

Abstract
Two genes, gusB and gusC, from a natural fecal isolate of Escherichia coli are shown to encode proteins responsible for transport of β-glucuronides with synthetic [14C]phenyl-1-thio-β-d-glucuronide as the substrate. These genes are located in the gus operon downstream of the gusA gene on the E. coli genome, and their expression is induced by a variety of β-d-glucuronides. Measurements of transport in right-side-out subcellular vesicles show the system has the characteristics of secondary active transport energized by the respiration-generated proton motive force. When the genes were cloned together downstream of the tac operator-promoter in the plasmid pTTQ18 expression vector, transport activity was increased considerably with isopropylthiogalactopyranoside as the inducer. Amplified expression of the GusB and GusC proteins enabled visualization and identification by N-terminal sequencing of both proteins, which migrated at ca. 32 kDa and 44 kDa, respectively. Separate expression of the GusB protein showed that it is essential for glucuronide transport and is located in the inner membrane, while the GusC protein does not catalyze transport but assists in an as yet unknown manner and is located in the outer membrane. The output of glucuronides as waste by mammals and uptake for nutrition by gut bacteria or reabsorption by the mammalian host is discussed.
Glucuronidation is the major conjugation process in mammals and other vertebrates (14, 15). Endogenous metabolic wastes, vitamins, steroid hormones, plant- and animal-derived secondary metabolites, xenobiotics, and pharmaceuticals are often conjugated with glucuronic acid. The reaction is catalyzed by many isomers of glucuronyltransferase in the lumen of the endoplasmic reticulum, with UDP-glucuronic acid (7, 8, 9, 14, 15). Once synthesized, those glucuronides are transported out of the mammalian cells, probably by ATP-dependent transport processes (39, 55). The glucuronides are then mostly excreted out of the body via the bile ducts into the intestine, through apocrine secretions, and through the bladder into the urinary tract. The conjugated compounds are typically much more water soluble than the respective aglycones and are often biochemically and biologically inactive. The glucuronidation and excretion of such compounds have thus often been described as detoxification.
In the intestine and on the skin, large populations of commensal and symbiotic microorganisms have access to these diverse glucuronides, and some have evolved the ability to metabolize these compounds. The enterobacterium Escherichia coli colonizes all known vertebrates. Most E. coli strains living in natural environments possess β-d-glucuronidase (EC 3.2.1.31) and are able to acquire glucuronides as nutrients (10, 13, 59). β-d-Glucuronidase is an intracellular hydrolase that catalyzes the hydrolysis of β-d-glucuronides within the cytoplasm of E. coli (31, 58). The glycone released can then be used as a carbon source. Almost any aglycone conjugated in a hemiacetal linkage to the C-1 hydroxyl of glucuronate in the β-configuration can be cleaved by β-d-glucuronidase (30, 65), except for some thio-β-d-glucuronides (58). β-d-Glucuronidase is encoded by the gusA (formerly uidA [65]) gene of the gus operon, located at 36.5 min on the E. coli genome (3, 31).
When glucuronides produced by vertebrates and excreted into the bile, including many conjugates of steroid hormones, pharmaceuticals, toxins, or xenobiotics, are deconjugated by microbial glucuronidases in the intestine, the apolar aglycone is released and can frequently be reabsorbed by the host (20). This phenomenon is referred to as enterohepatic circulation. Thus, a substantial proportion of the circulating hormones and xenobiotics are derived from the enterohepatic circulation and depend on the glucuronide metabolism of the enteric bacteria.
As glucuronide conjugates are polar molecules, there must be a specific method for their uptake into the E. coli cell. Stoeber (59) found that the uptake of glucuronides in a wild E. coli isolate was coordinately induced with β-d-glucuronidase by the addition of exogenous β-d-glucuronides. He also showed that different glucuronides were taken up with various affinities. Curiously, numerous subsequent studies on the induction and regulation of β-d-glucuronidase in E. coli failed to address the question of how the glucuronides are transported into E. coli, a critical feature in determining the access of inducers and substrates to the cell, and an important candidate for mechanisms of regulation (47). Thus, the nature of the bacterial glucuronide transport system remained obscure.
Here, we report our studies on the biochemical properties of the glucuronide transport system from a natural E. coli isolate, including, most importantly, the identification and localization of the encoded proteins.
MATERIALS AND METHODS
Strains and plasmids used.
E. coli strain CE1 is a natural isolate from feces of a female human vegetarian. The E. coli strains and plasmids used or generated in this work are listed in Tables 1 and 2.
TABLE 1.
Escherichia coli strains used in this study
| Strain | Relevant genotype | Source or reference |
|---|---|---|
| AR120 | λN99 cI+ Δgal nadA::Tn10 T11 Δ(cII uvrB) | 11 |
| CE1 | Natural isolate (human) | This work |
| NO2947 | Str (MC1061 Δlac recA56 Srl::Tn10) | M. Thomas |
| S⊘200 | F−purB68 Δ(manA-add)84 relA1 metB1 rpsL254 | E. coli Genetic Stock Centre, Yale University |
| KW1 | metB strA purB Δ(add-gus-man) hsdR hsdM+ | 53 |
TABLE 2.
Plasmids constructed and used in this study
| Plasmid | gus gene(s) | Promoter | Backbone | Reference |
|---|---|---|---|---|
| pLAFR3 | None | lac | 17 | |
| pAD284 | None | λ OLPL | pBR322 | 11 |
| pTTQ18 | None | tac | pUC18 | 47 |
| pKW219 | gusCE1 operon | gus | pLAFR3 | This work |
| pKW220 | gusK-12 operon | gus | pLAFR3 | This work |
| pRAJ289 | gusABCK-12 | tac | pTTQ18 | 53 |
| pWJL4 | gusBCCE1 | λ OLPL | pAD284 | This work |
| pWJL6 | gusBCE1 | λ OLPL | pAD284 | This work |
| pWJL11 | gusCCE1 | λ OLPL | pAD284 | This work |
| pWJL24 | gusBCCE1 | tac | pTTQ18 | This work |
| pWJL26 | gusBCE1 | tac | pTTQ18 | This work |
| pWJL31 | gusCCE1 | tac | pTTQ18 | This work |
Cloning the glucuronide transporter genes.
Restriction-digested PstI-HindIII fragments from the genomic DNA of an E. coli human fecal isolate, CE1, were shotgun cloned into the low-copy-number plasmid pLAFR3 (17) with E. coli KW1 (gus operon deleted, Table 1) as the host strain. Positive clones containing the entire gus operon from strain CE1 were identified when the host strain turned blue on Luria-Bertani (LB) agar plates containing tetracycline at 10 μg ml−1 and X-GlcA (5-bromo-4-chloro-3-indolyl β-d-glucuronide) at 50 μg ml−1 due to their abilities to cleave X-GlcA by cloned β-glucuronidase. One clone was designated plasmid pKW219 (Table 2) and is illustrated in Fig. 1, which shows the 9.0-kbp PstI-HindIII fragments containing the gus operon. The same steps were performed to create pKW220, which contains the gus operon from E. coli K-12.
