

Key Points
-
•
We prove in genotype-matched complement-mediated aHUS cohorts that C5 inhibition results in a statistical improvement in ESKD-free survival.
-
•
We demonstrate that biallelic pathogenic mutations in EXOSC3 cause eculizumab nonresponsive aHUS.
Visual Abstract
Abstract
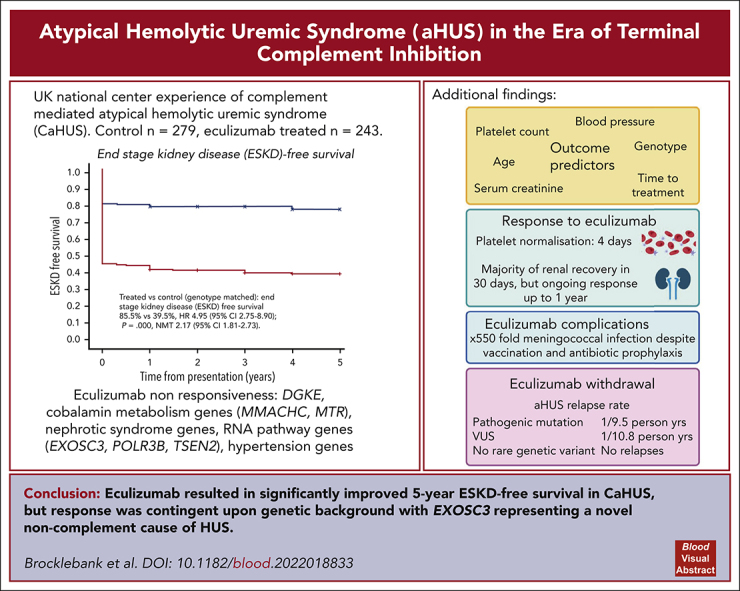
Historically, the majority of patients with complement-mediated atypical hemolytic uremic syndrome (CaHUS) progress to end-stage kidney disease (ESKD). Single-arm trials of eculizumab with a short follow-up suggested efficacy. We prove, for the first time to our knowledge, in a genotype matched CaHUS cohort that the 5-year cumulative estimate of ESKD-free survival improved from 39.5% in a control cohort to 85.5% in the eculizumab-treated cohort (hazard ratio, 4.95; 95% confidence interval [CI], 2.75-8.90; P = .000; number needed to treat, 2.17 [95% CI, 1.81-2.73]). The outcome of eculizumab treatment is associated with the underlying genotype. Lower serum creatinine, lower platelet count, lower blood pressure, and younger age at presentation as well as shorter time between presentation and the first dose of eculizumab were associated with estimated glomerular filtration rate >60 ml/min at 6 months in multivariate analysis. The rate of meningococcal infection in the treated cohort was 550 times greater than the background rate in the general population. The relapse rate upon eculizumab withdrawal was 1 per 9.5 person years for patients with a pathogenic mutation and 1 per 10.8 person years for those with a variant of uncertain significance. No relapses were recorded in 67.3 person years off eculizumab in those with no rare genetic variants. Eculizumab was restarted in 6 individuals with functioning kidneys in whom it had been stopped, with no individual progressing to ESKD. We demonstrated that biallelic pathogenic mutations in RNA-processing genes, including EXOSC3, encoding an essential part of the RNA exosome, cause eculizumab nonresponsive aHUS. Recessive HSD11B2 mutations causing apparent mineralocorticoid excess may also present with thrombotic microangiopathy.
In this month’s CME article, Brocklebank et al present detailed clinical characteristics and complete genetic analyses of a large United Kingdom cohort of patients with suspected complement-associated atypical hemolytic uremic syndrome (CaHUS) treated with eculizumab. The authors demonstrate superiority over historical therapies in genotype-matched patients with CaHUS, document that outcome after eculizumab cessation is genotype-dependent, and show that relapse is very unlikely if no pathogenic gene variant is present. The data also reveal a new causative genetic lesion for eculizumab-refractory CaHUS and provide a benchmark that should prove useful as additional options for CaHUS treatment emerge.
Medscape Continuing Medical Education online

In support of improving patient care, this activity has been planned and implemented by Medscape, LLC and the American Society of Hematology. Medscape, LLC is jointly accredited with commendation by the Accreditation Council for Continuing Medical Education (ACCME), the Accreditation Council for Pharmacy Education (ACPE), and the American Nurses Credentialing Center (ANCC), to provide continuing education for the healthcare team.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1.0 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Successful completion of this CME activity, which includes participation in the evaluation component, enables the participant to earn up to 1.0 MOC points in the American Board of Internal Medicine's (ABIM) Maintenance of Certification (MOC) program. Participants will earn MOC points equivalent to the amount of CME credits claimed for the activity. It is the CME activity provider's responsibility to submit participant completion information to ACCME for the purpose of granting ABIM MOC credit.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives; (2) study the education content; (3) take the post-test with a 75% minimum passing score and complete the evaluation at https://www.medscape.org/journal/blood; and (4) view/print certificate. For CME questions, see page 1406.
Disclosures
CME questions author Laurie Barclay, freelance writer and reviewer, Medscape, LLC, declares no competing financial interests.
Learning Objectives
Upon completion of this activity, participants will:
-
1.
Describe clinical and genetic characteristics of individuals with suspected complement-mediated atypical hemolytic uremic syndrome (CaHUS) treated with eculizumab, based on a national observational cohort study using data from the National Renal Complement Therapeutics Centre (NRCTC) in the United Kingdom
-
2.
Describe outcomes of individuals with suspected CaHUS treated with eculizumab, based on a national observational cohort study using NRCTC data
-
3.
Describe clinical implications of real-world experience of treating individuals with suspected CaHUS with eculizumab, based on a national observational cohort study using NRCTC data
Release date: October 19, 2023; Expiration date: October 19, 2024
Introduction
Complement-mediated atypical hemolytic uremic syndrome (CaHUS) is a rare kidney disease in which complement activation occurs on endothelial cell surfaces, resulting in thrombotic microangiopathy.1 It is characterized by the clinical presentation of thrombocytopenia, microangiopathic hemolytic anemia (MAHA), and acute kidney injury. In ∼50% of individuals, an inherited (CFH, CFI, CD46, CFB, and C3 mutations) or acquired (factor H autoantibodies [FHAA]) complement abnormality is identified.1,2 Historically, management comprised supportive care with or without plasma exchange, and outcomes were poor, with end-stage kidney disease (ESKD) or death occurring at first presentation in a high proportion of individuals, contingent upon the genetic background.3
Small, single-arm trials of eculizumab suggested its efficacy4 in CaHUS; however, its rarity restricted the ability to power clinical trials and published follow-up has been relatively short.4, 5, 6 In England, all cases of suspected CaHUS are referred to a single national specialized center, the National Renal Complement Therapeutics Centre (NRCTC) (http://www.atypicalhus.co.uk/),7 and a genotyped registry established in the preeculizumab era allows for comparison.8
In this observational cohort study, we report the real-world experience of treating individuals with suspected CaHUS with eculizumab in a national cohort, describe the response to treatment, and compare outcomes with those of a genotype-matched cohort who did not receive eculizumab. Here, we describe novel noncomplement genetic causes of aHUS.
Methods
Study population
All individuals who were referred to the NRCTC with suspected CaHUS were considered for the study (Figure 1). Renal transplantation–associated CaHUS cases were excluded.9 The control CaHUS cohort comprised individuals referred between 1995 and 2019 with a pathogenic mutation in a gene associated with CaHUS or positive FHAA not treated with eculizumab. The treated cohort comprised individuals referred between 2013 and July 2019 with suspected CaHUS who received eculizumab for native kidney disease (inclusion/exclusion criteria, supplemental Methods, available on the Blood website). Individuals referred before 2013 who were treated with eculizumab either as part of a clinical trial or on compassionate grounds were included in the treated cohort. FHAA aHUS management did not include immunosuppression. In the survival analysis, individuals who were treated with eculizumab at the time of relapse but not at the first presentation of aHUS (n = 17) were analyzed in the control cohort until the point at which they received eculizumab. The minimum follow-up duration was 12 months.
Figure 1.

