

Abstract
The EFSA Panel on Food Additives and Flavourings (FAF) was requested to evaluate the safety of the smoke flavouring Primary Product Scansmoke SEF7525 (SF‐004), for which a renewal application was submitted in accordance with Article 12(1) of Regulation (EC) No 2065/2003. This opinion refers to the assessment of data submitted on chemical characterisation, dietary exposure and genotoxicity of the Primary Product. Scansmoke SEF7525 is obtained from a tar produced from a mixture of red oak, white oak, maple, beech and hickory. Based on the compositional data, the Panel noted that the identified and quantified proportion of the solvent‐free fraction amounts to 32.6 weight (wt)%, thus the applied method does not meet the legal quality criterion that at least 50% of the solvent‐free fraction shall be identified and quantified. At the maximum proposed use levels, dietary exposure estimates calculated with Food Additive Intake Model (FAIM) ranged from 0.6 to 3.8 mg/kg body weight (bw) per day at the mean and from 1.1 to 10.1 mg/kg bw per day at the 95th percentile. Based on the available information on genotoxicity on 44 identified components, the Panel concluded that two substances in the Primary Product, styrene and benzofuran, raise a potential concern for genotoxicity. In addition, a potential concern for genotoxicity was identified for the unidentified part of the mixture. Considering that the exposure estimates for styrene and benzofuran are above the threshold of toxicological concern (TTC) value of 0.0025 kg/kg bw per day for DNA‐reactive mutagens and/or carcinogens and since further data are needed to clarify their potential genotoxicity, the Panel concluded that the potential safety concern for genotoxicity of the Primary Product cannot be ruled out.
Keywords: Scansmoke SEF7525, SF‐004, smoke flavouring Primary Product, genotoxicity, styrene, benzofuran
1. Introduction
1.1. Background and Terms of Reference as provided by the requestor
1.1.1. Background
Regulation (EC) No 2065/2003 1 establishes a procedure for the safety assessment and the authorisation of smoke flavouring primary products with a view to ensuring a high level of protection of human health and the effective functioning of the internal market. No smoke flavouring or any food where such a smoke flavouring is present (in or on) can be placed in the market if the smoke flavouring is not an authorised Primary Product or is not derived therefrom and if the conditions of use laid down in the authorisation in accordance with this Regulation are not adhered to (Article 4(2) of Regulation (EC) No 2065/2003).
Commission Implementing Regulation (EU) No 1321/2013 2 authorised 10 smoke flavouring primary products for a 10‐year period, due to expire on 31 December 2023.
The European Commission has received an application for the renewal of the authorisation of the smoke flavouring primary product Scansmoke SEF7525 (SF‐004) for a 10‐year period, in accordance with Article 12 of Regulation (EC) No 2065/2003.
1.1.2. Terms of Reference
The European Commission requests the European Food Safety Authority (EFSA) to evaluate the safety of the smoke flavouring primary product Scansmoke SEF7525 (SF‐004), for which a renewal application has been submitted, in accordance with Article 12(1) of Regulation (EC) No 2065/2003.
The safety assessment shall be carried‐out in two steps. Firstly, EFSA shall give a scientific opinion on the data included in the renewal application dossier related to the chemical characterisation, the genotoxicity and the dietary exposure to Scansmoke SEF7525 (SF‐004). Secondly, provided that the genotoxic concern can be ruled out in the first part of the evaluation, EFSA shall complete the rest of the safety assessment without delay upon submission of the relevant pending data from the applicant.
1.2. Interpretation of the Terms of Reference
In line with the terms of reference (see Section 1.1.2), the safety of the Primary Product will be assessed in two steps.
The current (first) opinion will address the chemical characterisation, genotoxicity and dietary exposure to the smoke flavouring Primary Product.
If in the first opinion, no concern for genotoxicity is raised, EFSA will issue a second opinion assessing the toxicity other than genotoxicity data, as required by the EFSA guidance for the preparation of applications on smoke flavouring Primary Products (EFSA FAF Panel, 2021).
1.3. Additional information
EFSA issued a previous opinion on the safety of this smoke flavouring Primary Product Scansmoke SEF7525 in 2009 (EFSA CEF Panel, 2009).
Following the safety assessment from EFSA, Scansmoke SEF7525 was authorised in the European Union and assigned the unique code ‘SF‐004’, according to Commission Implementing Regulation (EU) No 1321/2013, establishing the Union list of authorised smoke flavouring Primary Products, for a 10‐year period with effect from 1 January 2014.
The present opinion refers to an assessment of the data submitted by the authorisation holder for the renewal of the authorisation of Scansmoke SEF7525 (SF‐004) as a smoke flavouring Primary Product, in line with Article 12(1) of Regulation (EC) No 2065/2003.
2. Data and methodologies
2.1. Data
The present evaluation is based on the data provided by the applicant in the form of a technical dossier, submitted according to Article 12(1) of Regulation (EC) No 2065/2003 for the renewal of the authorisation of the smoke flavouring Primary Product Scansmoke SEF7525 (SF‐004).
In accordance with Article 38 of the Regulation (EC) No 178/2002 3 and taking into account the protection of confidential information and of personal data in accordance with Articles 39 to 39e of the same Regulation and of the Decision of the EFSA's Executive Director laying down practical arrangements concerning transparency and confidentiality, 4 the non‐confidential version of the dossier is published on Open.EFSA. 5
According to Art. 32c(2) of Regulation (EC) No 178/2002 and to the Decision of EFSA's Executive Director laying down the practical arrangements on pre‐submission phase and public consultations, EFSA carried out a public consultation on the non‐confidential version of the application from 27 October to 17 November 2022, for which no comments were received.
Additional information was sought from the applicant during the assessment process by requests from EFSA sent on 25 November 2022 and was subsequently provided (see Documentation provided to EFSA No. 2).
The Panel acknowledged the submission of data on toxicity other than genotoxicity by the applicant in the technical dossier (see Documentation provided to EFSA No. 1). As indicated in Section 1.2, the assessment of these data is outside the scope of the present opinion.
2.2. Methodologies
The safety assessment of the Primary Product Scansmoke SEF7525 was conducted in line with the requirements laid down in Regulation (EC) No 2065/2003 and following the principles of the EFSA guidance for the preparation of applications on smoke flavouring Primary Products (EFSA FAF Panel, 2021).
The principles described in the EFSA Guidance on transparency with regard to scientific aspects of risk assessment (EFSA Scientific Committee, 2009) as well as the relevant cross‐cutting guidance documents from the EFSA Scientific Committee published after the adoption of the guidance on smoke flavourings (EFSA FAF Panel, 2021), in particular the ‘Guidance on technical requirements for regulated food and feed product applications to establish the presence of small particles including nanoparticles’ (EFSA Scientific Committee, 2021a), were also considered during the risk assessment.
The uncertainty analysis was performed by checking whether standard or non‐standard sources of uncertainties are present, as outlined in the standard procedure described in Section 4.2 of the EFSA guidance on smoke flavouring and listed in Table G.1 therein (EFSA FAF Panel, 2021). Standard uncertainties are not discussed in detail in the present assessment. In case of the presence of non‐standard uncertainties, these are reported in the relevant sections of the opinion and their combined impact on the assessment was evaluated by the Panel (see Section 4).
3. Assessment
3.1. Technical data
3.1.1. Manufacturing process
3.1.1.1. Source materials for the Primary Product
The source material of Scansmoke SEF7525 is a tar obtained by the applicant from an external supplier as by‐product of the production of liquid smoke. According to a statement of the supplier, the tar is produced from a mixture of 30–40% red oak (Quercus rubra), 30–40% white oak (Quercus alba), 5–15% maple (Acer saccharum), 5–15% beech (Fagus grandifolia) and 5–15% hickory (Carya ovata) (Documentation provided to EFSA No. 1 and 2). The hardwoods are blended as sawdust, which is then dried and heated to generate smoke.
The applicant submitted a statement of the supplier informing that the trees from which the tar is produced were not treated with chemical substances (including pesticides) during the 6 months preceding the felling (Documentation provided to EFSA No. 2). The tar is subjected to an internal quality control procedure (i.e. FSSC 22000 consisting of ISO 22000:2018 and ISO TS 22002‐1:2009).
The Panel noted that no information was provided on the pyrolysis conditions applied to produce the tar used as source material for the Primary Product by the applicant. This creates a non‐standard uncertainty with respect to the manufacturing process of the Primary Product (see Section 2.2 of this opinion and Table G.1 of the EFSA guidance document on smoke flavouring (EFSA FAF Panel, 2021)).
3.1.1.2. Method of manufacture of the Primary Product
As described by the applicant, the Primary Product is obtained by (i) extracting the tar raw material with diethyl ether, (ii) subjecting the extracts to purification steps and (iii) combining the obtained fractions (SEF1 and SEF2) at a defined ratio. In the first step, the extraction of an aqueous suspension of the tar is performed under alkaline conditions (pH‐adjustment by addition of sodium hydroxide). The organic phase is subjected to evaporation to remove solvent and water and subsequently to vacuum distillation. After re‐dilution with diethyl ether, the obtained distillate is treated with active carbon to remove polycyclic aromatic hydrocarbons (PAHs), and SEF1 is obtained after evaporation of the solvent and a final filtration (1 mm). In the second step, the pH of the heavy phase remaining after the first extraction is lowered to a less alkaline pH (addition of sulphuric acid). SEF2 results from the extraction with diethyl ether, followed by treatment of the extract with active carbon, evaporation of the solvent and filtration (1 mm). The final Primary Product is obtained by mixing SEF1 and SEF2 at a fixed ratio and is subjected to physico‐chemical testing to verify that it meets the product specifications (Documentation provided to EFSA No. 2).
The applicant submitted a description of the manufacturing process, with information on the drying step, the range of temperatures during the pyrolysis and the extraction and distillation conditions.
3.1.2. Identity of the Primary Product
3.1.2.1. Trade name of the Primary Product
The trade name of the product is Scansmoke SEF7525.
3.1.2.2. Information on existing evaluation from other regulatory bodies and authorisations in non‐EU countries
The applicant indicated that the smoke flavouring Scansmoke SEF7525 has not been evaluated by other regulatory bodies other than EFSA (Documentation provided to EFSA No. 1).
Regarding the existing authorisations in non‐EU countries, the applicant stated that Scansmoke SEF7525 is currently authorised in the United Kingdom and in China (see Documentation provided to EFSA No. 3).
3.1.2.3. Description of the physical state and sensory characteristics
The applicant described the smoke flavouring Primary Product as a ‘viscous liquid of dark brown colour with a characteristic strong odour of smoke’ (Documentation provided to EFSA No. 1). The Primary Product has an average density (at 20°C) of 1.1475 g/mL, refractive index (at 20°C) ranging from 1.50 to 1.70 and a viscosity (at 25°C) ranging from 2,214 to 2,349 mPa·s. In an additional data request, EFSA requested the applicant to specify the parameters of pH and staining index of the Primary Product. These parameters were not provided since they were considered by the applicant as ‘not meaningful for the characterisation of this Primary Product’ (Documentation provided to EFSA No. 2). Considering the low water content of the Primary Product, the Panel accepted that no pH value was provided. Furthermore, the Panel agreed that the staining index is not fundamental for the safety assessment.
3.1.2.4. Chemical composition of the Primary Product
The compositional data provided by the applicant for six batches of the Primary Product in the original dossier and in response to the EFSA requests for additional information are summarised in Table 1 (Documentation provided to EFSA No. 1 and 2).
Table 1.
Overview on the compositional data provided for six batches of the Primary Product (Documentation provided to EFSA No. 1 and 2)
| Batch no. | Density (g/L) | Total volatiles (wt%) | Identified volatiles (wt%) | Unidentified volatiles (wt%) | Total non‐volatiles (wt%) (1) | Identified non‐volatiles (wt%) | Unidentified non‐volatiles (wt%) | Water (wt%) | Solvent‐free fraction (wt%) | Ident./quant. proportion of solvent‐free fraction (wt%) (2) , (a) | Ident./quant. Proportion of volatile fraction (wt%) (3) , (b) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 13390 # | 1,149 | 33 | 27 | 5.9 | 66.4 | – | 66.4 | 0.6 | 99.4 | 27.2 | 81.8 |
| 14185 | 1,144 | 38.4 | 31.5 | 6.9 | 61.2 | – | 61.2 | 0.4 | 99.6 | 31.6 | 82.0 |
| 14300 | 1,151 | 43 | 34 | 8.3 | 57 | – | 57 | 0.3 | 99.7 | 34.1 | 79.1 |
| 14301 | 1,151 | 45 | 37 | 8.1 | 55 | – | 55 | 0.4 | 99.6 | 37.1 | 82.2 |
| 14302 | 1,148 | 42 | 34 | 7.4 | 57.9 | – | 57.9 | 0.4 | 99.6 | 34.1 | 81.0 |
| 14303 | 1,145 | 39 | 31 | 7.9 | 60.7 | – | 60.7 | 0.5 | 99.5 | 31.2 | 79.5 |
| Average | 1,148 | 40.1 | 32.4 | 7.4 | 59.7 | 59.7 | 0.4 | 99.6 | 32.6 | 80.9 | |
| SD | 3.0 | 4.3 | 3.4 | 0.9 | 4.0 | – | 4.0 | 0.1 | 0.1 | 3.4 | 1.4 |
| RSD (%) | 0.3 | 10.6 | 10.5 | 12.1 | 6.7 | – | 6.7 | 23.8 | 0.1 | 10.5 | 1.7 |
wt: weight; SD: standard deviation; RSD: relative standard deviation.
Calculated as 100 ‐ water (wt%) – total volatiles (wt%).
Calculated as: ((identified volatiles + identified non‐volatiles)/solvent‐free fraction) * 100.
Calculated as: (identified volatiles/total volatiles) * 100.
Batch tested in the newly submitted genotoxicity studies.
Regulatory quality criterion for the applied method according to Regulation (EC) No 627/2006 6 : ≥ 50 (wt%).
Regulatory quality criterion for the applied method according to Regulation (EC) No 627/2006: ≥ 80 (wt%).
3.1.2.4.1. Chemical characterisation
The water content of the Primary Product was determined by the Karl Fischer titration method (Documentation provided to EFSA No. 1). The applicant also provided data on the contents of the major chemical classes, i.e. acids, phenols and carbonyls, in six batches of the Primary Product (Table 2) (Documentation provided to EFSA No. 1 and 2).
Table 2.
Chemical compositions reported for six batches of the Primary Product
| Batch no. | Average | SD | ||||||
|---|---|---|---|---|---|---|---|---|
| 13390 # | 14185 | 14300 | 14301 | 14302 | 14303 | |||
| Acids (wt%) (as acetic acid) | 0.7 | 1.5 | 1.0 | 1.5 | 0.9 | 0.5 | 1.0 | 0.4 |
| Phenols (wt%) (as syringol) | 9.5 | 7.5 | 9.2 | 9.9 | 8.8 | 8.1 | 8.8 | 0.9 |
| Carbonyls (wt%) (as furfural) | 4.7 | 4.9 | 4.4 | 4.5 | 4.5 | 5.4 | 4.7 | 0.4 |
| Water (wt%) | 0.6 | 0.4 | 0.3 | 0.4 | 0.4 | 0.5 | 0.4 | 0.1 |
wt: weight; SD:standard deviation.
Batch tested in the newly submitted genotoxicity studies.
The Panel acknowledges the explanation provided by the applicant that the difference between the average concentration of carbonyls in the current application and the average concentration (1.9 weight (wt)%) reported in the previous opinion (EFSA CEF Panel, 2009) is not due to changes in the manufacturing process but related to differences in the performance of the analyses that are based on the reaction of carbonyls with hydroxylamine (Documentation provided to EFSA No. 1). The method applied for this renewal application differs from the method applied previously regarding pH‐adjustment of the reaction mixture and the heating conditions. The contract laboratory was not able to implement and apply the former method to the Primary Product. The Panel considers the values for carbonyl contents provided in this application as reliable, albeit method‐specific.
Concentrations of arsenic, cadmium, lead and mercury were determined by inductively coupled plasma–mass spectrometry (ICP–MS) and were submitted to EFSA (Table 3) (Documentation provided to EFSA No. 1 and 2).
Table 3.
Toxic elements reported for six batches of the Primary Product
| Batch no. (mg/kg) | Average (mg/kg) | SD | ||||||
|---|---|---|---|---|---|---|---|---|
| 13390 # | 14185 | 14300 | 14301 | 14302 | 14303 | |||
| Arsenic (As) | < 0.1 (a) | < 0.1 (a) | < 0.1 (a) | < 0.1 (a) | < 0.1 (a) | < 0.1 (a) | < 0.1 (a) | – |
| Cadmium (Cd) | < 0.01 (a) | < 0.01 (a) | < 0.01 (a) | < 0.01 (a) | < 0.01 (a) | < 0.01 (a) | < 0.01 (a) | – |
| Lead (Pb) | < 0.05 (a) | < 0.05 (a) | < 0.05 (a) | < 0.05 (a) | < 0.05 (a) | < 0.05 (a) | < 0.05 (a) | – |
| Mercury (Hg) | < 0.005 (a) | < 0.005 (a) | < 0.005 (a) | < 0.005 (a) | < 0.005 (a) | < 0.005 (a) | < 0.005 (a) | – |
SD: standard deviation.
Batch tested in the newly submitted genotoxicity studies.
Value below the corresponding limit of quantification (LOQ).
3.1.2.4.2. Identification and quantification of the volatile fraction
Gas chromatography–mass spectrometry (GC–MS) and gas chromatography–flame ionisation detection (GC‐FID) were applied for identification and quantification of the constituents of the volatile fraction of the Primary Product. Individual volatile constituents were considered as identified if their chromatographic (i.e. retention times) and mass spectral data were in agreement with those of reference standards. Quantifications of the volatile constituents were based on compound specific response factors and the use of anthracene d‐10 as internal standard. The analytical methods employed to identify and quantify the volatile constituents were submitted to EFSA together with the validation parameters (Documentation provided to EFSA No. 1).
Overall, using this approach, 41 constituents were identified and quantified in six batches of the Primary Product (for batch numbers see Table 1) (Appendix A, Table A.1). The lowest concentration reported by the applicant was 0.05 wt% for 1‐(4‐hydroxy‐3‐ methoxyphenyl)‐2‐propanone (CAS no.: 2503‐46‐0). For three additional identified constituents, quantitative data were not provided, i.e. 1‐hydroxy‐2‐butanone (CAS no.: 5077‐7‐8), syringyl acetone (CAS no.: 19037‐58‐2) and 2‐hydroxy‐γ‐butyrolactone (CAS no.: 19444‐84‐9) (Documentation provided to EFSA No. 1). The 20 principal volatile constituents of the Primary Product are presented in Table 4.
Table 4.
