cloned into the retroviral expression vector pMSCV-IRES-GFP and transduced into MOLM-13 WT or BAX/BAK double knockout (DKO) cells using the packaging cell line HEK293T cells.
Mouse AML xenograft model
Irradiated NOD-SCID IL2Rγ (NSG)–null mice were IV injected with 1 × 105 OCI-AML3, OCI-AML3 combo R, OCI-AML3 BAX−/−, or OCI-AML3 BAK−/− cells, and nonirradiated NSG mice were IV injected with 1 × 105 MOLM13 WT or MOLM13 BAX/BAK DKO+BAXP168A cells, and dosing commenced on day 4 posttransplant for 4 weeks. Drug efficacy was determined by flow-cytometric analysis of human CD45+ cells.
Immunohistological studies
Sections were fixed in 3.7% formalin, decalcified, then embedded in paraffin and sectioned. Human CD45 was stained using established methods using antihuman CD45 monoclonal antibody (Dako, catalog number M0701) in a Dako Autostainer Plus.15
For detailed description of the methods, please refer to the supplemental materials, available on the Blood website.
Results
BAX mutations are implicated in adaptive resistance to venetoclax in patients with AML
To investigate mechanisms of adaptive resistance to venetoclax in AML, we studied paired diagnosis/treatment failure samples from 41 patients who either achieved initial remission but later relapsed (n = 34) or were primary refractory (n = 7) to venetoclax combined with either low-dose cytarabine or hypomethylating agent (lower intensity) or cytarabine/idarubicin (high intensity) (Figure 1A). In the current cohort, FLT3-ITD or TP53 abnormalities were detected at relapse in 12% and 29% of cases, respectively (Figure 1A; supplemental Figure 1A). Based on previous reports of BCL-2–binding groove variants (G101V or D103Y) in patients with CLL receiving venetoclax, we searched for but did not identify these BCL-2 variants in our AML cohort relapsing after or refractory to venetoclax treatment.16 A single BCL-2 variant of uncertain significance (BCL-2 G203S with a variant frequency of 22%) was detected in the chemotherapy cohort (supplemental Figure 1B).
Figure 1.

Venetoclax-based therapies drive selection of novel BAX variants in relapsed AML. (A) Patients with either primary refractory or relapsed AML divided according to whether venetoclax (VEN) was combined with lower intensity (hypomethylating agent or low-dose cytarabine [LDAC]) or high intensity (infusional cytarabine + idarubicin) chemotherapy. The venetoclax dose used is indicated, along with the FLT3 and TP53 mutation status and BM blast percentage at the time of treatment failure. The time spent in remission (months) is shown as a green bar. Treatment failure is shown as a pink-colored bar cap. Patients refractory to therapy spent no time in remission. No BAX variants were identified among patients with primary refractory disease. At relapse, 7 patients (ID number shown) were found to harbor ≥1 BAX variant (red star within pink cap) and the dominant predicted BAX protein change is shown. (B) The distribution of BAX variant allele frequencies (yellow circles) at relapse are shown in the venetoclax-resistant cohort (n = 41), along with a control population of patients with AML relapsing after prior intensive chemotherapy (n = 34). The predicted protein change is indicated for BAX variants with variant allele frequencies (VAFs) ≥5%. (C) Schematic overview of BAX variants. Protein domain structure of BAX and identified variants observed in patients treated with venetoclax or standard chemotherapy. The presence of four BCL-2 homology (BH) domains and the transmembrane domain (TM) are indicated along with the location of each of the nine alpha helices. The red bar indicates the position of the hydrophobic groove. Missense variants, frameshift/nonsense variants, and splice site variants are denoted by pink, green, and blue circles, respectively. (D-G) Fishplot representation of selected patients with BAX variants (also refer to supplemental Figure 1). Clonal architecture presented in fishplot format was inferred from bulk sequencing showing clonal dynamics of leukemic or preleukemic clones in serial samples from 4 venetoclax-treated patients with detected BAX mutations. The dominant BAX subpopulation is shown in red in each case. (D-E) BAX variants emerging with leukemic relapse. (F) A BAX mutation emerges in remission, suggesting presence in an expanded preleukemic population after suppression of the original diagnostic AML clone. (G) BAX mutation is present at diagnosis and persists in remission after venetoclax-based therapy, despite suppression of several AML-related mutations. Duration of therapy is denoted by the gray arrow, clinical status including first complete remission (CR1), CR2, or measurable residual disease (MRD). Available cytogenetic, BM blast information, and time elapsed to remission and relapse are indicated below each fishplot. ∗indicates time points verified by single-cell DNA sequencing. Freq, frequency.
Unbiased genome-wide in vitro CRISPR screens have previously identified BAX deficiency as a putative resistance mechanism to selective inhibitors of BCL-2 or MCL1.11,12,17 Although we have recently reported the selective outgrowth of BAX mutations in the myeloid compartment of patients with CLL treated with venetoclax,14 defective BAX as a tumor resistance mechanism to venetoclax in AML has yet to be reported. We therefore screened our venetoclax-treated cohort for pathogenic BAX variants and identified mutations in 7 of 41 patients (17%) at relapse, with no cases identified among patients with primary refractory disease (Figure 1A-B). Among patients with a BAX abnormality, 5 of 41 (12%) had a BAX VAF of ≥5% (median, 31%; range, 9.7% 48.6%). Three of the 5 patients with a dominant BAX variant also harbored a concurrent minor BAX clone (VAF range, 0.7%-2.9%), suggesting co-evolution of multiple clones and the potential for some clones to harbor multihit BAX abnormalities (supplemental Table 1A).