FIG. 1.

Cloning of the gus operon. A 7,740-bp PstI-HindIII fragment containing the gus operon from a natural E. coli fecal isolate (CE1) was cloned into the PstI and HindIII restriction sites (not shown) of low-copy-number plasmid pLAFR3, generating plasmid pKW219. The corresponding fragment of the gus operon from E. coli K-12 was also cloned to generate plasmid pKW220 (not shown). A BstYI fragment (3,368 bp) containing the gusBCCE1 genes was subcloned into plasmid pAD284 at the HpaI site, generating plasmid pWJL4, so that the gene expression is under the control of the λ OLPL promoter. Plasmids containing the gusBCE1 gene (pWJL6) and the gusCCE1 gene (pWJL11) were generated from plasmid pWJL4 by a combination of restriction digestions (see Materials and Methods for details). The base pair numbers are those listed in GenBank (accession number M14641), while the base pair numbers in brackets correspond to those listed in the genome sequence of E. coli MG1655 (3).
To make a gusBC subclone in an expression vector, the BstYI fragment (3,368 bp) containing the gusBCCE1 genes from plasmid pKW219 was blunt ended with DNA polymerase I Klenow fragment and then subcloned into the expression vector pAD284 (12) at its unique HpaI site downstream of the λOLPL operator-promoter to create pWJL4 (Fig. 1). This fragment contains the gusB gene with 102 bp of 5′-flanking region (partial coding region of the gusA gene), the gusC gene, and 542 bp of the gus operon terminator. The orientation of the insert was determined by restriction digestions with a combination of HindIII and EcoRV, HindIII and MluI, and HindIII and StuI (37).
Plasmid pWJL6 was constructed to enable expression of only the gusB gene. pWJL4 was partially digested with NcoI to linearize the plasmid at the beginning of the gusC gene, followed by partial digestion with PvuI at the end of the gusC gene, generating an 8,596-bp DNA fragment. This was blunt ended with T4 DNA polymerase I in the presence of deoxynucleoside triphosphates and religated to generate pWJL6 (8,594 bp) containing the gusBCE1 gene and the gus transcription terminator downstream of the λOLPL operator-promoter. Plasmid pWJL11 was constructed to express only the gusC gene. pWJL4 was digested with EcoRV followed by a partial digest with NruI. The resulting 8,327-bp fragment was religated to obtain pWJL11 (8,327 bp) (Table 2; details in reference 37).
The equivalent gusBC, gusB, and gusC clones were also constructed in the high-copy-number and high-expression vector pTTQ18 (57). The gusBC-containing BstYI fragment (3,368 bp) from E. coli CEI was inserted at the compatible BamHI site downstream of the tac promoter to create pWJL24 (7,927 bp), which has the gusBCCE1 reading frame in the correct orientation under the control of the tac promoter. To create a pTTQ18 clone containing only gusB, plasmid pWJL24 was digested with StuI, followed by partial digestion with NcoI to eliminate the gusC gene (Fig. 1). The recessive ends of a purified 6,550-bp fragment were blunted with T4 DNA polymerase in the presence of deoxynucleoside triphosphates. This was followed by recircularization of the DNA in the ligation reaction to create pWJL26 (6,550 bp), which has the gusB gene under the control of the tac promoter and contains the gus operon transcription terminator sequence. A pTTQ18 subclone for expressing only the gusC gene was constructed by partially digesting pWJL24 with SmaI, which occurs in the polylinker upstream of gusB, followed by partial digestion with EcoRV to eliminate the gusB gene (Fig. 1). The blunt ends of this DNA fragment were religated to form plasmid pWJL31 (6,460 bp), containing the gusC gene alone and the gus terminator sequence. All the subcloning procedures and expression experiments were carried out in the E. coli host strain NO2947 (Table 1).
Sequence determination of the gusB and gusC genes.
The gusBCCE1 and gusBCK12 genes were sequenced with the dideoxynucleotide termination method (53). Kyte-Doolittle hydropathy analysis (35) and Garnier-Robson secondary-structure prediction (18) were employed for the analysis of the deduced amino acid sequence of the GusB and GusC proteins.
Growth of bacterial strains and induction of gene expression in different plasmids.
For the induction of gene expression of the gus operon on plasmid pKW219 in E. coli strain KW1, cells growing in M9CA medium (43), containing 15 μg of hypoxanthine, 15 μg of adenine, and 20 μg of tetracycline per ml (not added to control host cell culture) and 20 mM glycerol instead of glucose were induced with 1 mM p-nitrophenyl-β-d-glucuronide for 40 min starting at log phase. For gene expression driven by the OLPL promoter on plasmid vector pAD284 in the E. coli λ lysogen strain AR120, cells growing in LB medium containing 50 μg of carbenicillin per ml (not added to control host cell culture) and harboring plasmid pWJL4, pWJL6, or pWJL11 were induced with 40 μg of nalidixic acid per ml for 4 h after mid-log phase. For gene expression driven by the tac promoter on plasmid vector pTTQ18 in E. coli strain NO2947, cells growing on M9CA medium containing 50 μg of carbenicillin per ml (not added to control host cell culture) and harboring plasmid pWJL24, pWJL26, or pWJL31 were induced with 1 mM isopropylthiogalactopyranoside (IPTG) for 14 h upon inoculation.
Assays for glucuronide transport in intact E. coli cells.
A nonmetabolizable glucuronide analogue, [14C]phenyl-thio-β-d-glucuronide (PTG, custom synthesis by Amersham) or [14C]-p-acetamidophenyl-β-d-glucuronide (APG, Sigma) were used as substrates for transport assays (24). Induced and uninduced E. coli cells were grown on medium supplemented with 20 mM glycerol (see below) and harvested and washed thoroughly with 150 mM KCl--5 mM morpholinoethanesulfonic acid (MES), pH 6.6. The cells were then resuspended in the same buffer to a concentration of 1.36 mg of dry cell mass per ml, and samples were energized with 20 mM glycerol for 3 min at 25°C under aerobic conditions. At time zero, 50 μM [14C]PTG was mixed with the cells. Aliquots were withdrawn at different times and immediately collected and washed on a nitrocellulose membrane (25 mm in diameter, 0.45-μm pore size). The amount of [14C]PTG inside the cells was determined by scintillation counting. An internal volume of 1 μl per mg of dry mass as defined by Ahmed and Booth (1) was assumed for the calculation of the accumulation of PTG inside the cells at steady state. Apparent Km and Vmax values were obtained by measuring uptake of [14C]PTG in triplicate at 15 s with concentrations from 7.8 to 1,000 μM and fitting the uptake data to the Michaelis-Menten equation with the least-squares method of Cleland (11).
Transport assay with right-side-out vesicles.