Twenty principal volatile constituents of the Primary Product (Documentation provided to EFSA No. 1 and2)
| CAS no | FL‐no | Chemical name (a) | Average concentration (wt%) | |
|---|---|---|---|---|
| Current application (b) | Former application (c) | |||
| 91‐10‐1 | 04.036 | 2,6‐dimethoxyphenol | 4.9 | 1.8 |
| 6638‐05‐7 | 04.053 | 4‐methyl‐2,6‐dimethoxyphenol (2,6‐dimethoxy‐4‐methylphenol) | 3.4 | 6.9 |
| 90‐05‐1 | 04.005 | 2‐methoxyphenol | 2.3 | 1.4 |
| 93‐51‐6 | 04.007 | 2‐methoxy‐4‐methylphenol (creosol/methylguaiacol) | 2.3 | 0.4 |
| 14059‐92‐8 | 04.052 | 4‐ethyl‐2,6‐dimethoxyphenol | 2.2 | 2.8 |
| 20675‐95‐0 | 04.055 (d) | 2,6‐dimethoxy‐4‐prop‐1‐enylphenol (2,6‐dimethoxy‐4‐(2‐propenyl)‐phenol, E) | 1.4 | |
| 105‐67‐9 | 04.066 | 2,4‐dimethylphenol | 1.3 | 1.4 |
| 2785‐89‐9 | 04.008 | 4‐ethylguaiacol (4‐ethyl‐2‐methoxyphenol) | 1.2 | 1.9 |
| 95‐48‐7 | 04.027 | 2‐methylphenol | 1.1 | 1.1 |
| 108‐95‐2 | 04.041 | phenol | 0.9 | 0.9 |
| 5932‐68‐3 | 04.004 (e) | isoeugenol (trans‐isoeugenol) | 0.9 | 1.2 |
| 6627‐88‐9 | 04.051 | 4‐allyl‐2,6‐dimethoxyphenol (2,6‐dimethoxy‐4‐(2‐propenyl)‐phenol) | 0.9 | |
| 97‐53‐0 | 04.003 | eugenol | 0.6 | 1.2 |
| 576‐26‐1 | 04.042 | 2,6‐dimethylphenol | 0.6 | |
| 26624‐13‐5 | 04.055 (d) | 2,6‐dimethoxy‐4‐prop‐1‐enylphenol(4‐propenyl‐2,6‐dimethoxyphenol, Z) | 0.6 | 1.1 |
| 1121‐05‐7 | – | 2,3‐dimethyl‐2‐cyclopenten‐1‐one | 0.6 | |
| 108‐39‐4 | 04.026 | 3‐methylphenol | 0.6 | 0.6 |
| 527‐60‐6 | 04.095 | 2,4,6‐trimethyl phenol | 0.6 | |
| 2785‐87‐7 | 04.049 | 2‐methoxy‐4‐propylphenol | 0.5 | |
| 98‐00‐0 | 13.019 | furfuryl alcohol (2‐furanmethanol) | 0.4 | |
CAS: Chemical Abstract Service; FL‐no: FLAVIS number; wt: weight.
In case a constituent of the Primary Product is an authorised flavouring substance (FL‐no), the assigned chemical name corresponds to the respective entry in the EU Union List of flavourings. Deviating chemical names reported by the applicant in the dossier are given in brackets, if applicable.
From the analysis of the six batches presented in Table 1.
From the data presented in the previous safety evaluation of the Primary Product (EFSA CEF Panel, 2009).
[FL‐no: 04.055] refers to the mixture of E/Z stereoisomers of 2,6‐dimethoxy‐4‐prop‐1‐enylphenol.
[FL‐no: 04.004] refers to the mixture of E/Z stereoisomers of isoeugenol.
The applicant reported 280 tentatively identified volatile constituents (Documentation provided to EFSA No. 1). The identification was considered as tentative when it was (solely) based on structural similarities to identified constituents or when the mass spectral data were only compared to a fragmentation mass spectral library rather than to those of a reference standard. In accordance with the EFSA Scientific Guidance on Smoke Flavourings (EFSA FAF Panel, 2021), EFSA considered these tentatively identified constituents as part of the unidentified fraction.
According to the information provided by the applicant (Documentation submitted to EFSA No. 2), the total volatile fraction of Scansmoke SEF7525 accounted on average for 40.1 wt% of the Primary Product. The proportion of identified and quantified volatiles amounted to approximately 81% of the total volatile fraction; thus, the applied methods meet the legal quality criterion that at least 80% by mass of the volatile fraction shall be identified and quantified (Regulation (EC) No 627/2006).
Following an additional data request from EFSA, the applicant commented on the fact that the current list of identified volatile constituents does not fully match the list of identified volatile constituents provided at the time of the previous EFSA assessment of Scansmoke SEF7525 (EFSA CEF Panel, 2009). The applicant emphasised that there were no changes in the manufacturing process and explained that the observed differences are mainly due to the fact that in contrast to the previous application, volatiles were only considered as identified if their chromatographic and mass spectrometric data matched those of reference standards (Documentation provided to EFSA No. 2). The Panel acknowledges this explanation. The Panel further noted that the applicant reported differences in the conditions of the gas chromatography (GC)‐based quantification approaches, e.g. limitation of the upper GC column temperature to 250 °C in the analysis performed for the present application (Documentation provided to EFSA No. 1 and 2). It is very likely that the use of more recent analytical techniques allowed the applicant to perform a more accurate characterisation of the volatile fraction. Although, in the current application, the portion of identified and quantified volatile components is lower than in the former application, the newly developed GC–MS method allowed better peak separation and shape, and enhanced the retention of components with high boiling point and polarity. For this reason, the characterisation performed here is more reliable than the characterisation performed in the previous application (EFSA CEF Panel, 2009), and the product evaluated in the present assessment does not fundamentally deviate from the product evaluated formerly (EFSA CEF Panel, 2009).
3.1.2.4.3. Characterisation of the non‐volatile fraction
In order to characterise the non‐volatile fraction of the Primary Product, the applicant employed the following approaches (Documentation provided to EFSA No. 1):
Heating of the Primary Product at 350°C for determination of the residual non‐volatile fraction. The residue remaining amounted on average to approximately 24 wt%.
Size exclusion chromatography was performed for an untreated sample of the Primary Product and for a sample obtained after evaporation of the Primary Product at 80°C. Using a calibration curve for a polystyrene standard, the weight‐average molecular mass was estimated to be 1.4 kDa for both samples.
A sample of the Primary Product, obtained after evaporation at 80°C, was subjected to alkaline oxidation using H2O2/NaOH followed by GC–MS analysis to identify volatile products resulting from degradation of the non‐volatile material. After this oxidation step, 58 volatile constituents (Documentation provided to EFSA No. 1) were tentatively identified by GC–MS analysis, based on comparisons of their mass spectra to those from an MS‐library. Full identifications and quantifications of the constituents were not performed. The range of detected volatile degradation products encompassed compound classes as described for the oxidative cracking of precipitated hardwood lignin by hydrogen peroxide (Xiang and Lee, 2000). However, the Panel noted that this investigation of the non‐volatile fraction was performed following a treatment with H2O2 under alkaline conditions. Owing to the chemical changes expected under these oxidative conditions, the detected degradation products do not necessarily represent monomers of the non‐volatile material. Thus, the data provided contribute to a characterisation of the non‐volatile fraction, but this part of the Primary Product cannot be considered as identified.
The Panel considered that the applicant could have used a more direct method to identify and quantify constituents in the non‐volatile fraction, e.g. high performance liquid chromatography coupled to mass spectrometry (HPLC–MS), as suggested in the EFSA guidance on smoke flavourings (EFSA FAF Panel, 2021).
3.1.2.4.4. Unidentified fraction
The unidentified fraction of the Primary Product amounts to approximately 67 wt% and comprises the unidentified volatile constituents and the unidentified non‐volatile fraction (i.e. the total non‐volatile fraction); for the individual values see Table 1.
3.1.2.4.5. Overall composition of the Primary Product
Based on the chemical analyses performed on the production batches of the Primary Product (Table 1), the overall composition of Scansmoke SEF7525 (wt% of Primary Product) is shown in Figure 1, whereas the composition (wt%) of the solvent‐free fraction is shown in Figure 2.
Figure 1.
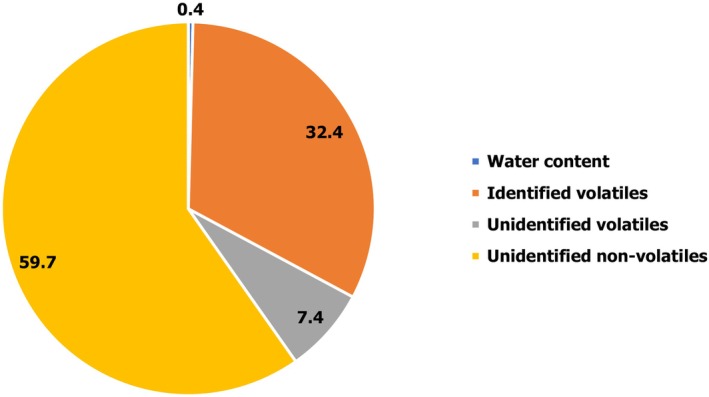
Overall composition of Scansmoke SEF7525 (wt% of Primary Product)
Figure 2.

Composition (wt%) of the solvent‐free fraction of Scansmoke SEF7525
Regarding the identified and quantified proportion of the volatile fraction, the applied methods comply with the legal requirement that at least 80 wt% of the volatile fraction shall be identified and quantified (Regulation (EC) No 627/2006). The Panel noted that for the investigated batches of the Primary Product, the identified and quantified proportion of the solvent‐free fraction is on average 32.6 wt%; thus, the applied methods do not meet the legal quality criterion that at least 50% by mass (wt%) of the solvent‐free fraction shall be identified and quantified, as specified in Regulation (EC) No 627/2006. This creates a non‐standard uncertainty with respect to the chemical composition of the Primary Product (see Section 2.2 of this opinion and Table G.1 of the EFSA guidance document on smoke flavourings (EFSA FAF Panel, 2021)).
3.1.2.5. Polycyclic aromatic hydrocarbons (PAHs)
Analytical data on the contents of 16 PAHs were provided for six batches of the Primary Product. The analysis meets the performance criteria as set in Regulation (EC) No 627/2006. The levels reported for the individual PAHs (Table 5) are consistently below the minimum required limits of quantification according to Regulation (EC) No 627/2006.
Table 5.
Concentrations of PAHs in the Primary Product, average from six batches (for batch numbers see Table 1) (Documentation provided to EFSA No. 1)
| PAH | Conc. range (μg/kg) | Average (μg/kg) | SD |
|---|---|---|---|
| benzo[a]anthracene (a) | 6.9–15.2 | 9.4 | 3.0 |
| chrysene (a) | 5.0–11.2 | 6.7 | 2.7 |
| benzo[b]fluoranthene (a) | 1.8–3.5 | 2.4 | 0.6 |
| benzo[k]fluoranthene | 0.9–1.04 | 1.0 | 0.2 |
| benzo[j]fluoranthene | 1.4–2.9 | 1.9 | 0.6 |
| benzo[a]pyrene (a) | 2.2–4.7 | 3.0 | 0.9 |
| indeno[123‐cd]pyrene | < 0.5 (b) –1.1 | 0.8 | 0.2 |
| dibenzo[a,h]pyrene | < 1 (b) | < 1 (b) | – |
| benzo[g,h,i]perylene | 1.0–2.8 | 1.6 | 0.7 |
| dibenzo[a,l]pyrene | < 1 (b) | < 1 (b) | – |
| dibenzo[a,i]pyrene | < 1 (b) | < 1 (b) | – |
| dibenzo[a,h]anthracene | < 0.5 (b) | < 0.5 (b) | – |
| dibenzo[a,e]pyrene | < 1 (b) | < 1 (b) | – |
| cyclopenta[cd]pyrene | 7.3–26.3 | 17.5 | 7.8 |
| 5‐methylchrysene | < 1 (b) | < 1 (b) | – |
| benzo[c]fluorene | 44.7–75.7 | 57.6 | 11.5 |
| PAH4 | 16.8–34.6 (c) | 21.4 | 4.2 (d) |
PAH: polycyclic aromatic hydrocarbon; SD: standard deviation.
PAHs printed in bold are included in the calculation of ‘PAH4’, which is used for the evaluation of the exposure to these contaminants (see Section 3.3.3.2).
Value below the corresponding Limit of Quantification (LOQ).
Values for range of PAH4 represent the PAH4 values for the individual batches.
Value calculated as the square root of the summed variances.
The levels of benzo[a]pyrene and benzo[a]anthracene are below their respective limits of 10 and 20 μg/kg as laid down in the Regulation (EC) No 2065/2003.
3.1.2.6. Batch‐to‐batch variability
The batch‐to‐batch variability of the 20 principal volatile constituents of the batches presented in Table 1 was investigated by GC–MS and GC‐FID. The Panel considered that the batch‐to‐batch variability of the six investigated production batches (Table 6), with production dates spanning 15 months, was acceptable, also taking into account information on the other measured parameters evaluated in this opinion. Information on the criteria underlying the selection of these batches was not provided.
Table 6.
Batch‐to‐batch variability of the Primary Product
| CAS no. | Chemical name | Batch no. (production date) | Average (wt%) | SD | RSD (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 13390 # (12‐07‐2021) | 14185 (16‐11‐2021) | 14300 (30‐09‐2020) | 14301 (14‐12‐2020) | 14302 (17‐08‐2020) | 14303 (30‐06‐2021) | |||||
| 91‐10‐1 | 2,6‐dimethoxyphenol | 4.1 | 4.1 | 5.5 | 6.7 | 5.3 | 4.1 | 4.9 | 1.1 | 21.4 |
| 6638‐05‐7 | 4‐methyl‐2,6‐dimethoxyphenol (2,6‐dimethoxy‐4‐ methylphenol) | 2.6 | 3.3 | 3.9 | 4.0 | 3.7 | 3.2 | 3.4 | 0.5 | 15.2 |
| 90‐05‐1 | 2‐methoxyphenol | 2.0 | 2.0 | 2.5 | 3.1 | 2.3 | 2.0 | 2.3 | 0.4 | 18.8 |
| 93‐51‐6 | 2‐methoxy‐4‐methylphenol (creosol/methylguaiacol) | 1.9 | 2.2 | 2.4 | 2.5 | 2.3 | 2.2 | 2.3 | 0.2 | 9.2 |
| 14059‐92‐8 | 4‐ethyl‐2,6‐dimethoxyphenol | 1.6 | 2.1 | 2.6 | 2.6 | 2.4 | 2.1 | 2.2 | 0.4 | 17.1 |
| 20675‐95‐0 | 2,6‐dimethoxy‐4‐(2‐propenyl)‐phenol, E | 1.0 | 1.3 | 1.7 | 1.2 | 2.1 | 1.4 | 1.4 | 0.4 | 27.2 |
| 105‐67‐9 | 2,4‐dimethylphenol | 1.1 | 1.4 | 1.3 | 1.2 | 1.3 | 1.4 | 1.3 | 0.1 | 9.1 |
| 2785‐89‐9 | 4‐ethylguaiacol (4‐ethyl‐2‐methoxyphenol) | 1.0 | 1.2 | 1.3 | 1.2 | 1.3 | 1.2 | 1.2 | 0.1 | 9.1 |
| 95‐48‐7 | 2‐methylphenol | 1.1 | 1.2 | 0.8 | 1.2 | 0.9 | 1.1 | 1.1 | 0.1 | 15.6 |
| 108‐95‐2 | phenol | 1.0 | 1.0 | 0.8 | 1.0 | 0.9 | 1.0 | 0.9 | 0.1 | 8.8 |
| 5932‐68‐3 | trans‐isoeugenol | 0.6 | 0.9 | 1.0 | 0.9 | 1.2 | 0.9 | 0.9 | 0.2 | 21.2 |
| 6627‐88‐9 | 4‐allyl‐2,6‐dimethoxyphenol (2,6‐dimethoxy‐4‐(2‐propenyl)‐phenol) | 0.6 | 0.8 | 1.1 | 1.0 | 1.1 | 0.8 | 0.9 | 0.2 | 22.2 |
| 97‐53‐0 | eugenol | 0.5 | 0.6 | 0.7 | 0.6 | 0.7 | 0.6 | 0.6 | 0.1 | 12.2 |
| 576‐26‐1 | 2,6‐dimethylphenol | 0.63 | 0.64 | 0.60 | 0.61 | 0.60 | 0.61 | 0.61 | 0.02 | 2.6 |
| 26624‐13‐5 | 4‐propenyl‐2,6‐dimethoxyphenol, Z | 0.51 | 0.63 | 0.68 | 0.43 | 0.68 | 0.62 | 0.59 | 0.1 | 16.9 |
| 1121‐05‐7 | 2,3‐dimethyl‐2‐cyclopenten‐1‐one | 0.51 | 0.60 | 0.59 | 0.61 | 0.60 | 0.58 | 0.58 | 0.04 | 6.6 |
| 108‐39‐4 | 3‐methylphenol | 0.61 | 0.63 | 0.43 | 0.60 | 0.55 | 0.63 | 0.58 | 0.08 | 13.2 |
| 527‐60‐6 | 2,4,6‐trimethyl phenol | 0.55 | 0.57 | 0.57 | 0.55 | 0.56 | 0.52 | 0.55 | 0.02 | 3.6 |
| 2785‐87‐7 | 2‐methoxy‐4‐propylphenol | 0.48 | 0.46 | 0.50 | 0.44 | 0.46 | 0.43 | 0.46 | 0.02 | 5.0 |
| 98‐00‐0 | furfuryl alcohol (2‐furanmethanol) | 0.46 | 0.40 | 0.33 | 0.38 | 0.41 | 0.38 | 0.39 | 0.04 | 10.3 |
wt: weight; SD: standard deviation; RSD: relative standard deviation.
Batch tested in the newly submitted genotoxicity studies.
3.1.2.7. Solubility and particle size
According to the applicant Scansmoke SEF7525 is a viscous liquid (Section 3.1.2.3) obtained from the combination of two individual fractions. One of the fractions (SEF1) is obtained by subjecting the ether extract of the alkalised tar material to vacuum distillation followed by treatment with active carbon and filtration (1 mm). However, in order to obtain the second fraction (SEF2) the ether extract of the acidified tar material is not subjected to distillation but only to treatment with active carbon and filtration, which is not sufficient to exclude the presence of particles. Therefore, EFSA requested the applicant to clarify the potential presence of small particles including nanoparticles, in line with the EFSA ‘Guidance on technical requirements for regulated food and feed product applications to establish the presence of small particles including nanoparticles’ (EFSA Scientific Committee, 2021a).