Patient selection. All individuals referred to the NRCTC with suspected aHUS were considered for study entry. Individuals referred between 1995 and 2012 (before the approval of eculizumab) and those not treated with eculizumab were retrospectively identified. Those with a pathogenic mutation in a gene associated with aHUS or a positive factor H autoantibody were included in the control cohort. Individuals referred to the National Service between 2013 and July 2019 with suspected aHUS who received eculizumab for native kidney disease were prospectively identified and included in the treated cohort. Recipients who had undergone kidney transplantation were excluded. Individuals referred after 2013 but who did not receive eculizumab, were included in the control cohort. THBD and VTN mutations have been reported in aHUS but were not detected in our cohort, and the PLG susceptibility variant c.1481C>T42 was identified in 1 individual who had compound heterozygous DGKE mutations. «Twenty-two individuals were treated with eculizumab between 2010 and 2012, before regulatory approval, either as part of a clinical trial or on compassionate grounds. ¥Excluding individuals treated preemptively during kidney transplantation or posttransplantation. In addition to eculizumab, treatment could have comprised supportive management, renal replacement therapy, and plasma exchange. ¢Management determined by the treating physician could have comprised supportive management, renal replacement therapy, and plasma exchange. ¤Of the 40 individuals, 24 recovered renal function, 15 developed/presented with ESKD, and 1 died. See supplemental Table 1 for genetic and clinical details. ꬸFor survival analysis, 17 individuals were analyzed in the control CaHUS group until the point at which they received eculizumab for relapse and were then analyzed in the treated CaHUS group. CFH, n = 8; CD46, n = 7; C3, n = 1; combined, n = 1. ‡For 2 individuals (nmd) in the treated CaHUS cohort and 13 individuals in the control CaHUS cohort (CFH, n = 5; CFHR1 hybrid, n = 1; CFI, n = 1; CD46, n = 4; C3, n = 1; and FHAA, n = 1), survival data were not available. ¶Thrombotic thrombocytopenic purpura is excluded before treatment with eculizumab: treatment is not commenced if ADAMTS13 <10%. Other secondary TMAs may be identifiable before the initial decision to commence eculizumab based on clinical history or initial laboratory testing: disseminated intravascular coagulation; malignancy-associated TMA; bone marrow transplantation–associated TMA; de novo TMA after solid organ transplantation; and drug-induced TMA. Some secondary TMAs may not be identified until further genetic or serological tests or kidney biopsies are available: Shiga toxin HUS (STEC-HUS); pneumococcal HUS; HIV; cobalamin C deficiency TMA; glomerular disease–associated TMA; and autoimmune disease–associated TMA. #No complement mutations and no FHAA were identified in any of those with a secondary cause identified. §PLG screened only for the susceptibility variant c.1481C>T rs4252128; identified in 1 individual (with compound heterozygous DGKE mutations). ꬸOther diagnoses, n = 4: renal biopsy specimen showed severe chronic damage, no TMA detected with a renal biopsy specimen; renal biopsy specimen showed acute tubular necrosis; no TMA detected with a renal biopsy specimen; died before definitive diagnosis made. ∗Analysis of variants in July 2020 classified those with definitive evidence of functional significance as pathogenic and those without as VUS. GN, glomerulonephritis; IgA, immunoglobulin A; PNH, paroxysmal nocturnal hemoglobinuria.
Genetic analysis for known aHUS-associated genes and autoantibody analysis were performed as previously described.10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 Whole-exome sequencing was performed on a research basis as described,21 the analysis was approved by the Northern and Yorkshire Multicenter Research Ethics Committee, and informed consent was obtained in accordance with the Declaration of Helsinki.
Outcomes
The primary outcome in the comparison between eculizumab-treated CaHUS with complement mutations or FHAA and control CaHUS cohorts with complement mutations or FHAA was the 5-year ESKD-free survival. The secondary end points are described in supplemental Methods.
Variant classification
The analysis of variants in July 2020 classified those with definitive evidence of functional significance as pathogenic and those without as variants of unknown significance (VUS). Mutations in CFH and CFI were subclassified as type 1 (low plasma levels) or type 2 (normal plasma levels but with functional consequences22,23), and mutations in CD46 were subclassified as type 1 (reduced cell surface expression) or type 2 (normal expression with reduced function).24
Statistical analysis
Renal survival was examined using Kaplan-Meier analysis and Cox regression. Significance tests for continuous variables were analysis of variance for normally distributed data and the Kruskal-Wallis test for data that were not normally distributed. Serial creatinine measurements were analyzed using the Friedman test. Multivariate analysis is described in the supplemental Methods. Statistical analyses were performed using the IBM Statistical Package for Social Sciences and R.25
Results
Study population
The control CaHUS cohort included 279 individuals, and the eculizumab-treated cohort included 243 individuals, of whom 192 had CaHUS and 51 had a subsequent diagnosis identified (Figure 1; supplemental Table 1). The participant characteristics are summarized in Table 1.
Table 1.
Participant characteristics
| Characteristic | Eculizumab-treated cohort (n = 243), n (%) | Control cohort (n = 279), n (%) |
|---|---|---|
| Age at first presentation, y | ||
| Median | 25 | 22 |
| Range | 0-80 | 0-79 |
| Sex | ||
| Male | 98 (41) | 135 (48) |
| Female | 145 (59) | 144 (52) |
| Plasma exchange | ||
| Yes | 130 (53) | 114 (38) |
| No | 94 (39) | 67 (23) |
| Data not available | 20 (8) | 116 (39) |
| Complement gene mutation | 90 (37) | 279 (100) |
| CFH | 33 (37∗) | 131 (47) |
| CFHR1:CFH hybrid | 1 (1∗) | 11 (4) |
| CFI | 6 (7∗) | 19 (7) |
| CD46 | 23 (26∗) | 61 (22) |
| C3 | 10 (11∗) | 24 (9) |
| CFB | 1 (1∗) | 0 |
| Factor H autoantibody positive | 11 (12∗) | 29 (10) |
| Combined mutation(s)/autoantibody | 5 (6∗) | 4 (1) |
| No complement mutation/autoantibody | 102 (42); 24 (10) with VUS | 0 |
| Not CaHUS | 51 (21) | NA |
| Eculizumab-treated CaHUS cohort (n = 192), n (%) | ||
| Trigger, n = 189† | 59 (31) | |
| Infection | 31 (16) | |
| Pregnancy | 18 (10) | |
| Other | 12 (6) | |
| None | 128 (68) | |
| Extrarenal manifestations, n = 191† | ||
| Neurological§ | 22 (12) | |
| Cardiac|| | 8 (4) | |
| Pancreatitis | 4 (2) | |
| Other¶ | 2 (1) | |
| None | 155 (81) | |
| Prodromal diarrhea, n = 180† | 45 (25) | |
|
Laboratory variables at presentation mean (range) |
||
| Serum creatinine (μmol/L), n = 187† | 488 (18-2435) | |
| eGFR (ml/min per 1.73 m2) | 21 (2-118) | |
| Hemoglobin (g/L), n = 175† | 80 (38-142) | |
| Platelet count (×109/L), n = 185† | 69 (4-329) | |
| Lactate dehydrogenase (IU), n = 177† | 2373 (290-19130) | |
| C-reactive protein (mg/L), n = 140† | 44 (0.5-309) | |
| Urine protein:creatinine ratio (mg/mmol), n = 90† | 1381 (0-17520) | |
| C3 (g/L) n = 182† | 0.93 (0.04-2.31) | |
| Presenting blood pressure | ||
| Adults: systolic blood pressure, mean (range), mmHg, n = 92† | 175 (100-262) (88% hypertensive‡) | |
| Children: % hypertensive, n = 51† | 30 (59%‡) | |
Percentage of those with CaHUS and a mutation or FHAA.
n refers to number of individuals for whom data were available.
Hypertension defined by >140mmHg systolic in adults and >95th centile in children, according to the fourth report on diagnosis, evaluation, and treatment of high blood pressure in children and adolescents.
Extrarenal manifestations: Neurological manifestations reported were seizures, n = 12; stroke, n = 7; hemiparesis, n = 3; agitation/confusion, n = 3; encephalopathy, n = 2; cortical blindness, n = 1; and reduced conscious level, n = 1 (in some individuals >1 was reported).
Cardiac manifestations were left ventricular systolic dysfunction, n = 5, and cardiomyopathy, n = 2.
Other manifestations reported were liver failure and retinopathy.