To determine if the selection of BAX variants at relapse was specific to venetoclax-based therapy, we examined a cohort of patients with AML relapsing after conventional intensive chemotherapy (Figure 1B; supplemental Table 1B-C). As expected of patients who were fit for intensive chemotherapy, the median age of this cohort was younger and less frequently had secondary/therapy-related AML. In the chemotherapy failure cohort, only 2 of 34 patients (6%) had a minor BAX variant, and in both cases, the BAX variant was very small (VAF, 0.73% and 3.5%). Analysis of publicly available data from The Cancer Genome Atlas (n = 200) and Beat AML (n = 622) cohorts failed to reveal the presence of BAX variants at baseline in AML, acknowledging the limited sensitivity of whole-exome sequencing. Thus, dominant BAX variants were not observed as a mechanism of treatment failure among patients receiving conventional chemotherapy.
The BAX variants that emerged at relapse were of two types. The first included frameshift, nonsense, or donor splice site abnormalities upstream of the BAX COOH-terminal domain, likely resulting in nonsense-mediated decay of the BAX messenger RNA, truncation, or a structurally altered form of BAX (Figure 1C). The second comprised missense abnormalities concentrated in, or just before, the BAX COOH-terminal domain (G166E, P168A, and W170S), suggesting perturbation of BAX protein function (Figure 1C). BAX translocation from the cytosol into the mitochondrial outer membrane (MOM), where it disrupts membrane integrity, depends critically on its COOH-terminal α9 helix.18 Insertion of this COOH-terminal transmembrane region (TMR) into the MOM is required for BAX oligomerization and MOM pore formation.19 In healthy cells, BAX is cytosolic and inert, with its TMR sequestered within a hydrophobic pocket on BAX. A BAX P168 hinge residue located just before the BAX TMR is crucial for TMR release from its hydrophobic pocket.19 Pertinently, BAX P168 missense variants (such as BAX P168A in patient CAL-035) and W170A (which cooperates with P168 to release the TMR) strongly impede apoptosis, likely by impairing release of the BAX TMR.19
Clonal dynamics of BAX variants associated with venetoclax resistance in AML
In two of the BAX clones detected at relapse after venetoclax-based therapy (Figure 1B), morphologic relapse coincided with the emergence of a relapse-specific and dominant BAX variant (Figure 1D-E). In CAL-021, a BAX variant (BAX R94Pfs∗5; VAF, 48.6%) emerged at relapse when leukemic blasts represented 21% of BM cells (Figure 1D; supplemental Figure 2A). In CAL-035, a relapse-specific BAX missense variant inactivating the critical TMR hinge residue (BAX P168A; VAF, 31.5%) emerged at relapse (with 17% BM blasts; Figure 2E; supplemental Figure 2B). In both cases, the BAX variant was not detected at diagnosis or at remission, emerging only at relapse after exposure to venetoclax in combination with chemotherapy.
Figure 2.

BAX variants are observed in leukemic cell populations. (A) Proportion of cells in CAL-021 at relapse identified as WT or containing leukemia-associated mutations (top). Clones with leukemia-containing mutations are listed in decreasing order. Schematic of mutation zygosity for each clone (bottom). (B) Two-dimensional UMAP plot of CAL-021 at relapse showing specific phenotypic populations based on antibody tags. CAL-021 cells clustered into four main blood cell compartments including myeloid and monocytic blast populations is shown (top left). The remaining 5 UMAP plots are colored according to the genotype of each individual DNA variant. (C) Proportion of each mutant clone in CAL-021 observed at relapse within specified blood cell lineages. (D) Proportion of cells in CAL-013 after one week of venetoclax treatment identified as WT or containing leukemia-associated mutations (top). Clones with leukemia-containing mutations are listed in decreasing order. Schematic of mutation zygosity for each clone (bottom). (E) Two-dimensional UMAP plot of CAL-013 after one week of venetoclax treatment according to specific phenotypic populations identified by antibody tags. Cells clustered into six major blood cell compartments, including myeloid and monocytic blast populations (top left). The remaining five UMAP plots are colored according to the genotype of each DNA variant detected. (F) Percentages of each mutant clone observed in CAL-013 after 1 week of venetoclax treatment in specified blood cell lineages. HET, heterozygous; HOM, homozygous; NK, natural killer.
In these two cases and two others with a dominant BAX variant at relapse (AML388 and CAL-013), venetoclax-based therapy had markedly repressed or extinguished the original AML-defining clone, with relapse derived from a rare ancestral population (Figure 1D-G). For example, in case CAL-021, NPM1 and NRAS variants were prominent at diagnosis but lost at relapse. Rather, recurrence was driven by an ancestral preleukemic clone containing DNMT3A and IDH1 variants, along with subclonal evolution of one dominant and three minor BAX variants (Figure 1D). In CAL-035, the original AML-defining clone harboring mutant IDH1, NRAS, RUNX1, or STAG2 was lost after venetoclax-based therapy, with relapse revealing the emergence of two new BAX variants (BAX P168A and BAX G166E) (Figure 1E).