The applicant submitted a study report based on dynamic light scattering (DLS) analysis (Documentation provided to EFSA No. 2). The DLS profiles of the Primary Product and of the Primary Product in seven selected solvents (i.e. water, propylene glycol, ethanol, acetic acid, cyclohexane, diethyl ether and medium chain triglycerides (MCT)) were submitted. The justification for selecting these solvents was not provided, although it can be noted that they span a polarity range and could be used as carriers of the Primary Product in foods. DLS profiles of four pure solvents (i.e. water, ethanol, propylene glycol and MCT) were also provided. As a positive control, the applicant added platinum nanoparticle clusters (Pt‐NP) to the pure solvents at a concentration of 15 μg/mL (Documentation provided to EFSA No. 2). The applicant stated that the solubility of the Primary Product in water and cyclohexane was poor, whereas its solubility in propylene glycol, ethanol, acetic acid and MCT was high. The applicant further stated that in one part of diethyl ether (1:1), Scansmoke SEF7525 was fully soluble, but an increase of diethyl ether led to precipitation. However, quantitative data on the solubility of the Primary Product in the selected solvents was not provided. Based on the analyses performed, the applicant concluded that the Primary Product is a true liquid, and consequently, does not contain small particles, including nanoparticles.
The Panel noted that the DLS profiles of the Primary Product and of the Primary Product in propylene glycol (1:5) exhibit one peak in the range of around 1 nm, which is likely to be an artefact due to the limitation of the DLS method. However, when the Primary Product is mixed with the other solvents, the DLS profiles show various peaks up to 10,000 nm, which do not correspond to the peaks observed in the DLS profiles of the four pure solvents for which data were submitted.
The Panel noted that the evidence provided is not sufficient to prove the absence of a fraction of small particles in the Primary Product since:
the presence of peaks in the DLS profiles of the Primary Product when mixed with the selected solvents is not explained by the applicant, and may indicate the presence of particulate matter, e.g. the DLS profile of the Primary Product in diethyl ether (1:1) shows various peaks from 10 to 10,000 nm;
the DLS profile of the pure diethyl ether was not provided for comparison with the DLS profile of the Primary Product in diethyl ether (1:1). In addition, the applicant claimed that the increase of diethyl ether in the mixture with the Primary Product would lead to precipitation. This contradicts the assumption that the Primary Product is fully soluble in diethyl ether;
the Pt‐NP do not represent an appropriate positive control, as they have different light scattering properties compared to the organic particles potentially present (if any) in the Primary Product. Moreover, since large nanoparticles produce very high DLS signal, this could have covered the signals belonging to smaller particles, thereby reducing the sensitivity of the testing. Since the size of the Pt‐NP was not specified, it is not possible to judge if the Pt‐NP positive control would have introduced DLS signalling bias.
Based on the above, the Panel concluded that more evidence is needed to exclude the presence of nanoparticles in the Primary Product. This creates a non‐standard uncertainty with respect to the solubility and particle size of the Primary Product (see Section 2.2 of this opinion and Table G.1 of the EFSA guidance document on smoke flavouring (EFSA FAF Panel, 2021)).
3.1.3. Specifications
The applicant provided the required product specification data and reported that the Primary Product Scansmoke SEF7525 is manufactured within its proposed specifications (Documentation provided to EFSA No. 1 and 2). Information on parameters relevant for the specifications has been compiled by the Panel in Table 7.
Table 7.
Relevant information for specifications of the Primary Product
| Specifications for Scansmoke SEF7525 as proposed by the applicant | Specifications as reported in in EFSA CEF Panel (2009) | Specifications as laid down in Regulation (EU) No 1321/2013 | |
|---|---|---|---|
| Description | Smoke flavouring Primary Product obtained from a combination of different wood species. | ||
| Source material | |||
|
Red oak (Quercus rubra; 35%), white oak (Quercus alba; 35%), maple (Acer saccharum; 10%), beech (Fagus grandifolia; 10%) and hickory (Carya ovata; 10%) | Red oak (Quercus rubra; 35%), white oak (Quercus alba; 35%), maple (Acer saccharum; 10%), beech (Fagus grandifolia; 10%) and hickory (Carya ovata; 10%) | |
| Identity parameters: | |||
|
|||
|
n.d. | ||
|
n.d. | ||
|
1.14–1.16 g/mL | ||
|
1.50–1.70 | ||
| Chemical composition: | |||
|
|||
|
0.43–1.64 wt% (0.07–0.27 mEq/g) (as acetic acid) |
0.09–0.25 mEq/g | 0.09–0.25 mEq/g (as acetic acid) |
|
4.0–6.0 wt% (as furfural) |
1.2–3.0 wt% | 1.2–3.0 wt% |
|
7.0–12.0 wt% (as syringol) |
8–12 wt% | 8–12 wt% |
|
0.3–0.9 wt% | 0.3–0.9 wt% | 0.3–0.9 wt% |
| 20 principal constituents of the volatile fraction | See Table 4 | ||
| Purity: | |||
|
< 10 μg/kg | < 10 μg/kg | |
|
< 20 μg/kg | < 20 μg/kg | |
|
|||
|
< 3 mg/kg | < 1 mg/kg | < 3 mg/kg |
|
< 1 mg/kg | < 0.025 mg/kg | < 1 mg/kg |
|
< 5 mg/kg | < 0.1 mg/kg | < 5 mg/kg |
|
< 1 mg/kg | < 0.01 mg/kg | < 1 mg/kg |
wt: weight; n.d.: not determined. The applicant did not determine staining index and pH of the Primary Product.
The Panel noted that in the specifications proposed by the applicant, the woods used as raw material are presented in a fixed percentage. However, the Primary Product is manufactured using the wood materials in the range of proportion described in Section 3.1.1.1 This is confirmed by the applicant following an additional data request from EFSA (Documentation provided to EFSA No. 2).
The Panel noted that the analytical data for the batches analysed indicated that actual concentrations of toxic elements and PAHs, reported in Tables 3 and 5, respectively, are lower than the currently proposed limits (Table 7), being the same as the limits laid down in the respective Regulations (i.e. Regulation (EU) No 1321/2013 for toxic elements and Regulation (EC) No 2065/2003 for benzo[a]pyrene and benzo[a]anthracene).
With regard to carbonyl compounds, the applicant informed that the difference between the concentration range of carbonyls in the current application and in the technical dossier evaluated by EFSA formerly (EFSA CEF Panel, 2009) is not due to changes in the manufacturing process but related to the measurement technique (Documentation provided to EFSA No. 1). In addition, based on the newly provided compositional data, the Panel considered that the proposed extension of the range of phenols and acids (see Table 7) is justified.
3.1.4. Stability and fate in food
Stability tests were performed for one of the batches of the Primary Product (batch no. 14185) listed in Table 1.
A stability test was performed with the batch stored for 6 months at temperature between 7 and 25°C, and approximately 60% of relative humidity. In addition, the batch was subjected to an accelerated stability test (forced ageing) at 40°C for 5.5 months. The storage stability of the Primary Product Scansmoke SEF7525 was assessed by monitoring the volatile constituents of the Primary Product; based on the observed relative standard deviations (on average approximately 10%), their concentrations were sufficiently stable under both storage conditions. On the basis of these data, the Panel considered the stability of the Primary Product upon storage under the intended conditions not to be of concern. No data on the stability of the Primary Product in commercial formulations or in the proposed food categories were provided.
3.2. Proposed uses and use levels
The applicant applied for a renewal of authorisation of the Primary Product Scansmoke SEF7525 for use in the food at the proposed maximum and expected typical use levels as presented in Table 8.
Table 8.
Proposed maximum and expected typical use levels of Primary Product (mg/kg) in food categories according to Annex II of Regulation (EC) No 1333/2008 7
| Food category number | Food category name | Restrictions/Exceptions | Proposed maximum use levels (mg/kg) (a) | Expected typical use levels (mg/kg) (a) |
|---|---|---|---|---|
| 1.4 | Flavoured fermented milk products including heat‐treated products | 2 | 2 | |
| 1.7 | Cheese and cheese products | 160 | 100 | |
| 1.8 | Dairy analogues, including beverage whiteners | 160 | 140 | |
| 2.2.2 | Other fat and oil emulsions including spreads as defined by Regulation (EC) No 1234/2007 and liquid emulsions | 50 | 20 | |
| 3 | Edible ices | 2 | 2 | |
| 4.1.3 | Frozen fruits and vegetables | Vegetables only | 50 | 45 |
| 4.2.2 | Fruits and vegetables in vinegar, oil, brine | 50 | 45 | |
| 4.2.3 | Canned or bottled fruits and vegetables | Vegetables only | 50 | 45 |
| 4.2.5.4 | Nut butters and nut spreads | 50 | 45 | |
| 5.1 | Cocoa and Chocolate products as covered by Directive 2000/36/EC | 2 | 2 | |
| 5.4 | Decorations, coatings and fillings, except fruit‐based fillings covered by category 4.2.4 | 80 | 75 | |
| 6.4.5 | Fillings of stuffed pasta (ravioli and similar) | 50 | 45 | |
| 6.5 | Noodles | 50 | 45 | |
| 6.6 | Batters | 50 | 45 | |
| 6.7 | Pre‐cooked or processed cereals | 50 | 45 | |
| 7.2 | Fine bakery wares | 80 | 25 | |
| 8.2 | Meat preparations as defined by Regulation (EC) No 853/2004 of the European Parliament and of the Council | 160 | 150 | |
| 8.3 | Meat products | 160 | 150 | |
| 9.2 | Processed fish and fishery products including crustaceans and molluscs | 160 | 150 | |
| 9.3 | Fish roe | 160 | 150 | |
| 10.2 | Processed eggs and egg products | 10 | 10 | |
| 12.2.2 | Seasonings and condiments | 180 | 120 | |
| 12.5 | Soups and broths | 50 | 45 | |
| 12.6 | Sauces | 50 | 45 | |
| 12.7 | Salads and savoury based sandwich spreads | 50 | 50 | |
| 12.9 | Protein products, excluding products covered in category 1.8 | 160 | 100 | |
| 14.1.4 | Flavoured drinks | 50 | 30 | |
| 14.1.5 | Coffee, coffee and chicory extracts, tea, herbal‐ and fruit‐infusions; coffee substitutes, coffee mixes and mixes for ‘hot beverages’ | 50 | 45 | |
| 14.2 | Alcoholic beverages, including alcohol‐free and low‐alcohol counterparts | 50 | 10 | |
| 15.1 | Potato‐, cereal‐, flour‐ or starch‐based snacks | 80 | 70 | |
| 15.2 | Processed nuts | 80 | 50 | |
| 16 | Desserts excluding products covered in category 1, 3 and 4 | 10 | 10 |
Use levels are provided for the foods as consumed.
The proposed maximum and expected typical use levels were used to assess the dietary exposure to this Primary Product (see Section 3.3.2). 7
3.3. Exposure
3.3.1. Food consumption data used for the exposure assessment
The food consumption data used for the exposure assessment are from the EFSA Comprehensive European Food Consumption Database. 8 This database contains food consumption data at the level of the individual consumer from the most recent national dietary surveys carried out in EU countries and includes the currently best available food consumption data across the EU. These data cover infants (from 0 weeks of age), toddlers (1–2 years), children (3–9 years), adolescents (10–17 years), adults (18–64 years) and the elderly (65 years and older). As these data were collected by different methodologies, direct country‐to‐country comparisons of exposure estimates based on these data may not be appropriate.
The dietary exposure to the Primary Product was calculated by the applicant and EFSA using Food Additive Intake Model (FAIM, version 2.1). The food consumption data in FAIM (version 2.1) used in the exposure assessment were based on the version of the Comprehensive Database that was published in July 2021. These data cover 42 dietary surveys carried out in 22 EU countries (Table 9).
Table 9.
Population groups and countries considered for the exposure estimates of the Primary Product obtained with FAIM
| Population | Age range | Countries with food consumption surveys covering more than 1 day |
|---|---|---|
| Infants | From 12 weeks up to and including 11 months of age | Bulgaria, Cyprus, Denmark, Estonia, Finland, France, Germany, Italy, Latvia, Portugal, Slovenia |
| Toddlers (a) | From 12 months up to and including 35 months of age | Belgium, Bulgaria, Cyprus, Denmark, Estonia, Finland, France, Germany, Hungary, Italy, Latvia, the Netherlands, Portugal, Slovenia, Spain |
| Children (b) | From 36 months up to and including 9 years of age | Austria, Belgium, Bulgaria, Cyprus, Czechia, Denmark, Estonia, Finland, France, Germany, Greece, Hungary, Italy, Latvia, the Netherlands, Portugal, Spain, Sweden |
| Adolescents | From 10 years up to and including 17 years of age | Austria, Belgium, Cyprus, Czechia, Denmark, Estonia, Finland, France, Germany, Greece, Hungary, Italy, Latvia, the Netherlands, Portugal, Romania, Slovenia, Spain, Sweden |
| Adults | From 18 years up to and including 64 years of age | Austria, Belgium, Croatia, Cyprus, Czechia, Denmark, Estonia, Finland, France, Germany, Greece, Hungary, Ireland, Italy, Latvia, the Netherlands, Portugal, Romania, Slovenia, Spain, Sweden |
| The elderly (b) | From 65 years of age and older | Austria, Belgium, Cyprus, Denmark, Estonia, Finland, France, Germany, Greece, Hungary, Ireland, Italy, Latvia, the Netherlands, Portugal, Romania, Slovenia, Spain, Sweden |
The term ‘toddlers’ in the Comprehensive Database (EFSA, 2011) corresponds to ‘young children’ (from 12 months up to and including 35 months of age) in Regulations (EC) No 1333/2008 and (EU) No 609/2013 9 .
In FAIM, the terms ‘children’ and ‘the elderly’ correspond, respectively, to ‘other children’ and the merge of ‘elderly’ and ‘very elderly’ in Comprehensive Database (EFSA, 2011).
The food consumption data from the Comprehensive Database in FAIM are codified according to the food categories as presented in Annex II, Part D, of Regulation (EC) No 1333/2008, which is the relevant regulation for the food categories of the smoke flavourings. 9
3.3.2. Exposure assessment to the Primary Product using FAIM
Using FAIM, dietary exposure to the Primary Product was calculated by multiplying the relevant use level for each food category with its respective consumption amount for each individual. This was done for all individuals in the surveys (i.e. the estimates are not based on consumers only). The exposures per food category were subsequently added and divided by the individual body weight (bw) (as registered in the consumption survey) to derive an individual total exposure per day expressed per kilogram bw. These exposure estimates were averaged over the number of survey days in the survey, resulting in an individual average exposure per day. Dietary surveys with only 1 day per subject were excluded as they are not considered adequate to assess repeated exposure. The calculations resulted in distributions of individual exposure per survey and population group. Based on these distributions, the mean and the 95th percentile of exposure were calculated per survey and population group. The 95th percentile of exposure was only calculated for those population groups with a sufficiently large sample size to obtain a reliable estimate (EFSA, 2011).
3.3.2.1. Exposure assessment using FAIM
The applicant provided estimates of dietary exposure to the Primary Product using FAIM, based on the proposed maximum (proposed maximum use level exposure assessment scenario) and expected typical use levels (expected typical use level exposure assessment scenario) (Documentation provided to EFSA No. 1). These estimates were re‐calculated by EFSA following a submission of updated uses and use levels from the applicant (Documentation provided to EFSA No. 2).
In FAIM, use levels were linked to the corresponding food categories according to the instructions provided for its use. 10 Furthermore, all foods belonging to the food categories (FC) were included in the assessment without applying the restrictions/exceptions as indicated in Table 8. This tool does not allow to include or exclude specific foods from the exposure assessment. See Annex A1 for the food categories and use levels considered in FAIM.
Exposure estimates using FAIM
In Table 10, the dietary exposure estimates of the Primary Product with FAIM are presented.
Table 10.
Summary of dietary exposure to the Primary Product from its proposed maximum and expected typical use levels as a smoke flavouring in six population groups and estimated with FAIM (minimum‐maximum across the dietary surveys in mg/kg body weight (bw) per day)
| Infants (12 weeks–11 months) (n = 11/9) | Toddlers (12–35 months) (n = 15/13) | Children (3–9 years) (n = 19/19) | Adolescents (10–17 years) (n = 21/20) | Adults (18–64 years) (n = 22/22) | The elderly (≥ 65 years) (n = 22/21) | |
|---|---|---|---|---|---|---|
| Proposed maximum use level exposure assessment scenario | ||||||
| Mean | 0.6–3.0 | 1.4–3.8 | 1.4–3.7 | 0.7–2.0 | 0.7–1.5 | 0.6–1.4 |
| 95th percentile | 1.7–10.1 | 2.5–9.4 | 2.3–8.6 | 1.4–4.5 | 1.3–3.3 | 1.1–2.2 |
| Expected typical use level exposure assessment scenario | ||||||
| Mean | 0.5–2.6 | 1.2–3.3 | 1.0–3.2 | 0.5–1.7 | 0.6–1.3 | 0.4–1.1 |
| 95th percentile | 1.5–9.1 | 2.2–8.2 | 1.8–7.6 | 1.0–3.9 | 1.0–2.9 | 0.8–2.0 |
n: number of surveys from which a mean/P95 could be calculated.
At the proposed maximum use levels, the mean exposure to the Primary Product from its use as a smoke flavouring ranged from 0.6 mg/kg bw per day in infants and the elderly to 3.8 mg/kg bw per day in toddlers. The 95th percentile of exposure to the Primary Product ranged from 1.1 mg/kg bw per day in the elderly to 10.1 mg/kg bw per day in infants.
At the expected typical use levels, the mean exposure to the Primary Product from its use as a smoke flavouring ranged from 0.4 mg/kg bw per day in the elderly to 3.3 mg/kg bw per day in toddlers. The 95th percentile of exposure to the Primary Product ranged from 0.8 mg/kg bw per day in the elderly to 9.1 mg/kg bw per day in infants.
Because the maximum and typical use levels were similar (Table 10), the exposure results of the two exposure scenarios were almost the same.
The Primary Product is requested for renewal of authorisation in 32 food categories (Table 8). For all these 32 food categories considered, it was assumed that 100% of the foods belonging to these food categories will contain the Primary Product at the proposed maximum or expected typical use levels. As it is unlikely that the Primary Product will be added to all foods and given the restrictions/exceptions for two food categories (Table 8), the Panel considered that the calculated exposure to the Primary Product using FAIM is an overestimation of the expected exposure in EU countries if this Primary Product is used at the proposed maximum or expected typical use levels.
Additionally, overall sources of standard uncertainties (Annex A6) also contributed to an overestimation of the exposure.
Detailed results per population group and survey are presented in Annexes A2 (Proposed maximum use level exposure assessment scenario) and A3 (Expected typical use level exposure assessment scenario).
