

Abstract
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are fatal neurodegenerative disorders that share pathological features, including the aberrant accumulation of ubiquitinated protein inclusions within motor neurons. Previously, we have shown that the sequestration of ubiquitin (Ub) into inclusions disrupts Ub homeostasis in cells expressing ALS-associated variants superoxide dismutase 1 (SOD1), fused in sarcoma (FUS) and TAR DNA-binding protein 43 (TDP-43). Here, we investigated whether an ALS/FTD-linked pathogenic variant in the CCNF gene, encoding the E3 Ub ligase Cyclin F (CCNF), also perturbs Ub homeostasis. The presence of a pathogenic CCNF variant was shown to cause ubiquitin-proteasome system (UPS) dysfunction in induced pluripotent stem cell-derived motor neurons harboring the CCNF S621G mutation. The expression of the CCNFS621G variant was associated with an increased abundance of ubiquitinated proteins and significant changes in the ubiquitination of key UPS components. To further investigate the mechanisms responsible for this UPS dysfunction, we overexpressed CCNF in NSC-34 cells and found that the overexpression of both wild-type (WT) and the pathogenic variant of CCNF (CCNFS621G) altered free Ub levels. Furthermore, double mutants designed to decrease the ability of CCNF to form an active E3 Ub ligase complex significantly improved UPS function in cells expressing both CCNFWT and the CCNFS621G variant and were associated with increased levels of free monomeric Ub. Collectively, these results suggest that alterations to the ligase activity of the CCNF complex and the subsequent disruption to Ub homeostasis play an important role in the pathogenesis of CCNF-associated ALS/FTD.
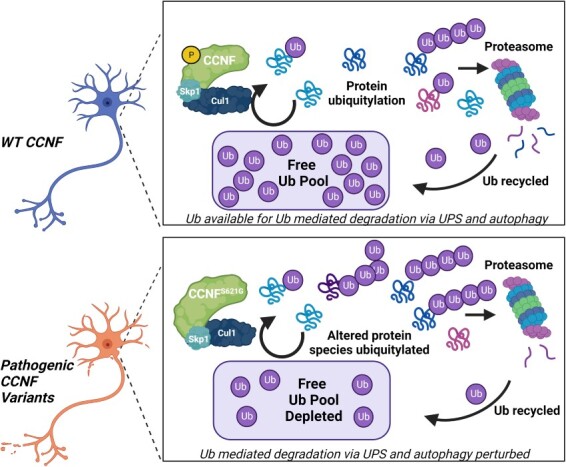
Graphical Abstract
Graphical Abstract.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder characterized by the progressive degeneration of motor neurons in the brain and spinal cord (1). This degeneration manifests as muscle atrophy and subsequent paralysis, typically eventuating in death within 3–5 years of diagnosis. Despite decades of dedicated research, the mechanisms of ALS pathogenesis remain unclear, and in most cases, the cause remains unknown (sporadic ALS). Approximately 10% of cases have an inherited genetic cause (familial ALS), with mutations in over 50 genes currently known to be associated with ALS (2). Up to 15% of ALS patients also develop signs of frontotemporal dementia (FTD) (3) and several genes, including C9ORF72, TDP-43, VCP, SQSTM1, OPTN, UBQLN2 and TBK1, contribute to the etiology of both ALS and FTD (4–6). Strikingly, the large majority of proteins encoded by these genes regulate aspects of intracellular protein degradation, one of three main functional pathways critical for maintaining proteome homeostasis (7). Mutations in VCP (8,9), SQSTM1 (10–12), UBQLN2 (13), OPTN (14,15), TBK1 (16,17), DNAJC7 (18) and CYLD (19) have all been associated with ALS/FTD, and all encode components of the ubiquitin-proteasome system (UPS) or autophagic machinery, implicating defective protein degradation in ALS and FTD. Adding to this growing list of protein degradation machinery genes associated with ALS/FTD, pathogenic variants in the CCNF gene, encoding the E3 ubiquitin ligase Cyclin F (CCNF), have been identified to cause ALS and FTD (20).
CCNF belongs to the F-box family of proteins, which are characterized by the presence of an F-box motif (21). This motif is crucial to the activity of CCNF as it binds to S-phase kinase-associated protein 1 (SKP1), facilitating interactions with Cullin 1 (CUL1) to form the SKP1-CUL1-F-box E3 ubiquitin ligase complex, mediating the ubiquitination of substrates. The ubiquitination of proteins occurs through a highly ordered three-step process in which ubiquitin (Ub) is first activated by an E1 Ub-activating enzyme and then transferred to an E2 conjugating enzyme, which binds to a specific E3 ligase, such as CCNF, to facilitate Ub attachment to a target substrate (22,23). In the final step, Ub is covalently bound to a lysine residue on a protein substrate, and by cycling through this step multiple times produces distinct poly-Ub chain linkages, which signal a range of essential cellular functions, including protein degradation, signal transduction, endocytosis, transcription and DNA repair (24,25).
In neurons, Ub regulates neuronal growth and survival by contributing to processes, such as synaptic plasticity and neurotransmission (26). Ub exists in a dynamic equilibrium between free Ub and poly-Ub conjugates in cells, with Ub availability controlled through the opposing actions of Ub ligases and deubiquitinating enzymes (DUBs) in addition to the regulation of synthesis and degradation rates (27,28). We have recently shown that the sequestration of Ub into insoluble protein aggregates also alters the cellular availability of Ub (29,30).
The accumulation of ubiquitinated protein inclusions is a hallmark pathology of many neurodegenerative diseases, including ALS and FTD (31). The main pathological constituent of these inclusions varies based upon the disease, whether the disease is sporadic or familial, and the genetics of the familial forms. Although the majority of sporadic ALS and FTD cases have inclusions that are immunoreactive for TDP-43 (32–34), SOD1 and FUS familial ALS cases are associated with the deposition of SOD1 and FUS, respectively, and show no immunoreactivity for TDP-43 (35). This variation is supported by the observation that TDP-43, SOD1 and FUS form distinct inclusions via different pathways in cells (36). Despite these differences, we have recently shown that SOD1, TDP-43 and FUS all co-aggregate with supersaturated proteins (supersaturation is a balance between protein concentration and solubility, i.e. supersaturated proteins have cellular concentrations that exceed their predicted solubility) (37). Consistent with a collapse in the proteostasis capacity of motor neurons, these supersaturated proteins are often associated with cellular quality control machinery, including molecular chaperones and components of the UPS and autophagy pathways (37). In fact, spinal motor neurons have been shown to be particularly susceptible to UPS stress (38), and have a reduced UPS capacity and metastable proteome, compared with oculomotor neurons that are resistant to degeneration in ALS (39,40). Together, this highlights the importance of UPS and Ub homeostasis in the pathogenesis of ALS/FTD.
The functional role of CCNF in the context of ALS/FTD is not yet fully understood. Initial studies have indicated that ALS-associated variants in CCNF cause UPS dysfunction (20) and increased K48 polyubiquitination, resulting from changes to the E3 ligase activity of the CCNF complex (41–43). Recently, we have also shown that the expression and aggregation of several ALS-associated proteins lead to UPS dysfunction and perturbed cellular Ub homeostasis (29,30), common features of ALS pathogenesis. Hence, we aimed to examine the effects of pathogenic CCNF on these pathways using induced pluripotent stem cell (iPSC)-derived motor neurons from a symptomatic CCNFS621G ALS patient or healthy control, Ub proteomics and mutants that disrupt the ability of CCNF to form an active Ub ligase complex, coupled with reporters of UPS function and Ub homeostasis. Our findings highlight the importance of Ub homeostasis in ALS.
Results
UPS activity is decreased in CCNFS621G motor neurons
Previous work showed that overexpression of CCNFS621G resulted in UPS dysfunction and alterations in the Ub-modified proteome in NSC-34 and Neuro-2A cells (20,42). Here, we sought to examine the effect of the CCNFS621G mutation on the UPS using iPSC-derived motor neurons harboring this mutation instead of an overexpression model. The iPSC lines have previously been characterized (Supplementary Material, Table S1) (44)) and were simultaneously differentiated to motor neurons using a protocol that has been well characterized (38). HB9 and Islet 1 staining were quantified in 75–95% of cells (Supplementary Material, Fig. S1), consistent with our previous findings (38). First, we examined the flux of substrate through the UPS in iPSC-derived motor neurons differentiated from a symptomatic ALS patient heterozygous for the CCNFS621G variant, and a healthy donor homozygous for CCNFWT (Fig. 1A, Supplementary Material, Table S1) by monitoring the degradation of a fluorescent substrate containing a CL1 degron sequence (GFPu). As GFPu passes through the UPS, including both the ubiquitination and proteasomal degradation steps, it acts as a reporter of UPS efficiency. Our previous work suggests that pathogenic variants of CCNF result in the accumulation of GFPu in the absence of proteasome inhibition (20). GFPu expression was calculated as GFP total integrated intensity (GCUxμm2) normalized to the CCNFS621G cell line under MG132-induced proteasomal stress, demonstrating maximal inhibition of the UPS. There was no significant difference between the maximal inhibition of MG132-treated CCNF wild-type (WT) and CCNFS621G. However, CCNFWT under basal conditions (endogenous stress) had significantly reduced GFP intensity compared with the CCNFS621G under endogenous stress (45% reduction, P < 0.05), suggesting impaired GFPu degradation and UPS activity in CCNFS621G motor neurons (Fig. 1B). CCNFWT under endogenous stress also had a significant reduction in relative GFP intensity compared with the CCNFWT and CCNFS621G lines in the presence of 10 μM MG132 (P < 0.05 and P < 0.01, respectively) (Fig. 1B). There were no significant differences in GFP intensity between CCNFS621G under endogenous stress and CCNFWT or CCNFS621G under proteasomal stress conditions, indicating the UPS system in the CCNFS621G is dysfunctional under basal conditions. Taken together, these findings suggest that the UPS is impaired in CCNFS261G cells, represented by the impaired degradation of GFPu, with relative GFP intensity levels comparable to both cell lines under UPS inhibition (Fig. 1B, Supplementary Material, Fig. S2).