Genetic analysis
A total of 90 out of 192 (47%) of the eculizumab-treated CaHUS cohort had an inherited (n = 76) or acquired (n = 11) complement abnormality or both (n = 3). An additional 24 of 192 (12.5%) patients had a VUS. The control CaHUS cohort comprised inherited (n = 250) or acquired (n = 29) complement abnormalities or both (n = 1) (Figure 1). The genetic and outcome data for each participant are reported in supplemental Table 2 (control cohort) and supplemental Table 3 (eculizumab-treated cohort). The genetic variants are demonstrated in supplemental Figure 1. CFH mutations were the most common and were predominantly type 2 C-terminal (n = 77, 47%) or type 1 (n = 56, 34%). CFI mutations were predominantly type 1 (n = 21, 84%). Type 1 mutations comprised 94% (n = 79) of the CD46 mutations. Most of the CaHUS mutations were heterozygous. Homozygous mutations were infrequently identified in CFH (n = 3; 1.8%), CD46 (n = 7; 8.3%), CFI (n = 1; 4%), and C3 (n = 1; 2.5%), whereas compound heterozygous mutations were identified in CFH (n = 1; 0.6%), CD46 (n = 3; 3.6%), and CFI (n = 1; 4%). No mutations were detected in THBD.
The proportion of individuals with a family history of aHUS was 10.1% in the treated cohort and 38.9% in the control cohort (supplemental Figure 2). The genetic causes of 68 pedigrees with suspected familial thrombotic microangiopathy (TMA) who were referred to our center are summarized in supplemental Figure 2.
In 51 individuals who commenced eculizumab, a diagnosis other than CaHUS was ultimately made (Figure 1). Of these, 8 were found to have a recognized genetic cause of TMA: 6 DGKE, 1 MMACHC, and 1 MTR. Four individuals had pathogenic variants in nephrotic syndrome-associated genes (INF2, LMX1B, NPHS2, and ACTN4), 1 had pathogenic variants in HSD11B2, 1 had pathogenic variants in AGXT (primary hyperoxaluria), and 3 had pathogenic variants in RNA pathway genes (EXOSC3 and POLR3B). Two individuals had inherited red cell abnormalities (DHFR and G6PD) mimicking aHUS. Screening of the control cohort also revealed several rare genetic variants of noncomplement genes (ADAMTS13, DGKE, TSEN2, INF2, G6PD, COL4A5, CUBN, RNU4ATAC, and GNE).
Clinical characteristics
The clinical and laboratory data for the eculizumab-treated cohort according to mutation type are reported in Table 2. The incidence rate of CaHUS was 0.41 per million per year. In adults, the mean systolic blood pressure was 174 mmHg, and there was no statistical difference in blood pressure between mutation types (supplemental Figure 3Ai). Fifty-nine percent of children were hypertensive at presentation (supplemental Figure 3Aii). There was no difference in proteinuria between the mutation groups (supplemental Figure 3B). C-reactive protein (CRP) was statistically significantly lower in the CaHUS with mutation and FHAA groups than in the group without CaHUS (P = .009; Table 2; supplemental Figure 3C). The presenting complement levels are described (supplemental Figure 3Di-iii).
Table 2.
Characteristics of the eculizumab-treated cohort
| Mutation/autoantibody |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| All CaHUS | CFH | CFHR1 | CFI | CD46 | C3 | CFB | FHAA | Combined | Nmd | Not CaHUS | |
| n = 243 | 192 | 33 | 1 | 6 | 23 | 10 | 1 | 11 | 5 | 102 | 51 |
| Incidence rate (per million per y) | 0.41 | 0.06 | 0.003 | 0.01 | 0.04 | 0.02 | 0.003 | 0.02 | 0.008 | 0.24 | NR |
| Age at presentation, y; median (range) | 25 (0-80) | 21 (1-50) | NR 25 |
42 (4-67) | 20 (1-47) | 16 (1-51) | NR 1 |
16 (1-69) | 16 (2-28) | 33 (1-80) | 23 (1-76) |
| Sex (% female) | 61 | 70 | NR (F) | 71 | 52 | 70 | NR (F) | 36 | 100 | 59 | 51 |
| Trigger, n (%) | 59 (31.1) | 13 (39) | 0 | 1 (17) | 7 (30) | 1 (10) | 1 | 6 (55) | 0 | 30 (29) | NR |
| Infection | 29 (15.1) | 4 | 0 | 1 | 4 | 1 | 0 | 5 | 0 | 14 | NR |
| Pregnancy | 18 (9.5) | 5 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 12 | NR |
| Diarrheal prodrome, n (%) | 45 (25) | 8 (24) | 0 | 0 | 1 (4) | 4 (40) | 0 | 1 (9) | 1 (20) | 30 (29) | 12 (27) |
| Extrarenal manifestations, n (%) | 54 (19) | 4 (12) | 0 | 1 (17) | 3 (13) | 1 (10) | 0 | 2 (18) | 0 | 25 (24) | 18 (40) |
| Neurological | 22 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 21 | 13 |
| Cardiac | 8 | 4 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 2 |
| Pancreatitis | 4 | 0 | 0 | 0 | 2 | 0 | 0 | 1 | 0 | 1 | 1 |
| Platelet count at presentation, ×109/L; mean (range) | 69 (4-329) | 104 (31-329) | NR 35 |
76 (25-112) | 43 (9-116) | 60 (19-157) | NR 112 |
53 (6-96) | 67 (26-109) | 65 (4-197) | 73 (12-336) |
| Creatinine, μmol/L; mean (range) | 488 (18-2435) | 660 (50-1989) | NR 630 |
366 (86-652) | 470 (79-1377) | 465 (37-1709) | NR 59 |
292 (61-1131) | 625 (94-1172) | 466 (18-2435) | 386 (19-1859) |
| CRP, mg/L; mean (range) | 44 (0.5-309) | 31 (1-246) | NA | 38 (5-160) | 34 (3-182) | 45 (1-136) | NR 13 |
10 (1-34) | 18 (7-28) | 48 (0.5-309) | 83 (1-366) |
| C3, g/L; mean (range) | 0.93 (0.06-2.31) | 0.78 (0.06-2.31) | NR 0.94 |
0.88 (0.63-1.09) | 1.02 (0.55-1.54) | 0.64 (0.08-1.02) | NR 0.85 |
0.75 (0.06-1.23) | 1.07 (0.28-1.67) | 1.0 (0.22-1.99) | 1.0 (0.36-1.79) |
| Adult systolic blood pressure, mmHg; mean (range) | 174 | 180 (117-229) | NR | 194 (157-230) | 165 (110-220) | 168 (145-180) | NR | 152 (144-160) | 147 (140-153) | 176 (137-262) | 167 (165-270) |
| Children; % hypertensive | 59 | 67 | NR | NR | 67 | 40 | NR | 38 | NR | 62 | 70 |
CRP, C-reactive protein; NR, not relevant (missing or insufficient data).
The median age at presentation of the eculizumab-treated CaHUS cohort was 25 years (range, 0-80 years), and that for the control cohort was 22 years (range, 0-79) (Table 1). The age at presentation according to mutation type is presented in Table 2 and supplemental Figure 4. The peak presentation is in the first 4 years of life, with another peak during early adulthood.
A statistically significant difference was observed in the age of presentation between FHAA-associated CaHUS, which predominantly presents in childhood, CaHUS with no mutation detected (P = .001), and CFI mutation-associated CaHUS (P = .008). There was a statistically significant increase in incidence during the years of childbearing potential for females compared with that in males (supplemental Figure 4J-K).
In the eculizumab-treated CaHUS cohort, a trigger was identified in 31%, most commonly, infection (16%) and pregnancy (10%) (Table 2; supplemental Figure 5). The trigger identified by mutation type is shown in supplemental Figure 5A. A nonbloody diarrheal prodrome (Shiga toxin–producing Escherichia coli [STEC] polymerase chain reaction–negative) was recorded in 25% of those with CaHUS (Table 2; supplemental Figure 5B). Extrarenal manifestations (Table 2; supplemental Figure 5C-D) were reported in 19% of the cohort, more frequently in children (23%) than in adults (16%), most commonly, neurological (11.6%; adult 10%, and children 15%) and cardiac (4.2%). The reported prodromal symptoms were numerous and variable (supplemental Figure 5E-F). Fewer presentations occurred during the summer months (supplemental Figure 5G).
Primary outcome: long-term prognosis
The 5-year cumulative estimate (Kaplan-Meier) of ESKD-free survival was 39.5% in the control CaHUS cohort and 85.5% in the eculizumab-treated CaHUS with a mutation or FHAA cohort subgroup (hazard ratio [HR]. 4.95 [95% confidence interval [CI], 2.75-8.90]; P = .000; number needed to treat [NNT], 2.17 [95% CI, 1.81-2.73]; Table 3; Figure 2A).
