

Abstract
The extracts of Aquilaria crassna pericarp were investigated on the MDA-MB-468, a breast cancer cell line, at desired concentration (1–50 μg/mL). The results showed that the dichloromethane (DCM) extract exhibited the strongest toxicity and was carried out subsequently. A total of nine compounds were isolated from the DCM extract using column chromatography and recrystallization, of which their structures were determined. Intriguingly, in addition to the previously reported compounds, neocucurbitacin A, a cucurbitacin triterpenoid aglycone with a lactone in ring A, was reported for the first time in the Aquilaria genus. Among the isolated compounds, cucurbitacin E highly inhibited MDA-MB-468 cell growth in a dose-dependent manner. Owing to binding abilities with the SH2 domain in the molecular docking study, cucurbitacin E, neocucurbitan A, neocucurbitan B, and cucurbitacin E 2-O-β-d-glucopyranoside act as STAT3 inhibitors and are suitable for further research. This study suggests thatAquilaria crassnafruits could serve as a promising source of natural compounds with potential anticancer effects, particularly against breast cancer.
Introduction
Aquilaria crassna Piere ex Lecomte (hereafter referred to asA. crassna) is a tropical and subtropical tree, belonging to the Thymelaeaceae family. This species is distributed in the rainforests of Cambodia, Laos, Thailand, and Vietnam. The tree is best known for its resinous heartwood, which is used for medicinal and aromatic purposes.1 However, natural disasters and excessive exploitation have led to a considerable decrease in the natural populations of theAquilaria genus, as well as the number of A. crassna trees.2 Therefore, cultivation of agarwood trees is encouraged to reduce the harvest from wild populations. In Vietnam, A. crassna is primarily being cultivated in southern Vietnam, including in the provinces of An Giang, Kien Giang, and Binh Phuoc.3
Plant materials derived from A. crassna have shown a range of pharmacological activities, including inhibition of inflammatory cytokines (TNF-α and IL-1α),4 as well as possessing cardioprotective,5 anticancer,6 and antioxidant properties.7 Apart from its heartwood and the essential oil, which are highly valuable in both economic and pharmaceutical benefits, the leaves of A. crassna have been used traditionally for the treatments of various disorders such as antipyretic, analgesic, antioxidative properties, neuroprotection, anti-inflammation, antiaging, as well as anticancer activities.8,9
Breast cancer is a significant public health concern worldwide. According to the Global Cancer Project (GLOBOCAN 2020), it is the most commonly diagnosed cancer globally, and its incidence has been increasing over the past few decades.10 Triple-negative breast cancer cells lack the expression of the estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2), making them particularly challenging to target therapeutically. These cells are highly invasive and often show a relatively low response to existing treatments.11 MDA-MB-468 is well-known for triple-negative breast cancer cells, which is an aggressive breast cancer cell line used in research to better understand chemotherapy resistance and develop more effective treatments.12 It serves as a critical model for studying basal-like breast cancer, which is associated with a more challenging prognosis.13 Due to its high malignancy level, MDA-MB-468 is commonly used for testing anticancer agents, especially those derived from natural sources. In recent years, there has been growing interest in studying the anticancer properties of extracts from various parts of Aquilaria species. Aquilaria sinensis, in particular, has been extensively investigated for its cytotoxic activities, with studies focusing on its chemical constituents.14 Cucurbitacin triterpenoids found in the fruits and flowers of A. sinensis have been identified as the main constituents contributing to the cytotoxic activities against different cancer cell lines.15−17 Recent studies have shown that the extract of A. crassna’s agarwood induces apoptosis and inhibits cancer angiogenesis in breast cancer models, specifically MDA-MB-231 and MCF-7 cells.18 The leaf extract of A. crassna has been reported for its medicinal activities, including enhancing cancer-immune responses.19 However, studies on cucurbitacins from A. crassna or its fruits and their anticancer activity remain sparse. Therefore, in this study, we investigated the chemical components and cytotoxic effects of fruit pericarp extracts and isolated compounds on the breast cancer cell line MDA-MB-468.
Results and Discussion
Cytotoxic Effects of MeOH Extracts and Other Extracts
Extracts were tested at five designed concentrations. The results showed that the activity decreased in the following order: DCM ∼ n-hexane > EtOAc > n-BuOH > MeOH. Among the five extracts, nonpolar extract (DCM) appeared to show strong activity, even at a concentration of 1 μg/mL giving over 96% of inhibition (Table 1). Therefore, the DCM was further investigated.
Table 1. Cytotoxic Effects of Extracts from Aquilaria crassna Pericarpa.
| concentration (μg/mL) | percentage
of viable cells (%) |
|||||||
|---|---|---|---|---|---|---|---|---|
| n-hexane | DCM | EtOAc | n-BuOH | MeOH | DMSO | ACT-D | DMEM | |
| 50 | 5.33 ± 1.07*** | 0.59 ± 0.32*** | 6.24 ± 2.33** | 3.24 ± 0.27*** | 52.68 ± 8.14 | |||
| 25 | 4.46 ± 1.33*** | 0.40 ± 0.22*** | 3.85 ± 1.12*** | 3.16 ± 0.26*** | 81.00 ± 7.11 | 96.93 | 1.78 | 100.00 |
| 10 | 3.74 ± 0.30*** | 2.09 ± 0.78*** | 3.74 ± 0.57*** | 2.01 ± 0.11*** | 80.58 ± 10.74 | |||
| 5 | 2.52 ± 0.19*** | 3.72 ± 0.39*** | 6.62 ± 0.62** | 5.90 ± 0.37** | 91.14 ± 4.42 | |||
| 1 | 4.20 ± 0.41*** | 4.1 ± 0.19*** | 76.69 ± 9.87* | 79.30 ± 5.37 | 90.54 ± 8.6 | |||
ACT-D: positive control actinomycin D, the data are presented with mean ± standard deviation of three replications. P-values < 0.05 (*); 0.01 (**) or 0.001 (***) statistical significance compared with control group treated with medium containing 0.5% DMSO.
Chemical Extraction and Isolation
1.3 kg portion ofA. crassna fruit pericaps was extracted with MeOH solvent and separated into different fractions. The DCM fraction was further isolated to obtain 9 compounds. The isolation results are summarized in Scheme 1.
Scheme 1. Extraction and Chromatographic Separation of Aqualaria crassna Fruit Pericarp.

Structure Elucidation of Isolated Compounds
Cucurbitacin Derivatives
Compound 3 was isolated fromA. crassnaas a white amorphous powder, slightly soluble in MeOH, soluble in DCM, and exhibits maximum absorbance (λmax) at 243 nm in methanol. The MS (ESI–) spectrum showed the fragment m/z [M – H]− = 555.35, suggesting the molecular formula C32H44O8 with 11 degrees of unsaturation. The 1H NMR spectroscopic data in pyridine (Table 3) of 3 showed the presence of 9 methyl group signals (δH 1.59; 1.11; 1.19; 1.44; 1.88; 1.29; 1.50; 1.53; 1.7, and 0.83 ppm); 2 proton signals belong to the olefin group (δH 7.38 and 7.35 ppm) which were in the trans configuration (J = 16 Hz); 3 signals of the –OH group (δH 10.6; 6.22; 6.36 ppm). Analysis of the 13C NMR spectroscopic data (Table 2) indicated that 3 had 32 carbons with 9 methyl groups; 3 methylene groups; 6 downfield methine groups δC 100–150 ppm of 6 olefinic carbon; 3 carbonyl groups at δC 199.0, 204.4, and 213.8, 1 ester group at δC 169.8 ppm. These data suggest that 3 is a four-ring triterpene that shares the main features of the cucurbitacins. According to the HMBC spectroscopic experiment and comparison with the spectral data in ref (20), 3 was cucurbitacin E.