Main food category contributing to exposure to the Primary Product using FAIM
Under the conservative assumptions mentioned above, the main food categories contributing to the total mean exposure to the primary product for both exposure scenarios contributing to at least 30% to the total mean exposure in at least one population group in one survey, listed in order of the number of the FCs, are:
FC 01.7.1 Unripened cheese excluding products falling in category 16.
FC 04.1 Unprocessed fruit and vegetables.
FC 12.5 Soups and broths.
FC 14.1.4.1 Flavoured drinks with sugar.
FC 14.1.5.1 Coffee, coffee extracts.
FC 14.1.5.2 Other.
Considering the conservative nature of the underlying assumption that 100% of the foods within the food categories (Table 8) contain the Primary Product, the Panel emphasises that the main food categories listed here may not reflect the food categories that contribute most to the exposure in real life.
Detailed results of the contributing food categories are presented in Annexes A4 (proposed maximum use level exposure assessment scenario) and A5 (expected typical use level exposure assessment scenario).
3.3.3. Anticipated exposure to impurities in the Primary Product
The potential exposure to the impurities arsenic, lead, cadmium, mercury and PAHs (as PAH4) from the use of the Primary Product can be calculated by assuming that they are present in the Primary Product up to a limit value and then by calculating pro‐rata to the estimates of exposure to the Primary Product itself.
With regard to the dietary exposure to the Primary Product, the Panel considered the highest mean and the highest 95th percentile exposure estimates resulting from the exposure assessment using FAIM among the different population groups, i.e. 3.8 mg/kg bw per day for toddlers and 10.1 mg/kg bw per day for infants, respectively (Table 10).
The level of the impurities in the Primary Product combined with the estimated exposure to the Primary Product (Table 10) can be used to estimate the exposure to these impurities. This exposure can then be compared with reference points (RP, i.e. lower limit of the benchmark dose (BMDL) for arsenic, lead and PAH4) or health‐based guidance values (HBGV, i.e. tolerable weekly intake (TWI) for cadmium and mercury) for the undesirable impurities present in the Primary Product (Table 11).
Table 11.
Reference points/health‐based guidance values for the impurities potentially present in the Primary Product
| Impurity/constituent/HBGV/RP | Basis/Reference |
|---|---|
| Arsenic (As)/0.3–8 μg/kg bw per day (BMDL01) | The reference point is based on a range of benchmark dose lower confidence limit (BMDL01) values between 0.3 and 8 μg/kg body weight (bw) per day identified for cancers of the lung, skin and bladder, as well as skin lesions. MOE should be at least 10,000 if the reference point is based on carcinogenicity in animal studies. However, as the BMDL for As is derived from human studies, an interspecies extrapolation factor (i.e. 10) is not needed, i.e. a MOE of 1,000 would be sufficient (EFSA CONTAM Panel, 2009a; EFSA Scientific Committee, 2012). |
| Cadmium (Cd)/2.5 μg/kg bw per week (TWI) | The derivation of the reference point is based on a meta‐analysis to evaluate the dose–response relationship between selected urinary cadmium and urinary beta‐2‐microglobulin as the biomarker of tubular damage recognised as the most useful biomarker in relation to tubular effects. A group‐based BMDL5 of 4 μg Cd/g creatinine for humans was derived. A chemical specific adjustment factor of 3.9 was applied to account for human variability in urinary cadmium within each dose‐subgroup in the analysis resulting in a reference point of 1.0 μg Cd per g creatinine. In order to remain below 1 μg Cd/g creatinine in urine in 95% of the population by age 50. The average daily dietary cadmium intake should not exceed 0.36 μg Cd/kg bw. Corresponding to a weekly dietary intake of 2.5 μg Cd/kg bw (EFSA CONTAM Panel, 2009b). |
| Lead (Pb)/0.5 μg/kg bw per day (BMDL01) | The reference point is based on a study demonstrating perturbation of intellectual development in children with the critical response size of 1 point reduction in IQ. The EFSA CONTAM Panel mentioned that a 1‐point reduction in IQ is related to a 4.5% increase in the risk of failure to graduate from high school and that a 1‐point reduction in IQ in children can be associated with a decrease of later productivity of about 2%. A risk cannot be excluded if the exposure exceeds the BMDL01 (MOE lower than 1) (EFSA CONTAM Panel, 2010). |
| Mercury (Hg)/4 μg/kg bw per week (TWI) | The HBGV was set using kidney weight changes in male rats as the pivotal effect. Based on the BMDL10 of 0.06 mg/kg bw per day, expressed as mercury, and an uncertainty factor of 100 to account for inter and intra species differences, with conversion to a weekly basis and rounding to one significant figure, a TWI for inorganic mercury of 4 μg/kg bw per week, expressed as mercury was established (EFSA CONTAM Panel, 2012). |
| PAH4/340 μg/kg bw per day (BMDL10) | Polycyclic aromatic hydrocarbons (PAHs) are considered genotoxic and carcinogenic. The reference point is based on a carcinogenicity study by Culp et al. (1998), as reported by the EFSA CONTAM Panel (2008), who concluded that PAH4 (i.e. the sum of benzo[a]anthracene, benzo[a]pyrene, benzo[b]fluoranthene and chrysene) is a suitable indicator for the occurrence and toxicity of PAHs in food. The MOE should be at least 10,000 (EFSA CONTAM Panel, 2008). |
HBGV: health‐based guidance value; RP: reference point; BMDL01: lower confidence limit of the benchmark dose associated with a 1% extra risk for tumours (EFSA Scientific Committee, 2017a); BMDL10: lower confidence limit of the benchmark dose associated with a 10% extra risk for tumours (EFSA Scientific Committee, 2017a); TWI: tolerable weekly intake; MOE: margin of exposure.
The risk assessment of the undesirable impurities helps to determine whether there could be a possible health concern if these impurities were present at their limit values in the Primary Product. The assessment is performed by calculating the MOE by dividing the reference point (i.e. BMDL, Table 11) by the exposure estimate for an impurity (Table 10), or by estimating the contribution of the exposure to an impurity due to the use of Primary Product to the HBGV (expressed as percentage of the HBGV).
3.3.3.1. Toxic elements
The results of the analysis of arsenic, cadmium, lead and mercury in six batches of the Primary Product were reported (Table 3).
The applicant proposed maximum limits for these toxic elements, which are the same as the limits in the current EU specifications (Table 7). The Panel noted that the actual measured levels of the toxic elements in commercial samples of the Primary Product were substantially lower than these limits.
The Panel assessed the risk that would result if these toxic elements were present in the Primary Product according to two concentration scenarios: (i) at the current limits in the EU specifications and (ii) at the reported LOQ multiplied by a factor of 10, since the analytical data were below the LOQs (see Table 3); this to account for standard variability with respect to representativeness, homogeneity and analytical measurement.
The outcome of the risk assessment for the two concentration scenarios and based on the highest mean and the highest 95th percentile exposure estimates among the different population groups (see Section 3.3.2) is presented in Table 12.
Table 12.
Risk assessment for four toxic elements present in the Primary Product according to two concentration scenarios, using the reference points/health‐based guidance values as provided in Table 11
| Exposure to Scansmoke SEF7525 (mg/kg bw/day) | (i) Considering the presence of toxic elements at the current EU specifications limits for Scansmoke SEF7525 | |||
|---|---|---|---|---|
| MOE for As at 3 mg/kg | % of the TWI for Cd at 1 mg/kg | MOE for Pb at 5 mg/kg | % of the TWI for Hg at 1 mg/kg | |
| 3.8 (a) | 26.3–701.8 | 1.1 | 26.3 | 0.7 |
| 10.1 (b) | 9.9–264 | 2.8 | 9.9 | 1.8 |
| (ii) Considering the presence of toxic elements at the reported LOQs multiplied by a factor of 10 | ||||
| MOE for As at 1 mg/kg | % of the TWI for Cd at 0.1 mg/kg | MOE for Pb at 0.5 mg/kg | % of the TWI for Hg at 0.05 mg/kg | |
| 3.8 (a) | 78.9–2105 | 0.1 | 263.2 | 0.03 |
| 10.1 (b) | 29.7–792.1 | 0.3 | 99.0 | 0.1 |
bw: body weight; MOE: margin of exposure; TWI: tolerable weekly intake; LOQ: limit of quantification.
Highest mean exposure level among the different population groups (proposed maximum use level exposure assessment scenario – toddlers (Table 10)).
Highest 95th percentile exposure level among the different population groups (proposed maximum use level exposure assessment scenario – infants (Table 10)).
When considering the current limits of the EU specifications (scenario (i) in Table 12), the Panel concluded that for arsenic the ranges of the calculated MOE values were insufficient, i.e. below the target value of 1,000 (Table 11). For the other three toxic elements (cadmium, lead and mercury), the EU current specifications limit values do not give rise to safety concerns.
When considering the LOQs multiplied by a factor of 10 (scenario (ii) in Table 12), the Panel concluded that for arsenic (a) the lower end of the range for the highest mean and (b) the range for the highest 95th percentile of the calculated MOE values were insufficient, i.e. below the target value of 1,000 (Table 11). The presence of the other toxic elements in the Primary Product does not give rise to concern.
Overall, the Panel considered that the limits in the EU specifications for arsenic, cadmium, lead and mercury should be established based on actual levels in the commercial Primary Product. If the European Commission decides to revise the current limits in the EU specifications, the estimated exposure to the toxic elements as described above could be considered.
3.3.3.2. Polycyclic aromatic hydrocarbons (PAHs)
The results of the analysis of 16 PAHs were reported by the applicant for six batches of the Primary Product (Table 5).
The proposed limits for two of these PAHs (i.e. benzo[a]pyrene and benzo[a]anthracene) are below their respective limits of 10 and 20 μg/kg as laid down in Regulation (EC) No 2065/2003. However, the Panel noted that the actual measured levels for benzo[a]pyrene and benzo[a]anthracene in the Primary Product (Table 5) were lower than the current limits in Regulation (EC) No 2065/2003.
According to the data submitted by the applicant, the Panel considered the maximum reported level of PAH4 in the Primary Product, i.e. 34.6 μg/kg (Table 5). Based on this level, the Panel assessed the risk that would result if PAH4 were present in the Primary Product: (i) at the specifications limits for the sum of benzo[a]pyrene and benzo[a]anthracene in the Primary Product, i.e. 30 μg/kg, as proposed by the applicant (Table 7) and setting the concentration of the other two members of PAH4 (chrysene and benzo[b]fluoranthene) at zero for the purpose of this concentration scenario, and also (ii) at the maximum reported level of PAH4 in six batches of the Primary Product (Table 5). The outcome of the risk assessment for the two concentration scenarios and based on the highest mean and the highest 95th percentile FAIM exposure estimates among the different population groups (see Section 3.3.2) is presented in Table 13.
Table 13.
Risk assessment for PAH4, i.e. benzo[a]anthracene, benzo[a]pyrene, benzo[b]fluoranthene and chrysene in the Primary Product according to two concentration scenarios, using the reference points/health‐based guidance values as provided in Table 11
| Exposure to Scansmoke SEF7525 (mg/kg bw/day) | MOE for PAH4 |
|---|---|
| (i) Considering the presence of PAH4 at the sum of the specifications limits for benzo[a]pyrene and benzo[a]anthracene in Scansmoke SEF7525 (30 μg/kg) | |
| 3.8 (a) | 2.98 × 106 |
| 10.1 (b) | 1.12 × 106 |
| (ii) Considering the presence of PAH4 at their maximum reported level in Scansmoke SEF7525 (34.6 μg/kg) | |
| 3.8 (a) | 2.59 × 106 |
| 10.1 (b) | 9.73 × 105 |
bw: body weight: MOE: margin of exposure.
Highest mean exposure level among the different population groups (proposed maximum use level exposure assessment scenario – toddlers (Table 10)).
Highest 95th percentile exposure level among the different population groups (proposed maximum use level exposure assessment scenario – infants (Table 10)).
The Panel noted that the resulting MOEs for PAH4 were far above the target value of 10,000 for both concentration scenarios and both exposure estimates of the Primary Product (EFSA Scientific Committee, 2012) (Table 11).
Furthermore, the Panel noted that at the highest proposed maximum use level of the Primary Product in any of the food categories, i.e. 160 mg/kg food (Table 8), and the maximum reported level of PAH4 in the Primary product, i.e. 34.6 μg/kg, the concentration of PAH4 in food would be 6.2 × 10−3 μg/kg food, which is far below the lowest maximum level (ML) of these contaminants in any of the foodstuffs as listed in Regulation (EU) 2023/915 11 (i.e. 1 μg PAH4/kg food).
3.4. Genotoxicity data
The present evaluation is conducted in line with the applicable EFSA guidance on smoke flavourings (EFSA FAF Panel, 2021) which encompasses all the EFSA guidance documents on genotoxicity (EFSA Scientific Committee, 2011, 2017b, 2019, 2021b). These documents were not available at the time when the smoke flavourings were evaluated previously by the CEF Panel. In addition, for the assessment of the renewal applications, the reliability and relevance of all submitted genotoxicity studies were evaluated by the FAF Panel (see Sections 3.4.1 and 3.4.2) based on the criteria described in Appendix C.
3.4.1. Genotoxicity assessment of the individual components
The 44 identified components of Scansmoke SEF7525 (SF‐004) (i.e. 41 identified and quantified components as listed in the Appendix A, Table A.1 plus 3 identified, but not quantified, components as described in Section 3.1.2.4.2) were evaluated individually for genotoxicity considering first the data available from the literature as provided by the applicant and then, in the absence of relevant information from the literature, considering the in silico information/data submitted by the applicant, when available, supplemented by in silico data generated by EFSA (see Annex B).
Out of the 41 identified and quantified components, the applicant reported that 31 were already evaluated by EFSA and/or JECFA/CoE as flavouring substances and relied on these assessments to conclude on their genotoxic potential. For the remaining 10 components, the applicant's conclusions were based on literature search, in silico analysis, applying (Q)SAR models, i.e. Derek Nexus (version 6.1.0) 12 and Organisation for Economic Co‐operation and Development (OECD) quantitative structure–activity relationship (QSAR) Toolbox v. 4.5 13 and read‐across.
The following models, as implemented in Derek Nexus, were considered by the applicant (Documentation provided to EFSA No. 1):
Chromosome damage in vitro;
Chromosome damage in vivo;
Photo‐induced chromosome damage in vitro;
Mutagenicity in vitro;
Mutagenicity in vivo;
Photomutagenicity in vitro;
Non‐specific genotoxicity in vitro;
Non‐specific genotoxicity in vivo;
Photo‐induced non‐specific genotoxicity in vitro;
Photo‐induced non‐specific genotoxicity in vivo.
Alerts were reported for Mutagenicity in vitro and in vivo and for Chromosome damage in vitro and in vivo, but not for any of the other models.
The following profilers as implemented in OECD QSAR Toolbox v. 4.5 13 were also considered by the applicant to complement the in silico analysis:
DNA alerts for AMES, Chromosomal Aberrations (CA) and Micronucleus (MN) by OASIS;
DNA binding by OASIS;
DNA binding by OECD;
Protein binding alerts for chromosomal aberration by OASIS;
In vitro mutagenicity (Ames test) alerts by ISS;
In vivo mutagenicity (Micronucleus) alerts by ISS.
A short summary of the data available from the literature as submitted by the applicant and of the overall conclusions from the applicant on the genotoxicity of the individual components, including the in silico analysis, when available, is reported in Annex B of this opinion (see columns ‘G’ and ‘I’). The complete set of information from the applicant is available under the section ‘Genotoxicity’ of the technical dossier (see Documentation provided to EFSA No. 1).
In line with the EFSA guidance on smoke flavourings (EFSA FAF Panel, 2021), the Panel conducted a (Q)SAR analysis for all the 44 identified components of the Primary Product using the same six profilers available in the OECD QSAR Toolbox v. 4.5, as listed above.
As described in column ‘K’ of Annex B, reporting the EFSA's conclusions on the genotoxicity of the components of the Primary Product based on the available data, the individual structural alerts identified by the six profilers may have different positive predictivity (i.e. rate of positives to the total number of substances with the alert) for the genotoxicity of the target substance. The concepts of the alerts are described by the European Chemicals Agency (ECHA, 2008) and the predictivities of the individual alerts are documented by Benigni et al. (2008 and 2009). When necessary, the application of profilers was followed by an expert review (e.g. check of close analogues/structurally related substances).
Overall, regarding the genotoxicity assessment of the individual components of the Primary Product the Panel noted that:
genotoxicity data were available from the literature for all the 44 identified components of the Primary Product, either on the substance or on structurally related substances. Based on these (often‐limited) data, the Panel concluded for 42 substances that these data did not indicate a concern for genotoxicity.
for two components, i.e. styrene (CAS No. 100‐42‐5; former [FL‐no: 01.015]) and benzofuran (CAS No. 271‐89‐6), the Panel identified a potential concern for genotoxicity for which additional data would be needed to reach a final conclusion on the genotoxic potential of these substances (see Annex B and Appendix B).
The Panel investigated if the potential concern for genotoxicity for styrene and benzofuran could be ruled out by application of the threshold of toxicological concern (TTC) approach for DNA‐reactive mutagens and/or carcinogens (EFSA Scientific Committee, 2019). For this purpose, the Panel calculated the exposure to each of these components by multiplying the estimated exposure to the Primary Product (proposed maximum use level exposure assessment scenario, estimated with FAIM – Table 10) by the average content of these components in the Primary Product (see Appendix A).
The obtained exposure estimates were compared with the TTC value of 0.0025 μg/kg bw per day for DNA‐reactive mutagens and/or carcinogens. All exposure estimates were at least a factor of 4,440 above this TTC value (see Table 14) and therefore the application of the TTC approach could not rule out the (potential) concern for genotoxicity for these components.
Table 14.
Dietary exposure in μg/kg body weight (bw) per day to the two individual components for which a potential concern for genotoxicity has been identified (see Appendix B), based on the proposed maximum use level exposure assessment scenario using FAIM (Table 10)
| CAS No. | Chemical name | Average content in the Primary Product (wt%) | Exposure | Infants (12 weeks‐11 months) | Toddlers (12–35 months) | Children (3–9 years) | Adolescents (10–17 years) | Adults (18–64 years) | The elderly (≥ 65 years) | Ratio between the highest exposure estimate and TTC |
|---|---|---|---|---|---|---|---|---|---|---|
| Components for which a potential concern for genotoxicity is identified | ||||||||||
| 100‐42‐5 | Styrene | 0.15 | Mean 95th percentile | 0.9–4.5 | 2.1–5.7 | 2.1–5.6 | 1.1–3.0 | 1.1–2.3 | 0.9–2.1 | 6.08 × 103 |
| 2.6–15.2 | 3.8–14.1 | 3.5–12.9 | 2.1–6.8 | 2.0–5.0 | 1.7–3.3 | |||||
| 271‐89‐6 | Benzofuran | 0.11 | Mean 95th percentile | 0.7–3.3 | 1.5–4.2 | 1.5–4.1 | 0.8–2.2 | 0.8–1.7 | 0.7–1.5 | 4.44 × 103 |
| 1.9–11.1 | 2.8–10.3 | 2.5–9.5 | 1.5–5.0 | 1.4–3.6 | 1.2–2.4 | |||||
wt: weight; TTC: threshold of toxicological concern.