Table 3.
Long-term outcomes
| Outcome | Eculizumab-treated cohort (%) | Control cohort (%) | P value |
|---|---|---|---|
| Five-year ESKD-free survival | |||
| Total CaHUS | 78 | 39.5 | .000 |
| Total CaHUS with mutation or FHAA | 85.5 | 39.5 | .000 |
| CFH mutation | 78 | 17 | .000 |
| CFHR1 hybrid | NR∗ | 15 | na |
| CFI mutation | 83.5 | 16 | .004 |
| CD46 mutation | 95.6 | 87.5 | .274 |
| C3 mutation | 80 | 25 | .007 |
| CFB mutation | NR∗ | NR | na |
| Factor H autoantibody | 100 | 71 | .057 |
| Combined mutation(s)/autoantibody | 75 | 20 | .005 |
| VUS | 79 | NR | na |
| No mutation | 65 | NR | na |
| Not CaHUS | 57 | NR | na |
| Duration of follow-up, median (range) d | 1514 (2-3720) = 4.2 y | ||
| Renal function at last follow-up, number (%) n = 153 CaHUS | |||
| CKD stage G1 | 52 (34) | ||
| CKD stage G2 | 30 (20) | ||
| CKD stage G3a | 14 (9) | ||
| CKD stage G3b | 9 (6) | ||
| CKD stage G4 | 10 (7) | ||
| CKD stage G5 | 3 (2) | ||
| CKD stage G5D | 23 (14) | ||
| Underwent transplantation | 13 (9) | ||
| Preemptive eculizumab | 10 of 13 | ||
| Functioning transplantation | 12 of 13 |
P value refers to the log-rank (Mantel-Cox) comparison.
CKD based on Kidney Disease Improving Global Outcomes grade (G).
na, not applicable; NR (not relevant; insufficient number).
Single patient free from ESKD.
Figure 2.


Five-year cumulative estimates (Kaplan-Meier) of end-stage kidney disease–free survival. HRs and 95% CIs calculated using the Cox proportional hazard regression model, P values calculated using the log-rank test, and the NNT are shown where appropriate. (A) Treated vs control, CaHUS with mutation or FHAA. (B) Children vs adults, CaHUS with mutation or FHAA. (C) Control group based on the mutation type. (D) Treated group, based on the mutation type. (E) Treated vs control, individuals with CFH mutation subgroup. (F) Treated vs control, individuals with CD46 mutation subgroup.
ESKD-free survival was worse in individuals presenting for the first time as adults than in those presenting from childhood. For those with CaHUS and a mutation or autoantibody in the control group HR for adults vs children was 1.73 (95% CI, 1.25-2.40; P = .000), and in the treated group HR for adults vs children, it was 7.99 (95% CI, 1.03-61.93; P = .013; Figure 2B).
Prognosis varied according to mutation type in the control cohort, with a worse 5-year ESKD-free survival observed for those with mutations in CFH (17%), CFHR1:CFH hybrid (15%), CFI (16%), and C3 (25%) than for those with a mutation in CD46 (87.5%) or FHAA (71%) (Table 3; Figure 2C).
In the treated cohort, the 5-year cumulative estimate (Kaplan-Meier) of ESKD-free survival was 78% for individuals with a CFH mutation and 95.6% for those with a CD46 mutation (Table 3; Figure 2D).
For individuals with a CFH mutation, ESKD-free survival was significantly better in the eculizumab-treated group (HR, 4.58 [95% CI, 2.12-9.91]; P = .000; NNT, 1.64 [95% CI, 1.30-2.22]; Figure 2E). For individuals with a CD46 mutation, ESKD-free survival was not significantly better in the eculizumab-treated group (P = .274; Figure 2F). ESKD-free survival was significantly better in the eculizumab-treated group for individuals with a CFI mutation (HR, 5.75 [95% CI, 0.75-44.24]; P = .004; NNT, 1.48 [95% CI, 0.98-3.02]) and for individuals with a C3 mutation (HR, 4.48 [95% CI, 1.06-19.88]; P = .007; NNT, 1.82 [95% CI, 1.22-3.55]), but there was no statistically significant difference for individuals with FHAA (P = .061; supplemental Figure 6F-I).
A subanalysis of the CFH pathogenic variant groups revealed that there was a significantly poorer outcome in the 5-year cumulative estimate (Kaplan-Meier) of ESKD-free survival in the type 2 C-terminal/CFH hybrid mutations in the control group than in the type 1 CFH mutations (P = .049; supplemental Figure 6E).
The outcome data for all individuals in the eculizumab-treated cohort are presented in supplemental Tables 4 and 5. Twenty-one treated individuals died during the study period: 9 with CaHUS and ESKD, 7 with CaHUS and functioning kidneys, and 5 who did not have CaHUS (supplemental Table 5). For the eculizumab-treated CaHUS cohort, renal prognosis based on the estimated glomerular filtration rate (eGFR) range is presented in Figure 3A (at 6 months) and supplemental Figure 7 (at the most recent follow-up). At presentation, 54% had an eGFR of <15, 8% had an eGFR of ≥60, and at 6 months after presentation, 21% were on dialysis, 1% had chronic kidney disease (CKD) stage 5 (eGFR of <15, nondialysis), and 51% had an eGFR of ≥60 (CKD stage 1-2).
Figure 3.

Hematological and renal responses to eculizumab. (A) Renal function 6 months after eculizumab commenced in the treated CaHUS cohort compared with that at presentation. Not available for 6 individuals; 4 individuals died. (B) Hematological response to eculizumab, defined by the number of days from the first dose of eculizumab to platelet normalization (>150 × 109/L). Median, interquartile range, 1.5× interquartile range, and outliers are shown. There were no statistically significant differences between the mutation types. Three extreme outliers are not shown in the chart: 70 and 90 days (CFH) and 96 days (CD46). Twenty-nine percent already had normal platelets at the time of the first eculizumab dose. In 11 individuals, the platelet count did not normalize; 8 out of 11 had no mutation detected, 2 had VUS (1 had hypersplenism), and 1 had FHAA (and von Willebrand Disease). Of the 17 individuals with a response time of >2 weeks (range, 15-96 days), 3 had a CFH mutation, 1 had a CFI mutation, 2 had a CD46 mutation, 3 had a C3 mutation, 1 had FHAA, 4 had a VUS, and 3 had no mutation detected; 4 out of 17 developed ESKD, 2 of whom died. Fifty-eight percent received plasma exchange before the first dose of eculizumab (supplemental Figure 8B). The subgroup analysis of hematological response in those who received eculizumab but not plasma exchange is shown in supplemental Figure 8C, the median time to platelet normalization in individuals with CaHUS was 5 days. (C) Renal response to eculizumab in the treated CaHUS cohort. Changes in creatinine as a percentage of creatinine in patients with CaHUS (this includes only those who recovered renal function and not those who remained dialysis-dependent) at 1 week, 2 weeks, 1 month, 3 months, 6 months, and 12 months after commencing eculizumab treatment. The complete data set is available for n = 101. Solid circles with a connecting line, mean; bars, 95% CI; x, median. A Wilcoxon Signed Ranks Test demonstrated that there was a statistically significant difference in the mean change in creatinine between all the time points recorded (P value shown). (D) Renal response to eculizumab in the CaHUS cohort. The mean change in creatinine as a percentage of creatinine in patients with CaHUS by mutation type (this includes only those who recovered renal function, not those who remained dialysis-dependent) at 1 week, 2 weeks, 1 month, 3 months, 6 months, and 12 months after commencing eculizumab treatment. (E) Recovery from dialysis dependency in the CaHUS cohort. The proportion of patients with CaHUS on dialysis in the first year after commencing eculizumab. At presentation, 66.7% received dialysis, and at 6 months, 65.7% became dialysis-independent, with 22.9% remaining on dialysis. (F) Recovery from dialysis dependency in the CaHUS cohort. The proportion of patients with CaHUS on dialysis for the first year after commencing eculizumab according to mutation type.
Secondary outcomes: response to treatment
For the eculizumab-treated CaHUS cohort, the response to treatment is presented in Figure 3. The eGFR and platelet count at presentation based on the mutation type are summarized in supplemental Figure 8A. The median time to platelet normalization for individuals with CaHUS was 4 days, although the range was beyond 28 days and there was no statistically significant difference between the mutation groups Figure 3B.