Table 3. 1H NMR of Isolated Compounds in Pyridine.
| 3 | 9 | 7 | 8 | |
|---|---|---|---|---|
| position | cucurbitacin E | cucurbitacin E 2-O-β-d-glucopyranoside | neocucurbitan A | neocucurbitan B |
| 1 | 6.29 d (2.8) | 6.18 brs | 6.22 s | 6.25 s |
| 6 | 5.67 dt (5.1, 2.6) | 5.75 brs | 5.68 m | 5.67 d (4.6) |
| 7 | 2.24 m | 2.34 m | 2.10 dd (20.2, 5.1) | 2.09 dd (20, 5.1) |
| 1.97 m | 2.00 m | 2.31 ddt (20.3, 8.2, 3.8) | 2.3 ddd (20.3, 8, 3.2) | |
| 8 | 2.0 d (8.5) | 2.01 m | 2.04 d (8) | 2.02 d (8) |
| 10 | 3.75 m | 3.50 brs | ||
| 12 | 3.36 d (14.7) | 3.23 bd (14.0) | 2.91 d (14.6) | 2.89 d (14.6) |
| 2.93 d (14.7) | 2.72 d (14.5) | 3.07 d (14.6) | 2.99 d (14.7) | |
| 15 | 1.93 m | 1,65 d (13,1) | 1.63 d (13.0) | |
| 1.74 m | 1,91 m | 1.89 m | ||
| 16 | 5.10 td (7.7, 4.4) | 4,99 t (8,0) | 4.92 d (9.1) | |
| 17 | 3.00 d (7.1) | 2.46 d (6.7) | 2.94 d (7.0) | 2.92 d (9.0) |
| 18 | 1.19 s | 0.94 s | 1.17 s | 1.18 s |
| 19 | 1.11 s | 1.00 s | 1.30 s | 1.30 s |
| 21 | 1.70 s | 1.41 s | 1.67 s | 1.60 s |
| 23 | 7.35 d (16) | 6.48 d (15.6) | 7.35 d (15.7) | 7.54 d (15.0) |
| 24 | 7.38 d (16) | 7.02 d (15.6) | 7.41 d (15.7) | 7.47 d (15.3) |
| 26 | 1.53 s | 1.51 s | 1.56 s | 1.42 s |
| 27 | 1.50 s | 1.54 s | 1.52 s | 1.46 s |
| 28 | 1.59 s | 1.36 s | 1.31 s | 1.23 s |
| 29 | 1.29 s | 1.26 s | 1.25 s | 1.45 s |
| 30 | 1.44 s | 1.41 s | 1.46 s | 1.25 s |
| 32 | 1.88 s | 1.99 s | 1.88 s | |
| 1′ | 4.68 d (7.6) |
Table 2. 13C NMR Data of Isolated Compounds in Pyridine.
| 3 | 9 | 7 | 8 | |
|---|---|---|---|---|
| position | cucurbitacin E | cucurbitacin E 2-O-β-d-glucopyranoside | neocucurbitan A | neocucurbitan B |
| 1 | 115.8, CH | 121.3, CH | 134.3, CH | 134.4, CH |
| 2 | 147.3, C | 145.3, C | ||
| 3 | 199.0, C | 198.9, C | 172.6, C | 172.6, C |
| 4 | 48.6, C | 48.9, C | 42.6, C | 42.6, C |
| 5 | 137.7, C | 135.6, C | 132.7, C | 132.7, C |
| 6 | 120.5, CH | 121.3, CH | 123.8, CH | 123.8, CH |
| 7 | 23.9, CH2 | 23.7, CH2 | 25.0, CH2 | 25.0, CH2 |
| 8 | 42.1, CH | 41.3, CH | 42.4, CH | 42.3, CH |
| 9 | 49.3, C | 48.9, C | 47.9, C | 47.8, C |
| 10 | 35.1, CH | 35.4, CH | 117.7, C | 117.6, C |
| 11 | 213.8, C | 214.6, C | 211.9, C | 211.8, C |
| 12 | 49.3, CH2 | 49.0, CH2 | 50.4, CH2 | 50.5, CH2 |
| 13 | 48.6, C | 50.6, C | 48.9, C | 51.0, C |
| 14 | 51.0, C | 48.0, C | 51.0, C | 48.9, C |
| 15 | 46.5, CH2 | 45.5, CH2 | 45.7, CH2 | 45.8, CH2 |
| 16 | 70.7, CH | 71.2, CH | 70.4, CH | 70.2, CH |
| 17 | 60.0, CH | 58.2, CH | 59.8, CH | 59.3, CH |
| 18 | 20.6, CH3 | 20.2, CH3 | 20.4, CH3 | 20.4, CH3 |
| 19 | 20.1, CH3 | 20.0, CH3 | 25.1, CH3 | 25.2, CH3 |
| 20 | 79.8, C | 78.2, C | 79.6, C | 79.2, C |
| 21 | 25.6, CH3 | 24.0, CH3 | 25.4, CH3 | 25.4, CH3 |
| 22 | 204.4, C | 202.6, C | 204.2, C | 204.2,C |
| 23 | 122.5, CH | 120.4, CH | 122.4, CH | 120.8, CH |
| 24 | 150.2, CH | 152.0, CH | 150.3, CH | 155.7, CH |
| 25 | 79.7, C | 79.4, C | 79.8, C | 70.2, C |
| 26 | 26.2, CH3 | 25.9, CH3 | 26.2, CH3 | 29.7, CH3 |
| 27 | 26.5, CH3 | 26.5, CH3 | 26.6, CH3 | 30.0, CH3 |
| 28 | 18.4, CH3 | 19.9, CH3 | 18.1, CH3 | 18.0, CH3 |
| 29 | 28.0, CH3 | 27.9, CH3 | 28.5, CH3 | 22.4, CH3 |
| 30 | 20.8, CH3 | 18.2, CH3 | 22.3, CH3 | 28.5, CH3 |
| 31 | 169.8, C | 170.4, C | 169.8, C | |
| 32 | 21.7, CH3 | 22.0, CH3 | 21.7, CH3 | |
| 1′-Oglc | 100.5, CH | |||
| 2′ | 72.3, CH | |||
| 3′ | 75.8, CH | |||
| 4′ | 69.3, CH | |||
| 5′ | 76.7, CH | |||
| 6′ | 61.5, CH2 |
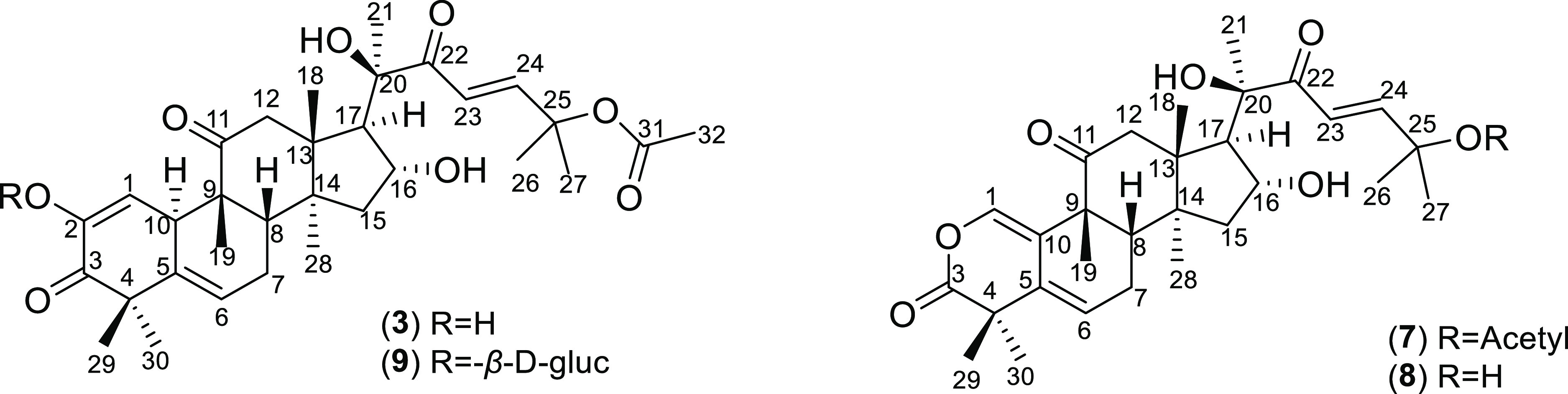
Compound 9 was isolated as a white, amorphous powder soluble in MeOH and DCM. MS (ESI–) showed a molecular ion at m/z 716.98, indicating the molecular formula C38H54O13 (Ω = 12) (Figure 1).