Although venetoclax-based therapy also suppressed the founding AML clones in AML388 and CAL-013, the linkage of resistance to mutant BAX was more complex. In AML388, venetoclax-based therapy led to suppression of RUNX1, PHF6, and BCORL1, but not preleukemic variants ASXL1, SRSF2, and ETV6, which persisted (Figure 1F). During remission, two new preleukemic variants arose, a dominant clone harboring both RUNX1 R207Q and BAX R94∗ and a minor population with RUNX1 R207Q and TP53 A161T (Figure 1F). Approximately 12 months later, the patient relapsed with 45% blasts. At relapse, two new TP53 mutant clones emerged (TP53 splice site variant and TP53 G145V) (Figure 1F). Single-cell sequencing confirmed the presence of two parallel clones, one harboring both TP53 variants (TP53 double-hit) and a mutually exclusive clone carrying the BAX R94∗ variant (Figure 1F; supplemental Figure 3B). In this case, compartmentally distinct mechanisms of venetoclax resistance evolved in parallel, mediated in leukemic blasts by biallelic mutant TP53 and in the preleukemic compartment by mutant BAX. In summary, during selective pressure imparted by venetoclax, BAX variants have the potential to mediate resistance in AML blasts, or alternatively promote the expansion of the preleukemic BM compartment, as reported previously.14
One limitation of bulk sequencing is that the association between mutations and the cell of origin is inferred. To clarify the phylogeny of gene variants within phenotypically defined hematopoietic BM populations, a multiomic single-cell approach was used. In CAL-021, single-cell DNA sequencing at relapse demonstrated DNMT3A R735C, DNMT3A R729Q, and IDH1 R132C in myeloid, monocytic, and NK cell compartments (Figure 2A-C). BAX R94Pfs∗5, along with a minor BAX donor splice site subclone, was confined to myeloid/monocytic blast cells, without substantial involvement of either T or NK lymphoid populations (Figure 2A-C; supplemental Figure 3A). Therefore, single-cell studies confirmed that therapeutic pressure exerted by venetoclax could select for the expansion of BAX variants in AML blasts as a potential mechanism of relapse.
For CAL-013, single-cell multiomic analysis was performed on BM infiltrated with 76% blasts after the patient had received 8 days of venetoclax monotherapy. The BAX E41Gfs∗33 variant was present in myeloid and monocytic blasts but not in B and T cells (supplemental Figure 3C; Figure 2E-F). The multiomic study revealed a clear association between BAX E41Gfs∗33 and variants NPM1 and FLT3 in myeloid/monocytic blasts (Figure 2F). These blasts initially displayed resistance to venetoclax (administered from diagnosis to day 8), with the expansion of AML blasts harboring BAX, NPM1, and FLT3 variants (Figure 1G). Upon addition of chemotherapy to venetoclax, these blasts were cleared, resulting in near eradication of variants NPM1 and FLT3 by day 53, amidst a persistent preleukemic BAX E41Gfs∗33 population. This example suggests that although BAX E41Gfs∗33 in AML was not able to be suppressed by venetoclax alone, the BAX variant appeared more susceptible to chemotherapy.
Acquired resistance to BCL-2 inhibitors in vitro and in vivo is associated with BAX deficiency
We next investigated the functional impact of BAX mutation/loss in response to BH3-mimetic and conventional cytotoxic therapies. The OCI-AML3 cell line has a naturally occurring BAX E41Gfs∗33 variant in exon 3, analogous to the BAX mutation in patient CAL-013 (Figure 1G). This line allowed us to study the fate of AML cells heterozygous for the BAX variant during exposure to BH3-mimetics targeting BCL-2 without the confounding effect of concomitant chemotherapy. Serial passage of OCI-AML3 cells with escalating doses of vehicle (dimethyl sulfoxide (DMSO), up to 3 μM) over a 3-month period demonstrated no change in the BAX E41Gfs∗33 burden (VAF, 41%) (Figure 3A). In contrast, escalating doses of a BCL-2i (S55746, up to 3 μM) resulted in a marked increase in the BAX E41Gfs∗33 burden (VAF, 95%) (Figure 3A) associated with BAX loss of heterozygosity (LOH) (supplemental Figure 4). Removal of the BCL-2i from culture for a further 6 weeks confirmed the change was stable, with the BAX variant remaining at high VAF.
Figure 3.

Acquired resistance to BH3-mimetics selects for BAX loss in vitro. (A) VAF of indicated variants in OCI-AML3 cells with acquired resistance to BH3-mimetics compared with dimethyl sulfoxide (DMSO) control. OCI-AML3 cells, which harbor a naturally occurring BAX E41Gfs∗33 abnormality were continuously passaged with progressively higher concentrations (maximum 3 μM) of BCL-2i resistant (BCL-2i-R), MCL1i resistant (MCL1i-R), or combined BCL-2i-R and MCL1i-R (combo R). Cells were then treated in the absence of drug(s) for 6 weeks and BAX targeted amplicon sequencing was performed and compared with DMSO control. (B) Immunoblot profiling of BCL-2 family proteins in OCI-AML3 cultures tolerant to 3 μM of drug (DMSO, BCL-2i-R, MCL1i-R, or combo R). (C-E) Sensitivity of BH3-mimetic resistant (tolerant to 3 μM) OCI-AML3 cells to various drugs and combinations. OCI-AML3 DMSO control, BCL-2i-R, MCL1i-R, or combo R cells were treated with the indicated drugs as single agents or in equimolar combination (0.001-10 μM). Sensitivity was expressed as the 50% lethal concentration (LC50 μM) determined by flow cytometry after 48 hour-exposure. Error bars are standard deviation (SD) of 3 independent experiments. ∗P < .05, ∗∗P <.01, and ∗∗∗P < .001.