Right-side-out vesicles of the induced E. coli strain AR120(pWJL4) expressing the GusBCCE1 proteins were prepared based on the method of Horne and Henderson (26). Transport assays were carried out in 40 mM potassium phosphate buffer (pH 6.6, unless indicated otherwise)-10 mM MgSO4-1.0 to 1.3 mg of protein/ml-50 μM [14C]APG or [14C]PTG gassing with oxygen. Proton motive force was generated by addition of 0.1 mM phenazine methosulfate and 20 mM ascorbate (32) as indicated.
Protein expression and identification.
For induction of gene expression with clones in plasmid vectors pAD284 and pTTQ18, see above. Total membrane proteins were prepared by the water lysis method of Henderson and Macpherson (25). Inner and outer membrane fractions were prepared with a French press and separated with sucrose gradient centrifugation (4). The OmpA and OmpC outer membrane proteins were used as markers to confirm separation of the two membranes. The amounts of protein in membrane samples were determined by the method of Schaffner and Weissmann (56). Membrane proteins were separated by sodium dodecyl sulfate--12% polyacrylamide gel electrophoresis (25) followed by staining (0.1% Coomassie blue R-250, 45% methanol, and 10% acetate) and then destaining (14% methanol and 10% acetate) procedures. Identification of the GusB and GusC proteins was achieved by N-terminal amino acid sequencing after transfer from sodium dodecyl sulfate-polyacrylamide gels to a polyvinylidene difluoride membrane (44). Induction of β-glucuronidase (GusA) activity was assayed by measuring the hydrolysis of added p-nitrophenyl-β-d-glucuronide (colorless) to the yellow p-nitrophenol.
RESULTS AND DISCUSSION
gusB and gusC genes of the gus operon confer glucuronide-inducible glucuronide transport activity when expressed together.
The host strain KW1 (gus deletion) harboring the gusCE1operon on plasmid pKW219 exhibited [14C]PTG transport activity (Fig. 2A), provided a glucuronide, in this case p-nitro-phenyl-β-d-glucuronide, was added during growth as inducer.Phenyl-β-d-glucuronide, 4-methylumbelliferyl-β-d-glucuro-nide, 5-bromo-4-chloro-3-indolyl-β-d-glucuronide, trypto-phanyl-β-d-glucuronide, o-aminophenyl-β-d-glucuronide, CN-umbelliferone-β-d-glucuronide, 8-hydroxyquinoline-β-d-glucuronide, phenolphthalein-β-d-glucuronide, estriol-3-β-d-glucuronide, estriol-17-β-d-glucuronide, estrone-3-β-d-glucuronide, testosterone-β-d-glucuronide, and pregnanediol glucuronide (3α,20α-dihydroxy-5β-pregnane-3-glucuronide)were also confirmed to be inducers, as indicated by the activity of the GusA, β-glucuronidase, activity (36, 64, 65), while naphthol AS-BI glucuronide was not (36). The initial rate of [14C]PTG transport (from a concentration of 50 μM) conferred by the gusCE1 operon was about 5.43 (± 0.41 standard deviation) nmol per min per mg of dry cell mass and the [14C]PTG accumulation at the steady state was 1.43 (standard deviation ± 0.27) nmol per mg of cells, which corresponds to a [PTG]in/[PTG]out gradient of about 28.6:1. We conclude that the gusBC genes encode a glucuronide-inducible glucuronide transport system.
FIG. 2.

[14C]-Phenyl-thio-β-d-glucuronide (PTG) transport in different host-vector systems. Each line represents curve fits of mean values of triplicate analyses at each of the time points; error bars are standard deviations. PTG was used at 50 μM. (A) Induction of the complete gusCE1 operon on plasmid pKW219 or the gusK-12 operon on plasmid pKW220. The host E. coli strain KW1 has the entire gus operon deleted. Cells were induced with 1 mM p-nitrophenyl-β-d-glucuronide for 40 min during logarithmic growth. Solid upward triangles, PTG uptake into cells with induced gusABCCE1 operon (line represents curve fits); open downward triangles, cells with uninduced gusABCCE1 operon; solid squares, cells with induced gusK-12 operon; solid circles, vector control [KW1(pLARF3)]; solid downward triangle, strain control (KW1). (B) Expression of the gusBCE1 and gusCCE1 genes under control of the λ OLPL promoter in the λ lysogen strain AR120. Strains were induced with 40 μg of nalidixic acid per ml for 3 h at mid-log phase. Solid squares, cells with induced gusBCCE1 genes on plasmid pWJL4; solid downward triangles, cells with uninduced gusBCCE1 genes on plasmid pWJL4; open squares, cells with induced gusBCE1 gene alone on plasmid pWJL6; open diamonds, cells with induced gusCCE1 gene alone on plasmid pWJL11; solid upward triangles, vector control [AR120(pAD284)]. (C) Expression of the gusBCE1 and gusCCE1 genes under control of the tac promoter. Strains of E. coli NO2947 containing the plasmids below were induced with 1 mM IPTG for 14 h after inoculation in minimal medium. Solid upward triangles, cells with induced gusBCCE1 genes on plasmid pWJL24; solid downward triangles, cells with uninduced gusBCCE1 genes on plasmid pWJL24; open upward triangles, cells with induced gusBCE1 gene alone on plasmid pWJL26; open diamonds, cells with induced gusCCE1 gene alone on plasmid pWJL31; solid circles, vector control [NO2947(pTTQ18)].
When a gus operon from E. coli K-12 was inserted instead, no transport activity was observed (Fig. 2A). This turned out to be the result of a Pro100Leu mutation in the GusB protein, and so the gus operons studied further all originated from the CE1 natural isolate.
tac and λ OL promoters amplify transport activities of the cloned gusBC genes.
Figure 2B shows the [14C]PTG transport activity of the gusBCCE1 genes driven by the nalidixic acid-induced λ PL promoter in pBR322-based plasmid pWJL4 in the host strain AR120 (46). The initial rate of 50 μM [14C]PTG transport was 14.9 (standard deviation ± 0.50) nmol per min per mg of dry cell mass, and accumulation of [14C]PTG at steady state (after 4 min of uptake) was more than 10.05 (standard deviation ± 0.47) nmol per mg of cells; thus, the PTG concentration inside the cells was about 200-fold higher than that outside ([PTG]in/[PTG]out = 201:1). The overall activity was higher than the activity from the genes cloned into the low-copy-number plasmid pKW219 (above), reflecting the expected amplification of expression of the gusB and gusC genes.
The rate and accumulation level of [14C]PTG transport activity were even higher with the IPTG-induced tac promoter in a pUC-based plasmid (57), pWJL24, in the host strain NO2947 (Fig. 2C), achieving initial rates of 22.65 (standard deviation ± 1.97) nmol per min per mg of dry mass and [14C]PTG and internal accumulation of 13.52 (standard deviation ± 0.97) nmol per mg of cells after 4 min, gradients approximately 270-fold higher than outside. The reasons for this enhanced expression were examined further and are explained below.