The lack of robust experimental data on genotoxicity for the two components listed in (ii) for which a potential concern for genotoxicity was identified is a non‐standard uncertainty with respect to the genotoxicity assessment of the individual components (see Section 2.2 of this opinion and Table G.1 of the EFSA guidance document on smoke flavouring (EFSA FAF Panel, 2021)). This uncertainty can only be addressed with additional genotoxicity data.
3.4.2. Genotoxicity assessment of the Primary Product (whole mixture)
The applicant resubmitted the genotoxicity studies on the Primary Product (whole mixture) that were already evaluated by the CEF Panel in 2009 (except the in vivo rat liver unscheduled DNA synthesis (UDS) assay), to investigate the genotoxicity of the unidentified fraction of the Primary Product, in line with the EFSA Scientific Committee statement on genotoxicity assessment of chemical mixtures (EFSA Scientific Committee, 2019): a bacterial reverse mutation test (Lab International Research Centre, 2005a), an in vitro mammalian cell gene mutation assay in mouse lymphoma cells (Lab International Research Centre, 2005b), an in vitro mammalian chromosomal aberration test (Lab International Research Centre, 2005c) and an in vivo MN assay in mouse bone marrow (Lab International Research Centre, 2005d).
The evaluation of these studies as described in the scientific opinion ‘Safety of smoke flavour Primary Product – Scansmoke SEF7525’ (EFSA CEF Panel, 2009) is reported in Section 3.4.2.1. For each study, comments and evaluation by the FAF Panel are reported. These studies are summarised in Tables D.1 and D.2 (Appendix D), where the evaluation of reliability and relevance are reported (according to the approach described in Appendix C).
Table D.1.
Summary of in vitro genotoxicity studies on Scansmoke SEF 7525 (SF‐004) including re‐evaluation of reliability and relevance by the FAF Panel (approach described in Appendix C)
| Name | Test system in vitro | Test object | Concentrations and test conditions | Result | Reliability/comments | Relevance of test system/relevance of the result | Reference |
|---|---|---|---|---|---|---|---|
| Scansmoke SEF 7525 | Bacterial Reverse Mutation test |
S. Typhimurium TA98, TA100, TA1535, TA1537 E. coli WP2 uvrA |
Experiment 1: 78.13–2500 μg/plate (+/−S9, plate incorporation) Experiment 2: 78.13, − 2500 μg/plate (+/−S9, pre‐incubation) |
Negative | Reliable without restrictions. Study performed according to OECD TG 471 and in compliance with GLP | High/High | LAB International Research Centre, 2005a |
| In vitro mammalian cell gene mutation test in mouse lymphoma cells | L5178Y TK+/− mouse lymphoma cells |
Experiment 1: 5–150 μg/mL (3 + 21 h, +S9) 5–40 μg/mL (3 + 21 h, −S9) Experiment 2: 5–150 μg/mL (3 + 21 h, +S9) 5–25 μg/mL (24 h −S9) |
Positive |
Reliable with restrictions (historical controls not provided). Study performed according to OECD TG 476 (applicable at that time, now OECD TG 490) and in compliance with GLP | High/Limited | LAB International Research Centre, 2005b | |
| In vitro mammalian chromosomal aberration test | Chinese hamster ovary cells (CHO‐KI cell line) |
Experiment 1: 1, 15, 30 μg/mL (4 + 20 h, +S9) 1, 15, 30 μg/mL (4 + 20 h, −S9) Experiment 2: 1, 5, 15 μg/mL (20 h, −S9) 1, 15, 30 μg/mL (4 + 20 h, +S9) |
Negative | Reliable with restrictions (insufficient number of cells scored). Study performed according to OECD TG 473 and in compliance with GLP | High/Limited | LAB International Research Centre, 2005c |
Table D.2.
Summary of in vivo genotoxicity studies on Scansmoke SEF 7525 (SF‐004) including re‐evaluation of reliability and relevance by the FAF Panel (approach described in Appendix C)
| Name | Test system in vivo | Test Object Route | Doses (mg/kg bw per day) | Result | Reliability/comments | Relevance of test system/relevance of the result | Reference |
|---|---|---|---|---|---|---|---|
| Scansmoke SEF 7525 | Micronucleus assay in bone marrow |
NMRI BR mice; M and F Oral |
500, 1,000 and 2,000 (a) |
Inconclusive (negative, but without demonstration of bone marrow exposure) |
Reliable with restrictions (no demonstration of bone marrow exposure; historical control data not reported). Study performed according to OECD TG 474 and in compliance with GLP. |
High/Low | LAB International Research Centre, 2005d |
bw: body weight; M: males; F: females.
One administration with sampling at: 24 h, 48 h and 72 h.
The Panel noted that the general compositional data of the product evaluated in 2009 do not fundamentally deviate from the product assessed in the current opinion. In addition, as stated by the applicant, the manufacturing process has not changed and the batch‐to‐batch variability was low both in the previous evaluation (EFSA CEF Panel, 2009) and in the current opinion (see Table 6 in Section 3.1.2.6). Therefore, the Panel considered the Primary Product that was evaluated in 2009 similar to the Primary Product evaluated in this opinion and that the batch used for the genotoxicity testing in the past can still be considered representative for the current product.
In addition, new genotoxicity studies were provided, which are described in Section 3.4.2.2 and summarised in Appendix E.
The batch used in these newly submitted genotoxicity studies (no. 13390) fell within the reported range of batch‐to‐batch variability and could be considered representative (see Section 3.1.3).
The Panel noted that information provided to confirm the absence of a fraction of small particles is not sufficient (see Section 3.1.2.7). Therefore, the conclusions reached for each of the genotoxicity studies described below is applicable only under the assumption that the material is covered by the conventional risk assessment and does not require a separate assessment regarding nanoscale properties.
3.4.2.1. Studies evaluated in the EFSA CEF Panel opinion (EFSA CEF Panel, 2009)
3.4.2.1.1. Bacterial reverse mutation test (Lab International Research Centre, 2005a)
‘The Primary Product did not induce gene mutations in a bacterial assay which was performed using Salmonella Typhimurium strains TA98, TA100, TA1535 and TA1537 and the Escherichia coli strain WP2 uvrA. The experiments have been carried out in the presence and absence of a metabolic activation system prepared from enzyme‐induced rat liver. Phenobarbitone and ß‐naphthoflavone were used for enzyme‐induction. The assay was performed in accordance with OECD guideline 471 (1997)’. (EFSA CEF Panel, 2009).
The FAF Panel agreed with this evaluation and considered the study to be reliable without restrictions and its result of high relevance.
3.4.2.1.2. In vitro mammalian cell gene mutation assay in mouse lymphoma cells (Lab International Research Centre, 2005b)
‘The Primary Product induced statistically significant and dose‐related increases in the mutant frequency in mouse lymphoma cells in the MLTK assay (performed in accordance with OECD guideline 476 (1997)) both in the absence (up to 3.3‐fold at 0.04 mg/ml resulting in a Relative Survival of 25%) and presence of metabolic activation (up to 3.9‐fold at 0.1 mg/ml resulting in a Relative Survival of 20%). The metabolic activation system was prepared from rat liver. It was not reported whether the liver enzymes have been induced, however, since the assay has been performed in the same laboratory as the bacterial assay it could be assumed that the metabolic activation system used was prepared from enzyme‐induced rat liver likewise. Both large and small colonies were induced with large colonies predominating at all concentrations in the absence of a metabolising system and at all, except the highest, concentrations in the presence of S9. This suggests that the genotoxic effects observed may be due to both clastogenicity and the induction of gene mutations in this assay’. (EFSA CEF Panel, 2009).
The FAF Panel agreed with the previous evaluation of the CEF Panel that the Primary Product gave clear positive results in all test conditions. Also, based on the most recent OECD TG 490 (OECD, 2016a) according to which the global evaluation factor has to be taken into account as an additional criterion, the FAF Panel considered the results in all test conditions as positive. Historical controls were not reported and therefore the Panel considered that the study is reliable with restrictions and its results are of limited relevance.
3.4.2.1.3. In vitro mammalian chromosomal aberration test (Lab International Research Centre, 2005c)
‘The Primary Product did not induce chromosomal aberrations in vitro in Chinese Hamster Ovary cells in an assay performed in accordance with OECD guideline 473 (1997). The experiments have been carried out in the presence and absence of a metabolic activation system. The type of S9‐fraction was not reported, however, since the assay has been performed in the same laboratory as the bacterial assay it could be assumed that the metabolic activation system used was prepared from enzyme‐induced rat liver likewise. Based on the results of a cytotoxicity assay the cells were exposed to the Primary Product at 1, 15 and 30 μg/mL with and without S9 mix during 4 hours in a first experiment and at 1, 5 and 15 μg/mL without S9 mix for 20 hours and at 1, 5 and 30 μg/mL with S9 mix for 4 hours in a second experiment. In experiment 1, chromosomal aberrations were induced at the highest concentration up to two‐fold compared to solvent control in the test without S9 and up to 2.4‐fold with S9. No clear dose‐response and no statistically significant differences between treated cells and solvent controls were observed. In experiment 2, chromosomal aberrations were induced up to 1.8‐fold with and without S9 mix, but the result was not dose‐related and not statistically significant. Although the mean aberration frequency of 6 % observed at 30 μg/mL in the presence of S9 mix in the first experiment was above the maximum value of the historical control data for the solvent control (4 %), this result was not reproducible in the second experiment where the mean percent aberrant cells was 3.5 % which was within the range of the historical control data obtained with the solvent control. Thus, the Panel agreed with the authors of the study report and considered the result negative’. (EFSA CEF Panel, 2009).
The FAF Panel agreed that the Primary Product did not show evidence of clastogenic activity, polyploidy or endoreduplication in this test. However, based on the most recent OECD TG 473 (OECD, 2016b) the study is considered as reliable with restrictions, because only 200 metaphases/concentration instead of 300 were scored. Based on this limitation, the relevance of the study results is limited.
3.4.2.1.4. In vivo bone marrow mouse micronucleus test (Lab International Research Centre, 2005d)
‘ In vivo, the Primary Product was tested in a mouse micronucleus assay performed in accordance with OECD guideline 474 (1997). It was administered orally by gavage to male and female mice at dose levels of 500, 1000 and 2000 mg/kg. The Primary Product induced a dose‐related slight increase in the percentage of micronucleated polychromatic erythrocytes (MCPE) 14 in bone marrow of male mice at 24 hours (up to 1.5‐fold compared to control) which was, however, not statistically significant. At 24 hours in females, there was no statistically significant increase nor were the results dose‐related. At 72 hours, the Primary Product induced a slight increase in females (1.4‐fold) which was not clearly dose‐related but statistically significant. However, in males there was only a marginal increase up to 1.2‐fold at 72 hours which was not statistically significant. Statistically significant effects were only observed at the later sampling time (72 hours) and not consistent with the lack of cytotoxicity indicated by the unchanged PCE/NCE ratio. Therefore, the CEF Panel concluded that this study is negative’. (EFSA CEF Panel, 2009).
Considering the current version of the OECD TG 474 (OECD, 2016c) according to which the exposure of the bone marrow to the test substance needs to be demonstrated, the FAF Panel noted that there was no indication of bone marrow toxicity that could be considered as evidence of bone marrow exposure. Since there were no clinical investigations reported in this study and since there were no other lines of evidence for systemic bioavailability that could be indicative of bone marrow exposure, the FAF Panel concluded that the result of this study was inconclusive. Moreover, no historical controls were reported.
It should also be noted that, according to the statement on genotoxicity assessment of chemical mixtures (EFSA Scientific Committee, 2019), even if there were demonstration of bone marrow exposure, the assessment of genotoxicity of mixtures in the bone marrow is limited by the fact that target tissue exposure to all potential genotoxic components cannot be demonstrated unequivocally.
Therefore, the Panel considered the study as reliable with restrictions and the study result of low relevance.
3.4.2.1.5. In vivo rat liver UDS assay
‘Scansmoke SEF7525 was also tested in vivo in an UDS assay which was performed in compliance with GLP. UDS was assessed in hepatocytes of Sprague Dawley rats following oral gavage administration of Scansmoke SEF7525 on two separate occasions (the second dose being administered 14 hours after the first dose and 2 hours before perfusion). This study design deviated slightly from the OECD guideline 486 (1997) with respect to dosing and sampling. However, the protocol was considered acceptable. Scansmoke SEF7525 was administered at dosages of 500, 1000 and 2000 mg/kg bw. Under the conditions of this study, Scansmoke SEF7525 did not induce UDS in vivo’. (EFSA CEF Panel, 2009).
The study report on the in vivo UDS assay was not submitted in the new dossier, because the applicant considered that the negative results observed in this assay do not contribute to the overall assessment of genotoxicity. The FAF Panel agreed with this consideration and confirmed that the results of a negative UDS study are of low relevance, based on the EFSA Scientific Committee opinion on the adequacy of the UDS assay to follow‐up positive results in the in vitro gene mutation tests (EFSA Scientific Committee, 2017b).
3.4.2.2. New genotoxicity studies
Based on the available data and on the requirements of the EFSA guidance on smoke flavouring Primary Products (EFSA FAF Panel, 2021) new genotoxicity studies were submitted: an in vitro MN test (BSRC, 2022a), an in vivo MN test (BSRC, 2022b) and an in vivo gene mutation test in transgenic rodents (BSRC, 2022c).
The stability of test article formulations (1 and 500 mg/mL) was confirmed in a separate study using a validated analytical method for the determination of 2,6‐dimethoxyphenol as a typical component of the Primary Product in propylene glycol (BSRC, 2022d). The Panel noted that the stability of the Primary Product in the test article formulations (within 8 days) was also confirmed by visual comparison of the HPLC chromatograms provided in the study report.
3.4.2.2.1. In vitro mammalian cell micronucleus test
An in vitro MN test was conducted using the human lymphoblast cell line (TK6 cells) treated with Scansmoke SEF 7525 (batch: 13390). The in vitro MN assay was carried out according to OECD TG 487 (OECD, 2016d) and GLP. The cytokinesis block micronucleus assay protocol was used. Cyclophosphamide, mitomycin C and colchicine were used as the positive controls. Dimethyl sulfoxide (DMSO) was used as the negative control (BSRC, 2022a).
Concentrations for the MN experiment were selected based on the results of a cytotoxicity range‐finding experiment carried out at a range of concentrations from 7.81 to 1000 μg/mL.
For the MN experiment, lymphoblasts were treated with Scansmoke SEF 7525 at concentrations ranging from 10 to 150 μg/mL in the 3 h + 24 h treatment in the presence of metabolic activation (S9‐mix 6.6% final concentration, inducing agents: phenobarbital and 5,6‐benzoflavone), at eight concentrations from 2.5 to 20 μg/mL in the 3 h + 24 h treatment in the absence of S9‐mix and at seven concentrations from 1.25 to 15 μg/mL in the 24 h treatment in the absence of S9‐mix. No precipitate of the test item was noted in any of the exposure conditions. Each test concentration and the positive controls were tested in duplicate cultures.
The replication index cytotoxicity data were used to select the concentrations for the MN analysis.
In the 3 h + 24 h treatment in the presence of S9‐mix, the following concentrations were chosen for MN analysis: 20, 75 and 150 μg/mL (cytotoxicity of 3.3%, 20.9% and 50.6%, respectively). A statistically significant increase in the frequency of bi‐nucleated cells with micronuclei (MNBN) was observed at 150 μg/mL (1.8%), which was outside the range of historical negative controls (0.28–1.44%).
In the 3 h + 24 h treatment in the absence of S9‐mix, the following concentrations were chosen for MN analysis: 5, 10 and 12.5 μg/mL (cytotoxicity of 11.7%, 22.6% and 51.2%, respectively). A statistically significant increase in the frequency of MNBN was observed at 12.5 μg/mL (2%), which was outside the range of historical negative controls (0.25–1.37%).
In the 24 h treatment in the absence of S9‐mix, the following concentrations were chosen for MN analysis: 2.5, 5 and 12.5 μg/mL (cytotoxicity of 10.8%, 28% and 50.4%, respectively). A statistically significant increase in the frequency of MNBN was observed at 12.5 μg/mL (2.7%), which was outside the range of historical negative controls (0.31–1.51%).
In all three test conditions, the top concentrations of Scansmoke SEF 7525 induced statistically significant increases in the frequency of micronucleated cells compared to the negative control. Furthermore, concentration‐dependent trends were also observed and the frequency of micronucleated cells at the top concentrations were greater than the acceptable ranges calculated from the historical negative control data.
The study authors concluded that Scansmoke SEF 7525 induced micronuclei in cultured mammalian cells in both the absence and presence of metabolic activation under the conditions in this study. The Panel agreed with this conclusion and evaluated the study as reliable without restrictions and the results of high relevance.
Results are summarised in Appendix E, Table E.1.
Table E.1.
Summary of new in vitro genotoxicity study on Scansmoke SEF 7525 (SF‐004)
| Name | Test system in vitro | Test object | Concentrations (a) and test conditions | Result | Reliability/comments | Relevance of test system/relevance of the result | Reference |
|---|---|---|---|---|---|---|---|
| Scansmoke SEF 7525 | Micronucleus assay | Human TK6 cell line |
20, 75, 150 μg/mL (3 + 24 h, +S9) 5, 10, 12.5 μg/mL (3 + 24 h, −S9) 2.5, 5, 12.5 μg/mL (24 h, −S9) |
Positive |
Reliable without restrictions. Study performed according to OECD TG 487 and in compliance with GLP. |
High/High | BSRC, 2022a |
The given concentrations are those for the cultures that were scored for micronuclei.
3.4.2.2.2. In vivo mammalian erythrocyte micronucleus test
Scansmoke SEF 7525 (batch no. 13390) was tested in a bone marrow micronucleus assay in mice which was performed in compliance with GLP and according to OECD TG 474 (OECD, 2016c) (BSRC, 2022b).
A dose range‐finding study was performed to identify the appropriate maximum dose level for the main test. Groups of three B6D2F1/Slc [SPF] male mice were treated twice at 24 h intervals by oral gavage at 500, 750, 1,000, 2,000 and 3,000 mg/kg bw per day. In the top dose group, there was one death and in the 2,000 mg/kg bw per day group there were no deaths, but two mice showed clinical signs. No mortality, no adverse reactions to treatment and no suppression of bw gain were observed in the other groups and therefore 2,000 mg/kg bw per day group was considered the maximum tolerated dose (MTD). The MTD was used as the highest dose level in the main study.