Figure 1.
Structure of isolated cucurbitacins.
The 1H NMR spectrum (Table 3) showed 8 methyl group signals of the aglycon (δH 1.54; 1.51; 1.41; 1.36; 1.26; 1.21; 0.96 and 0.94) and 1 methyl signal of the acetyl group (1.99 ppm); 2 olefin protons (δH 7.02 and 6.48) in trans configuration (J ∼ 16), which closely resembled compound 3. Additionally, the proton anomer of the sugar moiety δH 4.68 helped to predict β-d-glucopyranosyl. 13C NMR spectrum (Table 2) showed a total of 38 resonances corresponding to aglycon with 30 carbons, one hexose sugar moiety, and one acetyl group. The 13C NMR chemical shifts for the aglycon were assigned by comparison with those of compound 3 (cucurbitacin E). Compound 9 belonged to the cucurbitacin skeleton with an acetyl group attached to C25 and one molecule of β-d-glucopyranosyl. The structure of compound 9 was identified as cucurbitacin E 2-O-β-d-glucopyranoside. This result was supported by the comparison of the NMR spectroscopic data of the reported ref (21).
Compound 7 was obtained as white needles, easily soluble in MeOH and DCM. The UV spectrum gave a maximum absorption peak at 231 nm. This compound showed a molecular peak at m/z 565.00 [M + Na]+ in the ESI+ mass spectrum, suggesting the molecular formula C31H44O8 (Ω = 10). The 1H NMR spectrum (Table 3) showed the main features of neo-cucurbitacin skeleton with resonances for 9 methyl resonances at δH 1.17, 1.25, 1.30, 1.31, 1.46, 1.52, 1.56, 1.67, and 1.88 (methyl proton of acetyl group); 2 proton signals of the olefin group (δH 7.41 and 7.36) in the trans configuration (J ∼ 16); and 1 signal of the –OH group (δH 6.24). The 13C NMR spectrum (Table 2) also showed compatibility with the 1H NMR spectrum, which has 31 signals including 9 methyl groups; 3 methylene groups; δH of (172.7 and 169.8). Compound 7 was exclusively different from compound 3 in the A-ring signals because of the absence of C2. These data indicated that compound 7 was a lactone-type cucurbitacin that possessed a neocucurbitacin skeleton. A comparison with the spectral data in ref (22) helps to conclude that 7 was neocucurbitacin A (Figure 1).
Compound 8 was isolated as a white amorphous powder, easily soluble in MeOH, DCM, and exhibits maximum absorbance (λmax) at 231 nm in methanol. The MS (ESI–) spectrum showed the fragment m/z [M – H]− = 499.40, suggesting the molecular formula C29H40O7 with 10 degrees of unsaturation. The NMR spectroscopic data indicated that compound 8 was also a neocucurbitacin. The 1H NMR (Table 3) and 13C NMR (Table 2) spectrum indicated similar resonances to compound 7 except for the absence of two carbons of acetyl group that were attached at C25. Based on the HMBC spectroscopy and comparison with the spectral data in ref (15), 8 was deduced to be neocucurbitacin B (Figure 1).
Flavonoid Compounds
Compound 4 was obtained as yellow needles, slightly soluble in MeOH, soluble in DCM. The MS (ESI+) spectrum gives the fragment m/z [M + H]+ = 299.22, suggesting the molecular formula C17H14O5 (Ω = 11). The 13C NMR spectroscopic data in DMSO-d6 of 4 showed 15 signals, including two high-intensive signals, which indicated a symmetrical structure. These data corresponded to 17 carbons, including 15 carbons of the flavonoid skeleton and 2 methoxy groups. According to the HMBC spectrum, the two methoxy groups were attached at C7 and C4′. Thus, compound 4 was identified and confirmed as 5-hydroxy-7,4′-dimethoxyflavone by comparison with reported data.23
The MS (ESI–), NMR spectroscopic data in DMSO-d6 suggested that compound 5 was also a flavon derivative with two methoxy groups and two hydroxy groups. Based on HMBC data, the two methoxy groups were attached at C7 and C3′, meanwhile two hydroxy groups were linked at the C5 and C4′ positions. The compound 5 was identified as 5,4′-hydroxy-7,3′-dimethoxyflavone (velutin) by comparison of the physical and spectral data with those reported (Figure 2).24
Figure 2.

Structure of isolated flavonoids, chromone, and sterol.
Chromone Compound
Compound 2 was isolated as a white amorphous powder, slightly soluble in MeOH and was assigned the molecular formula C17H14O6 (Ω = 11) (MS (ESI–) m/z 265.00 [M – H]−). The UV spectrum indicated the maximum absorption peak at 326 nm in methanol. The 1H NMR spectrum in pyridine showed a set of typical ABX coupling systems at δH 7.47 (1H, d, J = 3.3 Hz, H-5), 7.48 (1H, dd, J = 2.6, 3.3 Hz, H-6), and 8.03 (1H, d, J = 2.6 Hz, H-8). Additionally, the 1H NMR features of the aromatic protons also suggested the presence of a nonsubstituted aromatic ring from the one doublet at δ 7.23 (2H, d, J = 7.4 Hz, H-2′ and H-6′) and two triplets at δ 7.28 (2H, d, J = 7.6 Hz, H-3′ and H-5′) and δ 7.20 (1H, d, J = 7.3, H-4′). The 13C NMR spectrum showed 15 resonances for 6 quaternary carbons, including one carbonyl group (δC 177.7), 2 methylene carbon, and 7 methine carbons including one pair of abnormally high signals δC 128.9 and 128.7, which were identified as the signal of four symmetric carbons on the phenyl ring without substituents. Based on these data, compound 3 was predicted to be a chromone derivative with a phenyl moiety attached via two carbons at C-2. From the HMBC interaction and comparison with the spectral data in ref (25), it was concluded that 2 is 7-hydroxy-2-(2-phenylethyl)chromon (Figure 2). The chromone compounds that are characteristic of agarwood are also found in the fruits.