The renal response to eculizumab in the CaHUS cohort is summarized in Figure 3C-F. The mean change in serum creatinine as a percentage of creatinine for individuals with recovered renal function is shown in Figure 3C (all CaHUS) and Figure 3D (based on the mutation type). Most renal recovery occurred within the first 30 days, although there was a statistically significant difference between all time points recorded up to 12 months after the first dose of eculizumab. The recovery from dialysis dependency is presented in Figure 3E (all CaHUS) and Figure 3F (based on the mutation type). Most individuals who recovered renal function having been dialysis-dependent at presentation did so within the first 30 days after starting eculizumab treatment, although recovery was observed for up to 9 months.
The factors associated with response to eculizumab in the treated CaHUS cohort were evaluated using multivariate analysis (supplemental Figure 9). Lower serum creatinine, lower platelet count, and younger age at presentation were associated with ESKD-free survival at 6 months. A lower serum creatinine, lower platelet count, younger age at presentation, and shorter time between presentation and the first dose of eculizumab were associated with eGFR >60 ml/min at 6 months. A lower platelet count and younger age at presentation were associated with a renal response (defined in supplemental Methods) at 6 months. High blood pressure at presentation was also associated with worse outcomes in all 3 analyses when the subgroup with blood pressure data were analyzed.
Stopping eculizumab
Of 243 individuals in the treated cohort who started eculizumab, 145 discontinued it during the study period (Figure 4) with 48 stopping because of an alternative diagnosis. In 29 cases, eculizumab was stopped because of a lack of renal recovery. For 49 individuals with CaHUS who stopped eculizumab because of patient or clinician choice or for a clinical reason (supplemental Table 4), the relapse rate was calculated. The median time to cessation of eculizumab in this group was 165 days (range, 7-1460 days).
Figure 4.

Eculizumab withdrawal. Reasons for restarting eculizumab in patients with functioning kidneys in whom it was stopped but restarted. Individuals who entered the SETS clinical trial are excluded. For 49 individuals with CaHUS who stopped eculizumab because of patient or clinician choice, the median time to relapse following eculizumab withdrawal was 244 days (range, 104-1095 days). ∞Two patients with CFH pathogenic mutations were noncompliant with eculizumab and died because of refusal to undergo dialysis. ¥One patient with CD46 VUS who moved overseas. ¤Three patients with no mutation detected were lost to the follow-up. ˆOne individual with pancreatitis and a pathogenic CD46 mutation had no renal involvement but no hematological improvement. Nmd, no mutation detected.
The relapse rate was 1 per 9.5 person years without eculizumab for those with a pathogenic mutation and 1 per 10.8 person years without eculizumab for those with a VUS, and there were no relapses recorded in 67.3 person years without eculizumab in those without a rare genetic variant in a complement gene. Eculizumab was restarted in 6 individuals with functioning kidneys in whom it had been stopped, with no individual progressing to ESKD. Five of the 6 individuals who relapsed upon cessation of eculizumab had no change in their eGFR upon reintroduction of eculizumab, whereas in 1 individual, the CKD stage progressed from stage 2 to stage 3. Of the 29 individuals with ESKD who stopped eculizumab because there was no recovery of renal function, the drug was restarted in 2: an individual with a CFH/CFHR1 hybrid gene because of hyperkalemia on dialysis with hematological evidence of MAHA and an individual with no mutation because of local clinician choice, owing to gastrointestinal symptoms with no evidence of hemolysis or thrombocytopenia.
Eculizumab side effects
The time on treatment and incidence of meningococcal infection in the eculizumab-treated cohort are summarized in supplemental Figure 10. Three individuals had meningococcal infection during the study period (incidence 550 per 100 000 person years, compared with a background national incidence of 1 per 100 00026; supplemental Figure 10); 2 were not adherent to prophylactic antibiotics, and 1 had a penicillin-resistant strain. All 3 survived without significant morbidity. One individual stopped eculizumab because of headaches, but no other adverse effects of the drug were reported.
Discussion
To our knowledge, this is the first description of the impact of eculizumab treatment on renal and patient survival in genotype-matched cohorts with CaHUS. CaHUS is rare (incidence, 0.41 per million per year in England), with a bimodal distribution of age at presentation.3 There is an equal ratio of male-to-female peak in early childhood and a female predominance in the years of childbearing potential, reflecting the triggering effect of pregnancy or the possibility of hormonal influence.27,28 A trigger was identified in 31% (59/189), a lower proportion than previously reported,3 principally, viral infections and pregnancy, with pregnancy triggering the CaHUS presentation in 15% of those in the treated cohort with CFH mutations. Nineteen percent with CaHUS were reported to have an extrarenal manifestation of TMA, which is a much lower proportion than has been reported in clinical trials.29,30
Historically, management comprised supportive care with or without plasma exchange, and outcomes have been poor.3 Clinical trials of eculizumab have been small, single-arm ones, with limited follow-up4, 5, 6 but suggested efficacy.31 In this national cohort observational cohort study, we report a significant improvement in 5-year ESKD-free survival in individuals with CaHUS with mutations or FHAA treated with eculizumab compared with those who were not (85% vs 39.5%). In keeping with previous reports,3,32 the outcome in the control cohort was contingent on the genetic background; thus, a genetically matched analysis was performed.
In the treated cohort, a difference remained among mutation subgroups, with a better 5-year ESKD-free survival observed in those with CD46 mutations (95.5%) compared with that in those with CFH mutations (78%), suggesting that previously observed differences in disease natural history are not completely abrogated by complement-inhibiting therapy, possibly reflecting late presentation. ESKD-free survival was the worst in those with no complement mutation (65.2%), which may represent the noncomplement-mediated disease in this heterogeneous group. For those with CFH, CFI, and C3 mutations, ESKD survival was significantly better in the treated cohort, but there was no statistically significant difference between the treated and control groups for those with a CD46 mutation or FHAA. Given that CD46- and FHAA-mediated aHUS are both complement-mediated, it may be considered surprising that there was no statistically significant improvement with eculizumab. For CD46, the lack of significant improvement likely reflects good outcomes before the availability of eculizumab. For FHAA, a smaller sample size affected the statistical power. These findings should not alter practice at initial presentation when the underlying etiology of CaHUS is not known, but in the case of CD46-mediated disease, it should provide reassurance if eculizumab withdrawal is to be considered.
In our treated CaHUS cohort, the median time to platelet normalization was less than 1 week after treatment. In individuals who did not require dialysis, most renal recovery occurred within the first 30 days, although a statistically significant improvement between time points was observed for up to 1 year. Similarly, in individuals who did require dialysis, most who recovered function did so within the first 30 days, although recovery continued to be observed for over 9 months. Given the difficulty in diagnosing CaHUS, failure to normalize platelets or recover function within these time frames should prompt a reevaluation of the diagnosis.
Factors associated with a response to eculizumab in those with CaHUS were evaluated using multivariate analysis. Lower serum creatinine, lower platelet count, and younger age at presentation as well as shorter time between presentation and the first dose of eculizumab were associated with a better response. An association between serum creatinine, a marker of renal insult, and outcome would be expected. A possible explanation for the association between a higher platelet count and worse outcome is that it could reflect chronicity and thus late presentation, with platelet consumption predicted to resolve when the disease progresses to the point of the kidneys no longer being perfused. The impact of age could be related to the degree of existing chronic damage, response to renal insult, and late presentation. Blood pressure at presentation was also associated with poor outcomes, which makes mechanistic sense as a marker of worse renal injury. A post hoc multivariate analysis of trial data found that treatment with eculizumab within 1 week of presentation resulted in better renal outcomes, as defined by the mean eGFR change from baseline at 1 year,33 and our data demonstrated that a shorter time interval from presentation to treatment with eculizumab was associated with eGFR >60 at 6 months. In our treated cohort, >50% had an eGFR >60 ml/min at 6 months; the trajectory of an AKI episode is a key outcome when considering the impact on quality of life, long-term morbidity, and prognosis.34
Of the 1956 individuals with suspected aHUS referred to our center, 243 were treated with eculizumab. Ninety of the 192 (47%) individuals with CaHUS had a pathogenic complement gene mutation or FHAA and only 5 had mutations in multiple complement genes or a mutation and FHAA. An additional 24 of the 192 (12.5%) had a VUS in a known aHUS complement gene. Among the 51 patients with an alternative diagnosis, 8 were found to have a recognized noncomplement genetic cause of TMA: 6 DGKE and 2 with disorders of cobalamin metabolism. Individuals with nephrotic syndrome genes presenting with an aHUS phenotype were also detected. TMA has previously been associated with nephrotic syndrome and is thought to be a secondary phenomenon.21,35 One patient who presented with features of aHUS, a renal biopsy specimen demonstrating predominant arteriolar TMA, and severe hypertension had recessive HSD11B2 mutations, a known cause of apparent mineralocorticoid excess (OMIM 218030), which likely resulted in a TMA secondary to hypertension. Given the response of HSB11B2 hypertension to spirolonactone,36 the genetic causes of hypertensive-driven TMA should be sought, especially in pediatric presentations.