Induced resistance of OCI-AML3 cells to BCL-2–targeted therapy (BCL-2i-R) was associated with acquired loss of BAX protein expression, whereas expression of other BCL-2 family members (BCL-2, BCL-XL, MCL1, BIM, and BAK) were unchanged (Figure 3B). In contrast, graduated adaptation to MCL1i was associated with increased expression of BCL-2 and BIM, without any change in BAX (Figure 3B). In another instance, however, acquired tolerance to an MCL1i (MCL1i-R) was found to be associated with the loss of BAX expression, suggesting multiple routes to MCL1i resistance are possible (supplemental Figure 5). Experimentally acquired resistance to combined inhibition of BCL-2 and MCL1 (combo R) also led to clonal enrichment of BAX E41Gfs∗33 (VAF, 90%) and loss of expressed BAX. In addition, BAK expression also appeared attenuated in combo R cells (Figure 3B). Further genomic analysis of cells rendered resistant to combined inhibitors of BCL-2 and MCL1, revealed the unexpected emergence of a BAK G126S variant, concurrent with the expanded BAX E41Gfs∗33 variant (Figure 3A; supplemental Figure 6). A review of clinical samples from patients at relapse after venetoclax-based therapy or conventional chemotherapy, but without exposure to an MCL1i, did not identify occurrence of pathogenic BAK variants (supplemental Figure 1A-B).
We next assessed whether induced resistance to BH3-mimetics targeting either BCL-2, MCL1, or both could be salvaged by combination with other BH3-mimetics or alternatively, by cytotoxic drugs commonly used for AML. The selective BCL-2i S55746 has comparable in vitro activity to venetoclax.15 Accordingly, induced resistance of OCI-AML3 cells to S55746 (BCL-2i-R) produced cross resistance to venetoclax (Figure 3C). Notably, the BCL-2i-R cells were also resistant to the MCL1i S63845, either alone or with dual MCL1/BCL-2 or triple MCL1/BCL-2/BCL-XL targeting using selective small molecule inhibitors (Figure 3C). Interestingly, BAX loss did not confer resistance to the cytotoxic drugs etoposide, cytarabine, idarubicin, or daunorubicin. OCI-AML3 cells rendered resistant to the MCL1i S63845 (MCL1-i-R) displayed only modest cross resistance to BCL-2i (Figure 3D). In contrast, OCI-AML3 cells with acquired resistance to combined BCL-2 and MCL1 inhibition (combo R) were cross resistant to BH3-mimetics targeting BCL-2 and/or MCL1, as well as combined targeting of BCL-2, BCL-XL, and MCL1, whereas sensitivity to conventional cytotoxic drugs remained intact (Figure 3E). Notably, these findings suggest that acquired resistance to BCL-2 targeting associated with BAX loss could render these cells cross resistant to other BH3-mimetics without markedly impairing the efficacy of standard cytotoxic drugs used to treat AML.
Efficacy of BH3-mimetics targeting BCL-2 and MCL1 is impaired by deficiency of BAX but not BAK
Apoptosis induced by BH3-mimetic agents typically depends on downstream effectors, BAX and BAK.20 Although the BAX frameshift variant in OCI-AML3 cells was selected during prolonged exposure to BCL-2i, we cannot rule out the possibility that other drug resistance mechanisms were co-acquired, as was evident in MCL1-iR cells. We therefore sought to validate the effect of BAX-deficient function by orthogonally deleting the gene by doxycycline-inducible CRISPR-Cas9 editing (Figure 4A).
Figure 4.