Groups of six or eight B6D2F1/Slc [SPF] male mice (eight animals for top dose group only) were treated via oral gavage with Scansmoke SEF 7525 at doses of 0 (propylene glycol used as vehicle control), 500, 1,000 and 2,000 mg/kg bw per day. Test item formulations were prepared prior to the first administration and stored for the second administration. A single administration of 25 mg/kg bw per day cyclophosphamide via oral gavage was used as positive control. The positive control was prepared just before use. Animals were dosed at 0 and 24 h, except the positive control group that was dosed only at 24 h.
In the top dose group, abnormal respiratory noise, prone position, decrease in locomotor activity, clonic convulsion, irregular respiration and chromaturia (purple) were observed. Abnormal respiratory noise was also observed in the 1000 mg/kg bw per day group. No clinical signs were observed in the low dose group. Also in the top dose group, the bw of one animal decreased by approximately 12% but generally there was no apparent suppression of bw gain observed in any of the treatment groups.
Twenty‐four hours after the final administration, femoral bone marrow was harvested and prepared for the MN analysis (three bone smears per animal) for five animals per group. A total of at least 500 polychromatic erythrocytes (PCE) and normochromatic erythrocytes (NCE) were scored to assess potential bone marrow toxicity. For MN analysis 4000 PCE per animal were scored for the presence of MN.
The vehicle control data were comparable with the laboratory's historical vehicle control data. Positive control data resulted in a statistically significant increase in micronucleated polychromatic erythrocytes (MNPCE), compared to the concurrent vehicle control, which was comparable with the laboratory's historical positive control data.
The Panel noted that one out of three animals died in the dose range‐finding study in the top dose group that received a dose of 3000 mg/kg bw per day. Therefore, the Panel considered the top dose of 2000 mg/kg bw per day in the main study appropriate.
In all three dose groups of mice treated with Scansmoke SEF 7525, there were no statistically significant increases in MNPCE frequency compared to the vehicle controls. Individual frequencies of MNPCE for all treated animals were consistent with historical vehicle control data. The proportions of PCE in counted erythrocytes were 61.4%, 60.7%, 56.7% and 51.7% at 0, 500, 1,000 and 2,000 mg/kg bw per day, respectively. These changes were statistically significant compared to negative control. The reduction in the proportion of PCE to total erythrocytes at the high dose was only 15%, but dose‐related, and can therefore be considered as limited evidence of bone marrow exposure. In addition, some clinical signs observed in the top dose group (i.e. decrease in locomotor activity, clonic convulsion and irregular respiration) can be considered as a line of evidence of systemic bioavailability that indicates bone marrow exposure. It should also be noted that, according to the statement on genotoxicity assessment of chemical mixtures (EFSA Scientific Committee, 2019), even in the case of lines of evidence of bone marrow exposure, the assessment of genotoxicity of mixtures in the bone marrow is limited by the fact that target tissue exposure to all potential genotoxic components cannot be demonstrated unequivocally.
Therefore, the Panel considered the study reliable without restriction and the negative result of limited relevance.
3.4.2.2.3. In vivo gene mutation assay in Muta™Mouse transgenic mice
Scansmoke SEF 7525 (batch no. 13390) was tested in a 14‐day dose range‐finding (non‐GLP) study in CD2F1/Slc mice (i.e. wild type Muta™Mouse), in order to determine the MTD and dose levels for the transgenic rodent (TGR) gene mutation assay using the same rodent strain (BSRC, 2022e).
Scansmoke SEF 7525 was administered via oral gavage (propylene glycol used as vehicle) to groups of CD2F1/Slc mice (three animals per sex per group) at dose levels of 500, 1,000 and 2,500 mg/kg bw per day. Animals were observed daily for clinical signs. All animals in the top dose group died or were sacrificed moribund on days 2–4 and one male and one female in the 1,000 mg/kg bw per day group were found to be moribund on days 6 and 8, respectively. At scheduled necropsy, thickening of the forestomach was found in one male and one female of the 1,000 mg/kg bw per day group. No signs of toxicity were observed in the 500 mg/kg bw per day group. No gender‐specific differences were observed. No body weight changes associated with Scansmoke SEF 7525 treatment or clinical signs of toxicity were observed at 500 mg/kg bw per day. Based on these results in which 750 mg/kg bw per day was considered to be the MTD, the study authors recommended doses of 250, 500 and 750 mg/kg bw per day for the in vivo gene mutation assay in Muta™Mouse (BSRC, 2022e).
In the in vivo gene mutation assay in Muta™Mouse (lacZ/GalE), Scansmoke SEF 7525 (batch no. 13390) was administered via oral gavage (propylene glycol used as vehicle) to four groups of male transgenic CD2‐LacZ80/HazfBR SPF mice (Muta™Mouse) (six to eight animals per group) at dose levels of 250, 500 and 750 mg/kg bw per day for 28 consecutive days (BSRC, 2022c). This study was performed according to OECD TG 488 (OECD, 2022) and in compliance with GLP. The treatment period was followed by a 3‐day manifestation period and then animals were sacrificed, and the liver, stomach and duodenum removed. N‐ethyl‐N‐nitrosourea (ENU) administered intraperitoneally at a dose of 100 mg/kg bw per day for two consecutive days followed by a 10‐day manifestation period was used as a concurrent positive control. Test item formulations were prepared on a weekly basis, 1 day prior to first use. The positive control was prepared just before use.
One animal treated at 250 mg/kg bw per day and one animal in the 500 mg/kg bw per day exhibited abnormal respiratory noise on day 15 and day 3, respectively: the animal from the 250 mg/kg bw per day group subsequently died on the same day following test item administration. In the 750 mg/kg bw per day group, there were no clinical signs, though the stomach weight and relative stomach weight were significantly increased compared to the negative control and in one animal white patches were observed in the liver. No differences in bw gain or food consumption were observed for any group.
Liver, duodenum and stomach samples from five animals per group (including controls) were processed for DNA isolation. For each DNA sample, the number of plaques from a single packaging was greater than 300,000 (i.e. more than the OECD recommended minimum of 125,000 plaques). Except for one animal in the lowest dose group, treatment with Scansmoke SEF 7525 did not increase the mutation frequency at the lacZ gene in liver, stomach or duodenum of Muta™Mouse mice. The authors attributed the increase in one animal as a result of clonal expansion since there was no dose–response. In line with the study authors, the Panel concluded that in this in vivo gene mutation assay in Muta™Mouse, Scansmoke SEF 7525 did not induce a statistically significant increase in mutation frequency in the liver, stomach or duodenum when tested up to the MTD. The Panel considered the study as reliable without restrictions and the results of high relevance.
Results of in vivo studies are summarised in Appendix E, Table E.2.
Table E.2.
Summary of new in vivo genotoxicity studies on Scansmoke SEF 7525 (SF‐004)
| Name | Test system in vivo | Test Object Route | Doses (mg/kg bw per day) | Result | Reliability/comments | Relevance of test system/relevance of the result | Reference |
|---|---|---|---|---|---|---|---|
| Scansmoke SEF 7525 | Micronucleus assay in bone marrow |
B6D2F1/Slc [SPF] mice; M gavage |
500, 1,000 and 2,000 (a) |
Negative | Reliable without restrictions. Study performed according to OECD TG 474 and in compliance with GLP. The highest dose tested was the MTD. | High/Limited (b) | BSRC, 2022b |
| Gene mutation assay in liver, stomach and duodenum |
Muta™Mouse (lacZ/GalE) CD2‐LacZ80/HazfBR SPF transgenic mice; M gavage |
250, 500 and 750 | Negative | Reliable without restrictions. Study performed according to OECD TG 488 and in compliance with GLP | High/High | BSRC, 2022c |
M: males.
the Primary Product was administered once daily on 2 consecutive days; sampling at 24 h after the last administration.
the reason for the limitation of the relevance is that, according to the statement on genotoxicity assessment of chemical mixtures (EFSA Scientific Commitee, 2019), even in the case of lines of evidence of bone marrow exposure, the assessment of genotoxicity of mixtures in the bone marrow is limited by the fact that target tissue exposure to all potential genotoxic components cannot be demonstrated unequivocally.
4. Discussion
The European Commission has requested the European Food Safety Authority (EFSA) to evaluate the safety of the smoke flavouring Primary Product Scansmoke SEF7525 (SF‐004), for which a renewal application has been submitted, in accordance with Article 12(1) of Regulation (EC) No 2065/2003.
The Primary Product is produced from a tar fraction derived of a hardwood mixture, i.e. 30–40% red oak (Q. rubra), 30–40% white oak (Q. alba), 5–15% maple (A. saccharum), 5–15% beech (F. grandifolia) and 5–15% hickory (C. ovata).
The production of the Primary Product occurs in three main stages: (i) extracting the tar raw material with diethyl ether at two pH values, (ii) subjecting the extracts to purification steps and (iii) combining the obtained fractions at a fixed ratio. The Panel considered the information provided on the manufacturing process as sufficient. The data demonstrated that the Primary Product is produced in the same way as the product evaluated formerly (EFSA CEF Panel, 2009).
The applicant provided compositional data for six batches of the Primary Product. The Panel noted that the applied methods meet the legal quality criterion that at least 80% by mass of the volatile fraction shall be identified and quantified (Regulation (EC) No 627/2006).
For the investigated batches, the identified and quantified proportion of the solvent‐free fraction was on average 32.6 wt% (range from 27.2 to 37.1 wt%). Thus, the applied methods do not meet the legal quality criterion that at least 50% of the solvent‐free fraction shall be identified and quantified (Regulation (EC) No 627/2006). The Panel noted that this is due to the fact that the portion of volatiles identified and quantified in the Primary Product is lower than previously reported in the submission underlying the former opinion on Scansmoke SEF7525 (EFSA CEF Panel, 2009). The applicant explained this difference by the more stringent criteria applied before a constituent is considered as identified and by differences in the upper GC column temperature (250°C). The gap between the total non‐volatiles, calculated by subtracting the content of water and the content of total volatiles from the total mass of the Primary Product (on average 59.7 wt%), and the residue remaining after heating the Primary Product at 350°C (on average 24 wt%) indicates that there is a substantial volatile portion of the Primary Product not amenable to the employed GC‐method because it does not elute from the GC column under the given analytical conditions.
Data provided for six batches of the Primary Product demonstrated that their batch‐to‐batch variability was sufficiently low (i.e. the observed relative standard deviations for the individual constituents was on average approximately 13%), based on the analytical data for the 20 principal volatile constituents and the chemical classes. The Panel considered that the data provided in the selected batches are representative of the Primary Product.
Based on the data provided, the Panel reckoned that the combined evidence is not conclusive to accept that Scansmoke SEF7525 is a true liquid, and further evidence is needed to exclude the presence of small particles including nanoparticles for the Primary Product. If based on additional evidence, the presence of small particles including nanoparticles cannot be eventually excluded in the Primary Product, a specific assessment at the nanoscale would be required, in line with the EFSA Scientific Committee Guidance on risk assessment of nanomaterials (EFSA Scientific Committee, 2021a).
The applicant proposed limits for four toxic elements (arsenic, cadmium, lead and mercury), which are the same as in the current EU specifications (Table 7). The Panel noted that the actual measured levels for these elements in six batches of the Primary Product (Table 3) were substantially lower than these limits. The Panel performed a risk assessment on the presence of these toxic elements in the Primary Product and concluded that, when considering the current limits of the EU specifications (scenario (i) in Table 12), the ranges of the calculated MOE values for arsenic were insufficient, i.e. below the target value of 1,000. For the other three toxic elements (cadmium, lead and mercury), their presence in the Primary Product up to the current limits in the EU specifications does not give rise to a safety concern. When considering the reported LOQs multiplied by a factor of 10 (scenario (ii) in Table 12), the Panel concluded that for arsenic (a) the lower end of the range for the highest mean and (b) the range for the highest 95th percentile of the calculated MOE values were still insufficient, i.e. below the target value of 1,000. In this scenario, the presence of the other toxic elements in the Primary Product does not give rise to concern.
The analytical procedure for the determination of 16 PAHs meets the performance criteria as set in Regulation (EC) No 627/2006. The levels of benzo[a]pyrene and benzo[a]anthracene were below the current limits in Regulation (EC) No 2065/2003. Based on the estimated exposure to the Primary Product and the maximum reported level of the PAH4 in the Primary Product (i.e. 34.6 μg/kg), an MOE of at least 9.73 × 105 could be calculated for the exposure to PAHs, which would be of low concern from a public health point of view and might be reasonably considered as a low priority for risk management actions (see EFSA Scientific Committee, 2012). The Panel noted that including a limit for PAH4 in the EU specifications would take better account of the presence of other PAHs than only the two PAHs benzo[a]pyrene and benzo[a]anthracene.
Overall, the Panel considered that limits in the EU specifications for the four toxic elements and PAH4 should be established based on actual levels in the Primary Product. If the European Commission decides to revise the limits already present and to include a limit for PAH4, the estimated exposure to the four toxic elements and PAH4 as presented in Sections 3.3.3.1 and 3.3.3.2 could be considered.
The Primary Product is requested to be authorised for use in 32 food categories. The Panel performed an exposure assessment for this Primary Product based on proposed maximum and expected typical use levels in these food categories using FAIM. At the maximum proposed use levels, mean exposure estimates to the Primary Product ranged from 0.6 mg/kg bw per day in infants and the elderly to 3.8 mg/kg bw per day in toddlers (Table 10). The 95th percentiles of exposure to the Primary Product ranged from 1.1 mg/kg bw per day in the elderly to 10.1 mg/kg bw per day in infants. At the expected typical use levels, the mean dietary exposure to the Primary Product estimates ranged from 0.4 mg/kg bw per day in the elderly to 3.3 mg/kg bw per day in toddlers and the 95th percentile exposure to the Primary Product estimates ranged from 0.8 mg/kg bw per day in the elderly to 9.1 mg/kg bw per day in infants (Table 10).
For all food categories considered, it was assumed that 100% of the foods belonging to these food categories will contain Scansmoke SEF7525 (SF004) at the proposed maximum use levels or the upper level of the range of expected typical use levels. As it is unlikely that the Primary Product will be added to all foods belonging to these food categories, the Panel considered that the calculated exposure to Scansmoke SEF7525 (SF004) is an overestimation of the expected exposure in EU countries, even if this Primary Product is used at the proposed maximum use levels or upper level of the range of expected typical use levels.
Regarding the genotoxicity data, the Panel conducted the evaluation in line with the currently applicable EFSA guidance on smoke flavourings (EFSA FAF Panel, 2021) which encompasses all the EFSA guidance documents on genotoxicity (EFSA Scientific Committee, 2011, 2017b, 2019, 2021b).
From the analysis of the available information on genotoxicity of the 44 individual components of the Primary Product, the Panel considered that:
for 42 individual components no concern for genotoxicity is identified (see Annex B);
for two components, i.e. styrene and benzofuran, a potential concern for genotoxicity is identified, for which additional data would be needed to reach a conclusion on the genotoxic potential of these substances.
The details of the genotoxicity data available on styrene and benzofuran are given and discussed in Appendix B.
The Panel investigated if the potential concern for genotoxicity of styrene and benzofuran could be ruled out by application of the TTC approach for DNA‐reactive mutagens and/or carcinogens (EFSA Scientific Committee, 2019). The obtained exposure estimates for styrene and benzofuran were compared with the TTC value of 0.0025 μg/kg bw per day for DNA‐reactive mutagens and/or carcinogens. Exposure estimates for both substances across the different population groups were all above this TTC value (Table 14) and therefore the application of the TTC approach could not rule out the potential concern for genotoxicity.
The Panel considered whether refined exposure estimates for the Primary Product (estimated by EFSA in line with the principles described in the guidance on smoke flavourings (EFSA FAF Panel, 2021)) could mitigate the concern for the genotoxic potential of these two components. However, taking into account:
the magnitude of the calculated ratios between the exposure estimates for styrene and benzofuran and the above mentioned TTC value (see Table 14);
the uses of the Primary Product and the nature of the restrictions/exceptions indicated by the applicant for the different food categories (see Table 9),
the Panel considered that a more refined exposure assessment will not reduce the exposure estimates for these two components to such an extent that they will be below the TTC value of 0.0025 μg/kg bw per day.
Overall, the Panel considered that further information on the genotoxicity of styrene and benzofuran would be needed to rule out the potential safety concern for genotoxicity of these substances. While for styrene conclusions on its genotoxic potential are expected from the CEP Panel, for benzofuran, appropriate genotoxicity studies conducted according to EFSA guidance (EFSA Scientific Committee, 2011, 2017b, 2021b) would be needed.
The Primary Product (whole mixture) was tested in in vitro and in vivo genotoxicity studies to investigate the genotoxicity of the unidentified fraction of the Primary Product, in line with the EFSA Scientific Committee statement on genotoxicity assessment of chemical mixtures (EFSA Scientific Committee, 2019).
The Primary Product did not induce gene mutations in bacterial cells, but it induced gene mutations in mammalian cells. In vivo, the Primary Product did not induce gene mutations in liver, stomach and duodenum of transgenic mice.
The Primary Product was not clastogenic in an in vitro chromosomal aberration test in CHO cells. However, the Primary Product induced micronuclei in TK6 cells, both in the absence and in the presence of metabolic activation. In vivo, the Primary Product did not induce micronuclei in mice bone marrow, but the study result was considered of limited relevance, because, according to the statement on genotoxicity assessment on chemical mixtures (EFSA Scientific Committee, 2019), even in the case of lines of evidence of bone marrow exposure, the assessment of genotoxicity of mixtures in the bone marrow is limited by the fact that target tissue exposure to all potential genotoxic components cannot be demonstrated unequivocally. Therefore, this study is not strong enough to alleviate concern for the whole mixture that is raised by the findings of chromosomal aberrations in the in vitro MN assay.
In principle, based on the EFSA Scientific Committee statement on genotoxicity assessment of chemical mixtures (EFSA Scientific Committee, 2019) as well as on the EFSA guidance on smoke flavourings (EFSA FAF Panel, 2021), if aneugenicity can be excluded, an in vivo Comet assay (OECD TG 489 (2016e)) at the site of contact and in the liver might also be considered appropriate to follow‐up the chromosomal aberrations observed in vitro. The studies at the site of contact allow investigation of genotoxic effects at the site where the exposure to the components is expected to be maximal.