Two children under 1 year with a background of pontocerebellar hypoplasia (OMIM: 614678) presented with MAHA, thrombocytopenia, and AKI and were commenced on eculizumab for presumed CaHUS (supplemental Figures 11-12). One child received a single dose of eculizumab; however, because of the underlying severity of their neurological disorder, palliative care was initiated, and no response to eculizumab was observed. The condition of the other child initially appeared to improve with eculizumab; however, they presented 4 months later with aHUS despite eculizumab treatment and died shortly thereafter. Both children were demonstrated to have biallelic pathogenic variants of EXOSC3, an essential component of the RNA exosome. The RNA exosome is a ubiquitously expressed complex responsible for processing and modifying all forms of intracellular RNA. In the absence of a normally functioning RNA exosome, there is an accumulation of RNA species, resulting in the activation of proapoptotic pathways secondary to ribosomal dysfunction.37 We hypothesized that this RNA accumulation induces ribosomal dysfunction in endothelial cells, resulting in apoptosis and eculizumab nonresponsive aHUS. Shiga toxin is a ribosomal toxin that causes ribosomal depurination, protein synthesis arrest, and upregulation of apoptosis,38 suggesting a common underlying mechanism of HUS. Additionally, 1 patient with biallelic mutations in POLR3B, encoding an essential subunit of RNA polymerase III, and 2 affected siblings with biallelic splice-site variants in TSEN2, encoding an essential subunit of the tRNA spliceosome, presented with aHUS. Together, these data suggest a previously unrecognized role for disorders in RNA processing in the pathogenesis of aHUS.
Two individuals had inherited red cell abnormalities (DHFR and G6PD), presenting with a clinical phenotype indistinguishable from CaHUS,39 and 1 had another genetic disorder (AGXT, primary hyperoxaluria), presenting with an aHUS phenotype.
Screening of the control cohort identified aHUS-associated complement genes, most commonly CFH (49%), as previously reported.40 In the prospectively identified CaHUS cohort, 10.1% were familial, predominantly those with a CFH or CD46 mutation. This is much lower than that in our control cohort (38.9%) and other historical ascertainment cohorts enriched for genetic studies,41 suggesting that penetrance is lower than previously reported.
In the control cohort, the next-generation sequencing studies also revealed several rare genetic variants in noncomplement genes (ADAMTS13, DGKE, TSEN2, INF2, G6PD, COL4A5, CUBN, RNU4ATAC, and GNE), demonstrating their utility in aHUS-like presentations, especially in eculizumab nonresponsive cases. In 15% of familial cases of TMA referred to our center, no mutations were identified, suggesting that other aHUS-associated genes remain undiscovered. We did not detect THBD mutations, in keeping with previous reports42,43 that failed to replicate the association between THBD and aHUS.44
Severe hypertension is common, although not universal, among patients with CaHUS,45 and clinical and pathological factors have not been shown to be helpful in distinguishing between severe hypertension that causes TMA and CaHUS associated with hypertension.46 In our treated CaHUS cohort, the mean systolic blood pressure in adults was 175 mmHg, with no statistical difference in blood pressure between mutation types, and 59% of the children were hypertensive, which is consistent with previous findings that presenting blood pressure is not helpful in diagnostic evaluation. Proteinuria has been reported to characterize the DGKE nephropathy phenotype.11,47 However, in this study, we observed proteinuria in all causes of TMA, and there was no difference between the mutation groups. Individuals with non-CaHUS had significantly higher CRP at presentation than those with CaHUS but we did not identify a diagnostically useful cutoff.
Eculizumab is currently licensed for lifelong use in individuals with CaHUS; however, meningococcal sepsis is 550-fold higher in these patients despite mandatory meningococcal vaccination and long-term prophylactic antibiotics. The STOPECU study,48 the first of a series of 3 prospective trials of eculizumab withdrawal in CaHUS,48, 49, 50 has recently reported that eculizumab withdrawal can be performed, although 2 patients had worse renal function, with 1 progressing to ESKD despite eculizumab reintroduction.48 The CUREiHUS study reported safe withdrawal in 18 patients with relapse in 4 patients with rapid eculizumab reintroduction, preventing any clinically significant consequences.50 Other real-world studies have described safe withdrawal of eculizumab.51,52 Of the 243 individuals in our treated cohort, 145 discontinued eculizumab primarily because an alternative diagnosis to CaHUS was ultimately made, although a significant number also stopped for clinical reasons or because they chose to do so. The relapse rates in those with a pathogenic variant or a VUS were 1 per 9.5 and 1 per 10.8 person years off treatment, whereas nobody without a rare complement genetic variant had a relapse in 67.3 person years off treatment. In both reported studies of eculizumab withdrawal in aHUS and herein, it is notable that relapse was not observed in those without complement mutations or FHAA.48,50,51 No individual with a relapse progressed to ESKD or had a marked decline in renal function in this study. Thus, patients may be able to discontinue eculizumab if prospective trials confirm this data that relapses can be recognized and treatment can be restarted promptly without adverse outcomes.
In summary, in this national observational cohort study, we report a significant improvement in 5-year ESKD-free survival in individuals with CaHUS treated with eculizumab compared with those who were not. Lower serum creatinine, lower platelet count, younger age at presentation, and shorter time between presentation and the first dose of eculizumab were associated with eGFR >60 ml/min at 6 months. The rate of meningococcal infection in the treated cohort was 550 times greater than the national rate. A large proportion of the treated cohort discontinued eculizumab and no relapses were observed in individuals without a rare complement genetic variant; however, given the observational study design, these findings should be considered preliminary, and eculizumab withdrawal trial data are required. We demonstrated that biallelic pathogenic mutations in the RNA-processing genes (EXOSC3, TSEN2, and POLR3B) cause aHUS. Our hypothesis that the accumulation of aberrant RNA species in the endothelium results in the activation of proapoptotic pathways secondary to ribosomal dysfunction requires further mechanistic studies.
Conflict-of-interest disclosure: S.J., E.K.S.W., N.S.S., and D.K. have received honoraria for consultancy work from Alexion Pharmaceuticals. S.J. has received honoraria from Novartis and was a member of the Alexion Global aHUS Registry Scientific Advisory Board until 2021. K.J.M. has received consultancy income from Freeline Therapeutics and MPM Capital and grant income from Gemini Therapeutics and Catalyst Biosciences. T.D. has received speaker and advisory board fees from Alexion. T.M.H. has received employment income and equity from Gyroscope Therapeutics. N.S.S. has provided consultancy for Novartis, AstraZeneca, and Roche. D.K. has received advisory board payments from Idorsia, Novartis, Chemocentryx, and Apellis. D.K., K.J.M., and T.M.H. are authors of patent applications referencing recombinant complement factor I production or formation of the C3b/FH/FI trimolecular complex. E.K.S.W. has received consultancy income from Alexion Pharmaceuticals, Biocryst, and Novartis. M.M. received honoraria for consultancy work from Alexion Pharmaceuticals and Novartis. The remaining authors declare no competing financial interests.
A complete list of the members of the the National Renal Complement Therapeutics Centre (NRCTC) aHUS Research Consortium appears in the supplemental Appendix.
Acknowledgments
The authors thank Christine Maville, Gemma Allen and Claire Turnbull for clinical support. The authors also thank the RaDaR aHUS rare disease working group.