Figure 1.
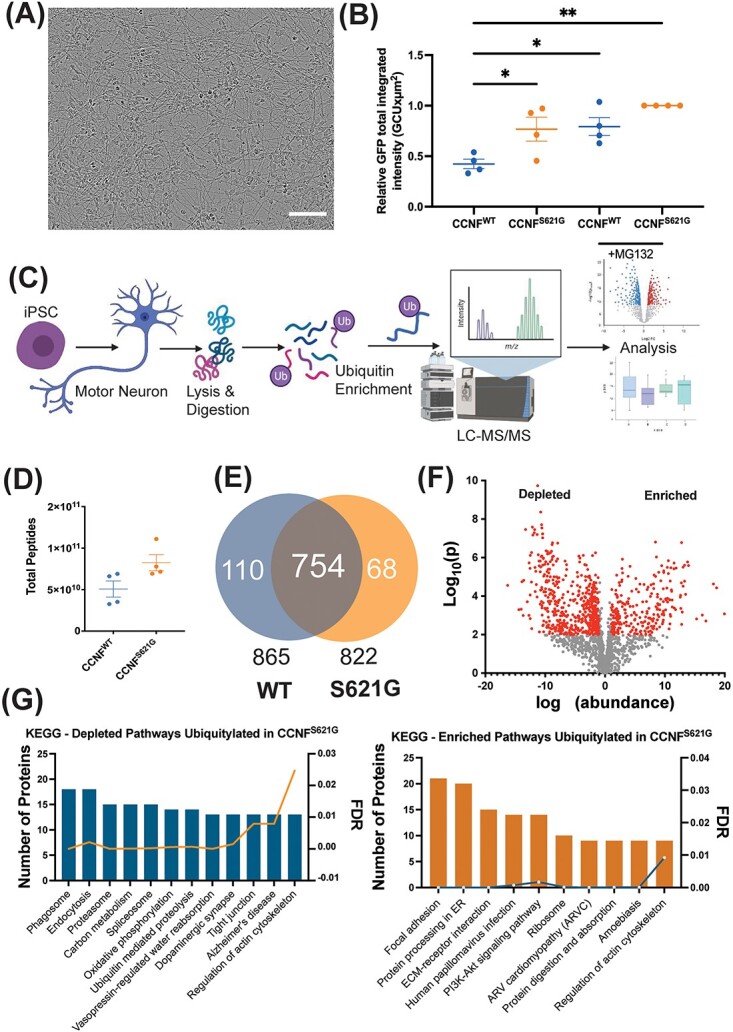
The effect of CCNFS621G on the ubiquitin-modified proteome of iPSC-derived motor neurons. (A) Representative image of iPSC-derived motor neurons differentiated from a symptomatic CCNFS621G ALS patient and a healthy donor. Cells were compared for UPS efficiency using: (B) GFPu degron assay under basal conditions and following proteasomal inhibition with 10 μM MG132. GFPu expression was calculated as GFP total integrated intensity (GCU × μm2). Data points show replicates, horizontal bars show mean and error bars denote the standard error of the mean, with each data point representing the average of 9 images across independent differentiations. Significant differences by one-way ANOVA with Tukey’s post hoc multiple comparisons tests: *P < 0.05, **P < 0.01. Data were normalized to MG132-treated CCNFS621G cells, demonstrating maximal inhibition of the UPS. There was no significant difference between the maximal inhibition of MG132-treated CCNF WT and CCNFS621G. (C) Schematic of the workflow, showing the derivation of patient-derived motor neuron cultures and ubiquitomics analysis. (D) Total peptide counts and (E) proteins were identified in the ubiquitome of iPSC-derived motor neurons derived from CCNFWT and CCNFS621G donors. (F) Volcano plot showing altered expression of individual proteins in the ubiquitome of CCNFWT and CCNFS621G expressing motor neurons. (G) Functional enrichment analysis of KEGG pathways enriched and depleted in CCNFWT and CCNFS621G expressing motor neurons.
Given the biochemical function of CCNF as an E3 Ub ligase, we postulated that this increased load on the UPS was likely because of the previously observed increase in ligase activity (42,43). Examination of the Ub-bound proteome (ubiquitome) using mass spectrometry provides an unbiased and systematic approach to characterize changes in the spectrum of ubiquitinated proteins in cells under different conditions. Hence, to better understand the functional role of CCNF in ALS, we next deployed quantitative proteomics (ubiquitomics) to profile changes in the ubiquitome of iPSC-derived motor neurons from a patient harboring the pathogenic CCNFS621G variant. Lysates of CCNFWT and CCNFS621G iPSC-derived motor neurons were subjected to affinity enrichment and label-free quantitative liquid chromatography–tandem mass spectrometry (LC–MS/MS) to identify differences in the cellular repertoire of Ub-modified proteins (Fig. 1C; Supplementary Material, Table S2; Supplementary Material, Fig. S3). We observed an increased total peptide count following ubiquitin-affinity matrix enrichment of ubiquitinated proteins and ubiquitin-binding proteins (the ubiquitome) from CCNFS621G cell lysates (Fig. 1D), indicating a greater abundance of ubiquitinated proteins in CCNFS621G motor neurons compared with CCNFWT motor neurons. We also observed different ubiquitome repertoires in CCNFS621G expressing motor neurons compared with CCNFWT motor neurons (Fig. 1E). Although there were 754 proteins present in common between CCNFS621G and CCNFWT motor neurons, we observed 110 proteins present uniquely in the CCNFWT motor neuron ubiquitome, and 68 proteins uniquely identified in CCNFS621G motor neurons. This indicates a shift in the spectrum of proteins targeted for ubiquitination by the CCNFS621G variant.
Quantitative analysis revealed a significant and widespread effect of the CCNFS621G variant on the motor neuron ubiquitome (Fig. 1F). We observed 256 proteins with significantly enriched abundance in the ubiquitome of CCNFS621G cells, and 545 proteins significantly depleted in CCNFS621G cells, compared with CCNFWT. KEGG pathway analysis of these differential ubiquitomes suggests the CCNFS621G variant perturbs a number of functional pathways in motor neurons. For example, proteins depleted from the ubiquitome of CCNFS621G cells were significantly over-represented in proteasome (hsa03050), spliceosome (03040) and Alzheimer’s disease-associated proteins (hsa05010) KEGG pathways (Fig. 1G, Supplementary Material, Table S3), whereas focal adhesion (hsa04510), protein processing in the endoplasmic reticulum (hsa04141) and the ribosome (hsa03010) were among the top KEGG pathways represented by proteins enriched in the ubiquitome of CCNFS621G motor neurons. Interestingly, distinct sets of proteins involved in the regulation of the actin cytoskeleton were found to be enhanced or depleted, indicating a significant effect of CCNFS621G on the motor neuron cytoskeleton.
Altered UPS regulation in CCNFS621G motor neurons
Consistent with the widespread dysregulation of Ub distribution observed in cells with pathogenic CCNF variants (42,43), and extensive cross-talk between components of the Ub conjugation system, we observed significant changes in the abundance of key enzymes at each level in the Ub conjugation hierarchy (E1, E2, E3 and DUBs) in the ubiquitome of CCNFS621G motor neurons (Fig. 2A). The ubiquitination of the E1 ubiquitin-activating enzyme (UBA1), E2 conjugating enzymes (e.g. UBE2O and UBE2N), E3 ligases (including NEDD4L and RNF25), DUBs (e.g. USP7 and USP11) and subunits of the 19S proteasome cap (e.g. PSMD1, 6, 7, 10, 12 and 13) was modified in CCNFS621G motor neurons (Fig. 2B). Notably, these components included CUL1 (a component of the SCF E3 ligase complex formed with CCNF), and UBR5, an E3 previously linked to neurodegeneration (45). Of particular interest are decreased abundance of the Ub activator UBA1 in the Ub-modified proteome of CCNFS621G motor neurons. Pathogenic UBA1 variants result in a rare form of childhood motor neuron disease spinal muscular atrophy (46). Decreased UBA1 signal in the ubiquitome indicates decreased conjugation of UBA1 to Ub and hence decreased UBA1 activity in these cells. Conversely, we observed an increased abundance of Ub in CCNFS621G motor neurons (Fig. 2C). These widespread effects are consistent with significant disruption or alterations to Ub homeostasis, or reprogramming, of the UPS in CCNFS621G motor neurons.
Figure 2.