However, in this case, the concern for genotoxicity for the Primary Product cannot be ruled out by an additional in vivo Comet assay performed on the whole mixture, since the exposure estimates for styrene and benzofuran are both above the TTC value of 0.0025 μg/kg bw per day for DNA‐reactive mutagens and/or carcinogens and since further information would be needed on these substances to clarify their genotoxicity. In fact, as outlined in the EFSA Scientific Committee statement on genotoxicity assessment on chemical mixtures (EFSA Scientific Committee, 2019), ‘if the mixture contains one or more chemical substances that are evaluated to be genotoxic in vivo via a relevant route of administration, the whole mixture raises concern about genotoxicity’.
The Panel concluded that, based on the available data, a potential concern for genotoxicity of Scansmoke SEF7525 cannot be ruled out.
5. Conclusions
In line with the ToR as provided by the European Commission, in the current opinion EFSA assessed the chemical characterisation, the genotoxicity and the dietary exposure to Scansmoke SEF7525 (SF‐004).
From all data available on characterisation, the Panel concluded that the Primary Product considered in this opinion is representative for the one authorised in Commission Implementing Regulation (EU) No 1321/2013 under the code name SF‐004. Nevertheless, the Panel concluded that the compositional data provided on the Primary Product were not adequate. The identified and quantified proportion of the solvent‐free fraction was on average 32.6 wt%. Thus, the Panel considered that the applied methods do not meet the legal quality criterion that at least 50% of the solvent‐free fraction shall be identified and quantified, as set in Regulation (EC) No 627/2006. The Panel concluded that the applicant has adequate control over the production process and that the Primary Product is sufficiently stable upon storage.
Based on the data submitted by the applicant, the Panel could not exclude the presence of small particles including nanoparticles and hence could not conclude if conventional risk assessment is sufficient or whether it needs to be complemented with nano‐specific considerations.
The Primary Product contains styrene and benzofuran, which are substances of potential concern for genotoxicity. For styrene further assessment of its genotoxic potential (including new data) is currently ongoing by the CEP Panel. For benzofuran, the available information is not sufficient to assess its genotoxicity, for which additional studies conducted according to EFSA guidance (EFSA Scientific Committee, 2011, 2017b, 2021b) would be needed. Furthermore, a potential concern for genotoxicity was also identified for the unidentified fraction of the mixture.
Considering that the exposure estimates for styrene and benzofuran are both above the TTC value of 0.0025 μg/kg bw per day for DNA‐reactive mutagens and/or carcinogens, the Panel concluded that the potential concern for genotoxicity of Scansmoke SEF7525 cannot be ruled out, until the genotoxic potential of both substances is clarified.
Overall, according to the EFSA guidance on smoke flavourings (EFSA FAF Panel, 2021), the safety of the smoke flavouring Primary Product Scansmoke SEF7525 has not been sufficiently demonstrated.
6. Documentation as provided to EFSA
Dossier “Application for renewal of an already authorised smoke flavouring ‐ Scansmoke SEF 7525”. Dossier number: SFL‐2021‐2372. June 2022. Submitted by Azelis Denmark A/S. 5
Additional data received on 10 February 2023, submitted by Azelis Denmark A/S in response to additional data request from EFSA sent on 25 November 2022.
Additional data received on 11 May 2023, submitted by Azelis Denmark A/S as spontaneous submission.
BSRC, 2022a. In Vitro Micronucleus (MNvit) Test of Scansmoke SEF 7525 in Human Lymphoblast Cell Line (TK6). BioSafety Research Centrer Inc., Japan. Experiment No. K100 (820‐013). March, 2022. Unpublished study report submitted by Azelis Denmark A/S.
BSRC, 2022b. Micronucleus Test of Scansmoke SEF 7525 in Mice. BioSafety Research Center Inc., Japan. Experiment No. K173 (820‐015). June, 2022. Unpublished study report submitted by Azelis Denmark A/S.
BSRC, 2022c. In Vivo Gene Mutation Assay of Scansmoke SEF 7525 in MutaMouse. BioSafety Research Center Inc., Japan. Experiment No. K099 (820‐012), June, 2022. Unpublished study report submitted by Azelis Denmark A/S.
BSRC, 2022d. Stability Study of Scansmoke SEF 7525 in Propylene Glycol. BioSafety Research Center Inc., Japan. Experiment No. K097 (820‐010). January, 2022. Unpublished study report submitted by Azelis Denmark A/S.
BSRC, 2022e. Dose Range‐finding Study for Transgenic Mouse Gene Mutation Assay of Scansmoke SEF 7525 [Non‐GLP]. BioSafety Research Center Inc., Japan. Experiment No. K098 (820‐011), March, 2022. Unpublished study report submitted by Azelis Denmark A/S.
Lab International Research Centre, 2005a. The testing of Scansmoke SEF 7525 with bacterial reverse mutation assay. Lab International Research Centre Hungary Ltd. Study Code 04/830‐007M. June 2005. Submitted by Azelis Denmark A/S.
Lab International Research Centre, 2005b. Testing of mutagenic effect of Scansmoke SEF 7525 by Mouse Lymphoma Assay. Lab International Research Centre Hungary Ltd. Study Code 04/830‐033E. June 2005. Submitted by Azelis Denmark A/S.
Lab International Research Centre, 2005c. Testing of Scansmoke SEF 7525 with in vitro mammalian chromosome aberration test. Lab International Research Centre Hungary Ltd. Study Code 04/830‐020C. June 2005. Submitted by Azelis Denmark A/S.
Lab International Research Centre, 2005d. Testing of mutagenic effect of test item Scansmoke SEF 7525 by mouse micronucleus test. Lab International Research Centre Hungary Ltd. Study Code 04/830‐013E. June 2005. Submitted by Azelis Denmark A/S.
Abbreviations
- BMDL
benchmark dose lower limit
- BSRC
Bioscience Research center
- bw
body weight
- CA
chromosomal aberration
- CAS
Chemical Abstract Service
- CEF
Panel on Food Contact Materials, Enzymes, Flavourings and Processing Aids
- CEP
Panel on Food Contact Materials, Enzymes and Processing Aids
- COE
Council of Europe
- CONTAM
Panel on Contaminants in the Food Chain
- DLS
dynamic light scattering
- DMSO
dimethyl sulfoxide
- ECHA
European Chemicals Agency
- EM
electron microscopy
- ENU
N‐ethyl‐N‐nitrosourea
- FAF
Panel on Food Additives and Flavourings
- FAIM
Food Additive Intake Model
- FC
food category
- FL‐no
FLAVIS number
- GC
gas chromatography
- GC–FID
gas chromatography–flame ionisation detection
- GC–MS
gas chromatography–mass spectrometry
- GLP
good laboratory practices
- HBVG
health‐based guidance values
- HPLC
high performance liquid chromatography
- ICP‐MS
inductively coupled plasma‐mass spectrometry
- IQ
intelligence quotient
- ISS
Istituto Superiore di Sanità
- JECFA
Joint FAO/WHO Expert Committee on Food Additives
- LD50
lethal dose 50
- LOQ
limit of quantification
- MCT
medium chain triglycerides
- MN
micronucleus
- MNPCE
micronucleated polychromatic erythrocytes
- MOE
margin of exposure
- MS
mass spectrometry
- MTD
maximum tolerated dose
- NCE
normochromatic erythrocytes
- OECD
Organisation for Economic Co‐operation and Development
- P95
95th percentile
- PAHs
polycyclic aromatic hydrocarbons
- PCE
polychromatic erythrocytes
- Pt‐NP
platinum nanoparticle clusters
- QSAR
quantitative structure–activity relationship
- RP
reference points
- RSD
relative standard deviation
- SD
standard deviation
- SF
smoke flavouring
- TG
test guideline
- TGR
transgenic rodent
- TK
thymidine kinase
- TR
technical requirements
- TTC
threshold of toxicological concern
- TWI
tolerable weekly intake
- UDS
unscheduled DNA synthesis
- wt
weight
Appendix A – Full list of identified and quantified constituents of smoke flavouring Primary Product (SF‐004)
Table A.1.
Compilation of the 41 identified and quantified volatile constituents in the Primary Product (Documentation provided to EFSA No. 1)
| CAS no. | FL‐no | Chemical name (a) | Average (b) (wt%) |
|---|---|---|---|
| 91‐10‐1 | 04.036 | 2,6‐dimethoxyphenol | 4.9 |
| 6638‐05‐7 | 04.053 | 4‐methyl‐2,6‐dimethoxyphenol (2,6‐dimethoxy‐4‐methylphenol) | 3.4 |
| 90‐05‐1 | 04.005 | 2‐methoxyphenol | 2.3 |
| 93‐51‐6 | 04.007 | 2‐methoxy‐4‐methylphenol (creosol/methylguaiacol) | 2.3 |
| 14059‐92‐8 | 04.052 | 4‐ethyl‐2,6‐dimethoxyphenol | 2.2 |
| 20675‐95‐0 | 04.055 (c) | 2,6‐dimethoxy‐4‐prop‐1‐enylphenol (2,6‐dimethoxy‐4‐(2‐propenyl)‐phenol, E) | 1.4 |
| 105‐67‐9 | 04.006 | 2,4‐dimethylphenol | 1.3 |
| 2785‐89‐9 | 04.008 | 4‐ethylguaiacol (4‐ethyl‐2‐methoxyphenol) | 1.2 |
| 95‐48‐7 | 04.027 | 2‐methylphenol | 1.1 |
| 108‐95‐2 | 04.041 | phenol | 0.9 |
| 5932‐68‐3 | 04.004 (d) | isoeugenol (trans‐isoeugenol) | 0.9 |
| 6627‐88‐9 | 04.051 | 4‐allyl‐2,6‐dimethoxyphenol (2,6‐dimethoxy‐4‐(2‐propenyl)‐phenol) | 0.9 |
| 97‐53‐0 | 04.003 | eugenol | 0.6 |
| 576‐26‐1 | 04.042 | 2,6‐dimethylphenol | 0.6 |
| 26624‐13‐5 | 4.055 (c) | 2,6‐dimethoxy‐4‐prop‐1‐enylphenol (4‐propenyl‐2,6‐dimethoxyphenol, Z) | 0.6 |
| 1121‐05‐7 | – | 2,3‐dimethyl‐2‐cyclopenten‐1‐one | 0.6 |
| 108‐39‐4 | 04.026 | 3‐methylphenol | 0.6 |
| 527‐60‐6 | 04.095 | 2,4,6‐trimethyl phenol | 0.6 |
| 2785‐87‐7 | 04.049 | 2‐methoxy‐4‐propylphenol | 0.5 |
| 98‐00‐0 | 13.019 | furfuryl alcohol (2‐furanmethanol) | 0.4 |
| 123‐07‐9 | 04.022 | 4‐ethylphenol | 0.35 |
| 2758‐18‐1 | 07.112 | 3‐methyl‐2‐cyclopenten‐1‐one | 0.32 |
| 64‐19‐7 | 08.002 | acetic acid | 0.30 |
| 5912‐86‐7 | 04.004 (d) | isoeugenol (cis‐isoeugenol) | 0.30 |
| 90‐00‐6 | 04.070 | 2‐ethylphenol | 0.29 |
| 4265‐25‐2 | – | 2‐methylbenzofuran | 0.28 |
| 95‐13‐6 | – | indene | 0.23 |
| 765‐70‐8 | 07.056 (e) | 3‐methylcyclopentan‐1,2‐dione (3‐methyl‐1,2‐cyclopentanedione) | 0.17 |
| 620‐17‐7 | 04.021 | 3‐ethylphenol | 0.17 |
| 526‐75‐0 | 04.065 | 2,3‐dimethylphenol | 0.16 |
| 100‐42‐5 | Former 01.015 (f) | vinylbenzene (styrene) | 0.15 |
| 95‐65‐8 | 04.048 | 3,4‐dimethylphenol | 0.14 |
| 123‐31‐9 | – | hydroquinone | 0.13 |
| 1195‐09‐1 | – | 2‐methoxy‐5‐methylphenol | 0.12 |
| 20736‐25‐8 | – | dihydrosyringenin | 0.12 |
| 271‐89‐6 | – | benzofuran | 0.11 |
| 6766‐82‐1 | 04.056 | 2,6‐dimethoxy‐4‐propylphenol | 0.11 |
| 431‐03‐8 | 07.052 | diacetyl (2,3‐butanedione) | 0.08 |
| 98‐86‐2 | 07.004 | acetophenone | 0.07 |
| 6443‐69‐2 | – | 1,2,3‐trimethoxy‐5‐methylbenzene | 0.06 |
| 2503‐46‐0 | – | 1‐(4‐hydroxy‐3‐methoxyphenyl)‐2‐propanone | 0.05 |
wt: weight.
In case a constituent of the Primary Product is an authorised flavouring substance (FL‐no), the assigned chemical name corresponds to the respective entry in the EU Union List of flavourings. Deviating chemical names reported by the applicant in the dossier are given in brackets, if applicable.
From the analysis of the six batches presented in Table 1.
[FL‐no: 04.055] refers to the mixture of E/Z stereoisomers of 2,6‐dimethoxy‐4‐prop‐1‐enylphenol.
[FL‐no: 04.004] refers to the mixture of E/Z stereoisomers of isoeugenol.
[FL‐no: 07.056] refers to the mixture of the tautomeric forms of 3‐methylcyclopentan‐1,2‐dione.
‘Former FL‐number’ refers to substances that were initially included in the evaluation programme but were not included or were removed/withdrawn from the Union List.
Appendix B – Genotoxicity data available on 2 individual components for which a potential concern for genotoxicity is identified
1.
The data in this Appendix are related to the two substances described in Section 3.4.1 for which a potential concern for genotoxicity has been identified, i.e. styrene (CAS No. 100‐42‐5; former [FL‐no: 01.015]) and benzofuran (CAS No. 271‐89‐6).
B.1. Styrene (CAS No. 100‐42‐5; former [FL‐no: 01.015])
The Panel considered that in line with the conclusions of the EFSA CEP Panel (EFSA CEP Panel, 2020), a concern for genotoxicity associated with oral exposure to styrene (former [FL‐no: 01.015]) cannot be excluded. A comprehensive evaluation of the reliability and relevance of all available experimental and human findings on styrene genotoxicity, with consideration of toxicokinetic aspects, would be required for an in‐depth assessment of the genotoxicity of styrene via oral exposure. Further assessment of the genotoxic potential of styrene is currently ongoing by the CEP Panel (see EFSA‐Q‐2023‐00365 15 ).
Although no structural alerts have been identified by EFSA in the (Q)SAR analysis on the substance itself, the high positive predictivity of the structural alerts identified for the metabolite styrene‐7,8‐oxide could indicate a potential concern for genotoxicity for styrene (see Annex B).
Conclusion: the Panel concluded that the available data raise a potential concern for the genotoxicity for this substance. Depending on the outcome of the ongoing assessment by the CEP Panel, additional data might be needed, since exposure to the substance exceeds the TTC for DNA‐reactive mutagens and/or carcinogens (Table 14).
B.2. Benzofuran (CAS No. 271‐89‐6)
Inconsistent experimental genotoxicity results are available for benzofuran (CAS No. 271‐89‐6), as submitted by the applicant (see Documentation provided to EFSA No. 1). Two reports on gene mutations provided negative results (NTP, 1989; Kirkland and Fowler, 2010), although the NTP study had some limitations. However, the Panel noted that the study conducted by Robbiano et al. (2004), reported positive results in vitro in Comet and in micronucleus (MN) assays in rat and human kidney cells and in vivo in Comet and in MN assays in kidney of rats. The protocols applied are only partially described and the study authors refer to previous publications for the in vitro and in vivo studies (Robbiano et al., 1997, 1999).
For the described methodology of the in vitro studies, the Panel noted some limitations, e.g. only 50 cells from two slides were scored, and tail length was measured instead of tail intensity for the in vitro comet assay and the treatment schedule in the in vitro MN assay was not fully consistent with the current OECD test guideline (TG) 487 (OECD, 2016d), especially since no information on the cell cycle was provided.
Also for the in vivo studies, the Panel noted some limitations; e.g. benzofuran was tested at a single oral dose only (1/2 LD50) without reporting any data on toxicity and the number of animals was insufficient both in the in vivo MN and comet assay (Robbiano et al., 2004). Furthermore, the MN assay in kidney is not a standardised method and the criteria applied to identify the cells to be scored for the micronucleated cell frequency were not reported. Regarding the in vivo comet assay, a suboptimal sampling time of 48 h after treatment was applied and tail length was measured instead of tail intensity, which deviates from the currently applicable OECD TG 489 (OECD, 2016e).
Another limitation of both in vitro and in vivo studies is the reporting of the results, which are only given graphically as a ratio of exposed/control values without details.
The Panel considered the in vitro studies and the in vivo comet assay as reliable with restrictions and of limited relevance. However, the limitations are not strong enough to dismiss these results which raise a potential concern for genotoxicity.
No structural alerts have been identified in the (Q)SAR analysis performed by EFSA on benzofuran itself. However, the high predictivity of the structural alerts identified for the metabolite benzofuran‐2,3‐epoxide also raises a potential concern for the genotoxicity of benzofuran.
Conclusion: the Panel concluded that the available data raise a potential concern for the genotoxicity for this substance. Additional data would be needed to evaluate the genotoxic potential of benzofuran, since exposure to the substance exceeds the TTC for DNA‐reactive mutagens and/or carcinogens (see Table 14).
Appendix C – Approach for assessing reliability and relevance of genotoxicity studies
1.
Evaluation of data quality for hazard/risk assessment includes evaluation of reliability of studies and relevance of study results (Klimisch et al., 1997; ECHA, 2011; EFSA Scientific Committee, 2011, 2017b, 2021b). Reliability is assessed using a scoring system based on published criteria (Klimisch et al., 1997) described in the following Section. In a second step, the relevance (high, limited or low) of study results is assessed based on several aspects (genetic endpoint, route of administration, status of validation of the assay, etc.) discussed in Section C.2, and also taking into account the assessment of the reliability of the study.
Only studies with acceptable relevance (high or limited) are considered in the weight of evidence approach (WoE). Genotoxicity studies evaluated as of low relevance are not further considered in the WoE.
C.1. Evaluation of reliability of results of genotoxicity studies – general considerations
The scoring system for reliability is based on the scoring system of Klimisch et al. (1997). Reliability is defined by Klimisch as ‘evaluating the inherent quality of a test report or publication relating to preferably standardised methodology and the way that the experimental procedure and results are described to give evidence of the clarity and plausibility of the findings’. In assigning the reliability score, the compliance with the OECD Test Guidelines (TGs) or standardised methodology and the completeness of the reporting should be considered.
The reliability scores are:
reliable without restriction;
reliable with restrictions;
reliability insufficient;
reliability cannot be evaluated.
(1) Reliable without Restriction ‘This includes studies or data from the literature or reports which were carried out or generated according to generally valid and/or internationally accepted testing guidelines (preferably performed according to GLP) or in which the test parameters documented are based on a specific (national) testing guideline (preferably performed according to GLP) or in which all parameters described are closely related/comparable to a guideline method’.