The research was supported/funded by the National Institute for Health and Care Research Newcastle Biomedical Research Centers at the Newcastle upon Tyne Hospitals NHS Foundation Trust. V.B. has received funding from the Northern Counties Kidney Research Fund and is a Medical Research Council/Kidney Research United Kingdom Clinical Research Training Fellow (MR/R000913/1). P.R.W. is funded by the Wellcome Trust: 4Ward North Academy (RES/0248/7836). K.J.M. is a Medical Research Council Clinical Research Training Fellow (MR/R001359/1). K.J.M. was also funded by the Northern Counties Kidney Research Fund, Newcastle Healthcare Charities, and a Kidney Research United Kingdom project grant (RP7/2015). E.K.S.W. has received funding from the Medical Research Council. D.K. was funded by the Wellcome Trust, Medical Research Council, Kidney Research United Kingdom, European Union’s Seventh Framework Programme (EURenOmics) FP7/2007-2013 (305608), and European Reference Network for Rare Kidney Diseases (ERKNet) (Project ID 739532). N.S.S. is funded by the National Institute for Health Research and Medical Research Council.
Authorship
Contribution: V.B. and D.K. designed the research, collected, analyzed, and interpreted the data, and drafted the manuscript; P.R.W. undertook genetic analysis and reviewed the manuscript; K.S.-J., T.D., E.K.M., M.M., E.K.S.W., S.J., and N.S.S. collected data and reviewed the manuscript; T.B. analyzed the data; and T.M.H., K.J.M., and V.W. analyzed the data and reviewed the manuscript.
Footnotes
∗V.B. and P.R.W. contributed equally to this study.
Data are available on request form the corresponding author, David Kavanagh, (david.kavanagh@newcastle.ac.uk).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Contributor Information
David Kavanagh, Email: david.kavanagh@newcastle.ac.uk.
NRCTC aHUS Research Consortium:
Richard Holt, Caroline Jones, Neil Ashman, Stanley Fan, Suzanne Forbes, Ravindra Rajakariar, Declan De Freitas, Colm Magee, Jennifer Hanko, David Milford, Mordi Muorah, Alexander J. Howie, Jyoti Baharani, Indranil Dasgupta, Russell Roberts, Adam MacDiarmaid-Gordon, Andrew Fry, Nicholas Torpey, Mary Waldron, Atif Awan, Tara Raftery, Laura Ratcliffe, Enric Vilar, Joel Newman, Dale Seviar, Stephen D. Marks, Lesley Rees, Faidra Veligratli, Aoife Waters, Paramit Chowdhury, James Pattison, Caroline Booth, Nicole Williams, Richard Corbett, Beena Amin, Matthew Pickering, Kate Bramham, Mohamed Al-Hamed, Anita Hill, Andrew Mooney, Eric Finlay, Amanda Newnham, Pallavi Yadav, Amrit Kaur, Nicholas Plant, Mohan Shenoy, Lye Wai Choong, Aileen Helps, Robert McLemon, Thalakunte Muniraju, Izhar Khan, Dana Kidder, Neal Padmanabhan, Zoe Cousland, Jonathan Price, Alison Taylor, Claudine Jennings, Sohail Ahmad, Paul Mead, Mona Aslam, Charlotte Bebb, Catherine Byrne, Alastair Ferraro, Martin Christian, Jonathan Evans, Jon Jin Kim, Andrew Lunn, Meeta Mallik, Phil Mason, David Mole, Janet Craze, Nicholas Sangala, Mark Uniacke, Ellon McGregor, Stewart Lambie, Nitin Bhandary, Oliver Flossmann, Mobin Mohteshamzadeh, Asanga Abeyaratne, Coralie Bingham, Rhian Clissold, Lucy Smyth, John Connolly, Ben Reynolds, John Neary, Joanne Bell, Sarah Roxburgh, Joyce Popoola, Rosie Donne, Nicholas Fardon, Arif Khwaja, Shareen Siddiqi, Gurinder Kumar, Steve Kardasz, Iain Moore, Didem Tez, Jamie Willows, Rachel Davison, John Harty, William Wong, Andrew J. Williams, Daniel Gale, Richard Coward, Carol Inward, Martin Mraz, Waqar Ayub, Andrew Short, David Lappin, Richard Baines, Girish Bommayya, Kris Houlberg, Imran Saif, Rodney Gilbert, Sian Griffin, Graham Smith, Judith Van Der Voort, Jayanthi Chandar, Michael Freundlich, Frank McCarroll, Germaine Wong, Paul Laboi, and Rebekah Molyneux
Supplementary Material
References
- 1.Brocklebank V, Wood KM, Kavanagh D. Thrombotic microangiopathy and the kidney. Clin J Am Soc Nephrol. 2018;13(2):300–317. doi: 10.2215/CJN.00620117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goodship TH, Cook HT, Fakhouri F, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a "kidney disease: improving global outcomes" (KDIGO) controversies conference. Kidney Int. 2017;91(3):539–551. doi: 10.1016/j.kint.2016.10.005. [DOI] [PubMed] [Google Scholar]
- 3.Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5(10):1844–1859. doi: 10.2215/CJN.02210310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368(23):2169–2181. doi: 10.1056/NEJMoa1208981. [DOI] [PubMed] [Google Scholar]
- 5.Licht C, Greenbaum LA, Muus P, et al. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int. 2015;87(5):1061–1073. doi: 10.1038/ki.2014.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greenbaum LA, Fila M, Ardissino G, et al. Eculizumab is a safe and effective treatment in pediatric patients with atypical hemolytic uremic syndrome. Kidney Int. 2016;89(3):701–711. doi: 10.1016/j.kint.2015.11.026. [DOI] [PubMed] [Google Scholar]
- 7.Sheerin NS, Kavanagh D, Goodship TH, Johnson S. A national specialized service in England for atypical haemolytic uraemic syndrome-the first year's experience. QJM. 2016;109(1):27–33. doi: 10.1093/qjmed/hcv082. [DOI] [PubMed] [Google Scholar]
- 8.Warwicker P, Goodship TH, Donne RL, et al. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int. 1998;53(4):836–844. doi: 10.1111/j.1523-1755.1998.00824.x. [DOI] [PubMed] [Google Scholar]
- 9.Glover EK, Smith-Jackson K, Brocklebank V, et al. Assessing the impact of prophylactic eculizumab on renal graft survival in atypical haemolytic uraemic syndrome. Transplantation. 2023;107(4):994–1003. doi: 10.1097/TP.0000000000004355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brocklebank V, Johnson S, Sheerin TP, et al. Factor H autoantibody is associated with atypical hemolytic uremic syndrome in children in the United Kingdom and Ireland. Kidney Int. 2017;92(5):1261–1271. doi: 10.1016/j.kint.2017.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brocklebank V, Kumar G, Howie AJ, et al. Long-term outcomes and response to treatment in DGKE nephropathy. Kidney Int. 2020;97(6):1260–1274. doi: 10.1016/j.kint.2020.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Challis RC, Araujo GS, Wong EK, et al. A de novo deletion in the regulators of complement activation cluster producing a hybrid complement factor H/complement factor H-related 3 gene in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2016;27(6):1617–1624. doi: 10.1681/ASN.2015010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fremeaux-Bacchi V, Miller EC, Liszewski MK, et al. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood. 2008;112(13):4948–4952. doi: 10.1182/blood-2008-01-133702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gleeson PJ, Wilson V, Cox TE, et al. Chromosomal rearrangement-A rare cause of complement factor I associated atypical haemolytic uraemic syndrome. Immunobiology. 2016;221(10):1124–1130. doi: 10.1016/j.imbio.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 15.Kavanagh D, Kemp EJ, Mayland E, et al. Mutations in complement factor I predispose to development of atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16(7):2150–2155. doi: 10.1681/ASN.2005010103. [DOI] [PubMed] [Google Scholar]
- 16.Kavanagh D, Kemp EJ, Richards A, et al. Does complement factor B have a role in the pathogenesis of atypical HUS? Mol Immunol. 2006;43(7):856–859. doi: 10.1016/j.molimm.2005.06.041. [DOI] [PubMed] [Google Scholar]