The other compounds were identified as genkwanin (1), stigmasterol (6) by comparison of spectroscopic data with those reported in the literature.26,27 The spectral analyses of all compounds are included in the Supporting Information: Figures S1–S37.
Evaluate the Cytotoxic Activity of Some Isolated Potential Substances
Cucurbitacins are a class of tetracyclic triterpenes that exhibit high cytotoxic potential. Solid data revealed that cucurbitacins can induce cell cycle arrest and apoptosis and cancer cell differentiation in cancer cells. Furthermore, they inhibited the invasion, metastasis, or angiogenesis ability of cancer cells. In addition, cucurbitacins showed an inhibitory effect on cancer cell proliferation. This effect was tested on multiple cancer cell lines including breast cancer, hepatocellular carcinoma, pancreatic cancer, acute and chronic leukemia, lymphoma, colon cancer, glioblastoma tumor, throat cancer, lung cancer, oral epidermoid carcinoma, cervical cancer, central nervous system tumor, and Burkitt’s tumor cell line.28
Many cucurbitacins such as curcubitacin B, cucurbitacin E, cucurbitacin L, 23,24-dihydro-cucurbitacin D, and 25-acetoxy-23,24-dihydro-cucurbitacin F were evaluated for their ability to inhibit cancer cell proliferation on cancer breast cell lines MCF-7, PC-3, NCI–N87, MG-63, MDA-MB-468, and MDA-MB-231.29 Cucurbitacin E isolated from the hulls of Aquilaria agallocha showed potential in cancer treatment due to highly cytotoxic activity on HT29 cells and P-388 leukemia cells.15 Cucurbitacin E 2-O-β-d-glucopyranoside isolated from the fruit of Citrullus colocynthis showed potent cytotoxic activity in vitro against the HepG2 cell line with an IC50 of 3.5 μM and in vivo cucurbitacin E 2-O-β-d-glucopyranoside has the ability to prolong survival, lifespan, and normalize biochemical parameters in Ehrlich cervical cancer tumor-bearing mice.21
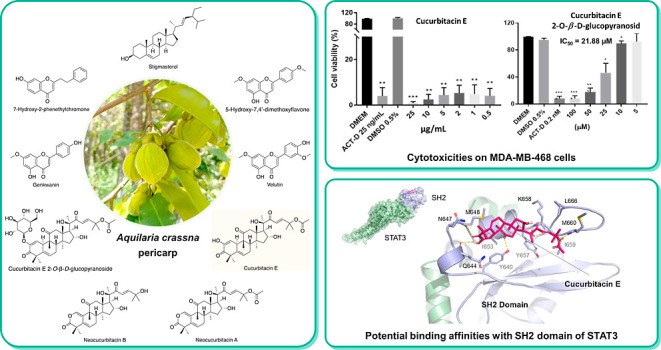
Hence, we investigated the cytotoxic activity of cucurbitacin E (3) and its glycoside (9) on MDA-MB-468 cells (Figure 3).
Figure 3.

Cytotoxic activity of cucurbitacin E (3) and cucurbitacin E 2-O-β-d-glucopyranoside (9) on MDA-MB-468 cell lines. P-values < 0.05 (*); 0.01 (**), or 0.001 (***) compared with control group treated with medium containing 0.5% DMSO.
Cucurbitacin E (3) illustrated the inhibition of the proliferation rate on breast cancer cells. Cucurbitacin E 2-O-β-d-glucopyranoside (9) showed moderate cytotoxic activity on MDA-MB-468 cells with IC50 = 21.88 μM. Meanwhile, its aglycon (3) exhibits significant activity, which inhibited cell growth by more than 90% at 0.5 μg/mL (∼0.89 nM). This result indicated that attaching a sugar moiety to a saponin structure could lead to reduced cell growth inhibitory activity compared to its aglycone. The reason probably is the polarity increase resulting in difficulty for the compounds to cross the phospholipid membrane.30 The cytotoxicities of compounds 3 and 9 are presented in the Supporting Information: Figure S38.
The triterpenoid compounds, including cucurbitacin and neocucurbitacin, are specific to the Cucurbitaceace family with strong cytotoxic activity on cancer cell lines as well as other biological activities. Neocucurbitacin A and B are two of the four known lactone-containing cucurbitacins to date and are considered difficult to isolate in nature. According to the literature, there are no reports of neocucurbtacin A for cytotoxic activity. Neocucurbitancin B isolated from the fruit of A. sinensis species exhibited cytotoxic activity against the liver cancer cell line SMMC-7721 and the human gastric cancer cell line SGC-7901 by MTT method, IC50 results, respectively, are 1.5 and 2.2 μg/mL.15 Therefore, neocucurbitacin A and B have been isolated with the potential for further research on cytotoxic effects on breast cancer cells.
The constitutive activation of STAT3 is frequently detected in human breast cancer cell lines as well as clinical breast cancer specimens, but not in normal breast epithelial cells.31,32 Activated STAT3 plays an important role in the oncogenesis of breast carcinoma, for instance, by stimulating cell proliferation, promoting tumor angiogenesis, and resisting apoptosis.33,34 Since constitutively active STAT3 is linked to several human cancers, STAT3 could be a desirable target for cancer treatment. As one of its principal domains, the SH2 transactivation region is responsible for interactions of STAT3 with tyrosine-phosphorylated receptors as well as its dimerization required for DNA binding and gene expression.35 The phosphorylation of STAT3 at Y705 leads to dimerization, nuclear translocation, recognition of STAT3-specific DNA binding elements, and activation of target gene transcription.35,36 In recent studies, cucurbitacin E (3) was reported to have an inhibition effect on the phosphorylation process of STAT3.29,37 Therefore, it would be intriguing to investigate the binding affinity of cucurbitacin E (3), relating to similar cucurbitacins 7–9 and other extracted compounds 1, 2, and 4–6 from A. crassna pericarp with the SH2 domain of STAT3.