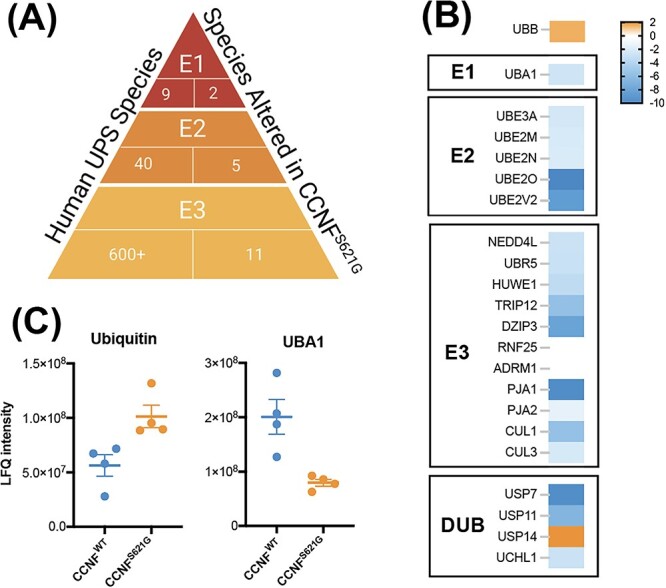
The ubiquitin-proteasome system is modified in CCNFS621G iPSC-derived motor neurons. (A) The number of key enzymes altered at each level of the Ub hierarchy in CCNFS621G motor neurons. (B) Relative abundance of UPS component proteins in the ubiquitome of CCNFS621G motor neurons compared with CCNFWT motor neurons. (C) Label-free quantitation shows an altered abundance of Ub and UBA1 in the ubiquitome of CCNFS621G motor neurons.
Disruption of CCNF ubiquitin ligase complex formation improves UPS function but does not significantly alter ubiquitin distribution in cells
We have previously shown that the expression of ALS-associated CCNF variants in NSC-34 cells caused significant accumulation of the UPS reporter GFPu compared with the expression of CCNFWT, indicating that these variants drive UPS dysfunction (20). To further explore the mechanism by which CCNF variants mediate UPS dysfunction, we co-transfected NSC-34 cells with GFPu and mCherry-CCNF constructs. Analysis of GFPu accumulation by flow cytometry revealed that overexpression of both CCNFWT and CCNFS621G led to significantly increased GFPu fluorescence compared with cells expressing mCherry alone (Fig. 3A), suggesting overexpression of CCNF or its increased activity overwhelms the UPS. However, as previously reported (20), GFPu accumulation was significantly higher in cells expressing CCNFS621G, compared with cells expressing CCNFWT. This effect was independent of CCNF expression levels (Supplementary Material, Fig. S4). Consistent with data that suggests overexpression of CCNF is toxic (47), overexpression of both CCNFWT and CCNFS621G variants led to a substantial accumulation of the UPS reporter when compared with the mCherry alone control. These data are consistent with the overactivity of CCNF, regardless of variant, causing dysfunction and subsequent cell death.
Figure 3.

Disrupting CCNF ubiquitin ligase complex formation improves UPS function but does not significantly alter the distribution of ubiquitin in NSC-34 cells. (A) UPS activity (GFPu fluorescence) was measured in NSC-34 cells co-transfected with mCherry-CCNF. Data represent mean GFPu fluorescence ± SEM (n = 3). One-way ANOVA with a Tukey’s multiple comparisons post-hoc test was used to determine statistical significance (**P < 0.01, ***P < 0.001). (B) Representative confocal images of NSC-34 cells co-transfected with mCherry-CCNF and GFP-Ub. Scale bars represent 10 μm. (C) FRAP analysis was performed on NSC-34 cells co-expressing mCherry-CCNF and GFP-Ub by photobleaching a region of interest in either the nucleus or cytoplasm and measuring the recovery of Ub fluorescence over 120 s. (D) Quantification of the proportion of mobile Ub in the nucleus and cytoplasm of cells expressing mCherry-CCNF. (E) Diffusion rates of GFP-Ub were measured in both the nucleus and cytoplasm of NSC-34 cells co-transfected with mCherry-CCNF. Data shown are means ± SEM combined from 3 independent experiments (n ≥ 13). Two-way ANOVA with Bonferroni post-hoc test was used to compare differences between cell populations (n.s. = not significant). Asterisks indicate a significant difference when compared with the mCherry control (*P ˂ 0.05, **P ˂ 0.01).
We next investigated the effect of disrupting the ability of CCNF to form an active Ub ligase complex by generating compound mutants containing an F-box mutation (LP/AA), in which the first two amino acids in the F-box domain of CCNF have been mutated to alanine. The LP/AA mutation disrupts the ability of CCNF to form an active Ub ligase complex with SKP1 and CUL1 (48). Accumulation of GFPu was significantly attenuated in cells expressing the compound F-box mutation (LP/AA). Both CCNFWT and CCNFS621G (CCNFWT(LP/AA) and CCNFS621G(LP/AA)) expression resulted in significantly less accumulation of GFPu when compared with cells expressing CCNFWT or CCNFS621G alone. These results suggest that the UPS dysfunction associated with CCNF overexpression is at least partially because of the formation of an active E3 Ub ligase complex.
To gain a more detailed understanding of the effects of CCNF mutations on Ub homeostasis, we examined the distribution of Ub in NSC-34 cells over-expressing various mCherry-CCNF variants using fluorescence recovery after photobleaching (FRAP). In cells co-expressing CCNFWT, CCNF was predominantly diffuse and localized to the nucleus, whereas GFP-Ub was mostly diffuse throughout the nucleus and cytoplasm, with some small GFP-Ub foci present in the cytoplasm (Fig. 3B). However, in cells expressing the CCNFS621G variants, large cytoplasmic aggregates of both CCNF and Ub formed, which coincided with the depletion of nuclear Ub (Fig. 3B). To determine both the cellular availability of mobile Ub and the rate of Ub diffusion, we bleached an ROI in either the nucleus or cytoplasm of cells co-expressing GFP-Ub and either mCherry-CCNFWT, mCherry-CCNFS621G, mCherry-CCNFWT(LP/AA), mCherry-CCNFS621G(LP/AA) or mCherry only and measured the recovery of GFP-Ub back into this region (Supplementary Material, Fig. S5). In cases where large cytoplasmic aggregates were present, an ROI was selected that contained only soluble material (Supplementary Material, Fig. S5). Recovery dynamics appeared broadly similar between cell populations, with the higher recovery of Ub in the cytoplasm compared with the nucleus (Fig. 3C), consistent with previous reports of greater Ub mobility in this compartment (27). Slightly lower levels of cytoplasmic recovery were observed in cells expressing CCNFS621G, in comparison to cells expressing CCNFWT or mCherry alone. However, no obvious differences were observed between cells expressing CCNF with the F-box mutation (CCNFWT(LP/AA) and CCNFS621G(LP/AA)) and cells expressing CCNFWT and CCNFS621G without the F-box mutation (Fig. 3C). Analysis of mobile Ub availability revealed that cells expressing CCNFS621G, CCNFWT(LP/AA) and CCNFS621G(LP/AA) had significantly lower levels of mobile Ub in the cytoplasm, in comparison to cells expressing mCherry alone (Fig. 3D). This is consistent with the presence of large Ub aggregates in the cytoplasm reducing the availability of mobile Ub (27). No significant differences in the amount of mobile Ub available to cells expressing CCNF with the F-box mutation (CCNFWT(LP/AA) and CCNFS621G(LP/AA)) and cells expressing CCNF without the F-box mutation (CCNFWT and CCNFS621G; Fig. 3D) were observed, suggesting that disrupting the ability of CCNF to form an active ligase complex does not alter Ub distribution in cells. In addition, no significant differences were observed in the mean half-life of recovery (T1/2) between cell populations (Fig. 3E), suggesting that although expression of CCNFS621G, CCNFWT(LP/AA) and CCNFS621G(LP/AA) decreased the amount of mobile Ub available to the cell, they did not significantly alter the kinetics of Ub diffusion.
The pool of free monomeric ubiquitin is increased in cells with disrupted CCNF ligase complex formation
Considering that disrupting the ability of CCNF to form an active ligase complex did not significantly alter the distribution of the total mobile Ub pool when measured by FRAP, we next examined the relative availability of the monomeric form of Ub specifically available to cells using fluorescence recovery after nuclear photobleaching (FRANP). FRANP relies on the nuclear pore acting as a molecular sieve, allowing only the passive diffusion of free monomeric Ub while preventing the diffusion of Ub conjugated in chains (27,29). We bleached the entire nucleus of cells co-expressing GFP-Ub and mCherry-CCNF (Fig. 4A, Supplementary Material, Fig. S6) and quantified the amount of relative monomeric GFP-Ub diffusing through the nuclear pore and into the nucleus (Fig. 4B). Significant decreases in the levels of free monomeric Ub were observed in cells expressing both CCNFWT and CCNFS621G compared with cells expressing mCherry control (Fig. 4C). Interestingly, in some cells, the expression of CCNFWT and CCNFS621G was also associated with the formation of cytosolic Ub aggregates and depletion of nuclear Ub (Fig. 4A). Disrupting the ligase activity of CCNF by introducing the F-box mutation (CCNFWT(LP/AA)), restored monomeric Ub availability to a level that was no longer significantly different from the mCherry control. However, there were no significant differences between the levels of free monomeric Ub observed in cells expressing CCNFS621G and CCNFS621G(LP/AA).