- 17.Richards A, Buddles MR, Donne RL, et al. Factor H mutations in hemolytic uremic syndrome cluster in exons 18-20, a domain important for host cell recognition. Am J Hum Genet. 2001;68(2):485–490. doi: 10.1086/318203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richards A, Kemp EJ, Liszewski MK, et al. Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc Natl Acad Sci U S A. 2003;100(22):12966–12971. doi: 10.1073/pnas.2135497100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kavanagh D, Pappworth IY, Anderson H, et al. Factor I autoantibodies in patients with atypical hemolytic uremic syndrome: disease-associated or an epiphenomenon? Clin J Am Soc Nephrol. 2012;7(3):417–426. doi: 10.2215/CJN.05750611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moore I, Strain L, Pappworth I, et al. Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood. 2010;115(2):379–387. doi: 10.1182/blood-2009-05-221549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Challis RC, Ring T, Xu Y, et al. Thrombotic microangiopathy in inverted formin 2-mediated renal disease. J Am Soc Nephrol. 2017;28(4):1084–1091. doi: 10.1681/ASN.2015101189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hallam TM, Cox TE, Smith-Jackson K, et al. A novel method for real-time analysis of the complement C3b:FH:FI complex reveals dominant negative CFI variants in age-related macular degeneration. Front Immunol. 2022;13:1028760. doi: 10.3389/fimmu.2022.1028760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kavanagh D, Richards A, Noris M, et al. Characterization of mutations in complement factor I (CFI) associated with hemolytic uremic syndrome. Mol Immunol. 2008;45(1):95–105. doi: 10.1016/j.molimm.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 24.Richards A, Kathryn Liszewski M, Kavanagh D, et al. Implications of the initial mutations in membrane cofactor protein (MCP; CD46) leading to atypical hemolytic uremic syndrome. Mol Immunol. 2007;44(1-3):111–122. doi: 10.1016/j.molimm.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 25.R Foundation for Statistical Computing; 2013. R Core Team. R. a language and environment for statistical computing.http://www.R-project.org/ [Google Scholar]
- 26.Public Health England Invasive meningococcal disease in England: annual laboratory confirmed reports for epidemiological year 2018/2019. Health Protection Report. 2019;13(38) [Google Scholar]
- 27.Fakhouri F, Scully M, Provot F, et al. Management of thrombotic microangiopathy in pregnancy and postpartum: report from an international working group. Blood. 2020;136(19):2103–2117. doi: 10.1182/blood.2020005221. [DOI] [PubMed] [Google Scholar]
- 28.Bruel A, Kavanagh D, Noris M, et al. Hemolytic uremic syndrome in pregnancy and postpartum. Clin J Am Soc Nephrol. 2017;12(8):1237–1247. doi: 10.2215/CJN.00280117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rondeau E, Scully M, Ariceta G, et al. The long-acting C5 inhibitor, ravulizumab, is effective and safe in adult patients with atypical hemolytic uremic syndrome naive to complement inhibitor treatment. Kidney Int. 2020;97(6):1287–1296. doi: 10.1016/j.kint.2020.01.035. [DOI] [PubMed] [Google Scholar]
- 30.Barbour T, Scully M, Ariceta G, et al. Long-term efficacy and safety of the long-acting complement c5 inhibitor ravulizumab for the treatment of atypical hemolytic uremic syndrome in adults. Kidney Int Rep. 2021;6(6):1603–1613. doi: 10.1016/j.ekir.2021.03.884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fakhouri F, Hourmant M, Campistol JM, et al. Terminal complement inhibitor eculizumab in adult patients with atypical hemolytic uremic syndrome: a single-arm, open-label trial. Am J Kidney Dis. 2016;68(1):84–93. doi: 10.1053/j.ajkd.2015.12.034. [DOI] [PubMed] [Google Scholar]
- 32.Caprioli J, Noris M, Brioschi S, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108(4):1267–1279. doi: 10.1182/blood-2005-10-007252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walle JV, Delmas Y, Ardissino G, Wang J, Kincaid JF, Haller H. Improved renal recovery in patients with atypical hemolytic uremic syndrome following rapid initiation of eculizumab treatment. J Nephrol. 2017;30(1):127–134. doi: 10.1007/s40620-016-0288-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chawla LS, Bellomo R, Bihorac A, et al. Acute kidney disease and renal recovery: consensus report of the acute disease quality initiative (ADQI) 16 workgroup. Nat Rev Nephrol. 2017;13(4):241–257. doi: 10.1038/nrneph.2017.2. [DOI] [PubMed] [Google Scholar]
- 35.Manenti L, Gnappi E, Vaglio A, et al. Atypical haemolytic uraemic syndrome with underlying glomerulopathies. A case series and a review of the literature. Nephrol Dial Transplant. 2013;28(9):2246–2259. doi: 10.1093/ndt/gft220. [DOI] [PubMed] [Google Scholar]
- 36.Dave-Sharma S, Wilson RC, Harbison MD, et al. Examination of genotype and phenotype relationships in 14 patients with apparent mineralocorticoid excess1. J Clin Endocrinol Metab. 1998;83(7):2244–2254. doi: 10.1210/jcem.83.7.4986. [DOI] [PubMed] [Google Scholar]
- 37.Muller JS, Burns DT, Griffin H, et al. RNA exosome mutations in pontocerebellar hypoplasia alter ribosome biogenesis and p53 levels. Life Sci Alliance. 2020;3(8) doi: 10.26508/lsa.202000678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tesh VL. Activation of cell stress response pathways by Shiga toxins. Cell Microbiol. 2012;14(1):1–9. doi: 10.1111/j.1462-5822.2011.01684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walsh PR, Johnson S, Brocklebank V, Salvatore J, Christian M, Kavanagh D. Glucose-6-phosphate dehydrogenase deficiency mimicking atypical hemolytic uremic syndrome. Am J Kidney Dis. 2018;71(2):287–290. doi: 10.1053/j.ajkd.2017.08.007. [DOI] [PubMed] [Google Scholar]
- 40.Ferreira VP, Herbert AP, Cortes C, et al. The binding of factor H to a complex of physiological polyanions and C3b on cells is impaired in atypical hemolytic uremic syndrome. J Immunol. 2009;182(11):7009–7018. doi: 10.4049/jimmunol.0804031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kavanagh D, Goodship TH, Richards A. Atypical hemolytic uremic syndrome. Semin Nephrol. 2013;33(6):508–530. doi: 10.1016/j.semnephrol.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bu F, Zhang Y, Wang K, et al. Genetic analysis of 400 patients refines understanding and implicates a new gene in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2018;29(12):2809–2819. doi: 10.1681/ASN.2018070759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fremeaux-Bacchi V, Fakhouri F, Garnier A, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol. 2013;8(4):554–562. doi: 10.2215/CJN.04760512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Delvaeye M, Noris M, De Vriese A, et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361(4):345–357. doi: 10.1056/NEJMoa0810739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cavero T, Arjona E, Soto K, et al. Severe and malignant hypertension are common in primary atypical hemolytic uremic syndrome. Kidney Int. 2019;96(4):995–1004. doi: 10.1016/j.kint.2019.05.014. [DOI] [PubMed] [Google Scholar]
- 46.Timmermans S, Werion A, Damoiseaux J, Morelle J, Reutelingsperger CP, van Paassen P. Diagnostic and risk factors for complement defects in hypertensive emergency and thrombotic microangiopathy. Hypertension. 2020;75(2):422–430. doi: 10.1161/HYPERTENSIONAHA.119.13714. [DOI] [PubMed] [Google Scholar]
- 47.Azukaitis K, Simkova E, Majid MA, et al. The phenotypic spectrum of nephropathies associated with mutations in diacylglycerol kinase epsilon. J Am Soc Nephrol. 2017;28(10):3066–3075. doi: 10.1681/ASN.2017010031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fakhouri F, Fila M, Hummel A, et al. Eculizumab discontinuation in children and adults with atypical hemolytic-uremic syndrome: a prospective multicenter study. Blood. 2021;137(18):2438–2449. doi: 10.1182/blood.2020009280. [DOI] [PubMed] [Google Scholar]
- 49.Dunn S, Brocklebank V, Bryant A, et al. Safety and impact of eculizumab withdrawal in patients with atypical haemolytic uraemic syndrome: protocol for a multicentre, open-label, prospective, single-arm study. BMJ Open. 2022;12(9) doi: 10.1136/bmjopen-2021-054536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bouwmeester RN, Duineveld C, Wijnsma KL, et al. Early eculizumab withdrawal in patients with atypical hemolytic uremic syndrome in native kidneys is safe and cost-effective: results of the CUREiHUS study. Kidney Int Rep. 2023;8(1):91–102. doi: 10.1016/j.ekir.2022.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ardissino G, Possenti I, Tel F, Testa S, Salardi S, Ladisa V. Discontinuation of eculizumab treatment in atypical hemolytic uremic syndrome: an update. Am J Kidney Dis. 2015;66(1):172–173. doi: 10.1053/j.ajkd.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 52.Merrill SA, Brittingham ZD, Yuan X, Moliterno AR, Sperati CJ, Brodsky RA. Eculizumab cessation in atypical hemolytic uremic syndrome. Blood. 2017;130(3):368–372. doi: 10.1182/blood-2017-02-770214. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.