BAX deficiency confers resistance to combined BCL-2 and MCL1 targeting in vitro. (A) BAX expression in CRISPR-Cas9–edited OCI-AML3 cells. Immunoblot demonstrating CRISPR-Cas9–induced BAX depletion in OCI-AML3 cells using guide RNAs (gRNAs) targeting BAX (gRNA-1.1 or gRNA-2.1) or a nontargeted (empty vector [EV]) control. Cells were treated with 5 μg/mL doxycycline for 72 hours to induce BAX loss. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. (B) Drug sensitivity of CRISPR-Cas9–edited OCI-AML3 cells. OCI-AML3 cells transduced with gRNA targeting BAX or a nontargeted control were treated with indicated drugs (0.001-10 μM for BH3-mimetics, 0.01-100 μM for cytarabine, and 0.001 to 1μM for idarubicin) and the LC50 determined by flow cytometry after 48-hour exposure. Error bars are SD of 2 independent experiments. ∗P < .05, ∗∗P <.01, ∗∗∗P < .001, and ∗∗∗∗P < .0001. (C) Survival of mice engrafted with BAX knockout cells in response to BH3-mimetic therapy. Irradiated NSG mice were transplanted with 105 OCI-AML3 nontargeted EV control or OCI-AML3 BAX knockout cells (gRNA 2.1). Dosing commenced on day 4 posttransplant and mice were randomly divided into cohorts of 6 mice and treated with vehicle or a combination of venetoclax (75 mg/kg, weekdays by gavage) and S63845 (25 mg/kg, IV weekly) for 4 weeks. Kaplan-Meier survival analysis (ethical end point) shows that combined treatment with venetoclax/S63845 significantly prolongs survival in OCI-AML3 EV but not OCI-AML3 BAX knockout cohorts (black bar indicates duration of treatment). (D) Survival of mice engrafted with BAK knockout cells in response to BH3-mimetic therapy. Irradiated NSG mice were transplanted with 105 OCI-AML3 nontargeted EV control or OCI-AML3 BAK knockout cells (gRNA 2.1). Dosing commenced on day 4 posttransplant and mice were randomly divided into cohorts of 6 mice and treated with vehicle or a combination of venetoclax (75 mg/kg, weekdays by gavage) and S63845 (25 mg/kg, IV weekly) for 4 weeks. Kaplan-Meier survival analysis (ethical end point) shows that combined treatment with venetoclax/S63845 significantly prolongs survival in both the OCI-AML3 EV and OCI-AML3 BAK knockout cohorts (black bar indicates duration of treatment). ns, not significant.
Short-term drug survival assays demonstrated that BAX deletion rendered OCI-AML3 cells resistant to BH3-mimetics targeting either BCL-2 or BCL-2/MCL1 in combination (Figure 4B), corroborating our findings in OCI-AML3 cells harboring BAX LOH and protein loss. Interestingly, BAX-deficient OCI-AML3 cells remained sensitive to cytarabine or anthracyclines, suggesting resistance conferred by BAX loss in OCI-AML3 cells was specific to BH3-mimetic drugs (Figure 4B).
We next explored whether BAX-deficient OCI-AML3 cells would compromise efficacy of BH3-mimetic drugs in a xenograft model of AML in vivo. As expected, a regimen cotargeting BCL-2 and MCL1 significantly improved the survival of mice engrafted with OCI-AML3 cells transduced with a nontargeting CRISPR-Cas9 EV, compared with vehicle control (Figure 4C). In contrast, combination BCL-2 and MCL1 targeting failed to improve the survival of mice engrafted with OCI-AML3 cells rendered BAX deficient by CRISPR-Cas9 editing. This response was specific to BAX deletion, as mice engrafted with BAK-deficient OCI-AML3 cells (supplemental Figure 7) had comparable survival to a cohort of mice engrafted with EV OCI-AML3 cells and treated with combined venetoclax and S63845 (Figure 4D). Thus, deficiency of BAX, but not BAK, contributed to BH3-mimetic drug resistance in a model of AML in vivo.
The BAX P168A variant is functionally resistant to venetoclax
In the cohort of patients with acquired resistance to venetoclax-based therapy, patient CAL-035 developed adaptive resistance in association with the emergence of a BAX P168A missense mutation at relapse (Figure 1E), suggesting this variant might impair BAX pro-apoptotic function. To determine the functional relevance of BAX P168A, MOLM-13 AML cells deficient in both BAX and BAK (DKO) were transduced with vectors expressing either WT BAX or BAX P168A and compared with parental MOLM-13 cells (Figure 5A). As expected, MOLM-13 BAX/BAK DKO cells were largely resistant to venetoclax-induced cell death (Figure 5B), whereas reexpression of WT BAX restored venetoclax sensitivity. BAX/BAK DKO cells reconstituted with BAX P168A, however, remained venetoclax-resistant, suggesting this variant abrogated BAX function in response to BH3-mimetics. To determine if BAX P168A functions in a dominant-negative manner, parental MOLM-13 cells were transduced with either BAX WT or BAX P168A and treated with venetoclax (Figure 5C). Comparable sensitivity to venetoclax was demonstrated, arguing against dominant-negative activity exerted by BAX P168A against endogenous BAX (Figure 5C; supplemental Figure 8). Interestingly, although MOLM-13 BAX/BAK DKO cells were relatively more resistant to cytotoxic drugs (cytarabine, idarubicin, and daunorubicin) than WT MOLM-13 cells, substantial antileukemic activity was still observed, consistent with additional modes of action associated with cytotoxic drugs (supplemental Figure 8).
Figure 5.