Figure 4.

The level of free monomeric ubiquitin is increased in cells with disrupted CCNF ligase complex formation. (A) The entire nucleus of NSC-34 cells co-expressing mCherry-CCNF and GFP-Ub was photobleached and (B) the recovery of nuclear Ub was monitored as a proportion of cytoplasmic fluorescence for 120 s. Scale bars represent 10 μm. (C) The percentage of mobile Ub in the nucleus at the final read was quantified as a proportion of cytoplasmic fluorescence. Data represent mean ± SEM (n ≥ 13 combined from 2 independent experiments). One-way ANOVA with a Tukey’s multiple comparisons post-test was used to determine statistical significance compared with mCherry control (*P ˂ 0.05, ***P ˂ 0.001). (D) NSC-34 cells transfected with mCherry-CCNF were fixed, permeabilized and stained for free Ub using the free Ub sensor tUI-HA 48 h post-transfection. Scale bars represent 10 μm. (E) Quantification of tUI-HA fluorescence in cells expressing mCherry-CCNF. Data shown are mean ± SD (n = 46 CCNFWT, n = 53 CCNFWT(LP/AA), n = 41 CCNFS621G, n = 45 CCNFS621G(LP/AA), n = 146 mCherry). Statistical significance was determined using one-way ANOVA with a Tukey’s multiple comparisons post-test (**P ˂ 0.01).
The data from the FRANP experiment relies on the co-expression of a Ub-tagged with GFP and does not provide information regarding the endogenously expressed Ub. Next, we analyzed the levels of endogenous free monomeric Ub in CCNF-expressing cells utilizing the free Ub probe tUI-HA. Previous work has demonstrated that tUI-HA has a high affinity and specificity for intracellular free Ub, facilitating the detection and quantification of free Ub in cells (49). Using confocal microscopy, we analyzed tUI-HA fluorescence in NSC-34 cells transiently transfected with mCherry-CCNF or mCherry control (Fig. 4D). Although there were no significant differences in tUI-HA fluorescence between cells expressing CCNFWT and CCNFS621G, we observed significant increases in tUI-HA fluorescence in cells expressing F-box mutant CCNF (CCNFWT(LP/AA) and CCNFS621G(LP/AA)) compared with CCNF variants that did not carry the F-box mutation (Fig. 4E). This suggests that disruption to the ligase activity of CCNF increases the free monomeric Ub pool in cells.
Discussion
The recent discovery of ALS and FTD-linked CCNF variants further expands a list of genes encoding and regulating components of the UPS implicated in these neurodegenerative conditions. Accordingly, UPS dysfunction is gaining prominence as a key pathological mechanism underlying ALS and FTD. In vitro expression of pathogenic CCNF variants has been shown to increase the cellular accumulation of ubiquitinated proteins, suggesting that pathogenic effects may be related to disrupted UPS function and Ub homeostasis, but these disease mechanisms are not fully understood. Hence, we performed detailed investigations into the effects of pathogenic CCNF variants on cellular Ub homeostasis, the maintenance of which is necessary for neuronal survival. We found a significant accumulation of the UPS reporter GFPu in CCNFS612G iPSC-derived motor neurons from a symptomatic ALS patient compared with healthy donor iPSC-derived motor neurons. A number of E3 ligases have been identified to facilitate CL1 degron degradation (50), however, it is currently unknown whether CCNF plays a direct role in targeting GFPu for degradation. Ubiquitome analysis indicated an intrinsic UPS dysfunction in the pathogenic CCNF variant motor neurons, along with a redistribution of Ub and significant changes in the abundance of key UPS components in the Ub-modified proteome. Disrupting the ability of CCNFS612G to form an active Ub ligase complex in an overexpression model restored cellular UPS function and increased the pool of free monomeric Ub. Together, these findings suggest that the UPS dysfunction and disruption of Ub homeostasis caused by pathogenic CCNF variants result from increased activity of the CCNF ligase complex.
The UPS is disturbed by pathogenic variant CCNFS216G
UPS impairment underlies many neurodegenerative diseases, including ALS, and overexpression of a number of ALS-associated proteins (including CCNF), can cause UPS dysfunction (20,29,30). Indeed, UPS dysfunction alone has been demonstrated to be sufficient in causing ALS pathology. Motor neuron-specific knockout of the proteasome subunit Rpt3 in mice resulted in the aggregation of ALS-associated proteins, and progressive motor neuron loss and locomotor dysfunction (51). Here, we show for the first time that iPSC-derived motor neurons harboring the CCNFS621G variant have compromised UPS function, leading to a reduced capacity to degrade a reporter protein targeted to the proteasome. These findings are consistent with previous demonstrations of UPS dysfunction of other ALS-causing genes, whereby GFPu accumulated to a significantly greater extent in ALS TDP-43 patient-derived fibroblasts compared with control fibroblasts (52).
The UPS is a key regulator of cellular processes that are fundamental to maintaining neuronal structure and function, including neurite length and neuronal viability (7,38,53). Our ubiquitomics analysis revealed significantly altered Ub distribution in neurons harboring the pathogenic CCNFS621G variant, with iPSC-derived motor neurons from an ALS patient containing a 2-fold greater abundance of ubiquitinated proteins relative to WT controls. Similar differences in K48-ubiquitination were reported between Neuro-2A cells expressing CCNFWT and CCNFS621G, with proteomic analysis revealing that the ubiquitinated proteins from CCNFS621G-expressing cells were associated with defective protein degradation via the autophagy pathway (42). Examination of the proteins differentially ubiquitinated in our CCNFS621G-expressing neurons also highlighted changes to cellular degradation pathways, including Ub-mediated proteolysis and the proteasome.
Furthermore, the abundance of key UPS components in the Ub-modified subset of the proteome, including UBA1, UBR5 and CUL1, were significantly altered in neurons harboring the pathogenic CCNFS621G variant. Previous ubiquitomics data identified UPS proteins at higher abundance in healthy donor-derived motor neurons, following their differentiation from iPSCs (38). This data highlighted the importance of UPS proteins in healthy motor neuron function (38). In this current study, some of these UPS proteins, including UBA1, E2 proteins UBE2N, UBE2O and the DUB USP7, were found at decreased abundance in CCNFS621G motor neurons, consistent with significant alterations to Ub homeostasis. UBA1 plays a central role in the UPS, with inhibition of UBA1 causing impaired neurite outgrowth and cell death (38). Dysregulation of UBA1 induces neuromuscular pathology in animal models of spinal muscular atrophy and systemic restoration of UBA1 rescues this pathology (54). Moreover, a number of the largest fold changes in ubiquitinated proteins in the CCNFS621G motor neurons (Table 1) were in ALS-associated proteins, including DCTN1, ENO2, INA, MAP6 and the more recently associated STMN2, further suggesting a direct role of CCNF in modulating the ALS protein network (55). Collectively, these studies reiterate the importance of the UPS in ALS/FTD pathogenesis. Based on the substantial ubiquitomic disruption in CCNFS621G motor neurons and the wide-ranging effects of Ub signaling in neurons, we predict that numerous cellular pathways will be affected in CCNFS621G motor neurons. Additionally, it remains unknown whether there is an increase in misfolded proteins in CCNFS621G motor neurons. Future studies should identify the impact of altered Ub homeostasis on protein misfolding and CCNFS621G motor neuron function.
Table 1.