The initial rates of transport measured at various [14C]PTG concentrations fit well to a rectangular hyperbola yielding an apparent Km of 198 ± 69 μM and an apparent Vmax of 29.2 ± 3.9 nmol per mg per min (Vmax/Km = 0.15) by the least-squares method of Cleland (11). [14C]APG was an even more effective substrate, yielding an apparent Km of 36 ± 4 μM and an apparent Vmax of 106 ± 5 nmol per mg per min (Vmax/Km = 2.94). These apparent Km values are considerably higher than those of 0.1 to 5 μM reported for primary active transport systems containing periplasmic binding proteins (4), but are within the range of values typical of secondary active transport systems (30 to 900 μM) (24).
When each is expressed on its own, the gusB gene confers reduced glucuronide transport activity, whereas the gusC gene confers no activity.
Figure 2B also shows that the induced gusBCE1 gene alone on plasmid pWJL6 did bring about [14C]PTG uptake, albeit at a lower rate and extent than when the gusCCE1 gene was coinduced with the gusBCE1 gene on plasmid pWJL4. The gusCCE1 gene on plasmid pWJL11, however, did not show any [14C]PTG uptake activity (Fig. 2B). The induced gusBCE1 gene conferred an initial rate for [14C]PTG transport of 1.60 (standard deviation ± 0.48) nmol per min per mg of dry cell mass, which is about 10-fold lower than that conferred by the induced gusBCCE1 genes, and after exposure for 4 min, accumulation reached 2.53 (standard deviation ± 0.15) nmol per mg of cells, a gradient of 50.5. Similar results were obtained (Fig. 2C) when the gusBCE1 and gusCCE1 genes were expressed separately under the control of the tac operator-promoter (Table 2); the induced gusBCE1 gene alone driven by the tac promoter conferred an initial rate of 2.67 nmol of [14C]PTG per min per mg of dry cell mass, which is also about 10-fold lower than that conferred by the induced gusBCCE1 genes driven by the same promoter, and after exposure for 4 min, accumulation reached 4.0 (standard deviation ± 0.67) nmol per mg of cells, corresponding to a gradient of 80. The gusCCE1 gene driven by the tac promoter failed to yield any transport, even though the GusC protein was expressed (see below).
Sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) of membranes from the induced strains and densitometry of the Coomassie blue-stained separated proteins (Fig. 3B, lanes 4, 5, and 6) showed that the levels of GusB and GusC proteins were not significantly different when expressed in plasmids carrying both (11.1% and 12.1% of total membrane proteins, respectively) or only one of the gusB and gusC genes (10.4% and 9.9% of total membrane proteins, respectively).
FIG. 3.


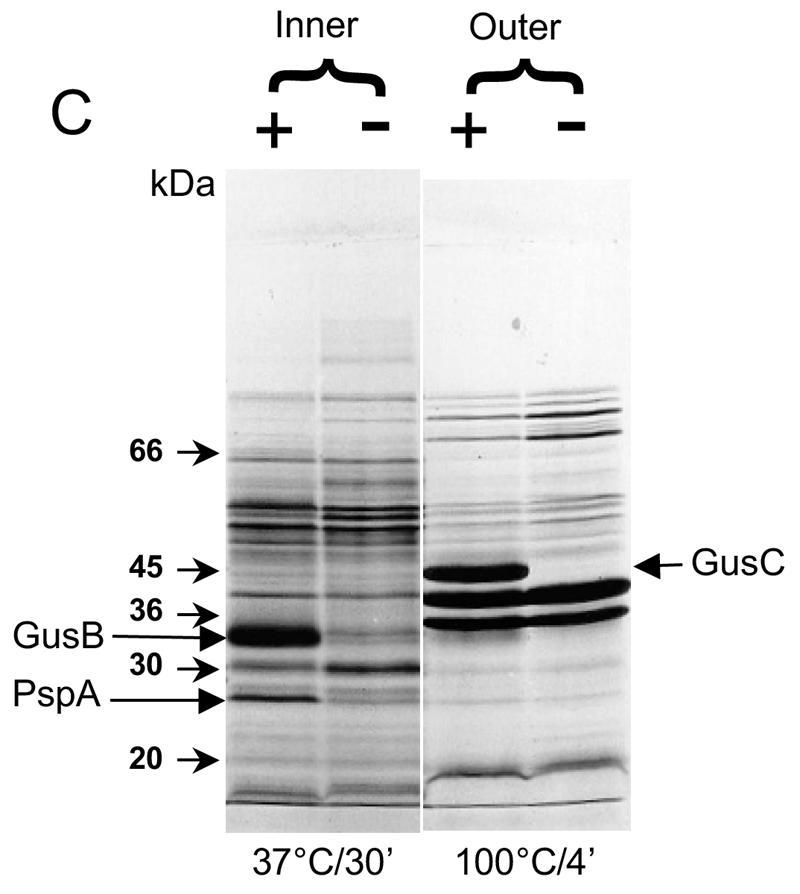
Appearance of putative GusB and GusC proteins in membrane preparations. (A) Total membrane proteins prepared from E. coli strain NO2947 harboring plasmids as indicated below were incubated in solubilization buffer at 37°C for 30 min; 45 μg of proteins was loaded onto each track of the SDS-12% bisacrylamide gel. Lane 1, protein size markers (in kilodaltons); lane 2, plasmid pWJL24 tac-gusBC induced; lane 3, pWJL24 tac-gusBC uninduced; lane 4, pTTQ18 induced control; lane 5, no plasmid in host cell control. (B) After solubilization conditions as shown, total membrane protein (45 μg) was loaded onto each track of the SDS-12% bisacrylamide gel. Lane B+C, pWJL24 tac-gusBCCE1 induced; lane B, pWJL26 tac-gusB CE1 induced; lane C, pWJL31 tac-gusCCE1 induced. Proteins in lanes 4, 5, and 6 were as same as those in lane 1, 2, and 3, respectively. (C) GusB occurs in the inner membrane and GusC in the outer membrane. Membrane fractionation was carried out by sucrose gradient and ultracentrifugationafter cell lysis by French press (20,000 lb/in2). Inner, inner membrane proteins solubilized at 37°C for 30 min; Outer, outer membrane proteins solubilized at 100°C for 4 min. +, proteins from NO2947(pWJL24) with induction of = tac-gusBCCE1; −, proteins from NO2947(pWJL24) without induction of tac-gusBCCE1.
From these data we conclude that the GusB protein, in contrast to GusC, is responsible for transport per se, though GusC is able to enhance the efficiency of transport by GusB in intact cells (see Concluding remarks).
Dependence of glucuronide transport on the proton motive force and its susceptibility to inhibitors.
Transport of [14C]APG or [14C]PTG into appropriately induced strains of E. coli containing the GusBCCE1 proteins was inhibited by uncoupling agents, e.g., m-chlorocyanocarbonyl phenylhydrazone (CCCP) and 2,4-dinitrophenol (DNP), which are protonophores that discharge a transmembrane electrochemical gradient of protons (data not shown), but this experiment does not distinguish between proton motive force-energized (secondary) and ATP-energized (primary) mechanisms in ATPase-positive strains (24). However, the use of right-side-out vesicles (26, 32) enables such a distinction, because the binding proteins and ATP necessary for primary active transport are eliminated.