(2) Reliable with Restrictions ‘This includes studies or data from the literature, reports (mostly not performed according to GLP), in which the test parameters documented do not totally comply with the specific testing guideline, but are sufficient to accept the data or in which investigations are described which cannot be subsumed under a testing guideline, but which are nevertheless well documented and scientifically acceptable’.
(3) Reliability Insufficient 16 ‘This includes studies or data from the literature/reports in which there are interferences between the measuring system and the test substance or in which organisms/test systems were used which are not relevant in relation to the exposure (…) or which were carried out or generated according to a method which is not acceptable, the documentation of which is not sufficient for an assessment and which is not convincing for an expert judgment’.
(4) Reliability cannot be evaluated 17 ‘This includes studies or data from the literature, which do not give sufficient experimental details, and which are only listed in short abstracts or secondary literature (books, reviews, etc.)’.
C.2. Evaluation of relevance of results of individual genotoxicity studies – general considerations
The relevance of the test system and test results are reported separately.
The relevance of the test systems (high, limited, low) is principally based on the following criteria:
Genetic endpoint: higher relevance is given to studies providing information on apical endpoints, i.e. gene mutations, structural and numerical chromosomal alterations. Supporting information may be obtained from indicator assays; exception is the in vivo Comet assay that is considered with high relevance when applied as follow‐up to a positive in vitro result (as recommended by the EFSA Scientific Committee (2011)).
Status of validation of the test system (e.g. (in order of decreasing relevance) availability of an OECD TG consolidated or in the course of development or internationally recommended protocol, validation at national level only).
The relevance of the study results (high, limited, low) are principally based on the following criteria:
Reliability of studies: the results of studies with reliability that are insufficient or which cannot be evaluated (see points 3–4 in Section C.1) are considered of low relevance.
Relevance of the test system.
Route of administration: higher relevance is given to oral vs intravenous or subcutaneous injection and inhalation exposure in case of in vivo studies. Lower relevance is given to studies using the intraperitoneal route, which is not physiological and not recommended by OECD TGs.
Biological relevance of the test results, considering: purity of the test substance; the metabolic capabilities of the test system; the bioavailability of the test substance, with particular consideration of the evidence of target tissue exposure in tests in vivo (negative results without evidence of target tissue exposure are considered as inconclusive and their relevance low); the interference of high cytotoxicity; the reproducibility of test results.
Appendix D – Genotoxicity studies on the Primary Product (whole mixture) evaluated by the CEF Panel (EFSA CEF Panel, 2009)
Appendix E – New genotoxicity studies on the Primary Product (whole mixture)
Annex A – Exposure assessment results
1.
-
–
Annex A1: Occurrence data per food category considered in FAIM, (mg/kg).
-
–
Annex A2: Total estimated exposure of Scansmoke SEF7525 (SF‐004) from its proposed maximum level exposure scenario per population group and survey: mean and 95th percentile (mg/kg bw per day).
-
–
Annex A3: Total estimated exposure of Scansmoke SEF7525 (SF‐004) from its expected typical exposure assessment scenario per population group and survey: mean and 95th percentile (mg/kg bw per day).
-
–
Annex A4: Main food categories contributing to exposure to Scansmoke SEF7525 (SF‐004) using the maximum level exposure assessment scenario (> 5% to the total mean exposure).
-
–
Annex A5: Main food categories contributing to exposure to Scansmoke SEF7525 (SF‐004) using the expected typical level exposure assessment scenario (> 5% to the total mean exposure).
-
–
Annex A6: Qualitative evaluation of the influence of standard uncertainties on the dietary exposure estimates of the Primary Product.
Annex A can be found in the online version of this output, in the ‘Supporting information’ section.
Annex B – Genotoxicity assessment of the identified components in the Primary Product
1.
Annex B can be found in the online version of this output, in the ‘Supporting information’ section.
Supporting information
Exposure assessment results
Genotoxicity assessment of the identified components in the Primary Product
Suggested citation: EFSA FAF Panel (EFSA Panel name on Food Additives and Flavourings) , Younes, M ., Aquilina, G ., Castle, L ., Degen G., Engel, K.‐H ., Fowler, P. J ., Frutos Fernandez, M. J ., Fürst, P ., Gundert‐Remy, U ., Gürtler, R ., Husøy, T ., Manco, M ., Moldeus, P ., Passamonti, S ., Shah, R ., Waalkens‐Berendsen, I ., Wright, M ., Benigni, R ., … Mennes, W . (2023). Scientific opinion on the renewal of the authorisation of Scansmoke SEF7525 (SF‐004) as a smoke flavouring Primary Product. EFSA Journal, 21(11), 1–50. 10.2903/j.efsa.2023.8366
Requestor European Commission
Question number EFSA‐Q‐2022‐00421
Correspondence fip@efsa.europa.eu
Panel members Maged Younes, Gabriele Aquilina, Laurence Castle, Gisela Degen, Karl‐Heinz Engel, Paul Fowler, Maria Jose Frutos Fernandez, Peter Fürst, Ursula Gundert‐Remy, Rainer Gürtler, Trine Husøy, Melania Manco, Wim Mennes, Peter Moldeus, Sabina Passamonti, Romina Shah, Ine Waalkens‐Berendsen and Matthew Wright.
Declarations of interest If you wish to access the declaration of interests of any expert contributing to an EFSA scientific assessment, please contact interestmanagement@efsa.europa.eu.
Acknowledgements The Panel wishes to thank the following for the support provided to this scientific output: Gabriele Gagliardi, Ana Rincon, Alkiviadis Stagkos‐Georgiadis, the members of the SCER Cross‐cutting WG nanotechnologies: Maria Bastos, Jacqueline Castenmiller, Francesco Cubadda, Mohammad Chaudhry, Roland Franz, David Gott, Maria Henriqueta Louro, Jan Mast, Alicja Mortensen, Agnes Oomen, Eveline Verleysen, Stefan Weigel. Furthermore, the Panel wishes to thank the following for the preparatory work performed for the genotoxicity assessment of the Primary Product: Arianna Bassan (Section 3.4.1), Manuela Pavan (Section 3.4.1) and Joanne Gartlon (Section 3.4.2).
EFSA may include images or other content for which it does not hold copyright. In such cases, EFSA indicates the copyright holder and users should seek permission to reproduce the content from the original source.
Adopted: 28 September 2023
Annexes A and B are available under the Supporting Information section .
Notes
Regulation (EC) No 2065/2003 of the European Parliament and of the Council of 10 November 2003 on smoke flavourings used or intended for use in or on foods. OJ L 309, 26.11.2003, pp. 1–8.
Commission Implementing Regulation (EU) No 1321/2013 of 10 December 2013 establishing the Union list of authorised smoke flavouring primary products for use as such in or on foods and/or for the production of derived smoke flavourings. OJ L 333, 12.12.2013, pp. 54–67.
Regulation (EC) No 178/2002 of the European Parliament and of the Council of 28 January 2002 laying down the general principles and requirements of food law, establishing the European Food Safety Authority and laying down procedures in matters of food safety. OJ L 31, 1.2.2002, pp. 1–48.
Decision available online: https://www.efsa.europa.eu/en/corporate-pubs/transparency-regulation-practical-arrangements
The non‐confidential version of the dossier, following EFSA's assessment of the applicant's confidentiality requests, is published on Open.EFSA and is available at the following link: https://open.efsa.europa.eu/dossier/SFL-2021-2372
Commission Regulation (EC) No 627/2006 of 21 April 2006 implementing Regulation (EC) No 2065/2003 of the European Parliament and of the Council as regards quality criteria for validated analytical methods for sampling, identification and characterisation of primary smoke products. OJ L 109, 22.4.2006, pp. 3–6.
Regulation (EC) No 1333/2008 of the European Parliament and of the Council of 16 December 2008 on food additives. OJ L 354, 31.12.2008, pp. 16–33.
Regulation (EU) No 609/2013 of the European Parliament and of the Council of 12 June 2013 on food intended for infants and young children, food for special medical purposes and total diet replacement for weight control and repealing Council Directive 92/52/EEC, Commission Directives 96/8/EC, 1999/21/EC, 2006/125/EC and 2006/141/EC, Directive 2009/39/EC of the European Parliament and of the Council and Commission Regulations (EC) No 41/2009 and (EC) No 953/2009. OJ L 181, 29.6.2013, pp. 35–54.
Link to FAIM instructions: https://www.efsa.europa.eu/sites/default/files/applications/FAIM-instructions.pdf
Commission Regulation (EU) 2023/915 of 25 April 2023 on maximum levels for certain contaminants in food and repealing Regulation (EC) No 1881/2006. OJ L 119, 5.5.2023, pp. 103–157.
MCPE is an erroneous abbreviation; it should read MNPCE.
Klimisch et al. (1997) used the term ‘Not reliable’ however, ‘Reliability Insufficient’ was considered more appropriate.
Klimisch et al. (1997) used the term ‘Not assignable’ however, ‘Reliability cannot be evaluated’ was considered more appropriate.
References
- Benigni R, Bossa C, Jeliazkova N, Netzeva T and Worth A, 2008. The Benigni / Bossa rulebase for mutagenicity and carcinogenicity ‐ a module of Toxtree. JRC Scientific and Technical Reports, OPOCE, Luxembourg, 70 pp. Available online: https://publications.jrc.ec.europa.eu/repository/handle/JRC43157
- Benigni R, Bossa C, Tcheremenskaia O and Worth A, 2009. Development of Structural Alerts for the In vivo Micronucleus Assay in Rodents. JRC Scientific and Technical Reports, OPOCE, Luxembourg, 42 pp. Available online: https://publications.jrc.ec.europa.eu/repository/handle/JRC52274
- Culp SJ, Gaylor DW, Sheldon WG, Goldstein LS and Beland FA, 1998. A comparison of the tumours induced by coal tar and benzo[a]pyrene in a 2‐year bioassay. Carcinogenesis, 19, 117–124. 10.1093/carcin/19.1.117 [DOI] [PubMed] [Google Scholar]
- ECHA (European Chemicals Agency) , 2008. Guidance for the implementation of REACH, Chapter R.6: QSARs and grouping of chemicals. ECHA, Helsink, 134 pp. Available online: https://echa.europa.eu/documents/10162/13632/information_requirements_r6_en.pdf/77f49f81-b76d-40ab-8513-4f3a533b6ac9
- ECHA (European Chemicals Agency) , 2011. Guidance on information requirements and chemical safety assessment. Chapter R.4: Evaluation of available information. Available online: https://echa.europa.eu/documents/10162/13643/information_requirements_r4_en.pdf/d6395ad2-1596-4708-ba86-0136686d205e
- EFSA (European Food Safety Authority) , 2011. Use of the EFSA Comprehensive European Food Consumption Database in Exposure Assessment. EFSA Journal 2011;9(3):2097, 34 pp. 10.2903/j.efsa.2011.2097 [DOI] [Google Scholar]
- EFSA CEF Panel (EFSA Panel on Food Contact Materials, Enzymes, Flavourings and Processing Aids) , 2009. Scientific Opinion on the safety of smoke flavour Primary Product – Scansmoke SEF7525. EFSA Journal 2009;7(5):1224, 24 pp. 10.2903/j.efsa.2009.1093 [DOI] [Google Scholar]
- EFSA CEP Panel (EFSA Panel on Food Contact Materials, Enzymes and Processing Aids) , 2020. Scientific Opinion on the assessment of the impact of the IARC Monograph Vol. 121 on the safety of the substance styrene (FCM No 193) for its use in plastic food contact materials. EFSA Journal 2020;18(10):6247, 23 pp. 10.2903/j.efsa.2020.6247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain) , 2008. Scientific opinion on polycyclic aromatic hydrocarbons in food. EFSA Journal 2008;6(8):724, 114 pp. 10.2903/j.efsa.2008.724 [DOI] [Google Scholar]
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain) , 2009a. Scientific Opinion on Arsenic in food. EFSA Journal 2009;7(10):1351, 199 pp. 10.2903/j.efsa.2009.1351 [DOI] [Google Scholar]
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain) , 2009b. Scientific Opinion on Cadmium in food. EFSA Journal 2009;7(3):980, 139 pp. 10.2903/j.efsa.2009.980 [DOI] [Google Scholar]
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain) , 2010. Scientific Opinion on Lead in food. EFSA Journal 2010;8(4):1570, 151 pp. 10.2903/j.efsa.2010.1570 [DOI] [Google Scholar]
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain) , 2012. Scientific Opinion on the risk for public health related to the presence of mercury and methylmercury in food. EFSA Journal 2012;10(12):2985, 241 pp. 10.2903/j.efsa.2012.2985 [DOI] [Google Scholar]
- EFSA FAF Panel (EFSA Panel on Food Additives and Flavourings) , 2021. Scientific Guidance for the preparation of applications on smoke flavouring Primary Products. EFSA Journal 2021;19(3):6435, 40 pp. 10.2903/j.efsa.2021.6435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2009. Guidance of the Scientific Committee on transparency in the scientific aspects of risk assessment carried out by EFSA. Part 2: general principles. EFSA Journal 2009;7(4):1051, 22 pp. 10.2903/j.efsa.2009.1051 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2011. Scientific Opinion on genotoxicity testing strategies applicable to food and feed safety assessment. EFSA Journal 2011;9(9):2379, 69 pp. 10.2903/j.efsa.2011.2379 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2012. Statement on the applicability of the Margin of Exposure approach for the safety assessment of impurities which are both genotoxic and carcinogenic in substances added to food/feed. EFSA Journal 2012;10(3):2578, 5 pp. 10.2903/j.efsa.2012.2578 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2017a. Update: Guidance on the use of the benchmark dose approach in risk assessment. EFSA Journal 2017;15(1):4658, 41 pp. 10.2903/j.efsa.2017.4658 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2017b. Scientific Opinion on the clarification of some aspects related to genotoxicity assessment. EFSA Journal 2017;15(12):5113, 25 pp. 10.2903/j.efsa.2017.5113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2019. Statement on the genotoxicity assessment of chemical mixtures. EFSA Journal 2019;17(1):5519, 11 pp. 10.2903/j.efsa.2019.5519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2021a. Guidance on technical requirements for regulated food and feed product applications to establish the presence of small particles including nanoparticles. EFSA Journal 2021;19(8):6769, 48 pp. 10.2903/j.efsa.2021.6769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2021b. Scientific Opinion on the guidance on aneugenicity assessment. EFSA Journal 2021;19(8):6770, 27 pp. 10.2903/j.efsa.2021.6770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland D and Fowler P, 2010. Further analysis of Ames‐negative rodent carcinogens that are only genotoxic in mammalian cells in vitro at concentrations exceeding 1 mM, including retesting of compounds of concern. Mutagenesis, 25(6), 539–553. 10.1093/mutage/geq041 [DOI] [PubMed] [Google Scholar]
- Klimisch HJ, Andreae M and Tillmann U, 1997. A systematic approach for evaluating the quality of experimental toxicological and ecotoxicological data. Regulatory Toxicology and Pharmacology, 25, 1–5. 10.1006/rtph.1996.1076 [DOI] [PubMed] [Google Scholar]
- NTP (National Toxicology Program) , 1989. Toxicology and Carcinogenesis Studies of Benzofuran (CAS No. 271‐89‐6) in F344/N Rats and B6C3F1 Mice (Gavage Studies). NTP Technical Report Series No.370, 189 pp. PMID: 12692651. Available online: https://pubmed.ncbi.nlm.nih.gov/12692651/ [PubMed]
- OECD (Organisation for Economic Co‐operation and Development) , 2016a. Test No. 490: In vitro Mammalian Cell Gene Mutation Tests Using the Thymidine Kinase Gene. OECD Guidelines for the Testing of Chemicals, Section 4, OECD Publishing, Paris, 21 pp 10.1787/9789264264908-en [DOI]
- OECD (Organisation for Economic Co‐operation and Development) , 2016b. Test No. 473: In vitro Mammalian Chromosomal Aberration Test. OECD Guidelines for the Testing of Chemicals, Section 4, OECD Publishing, Paris, 22 pp. 10.1787/9789264264649-en [DOI]
- OECD (Organisation for Economic Co‐operation and Development) , 2016c. Test No. 474: Mammalian Erythrocyte Micronucleus Test. Guidelines for the Testing of Chemicals, Section 4, OECD Publishing, Paris, 21 pp. 10.1787/9789264264762-en [DOI]
- OECD (Organisation for Economic Co‐operation and Development) , 2016d. Test No. 487: In Vitro Mammalian Cell Micronucleus Test. Guidelines for the Testing of Chemicals, Section 4, OECD Publishing, Paris, 29 pp 10.1787/9789264264861-en [DOI]
- OECD (Organisation for Economic Co‐operation and Development) , 2016e. Test No. 489: In Vivo Mammalian Alkaline Comet Assay. OECD Guidelines for the Testing of Chemicals, Section 4, OECD Publishing, Paris, 27 pp 10.1787/9789264264885-en [DOI]
- OECD (Organisation for Economic Co‐operation and Development) , 2022. Test No. 488: Transgenic Rodent Somatic and Germ Cell Gene Mutation Assays. OECD Guidelines for the Testing of Chemicals, Section 4, OECD Publishing, Paris, 29 pp 10.1787/9789264203907-en [DOI]
- Robbiano L, Mereto E, Migliazzi Morando A, Pastore P and Brambilla G, 1997. An in vivo micronucleus assay for detecting the clastogenic effect in rat kidney cells. Mutation Research/Genetic Toxicology and Environmental Mutagenesis, 390, 51–57. 10.1016/S0165-1218(96)00165-6 [DOI] [PubMed] [Google Scholar]
- Robbiano L, Carrozzino R, Porta Puglia C, Corbu C and Brambilla G, 1999. Correlation between induction of DNA fragmentation and micronuclei formation in kidney cells from rats and humans and tissue‐specific carcinogenic activity. Toxicology and Applied Pharmacology, 161, 153–159. 10.1006/taap.1999.8796 [DOI] [PubMed] [Google Scholar]
- Robbiano L, Baroni D, Carrozzino R, Mereto E and Brambilla G, 2004. DNA damage and micronuclei induced in rat and human kidney cells by six chemicals carcinogenic to the rat kidney. Toxicology, 204, 187–195. 10.1016/j.tox.2004.06.057 [DOI] [PubMed] [Google Scholar]
- Xiang Q and Lee YY, 2000. Oxidative cracking of precipitated hardwood lignin by hydrogen peroxide. Applied Biochemistry and Biotechnology, 84, 153–162. 10.1385/ABAB:84-86:1-9:153 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Exposure assessment results
Genotoxicity assessment of the identified components in the Primary Product