Top 30-fold changes from CCNF S621G
| Downregulated in S621G | Upregulated in S621G | ||||||
|---|---|---|---|---|---|---|---|
| Ubiquitylated protein | Fold change (log ratio) | log10pv | Ubiquitylated protein | Fold change (log ratio) | log10pv | ||
| 1 | ENO2 | −16.28 | 4.56 | 1 | MYH3 | 19.94 | 3.08 |
| 2 | ATAT1 | −14.39 | 3.30 | 2 | COL12A1 | 18.64 | 4.43 |
| 3 | GAP43 | −14.00 | 3.32 | 3 | COL14A1 | 18.11 | 4.63 |
| 4 | PRPF38A | −13.88 | 4.75 | 4 | COL3A1 | 16.86 | 2.71 |
| 5 | NPTX1 | −13.69 | 3.00 | 5 | RRBP1 | 16.26 | 3.01 |
| 6 | MAP6 | −13.59 | 4.82 | 6 | COL1A1 | 15.30 | 2.81 |
| 7 | CTNNA2 | −13.28 | 7.46 | 7 | CKM | 13.95 | 5.59 |
| 8 | INA | −12.99 | 3.71 | 8 | COL12A1 | 13.79 | 5.77 |
| 9 | CTNND2 | −12.71 | 3.00 | 9 | COL6A2 | 13.45 | 4.09 |
| 10 | STXBP1 | −12.69 | 5.72 | 10 | KBTBD10 | 13.29 | 5.60 |
| 11 | SCRIB | −12.53 | 5.44 | 11 | LOXL2 | 13.29 | 4.77 |
| 12 | BTBD17 | −12.52 | 4.06 | 12 | MYLPF | 13.05 | 5.50 |
| 13 | SEPT3 | −12.51 | 2.83 | 13 | C4A;C4B | 13.04 | 4.51 |
| 14 | GNG2 | −12.49 | 6.12 | 14 | LEPRE1 | 12.89 | 3.17 |
| 15 | C6orf174 | −12.30 | 4.73 | 15 | P4HA2 | 12.79 | 2.53 |
| 16 | PRKAR2B | −12.27 | 6.26 | 16 | EML4 | 12.79 | 6.78 |
| 17 | ABAT | −12.24 | 2.31 | 17 | PDLIM7 | 12.75 | 4.69 |
| 18 | DDR1 | −12.14 | 7.58 | 18 | MEST | 12.66 | 3.36 |
| 19 | BIRC6 | −12.06 | 7.17 | 19 | MYH8 | 12.62 | 4.04 |
| 20 | ITSN1 | −11.82 | 7.29 | 20 | DES | 12.57 | 3.95 |
| 21 | MLLT4 | −11.77 | 7.45 | 21 | ENO3 | 11.87 | 5.45 |
| 22 | KIF21A | −11.71 | 5.49 | 22 | NID1 | 11.78 | 3.30 |
| 23 | NEBL | −11.66 | 4.24 | 23 | TAGLN | 11.57 | 3.38 |
| 24 | GSK3B | −11.58 | 5.29 | 24 | UNC45B | 11.53 | 5.72 |
| 25 | EDC4 | −11.48 | 4.60 | 25 | MYL1 | 11.51 | 5.42 |
| 26 | STMN2 | −11.36 | 3.95 | 26 | SEPT5 | 11.22 | 3.44 |
| 27 | BCCIP | −11.35 | 7.20 | 27 | ACTN2 | 11.20 | 3.80 |
| 28 | EPB41L1 | −11.26 | 9.73 | 28 | VWA1 | 11.13 | 4.25 |
| 29 | DCTN1 | −11.16 | 6.15 | 29 | LAMB2 | 11.08 | 4.32 |
| 30 | GNL1 | −11.03 | 5.82 | 30 | COL16A1 | 10.99 | 5.47 |
Since the discovery of pathogenic CCNF variants in ALS/FTD, several studies have investigated the disease mechanisms of CCNFS621G, with a focus on the role of CCNF variants in ligase complex activity. Preliminary studies indicated that CCNFS621G may cause ALS through UPS dysfunction (20), with more recent studies drawing attention to the prevention of CCNFS621G phosphorylation, resulting in elevated Lys48-ubiquitination activity (41,42). Overexpression of CCNF has been shown to trigger an upregulation of cell death in zebrafish, with transient overexpression of variant CCNFS621G leading to abnormal axonal outgrowth and impaired motor function (47). Together, these results suggest that pathogenic CCNF variants are likely to cause a toxic gain of function. Complementing these findings, we show here that overexpression of CCNF was associated with impaired UPS function, as measured by the increased accumulation of the fluorescent UPS reporter GFPu. Interestingly, this impairment was partially rescued by disrupting the ability of CCNF to form an active Ub ligase complex, suggesting that the UPS dysfunction observed with CCNF overexpression results from changes in the ubiquitination activity of the CCNF complex. More specifically, these changes are likely to result from alterations to the ubiquitination kinetics of CCNF, as the F-box variants are still capable of binding substrate but decrease the ubiquitination of substrates (48). Furthermore, it remains possible that mutant CCNF sequesters SCF components in aggregates rendering the SCF complex inactive. Thus, inhibiting the SCF complex even further may aggravate the toxic effect of the mutant CCNF. The potential for the therapeutic application of CCNF will need to be tested in future research.
Ubiquitin homeostasis is disturbed by pathogenic variant CCNFS216G
The accumulation of ubiquitinated protein inclusions is a major pathological feature of both ALS and FTD (56), and abnormal Ub homeostasis is proposed to play a role in the pathogenesis of ALS. We have previously shown that the expression and aggregation of mutant ALS-associated proteins SOD1, TDP-43 and FUS cause Ub dyshomeostasis and depletion of the free Ub pool in NSC-34 cells (29,30). Here, we show that expression of a pathogenic CCNF variant also perturbs Ub homeostasis, with NSC-34 cells expressing CCNFS621G exhibiting large cytosolic aggregates and decreased levels of mobile Ub (specifically, free monomeric Ub) in comparison to controls. This depletion is consistent with previous findings that the pathogenic CCNFS621G variant causes an accumulation of ubiquitinated proteins through increased activity of the CCNF complex (41,42).
Although disrupting the ability of CCNF to form an active E3 ligase complex did not seem to significantly alter the distribution of the entire mobile Ub pool, disrupting the ligase activity of CCNF did significantly improve the availability of free monomeric Ub to cells. Ub exists in a dynamic equilibrium in cells, and the supply of free Ub must be preserved above threshold levels for the maintenance of the capacity to respond to different cellular stresses (57,58). Depletion of free Ub through modulation of the UBB gene results in a neurodegenerative phenotype in mice (59). Strikingly, compensatory expression of Ub through the UBC gene is upregulated in neurons from UBB deficient mice in an attempt to maintain levels of free Ub and protect against neuronal dysfunction (60). In fact, multiple studies have shown that modulation of the free Ub pool improves neuronal function and survival. Overexpression of Ub rescued the toxicity induced by Ub depletion in yeast (57), and restored free Ub levels in the motor neurons of ataxia mice, improving neurological symptoms (61,62). The UPS, and E3 Ub ligases in particular, may thus offer promise as molecular targets for neurodegenerative drug discovery. A number of small-molecule inhibitors acting on E3 Ub ligase complexes have already been identified as promising drug targets in cancer (63), and considering that free Ub levels were improved by altering the E3 ligase activity of the CCNF complex, modulating this activity may represent a potential therapeutic avenue for the treatment of CCNF-associated ALS. This concept will need to be tested in future research.
In conclusion, we showed that the ALS/FTD-linked variant CCNFS621G causes UPS dysfunction and disruption of Ub homeostasis in cellular models. These pathologies were partially alleviated by disrupting the ability of CCNF to form an active E3 ligase complex and are consistent with previous findings that suggest that the UPS dysfunction and perturbations to Ub homeostasis caused by CCNF result from changes to the E3 ligase activity of the CCNF complex. Together with our observations from other cellular models of ALS, these data highlight the importance of Ub homeostasis in the development of neurodegenerative diseases such as ALS/FTD.
Materials and Methods
Plasmids
pmCherry-C1 constructs containing CCNFWT and CCNFS621G were generated as described previously (20). CCNF F-box variants CCNFWT(LP/AA) and CCNFS621G(LP/AA) were obtained from Roger Chung (Macquarie University). GFP–Ub (Addgene plasmid 11 928, deposited by Nico Dantuma) (27) was acquired from Addgene and the GFPu construct was obtained from Ron Kopito (Stanford University) (64).
Antibodies
The following antibodies were used in this study: rabbit polyclonal anti-HA tag antibody (ab9110, Abcam; 1:1000 dilution), rabbit IgG polyclonal isotype control (ab171870, Abcam), Alexa Fluor 488-conjugated goat anti-rabbit-IgG secondary antibody (A11008, Invitrogen, USA; 1:1000 dilution).
Expression and purification of tUI-HA
Expression and purification of tUI-HA were carried out as reported by Choi et al. (49) with minor alterations, as described in Farrawell et al. (30). Briefly, the tUI-HA plasmid was transformed into chemically competent Rosetta BL21(DE3) pLysS cells for protein expression. The dissociated protein was checked for purity via reducing SDS-PAGE before being dialyzed into phosphate-buffered saline (PBS). The concentration of the tUI-HA sensor was determined using UV/VIS spectroscopy (POLARStar Nano, BMG Labtech) at 280 nm using an extinction coefficient of 22 920 M−1 cm−1 to be 3 mg/ml (65).
Cell line culture and transfection
Neuroblastoma × spinal cord hybrid NSC-34 cells (66) were maintained in Dulbecco's Modified Eagle's Medium/Ham's Nutrient Mixture F12 (DMEM/F12) supplemented with 10% fetal bovine serum (FBS, Gibco, Australia). Cells were maintained at 37°C in a humidified incubator with 5% atmospheric CO2. Cells were plated onto 8-well μslides or multi-well plates 24 h before transfection. Cells were transfected using Lipofectamine 3000 (Invitrogen) or TransIT-X2 transfection reagent (Mirus Bio, USA) according to the manufacturer's instructions. For co-transfections, the amount of DNA was divided equally between constructs.
iPSC culture and iPSC-derived motor neuron differentiation
All iPSC-based assays were conducted in accordance with the Human Research Ethics Application (HE13/272), in conjunction with a Materials Transfer Agreement (MTA; Ethics HREC/11/CRGH/179). The iPSCs were generated from a healthy 57-year-old male and a 59-year-old male who had symptomatic ALS at the time of collection (44). Cells were cultured under normoxic conditions (37°C, 5% CO2). All iPSC lines were generated, characterized and maintained as previously described (44). The iPSC-derived motor neurons were differentiated as per Bax et al. (38). Replicates shown are from individual differentiations.