The respiratory substrate ascorbate plus phenazine methosulfate did energize [14C]PTG or [14C]APG transport (Fig. 4) when added to right-side-out vesicles prepared from bacteria induced for GusB and GusC. A similar result was obtained with right-side-out vesicles prepared from cells induced only for GusB (data not shown). Glycerol was inactive, which excluded transport into contaminating intact cells. Also, the energized transport was strongly dependent on the pH, with an optimum at pH 6.0 to 6.5 (Fig. 4B). Both observations indicated that transport was driven by the proton motive force. Furthermore, the uncoupling agents CCCP and DNP inhibited respiration-energized [14C]PTG transport (Table 3). Neither valinomycin, which discharges only the transmembrane electrical potential ΔΨ, nor nigericin, which discharges the pH gradient ΔpH, completely abolished [14C]PTG transport at pH 6.6 (Table 3). However, the two in combination completely prevented transport (Table 3).
FIG. 4.

Energization of transport of glucuronides into right-side-out (RSO) subcellular vesicles. (A) Subcellular vesicles were prepared from E. coli NO2947(pWJL24) grown on minimal medium and induced with IPTG (see text). After incubation for 3 min with additions as below, 50 μM [14C]APG was added and transport was measured. Triangles, no addition; squares, plus 20 mM glycerol; circles, plus 20 mM ascorbate and 0.1 mM phenazine methosulfate. Similar results were obtained when 50 μM [14C]PTG was the substrate (not shown). The pH was 6.6. Points are means of duplicate measurement. (B) Samples of right-side-out vesicles were resuspended in 40 mM potassium phosphate buffer at the indicated pH. After 3 min of incubation, the initial rate of transport of 50 μM [14C]APG was measured with 20 mM ascorbate and 0.1 mM phenazine methosulfate for the respiratory substrate. Points are means of duplicates (15-s time point) expressed as a percentage of the highest value of 1.87 nmol/mg.
TABLE 3.
Effects of ionophores on glucuronide transport into subcellular vesiclesa
| Addition (concn) | Activity (%) |
|---|---|
| None | 100.0 |
| DNP (2 mM) | 10.9 |
| m-CCCP (5 μM) | 8.2 |
| Valinomycin (5 μM) | 27.2 |
| Nigericin (5 μM) | 48.8 |
| Valinomycin (5 μM) + nigericin (5 μM) | 9.3 |
Subcellular vesicles were prepared from E. coli NO2947(pWJL24) grown on minimal medium and induced with IPTG. After incubation for 2.5 min with or without additions as indicated, 20 mM ascorbate and 0.1 mM phenazine methosulfate were added, followed by 50 μM [14C]APG at 3.0 min. Initial rates of transport were measured in duplicate (15-s time point). The concentrations of ionophore used were shown to be maximal for effectiveness in separate dose-response measurements (not shown). All data were normalized to controls without ionophore performed with the same batch of vesicles.
Uptake of [14C]APG into right-side-out vesicles was not inhibited by 5 mM melibiose or lactose, which are substrates for homologous transport proteins (51, 66), or by d-glucose, d-galactose, or maltose, which are substrates for nonhomologous transporters important for growth and metabolism of E. coli. N-Ethylmaleimide (830 μM) and forskolin (130 μM) (commonly used as inhibitors for other bacterial secondary active transport systems) did not inhibit APG transport in the right-side-out vesicles (data not shown).
These results establish that glucuronide uptake occurs by a substrate-specific secondary active transport mechanism energized by either the ΔΨ or ΔpH component of the proton motive force across the cell membrane.
Amplified expression and identification of the GusB and GusC proteins.
Based on the DNA sequence obtained in this work, the gusB gene from E. coli CE1 is predicted to encode 457 amino acid residues with a theoretical size of 49,982 kDa (36). The gusC gene encodes 420 amino acids with an N-terminal sequence of 23 amino acids that strongly indicates it is exported from the cell; the putative leader sequence would then be removed to yield a mature GusC protein of about 44,220 Da.
When membrane preparations were solubilized at 37°C, a protein of ca. 32 kDa was seen in membranes from induced but not uninduced E. coli strain NO2947 carrying tac-inducible gusB and gusC genes on plasmid pWJL24 (gusB+ gusC+) (Fig. 3A). The protein was also not visible in the IPTG-induced host strain containing plasmid pTTQ18 without the gus genes or in the IPTG-induced host strain without a plasmid (Fig. 3A). Notably, it was still present in membranes from induced cells when the plasmid carried only the gusB gene and was absent when the plasmid used contained only the gusC gene (Fig. 3B). When the protein was electroeluted from the gel onto a polyvinylidene difluoride membrane, its N-terminal sequence was found to be M N Q Q L S X R T I V G Y X L G X V (where the amino acids at positions X could not be unambiguously identified). This corresponds to the amino acid sequence of the GusB protein predicted from the DNA sequence of the gusB gene.
When the membranes were dissolved at 100°C, the protein identified as GusB was actually still visible, but now a second IPTG-inducible protein of about 44 kDa appeared (Fig. 3B). This protein was present when the plasmid carried the gusC gene but absent when it carried the gusB gene only (Fig. 3B). Electroelution and N-terminal sequencing yielded A Q L A D D E A G L R I R L K N E L R R, which fits precisely the amino acids in GusC after the predicted signal peptidase cleavage site. Although the predicted molecular mass of the hydrophobic GusB protein is 49,982 Da, it migrated with an apparent molecular mass of 32 kDa in SDS-PAGE. This anomalous migration is very common for integral membrane transport proteins and is perhaps due to only partial unfolding, even in SDS (63). By contrast, the migration of the hydrophilic GusC protein of about 44 kDa fits well with the predicted molecular mass of 44,420 Da for the mature protein.
The preparations used for SDS-PAGE described so far contain both inner and outer membranes. However, these can be reasonably well separated by density gradient ultracentrifugation of French press membrane vesicles, as first described by Miura and Mizushima (45). Allowing for some slight cross-contamination of the membrane fractions (<10%), the GusB protein was associated with the inner membrane (Fig. 3C), while the GusC protein was clearly associated with the outer membrane (Fig. 3C). This result was obtained from two independent cultures.
When the gusB gene was expressed under control of the λOLPL promoter, either with gusC in pWJL4or on its own in pWJL6 (Fig. 1), the transport activity of the host strain was increased (Fig. 3B), but this was not accompanied by a sufficient increase in expression for satisfactory detection of the GusB protein in Coomassie blue-stained SDS-PAGE gels (Fig. 5). There was no evidence in any of our constructs that GusB was expressed in inclusion bodies that were not retained in the membrane preparation. However, amplified expression of the gusC gene did occur to a high level (Fig. 5), comparable to that obtained when expressed under control of the tac promoter (Fig. 5). It should be noted that total membrane proteins were incubated with solubilization buffer at 100°C for 4 min before electrophoresis. The unexpected failure of the upstream gene, gusB, to achieve amplified expression and/or insertion of the protein in the membrane when the downstream gusC gene did so is reminiscent of a similar failure of amplified expression of fucP in a T7 vector when the downstream fucI (l-fucose isomerase) gene was overexpressed (15). Note that in both cases, measurements of transport activities showed that the genes were inducibly expressed (15) (Fig. 2B).