The BAX P168A variant impairs BAX translocation and confers resistance to venetoclax in vitro and in vivo. (A) Expression of BAX P168A in BAX/BAK DKO MOLM-13 cells. Western blot showing BAX expression levels in MOLM-13 WT, BAX/BAK DKO (−) cells, or BAX/BAK DKO engineered to express BAX WT or BAX P168A upon transduction with retroviral expression constructs. (B) Sensitivity of BAX/BAK DKO plus BAX P168A cells to venetoclax. The indicated MOLM-13 cell lines (BAX/BAX DKO, BAX/BAX DKO + BAX WT, BAX/BAX DKO + BAX P168A, and parental MOLM13 WT) were treated with increasing concentrations of venetoclax (0.05-5 μM) for 24 hours and cell viability determined by annexin V/4′,6-diamidino-2-phenylindole staining and flow cytometry. Data are means ± standard error of the mean of 3 independent experiments. (C) BAX P168A does not have dominant-negative activity. BAX WT or BAX P168A was retrovirally expressed in MOLM-13 WT cells and cells treated with 500 nM venetoclax for 24 hours. Cell viability was determined by annexin V/4′,6-diamidino-2-phenylindole staining and flow cytometry. Data are means ± standard error of the mean of 2 independent experiments. (D) The BAX P168A mutation reduces MOM translocation and integration of BAX. MOLM-13 DKO cells expressing BAX WT or BAX P168A were pre-incubated with 25 μM Q-VD-OPh for 1 hour and then treated with 500 nM venetoclax. After 5 hours, cells were subjected to carbonate extraction and fractions run on sodium dodecyl sulfate polyacrylamide gel electrophoresis and immunoblotted for BAX. The results are representative of 3 independent experiments. (E-F) MOLM-13 BAX P168A cells are resistant to venetoclax in vivo. Irradiated NSG mice were transplanted with 5 × 105 MOLM13 WT or MOLM13 BAX/BAK DKO cells expressing BAX P168A. On day 4 posttransplant, cohorts of 3 mice were treated with vehicle or venetoclax 75 mg/kg gavage weekdays for 2 weeks. Cohorts were euthanized and human CD45+ cells in BM and peripheral blood enumerated by flow cytometry. Error bars represent SD of 3 mice per treatment arm.
The BAX P168 residue is critical for COOH-terminal function.19 Concordant with BAX P168A impairing BAX activation by BH3-mimetics, venetoclax induced BAX translocation and stable insertion into the MOM in MOLM-13 BAX/BAK DKO cells expressing WT BAX but not BAX P168A (Figure 5D). We surmise that BAX P168A “locks” the BAX COOH-terminal tail in its hydrophobic pocket, preventing its release and translocation/association of BAX to the MOM in response to the apoptotic stimulus induced by venetoclax.19 To assess the functional effect of BAX P168A on venetoclax response in vivo, cohorts of mice engrafted with MOLM-13 or DKO plus BAX P168A were treated with either vehicle or venetoclax. As expected, 10 days of venetoclax therapy reduced MOLM-13 cell burden in the peripheral blood and BM, whereas it failed to suppress the burden of MOLM-13 DKO plus BAX P168A cells in either compartment in vivo (Figure 5E-F; supplemental Figure 9). These findings confirm that BAX P168A mediates functional resistance to venetoclax-based therapy and support the emergence of BAX P168A in patient CAL-035 as the mechanism of its resistance to venetoclax-based therapy (Figure 1E).
Discussion
With an increasing number of countries approving venetoclax for AML treatment, the number of patients receiving this therapy worldwide is likely to increase substantially. Following on from our previous work describing FLT3-ITD and TP53 abnormalities in adaptive resistance to venetoclax in AML, we now document the emergence of pathogenic BAX variants in ∼17% of patients relapsing after venetoclax-based therapy.7,8 These BAX variants included frameshift abnormalities disrupting BAX protein expression or missense variants in the COOH-terminal region impairing BAX pro-apoptotic function. These lesions seldom overlapped with either FLT3-ITD or TP53 abnormalities, and, in several cases, the progressing BAX mutant clone arose from a clonal population distinct from the original AML clone characterized at diagnosis. In contrast, BAX variants were rare and of very low VAF (<5%) in a patient cohort relapsing after conventional chemotherapy without previous venetoclax exposure.
In CLL, which is clinically sensitive to single-agent venetoclax, on-target BCL-2 mutations have been identified as a relatively common pathway to treatment resistance.16 In AML, where venetoclax-based combination therapy is the norm, more diverse patterns of resistance have emerged.8,10,13,21,22 Our studies now highlight the potential for BAX mutants to emerge at AML relapse upon exposure to BH3-mimetics such as venetoclax. In some cases, the emerging BAX variant was not evident at the time venetoclax-based therapy was commenced. Therefore, BAX variants should be considered at relapse, especially when more common causes of venetoclax-mediated resistance, such as FLT3 or TP53 variants, have been excluded. In other cases, BAX variants were present in preleukemic cells, as exemplified by emergence (Figure 1F) or persistence (Figure 1G) in remission after venetoclax-based therapy, analogous to recent reports of preleukemic BAX variants emerging after venetoclax therapy for CLL.14 Similar to OCI-AML3 cells with acquired resistance to BCL-2i associated with the emergence of BAX E41Gfs∗33 LOH and loss of BAX expression, it remains to be seen whether patients with preleukemic heterozygous BAX variants receiving long-term venetoclax will evolve BAX subclones deficient in expressed BAX.