Ubiquitin proteomics
Ubiquitin proteomic analysis was performed on cell lysates as previously described (38,67). Briefly, 500 μg of total protein was used per sample to immunopurify mono- and poly-ubiquitinated proteins using a specialized ubiquitin affinity matrix (VIVAbind Ubiquitin Kit, VIVA Bioscience, Exeter, UK). After substantial washing to remove residual detergent, beads were digested for 30 min at 27°C, then reduced with 1 mM DTT and left to digest overnight at room temperature (RT) with sequencing-grade trypsin (5 μg/ml, Promega, Madison, WI, USA). Samples were alkylated with 5 mg/ml iodoacetamide and protease digestion terminated with trifluoroacetic acid. Trypsinized eluents were collected after brief centrifugation then purified and desalted using self-packed tips with 6 layers of C18 Empore disks (Pacific Laboratory Products), then dried in a SpeedVac. Samples were then resuspended in 12 μl 5% formic acid, 2% acetonitrile and stored at −80°C. Five microliters of each digested peptide sample were loaded and fractionated along a custom C18 column and introduced by nanoelectrospray into an LTQ Orbitrap Velos Pro coupled to an Easy-nLC HPLC (Thermo Fisher). Tandem mass spectrometry data was collected for the ten most abundant ions per scan over a 140 min time gradient and fragmented within the linear ion trap using higher-energy collisional dissociation. The order of data collection was randomized to interchange between biological conditions with bovine serum albumin (BSA) run between each sample to minimize temporal bias. MS/MS data were searched using MaxQuant (v1.2.7.4, Max Planck Institute of Biochemistry, Martinsried, Germany) (68) against the Uniprot human database (release 2016_04). A false discovery rate of 1% was tolerated for protein, peptide, and sites, and one missed cleavage was allowed. Data were filtered for contaminants, reverse hits, proteins only identified by site and number of unique peptides (≥2). Statistical and functional analysis was performed as previously described (38,67).
UPS activity
UPS activity was measured in NSC-34 cells expressing mCherry-CCNF using the UPS reporter GFPu, as described previously (20). Briefly, cells were harvested 48 h post-transfection and GFPu fluorescence was analyzed by flow cytometry on a Becton Dickinson Biosciences LSRFortessa X-20 analytical flow cytometer. UPS activity in iPSC motor neuron precursors was similarly assessed using GFPu. On day 21 of differentiation, cells differentiated in a 96-well plate were transfected with Lipofectamine LTX, according to the manufacturer’s protocol. Briefly, liposomes encapsulating a 2:1 ratio of GFPu and mCherry (as a transfection confirmation control) plasmids were added dropwise and incubated with the precursors for 60 h, imaging in-incubator with an Incucyte S3 automated fluorescent microscope (Essen BioScience, USA). Proteasome inhibitor 10 μM MG132 was included in this incubation period to examine the sensitivity of the precursors to UPS stress. Image analysis was performed on cells treated with or without MG132 by filtering cells with mCherry expression and quantifying the GFP expression. GFPu expression was calculated as GFP total integrated intensity (GCU × μm2). Data were obtained from the mean of 9 images per replicate, and the mean and standard error of the mean were calculated from four independent replicates.
Immunofluorescence
Free Ub levels were measured in NSC-34 cells expressing mCherry-CCNF using the high-affinity free Ub sensor tUI-HA, as described previously (30). Briefly, transfected cells in 8-well μslides were fixed with 4% paraformaldehyde before permeabilization with 0.1% Triton X-100 (TX-100) in PBS. Cells were then blocked with blocking buffer (10% FBS, 2% BSA, 0.1% TX-100 in PBS) for 1 h at RT and incubated with the tUI-HA probe diluted 1:400 in blocking buffer for 30 min at RT. Cells were incubated with anti-HA antibody in blocking buffer for 1 h at RT and washed three times with PBS before incubating with Alexa Fluor 488-conjugated secondary antibody in blocking buffer for 1 h at RT. Finally, cells were counterstained with Hoechst 33342 nucleic acid stain (diluted 1:5000 in PBS) for 5 min at RT and imaged on the SP8 confocal microscope.
Confocal microscopy
FRAP and FRANP experiments were performed on NSC-34 cells 48 h post-transfection using the LASAF FRAP application wizard on the Leica SP5 confocal microscope as described previously (29). Briefly, using the 63× objective and a scan speed of 700 Hz, five pre-bleach images were acquired before the region of interest (ROI) was bleached over five frames with the 488 laser power set to 100%. Recovery of GFP-Ub into the ROI was monitored over 120 s with the laser power set at 20%. For FRAP experiments the ROI was ~ 2 μm in diameter.
To quantify tUI-HA staining, single slice 12-bit images of transfected NSC-34 cells were acquired on the Leica SP8 confocal microscope using the 40× (1.3 numerical aperture) oil immersion objective, one line and frame average with a scan speed of 400 Hz. Images were subsequently processed using ImageJ software (69). Firstly, images from the mCherry channel were thresholded to remove background and the magic wand tool was used to select cells based on fluorescence. Segmentation of cells was performed manually. Regions of interest were applied to the tUI-HA images (green channel) before the mean tUI-HA fluorescence intensity was measured.
Statistics
All statistical analysis was performed using GraphPad Prism software version 5.00 for Windows unless stated.
Conflict of Interest statement. None declared.
Supplementary Material
Contributor Information
Natalie E Farrawell, Molecular Horizons and School of Chemistry and Molecular Bioscience, University of Wollongong, Wollongong, New South Wales 2522, Australia; Illawarra Health and Medical Research Institute, Wollongong, New South Wales 2522, Australia.
Monique Bax, Molecular Horizons and School of Chemistry and Molecular Bioscience, University of Wollongong, Wollongong, New South Wales 2522, Australia; Illawarra Health and Medical Research Institute, Wollongong, New South Wales 2522, Australia.
Luke McAlary, Molecular Horizons and School of Chemistry and Molecular Bioscience, University of Wollongong, Wollongong, New South Wales 2522, Australia; Illawarra Health and Medical Research Institute, Wollongong, New South Wales 2522, Australia.
Jessie McKenna, School of Medical Sciences, University of New South Wales, Sydney, New South Wales 2052, Australia.
Simon Maksour, Molecular Horizons and School of Chemistry and Molecular Bioscience, University of Wollongong, Wollongong, New South Wales 2522, Australia; Illawarra Health and Medical Research Institute, Wollongong, New South Wales 2522, Australia.
Dzung Do-Ha, Molecular Horizons and School of Chemistry and Molecular Bioscience, University of Wollongong, Wollongong, New South Wales 2522, Australia; Illawarra Health and Medical Research Institute, Wollongong, New South Wales 2522, Australia.
Stephanie L Rayner, Centre for Motor Neuron Disease Research, Department of Biomedical Sciences, Faculty of Medicine, Health and Human Sciences, Macquarie University, Sydney 2109, New South Wales, Australia.
Ian P Blair, Centre for Motor Neuron Disease Research, Department of Biomedical Sciences, Faculty of Medicine, Health and Human Sciences, Macquarie University, Sydney 2109, New South Wales, Australia.
Roger S Chung, Centre for Motor Neuron Disease Research, Department of Biomedical Sciences, Faculty of Medicine, Health and Human Sciences, Macquarie University, Sydney 2109, New South Wales, Australia.
Justin J Yerbury, Molecular Horizons and School of Chemistry and Molecular Bioscience, University of Wollongong, Wollongong, New South Wales 2522, Australia; Illawarra Health and Medical Research Institute, Wollongong, New South Wales 2522, Australia.
Lezanne Ooi, Molecular Horizons and School of Chemistry and Molecular Bioscience, University of Wollongong, Wollongong, New South Wales 2522, Australia; Illawarra Health and Medical Research Institute, Wollongong, New South Wales 2522, Australia.
Darren N Saunders, Molecular Horizons and School of Chemistry and Molecular Bioscience, University of Wollongong, Wollongong, New South Wales 2522, Australia; School of Medical Sciences, University of Sydney, Sydney 2006, Australia.
Funding
National Health and Medical Research Council of Australia grants (APP1107644, APP1095215 and APP1176913); National Health and Medical Research Council of Australia Boosting Dementia Research Leadership Fellowship (APP1135720 to L.O.); Australian Government Research Training Program Stipend Scholarship (matching scholarship to M.B.); Rotary Club of Cronulla and Rotary Health Australia (to M.B.); Investigator grant from the National Health and Medical Research Council of Australia (grant 1194872 to J.J.Y.); Motor Neuron Disease Research Institute of Australia (Betty Laidlaw Prize to J.J.Y. and grant to L.O., J.Y.Y and I.B.).
Data availability
Data are available upon request.