FIG. 5.

Expression of the GusB and GusC proteins in tac and OLPL promoter systems. Cells containing the plasmids indicated below were grown and membrane preparations were made; 1.0 mM IPTG was included for induction of the tacpromoter and 40 μg of nalidixic acid per ml for induction of the OLPL promoter where indicated. Total membrane protein was solubilized at 100°C for 4 min and 45 μg was loaded onto each track of the SDS--12% bisacrylamide gel. Lane 1, tac-gusBCCE1 induced; lane 2, tac-gusBCCE1 uninduced; lane 3, vector pTTQ18 plus IPTG; lane 4, OLPL gusBCCE1 induced; lane 5, OLPL gusBCCE1 uninduced; lane 6, OLPL gusBCE1 induced; lane 8, OLPL gusCCE1 induced; lane 10, vector pAD284 plus nalidixic acid.
In at least three independent cultures of the strains expressing the Gus proteins under control of the tac promoter, the level of GusB or GusC protein detected in comparably prepared membranes was not significantly changed when the original plasmid constructs contained both gusB and gusC or gusB or gusC alone (see, for example, Fig. 3B and 5). Thus, the ability of GusC to enhance transport of [14C]PTG, even though it did not catalyze transport itself, is likely due to its direct interaction with GusB rather than a change in the copy number of either. Since GusB is not sufficiently expressed under the control of the λPL promoter to be visualized in an SDS-PAGE gel, this conclusion cannot be verified in host strain AR120, though the observation that GusC enhances transport in this system indicates that some GusB is present.
A protein of about 25 kDa was always expressed from chromosomal DNA of the E. coli host strain when the GusCCE1 protein was overproduced (Fig. 3B, 3C, and 5). Electroelution from the inner membrane fraction and N-terminal sequencing revealed that this was an osmotic shock protein, PspA (5). Our experiments showed that extended expression of the gusCCE1 gene on its own caused cessation of growth and, eventually, cell death (36, 37), so secretion of the highly expressed GusCCE1 protein was, presumably, the cause of the shock for the E. coli strain.
Concluding remarks.
The results that we obtained show that the product of the gusB gene is responsible for transport of β-d-glucuronides across the inner membrane of E. coli and that such transport is energized by the proton motive force. By contrast, the product of the gusC gene is an outer membrane protein that can enhance glucuronide uptake but does not transport glucuronide. We have found no indications that GusC acts as a binding protein or interacts directly with GusB, with permeabilized cells, subcellular vesicles, or the purified proteins; there is a significant level of sequence homology of GusC (22.4% amino acid identity with a global alignment) to a porin-like protein of Pseudomonas aeruginosa (27) (accession number AAG03552.1). This may indicate that it acts to facilitate transfer of higher-molecular-weight glucuronides across the outer membrane of E. coli. A recent search showed that sequence homologues of GusC are not commonly found in the available genomes of bacteria (or other organisms). Its occurrence in E. coli EO 157, and perhaps other pathogenic E. coli species, indicates that glucuronides selectively toxic for bacteria might be of therapeutic value, especially as. no significant homologues of the GusB/GusC transport system appeared in the human or other sequenced eukaryotic genomes.
Homologues of GusB are found in various bacteria, but to date those characterized have sugar substrates other than glucuronides (50, 66). The data presented in this work and from Stoeber (59) show that a variety of glucuronides can induce expression of the gus operon. As the assay used for gus operon induction, activity of GusA, requires transport of these glucuronides across the membrane, it can be assumed that glucuronide substrates that act as inducers are transported by GusB. This supports the conclusion that E. coli plays a central role in the enterohepatic circulation of hormones and other aglycones of glucuronides.
The success of the tac promoter constructs in achieving high levels of GusB and GusC expression is an important observation for future cloning and amplified expression of other membrane proteins (48) in bacteria. It is not obvious why the λ PL promoter construct was ineffective for GusB when expression of the downstream gene encoding the GusC protein was increased, although the λ PL promoter has been used successfully for the expression of the arabinose, xylose, and fucose membrane transport proteins (19, 42). In this context, the failure of T7 promoters for the amplified expression of some membrane transport proteins but not others is also noteworthy. Work is in progress to understand these phenomena and devise reliable constructs for expression of transporters and other membrane proteins in bacteria.
Acknowledgments
We thank the following for financial support: Enichem Americas; Darwin College and the Rayne Foundation of the University of Cambridge; Human Frontier Programme; European Union; CAMBIA Biosystems LLC; Ajinomoto Co; BBSRC; and the CVCP United Kingdom for an Overseas Research Studentship to H.X.
We are also grateful to: Jeff Keen for his assistance in N-terminal amino acid sequencing; Li-Sha Ma, Jorge Mayer, and Sabine Eckert for invaluable discussions and suggestions in writing this paper; Nicholas G. Rutherford for his assistance in preparation of right-side-out vesicles; B. Poolman for helpful suggestions; and John O'Reilly for all-round technical expertise and help.
REFERENCES
- 1.Ahmed, S., and I. R. Booth. 1981. Quantitative measurements of the proton-motive force and its relation to steady state lactose accumulation in Escherichia coli. Biochem. J. 200:573-581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banhegyi, G., L. Braun, P. Marcolongo, M. Csala, R. Fulcer, J. Mandl, and A. Benedetti. 1996. Evidence for an UDP-glucuronic acid/phenol glucuronide antiport in rat liver microsomal vesicles. Biochem. J. 315:171-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blattner, F. R., G. Plunkett, C. A. Bloch, N. T. Perna, V. Burland, M. Riley, J. Collado-Vides, J. D. Glasner, C. K. Rode, G. F. Mayhew, J. Gregor, N. W. Davis, H. A. Kirkpatrick, M. A. Goeden, D. J. Rose, B. Mau, and Y. Shao. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453-1474. [DOI] [PubMed] [Google Scholar]
- 4.Boos, W., and J. M. Lucht. 1996. Periplasmic binding protein-dependent ABC transporters, p. 1175-1209. In F. C. Neidhardt et al. (ed.) Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. ASM Press, Washington, D.C.