Molecular docking results of nine phytochemicals and the reference ligand SI-109 into the SH2 domain of STAT3 were shown in Table 4 and Figure 4. Compound SI-109 exhibited superior binding affinity via its binding energy being −12.89 kcal mol–1 and its hydrogen bonds with several key residues in the SH2 domain. The rmsd value between the top docking pose and the cocrystallized conformation was below 2 Å, demonstrating that the docking method used and related parameters could reproduce the experimental data and could be applicable to evaluate the binding affinity of compound 1–9.38 In general, except for chromone derivative 2, cucurbitacins (3, 7, 8, and 9), flavones (1, 4, and 5), and stigmasterol 6 could occupy the same pockets shaped by V637-T641, Q643Q644, M648, I653, Y657-M660, and L666 as the glutamine, diphenylmethyl, and [8,5] bicyclic moieties of SI-109. Among those, cucurbitacins exhibited the strongest binding abilities with the SH2 domain (ΔG −7.91 to −7.76 kcal mol–1), in contrast with flavones (ΔG −5.56 to −5.15 kcal mol–1). Both these phytochemical groups could mostly form hydrogen bonds with Y640, Q644, and K658 and, in addition, with N647 and Y657 in the case of cucurbitacins, that were similar to the case of the SI-109 inhibitor. Besides, several other residues were also involved in hydrogen bonds with cucurbitacins, such as S636, E638 (e.g., cucurbitacin E 2-O-β-d-glucopyranoside 9), I653 (e. g., neocucurbitans 7, 8), G656 (e.g., cucurbitacins 7–9), and M660 (e.g., cucurbitacin E 3). Interestingly, cucurbitacin E 3, with its binding energy being slightly lower than that of its glucopyranoside 9, suggested that it could bind into the STAT3′s SH2 domain more efficiently. The combination of the better binding affinity and absorption ability of cucurbitacin E 3 than its glucopyranoside 9 could be the major reasons for its stronger inhibitory effect on breast cancer cell growth experimentally. Regarding stigmasterol 6, although its binding energy was comparable to those of the cucurbitacins (ΔG −7.90 kcal mol–1), this compound could form only a hydrogen bond with K658, suggesting that hydrophobic contacts played a dominant role in its binding affinity. Meanwhile, compound 2 was predicted to bind into a distinct hydrophobic pocket shaped by T527-K531, W564, and F588-E612, where it could form hydrogen bonds with R609 and F610 (ΔG −6.95 kcal mol–1). In the combination of binding affinities, binding sites of isolated compounds and their nonbonded interactions formed, we suggested that the cucurbitacin group (3, 7, 8, and 9) might be potential STAT3 inhibitors via their binding with the SH2 domain. The observed limitation of cucurbitacins was that they exhibited less effective binding abilities than the reference compound SI-109 due to the lack of the extensive hydrogen bonding network such as that of the phosphoric acid moiety of SI-109 with R609, S611, E612, and S613. This observation could pave the way for future studies in structural modification to develop new derivatives of these cucurbitacins as potent STAT3 inhibitors in which their binding affinity to the SH2 domain could be enhanced via increasing hydrogen bonding interactions with the aforementioned amino acids (Supporting Information: Figures S39–S69).
Table 4. Binding Energies (ΔG, kcal mol–1) and Non-Bonded Interactions between Nine Compounds 1–9 Isolated from A. crassna Pericarp and the SH2 Domain of STAT3 (PDB ID: 6NJS), Compared to SI-109 Inhibitor.
| compounds | ΔG (kcal mol–1) | residues
involved in |
|
|---|---|---|---|
| hydrogen bonds | hydrophobic contacts | ||
| genkwanin (1) | –5.56 | Q644, N647, G656, K658 | V637, Y640, Q644, M648, I653, Y657, K658 |
| 7-hydroxy-2-phenethylchromone (2) | –6.95 | R609, F610 | T527, L528, E530, K531, W564, F588, I589, S590, F610, S611, E612 |
| cucurbitacin E (3) | –7.82 | Y640, Q644, N647, Y657, K658, M660 | Y640, Q644, M648, I653, Y657, K658, I659, L666 |
| 5-hydroxy-7,4′-dimethoxyflavone (4) | –5.51 | Y640, K658 | V637, E638, P639, Y640, Q644, M648, I653, Y657 |
| velutin (5) | –5.15 | Y640, Q644, K658 | V637, E638, P639, Y640, M648, I653, Y657, I659 |
| stigmasterol (6) | –7.90 | K658 | Y640, T641, Q643, Q644, N647, M648, I653, Y657 |
| neocucurbitacin A (7) | –7.91 | Q644, N647, I653, G656, Y657, K658 | V637, E638, Q644, N647, M648, I653, Y657, K658, I659 |
| neocucurbitacin B (8) | –7.77 | Y640, Q644, I653, G656, Y657 | Y640, Q644, N647, M648, I653, Y657, K658, I659, M660, L666 |
| cucurbitacin E 2-O-β-d-Glc (9)a | –7.76 | S636, E638, Y640, Q644, N647, G656, K658 | W623, V637, Y640, Q644, M638, I653, Y657, K658, I659 |
| SI-109 (rmsd = 1.48 Å)39 | –12.89 | R609, S611, E612, S613, S636, E638, Y640, Q644 | W623, Q635, V637, P639, Y640, I653, Y657, K658, I659 |
Abbreviation: Glc = glucopyranoside.
Figure 4.

Docking results of nine isolated compounds from A. crassna pericarp in the SH2 domain of STAT3. Binding mode and hydrogen bonding between the top docking pose of nine phytochemicals 1–9 (shown in different colored sticks) at the SH2 domain (shown in light-purple cartoon and surface) of STAT3 (shown in light-green cartoon and surface), in comparison with the reference compound SI-109 (shown in white sticks).39 Residues within 4 Å surrounding the ligands were shown in sticks. Hydrogen bonds were indicated by yellow dashed lines, while heteroatoms were indicated by different colors such as oxygen (red), nitrogen (blue), fluorine (cyan), sulfur (yellow), and phosphorus (orange). Abbreviation: Glc = glucopyranoside.
Conclusion
In this study, bioassay-guided fractionation and purification were used to isolate the cytotoxic compounds of the extract from A. crassna fruits. From the active fraction (DCM), nine known compounds were isolated and identified, including genkwanin (1), 7-hydroxy-2-phenethylchromone (2), cucurbitacin E (3), 5-hydroxy-7,4′-dimethoxyflavone (4), velutin (5), stigmasterol (6), neocucurbitacin A (7), neocucurbitacin B (8), and cucurbitacin E 2-O-β-d-glucopyranoside (9). Compound 7 was isolated from the genus Aquilaria for the first time, while compounds 2, 3, 4, 5, 8, and 9 were first reported in A. crassna. Compounds 3 and 9 were investigated for cytotoxicity against breast cancer cell lines MDA-MB-468. Compound 9 showed moderate cytotoxic activity on MDA-MB-468 cells (IC50 = 21.88 μM). Meanwhile, its aglycon (compound 3) exhibited stronger cytotoxic activity, which inhibited cell growth by more than 90% at 0.89 nM.
Moreover, molecular docking studies of nine phytochemicals and the reference ligand SI-109 into the SH2 domain of STAT3 illustrated that cucurbitacins 3, 7, 8, and 9 may potentially act as STAT3 inhibitors due to their binding abilities with the SH2 domain.
In conclusion, the limited research on A. crassna suggests that there is much potential for further exploration of its phytochemical composition and biological activity. With the possibility of discovering new compounds, including novel derivatives of cucurbitacins with potent effects on desirable targets for cancer treatment, A. crassna’s fruits are an abundant source of raw materials for future synthesis and structural modification.
Materials and Methods
Plant Materials
The fruits of A. crassna were collected at the farm of Tien Phuoc Co. Ltd. at Dong Tam ward, Dong Phu district, Binh Phuoc province, Vietnam, in June 2018. A reference voucher specimen was deposited in the Department of Pharmacognosy, Faculty of Pharmacy, University of Medicine and Pharmacy, Ho Chi Minh City, Vietnam (UMP-2018-AQf-001). The fruits were freshly collected and oven-dried at 50 °C in 3 days, and then the seeds were removed to obtain pericarps. Then, the pericarps were powdered before extraction.