References
- 1. Hardiman, O., Al-Chalabi, A., Chio, A., Corr, E.M., Logroscino, G., Robberecht, W., Shaw, P.J., Simmons, Z. and van den Berg, L.H. (2017) Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers, 3, 17085. [DOI] [PubMed] [Google Scholar]
- 2. Mejzini, R., Flynn, L.L., Pitout, I.L., Fletcher, S., Wilton, S.D. and Akkari, P.A. (2019) ALS genetics, mechanisms, and therapeutics: Where are we now? Front. Neurosci., 13, 1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ringholz, G.M., Appel, S.H., Bradshaw, M., Cooke, N.A., Mosnik, D.M. and Schulz, P.E. (2005) Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology, 65, 586–590. [DOI] [PubMed] [Google Scholar]
- 4. Hardy, J. and Rogaeva, E. (2014) Motor neuron disease and frontotemporal dementia: sometimes related, sometimes not. Exp. Neurol., 262 Pt B, 75–83. [DOI] [PubMed] [Google Scholar]
- 5. Nguyen, H.P., Van Broeckhoven, C. and van der Zee, J. (2018) ALS genes in the genomic era and their implications for FTD. Trends Genet., 34, 404–423. [DOI] [PubMed] [Google Scholar]
- 6. Abramzon, Y.A., Fratta, P., Traynor, B.J. and Chia, R. (2020) The overlapping genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Front. Neurosci., 14, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yerbury, J.J., Farrawell, N.E. and McAlary, L. (2020) Proteome homeostasis dysfunction: a unifying principle in ALS pathogenesis. Trends Neurosci., 43, 274–284. [DOI] [PubMed] [Google Scholar]
- 8. Johnson, J.O., Mandrioli, J., Benatar, M., Abramzon, Y., Van Deerlin, V.M., Trojanowski, J.Q., Gibbs, J.R., Brunetti, M., Gronka, S., Wuu, J. et al. (2010) Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron, 68, 857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Watts, G.D., Wymer, J., Kovach, M.J., Mehta, S.G., Mumm, S., Darvish, D., Pestronk, A., Whyte, M.P. and Kimonis, V.E. (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet., 36, 377–381. [DOI] [PubMed] [Google Scholar]
- 10. Fecto, F., Yan, J., Vemula, S.P., Liu, E., Yang, Y., Chen, W., Zheng, J.G., Shi, Y., Siddique, N., Arrat, H. et al. (2011) SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol., 68, 1440–1446. [DOI] [PubMed] [Google Scholar]
- 11. Rubino, E., Rainero, I., Chio, A., Rogaeva, E., Galimberti, D., Fenoglio, P., Grinberg, Y., Isaia, G., Calvo, A., Gentile, S. et al. (2012) SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology, 79, 1556–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Le Ber, I., Camuzat, A., Guerreiro, R., Bouya-Ahmed, K., Bras, J., Nicolas, G., Gabelle, A., Didic, M., De Septenville, A., Millecamps, S. et al. (2013) SQSTM1 mutations in French patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol., 70, 1403–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deng, H.X., Chen, W., Hong, S.T., Boycott, K.M., Gorrie, G.H., Siddique, N., Yang, Y., Fecto, F., Shi, Y., Zhai, H. et al. (2011) Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature, 477, 211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Maruyama, H., Morino, H., Ito, H., Izumi, Y., Kato, H., Watanabe, Y., Kinoshita, Y., Kamada, M., Nodera, H., Suzuki, H. et al. (2010) Mutations of optineurin in amyotrophic lateral sclerosis. Nature, 465, 223–226. [DOI] [PubMed] [Google Scholar]
- 15. Kamada, M., Izumi, Y., Ayaki, T., Nakamura, M., Kagawa, S., Kudo, E., Sako, W., Maruyama, H., Nishida, Y., Kawakami, H. et al. (2014) Clinicopathologic features of autosomal recessive amyotrophic lateral sclerosis associated with optineurin mutation. Neuropathology, 34, 64–70. [DOI] [PubMed] [Google Scholar]
- 16. Cirulli, E.T., Lasseigne, B.N., Petrovski, S., Sapp, P.C., Dion, P.A., Leblond, C.S., Couthouis, J., Lu, Y.F., Wang, Q.L., Krueger, B.J. et al. (2015) Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science, 347, 1436–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pottier, C., Bieniek, K.F., Finch, N., van de Vorst, M., Baker, M., Perkersen, R., Brown, P., Ravenscroft, T., van Blitterswijk, M., Nicholson, A.M. et al. (2015) Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol., 130, 77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Farhan, S.M.K., Howrigan, D.P., Abbott, L.E., Klim, J.R., Topp, S.D., Byrnes, A.E., Churchhouse, C., Phatnani, H., Smith, B.N., Rampersaud, E. et al. (2019) Exome sequencing in amyotrophic lateral sclerosis implicates a novel gene, DNAJC7, encoding a heat-shock protein. Nat. Neurosci., 22, 1966–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dobson-Stone, C., Hallupp, M., Shahheydari, H., Ragagnin, A.M.G., Chatterton, Z., Carew-Jones, F., Shepherd, C.E., Stefen, H., Paric, E., Fath, T. et al. (2020) CYLD is a causative gene for frontotemporal dementia – amyotrophic lateral sclerosis. Brain, 143, 783–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Williams, K.L., Topp, S., Yang, S., Smith, B., Fifita, J.A., Warraich, S.T., Zhang, K.Y., Farrawell, N., Vance, C., Hu, X. et al. (2016) CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat. Commun., 7, 11253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bai, C., Sen, P., Hofmann, K., Ma, L., Goebl, M., Harper, J.W. and Elledge, S.J. (1996) SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell, 86, 263–274. [DOI] [PubMed] [Google Scholar]
- 22. Ciechanover, A. (1994) The ubiquitin-proteasome proteolytic pathway. Cell, 79, 13–21. [DOI] [PubMed] [Google Scholar]
- 23. Pickart, C.M. (2001) Mechanisms underlying ubiquitination. Annu. Rev. Biochem., 70, 503–533. [DOI] [PubMed] [Google Scholar]
- 24. Hershko, A. and Ciechanover, A. (1998) The ubiquitin system. Annu. Rev. Biochem., 67, 425–479. [DOI] [PubMed] [Google Scholar]
- 25. Chen, Z.J. and Sun, L.J. (2009) Nonproteolytic functions of ubiquitin in cell signaling. Mol. Cell, 33, 275–286. [DOI] [PubMed] [Google Scholar]
- 26. Bingol, B. and Sheng, M. (2011) Deconstruction for reconstruction: the role of proteolysis in neural plasticity and disease. Neuron, 69, 22–32. [DOI] [PubMed] [Google Scholar]
- 27. Dantuma, N.P., Groothuis, T.A., Salomons, F.A. and Neefjes, J. (2006) A dynamic ubiquitin equilibrium couples proteasomal activity to chromatin remodeling. J. Cell Biol., 173, 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Groothuis, T.A., Dantuma, N.P., Neefjes, J. and Salomons, F.A. (2006) Ubiquitin crosstalk connecting cellular processes. Cell Div., 1, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Farrawell, N.E., Lambert-Smith, I., Mitchell, K., McKenna, J., McAlary, L., Ciryam, P., Vine, K.L., Saunders, D.N. and Yerbury, J.J. (2018) SOD1(A4V) aggregation alters ubiquitin homeostasis in a cell model of ALS. J. Cell Sci., 131, jcs209122, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Farrawell, N.E., McAlary, L., Lum, J.S., Chisholm, C.G., Warraich, S.T., Blair, I.P., Vine, K.L., Saunders, D.N. and Yerbury, J.J. (2020) Ubiquitin homeostasis is disrupted in TDP-43 and FUS cell models of ALS. iScience, 23, 101700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Forman, M.S., Trojanowski, J.Q. and Lee, V.M. (2004) Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat. Med., 10, 1055–1063. [DOI] [PubMed] [Google Scholar]
- 32. Neumann, M., Sampathu, D.M., Kwong, L.K., Truax, A.C., Micsenyi, M.C., Chou, T.T., Bruce, J., Schuck, T., Grossman, M., Clark, C.M. et al. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science, 314, 130–133. [DOI] [PubMed] [Google Scholar]
- 33. Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., Mann, D., Tsuchiya, K., Yoshida, M., Hashizume, Y. et al. (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun., 351, 602–611. [DOI] [PubMed] [Google Scholar]
- 34. Mackenzie, I.R. (2007) The neuropathology of FTD associated with ALS. Alzheimer Dis. Assoc. Disord., 21, S44–S49. [DOI] [PubMed] [Google Scholar]
- 35. Turner, M.R., Hardiman, O., Benatar, M., Brooks, B.R., Chio, A., de Carvalho, M., Ince, P.G., Lin, C., Miller, R.G., Mitsumoto, H. et al. (2013) Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol., 12, 310–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Farrawell, N.E., Lambert-Smith, I.A., Warraich, S.T., Blair, I.P., Saunders, D.N., Hatters, D.M. and Yerbury, J.J. (2015) Distinct partitioning of ALS associated TDP-43, FUS and SOD1 mutants into cellular inclusions. Sci. Rep., 5, 13416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ciryam, P., Lambert-Smith, I.A., Bean, D.M., Freer, R., Cid, F., Tartaglia, G.G., Saunders, D.N., Wilson, M.R., Oliver, S.G., Morimoto, R.I. et al. (2017) Spinal motor neuron protein supersaturation patterns are associated with inclusion body formation in ALS. Proc. Natl. Acad. Sci. U.S.A., 114, E3935–E3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bax, M., McKenna, J., Do-Ha, D., Stevens, C.H., Higginbottom, S., Balez, R., Cabral-da-Silva, M.E.C., Farrawell, N.E., Engel, M., Poronnik, P. et al. (2019) The ubiquitin proteasome system is a key regulator of pluripotent stem cell survival and motor neuron differentiation. Cell, 8, 581, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brockington, A., Ning, K., Heath, P.R., Wood, E., Kirby, J., Fusi, N., Lawrence, N., Wharton, S.B., Ince, P.G. and Shaw, P.J. (2013) Unravelling the enigma of selective vulnerability in neurodegeneration: motor neurons resistant to degeneration in ALS show distinct gene expression characteristics and decreased susceptibility to excitotoxicity. Acta Neuropathol., 125, 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yerbury, J.J., Ooi, L., Blair, I.P., Ciryam, P., Dobson, C.M. and Vendruscolo, M. (2019) The metastability of the proteome of spinal motor neurons underlies their selective vulnerability in ALS. Neurosci. Lett., 704, 89–94. [DOI] [PubMed] [Google Scholar]