- 5.Brissette, J. L., L. Weiner, T. L. Ripmaster, and P. Model. 1991. Characterization and sequence of the Escherichia coli stress-induced psp operon. J. Mol. Biol. 220:35-48. [DOI] [PubMed] [Google Scholar]
- 6.Büchel, D. E., B. Gronenborn, and B. Müller-Hill. 1980. Sequence of the lactose permease gene. Nature 283:541-545. [DOI] [PubMed] [Google Scholar]
- 7.Burchell, B., and M. W. Coughtrie. 1989. UDP-glucuronosyltransferases. Pharmacol. Ther. 43:261-289. [DOI] [PubMed] [Google Scholar]
- 8.Burchell, B. 1991. Turning on and turning off the sense of smell. Nature 350:16-17. [DOI] [PubMed] [Google Scholar]
- 9.Burchell, B., M. W. Coughtrie, and P. L. Jansen. 1994. Function and regulation of UDP-glucuronosyltransferase genes in health and liver disease: report of the Seventh International Workshop on Glucuronidation, September 1993, Pitlochry, Scotland. Hepatology 20:1622-1630. [DOI] [PubMed] [Google Scholar]
- 10.Chang, G. W., J. Brill, and R. Lum. 1989. Proportion of β-d-glucuronidase-negative Escherichia coli in human fecal samples. Appl. Environ. Microbiol. 55:335-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cleland, W. W. 1967. The statistical analysis of enzyme kinetic data. Adv. Enzymol. 29:1-32. [DOI] [PubMed] [Google Scholar]
- 12.Das, A. 1990. Overproduction of proteins in Escherichia coli: vectors, hosts, and strategies. Methods Enzymol. 182:93-112. [DOI] [PubMed] [Google Scholar]
- 13.Delisle, G. J., and A. Ley. 1989. Rapid detection of Escherichia coli in urine samples by a new chromogenic β-glucuronidase assay. J. Clin. Microbiol. 27:778-779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dutton, G. J. 1966. The Biosynthesis of glucuronides, p. 185-299. In G. J. Dutton (ed.), Glucuronic acid, free and combined. Academic Press, New York, N.Y.
- 15.Dutton, G. J. 1980. Glucuronidation of drugs and other compounds. CRC Press, Boca Raton, Fla.
- 16.Edgerton, M. D., E. Allet, J.-C. Pechere, and T. Kohler. 1997. Direct submission to GenBank. Accession no. AF03389.
- 17.Friedman, A. M., S. R. Long, S. E. Brown, W. J. Buikema, and F. M. Ausubel. 1982. Construction of a broad host range cosmid cloning vector and its use in the genetic analysis of Rhizobium mutants. Gene 18:289-296. [DOI] [PubMed] [Google Scholar]
- 18.Garnier, J., D. J. Osguthorpe, and B. Robson. 1978. Analysis of accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J. Mol. Biol. 120:97-120. [DOI] [PubMed] [Google Scholar]
- 19.Gunn, F. J., C. G. Tate, and P. J. F. Henderson. 1994. Identification of a novel sugar-H+ symport protein, FucP, for transport of L-fucose into Escherichia coli. Mol. Microbiol. 12:799-810. [DOI] [PubMed] [Google Scholar]
- 20.Hazenberg, M. P., W. W. de Herder, and T. J. Visser. 1988. Hydrolysis of iodothyronine conjugates by intestinal bacteria. FEMS. Microbiol. Rev. 54:9-16. [DOI] [PubMed] [Google Scholar]
- 21.Henderson, P. J. F. 1986. Active transport of sugars into Escherichia coli, p. 409-460. In M. J. Morgan (ed.), Carbohydrate metabolism in cultured cells. Plenum Press, New York, N.Y.
- 22.Henderson, P. J. F. 1991. Sugar transport proteins. Curr. Opin. Struct. Biol. 1:590-601. [Google Scholar]
- 23.Henderson P. J. F. H., S. A. Baldwin, M. T. Cairns, B. M. Charalambous, H. C. Dent, F. Gunn, W.-J. Liang, V. A. Lucas, G. E. Martin, T. P. McDonald, B. J. McKeown, J. A. R. Muiry, K. R. Petro, P. E. Roberts, K. P. Shatwell, G. Smith, and C. Tate. 1992. Sugar-cation symport systems in bacteria. Int. Rev. Cytol. 137A:149-208. [PubMed] [Google Scholar]
- 24.Henderson, P. J. F., Giddens, R. A., and M. C. Jones-Mortimer. 1977. The transport of galactose, glucose and their molecular analogues by Escherichia coli K-12. Biochem. J. 162:309-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henderson, P. J. F., and A. J. S. Macpherson. 1986. Assay, genetics, proteins, and reconstitution of proton-linked galactose, arabinose, and xylose transport systems of Escherichia coli. Methods Enzymol. 125:387-429. [DOI] [PubMed] [Google Scholar]
- 26.Horne, P., and P. J. F. Henderson. 1983. The association of proton movement with galactose transport into subcellular membrane vesicles of Escherichia coli. Biochem. J. 210:699-705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang, H., R. J. Siehnel, F. Bellido, E. Rawling, and R. E. Hancock. 1992. Analysis of two gene regions involved in the expression of the imipenem-specific, outer membrane porin protein OprD of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 76:267-273. [DOI] [PubMed] [Google Scholar]
- 28.Jedlitschky, G., I. Leier, U. Buchholz, K. Barnouin, G. Kurz, and D. Keppler. 1996. Transport of glutathione, glucuronate, and sulfate conjugates by the MRP gene-encoded conjugate export pump. Cancer Res. 56:988-994. [PubMed] [Google Scholar]
- 29.Jefferson, R. A. 1989a. The GUS reporter gene system. Nature 342:837-838. [DOI] [PubMed] [Google Scholar]
- 30.Jefferson, R. A. 1989b. The GUS gene fusion system, a laboratory and reference manual. Nuovo Crai SpA, Ricerca Central del Gruppo SME.
- 31.Jefferson, R. A., S. M. Burgess, and D. Hirsh. 1986. β-Glucuronidase from Escherichia coli as a gene fusion marker. Proc. Natl. Acad. Sci. USA 86:8447-8451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaback, H. R. 1972. Transport across isolated bacterial cytoplasmic membranes. Biochim. Biophys. Acta 265:367-416. [DOI] [PubMed] [Google Scholar]
- 33.Klapper, M. H. 1977. The independent distribution of amino acid near neighbor pairs into polypeptides. Biochem. Biophys. Res. Commun. 78:1018-1024. [DOI] [PubMed] [Google Scholar]
- 34.Knol, J., L. Veenhoff, W. J. Liang, P. J. F. Henderson, G. Leblanc, and B. Poolman. 1996. Unidirectional reconstitution into detergent-destabilized liposomes of the purified lactose transport system of Streptococcus thermophilus. J. Biol. Chem. 271:15358-15366. [DOI] [PubMed] [Google Scholar]
- 35.Kyte, J., and R. F. Doolittle. 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157:105-132. [DOI] [PubMed] [Google Scholar]
- 36.Liang, W. J. 1989. Studies on the glucuronide operon of Escherichia coli. M.Phil. thesis. University of Cambridge, Cambridge, United Kingdom.
- 37.Liang, W. J. 1992. The glucuronide transport system of Escherichia coli. Ph.D. thesis. University of Cambridge, Cambridge, England.
- 38.Lin, E. C. C. 1987. Dissimilatory pathways for sugars, polyols, and carboxylates, p. 244-284. In F. C. Neidhardt et al. (ed.), Escherichia coli and Salmonella typhimurium: cellular and molecular biology. ASM Press, Washington, D.C.