Cell Culture
MDA-MB-468 cells (ATCC HTB-132) are a p16-positive human BLBC cell line isolated from metastatic pleural effusion from a 51 year-old African American female. MDA-MB-468 (ATCC HTB-132; p16+/+, p53+/+, p21+/+, and RB-null) and HMF cells were purchased from ATCC (USA). Breast cancer MDA-MB-468 cells and human mammary fibroblasts (HMF) cells were cultured as monolayers at a density of 10,000 and 2500 cells/cm2, respectively, in T25/T75 cm2 flasks (Corning, UK) and maintained in Dulbecco’s modified eagles medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) (Biosera, UK), 2 mM l-glutamine (Life Technologies, UK), and 1 mM Sodium Pyruvate without antibiotics at 37 °C with 5% CO2. MDA-MB-468 cells were passed every 7 days, and growth medium was changed every 2–3 days.
Chemicals
Thin layer chromatography (TLC) was performed using silica gel F254 (40–63 μm, Merck). Spots were detected by UV 254 and 365 nm, then heated after spraying with 10% H2SO4 in ethanol. Column chromatography (CC) was performed with silica gel (200–300 mesh). Silica gel 60, 40–63 μm (230–400 mesh ASTM), for column chromatography, was purchased from Scharlau (Barcelona, Spain). Other chemicals were purchased from Labscan (Thailand).
ESI low-resolution LC/MS data were obtained using an Agilent Technologies 6130 Quadrupole mass spectrometer (Agilent Technologies, Santa Clara, CA, USA) coupled with an Agilent Technologies 1200 series high-performance liquid chromatography (HPLC) instrument using a reversed-phase C18 column (Phenomenex Luna, 100 × 4.6 mm; 3.5 μm).
1H, 13C NMR, and 2D NMR spectra were conducted on a Bruker Avance 800 MHz spectrometer (Bruker, Billerica, MA, USA) at the Research Institute of Pharmaceutical Sciences, Seoul National University (Korea).
Cytotoxicity Assay
The cytotoxicity effect of extracts and compounds was determined as per the described method.40
Sample Preparation
The powder of A. crassna pericarps (100 g) was extracted by ultrasonic extractor with 500 mL of MeOH at 45 °C for 90 min. After filtering to collect the extract, the residue was extracted three consecutive times (300 mL × 3 times).
All extracts were combined and evaporated in vacuo to obtain concentration, termed the MeOH extract. The MeOH extract was separated by a liquid–liquid distribution with different solvents to yield n-hexane, dichloromethane (DCM), ethyl acetate (EtOAc), and n-butanol (n-BuOH) extracts. The extracts were lyophilized to remove organic solvents completely and stored in colored vials at 0–4 °C.
Extracts were dissolved in DMEM with 0.5% dimethyl sulfoxide (DMSO) to obtain a concentration of 50; 25; 10, 5; 1 μg/mL.
Purified compounds were mixed in DMEM containing 0.5% DMSO to obtain a concentration of 25; 10; 5; 2; 1; and 0.5 μg/mL.
Experiments
In the experiment, 10 μL of the sample was added to each well of a 384-well plate followed by adding 60 μL of cells at a density of 1.7 × 104 cells per well. The plate was then incubated for 48 h, and the medium was changed by withdrawing 50 μL of the old medium and adding 70 μL of new culture. The plate was incubated again for 72 h. Cells were then fixed and stained by replacing 80 μL of old medium, adding 40 μL of mixture SYTOX (1:10,000) and Hoechst (1:10,000) and continuously incubated for 30 min at 37 °C before counting the nuclei of live and dead cells by fluorescence microscopy (IN Cell Analyzer 2200, GE Healthcare, UK). Actinomycin D (ActD) 0.0025 μg/mL (∼0.2 nM) was used as the positive control.
The percentage of viable cells was calculated by the formula
Tt/c: Number of living and dead cell nuclei (colorized with Hoechst reagent) of the sample (t)/negative control (c).
Ct/c: Number of dead cell nuclei (staining with Sytox reagent) of the test sample (t)/negative control (c).
To quantify survival, cells were stained with 40 mL of 1.62 μM Hoechst 33,342 (Invitrogen, UK) and Sytox for 30 min and imaged using the IN Cell 2200 (GE, UK).
Chemical Extraction and Isolation
The dry pericarp powder (1.3 kg) was extracted by percolating with methanol (30 L) to yield an extract. Then, the extract was evaporated in vacuo to obtain MeOH extract (116 g), which was dissolved in water (800 mL), cooled down to remove chlorophyll, and then partitioned with DCM (6 L) to obtain DCM extract (30 g). The remaining aqueous extract was extracted with EtOAc (4 L) and n-BuOH (4 L), to yield the EtOAc residue (1.46 g), EtOAc extract (8.08 g), n-BuOH residue (1.48 g), and n-BuOH extract (7.74 g), respectively (Scheme 1).
The DCM extract (30 g) was chromatographed (silica gel, 0.045–0.063 mm), Merck, using a gradient of n-hexane, n-hexane–DCM, DCM, and DCM–EtOAc, to give 7 fractions (D1–D7).
Fraction D5 (2.9 g) was chromatographed on a silica gel column, using n-hexane–EtOAc as an eluent, to obtain 18 fractions (D5.1-D5.18). Fraction D5.7 and D5.10 were recrystallized in appropriate solvents to yield compounds 1 (32 mg), 3 (90.6 mg), respectively. Fraction D5.9 was subjected through a silica gel column and eluted with DCM-EtOAc to obtain compound 2 (4 mg).
Fraction D6 (12 g) was chromatographed using DCM–EtOAc as an eluent to obtain 25 fractions. By using recrystallizing in suitable solvents, compound 4 (1.6 mg) and 5 (1.2 mg) from fraction D6.2, compound 6 (4.0 mg) from fraction D6.5, compound 7 (15.8 mg) from fraction D6.10, compound 8 (27 mg) from fraction D6.22, and compound 9 (8 mg) from fraction D6.25 were yielded (Scheme 1).
Molecular Docking
Isolated compounds 1–9 and SI-109 (CAS 2429877-30-3)—as a reference ligand—were used in molecular docking studies to examine their binding affinities with the Src homology 2 (SH2) domain of the signal transducer and activator of transcription 3 (STAT3).39 Briefly, ligand structures were sketched by ChemDrawBio 2018 (*.sdf), then they were added all hydrogen atoms and protonated at pH 7.4, followed by their geometry optimization by Avogadro (*.mol2).41 On the other hand, the structure of STAT3 was retrieved from RSCB Protein Data Bank (PDB ID: 6NJS).39 All missing loops in the structure were modeled, energy minimized before removing all water molecules by using Chimera 1.16 and Modeler.42,43 In AutoDock Tools 1.5.6, ligands were assigned the number of active torsions with the fewest atoms moving while the protein was added all hydrogens, then they were saved as appropriate format (*.pdbqt).44 Molecular docking studies were conducted by applying AutoDock 4.2 with Lamarckian Genetic Algorithm (LA-GA).44,45 Each ligand was searched for possible conformations within a 30.0 × 37.5 × 37.5 Å3 box covering the entire surface of the STAT3 SH2 domain (residues 580–670), with the x-, y-, and z-coordinates of the center point being of 11.5, 51.5, and −1.5, respectively. There was a total of 500 feasible conformations obtained with a maximum of 2,500,000 evaluations per conformation. Docking results were clustered with a rmsd cutoff value being of 2.0 Å. The best binding pose was selected on the basis of lowest binding energy (ΔG, kcal mol–1) from the top cluster to be analyzed and visualized by using PyMOL 2.3.4.46
Statistical Analysis
Data were expressed as mean ± standard deviation. Data were analyzed by Dunnett-test: One-way ANOVA was performed using Graphpad Prism 8 software. The P-value < 0.05 was considered statistically significant. The P-values < 0.05 (*); 0.01 (**) or 0.001 (***) represents the level of statistical significance. The experiment was repeated three independent times.