In model studies, we demonstrated that BAX loss of function also impaired the efficacy of venetoclax when combined with other BH3-mimetics, such as MCL1 inhibitors. Unexpectedly, in contrast to BH3-mimetics targeting BCL-2, BAX deficiency did not impair cytotoxic drug efficacy. In addition, BAX/BAK-deficient cells conferred partial but not complete resistance to the antileukemic actions of cytotoxic drugs, consistent with mechanisms of action that are not BAX/BAK dependent.23 Conventional cytotoxic therapies may therefore play a role in preventing or salvaging venetoclax-resistant AML cells harboring defects in BAX. In contrast, our findings suggest that combining BH3-mimetics to target BCL-2 and MCL1 concurrently may not be sufficient to abrogate the selection of clinically detrimental BAX variants.15,24,25 Indeed, the combined targeting of BCL-2 and MCL1 in cells with a preexisting BAX defect could favor selection of BAK variants with impaired BAK function, the latter a consequence of MCL1 selective inhibitors.26 Therefore, in AML regimens incorporating BH3-mimetics, continued inclusion of chemotherapy may play a useful role in mitigating the selection of BAX and BAK variants as mediators of therapeutic resistance.
Previous studies have demonstrated the essential role of BAX and BAK in facilitating apoptosis induced by BH3-only proteins.27 Our studies suggest that, in certain AML contexts, deficiency of BAX alone may be sufficient to impair BH3-mimetic drug activity. Unexpectedly, we found that venetoclax-associated BAX resistance was not circumvented by combination with BH3-mimetics targeting complementary prosurvival members, suggesting that pathogenic loss of BAX function may, in some contexts, also result in resistance to other BH3-mimetics. With increasing use of venetoclax in AML treatment, the emergence of BAX variants in both the leukemic and preleukemic compartments should be considered when exploring the cause of adaptive resistance to BH3-mimetic drugs. Together with the recent discovery that BAX variants emerge in the myeloid compartment of patients with CLL treated with venetoclax,14 our current work highlights the importance of BAX loss-of-function defects as a potential hurdle to the long-term success of venetoclax and potentially its combination with other BH3-mimetics in the treatment of myeloid neoplasms, including AML.
Conflict-of-interest disclosure: S.B. is an employee of Servier. A.W.R., D.C.S.H., A.H.W., N.S.A., and D.M.M are current employees, whereas M.A.D. is a former employee of the Walter and Eliza Hall Institute of Medical Research, which has received milestone and royalty payments related to venetoclax. A.W.R is an inventor on a patent related to venetoclax assigned to AbbVie and Genentech. D.C.S.H. has received research funding from Genentech. A.W.R., D.C.S.H., and A.H.W. have received research funding from Servier and AbbVie. The remaining authors declare no competing financial interests.
Acknowledgments
This work was supported by fellowships and grants from the Australian National Health and Medical Research Council (Program grant 1016701) (D.C.S.H. and J.M.A.), (1113577 and 1079560) (A.W.R.), (1156024) (D.C.S.H.), (Investigator grant 1176175) (F.C.B), (1174902) (A.W.R.), (1162809) (A.H.W.), (Senior Research Fellowship 1156095) (M.J.H.), Metcalf Family Fellowship (A.H.W.); Leukemia and Lymphoma Society, USA, Specialized Center of Research (grant 7015-18) (A.W.R., A.H.W., J.M.A., J.HA., and D.C.S.H.); Victorian Cancer Agency MCRF Fellowship (19011) (D.M.M.) and (grant 15018) (A.W.R. and A.H.W.); Cancer Council Victoria (grants-in-aid 1141740) (D.M.M.); Medical Research Future Fund (grant 1141460) (A.H.W.); Tour de Cure Foundation (RSP-212-2020) (D.M.M); Snowdome Foundation (P.B.); and the Australian Cancer Research Foundation.
This work was made possible through Victorian State Government Operational Infrastructure Support and the Australian Government National Health and Medical Research Council Independent Research Institute Infrastructure Support Scheme.
Authorship
Contribution: D.M.M., F.C.B., and A.H.W. designed the research; D.M.M., F.C.B., M.A.D., G.P., N.S.A., V.L., E.T., T.M., C.C.C., S.M., and M.C. performed the research; E.T., L.K., S.B., M.C., M.S., M.J.H., G.L., J.S., J.M.A., P.B., and D.C.S.H. contributed vital new reagents or analytical tools; D.M.M., F.C.B., C.C.C., E.T., P.B., A.W.R., D.C.S.H., and A.H.W. analyzed and interpreted data; D.M.M. and F.C.B. performed the statistical analysis; A.H.W., D.M.M., A.W.R., D.C.S.H., and F.C.B. wrote the manuscript; and all authors read and approved the manuscript.