- 41. Lee, A., Rayner, S.L., De Luca, A., Gwee, S.S.L., Morsch, M., Sundaramoorthy, V., Shahheydari, H., Ragagnin, A., Shi, B., Yang, S. et al. (2017) Casein kinase II phosphorylation of cyclin F at serine 621 regulates the Lys48-ubiquitylation E3 ligase activity of the SCF((cyclin F)) complex. Open Biol., 7, 170058, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee, A., Rayner, S.L., Gwee, S.S.L., De Luca, A., Shahheydari, H., Sundaramoorthy, V., Ragagnin, A., Morsch, M., Radford, R., Galper, J. et al. (2018) Pathogenic mutation in the ALS/FTD gene, CCNF, causes elevated Lys48-linked ubiquitylation and defective autophagy. Cell. Mol. Life Sci., 75, 335–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cheng, F., De Luca, A., Hogan, A.L., Rayner, S.L., Davidson, J.M., Watchon, M., Stevens, C.H., Munoz, S.S., Ooi, L., Yerbury, J.J. et al. (2021) Unbiased label-free quantitative proteomics of cells expressing amyotrophic lateral sclerosis (ALS) mutations in CCNF reveals activation of the apoptosis pathway: a workflow to screen pathogenic gene mutations. Front. Mol. Neurosci., 14, 627740. 10.3389/fnmol.2021.627740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bax, M., Balez, R., Munoz, S.S., Do-Ha, D., Stevens, C.H., Berg, T., Cabral-da-Silva, M.C., Engel, M., Nicholson, G., Yang, S. et al. (2019) Generation and characterization of a human induced pluripotent stem cell line UOWi005-a from dermal fibroblasts derived from a CCNF(S621G) familial amyotrophic lateral sclerosis patient using mRNA reprogramming. Stem Cell Res., 40, 101530. [DOI] [PubMed] [Google Scholar]
- 45. Koyuncu, S., Saez, I., Lee, H.J., Gutierrez-Garcia, R., Pokrzywa, W., Fatima, A., Hoppe, T. and Vilchez, D. (2018) The ubiquitin ligase UBR5 suppresses proteostasis collapse in pluripotent stem cells from Huntington's disease patients. Nat. Commun., 9, 2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lambert-Smith, I.A., Saunders, D.N. and Yerbury, J.J. (2020) The pivotal role of ubiquitin-activating enzyme E1 (UBA1) in neuronal health and neurodegeneration. Int. J. Biochem. Cell Biol., 123, 105746. [DOI] [PubMed] [Google Scholar]
- 47. Hogan, A.L., Don, E.K., Rayner, S.L., Lee, A., Laird, A.S., Watchon, M., Winnick, C., Tarr, I.S., Morsch, M., Fifita, J.A. et al. (2017) Expression of ALS/FTD-linked mutant CCNF in zebrafish leads to increased cell death in the spinal cord and an aberrant motor phenotype. Hum. Mol. Genet., 26, 2616–2626. [DOI] [PubMed] [Google Scholar]
- 48. D'Angiolella, V., Donato, V., Vijayakumar, S., Saraf, A., Florens, L., Washburn, M.P., Dynlacht, B. and Pagano, M. (2010) SCF(Cyclin F) controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature, 466, 138–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Choi, Y.S., Bollinger, S.A., Prada, L.F., Scavone, F., Yao, T. and Cohen, R.E. (2019) High-affinity free ubiquitin sensors for quantifying ubiquitin homeostasis and deubiquitination. Nat. Methods, 16, 771–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stefanovic-Barrett, S., Dickson, A.S., Burr, S.P., Williamson, J.C., Lobb, I.T., van den Boomen, D.J., Lehner, P.J. and Nathan, J.A. (2018) MARCH6 and TRC8 facilitate the quality control of cytosolic and tail-anchored proteins. EMBO Rep., 19, e45603, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tashiro, Y., Urushitani, M., Inoue, H., Koike, M., Uchiyama, Y., Komatsu, M., Tanaka, K., Yamazaki, M., Abe, M., Misawa, H. et al. (2012) Motor neuron-specific disruption of proteasomes, but not autophagy, replicates amyotrophic lateral sclerosis. J. Biol. Chem., 287, 42984–42994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yang, S., Zhang, K.Y., Kariawasam, R., Bax, M., Fifita, J.A., Ooi, L., Yerbury, J.J., Nicholson, G.A. and Blair, I.P. (2015) Evaluation of skin fibroblasts from amyotrophic lateral sclerosis patients for the rapid study of pathological features. Neurotox. Res., 28, 138–146. [DOI] [PubMed] [Google Scholar]
- 53. Yerbury, J.J., Ooi, L., Dillin, A., Saunders, D.N., Hatters, D.M., Beart, P.M., Cashman, N.R., Wilson, M.R. and Ecroyd, H. (2016) Walking the tightrope: proteostasis and neurodegenerative disease. J. Neurochem., 137, 489–505. [DOI] [PubMed] [Google Scholar]
- 54. Powis, R.A., Karyka, E., Boyd, P., Come, J., Jones, R.A., Zheng, Y., Szunyogova, E., Groen, E.J., Hunter, G., Thomson, D. et al. (2016) Systemic restoration of UBA1 ameliorates disease in spinal muscular atrophy. JCI Insight, 1, e87908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Krus, K.L., Strickland, A., Yamada, Y., Devault, L., Schmidt, R.E., Bloom, A.J., Milbrandt, J. and DiAntonio, A. (2022) Loss of Stathmin-2, a hallmark of TDP-43-associated ALS, causes motor neuropathy. Cell Rep., 39, 111001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ling, S.C., Polymenidou, M. and Cleveland, D.W. (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron, 79, 416–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hanna, J., Leggett, D.S. and Finley, D. (2003) Ubiquitin depletion as a key mediator of toxicity by translational inhibitors. Mol. Cell. Biol., 23, 9251–9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Park, C.W. and Ryu, K.Y. (2014) Cellular ubiquitin pool dynamics and homeostasis. BMB Rep., 47, 475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ryu, K.Y., Garza, J.C., Lu, X.Y., Barsh, G.S. and Kopito, R.R. (2008) Hypothalamic neurodegeneration and adult-onset obesity in mice lacking the Ubb polyubiquitin gene. Proc. Natl. Acad. Sci. U.S.A., 105, 4016–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Park, C.W., Ryu, H.W. and Ryu, K.Y. (2012) Locus coeruleus neurons are resistant to dysfunction and degeneration by maintaining free ubiquitin levels although total ubiquitin levels decrease upon disruption of polyubiquitin gene Ubb. Biochem. Biophys. Res. Commun., 418, 541–546. [DOI] [PubMed] [Google Scholar]
- 61. Chen, P.C., Bhattacharyya, B.J., Hanna, J., Minkel, H., Wilson, J.A., Finley, D., Miller, R.J. and Wilson, S.M. (2011) Ubiquitin homeostasis is critical for synaptic development and function. J. Neurosci., 31, 17505–17513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vaden, J.H., Bhattacharyya, B.J., Chen, P.C., Watson, J.A., Marshall, A.G., Phillips, S.E., Wilson, J.A., King, G.D., Miller, R.J. and Wilson, S.M. (2015) Ubiquitin-specific protease 14 regulates c-Jun N-terminal kinase signaling at the neuromuscular junction. Mol. Neurodegener., 10, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Skaar, J.R., Pagan, J.K. and Pagano, M. (2014) SCF ubiquitin ligase-targeted therapies. Nat. Rev. Drug Discov., 13, 889–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bence, N.F., Sampat, R.M. and Kopito, R.R. (2001) Impairment of the ubiquitin-proteasome system by protein aggregation. Science, 292, 1552–1555. [DOI] [PubMed] [Google Scholar]
- 65. Walker, J.M. (2005) The Proteomics Protocols Handbook, Springer. https://link.springer.com/book/10.1385/1592598900.
- 66. Cashman, N.R., Durham, H.D., Blusztajn, J.K., Oda, K., Tabira, T., Shaw, I.T., Dahrouge, S. and Antel, J.P. (1992) Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev. Dyn., 194, 209–221. [DOI] [PubMed] [Google Scholar]
- 67. Nagarajan, S.R., Brandon, A.E., McKenna, J.A., Shtein, H.C., Nguyen, T.Q., Suryana, E., Poronnik, P., Cooney, G.J., Saunders, D.N. and Hoy, A.J. (2017) Correction: insulin and diet-induced changes in the ubiquitin-modified proteome of rat liver. PLoS One, 12, e0184610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cox, J., Neuhauser, N., Michalski, A., Scheltema, R.A., Olsen, J.V. and Mann, M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res., 10, 1794–1805. [DOI] [PubMed] [Google Scholar]
- 69. Schneider, C.A., Rasband, W.S. and Eliceiri, K.W. (2012) NIH image to ImageJ: 25 years of image analysis. Nat. Methods, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available upon request.