Acknowledgments
This study was supported by the University of Medicine and Pharmacy at Ho Chi Minh city (2019). We would also like to extend our thanks to Professor Jeong Hill Park from Seoul National University, Korea, for his valuable assistance in supporting NMR instrument.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c04656.
1D and 2D NMR, MS spectra of compounds 1–9; molecular docking to examine binding affinities of compounds 1–9 and SI-109 with the Src homology 2 (SH2) domain of STAT3 (PDF)
The authors declare no competing financial interest.
Special Issue
Published as part of the ACS Omegavirtual special issue “Phytochemistry”.
Supplementary Material
References
- LeeMohamed S. Y. R.The Origin and Domestication of Aquilaria, an Important Agarwood-Producing Genus. In Agarwood: Science Behind the Fragrance; Mohamed R., Ed.; Springer Singapore, 2016; pp 1–20. 10.1007/978-981-10-0833-7_1. [DOI] [Google Scholar]
- Lin Y.; Feng T.; Dai J.; Liu Q.; Cai Y.; Kuang J.; Wang Z.; Gao X.; Liu S.; Zhu S. DNA barcoding identification of IUCN Red listed threatened species in the genus Aquilaria (Thymelaeaceae) using machine learning approaches. Phytochem. Lett. 2023, 55, 105–111. 10.1016/j.phytol.2023.04.007. [DOI] [Google Scholar]
- Son H. N.Final Report of the Project “Assessment of the Current Situation and Sustainable Development of the Plant Do Tram Aquilaria spp.” of the Ministerium of Agriculture and Rural Development of the Socialist Republic of Vietnam, Implemented 2007–2010, Non-Timber Forest Product Research Center; Vietnamese Academy of Forest Sciences: Hanoi, Vietnam, 2010.
- Peng D.-Q.; Yu Z.-X.; Wang C.-H.; Gong B.; Liu Y.-Y.; Wei J.-H. Chemical Constituents and Anti-Inflammatory Effect of Incense Smoke from Agarwood Determined by GC-MS. Int. J. Anal. Chem. 2020, 2020, 1–19. 10.1155/2020/4575030. [DOI] [Google Scholar]
- Jermsri P.; Jiraviriyakul A.; Unajak S.; Kumphune S. Effect of Aquilaria crassna crude extract on simulated ischemia induced cardiac cell death. Int. J. Pharma Bio Sci. 2012, 3 (3), 604–613. [Google Scholar]
- Dahham S. S.; Ahamed M. B. K.; Saghir S. M.; Alsuede F. S.; Iqbal M. A.; Majid A. Bioactive essential oils from Aquilaria crassna for cancer prevention and treatment. GJPAAS 2014, 4, 26–31. 10.13140/2.1.3315.7125. [DOI] [Google Scholar]
- Tay P. Y.; Tan C. P.; Abas F.; Yim H. S.; Ho C. W. Assessment of Extraction Parameters on Antioxidant Capacity, Polyphenol Content, Epigallocatechin Gallate (EGCG), Epicatechin Gallate (ECG) and Iriflophenone 3-C-β-Glucoside of Agarwood (Aquilaria crassna) Young Leaves. Molecules 2014, 19 (8), 12304–12319. 10.3390/molecules190812304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattayasai J.; Bantadkit J.; Aromdee C.; Lattmann E.; Airarat W. Antipyretic, analgesic and anti-oxidative activities of Aquilaria crassna leaves extract in rodents. J. Ayurveda Integr. Med. 2012, 3 (4), 175. 10.4103/0975-9476.104427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wongwad E.; Pingyod C.; Saesong T.; Waranuch N.; Wisuitiprot W.; Sritularak B.; Temkitthawon P.; Ingkaninan K. Assessment of the bioactive components, antioxidant, antiglycation and anti-inflammatory properties of Aquilaria crassna Pierre ex Lecomte leaves. Ind. Crops Prod. 2019, 138, 111448. 10.1016/j.indcrop.2019.06.011. [DOI] [Google Scholar]
- Sung H.; Ferlay J.; Siegel R. L.; Laversanne M.; Soerjomataram I.; Jemal A.; Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. Ca-Cancer J. Clin. 2021, 71 (3), 209–249. 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- Foulkes W. D.; Smith I. E.; Reis-Filho J. S. Triple-Negative Breast Cancer. N. Engl. J. Med. 2010, 363 (20), 1938–1948. 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- Shokrollahi Barough M.; Hasanzadeh H.; Barati M.; Pak F.; Kokhaei P.; Rezaei-Tavirani M. Apoptosis/Necrosis Induction by Ultraviolet, in ER Positive and ER Negative Breast Cancer Cell Lines. Iran. J. Cancer Prev. 2015, 8 (6), e4193 10.17795/ijcp-4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. L.; Porter W.; Burghardt R.; Safe S. H. Mechanism of inhibition of MDA-MB-468 breast cancer cell growth by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Carcinogenesis 1997, 18 (5), 925–933. 10.1093/carcin/18.5.925. [DOI] [PubMed] [Google Scholar]
- Li W.; Chen H.-Q.; Wang H.; Mei W.-L.; Dai H.-F. Natural products in agarwood and Aquilaria plants: chemistry, biological activities and biosynthesis. Nat. Prod. Rep. 2021, 38 (3), 528–565. 10.1039/D0NP00042F. [DOI] [PubMed] [Google Scholar]
- Mei W.-L.; Lin F.; Zuo W.-J.; Wang H.; Dai H.-F. Cucurbitacins from fruits of Aquilaria sinensis. Chin. J. Nat. Med. 2012, 10 (3), 234–237. 10.3724/SP.J.1009.2012.00234. [DOI] [Google Scholar]
- Yang J.; Hu D. B.; Xia M. Y.; Luo J. F.; Li X. Y.; Wang Y. H. Bioassay-guided isolation of cytotoxic constituents from the flowers of Aquilaria sinensis. Nat. Prod. Bioprospect. 2022, 12 (1), 11. 10.1007/s13659-022-00334-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Extracts of Aquilaria hulls and use thereof in the treatment of cancer. U.S. Patent 2011/0160152 A1, 2012.