Footnotes
Data are available on request from the corresponding author, Andrew H. Wei (wei.a@wehi.edu.au).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Supplementary Material
References
- 1.Döhner H, Wei AH, Löwenberg B. Towards precision medicine for AML. Nat Rev Clin Oncol. 2021;18:1–14. doi: 10.1038/s41571-021-00509-w. [DOI] [PubMed] [Google Scholar]
- 2.Adams JM, Cory S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018;25(1):27–36. doi: 10.1038/cdd.2017.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konopleva M, Pollyea DA, Potluri J, et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 2016;6(10):1106–1117. doi: 10.1158/2159-8290.CD-16-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DiNardo C, Jonas B, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617–629. doi: 10.1056/NEJMoa2012971. [DOI] [PubMed] [Google Scholar]
- 5.Wei AH, Montesinos P, Ivanov V, et al. Venetoclax plus LDAC for patients with untreated AML ineligible for intensive chemotherapy: phase 3 randomized placebo-controlled trial. Blood. 2020;135(24):2137–2145. doi: 10.1182/blood.2020004856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DiNardo CD, Lachowiez CA, Takahashi K, et al. Venetoclax combined with FLAG-IDA induction and consolidation in newly diagnosed and relapsed or refractory acute myeloid leukemia. J Clin Oncol. 2021;39(25):2768–2778. doi: 10.1200/JCO.20.03736. JCO. 20.03736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chua CC, Roberts AW, Reynolds J, et al. Chemotherapy and Venetoclax in Elderly Acute Myeloid Leukemia Trial (CAVEAT): a phase Ib dose-escalation study of venetoclax combined with modified intensive chemotherapy. J Clin Oncol. 2020;38(30):3506–3517. doi: 10.1200/JCO.20.00572. [DOI] [PubMed] [Google Scholar]
- 8.DiNardo CD, Tiong IS, Quaglieri A, et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood. 2020;135(11):791–803. doi: 10.1182/blood.2019003988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chyla B, Daver N, Doyle K, et al. Genetic biomarkers of sensitivity and resistance to venetoclax monotherapy in patients with relapsed acute myeloid leukemia. Am J Hematol. 2018;93(8):E202–E205. doi: 10.1002/ajh.25146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pei S, Pollyea DA, Gustafson A, et al. Monocytic subclones confer resistance to venetoclax-based therapy in patients with acute myeloid leukemia. Cancer Discov. 2020;10(4):536–551. doi: 10.1158/2159-8290.CD-19-0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nechiporuk T, Kurtz SE, Nikolova O, et al. 7th. Vol. 9. Cancer discovery; 2019. The TP53 apoptotic network is a primary mediator of resistance to BCL2 inhibition in AML cells; pp. 910–925. CD-19-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen X, Glytsou C, Zhou H, et al. 7th. Vol. 9. Cancer discovery; 2019. Targeting mitochondrial structure sensitizes acute myeloid leukemia to venetoclax treatment; pp. 890–909. CD-19-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thijssen R, Diepstraten ST, Moujalled D, et al. Intact TP-53 function is essential for sustaining durable responses to BH3-mimetic drugs in leukemias. Blood. 2021;137(20):2721–2735. doi: 10.1182/blood.2020010167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blombery P, Lew TE, Dengler MA, et al. Clonal hematopoiesis, myeloid disorders and BAX-mutated myelopoiesis in patients receiving venetoclax for CLL. Blood. 2021;139(8):1198–1207. doi: 10.1182/blood.2021012775. [DOI] [PubMed] [Google Scholar]
- 15.Moujalled D, Pomilio G, Ghiurau C, et al. 4th. Vol. 33. Leukemia; 2019. Combining BH3-mimetics to target both BCL-2 and MCL1 has potent activity in pre-clinical models of acute myeloid leukemia; pp. 905–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blombery P, Anderson MA, Gong J-n, et al. Acquisition of the recurrent Gly101Val mutation in BCL2 confers resistance to venetoclax in patients with progressive chronic lymphocytic leukemia. Cancer Discov. 2019;9(3):342–353. doi: 10.1158/2159-8290.CD-18-1119. [DOI] [PubMed] [Google Scholar]
- 17.Thijssen R, Diepstraten ST, Moujalled D, et al. Intact TP53 function is essential for sustaining durable responses to BH3-mimetic drugs in leukemias. Blood. 2021;137(20):2721–2735. doi: 10.1182/blood.2020010167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suzuki M, Youle RJ, Tjandra N. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell. 2000;103(4):645–654. doi: 10.1016/s0092-8674(00)00167-7. [DOI] [PubMed] [Google Scholar]
- 19.Schinzel A, Kaufmann T, Schuler M, Martinalbo J, Grubb D, Borner C. Conformational control of Bax localization and apoptotic activity by Pro168. J Cell Biol. 2004;164(7):1021–1032. doi: 10.1083/jcb.200309013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Delft MF, Wei AH, Mason KD, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10(5):389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones CL, Stevens BM, D'Alessandro A, et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell. 2018;34(5):724–740.e724. doi: 10.1016/j.ccell.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stevens BM, Jones CL, Pollyea DA, et al. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat Cancer. 2020;1(12):1176–1187. doi: 10.1038/s43018-020-00126-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Gao W, Shi X, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547(7661):99–103. doi: 10.1038/nature22393. [DOI] [PubMed] [Google Scholar]
- 24.Kotschy A, Szlavik Z, Murray J, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538(7626):477–482. doi: 10.1038/nature19830. [DOI] [PubMed] [Google Scholar]
- 25.Caenepeel S, Brown SP, Belmontes B, et al. AMG 176, a selective MCL1 inhibitor, is effective in hematologic cancer models alone and in combination with established therapies. Cancer Discov. 2018;8(12):1582–1597. doi: 10.1158/2159-8290.CD-18-0387. [DOI] [PubMed] [Google Scholar]
- 26.Dewson G, Kratina T, Sim HW, et al. To trigger apoptosis, Bak exposes its BH3 domain and homodimerizes via BH3: groove interactions. Mol Cell. 2008;30(3):369–380. doi: 10.1016/j.molcel.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 27.Cheng E, Wei MC, Weiler S, et al. BCL-2, BCL-XL sequester BH3 domain-only molecules preventing BAX-and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8(3):705–711. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.