- 39.Loe, D. W., K. C. Almquist, S. P. C. Cole, and R. G. Deeley. 1996. ATP-dependent 17β-estradiol 17-(β-d-glucuronide) transport by multidrug resistance protein (MRP) J. Biol. Chem. 27:9683-9689. [DOI] [PubMed] [Google Scholar]
- 40.Lopilato, J., T. Tsuchiya, and T. H. Wilson. 1978. Role of Na+ and Li+ in thiomethylgalactoside transport by the melibiose transport system of Escherichia coli. J. Bacteriol. 134:147-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu, Z., and E. C. C. Lin. 1989. The nucleotide sequence of Escherichia coli genes for L-fucose dissimilation. Nucleic Acids Res. 17:4883-4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maiden, M. C., J. Jones, M. C. Mortimer, and P. J. Henderson. 1988. The cloning, DNA sequence and amplified expression of the gene araE for arabinose-proton symport in Escherichia coli. J. Biol. Chem. 263:8003-8010. [PubMed] [Google Scholar]
- 43.Maniatis, T., E. F. Fritsch, and J. Sambrook (ed.). 1982. Molecular cloning, a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 44.Matsudaira, P. 1987. Sequence from picomole quantities of proteins electroblotted onto polyvinylidene difluoride membranes. J. Biol. Chem. 262:10035-10038. [PubMed] [Google Scholar]
- 45.Miura, T., and S. Mizushima. 1968. Separation by density gradient centrifugation of two types of membranes from spheroplast membrane of Escherichia coli K-12. Biochim. Biophys. Acta 150:159-161. [DOI] [PubMed] [Google Scholar]
- 46.Mott, J. E., R. A. Grant, Y.-S. Ho, and T. Platt. 1985. Maximizing gene expression from plasmid vectors containing the λPL promoter: strategies for overproducing transcription termination factor ρ. Proc. Natl. Acad. Sci. USA 82:88-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Novel, M., and G. Novel. 1976. Regulation of β-glucuronidase synthesis in Escherichia coli K-12: constitutive mutants specifically derepressed for uidA expression. J. Bacteriol. 127:406-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paulsen, I. T., L. Nguyen, M. K. Sliwinski, R. Rabus, and M. H. Saier, Jr. 2000. Microbial genome analyses: comparative transport capabilities in eighteen prokaryotes. J. Mol. Biol. 301:75-100. [DOI] [PubMed] [Google Scholar]
- 49.Platt, T. 1986. Transcription termination and the regulation of gene expression. Annu. Rev. Biochem. 55:339-372. [DOI] [PubMed] [Google Scholar]
- 50.Poolman, B., J. Knol, C. van der Does, P. J. F. Henderson, W.-J. Liang, G. Leblance, T. Pourcher, and I. Mus-Veteau. 1996. Cation and sugar selectivity determinants in a novel family of transport proteins. Mol. Microbiol. 19:911-922. [DOI] [PubMed] [Google Scholar]
- 51.Poolman, B., T. J. Royer, S. E. Mainzer, and B. F. Schmidt. 1989. Lactose transport system of Streptococcus thermophilus: a hybrid protein with homology to the melibiose carrier and enzyme III of phosphoenolpyruvate-dependent phosphotransferase systems. J. Bacteriol. 171:244-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rosenberg, M., and D. Court. 1979. Regulatory sequences involved in the promotion and termination of RNA transcription. Annu. Rev. Genet. 13:319-353. [DOI] [PubMed] [Google Scholar]
- 53.Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 74:5463-5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sasatsu, M., T. K. Misra, L. Chu, R. Laddaga, and S. Silver. 1985. Cloning and DNA sequence of a plasmid-determined citrate utilization system in Escherichia coli. J. Bacteriol. 164:983-993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saxena, M., and G. B. Henderson. 1996. MOAT4, a novel multispecific organic-anion transporter for glucuronides and mercapturates in mouse L1210 cells and human erythrocytes. Biochem. J. 320:273-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schaffner, W., and C. Weissmann. 1973. A rapid, sensitive and specific method for the determination of protein in dilute solution. Anal. Biochem. 56:502-514. [DOI] [PubMed] [Google Scholar]
- 57.Stark, M. J. R. 1987. Multicopy expression vectors carrying the lac repressor gene for regulated high-level expression of genes in Escherichia coli. Gene 51:255-267. [DOI] [PubMed] [Google Scholar]
- 58.Stoeber, F. 1957. Sur la biosynthése induite de la β-glucuronidase chez Escherichia coli. C. R. Acad. Sci. 244:950-952. [PubMed] [Google Scholar]
- 59.Stoeber, F. 1961. Etudes des proprietes et de la biosynthese de la glucuronidase et de la glucuronide-permease chez Escherichia coli. Theses de Docteur es Sciences, University of Paris, Paris, France.
- 60.Tate, C. G., J. A. Muiry, and P. J. F. Henderson. 1992. Mapping, cloning, expression, and sequencing of the rhaT gene, which encodes a novel L-rhamnose-H+ transport protein in Salmonella typhimurium and Escherichia coli. J. Biol. Chem. 267:6923-6932. [PubMed] [Google Scholar]
- 61.Tsuchiya, T., K. Takeda, and T. H. Wilson. 1980. H+-substrate cotransport by the melibiose membrane carrier in Escherichia coli. Membr. Biochem. 3:131-146. [DOI] [PubMed] [Google Scholar]
- 62.Wada, K.-N., S.-I. Aota, R. Tsuchiya, F. Ishibashi, T. Gojobori, and T. Ikemura. 1990. Codon usage tabulated from GenBank genetic sequence data. Nucleic Acids Res. 18:2367-2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ward, A., N. M. Sanderson, J. O'Reilly, N. G. Rutherford, B. Poolman, and P. J. F. Henderson. 2000. The amplified expression, identification, purification, assay and properties of histidine-tagged bacterial membrane transport proteins, p. 141-166. In Membrane transport-a practical approach. Blackwell Press, Oxford, United Kingdom.
- 64.Wilson, K. J., S. G. Hughes,, and R. A. Jefferson. 1992. The Escherichia coli gus operon: induction and expression of the gus operon in E. coli and the occurrence and use of GUS in other bacteria, p. 7-23. In S. Gallagher (ed.), GUS protocols: using the GUS gene as a reporter of gene expression. Academic Press, New York, N.Y.
- 65.Wilson, K. J., A. Sessitsch, J. Corbo, K. E. Giller, A. D. L. Akkermans, and R. A. Jefferson. 1995. β-glucuronidase (GUS) transposons for ecological and genetic studies of rhizobia and other Gram-negative bacteria. Microbiology 141:1691-1705. [DOI] [PubMed] [Google Scholar]
- 66.Yazyu, H., S. Shiota-Niiya, T. Shimamoto, H. Kanazawa, M. Futai, and T. Tsuchiya. 1984. Nucleotide sequence of the melB gene and characteristics of deduced amino acid sequence of the melibiose carrier in Escherichia coli. J. Biol. Chem. 259:4320-4326. [PubMed] [Google Scholar]