- Jang H. M.; Lee J. H.; Kim J. H.; Park G.; Jeon J. H. In vitro anticancer effect of Aquilaria crassna extract on human mammary gland cancer cells. Int. J. Biosci. 2020, 16, 187–192. [Google Scholar]
- Pho-on P.; Sudsaward S.; Wongwad E.; Ingkaninan K.; Khunchai S. Aquilaria crassna leaf extract selectively upregulated calreticulin surface expression, a pro-phagocytotic signal, in triple-negative breast cancer cells. NUJST 2023, 31 (2), 42–55. 10.14456/nujst.2023.14. [DOI] [Google Scholar]
- Kong Y.; Chen J.; Zhou Z.; Xia H.; Qiu M.-H.; Chen C. Cucurbitacin E induces cell cycle G2/M phase arrest and apoptosis in triple negative breast cancer. PLoS One 2014, 9 (7), e103760 10.1371/journal.pone.0103760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyad S.-E. N.; Abdel-Lateff A.; Alarif W. M.; Patacchioli F. R.; Badria F. A.; Ezmirly S. T. In vitro and in vivo study of cucurbitacins-type triterpene glucoside from Citrullus colocynthis growing in Saudi Arabia against hepatocellular carcinoma. Environ. Toxicol. Pharmacol. 2012, 33 (2), 245–251. 10.1016/j.etap.2011.12.010. [DOI] [PubMed] [Google Scholar]
- Kawahara N.; Kurata A.; Hakamatsuka T.; Sekita S.; Satake M. Two Novel Cucurbitacins, Neocucurbitacins A and B, from the Brazilian Folk Medicine “Buchinha”(Luffa operculata) and Their Effect on PEBP2αA and OCIF Gene Expression in a Human Osteoblast-Like Saos-2 Cell Line. Chem. Pharm. Bull. 2001, 49 (10), 1377–1379. 10.1248/cpb.49.1377. [DOI] [PubMed] [Google Scholar]
- Arega E. Phytochemical studies of the ethyl acetate extract of the fruit of piper capense. J. Phytopharmacother. Nat. Prod. 2018, 04, 148–152. 10.4172/2472-0992.1000148. [DOI] [Google Scholar]
- Jung S.-H.; Kim J.; Eum J.; Choe J. W.; Kim H. H.; Kee Y.; Lee K. Velutin, an aglycone extracted from Korean mistletoe, with improved inhibitory activity against melanin biosynthesis. Molecules 2019, 24 (14), 2549. 10.3390/molecules24142549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L.; Qiao L.; Xie D.; Yuan Y.; Chen N.; Dai J.; Guo S. 2-(2-Phenylethyl) chromones from Chinese eaglewood. Phytochemicals 2012, 76, 92–97. 10.1016/j.phytochem.2011.11.017. [DOI] [PubMed] [Google Scholar]
- Isaev I.; Agzamova M.; Isaev M. Genkwanin and iridoid glycosides from Leonurus turkestanicus. Chem. Nat. Compd. 2011, 47, 132–134. 10.1007/s10600-011-9857-9. [DOI] [Google Scholar]
- Jain P. S.; Bari S. B. Isolation of Lupeol, Stigmasterol and Campesterol from Petroleum Ether Extract of Woody Stem of Wrightia tinctoria. Asian J. Plant Sci. 2010, 9, 163–167. 10.3923/ajps.2010.163.167. [DOI] [Google Scholar]
- Chen X. B. J.; Bao J.; Guo J.; Ding Q.; Lu J.; Huang M.; Wang Y. Biological activities and potential molecular targets of cucurbitacins: a focus on cancer. Anticancer Drugs 2012, 23 (8), 777–787. 10.1097/cad.0b013e3283541384. [DOI] [PubMed] [Google Scholar]
- Huang W. W.; Yang J. S.; Lin M. W.; Chen P. Y.; Chiou S. M.; Chueh F. S.; Lan Y. H.; Pai S. J.; Tsuzuki M.; Ho W. J.; Chung J. G. Cucurbitacin E Induces G(2)/M Phase Arrest through STAT3/p53/p21 Signaling and Provokes Apoptosis via Fas/CD95 and Mitochondria-Dependent Pathways in Human Bladder Cancer T24 Cells. J. Evidence-Based Complementary Altern. Med. 2012, 2012, 1–11. 10.1155/2012/952762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowe C. L.; Mokhtarzadeh L.; Venkatesan P.; Babu S.; Axelrod H. R.; Sofia M. J.; Kakarla R.; Chan T. Y.; Kim J. S.; Lee H. J.; et al. Design of compounds that increase the absorption of polar molecules. Proc. Natl. Acad. Sci. U.S.A. 1997, 94 (22), 12218–12223. 10.1073/pnas.94.22.12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia R.; Yu C. L.; Hudnall A.; Catlett R.; Nelson K. L.; Smithgall T.; Fujita D. J.; Ethier S. P.; Jove R. Constitutive activation of Stat3 in fibroblasts transformed by diverse oncoproteins and in breast carcinoma cells. Cell Growth Differ. 1997, 8 (12), 1267–1276. [PubMed] [Google Scholar]
- Dechow T. N.; Pedranzini L.; Leitch A.; Leslie K.; Gerald W. L.; Linkov I.; Bromberg J. F. Requirement of matrix metalloproteinase-9 for the transformation of human mammary epithelial cells by Stat3-C. Proc. Natl. Acad. Sci. U.S.A. 2004, 101 (29), 10602–10607. 10.1073/pnas.0404100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catlett-Falcone R.; Landowski T. H.; Oshiro M. M.; Turkson J.; Levitzki A.; Savino R.; Ciliberto G.; Moscinski L.; Fernández-Luna J. L.; Nuñez G.; et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 1999, 10 (1), 105–115. 10.1016/S1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- Alas S.; Bonavida B. Inhibition of constitutive STAT3 activity sensitizes resistant non-Hodgkin’s lymphoma and multiple myeloma to chemotherapeutic drug-mediated apoptosis. Clin. Cancer Res. 2003, 9 (1), 316–326. [PubMed] [Google Scholar]
- Zhong Z.; Wen Z.; Darnell J. E. Stat3: a STAT Family Member Activated by Tyrosine Phosphorylation in Response to Epidermal Growth Factor and Interleukin-6. Science 1994, 264 (5155), 95–98. 10.1126/science.8140422. [DOI] [PubMed] [Google Scholar]
- Darnell J. E. Jr.; Kerr l. M.; Stark G. R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264 (5164), 1415–1421. 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- Brouwer I. J.; Out-Luiting J. J.; Vermeer M. H.; Tensen C. P. Cucurbitacin E and I target the JAK/STAT pathway and induce apoptosis in Sézary cells. Biochem. Biophys. Rep. 2020, 24, 100832. 10.1016/j.bbrep.2020.100832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer R.; Jain A. N. Surflex-Dock: Docking benchmarks and real-world application. J. Comput.-Aided Mol. Des. 2012, 26 (6), 687–699. 10.1007/s10822-011-9533-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai L.; Zhou H.; Xu R.; Zhao Y.; Chinnaswamy K.; McEachern D.; Chen J.; Yang C.-Y.; Liu Z.; Wang M.; et al. A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo. Cancer Cell 2019, 36 (5), 498–511.e17. 10.1016/j.ccell.2019.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M.; Gammon L.; Dreger S.; Koh J.; Garbe J. C.; Stampfer M. R.; Philpott M.; Jones L.; Bishop C. L. Ribosomal stress-induced senescence as a novel pro-senescence strategy for p16 positive basal-like breast cancer. bioRxiv 2018, 469445. 10.1101/469445. [DOI] [Google Scholar]
- Hanwell M. D.; Curtis D. E.; Lonie D. C.; Vandermeersch T.; Zurek E.; Hutchison G. R. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminf. 2012, 4 (1), 17. 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25 (13), 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Sali A.; Blundell T. L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234 (3), 779–815. 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock 4 and AutoDock Tools 4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30 (16), 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Goodsell D. S.; Halliday R. S.; Huey R.; Hart W. E.; Belew R. K.; Olson A. J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19 (14), 1639–1662. . [DOI] [Google Scholar]
- PyMOL Molecular Graphics System, 2021; Schrodinger, LLC; (accessed June 06, 2021).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.